The Action Potential
BOX 4.1 BRAIN FOOD: Methods of Recording Action Potentials
Optogenetics: Controlling Neural Activity with Light
BOX 4.2 PATH OF DISCOVERY: The Discovery of the Channelrhodopsins, by Georg Nagel
BOX 4.5 OF SPECIAL INTEREST: Multiple Sclerosis, a Demyelinating Disease
BOX 4.6 OF SPECIAL INTEREST: The Eclectic Electric Behavior of Neurons
Now we come to the signal that conveys information over distances in the nervous system—the action potential. As we saw in Chapter 3, the inside of the neuronal membrane at rest is negatively charged in relation to the outside. The action potential is a rapid reversal of this situation such that, for an instant, the inside of the membrane becomes positively charged in relation to the outside. The action potential is also often called a spike, a nerve impulse, or a discharge.
The action potentials generated by a patch of membrane are all similar in size and duration, and they do not diminish as they are conducted down the axon. Keep in mind the big picture: The frequency and pattern of action potentials constitute the code used by neurons to transfer information from one location to another. In this chapter, we discuss the mechanisms that are responsible for the action potential and how it propagates down the axonal membrane.
Action potentials have certain universal properties, features that are shared by axons in the nervous systems of every animal, from a squid to a college student. Let’s begin by exploring some of these properties. What does the action potential look like? How is it initiated? How rapidly can a neuron generate action potentials?
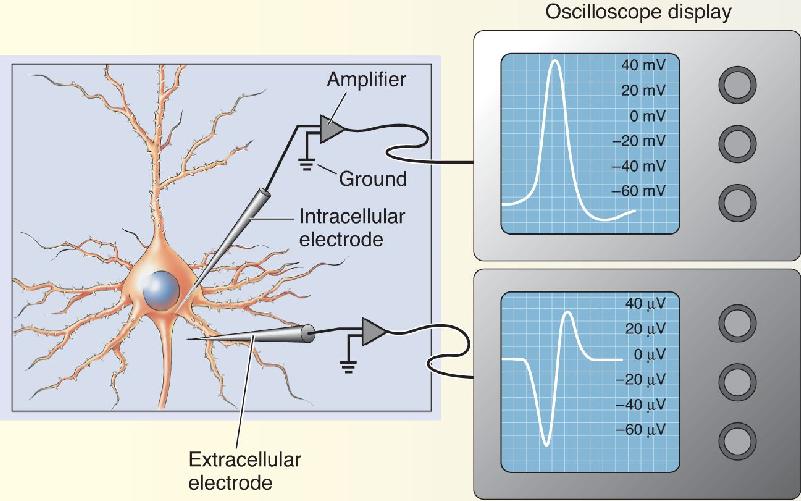
In Chapter 3, we saw that the membrane potential, Vm, can be determined by inserting a microelectrode in the cell. A voltmeter is used to measure the electrical potential difference between the tip of this intracellular microelectrode and another placed outside the cell. When the neuronal membrane is at rest, the voltmeter reads a steady potential difference of about –65 mV. During the action potential, however, the membrane potential briefly becomes positive. Because this occurs so rapidly—100 times faster than the blink of an eye—a special type of voltmeter, called an oscilloscope, is used to study action potentials. The oscilloscope records the voltage as it changes over time (Box 4.1).
Methods for studying nerve impulses may be broadly divided into two types: intracellular and extracellular (Figure A). Intracellular recording requires impaling the neuron or axon with a microelectrode. The small size of most neurons makes this method challenging, which is why so many early studies of action potentials were performed on the neurons of invertebrates, which can be 50–100 times larger than mammalian neurons. Fortunately, recent technical advances have made even the smallest vertebrate neurons accessible to intracellular recording methods, and these studies have confirmed that much of what was learned in invertebrates is directly applicable to humans.

The goal of intracellular recording is simple: to measure the potential difference between the tip of the intracellular electrode and another electrode placed in the solution bathing the neuron (electrically continuous with the earth, and thus called ground). The intracellular electrode is filled with a concentrated salt solution (often KCl) having a high electrical conductivity. The electrode is connected to an amplifier that compares the potential difference between this electrode and ground. This potential difference can be displayed using an oscilloscope. Early oscilloscopes worked by sweeping a beam of electrons from left to right across a phosphor screen. Vertical deflections of this beam show changes in voltage. Oscilloscopes today take a digital record of voltage across time, but the principle is the same. It is really just a sophisticated voltmeter that can record rapid changes in voltage (such as an action potential).
As we shall see, the action potential is characterized by a sequence of ionic movements across the neuronal membrane. These electrical currents can be detected, without impaling the neuron, by placing an electrode near the membrane. This is the principle behind extracellular recording. Again, we measure the potential difference between the tip of the recording electrode and ground. The electrode can be a fine glass capillary filled with a salt solution, but it is often simply a thin insulated metal wire. Normally, in the absence of neural activity, the potential difference between the extracellular recording electrode and ground is zero. However, when the action potential arrives at the recording position, positive charges flow away from the recording electrode into the neuron. Then, as the action potential passes by, positive charges flow out across the membrane toward the recording electrode. Thus, the extracellular action potential is characterized by a brief, alternating voltage difference between the recording electrode and ground. (Notice the different scale of the voltage changes produced by the action potential recorded with intracellular and extracellular recordings.) These changes in voltage can be seen using an oscilloscope, but they can also be heard by connecting the output of the amplifier to a loudspeaker. Each impulse makes a distinctive “pop” sound. Indeed, recording the activity of an active sensory nerve sounds just like popping popcorn.
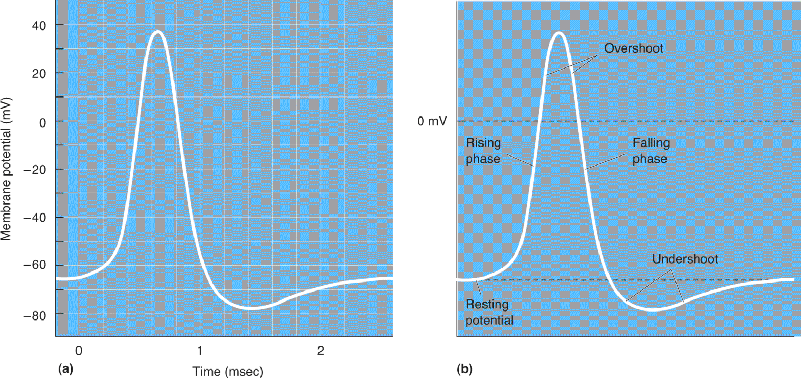
An action potential, as it would appear on the display of an oscilloscope, is shown in Figure 4.1. This graph represents a plot of membrane potential versus time. Notice that the action potential has certain identifiable parts. The first part, called the rising phase, is characterized by a rapid depolarization of the membrane. This change in membrane potential continues until Vm reaches a peak value of about 40 mV. The part of the action potential where the inside of the neuron is positively charged with respect to the outside is called the overshoot. The falling phase of the action potential is a rapid repolarization until the inside of the membrane is actually more negative than the resting potential. This last part of the falling phase is called the undershoot, or after-hyperpolarization. Finally, there is a gradual restoration of the resting potential. From beginning to end, the action potential lasts about 2 milliseconds (msec).

FIGURE 4.1 An action potential. (a) An action potential displayed by an oscilloscope. (b) The parts of an action potential. Description
In Chapter 3, we said that breaking of the skin by a thumbtack was sufficient to generate action potentials in a sensory nerve. Let’s continue that example to see how an action potential begins.

The perception of sharp pain when a thumbtack enters your foot is caused by the generation of action potentials in certain nerve fibers in the skin. (We’ll learn more about pain in Chapter 12.) The membrane of these fibers has a type of gated sodium channel that opens when the nerve ending is stretched. The chain of events therefore begins this way: (1) The thumbtack enters the skin, (2) the membrane of the nerve fibers in the skin is stretched, (3) and Na+-permeable channels open. Because of the large concentration gradient and the negative charge of the inside of the membrane, Na+ crosses the membrane through these channels. The entry of Na+ depolarizes the membrane; that is, the cytoplasmic (inside) surface of the membrane becomes less negative. If this depolarization, called a generator potential, achieves a critical level, the membrane will generate an action potential. The critical level of depolarization that must be reached in order to trigger an action potential is called threshold. Action potentials are caused by depolarization of the membrane beyond threshold.
The depolarization that causes action potentials arises in different ways in different neurons. In our previous example, depolarization was caused by the entry of Na+ through specialized ion channels that were sensitive to membrane stretching. In interneurons, depolarization is usually caused by Na+ entry through channels that are sensitive to neurotransmitters released by other neurons. In addition to these natural routes, neurons can be depolarized by injecting electrical current through a microelectrode, a method commonly used by neuroscientists to study action potentials in different cells.
Generating an action potential by depolarizing a neuron is something like taking a photograph by pressing the shutter button on an old-fashioned camera. Applying pressure on the button has no effect until it increases to the point of crossing a threshold, and then “click”—the shutter opens and one frame of film is exposed. Increasing depolarization of a neuron similarly has no effect until it crosses threshold, and then “pop”—one action potential. For this reason, action potentials are said to be “all-or-none.”
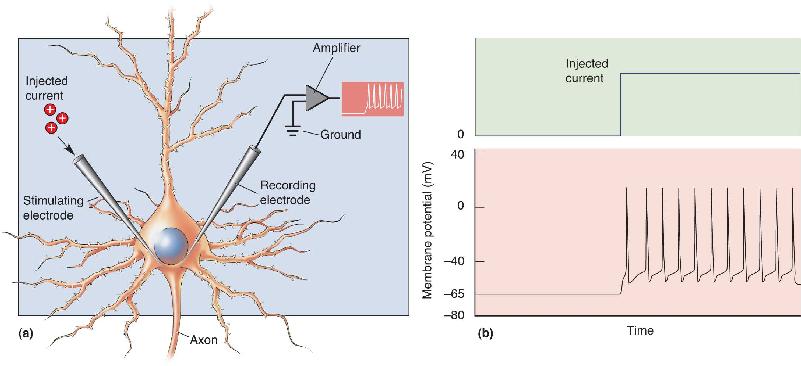
Earlier we likened the generation of an action potential by depolarization to taking a photograph by pressing the shutter button on a camera. But what if the camera is one of those fancy ones used by fashion and sports photographers where continued pressure on the button causes the camera to shoot frame after frame? The same thing is true for a neuron. If, for example, we pass continuous depolarizing current into a neuron through a microelectrode, we generate not one but many action potentials in succession (Figure 4.2).

FIGURE 4.2 The effect of injecting positive charge into a neuron. (a) The axon hillock is impaled by two electrodes, one for recording the membrane potential relative to ground and the other for stimulating the neuron with electrical current. (b) When electrical current is injected into the neuron (top trace), the membrane is depolarized sufficiently to fire action potentials (bottom trace). Description
The rate of action potential generation depends on the magnitude of the continuous depolarizing current. If we pass enough current through a microelectrode to depolarize just to threshold, but not far beyond, we might find that the cell generates action potentials at a rate of something like one per second, or one hertz (1 Hz). If we crank up the current a little bit more, however, we will find that the rate of action potential generation increases, say, to 50 impulses per second (50 Hz). Thus, the firing frequency of action potentials reflects the magnitude of the depolarizing current. This is one way that stimulation intensity is encoded in the nervous system (Figure 4.3).

FIGURE 4.3 The dependence of action potential firing frequency on the level of depolarization. Description
Although firing frequency increases with the amount of depolarizing current, there is a limit to the rate at which a neuron can generate action potentials. The maximum firing frequency is about 1000 Hz; once an action potential is initiated, it is impossible to initiate another for about 1 msec. This period of time is called the absolute refractory period. In addition, it can be relatively difficult to initiate another action potential for several milliseconds after the end of the absolute refractory period. During this relative refractory period, the amount of current required to depolarize the neuron to action potential threshold is elevated above normal.
Optogenetics: Controlling Neural Activity with Light. As we have discussed, action potentials are caused by the depolarization of the membrane beyond a threshold value, as occurs naturally in neurons by the opening of ion channels that allow Na+ to cross the membrane. To artificially control neuronal firing rates, neuroscientists historically have had to use microelectrodes to inject electrical current. This limitation was recently overcome with a revolutionary new approach called optogenetics, which introduces into neurons foreign genes that express membrane ion channels that open in response to light.
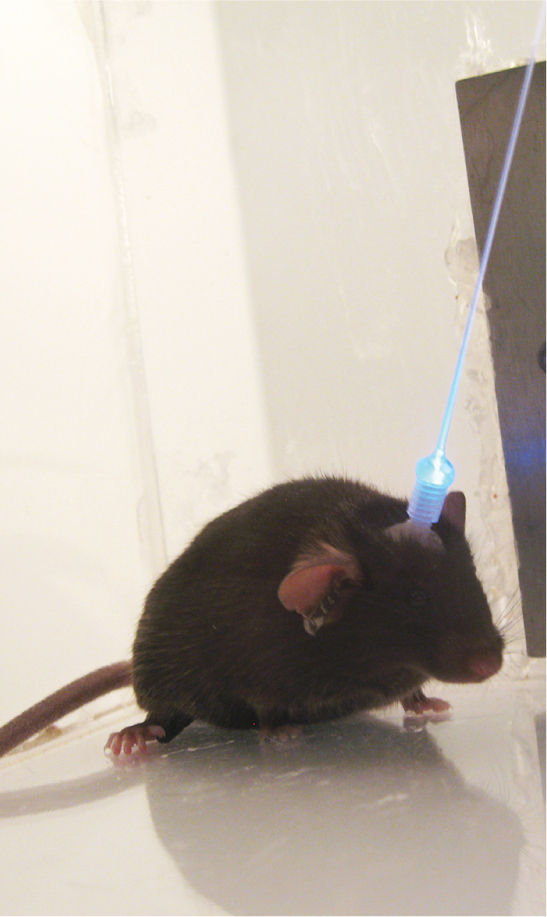
In Chapter 9, we will discuss how light energy is absorbed by proteins called photopigments to generate the neural responses in our retinas that ultimately give us sight. Of course, sensitivity to light is a property of many organisms. In the course of studying light responses in a green alga, researchers working in Frankfurt, Germany, characterized a photopigment they called channelrhodopsin-2 (ChR2). By introducing the ChR2 gene into mammalian cells, they showed that it encodes a light-sensitive cation channel that is permeable to Na+ and Ca2+ (Box 4.2). The channel opens rapidly in response to blue light, and in neurons the inward flow of cations is sufficient to produce depolarization beyond threshold for action potentials. The enormous potential of optogenetics was subsequently demonstrated by researchers in the United States who showed that the behavior of rats and mice could be dramatically influenced by shining blue light onto neurons in which the ChR2 gene was introduced (Figure 4.4). Newer additions to the “optogenetic toolkit” available to researchers include halorhodopsin, a protein derived from single-cell microbes that will inhibit neurons in response to yellow light.

FIGURE 4.4 Optogenetic control of neural activity in a mouse brain. The gene encoding channelrhodopsin-2 was introduced into neurons of this mouse’s brain using a virus. The firing of these neurons can now be controlled with blue light delivered via an optic fiber. (Source: Courtesy of Dr. Ed Boyden, Massachusetts Institute of Technology.)
When I returned in 1992 to the Max Planck Institute of Biophysics in Frankfurt, Germany, from my post-doctoral studies at Yale and Rockefeller University, I was mostly interested in the mechanisms that establish ion gradients across cell membranes. Ernst Bamberg, the director of my department, convinced me to undertake a novel approach in studying microbial rhodopsins—proteins that transport ions across membranes when they absorb light energy. We expressed the gene for bacteriorhodopsin in frog eggs (oocytes), and measured its light-activated electrical current with microelectrodes. In 1995, we demonstrated that illumination of bacteriorhodopsin triggered proton (H+) pumping across the oocyte membrane. We then went on in 1996 to study the light-activated chloride pump halorhodopsin with this new technique.
We also received DNA for chlamyopsin-1 and -2, proposed to be photoreceptor proteins in the green alga Chlamydomonas reinhardtii, from Peter Hegemann at the University of Regensburg. Unfortunately, like all the other labs who received this DNA, we were unable to observe any light-induced electrical signals. Nevertheless, I agreed to test the function of a new presumed rhodopsin from Chlamydomonas when Peter called me, announcing that they had found a “real light-gated calcium channel,” which he wanted to name chlamyrhodopsin-3. Although this new protein had not been purified, “chlamyopsin-3” was detected in a data bank of DNA sequences from Chlamydomonas, produced at the research center in Kazusa, Japan, and showed similarities to bacteriorhodopsin. This made it an interesting candidate for the long-sought rhodopsin in Chlamydomonas. Peter requested the DNA from Japan, and I then expressed it in oocytes. Our initial experiments, however, were disappointing because removal or addition of calcium to the oocyte bath solution made no difference to the light-activated electrical current, as would be expected if it actually were a Ca2+ permeable channel. The photocurrent itself was rather weak and did not seem to be influenced by any change in the ionic concentrations in the bath solution.
As I still liked the idea of a directly light-gated ion channel, which most other researchers in the field rejected, I continued to test different bath solutions. One evening, I got a stunningly large inward light-activated current with a solution designed to inhibit calcium currents. It turned out, however, that the solution I used was badly buffered; in fact, it was quite acidic with too much H+! But this was a breakthrough as I now had good evidence for an inward-directed light-dependent H+ conductance. Then, by acidifying the oocyte (that is, increasing the H+ concentration of the oocyte interior relative to the outside), I found I was able to reliably generate outward-directed light-activated currents as well. It soon became clear that we have with chlamyrhodopsin–3 a light-gated proton channel; therefore, I proposed to my colleagues Peter Hegemann and Ernst Bamberg to call this new protein channelrhodopsin-1. Further experiments revealed that other monovalent cations can also permeate channelrhodopsin-1. The small photocurrents we observed initially are now understood to be due to poor expression of channelrhodopsin-1 in oocytes.
Tantalized by this new finding, we prepared a manuscript (published in 2002) and applied for a patent describing the use of light-gated ion channels for noninvasive manipulation of cells and even living organisms. I next studied the closely related algal protein channelrhodopsin-2, and everything became so much easier as photocurrents were now really large and easy to analyze. Channelrhodopsin-2 (chop2), 737 amino acids long in its native form, could be shortened to 310 amino acids and attached to yellow fluorescent protein (YFP) to allow visualization of protein expression. After we published the superior features of chop2 in 2003, requests for the DNA started coming in, and we ourselves looked for collaborations with neurobiologists. One of our first “victims” was Alexander Gottschalk at the nearby University of Frankfurt, as he worked with the small translucent nematode worm Caenorhabditis elegans (C. elegans). Unfortunately, I made an error in preparing the DNA such that that the worms, although nicely labeled by YFP, did not react to light. Once I realized my mistake and got chop2–YFP into C. elegans muscle cells, we were amazed how easily these little worms could be induced to contract simply by illumination with blue light. At about the same time (April 2004) Karl Deisseroth at Stanford University asked for the DNA and advice on its use in a collaboration, which I happily accepted. Karl quickly demonstrated the power of channelrhodopsin-2 in mammalian neurons. His exciting work with Ed Boyden and Feng Zhang attracted a lot of attention, prompting many requests for the DNA to express this protein in the brain. Many colleagues from Europe only then realized that channelrhodopsins were first characterized in Frankfurt.
The success and ease of application of channelrhodopsin-2 led Karl and Alexander to wonder if there are other rhodopsins that might be used for light-induced inhibition of neuronal activity. We told them about bacteriorhodopsin and halorhodopsin, the light-activated proton export and chloride import pumps, respectively. Both pumps render the cell interior more negative (i.e., they are light-activated hyperpolarizers). We recommended halorhodopsin from the microbe Natronomonas pharaonis as a light-activated hyperpolarizer. We took advantage of what we had learned back in 1996: that halorhodopsin had a high affinity for chloride and that its expression was stable in animal cells.

Figure A Schematic drawings of channelrhodopsin-2 and halorhodopsin in the plasma membrane. Below, the effect of blue and yellow light on membrane potential, mediated by channelrhodopsin-2 and halorhodopsin, respectively.
As it turned out, light activation of the chloride pump halorhodopsin is sufficient to inhibit action potential firing in mammalian neurons and to inhibit muscle contraction of the nematode C. elegans. Ironically, these neurobiological experiments with halorhodopsin (and the same applies for bacteriorhodopsin) could have been done several years earlier, but only the discovery and application of channelrhodopsin-2 encouraged their use and helped create a new field, now called optogenetics. Many neurobiologists are now using these tools, and a few groups, including ourselves, are engaged in further improving and expanding the existing optogenetic tool box.
Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E. 2003. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proceedings of the National Academy of Sciences of United States of America 100:13940–13945.
Understanding how behaviors arise, of course, requires understanding how action potentials arise and propagate through the nervous system. We now take a look at how the movement of ions through the neuron’s own specialized protein channels causes a neural signal with these interesting properties.
The action potential is a dramatic redistribution of electrical charge across the membrane. Depolarization of the cell during the action potential is caused by the influx of sodium ions across the membrane, and repolarization is caused by the efflux of potassium ions. Let’s apply some of the concepts introduced in Chapter 3 to help us understand how ions are driven across the membrane, and how these ionic movements affect the membrane potential.
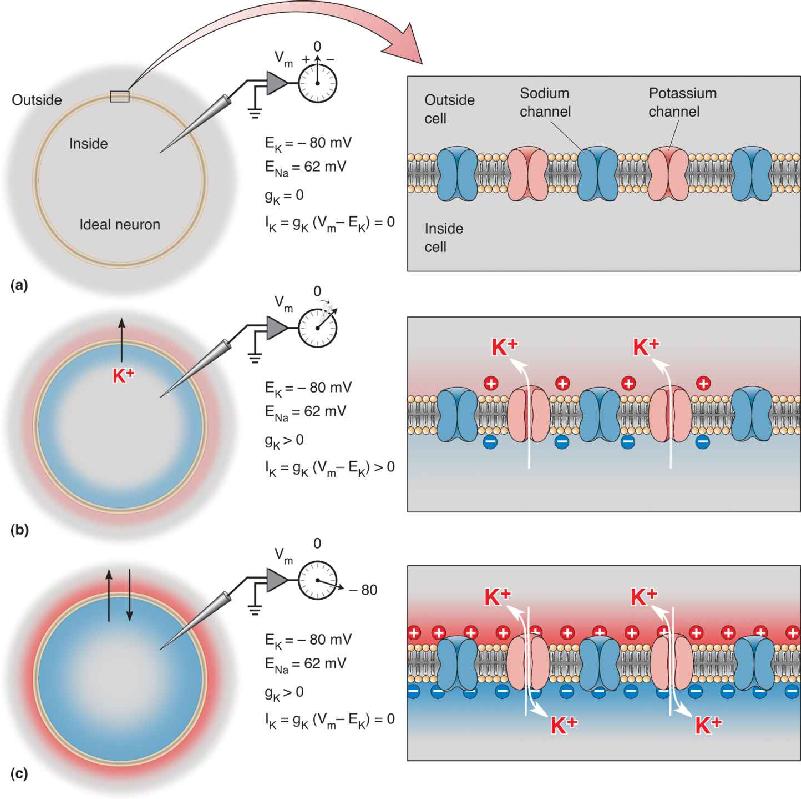
Consider the idealized neuron illustrated in Figure 4.5. The membrane of this cell has three types of protein molecules: sodium-potassium pumps, potassium channels, and sodium channels. The pumps work continuously to establish and maintain concentration gradients. As in all our previous examples, we’ll assume that K+ is concentrated twentyfold inside the cell and that Na+ is concentrated tenfold outside the cell. According to the Nernst equation, at 37 °C, EK = –80 mV and ENa = 62 mV. Let’s use this cell to explore the factors that govern the movement of ions across the membrane.

FIGURE 4.5 Membrane currents and conductances. Here is an idealized neuron with sodium-potassium pumps (not shown), potassium channels, and sodium channels. The pumps establish ionic concentration gradients so that K+ is concentrated inside the cell and Na+ is concentrated outside the cell. (a) Initially, we assume that all channels are closed and the membrane potential equals 0 mV. (b) Now we open the potassium channels, and K+ flows out of the cell. This movement of K+ is an electrical current, IK, and it flows as long as the membrane conductance to K+, gK, is greater than zero, and the membrane potential is not equal to the potassium equilibrium potential. (c) At equilibrium, there is no net potassium current because, although gK >0, the membrane potential at equilibrium equals EK. At equilibrium, an equal number of K+ enters and leaves. Description
We begin by assuming that both the potassium channels and the sodium channels are closed, and that the membrane potential, Vm, is equal to 0 mV (Figure 4.5a). Now let’s open the potassium channels only (Figure 4.5b). As we learned in Chapter 3, K+ will flow out of the cell, down the concentration gradient, until the inside becomes negatively charged, and Vm = EK (Figure 4.5c). Here we want to focus our attention on the movement of K+ that took the membrane potential from 0 mV to –80 mV. Consider these three points:
- The net movement of K+ across the membrane is an electrical current. We can represent this current using the symbol IK.
- The number of open potassium channels is proportional to an electrical conductance. We can represent this conductance by the symbol gK.
- Membrane potassium current, IK, will flow only as long as Vm ≈ EK. The driving force on K+ is defined as the difference between the real membrane potential and the equilibrium potential, which can be written as Vm – EK.
There is a simple relationship between the ionic driving force, ionic conductance, and the amount of ionic current that will flow. For K+, this may be written:
If this sounds familiar, that is because it is simply an expression of Ohm’s law, I = gV, which we learned about in Chapter 3.
Now let’s take another look at our example. Initially we began with Vm = 0 mV and no ionic membrane permeability (see Figure 4.5a). There is a large driving force on K+ because Vm ≠ EK; in fact, (Vm – EK) = 80 mV. However, because the membrane is impermeable to K+, the potassium conductance, gK, equals zero. Consequently, IK = 0. Potassium current flows only when the membrane has open potassium channels and therefore gK > 0. Now K+ flows out of the cell—as long as the membrane potential differs from the potassium equilibrium potential (see Figure 4.5b). Notice that the current flow is in the direction that takes Vm toward EK. When Vm = EK, the membrane is at equilibrium, and no net current will flow. In this condition, although there is a large potassium conductance, gK, there is no longer any net driving force on the K+ (Figure 4.5c).
Let’s pick up the action where we left off in the last section. The membrane of our ideal neuron is permeable only to K+, and Vm = EK = –80 mV. What’s happening with the Na+ concentrated outside the cell? Because the membrane potential is so negative relative to the sodium equilibrium potential, there is a very large driving force on Na+ ([Vm – ENa] = [–80 mV – 62 mV] = –142 mV). Nonetheless, there can be no net Na+ current as long as the membrane is impermeable to Na+. But now let’s open the sodium channels and see what happens to the membrane potential.
At the instant we change the ionic permeability of the membrane, gNa is high, and, as we discussed earlier, there is a large driving force pushing on Na+. Thus, we have what it takes to generate a large sodium current, INa, across the membrane. Na+ passes through the membrane sodium channels in the direction that takes Vm toward ENa; in this case, the sodium current, INa, is inward across the membrane. Assuming the membrane permeability is now far greater to sodium than it is to potassium, this influx of Na+ depolarizes the neuron until Vm approaches ENa, 62 mV.
Notice that something remarkable happened here. Simply by switching the dominant membrane permeability from K+ to Na+, we were able to rapidly reverse the membrane potential. In theory, then, the rising phase of the action potential could be explained if, in response to depolarization of the membrane beyond threshold, membrane sodium channels opened. This would allow Na+ to enter the neuron, causing a massive depolarization until the membrane potential approached ENa.
How could we account for the falling phase of the action potential? Simply assume that sodium channels quickly close and the potassium channels remain open, so the dominant membrane ion permeability switches back from Na+ to K+. Then K+ would flow out of the cell until the membrane potential again equals EK.
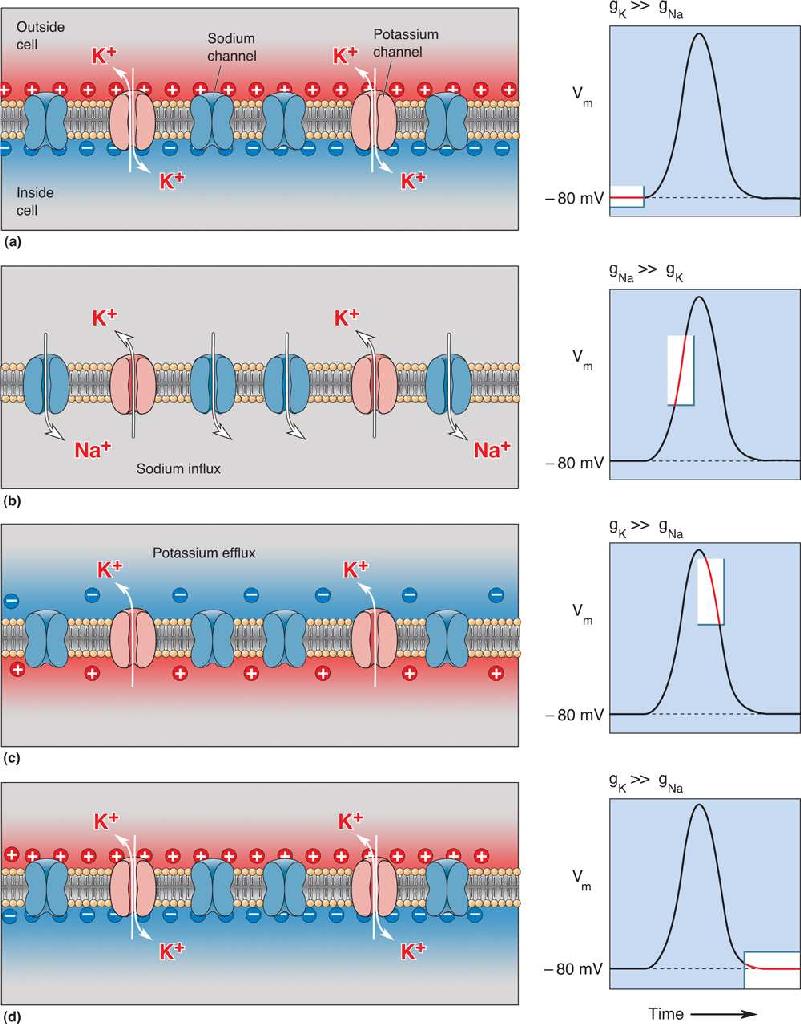
Our model for the ins and outs, ups and downs of the action potential in an idealized neuron is shown in Figure 4.6. The rising phase of the action potential is explained by an inward sodium current, and the falling phase is explained by an outward potassium current. The action potential therefore could be accounted for simply by the movement of ions through channels that are gated by changes in the membrane potential. If you understand this concept, you understand a lot about the ionic basis of the action potential. What’s left now is to see how this actually happens—in a real neuron.

FIGURE 4.6 Flipping the membrane potential by changing the relative ionic permeability of the membrane. (a) The membrane of the idealized neuron, introduced in Figure 4.4. We begin by assuming that the membrane is permeable only to K+ and that Vm = EK. (b) We now stipulate that the membrane sodium channels open so that gNa >> gK. There is a large driving force on Na+, so Na+ rushes into the cell, taking Vm toward ENa. (c) Now we close the sodium channels so that gK >> gNa. Because the membrane potential is positive, there is a large driving force on K+. The efflux of K+ takes Vm back toward EK. (d) The resting state is restored where Vm = EK. Description
Let’s quickly review our theory of the action potential. When the membrane is depolarized to threshold, there is a transient increase in gNa. The increase in gNa allows the entry of Na+, which depolarizes the neuron. And the increase in gNa must be brief in duration to account for the short duration of the action potential. Restoring the negative membrane potential would be further aided by a transient increase in gK during the falling phase, allowing K+ to leave the depolarized neuron faster.
Testing this theory is simple enough in principle. All one has to do is measure the sodium and potassium conductances of the membrane during the action potential. In practice, however, such a measurement proved to be quite difficult in real neurons. The key technical breakthrough came with a device called a voltage clamp, invented by the American physiologist Kenneth C. Cole and used in decisive experiments performed by Cambridge University physiologists Alan Hodgkin and Andrew Huxley around 1950. The voltage clamp enabled Hodgkin and Huxley to “clamp” the membrane potential of an axon at any value they chose. They could then deduce the changes in membrane conductance that occur at different membrane potentials by measuring the currents that flowed across the membrane. In an elegant series of experiments, Hodgkin and Huxley showed that the rising phase of the action potential was indeed caused by a transient increase in gNa and an influx of Na+, and that the falling phase was associated with an increase in gK and an efflux of K+. Their accomplishments were recognized with the Nobel Prize in 1963.
To account for the transient changes in gNa, Hodgkin and Huxley proposed the existence of sodium “gates” in the axonal membrane. They hypothesized that these gates are “activated” (opened) by depolarization above threshold and “inactivated” (closed and locked) when the membrane acquires a positive membrane potential. These gates are “deinactivated” (unlocked and enabled to be opened again) only after the membrane potential returns to a negative value.
It is a tribute to Hodgkin and Huxley that their hypotheses about membrane gates came more than 20 years before the direct demonstration of voltage-gated channel proteins in the neuronal membrane. We have a new understanding of gated membrane channels, thanks to two more recent scientific breakthroughs. First, new molecular biological techniques have enabled neuroscientists to determine the detailed structure of these proteins. Second, new neurophysiological techniques have enabled neuroscientists to measure the ionic currents that pass through single channels. We will now explore the action potential from the perspective of these membrane ion channels.
The voltage-gated sodium channel is aptly named. The protein forms a pore in the membrane that is highly selective to Na+, and the pore is opened and closed by changes in membrane voltage.
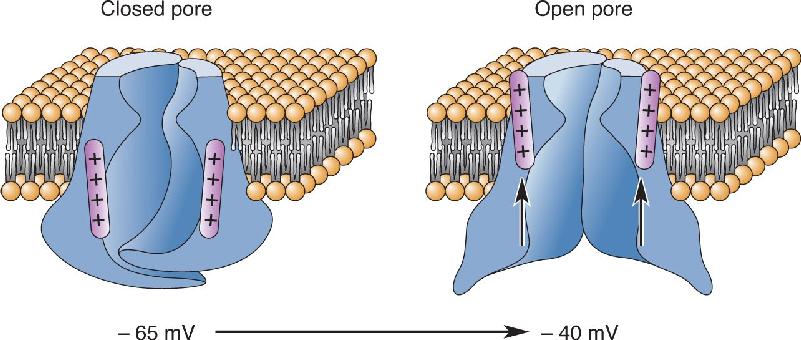
Sodium Channel Structure. The voltage-gated sodium channel is created from a single long polypeptide. The molecule has four distinct domains, numbered I–IV; each domain consists of six transmembrane alpha helices, numbered S1–S6 (Figure 4.7). The four domains clump together to form a pore between them. The pore is closed at the negative resting membrane potential. When the membrane is depolarized to threshold, however, the molecule twists into a configuration that allows the passage of Na+ through the pore (Figure 4.8).

FIGURE 4.7 The structure of the voltage-gated sodium channel. (a) A depiction of how the sodium channel polypeptide chain is believed to be woven into the membrane. The molecule consists of four domains, I–IV. Each domain consists of six alpha helices (represented by the blue and purple cylinders), which pass back and forth across the membrane. (b) An expanded view of one domain, showing the voltage sensor of alpha helix S4 and the pore loop (red), which contributes to the selectivity filter. (c) A view of the molecule showing how the domains may arrange themselves to form a pore between them. (Source: Adapted from Armstrong and Hille, 1998, Fig. 1.) Description

FIGURE 4.8 A hypothetical model for changing the configuration of the sodium channel by depolarizing the membrane. Description
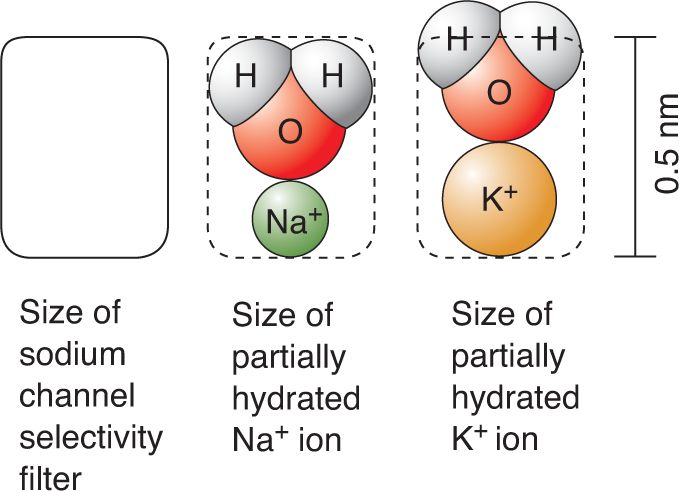
Like the potassium channel, the sodium channel has pore loops that are assembled into a selectivity filter. This filter makes the sodium channel twelve times more permeable to Na+ than it is to K+. Apparently, the Na+ ions are stripped of most, but not all, of their associated water molecules as they pass into the channel. The retained water serves as a sort of molecular chaperone for the ion, and is necessary for the ion to pass the selectivity filter. The ion–water complex can then be used to select Na+ and exclude K+ (Figure 4.9).

FIGURE 4.9 Dimensions of the sodium channel selectivity filter. Water accompanies the ions as they pass through the channel. Hydrated Na+ fits; hydrated K+ does not. (Source: Adapted from Hille, 1992, Figs. 5, 6.) Description
The sodium channel is gated by a change in voltage across the membrane. It has now been established that the voltage sensor resides in segment S4 of the molecule. In this segment, positively charged amino acid residues are regularly spaced along the coils of the helix. Thus, the entire segment can be forced to move by changing the membrane potential. Depolarization twists S4, and this conformational change in the molecule causes the gate to open.
Functional Properties of the Sodium Channel. Research performed around 1980 at the Max Planck Institute in Goettingen, Germany, revealed the functional properties of the voltage-gated sodium channel. A new method was used, called the patch clamp, to study the ionic currents passing through individual ion channels (Box 4.3). The patch-clamp method entails sealing the tip of an electrode to a very small patch of neuronal membrane. This patch can then be torn away from the neuron, and the ionic currents across it can be measured as the membrane potential is clamped at any value the experimenter selects. With luck, the patch will contain only a single channel, and the behavior of this channel can be studied. Patch clamping enabled investigation of the functional properties of the voltage-gated sodium channel.
The very existence of voltage-gated channels in the neuronal membrane was mere conjecture until methods were developed to study individual channel proteins. A revolutionary new method, the patch clamp, was developed by German neuroscientists Bert Sakmann and Erwin Neher in the mid-1970s. In recognition of their contribution, Sakmann and Neher were awarded the 1991 Nobel Prize.
Patch clamping enables one to record ionic currents through single channels (Figure A). The first step is gently lowering the fire-polished tip of a glass recording electrode, 1–5 μm in diameter, onto the membrane of the neuron (part a), and then applying suction within the electrode tip (part b). A tight seal forms between the walls of the electrode and the underlying patch of membrane. This “gigaohm” seal (so named because of its high electrical resistance: >109 Ω) leaves the ions in the electrode only one path to take, through the channels in the underlying patch of membrane. If the electrode is then withdrawn from the cell, the membrane patch can be torn away (part c), and ionic currents can be measured as steady voltages are applied across the membrane (part d).

With a little luck, one can resolve currents flowing through single channels. If the patch contains a voltage-gated sodium channel, for example, then changing the membrane potential from –65 to –40 mV will cause the channel to open, and current (I) will flow through it (part e). The amplitude of the measured current at a constant membrane voltage reflects the channel conductance, and the duration of the current reflects the time the channel is open.
Patch-clamp recordings reveal that most channels flip between two conductance states that can be interpreted as open or closed. The time they remain open can vary, but the single-channel conductance value stays the same and is therefore said to be unitary. Ions can pass through single channels at an astonishing rate—well over a million per second.
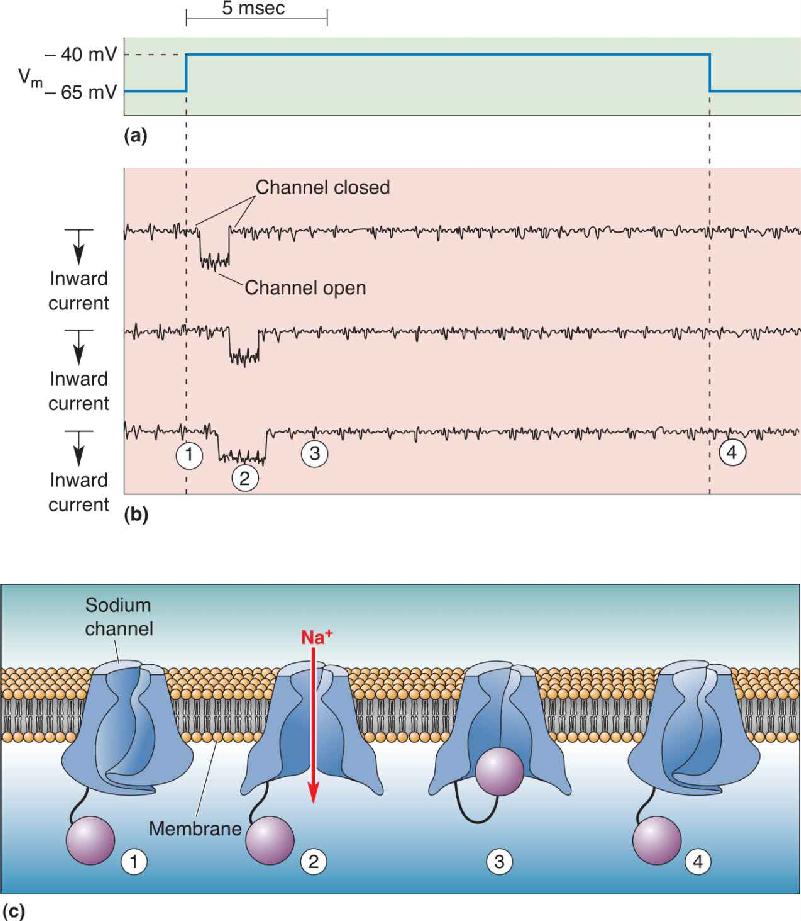
Changing the membrane potential of a patch of axonal membrane from –80 to –65 mV has little effect on the voltage-gated sodium channels. They remain closed because depolarization of the membrane has not yet reached threshold. Changing the membrane potential from –65 to –40 mV, however, causes these channels to pop open. As shown in Figure 4.10, voltage-gated sodium channels have a characteristic pattern of behavior:

FIGURE 4.10 The opening and closing of sodium channels upon membrane depolarization. (a) This trace shows the electrical potential across a patch of membrane. When the membrane potential is changed from –65 to –40 mV, the sodium channels pop open. (b) These traces show how three different channels respond to the voltage step. Each line is a record of the electrical current that flows through a single channel. ➀ At –65 mV, the channels are closed, so there is no current. ➁ When the membrane is depolarized to –40 mV, the channels briefly open and current flows inward, represented by the downward deflection in the current traces. Although there is some variability from channel to channel, all of them open with little delay and stay open for less than 1 msec. Notice that after they have opened once, they close and stay closed as long as the membrane is maintained at a depolarized Vm. ➂ The closure of the sodium channel by steady depolarization is called inactivation. ➃ To deinactivate the channels, the membrane must be returned to –65 mV again. (c) A model for how changes in the conformation of the sodium channel protein might yield its functional properties. ➀ The closed channel ➁ opens upon membrane depolarization. ➂ Inactivation occurs when a globular portion of the protein swings up and occludes the pore. ➃ Deinactivation occurs when the globular portion swings away and the pore closes by movement of the transmembrane domains. Description
- They stay open for about 1 msec and then close (inactivate).
- They cannot be opened again by depolarization until the membrane potential returns to a negative value near threshold.
A hypothetical model for how conformational changes in the voltage-gated sodium channel could account for these properties is illustrated in Figure 4.10c.
A single channel does not an action potential make. The membrane of an axon may contain thousands of sodium channels per square micrometer (μm2), and the concerted action of all these channels is required to generate what we measure as an action potential. Nonetheless, it is interesting to see how many of the properties of the action potential can be explained by the properties of the voltage-gated sodium channel. For example, the fact that single channels do not open until a critical level of membrane depolarization is reached explains the action potential threshold. The rapid opening of the channels in response to depolarization explains why the rising phase of the action potential occurs so quickly. And the short time the channels stay open before inactivating (about 1 msec) partly explains why the action potential is so brief. Furthermore, inactivation of the channels can account for the absolute refractory period: Another action potential cannot be generated until the channels are activated.
There are several different sodium channel genes in the human genome. Differences in the expression of these genes among neurons can give rise to subtle but important variations in the properties of the action potential. Recently, single amino acid mutations in the extracellular regions of one sodium channel have been shown to cause a common inherited disorder in human infants known as generalized epilepsy with febrile seizures. Epileptic seizures result from explosive, highly synchronous electrical activity in the brain. (Epilepsy is discussed in detail in Chapter 19.) The seizures in this disorder occur in response to fever (febrile is from the Latin word for “fever”). They usually occur only in early childhood, between 3 months and 5 years of age. Although precisely how the seizures are triggered by an increase in brain temperature is not clear, the mutations’ effects include slowing the inactivation of the sodium channel, prolonging the action potential. Generalized epilepsy with febrile seizures is a channelopathy, a human genetic disease caused by alterations in the structure and function of ion channels.
The Effects of Toxins on the Sodium Channel. Researchers at Duke University discovered in the 1960s that a toxin isolated from the ovaries of the puffer fish (Figure 4.11) could selectively block the sodium channel. Tetrodotoxin (TTX) clogs the Na+-permeable pore by binding tightly to a specific site on the outside of the channel. TTX blocks all sodium-dependent action potentials and therefore is usually fatal if ingested. Nonetheless, puffer fish are considered a delicacy in Japan. Sushi chefs licensed by the government train for years to prepare puffer fish in such a way that eating them causes only numbness around the mouth. Talk about adventuresome eating!
FIGURE 4.11 The puffer fish, source of TTX. (Source: Courtesy of Dr. Toshio Narahashi, Duke University.)
TTX is one of a number of natural toxins that interfere with the function of the voltage-gated sodium channel. Another channel-blocking toxin is saxitoxin, produced by dinoflagellates of the genus Gonyaulax. Saxitoxin is concentrated in clams, mussels, and other shellfish that feed on these marine protozoa. Occasionally the dinoflagellates bloom, causing what is known as a “red tide.” Eating shellfish at these times can be fatal because of the unusually high concentration of the toxin.
In addition to the toxins that block sodium channels, certain compounds interfere with nervous system function by causing the channels to open inappropriately. In this category is batrachotoxin, isolated from the skin of a species of Colombian frog. Batrachotoxin causes the channels to open at more negative potentials and to stay open much longer than usual, thus scrambling the information encoded by the action potentials. Toxins produced by lilies (veratridine) and buttercups (aconitine) have a similar mechanism of action. Sodium channel inactivation is also disrupted by toxins from scorpions and sea anemones.
What can we learn from these toxins? First, the different toxins disrupt channel function by binding to different sites on the protein. Information about toxin binding and its consequences have helped researchers deduce the three-dimensional structure of the sodium channel. Second, the toxins can be used as experimental tools to study the consequences of blocking action potentials. For example, as we shall see in later chapters, TTX is commonly used in experiments that require blocking impulses in a nerve or muscle. The third and most important lesson from studying toxins? Be careful what you put in your mouth!
Hodgkin’s and Huxley’s experiments indicated that the falling phase of the action potential was explained only partly by the inactivation of gNa. They found there was also a transient increase in gK that functioned to speed the restoration of a negative membrane potential after the spike. They proposed the existence of membrane potassium gates that, like sodium gates, open in response to depolarization of the membrane. Unlike sodium gates, however, potassium gates do not open immediately upon depolarization; it takes about 1 msec for them to open. Because of this delay, and because this potassium conductance serves to rectify, or reset, the membrane potential, they called this conductance the delayed rectifier.
We now know that there are many different types of voltage-gated potassium channels. Most of them open when the membrane is depolarized and function to diminish any further depolarization by giving K+ a path to leave the cell across the membrane. The known voltage-gated potassium channels have a similar structure. The channel proteins consist of four separate polypeptide subunits that come together to form a pore between them. Like the sodium channel, these proteins are sensitive to changes in the electrical field across the membrane. When the membrane is depolarized, the subunits are believed to twist into a shape that allows K+ to pass through the pore.
We can now use what we’ve learned about ions and channels to explain the key properties of the action potential (Figure 4.12):

FIGURE 4.12 The molecular basis of the action potential. (a) The membrane potential as it changes in time during an action potential. The rising phase of the action potential is caused by the influx of Na+ through hundreds of voltage-gated sodium channels. The falling phase is caused by sodium channel inactivation and the efflux of K+ through voltage-gated potassium channels. (b) The inward currents through three representative voltage-gated sodium channels. Each channel opens with little delay when the membrane is depolarized to threshold. The channels stay open for no more than 1 msec and then inactivate. (c) The summed Na+ current flowing through all the sodium channels. (d) The outward currents through three representative voltage-gated potassium channels. Voltage-gated potassium channels open about 1 msec after the membrane is depolarized to threshold and stay open as long as the membrane is depolarized. The high potassium permeability causes the membrane to hyperpolarize briefly. When the voltage-gated potassium channels close, the membrane potential relaxes back to the resting value, around –65 mV. (e) The summed K+ current flowing through all the potassium channels. (f) The net transmembrane current during the action potential (the sum of parts c and e). Description
- Threshold. Threshold is the membrane potential at which enough voltage-gated sodium channels open so that the relative ionic permeability of the membrane favors sodium over potassium.
- Rising phase. When the inside of the membrane has a negative electrical potential, there is a large driving force on Na+. Therefore, Na+ rushes into the cell through the open sodium channels, causing the membrane to rapidly depolarize.
- Overshoot. Because the relative permeability of the membrane greatly favors sodium, the membrane potential goes to a value close to ENa, which is greater than 0 mV.
- Falling phase. The behavior of two types of channels contributes to the falling phase. First, the voltage-gated sodium channels inactivate. Second, the voltage-gated potassium channels finally open (triggered to do so 1 msec earlier by the depolarization of the membrane). There is a great driving force on K+ when the membrane is strongly depolarized. Therefore, K+ rushes out of the cell through the open channels, causing the membrane potential to become negative again.
- Undershoot. The open voltage-gated potassium channels add to the resting potassium membrane permeability. Because there is very little sodium permeability, the membrane potential goes toward EK, causing a hyperpolarization relative to the resting membrane potential until the voltage-gated potassium channels close again.
- Absolute refractory period. Sodium channels inactivate when the membrane becomes strongly depolarized. They cannot be activated again, and another action potential cannot be generated, until the membrane potential becomes sufficiently negative to deinactivate the channels.
- Relative refractory period. The membrane potential stays hyperpolarized until the voltage-gated potassium channels close. Therefore, more depolarizing current is required to bring the membrane potential to threshold.
We’ve seen that channels and the movement of ions through them can explain the properties of the action potential. But it is important to remember that the sodium-potassium pump also is working quietly in the background. Imagine that the entry of Na+ during each action potential is like a wave coming over the bow of a boat making way in heavy seas. Like the continuous action of the boat’s bilge pump, the sodium-potassium pump works all the time to transport Na+ back across the membrane. The pump maintains the ionic concentration gradients that drive Na+ and K+ through their channels during the action potential.
To transfer information from one point to another in the nervous system, it is necessary that the action potential, once generated, be conducted down the axon. This process is like the burning of a fuse. Imagine you’re holding a firecracker with a burning match held under the end of the fuse. The fuse ignites when it gets hot enough (beyond some threshold). The tip of the burning fuse heats up the segment of fuse immediately ahead of it until it ignites. In this way, the flame steadily works its way down the fuse. Note that the fuse lit at one end only burns in one direction; the flame cannot turn back on itself because the combustible material just behind it is spent.
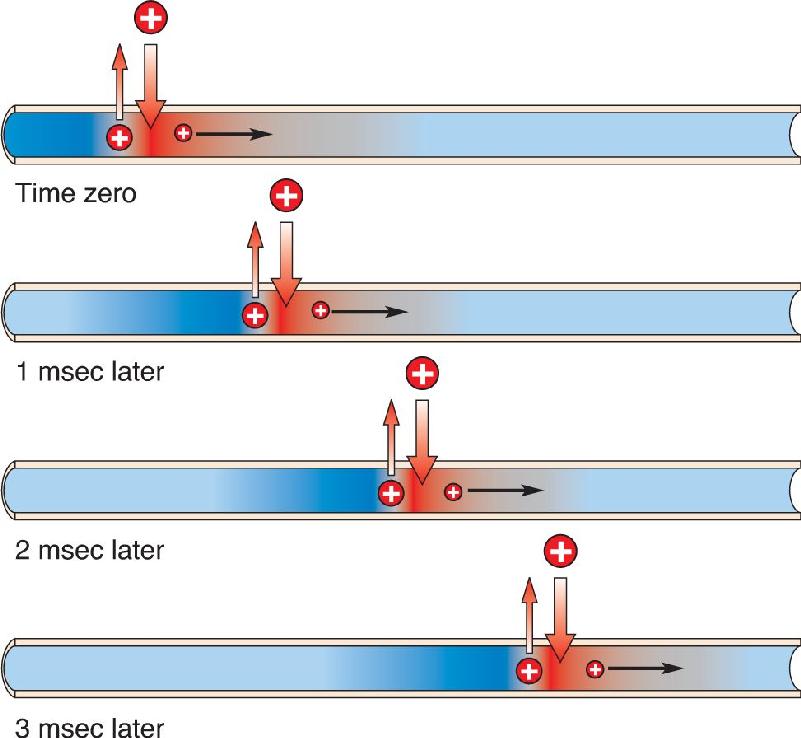
Propagation of the action potential along the axon is similar to the propagation of the flame along the fuse. When a patch of axonal membrane is depolarized sufficiently to reach threshold, voltage-gated sodium channels pop open, and the action potential is initiated. The influx of positive charge spreads inside the axon to depolarize the adjacent segment of membrane, and when it reaches threshold, the sodium channels in this patch of membrane also pop open (Figure 4.13). In this way, the action potential works its way down the axon until it reaches the axon terminal, thereby initiating synaptic transmission (the subject of Chapter 5).

FIGURE 4.13 Action potential conduction. The entry of positive charge during the action potential causes the membrane just ahead to depolarize to threshold. Description
An action potential initiated at one end of an axon propagates only in one direction; it does not turn back on itself. This is because the membrane just behind it is refractory, due to inactivation of the sodium channels. Normally, action potentials conduct only in one direction, from the soma to the axon terminal; this is called orthodromic conduction. But, just like the fuse, an action potential can be generated by depolarization at either end of the axon and can therefore propagate in either direction. Backward propagation, elicited experimentally, is called antidromic conduction. Note that because the axonal membrane is excitable (capable of generating action potentials) along its entire length, the impulse will propagate without decrement. The fuse works the same way because it is combustible along its entire length. Unlike the fuse, however, the axon can regenerate its firing ability.
Action potential conduction velocities vary, but 10 m/sec is a typical rate. Remember, from start to finish the action potential lasts about 2 msec. From this, we can calculate the length of membrane that is engaged in the action potential at any instant in time:
Therefore, an action potential traveling at 10 m/sec occurs over a 2 cm length of axon.
Remember that the inward Na+ current during the action potential depolarizes the membrane just ahead. When this patch of membrane reaches threshold, the voltage-gated sodium channels will open, and the action potential will “burn” on down the membrane. The speed with which the action potential propagates down the axon depends on how far the depolarization ahead of the action potential spreads, which in turn depends on certain physical characteristics of the axon.
Imagine that the influx of positive charge into an axon during the action potential is like turning on the water to a leaky garden hose. There are two paths the water can take: one, down the inside of the hose; the other, through the perforated wall of the hose. How much water goes along each path depends on their relative resistance; most of the water will take the path of least resistance. If the hose is narrow and the leaks are numerous and large, most of the water will flow out through the leaks. If the hose is wide and the leaks are few and tiny, most of the water will flow down the inside of the hose. The same principles apply to positive current spreading down the axon ahead of the action potential.
There are two paths that positive charge can take: down the inside of the axon, or across the axonal membrane. If the axon is narrow and there are many open membrane pores, most of the current will flow out across the membrane. If the axon is wide and there are few open membrane pores, most of the current will flow down inside the axon. The farther the current goes down the axon, the farther ahead of the action potential the membrane will be depolarized, and the faster the action potential will propagate. As a rule, therefore, action potential conduction velocity increases with increasing axonal diameter.
As a consequence of this relationship between axonal diameter and conduction velocity, neural pathways that are especially important for survival have evolved unusually large axons. An example is the giant axon of the squid, which is part of a pathway that mediates an escape reflex in response to strong sensory stimulation. The squid giant axon can be 1 mm in diameter, so large that originally it was thought to be part of the animal’s circulatory system. Neuroscience owes a debt to British zoologist J. Z. Young, who in 1939 called attention to the squid giant axon as an experimental preparation for studying the biophysics of the neuronal membrane. Hodgkin and Huxley used this preparation to elucidate the ionic basis of the action potential, and the giant axon continues to be used today in a wide range of neurobiological studies.
It is interesting to note that axonal size and the number of voltage-gated channels in the membrane also affect axonal excitability. Smaller axons require greater depolarization to reach action potential threshold and are more sensitive to being blocked by local anesthetics (Box 4.4).
Although you’ve tried to tough it out, you just can’t take it anymore. You finally give in to the pain of the toothache and head for the dentist. Fortunately, the worst part of having a cavity filled is the pinprick in the gum caused by the injection needle. Then your mouth becomes numb and you daydream while the dentist drills and repairs your tooth. What was injected, and how did it work?
Local anesthetics are drugs that temporarily block action potentials in axons. They are called “local” because they are injected directly into the tissue where anesthesia—the absence of sensation—is desired. Small axons, firing a lot of action potentials, are most sensitive to conduction block by local anesthetics.
The first local anesthetic introduced into medical practice was cocaine. The chemical was isolated from the leaves of the coca plant in 1860 by the German physician Albert Niemann. According to the custom of the pharmacologists of his day, Niemann tasted the new compound and discovered that it caused his tongue to go numb. It was soon learned that cocaine also had toxic and addictive properties. (The mind-altering effect of cocaine was studied by another well-known physician of that era, Sigmund Freud. Cocaine alters mood by a mechanism distinct from its local anesthetic action, as we shall see in Chapter 15.)
The search for a suitable synthetic anesthetic as a substitute for cocaine led to the development of lidocaine, which is now the most widely used local anesthetic. Lidocaine can be dissolved into a jelly and smeared onto the mucous membranes of the mouth (and elsewhere) to numb the nerve endings (called topical anesthesia); it can be injected directly into a tissue (infiltration anesthesia) or a nerve (nerve block); it can even be infused into the cerebrospinal fluid bathing the spinal cord (spinal anesthesia), where it can numb large parts of the body.
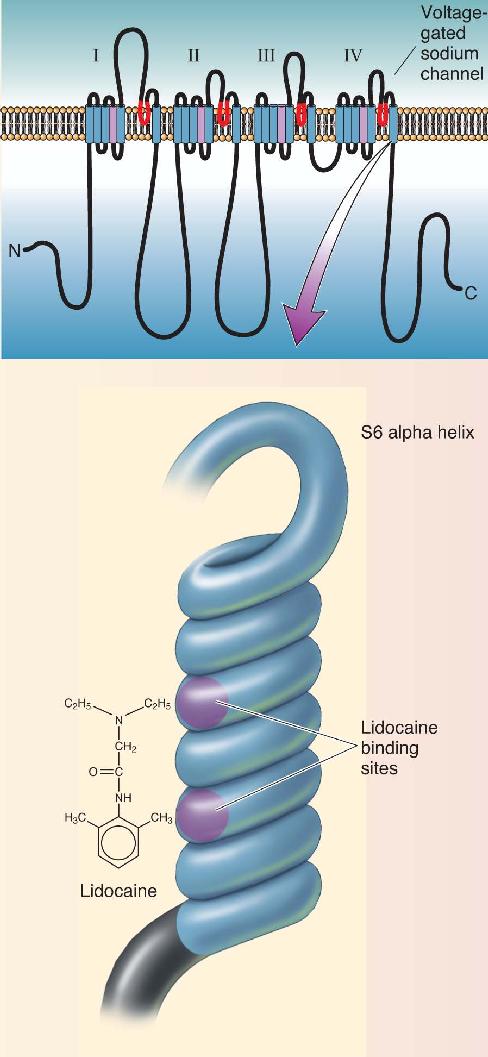
Lidocaine and other local anesthetics prevent action potentials by binding to the voltage-gated sodium channels. The binding site for lidocaine has been identified as the S6 alpha helix of domain IV of the protein (Figure A). Lidocaine cannot gain access to this site from the outside. The anesthetic first must cross the axonal membrane and then pass through the open gate of the channel to find its binding site inside the pore. This explains why active nerves are blocked faster (the sodium channel gates are open more often). The bound lidocaine interferes with the flow of Na+ that normally results from depolarizing the channel.

Figure A Lidocaine’s mechanism of action. (Source: Adapted from Hardman, et al., 1996, Fig. 15–3.) Description
Smaller axons are affected by local anesthetics before larger axons because their action potentials have less of a safety margin; more of the voltage-gated sodium channels must function to ensure that the action potential doesn’t fizzle out as it conducts down the axon. This increased sensitivity of small axons to local anesthetics is fortuitous in clinical practice. As we will discover in Chapter 12, it is the smaller fibers that convey information about painful stimuli—like a toothache.
The good thing about fat axons is that they conduct action potentials faster; the bad thing about them is that they take up a lot of space. If all the axons in your brain were the diameter of a squid giant axon, your head would be too big to fit through a barn door. Fortunately, vertebrates evolved another solution for increasing action potential conduction velocity: wrapping the axon with insulation called myelin (see Chapter 2). The myelin sheath consists of many layers of membrane provided by glial support cells—Schwann cells in the peripheral nervous system (outside the brain and spinal cord) and oligodendroglia in the central nervous system. Just as wrapping a leaky garden hose with duct tape facilitates water flow down the inside of the hose, myelin facilitates current flow down the inside of the axon, thereby increasing action potential conduction velocity (Box 4.5).
The critical importance of myelin for the normal transfer of information in the human nervous system is revealed by the neurological disorder known as multiple sclerosis (MS). Victims of MS often complain of weakness, lack of coordination, and impaired vision and speech. The disease is capricious, usually marked by remissions and relapses that occur over a period of many years. Although the precise cause of MS is still poorly understood, the cause of the sensory and motor disturbances is now quite clear. MS attacks the myelin sheaths of bundles of axons in the brain, spinal cord, and optic nerves. The word sclerosis is derived from the Greek word for “hardening,” which describes the lesions that develop around bundles of axons, and the sclerosis is multiple because the disease attacks many sites in the nervous system at the same time.
Lesions in the brain can now be viewed noninvasively using new methods such as magnetic resonance imaging (MRI). However, neurologists have been able to diagnose MS for many years by taking advantage of the fact that myelin serves the nervous system by increasing the velocity of axonal conduction. One simple test involves stimulating the eye with a checkerboard pattern and measuring the elapsed time until an electrical response is noted at the scalp over the part of the brain that is a target of the optic nerve. People who have MS characteristically have a marked slowing of the conduction velocity of their optic nerve.
Another demyelinating disease called Guillain–Barré syndrome attacks the myelin of the peripheral nerves that innervate muscle and skin. This disease may follow minor infectious illnesses and inoculations, and it appears to result from an anomalous immunological response against one’s own myelin. The symptoms stem directly from the slowing and/or failure of action potential conduction in the axons that innervate the muscles. This conduction deficit can be demonstrated clinically by stimulating peripheral nerves electrically through the skin and measuring the time it takes to evoke a response (a twitch of a muscle, for instance). Both MS and Guillain–Barré syndrome are characterized by a profound slowing of the response time because saltatory conduction is disrupted.
The myelin sheath does not extend continuously along the entire length of the axon. There are breaks in the insulation where ions cross the membrane to generate action potentials. Recall from Chapter 2 that these breaks in the myelin sheath are the nodes of Ranvier (Figure 4.14). Voltage-gated sodium channels are concentrated in the membrane of the nodes. The distance between nodes is usually 0.2–2.0 mm, depending on the size of the axon (fatter axons have larger internodal distances).

FIGURE 4.14 The myelin sheath and node of Ranvier. The electrical insulation provided by myelin helps speed action potential conduction from node to node. Voltage-gated sodium channels are concentrated in the axonal membrane at the nodes of Ranvier. Description
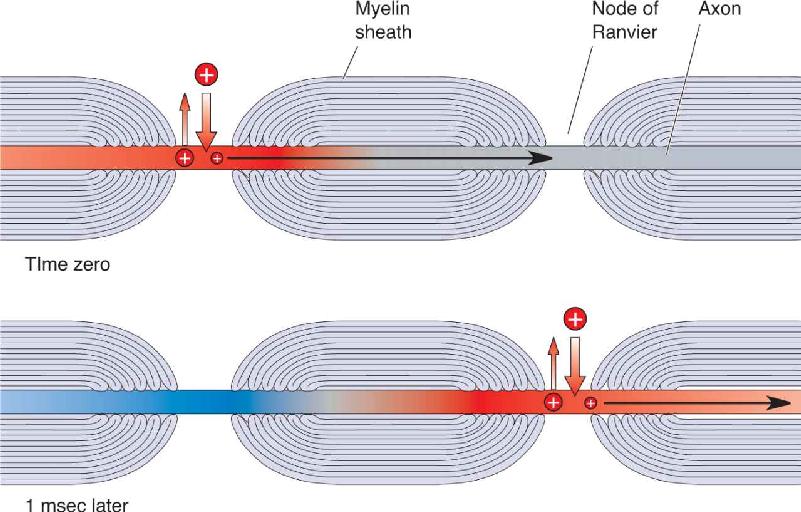
Imagine that the action potential traveling along the axon membrane is like you traveling down a sidewalk. Action potential conduction without myelin is like walking down the sidewalk in small steps, heel-to-toe, using every inch of the sidewalk to creep along. Conduction with myelin, in contrast, is like skipping down the sidewalk. In myelinated axons, action potentials skip from node to node (Figure 4.15). This type of action potential propagation is called saltatory conduction (from the Latin meaning “to leap”).

FIGURE 4.15 Saltatory conduction. Myelin allows current to spread farther and faster between nodes, thus speeding action potential conduction. Compare this figure with Figure 4.12. Description
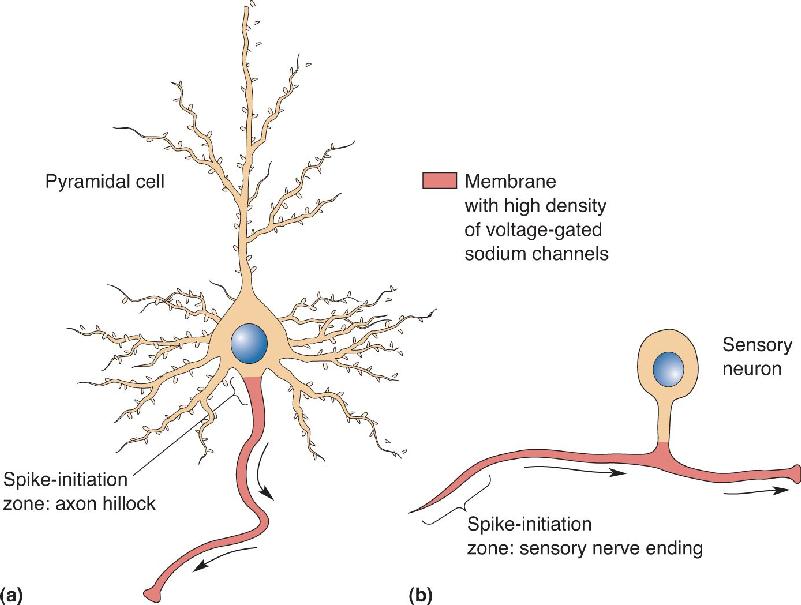
Action potentials of the type discussed in this chapter are a feature mainly of axons. As a rule, the membranes of dendrites and neuronal cell bodies do not generate sodium-dependent action potentials because they have very few voltage-gated sodium channels. Only membrane that contains these specialized protein molecules is capable of generating action potentials, and this type of excitable membrane is usually found only in axons. Therefore, the part of the neuron where an axon originates from the soma, the axon hillock, is often also called the spike-initiation zone. In a typical neuron in the brain or spinal cord, the depolarization of the dendrites and soma caused by synaptic input from other neurons leads to the generation of action potentials if the membrane of the axon hillock is depolarized beyond threshold (Figure 4.16a). In most sensory neurons, however, the spike-initiation zone occurs near the sensory nerve endings, where the depolarization caused by sensory stimulation leads to the generation of action potentials that propagate along the sensory nerves (Figure 4.16b).

FIGURE 4.16 The spike-initiation zone. Membrane proteins specify the function of different parts of the neuron. Depicted here are (a) a cortical pyramidal neuron and (b) a primary sensory neuron. Despite the diversity of neuronal structure, the axonal membrane can be identified at the molecular level by its high density of voltage-gated sodium channels. This molecular distinction enables axons to generate and conduct action potentials. The region of membrane where action potentials are normally generated is called the spike-initiation zone. The arrows indicate the normal direction of action potential propagation in these two types of neuron. Description
In Chapter 2, we learned that axons and dendrites differ in their morphology. We now see that they are functionally different, and that this difference in function is specified at the molecular level by the type of protein in the neuronal membrane. Differences in the types and density of membrane ion channels can also account for the characteristic electrical properties of different types of neurons (Box 4.6).
Neurons are not all alike; they vary in shape, size, gene expression, and connections. Neurons also differ from one another in their electrical properties. A few examples of the diverse behavior of neurons are shown in Figure A.

Figure A The diverse behavior of neurons. (Source: Adapted from Agmon and Connors, 1992.) Description
The cerebral cortex has two major types of neurons as defined by morphology: aspinous stellate cells and spiny pyramidal cells. A stellate cell typically responds to a steady depolarizing current injected into its soma by firing action potentials at a relatively steady frequency throughout the stimulus (part a). However, most pyramidal cells cannot sustain a steady firing rate. Instead, they fire rapidly at the beginning of the stimulus and then slow down, even if the stimulus remains strong (part b). This slowing over time is called adaptation, a very common property among excitable cells. Another firing pattern is the burst, a rapid cluster of action potentials followed by a brief pause. Some cells, including a particular subtype of large pyramidal neuron in the cortex, respond to a steady input with rhythmic, repetitive bursts (part c). Variability of firing patterns is not unique in the cerebral cortex. Surveys of many areas of the brain suggest that neurons have as large an assortment of electrical behaviors as morphologies.
What accounts for the diverse behavior of different types of neurons? Ultimately, each neuron’s physiology is determined by the properties and numbers of ion channels in its membrane. There are many more types of ion channels than the few described in this chapter, and each has distinctive properties. For example, some potassium channels activate only very slowly. A neuron with a high density of these will show adaptation because during a prolonged stimulus, more and more of the slow potassium channels will open, and the outward currents they progressively generate will tend to hyperpolarize the membrane. When you realize that a single neuron may have more than a dozen types of ion channels, the source of diverse firing behavior becomes clear. It is the complex interactions among multiple ion channels that create the eclectic electric signature of each class of neuron.
Let’s return briefly to the example in Chapter 3 of stepping on a thumbtack. The breaking of the skin caused by the tack stretches the sensory nerve endings of the foot. Special ion channels that are sensitive to the stretching of the membrane open and allow positively charged sodium ions to enter the ends of the axons in the skin. This influx of positive charge depolarizes the membrane to threshold, and the action potential is generated. The positive charge that enters during the rising phase of the action potential spreads along the axon and depolarizes the membrane ahead to threshold. In this way, the action potential is continuously regenerated as it sweeps like a wave along the sensory axon. We now come to the step where this information is distributed to and integrated by other neurons in the central nervous system. This transfer of information from one neuron to another is called synaptic transmission, the subject of the next two chapters.
It should come as no surprise that synaptic transmission, like the action potential, depends on specialized proteins in the neuronal membrane. Thus, a picture begins to emerge of the brain as a complicated mesh of interacting neuronal membranes. Consider that a typical neuron with all its neurites has a membrane surface area of about 250,000 μm2. The surface area of the 85 billion neurons that make up the human brain comes to 21,250 m2—roughly the size of three soccer fields. This expanse of membrane, with its myriad specialized protein molecules, constitutes the fabric of our minds.
1. Define membrane potential (Vm) and sodium equilibrium potential (ENa). Which of these, if either, changes during the course of an action potential?
2. What ions carry the early inward and late outward currents during the action potential?
3. Why is the action potential referred to as “all-or-none”?
4. Some voltage-gated K+ channels are known as delayed rectifiers because of the timing of their opening during an action potential. What would happen if these channels took much longer than normal to open?
5. Imagine we have labeled tetrodotoxin (TTX) so that it can be seen using a microscope. If we wash this TTX onto a neuron, what parts of the cell would you expect to be labeled? What would be the consequence of applying TTX to this neuron?
6. How does action potential conduction velocity vary with axonal diameter? Why?
Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. 2005. Millisecond-timescale, genetically targeted optical control of neural activity. Nature Neuroscience 8:1263–1268.
Hille B. 1992. Ionic Channels of Excitable Membranes, 2nd ed. Sunderland, MA: Sinauer.
Hodgkin A. 1976. Chance and design in electrophysiology: an informal account of certain experiments on nerves carried out between 1942 and 1952. Journal of Physiology (London) 263:1–21.
Kullmann DM, Waxman SG. 2010. Neurological channelopathies: new insights into disease mechanisms and ion channel function. Journal of Physiology (London) 588:1823–1827.
Neher E. 1992. Nobel lecture: ion channels or communication between and within cells. Neuron 8:605–612.
Neher E, Sakmann B. 1992. The patch clamp technique. Scientific American 266:28–35.
Nicholls J, Martin AR, Fuchs PA, Brown DA, Diamond ME, Weisblat D. 2011. From Neuron to Brain, 5th ed. Sunderland, MA: Sinauer.
Additional figures