Neurotransmitter Systems
Localization of Transmitters and Transmitter-Synthesizing Enzymes
BOX 6.1 PATH OF DISCOVERY: Finding Opiate Receptors, by Solomon H. Snyder
Other Neurotransmitter Candidates and Intercellular Messengers
BOX 6.3 OF SPECIAL INTEREST: This Is Your Brain on Endocannabinoids
BOX 6.4 OF SPECIAL INTEREST: Exciting Poisons: Too Much of a Good Thing
The Basic Structure of G-Protein-Coupled Receptors
DIVERGENCE AND CONVERGENCE IN NEUROTRANSMITTER SYSTEMS
Normal functions of the human brain require an orderly set of chemical reactions. As we have seen, some of the brain’s most important chemical reactions are those associated with synaptic transmission. Chapter 5 introduced the general principles of chemical synaptic transmission, using a few specific neurotransmitters as examples. In this chapter, we will explore in more depth the variety and elegance of the major neurotransmitter systems.
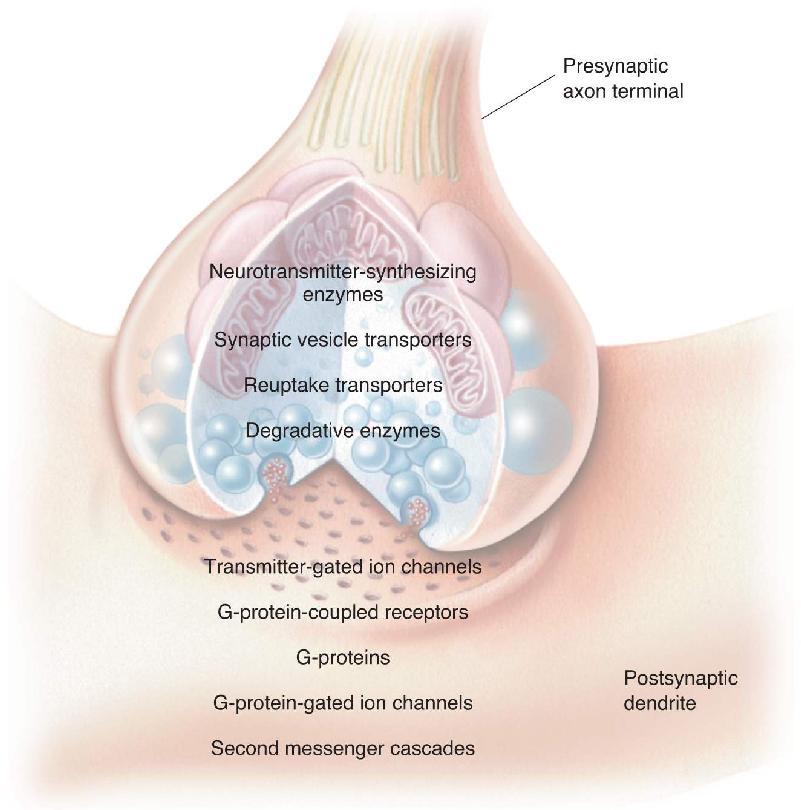
Neurotransmitter systems begin with neurotransmitters. In Chapter 5, we discussed the three major classes of neurotransmitters: amino acids, amines, and peptides. Even a partial list of the known transmitters, such as that appearing in Table 5.1, has more than 20 different molecules. Each of these molecules can define a particular transmitter system. In addition to the molecule itself, a neurotransmitter system includes all the molecular machinery responsible for transmitter synthesis, vesicular packaging, reuptake and degradation, and transmitter action (Figure 6.1).

FIGURE 6.1 Elements of neurotransmitter systems. Description
The first molecule positively identified as a neurotransmitter by Otto Loewi in the 1920s was acetylcholine, or ACh (see Box 5.1). To describe the cells that produce and release ACh, British pharmacologist Henry Dale introduced the term cholinergic. (Dale shared the 1936 Nobel Prize with Loewi in recognition of his neuropharmacological studies of synaptic transmission.) Dale termed the neurons that use the amine neurotransmitter norepinephrine (NE) noradrenergic. (NE is known as noradrenaline in United Kingdom.) The convention of using the suffix -ergic continued when additional transmitters were identified. Therefore, today we speak of glutamatergic synapses that use glutamate, GABAergic synapses that use GABA, peptidergic synapses that use peptides, and so on. These adjectives are also used to identify the various neurotransmitter systems. For example, ACh and all the molecular machinery associated with it are collectively called the cholinergic system.
With this terminology in hand, we can begin our exploration of the neurotransmitter systems. We start with a discussion of the experimental strategies that have been used to study transmitter systems. Then we will look at the synthesis and metabolism of specific neurotransmitters and explore how these molecules exert their postsynaptic effects. In Chapter 15, after we have learned more about the structural and functional organization of the nervous system, we’ll take another look at specific neurotransmitter systems in the context of their individual contributions to the regulation of brain function and behavior.
The first step in studying a neurotransmitter system is usually identifying the neurotransmitter. This is no simple task; the brain contains uncountable different chemicals. How can we decide which few chemicals are used as transmitters?
Over the years, neuroscientists have established certain criteria that must be met for a molecule to be considered a neurotransmitter:
- The molecule must be synthesized and stored in the presynaptic neuron.
- The molecule must be released by the presynaptic axon terminal upon stimulation.

- The molecule, when experimentally applied, must produce a response in the postsynaptic cell that mimics the response produced by the release of neurotransmitter from the presynaptic neuron.
Let’s start by exploring some of the strategies and methods that are used to satisfy these criteria.
Localization of Transmitters and Transmitter-Synthesizing Enzymes
The scientist often begins with little more than a hunch that a particular molecule may be a neurotransmitter. This idea may be based on observing that the molecule is concentrated in brain tissue or that the application of the molecule to certain neurons alters their action potential firing rate. Whatever the inspiration, the first step in confirming the hypothesis is to show that the molecule is, in fact, localized in, and synthesized by, particular neurons. Many methods have been used to satisfy this criterion for different neurotransmitters. Two of the most important techniques used today are immunocytochemistry and in situ hybridization.
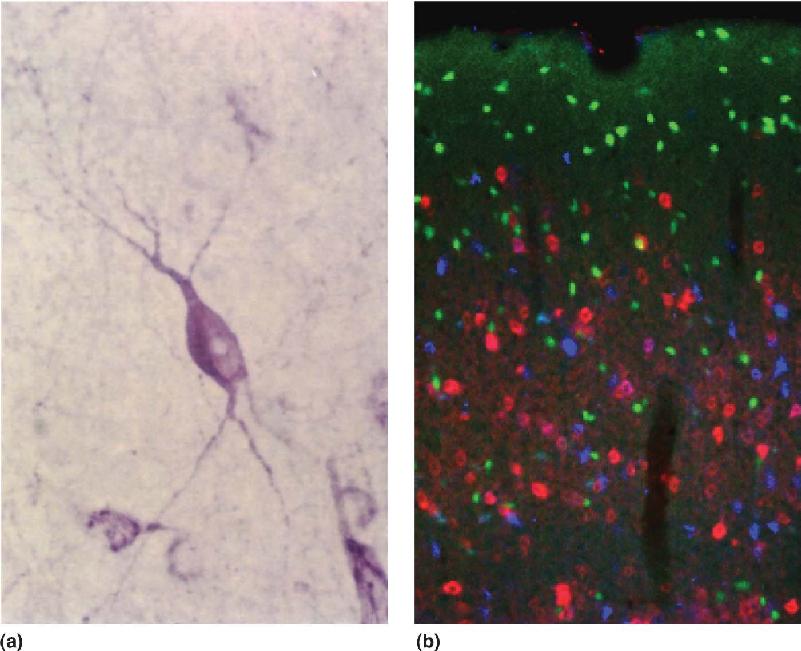
Immunocytochemistry. The method of immunocytochemistry is used to anatomically localize particular molecules to particular cells. When the same technique is applied to thin sections of tissue, including brain, it is often referred to as immunohistochemistry. The principle behind the method is quite simple (Figure 6.2). Once the neurotransmitter candidate has been chemically purified, it is injected under the skin or into the bloodstream of an animal where it stimulates an immune response. (Often, to evoke or enhance the immune response, the molecule is chemically coupled to a larger molecule.) One feature of the immune response is the generation of large proteins called antibodies. Antibodies can bind tightly to specific sites on the foreign molecule, also known as the antigen—in this case, the transmitter candidate. The best antibodies for immunocytochemistry bind very tightly to the transmitter of interest and bind very little or not at all to other chemicals in the brain. These specific antibody molecules can be recovered from a blood sample of the immunized animal and chemically tagged with a colorful marker that can be seen with a microscope. When these labeled antibodies are applied to a section of brain tissue, they will color just those cells that contain the transmitter candidate (Figure 6.3a). By using several different antibodies, each labeled with a different marker color, it is possible to distinguish several types of cells in the same region of the brain (Figure 6.3b).

FIGURE 6.2 Immunohistochemistry. This method uses labeled antibodies to identify the location of molecules within cells. (a) The molecule of interest (a neurotransmitter candidate) is injected into an animal, causing an immune response and the generation of antibodies. (b) Blood is withdrawn from the animal, and the antibodies are isolated from the serum. (c) The antibodies are tagged with a visible marker and applied to sections of brain tissue. The antibodies label only those cells that contain the neurotransmitter candidate. (d) A close-up of the complex that includes the neurotransmitter candidates, an antibody, and its visible marker. Description

FIGURE 6.3 Immunohistochemical localization of proteins in neurons. (a) A neuron in the cerebral cortex labeled with antibodies that bind to a peptide neurotransmitter. (Source: Courtesy of Dr. Y. Amitai and S. L. Patrick.) (b) Three distinct types of neurons in the cerebral cortex, each labeled with a different antibody tagged with a differently colored fluorescent marker (green, red, and blue). (Source: Courtesy of Dr. S.J. Cruikshank and S.L. Patrick.) The image in a is shown at a higher magnification than that in b.
Immunocytochemistry can be used to localize any molecule for which a specific antibody can be generated, including the synthesizing enzymes for transmitter candidates. Demonstration that the transmitter candidate and its synthesizing enzyme are contained in the same neuron—or better yet, in the same axon terminal—can help satisfy the criterion that the molecule be localized in, and synthesized by, a particular neuron.
In Situ Hybridization. The method known as in situ hybridization is also useful for confirming that a cell synthesizes a particular protein or peptide. Recall from Chapter 2 that proteins are assembled by the ribosomes according to instructions from specific mRNA molecules. There is a unique mRNA molecule for every polypeptide synthesized by a neuron. The mRNA transcript consists of the four different nucleic acids linked together in various sequences to form a long strand. Each nucleic acid has the unusual property that it will bind most tightly to one other complementary nucleic acid. Thus, if the sequence of nucleic acids in a strand of mRNA is known, it is possible to construct in the lab a complementary strand that will stick, like a strip of Velcro, to the mRNA molecule. The complementary strand is called a probe, and the process by which the probe bonds to the mRNA molecule is called hybridization (Figure 6.4). In order to see if the mRNA for a particular peptide is localized in a neuron, we chemically label the appropriate probe so it can be detected, apply it to a section of brain tissue, allow time for the probes to stick to any complementary mRNA strands, then wash away all the extra probes that have not stuck. Finally, we search for neurons that contain the label.

FIGURE 6.4 In situ hybridization. Strands of mRNA consist of nucleotides arranged in a specific sequence. Each nucleotide will stick to one other complementary nucleotide. In the method of in situ hybridization, a synthetic probe is constructed containing a sequence of complementary nucleotides that will allow it to stick to the mRNA. If the probe is labeled, the location of cells containing the mRNA will be revealed.
To visualize labeled cells after in situ hybridization, the probes can be chemically tagged in several ways. A common approach is to make them radioactive. Because we cannot see radioactivity, hybridized probes are detected by laying the brain tissue on a sheet of special film that is sensitive to radioactive emissions. After exposure to the tissue, the film is developed like a photograph, and negative images of the radioactive cells are visible as clusters of small white dots (Figure 6.5). It is also possible to use digital electronic imaging devices to detect the radioactivity. This technique for viewing the distribution of radioactivity is called autoradiography. An alternative is to label the probes with brightly colorful fluorescent molecules that can viewed directly with an appropriate microscope. Fluorescence in situ hybridization is also known as FISH.

FIGURE 6.5 In situ hybridization of the mRNA for a peptide neurotransmitter in neurons, visualized with autoradiography. Only neurons with the proper mRNA are labeled, visible here as clusters of white dots. (Source: Courtesy of Dr. S. H. C. Hendry.)
In summary, immunocytochemistry is a method for viewing the location of specific molecules, including proteins, in sections of brain tissue. In situ hybridization is a method for localizing specific mRNA transcripts for proteins. Together, these methods enable us to see whether a neuron contains and synthesizes a transmitter candidate and molecules associated with that transmitter.
Once we are satisfied that a transmitter candidate is synthesized by a neuron and localized to the presynaptic terminal, we must show that it is actually released upon stimulation. In some cases, a specific set of cells or axons can be stimulated while taking samples of the fluids bathing their synaptic targets. The biological activity of the sample can then be tested to see if it mimics the effect of the intact synapses, and then the sample can be chemically analyzed to reveal the structure of the active molecule. This general approach helped Loewi and Dale identify ACh as a transmitter at many peripheral synapses.
Unlike the peripheral nervous system (PNS), the nervous system outside the brain and spinal cord studied by Loewi and Dale, most regions of the central nervous system (CNS) contain a diverse mixture of intermingled synapses using different neurotransmitters. Until recently, this often made it impossible to stimulate a single population of synapses containing only a single neurotransmitter. Researchers had to be content with stimulating many synapses in a region of the brain and collecting and measuring all the chemicals that were released. One way to do this is to use brain slices that are kept alive in vitro. To stimulate release, the slices are bathed in a solution containing a high K+ concentration. This treatment causes a large membrane depolarization (see Figure 3.19), thereby stimulating transmitter release from the axon terminals in the tissue. Because transmitter release requires the entry of Ca2+ into the axon terminal, it must also be shown that the release of the neurotransmitter candidate from the tissue slice after depolarization occurs only when Ca2+ ions are present in the bathing solution. New methods such as optogenetics (see Box 4.2) now make it possible to activate just one specific type of synapse at a time. Genetic methods are used to induce one particular population of neurons to express light-sensitive proteins, and then those neurons can be stimulated with brief flashes of light that have no effect on the surrounding cells. Any transmitters released are likely to have come from the optogenetically selected type of synapse.
Even when it has been shown that a transmitter candidate is released upon depolarization in a calcium-dependent manner, we still cannot be sure that the molecules collected in the fluids were released from the axon terminals; they may have been released as a secondary consequence of synaptic activation. These technical difficulties make the second criterion—that a transmitter candidate must be released by the presynaptic axon terminal upon stimulation—the most difficult to satisfy unequivocally in the CNS.
Establishing that a molecule is localized in, synthesized by, and released from a neuron is still not sufficient to qualify it as a neurotransmitter. A third criterion must be met: The molecule must evoke the same response as that produced by the release of the naturally occurring neurotransmitter from the presynaptic neuron.
To assess the postsynaptic actions of a transmitter candidate, a method called microiontophoresis is sometimes used. Most neurotransmitter candidates can be dissolved in solutions that will cause them to acquire a net electrical charge. A glass pipette with a very fine tip, just a few micrometers across, is filled with the ionized solution. The tip of the pipette is carefully positioned next to the postsynaptic membrane of the neuron, and the transmitter candidate is ejected in very small amounts by passing electrical current through the pipette. Neurotransmitter candidates can also be ejected from fine pipettes with pulses of high pressure. A microelectrode in the postsynaptic neuron can be used to measure the effects of the transmitter candidate on the membrane potential (Figure 6.6).

FIGURE 6.6 Microiontophoresis. This method enables a researcher to apply drugs or neurotransmitter candidates in very small amounts to the surface of neurons. The responses generated by the drug can be compared to those generated by synaptic stimulation.
If iontophoretic or pressure application of the molecule causes electrophysiological changes that mimic the effects of transmitter released at the synapse, and if the other criteria of localization, synthesis, and release are met, then the molecule and the transmitter are usually considered to be the same chemical.
Each neurotransmitter exerts its postsynaptic effects by binding to specific receptors. As a rule, no two neurotransmitters bind to the same receptor; however, one neurotransmitter can bind to many different receptors. Each of the different receptors a neurotransmitter binds to is called a receptor subtype. For example, in Chapter 5, we learned that ACh acts on two different cholinergic receptor subtypes: One type is present in skeletal muscle, and the other is in heart muscle. Both subtypes are also present in many other organs and within the CNS.
Researchers have tried almost every method of biological and chemical analysis to study the different receptor subtypes of the various neurotransmitter systems. Three approaches have proved to be particularly useful: neuropharmacological analysis of synaptic transmission, ligand-binding methods, and molecular analysis of receptor proteins.
Neuropharmacological Analysis. Much of what we know about receptor subtypes was first learned using neuropharmacological analysis. For instance, skeletal muscle and heart muscle respond differently to various cholinergic drugs. Nicotine, derived from the tobacco plant, is a receptor agonist in skeletal muscle but has no effect in the heart. On the other hand, muscarine, derived from a poisonous species of mushroom, has little or no effect on skeletal muscle but is an agonist at the cholinergic receptor subtype in the heart. (Recall that ACh slows the heart rate; muscarine is poisonous because it causes a precipitous drop in heart rate and blood pressure.) Thus, two ACh receptor subtypes can be distinguished by the actions of different drugs. In fact, the receptors were given the names of their agonists: nicotinic ACh receptors in skeletal muscle and muscarinic ACh receptors in the heart. Nicotinic and muscarinic receptors also exist in the brain, and some neurons have both types of receptors.
Another way to distinguish receptor subtypes is to use selective antagonists. The South American arrow-tip poison curare inhibits the action of ACh at nicotinic receptors (thereby causing paralysis), and atropine, derived from belladonna plants (also known as deadly nightshade), antagonizes ACh at muscarinic receptors (Figure 6.7). (The eye drops an ophthalmologist uses to dilate your pupils are related to atropine.)

FIGURE 6.7 The neuropharmacology of cholinergic synaptic transmission. Sites on transmitter receptors can bind either the transmitter itself (ACh), an agonist that mimics the transmitter, or an antagonist that blocks the effects of the transmitter and agonists.
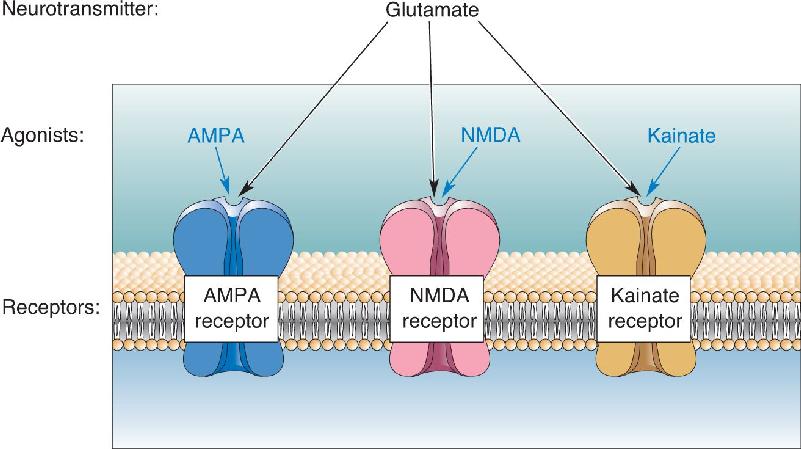
Different drugs were also used to distinguish several subtypes of glutamate receptors, which mediate much of the synaptic excitation in the CNS. Three subtypes are AMPA receptors, NMDA receptors, and kainate receptors, each named for a different chemical agonist. (AMPA stands for α-amino-3-hydroxy-5-methyl-4-isoxazole propionate, and NMDA stands for N-methyl-D-aspartate.) The neurotransmitter glutamate activates all three receptor subtypes, but AMPA acts only at the AMPA receptor, NMDA acts only at the NMDA receptor, and so on (Figure 6.8).

FIGURE 6.8 The neuropharmacology of glutamatergic synaptic transmission. There are three main subtypes of glutamate receptors, each of which binds glutamate and each of which is activated selectively by a different agonist.
Similar pharmacological analyses were used to split the NE receptors into two subtypes, α and β, and to divide GABA receptors into GABAA and GABAB subtypes. The same can be said for virtually all the neurotransmitter systems. Thus, selective drugs have been extremely useful for categorizing receptor subclasses (Table 6.1). In addition, neuropharmacological analysis has been invaluable for assessing the contributions of neurotransmitter systems to brain function.
Neurotransmitters, Some Receptors, and Their Pharmacology

Ligand-Binding Methods. As we said, the first step in studying a neurotransmitter system is usually identifying the neurotransmitter. However, with the discovery in the 1970s that many drugs interact selectively with neurotransmitter receptors, researchers realized that they could use these compounds to begin analyzing receptors even before the neurotransmitter itself had been identified. The pioneers of this approach were Solomon Snyder and his then student Candace Pert at Johns Hopkins University, who were interested in studying compounds called opiates (Box 6.1). Opiates are a class of drugs, derived from the opium poppy, that are both medically important and commonly abused. Opioids are the broader class of opiate-like chemicals, both natural and synthetic. Their effects include pain relief, euphoria, depressed breathing, and constipation.
Like so many events in science, identifying the opiate receptors was not simply an intellectual feat accomplished in an ethereal pursuit of pure knowledge. Instead, it all began with President Nixon and his “war on drugs” in 1971, at the height of very well-publicized use of heroin by hundreds of thousands of American soldiers in Vietnam. To combat all this, Nixon appointed as czar of drug abuse research Dr. Jerome Jaffe, a psychiatrist who had pioneered in methadone treatment for heroin addicts. Jaffe was to coordinate the several billions of federal dollars in agencies ranging from the Department of Defense to the National Institutes of Health.
Jerry, a good friend, pestered me to direct our research toward the “poor soldiers” in Vietnam. So I began wondering how opiates act. The notion that drugs act at receptors, specific recognition sites, had been appreciated since the turn of the century. In principle, one could identify such receptors simply by measuring the binding of radioactive drugs to tissue membranes. However, countless researchers had applied this strategy to opiates with no success.
About this time, a new Johns Hopkins faculty member, Pedro Cuatrecasas, located his laboratory adjacent to mine, and we became fast friends. Pedro had recently attained fame for his discovery of receptors for insulin. His success depended upon seemingly simple but important technical advances. Past efforts to identify receptors for hormones had failed because hormones can bind to many nonspecific sites, comprising proteins, carbohydrates, and lipids. The numbers of these nonspecific sites would likely be millions of times greater than the number of specific receptors. To identify the “signal” of insulin receptors binding above the “noise” of nonspecific interactions, Pedro developed a simple filtration assay. Since insulin should adhere more tightly to its receptors than to nonspecific sites, he incubated liver membranes with radioactive insulin and poured the mixture over filters attached to a vacuum that rapidly sucked away the incubation fluid, leaving the membrane with attached insulin stuck to the filters. He then “washed” the filters with large volumes of saline, but did this very rapidly so as to preserve insulin bound to receptors while washing away nonspecific binding.
Despite Pedro’s proximity, it did not immediately occur to me that the insulin success could be transferred to the opiate receptor problem. Instead, I had read a paper on nerve growth factor, showing that its amino acid sequence closely resembled that of insulin. Pedro and I soon collaborated in a successful search for the nerve growth factor receptor. Only then did I marshal the courage to extend this approach from proteins such as insulin and nerve growth factor to much smaller molecules such as opiates. Candace Pert, a graduate student in my laboratory, was eager to take on a new research project. We obtained a radioactive drug and monitored its binding to brain membranes using Pedro’s magic filter machine. The very first experiment, which took only about two hours, was successful.
Within a few months, we were able to characterize many features of opiate receptors. Knowing the exact sites where receptors are concentrated in the brain explained all the major actions of opiates, such as euphoria, pain relief, depression of breathing, and pupillary constriction. The properties of opiate receptors resembled very much what one would expect for neurotransmitters. Accordingly, we used similar approaches to search for receptors for neurotransmitters in the brain, and within a few years had identified receptors for most of them.
These findings raised an obvious question: Why do opiate receptors exist? Humans were not born with morphine in them. Might the opiate receptor be a receptor for a new transmitter that regulates pain perception and emotional states? We and other groups attempted to isolate the hypothesized, normally occurring, morphine-like neurotransmitters. John Hughes and Hans Kosterlitz in Aberdeen, Scotland, were the first to succeed. They isolated and obtained the chemical structures of the first “endorphins,” which are called the enkephalins. In our own laboratory, Rabi Simantov and I obtained the structure of the enkephalins soon after the published success of the Scottish group.
From the first experiments identifying opiate receptors until the isolation of the enkephalins, only three years elapsed—an interval of frantic, exhilarating work that changed profoundly how we think about drugs and the brain.
The question Snyder and Pert originally set out to answer was how heroin, morphine, and other opiates exert their effects on the brain. They and others hypothesized that opiates might be agonists at specific receptors in neuronal membranes. To test this idea, they radioactively labeled opiate compounds and applied them in small quantities to neuronal membranes that had been isolated from different parts of the brain. If appropriate receptors existed in the membrane, the labeled opiates should bind tightly to them. This is just what they found. The radioactive drugs labeled specific sites on the membranes of some, but not all, neurons in the brain (Figure 6.9). Following the discovery of opioid receptors, the search was on to identify endogenous opioids, or endorphins, the naturally occurring neurotransmitters that act on these receptors. Two peptides called enkephalins were soon isolated from the brain, and they eventually proved to be opioid neurotransmitters.

FIGURE 6.9 Opiate receptor binding to a slice of rat brain. Special film was exposed to a brain section that had radioactive opiate receptor ligands bound to it. The dark regions contain more receptors. (Source: Snyder, 1986, p. 44.)
Any chemical compound that binds to a specific site on a receptor is called a ligand for that receptor (from the Latin meaning “to bind”). The technique of studying receptors using radioactively or nonradioactively labeled ligands is called the ligand-binding method. Notice that a ligand for a receptor can be an agonist, an antagonist, or the chemical neurotransmitter itself. Specific ligands were invaluable for isolating neurotransmitter receptors and determining their chemical structure. Ligand-binding methods have been enormously important for mapping the anatomical distribution of different neurotransmitter receptors in the brain.
Molecular Analysis. There has been an explosion of information about neurotransmitter receptors in recent decades, thanks to modern methods for studying protein molecules. Information obtained with these methods has enabled us to divide the neurotransmitter receptor proteins into two groups: transmitter-gated ion channels and G-protein-coupled (metabotropic) receptors (see Chapter 5).
Molecular neurobiologists have determined the structure of the polypeptides that make up many proteins, and these studies have led to some startling conclusions. Receptor subtype diversity was expected from the actions of different drugs, but the breadth of the diversity was not appreciated until researchers determined how many different polypeptides could serve as subunits of functional receptors.
Consider as an example the GABAA receptor, a transmitter-gated chloride channel. Each channel requires five subunits (similar to the ACh-gated ion channel, Figure 5.14), and there are five major classes of subunit proteins, designated α, β, γ, δ, and ρ. At least six different polypeptides (designated α1–6) can substitute for one another as an α subunit. Four different polypeptides (designated β1–4) can substitute as a β subunit, and four different polypeptides (γ1–4) can be used as a γ subunit. Although this is not the complete tally, let’s make an interesting calculation. If it takes five subunits to form a GABAA receptor-gated channel and there are 15 possible subunits to choose from, then there are 151,887 possible combinations and arrangements of subunits. This means there are at least 151,887 potential subtypes of GABAA receptors!
It is important to recognize that the vast majority of the possible subunit combinations are never manufactured by neurons, and even if they were, they would not work properly. Nonetheless, it is clear that receptor classifications like those appearing in Table 6.1, while still useful, seriously underestimate the diversity of receptor subtypes in the brain.
Research using methods such as those discussed previously has led to the conclusion that the major neurotransmitters are amino acids, amines, and peptides. Evolution is conservative and opportunistic, and it often puts common and familiar things to new uses. This also seems true about the evolution of neurotransmitters. For the most part, they are similar or identical to the basic chemicals of life, the same substances that cells in all species, from bacteria to giraffes, use for metabolism. Amino acids, the building blocks of protein, are essential to life. Most of the known neurotransmitter molecules are either (1) amino acids, (2) amines derived from amino acids, or (3) peptides constructed from amino acids. ACh is one exception, but it is derived from acetyl CoA, a ubiquitous product of cellular respiration in mitochondria, and choline, which is important for fat metabolism throughout the body.
Amino acid and amine transmitters are generally each stored in and released by different sets of neurons. The convention established by Dale classifies neurons into mutually exclusive groups by neurotransmitter (cholinergic, glutamatergic, GABAergic, and so on). The idea that a neuron has only one neurotransmitter is often called Dale’s principle. Many peptide-containing neurons violate Dale’s principle because these cells usually release more than one neurotransmitter: an amino acid or amine and a peptide. When two or more transmitters are released from one nerve terminal, they are called co-transmitters. Many examples of neurons with co-transmitters have been identified in recent years, including some that release two small transmitters (e.g., GABA and glycine). Nonetheless, most neurons seem to release only a single amino acid or amine neurotransmitter, which can be used to assign them to distinct, nonoverlapping classes. Let’s take a look at the biochemical mechanisms that differentiate these neurons.
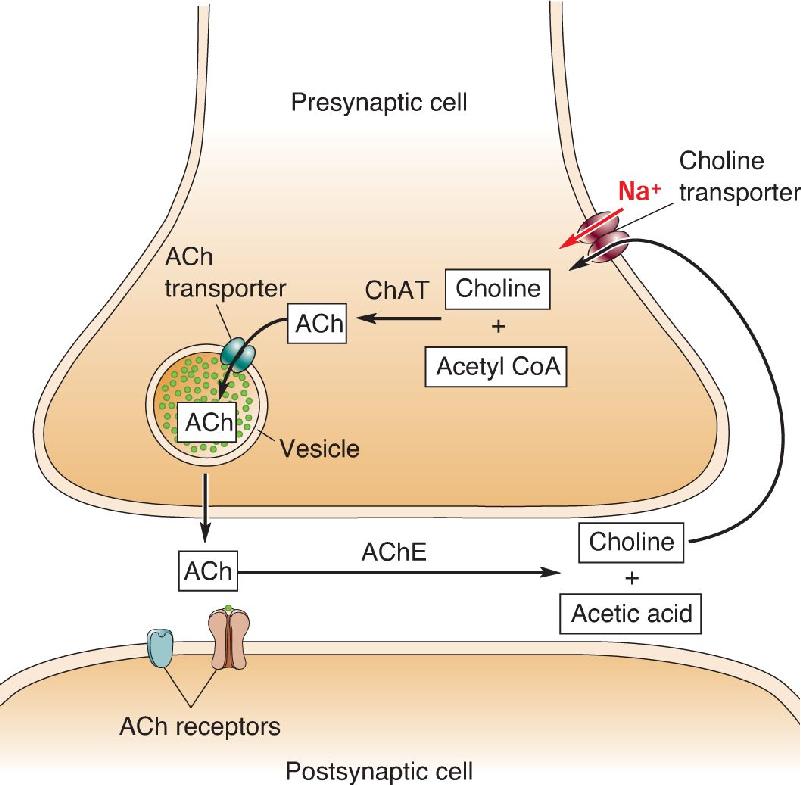
Acetylcholine (ACh) is the neurotransmitter at the neuromuscular junction and is therefore synthesized by all the motor neurons in the spinal cord and brain stem. Other cholinergic cells contribute to the functions of specific circuits in the PNS and CNS, as we will see in Chapter 15.
ACh synthesis requires a specific enzyme, choline acetyltransferase (ChAT) (Figure 6.10). Like nearly all presynaptic proteins, ChAT is manufactured in the soma and transported to the axon terminal. Only cholinergic neurons contain ChAT, so this enzyme is a good marker for cells that use ACh as a neurotransmitter. Immunohistochemistry with ChAT-specific antibodies, for example, can be used to identify cholinergic neurons. ChAT synthesizes ACh in the cytosol of the axon terminal, and the neurotransmitter is concentrated in synaptic vesicles by the actions of a vesicular ACh transporter (Box 6.2).

Neurotransmitters may lead an exciting life, but the most mundane part would seem to be the steps that recycle them back from the synaptic cleft and eventually into a vesicle. Where synapses are concerned, the exotic proteins of exocytosis and the innumerable transmitter receptors get most of the publicity. Yet, the neurotransmitter transporters are very interesting for at least two reasons: They succeed at an extraordinarily difficult job, and they are the molecular site at which many important psychoactive drugs act.
The hard job of transporters is to pump transmitter molecules across membranes so effectively that they become highly concentrated in very small places. There are two general types of neurotransmitter transporters. One type, the neuronal membrane transporter, shuttles transmitter from the extracellular fluid, including the synaptic cleft, and concentrates it up to 10,000 times higher within the cytosol of the presynaptic terminal. A second type, the vesicular transporter, then crams transmitter into vesicles at concentrations that may be 100,000 times higher than in the cytosol. Inside cholinergic vesicles, for example, ACh may reach the incredible concentration of 1000 mM, or 1 molar—in other words, about twice the concentration of salt in seawater!
How do transporters achieve such dramatic feats of concentration? Concentrating a chemical is like carrying a weight uphill; both are extremely unlikely to occur unless energy is applied to the task. Recall from Chapter 3 that ion pumps in the plasma membrane use ATP as their source of energy to transport Na+, K+, and Ca2+ against their concentration gradients. These ion gradients are essential for setting the resting potential and for powering the ionic currents that underlie action and synaptic potentials. Similarly, membranes of synaptic vesicles have pumps that use ATP to fuel the transport of H+ into vesicles. Notice that once ionic gradients are established across a membrane, they can themselves be tapped as sources of energy. Just as the energy spent in pulling up the weights on a cuckoo clock can be reclaimed to turn the gears and hands of the clock (as the weights slowly fall down again), transporters use transmembrane gradients of Na+ or H+ as an energy source for moving transmitter molecules up steep concentration gradients. The transporter lets one transmembrane gradient, that of Na+ or H+, run down a bit in order to build up another gradient, that of the transmitter.
The transporters themselves are large proteins that span membranes. There can be several transporters for one transmitter (e.g., at least four subtypes are known for GABA). Figure A shows how they work. Plasma membrane transporters use a cotransport mechanism, carrying two Na+ ions along with one transmitter molecule. By contrast, vesicular membrane transporters use a countertransport mechanism that trades a transmitter molecule from the cytosol for a H+ from inside the vesicle. Vesicle membranes have ATP-driven H+ pumps that keep their contents very acidic, or high in protons (i.e., H+ ions).
Figure A Neurotransmitter transporters. Description
What is the relevance of all this to drugs and disease? Many psychoactive drugs, such as amphetamines and cocaine, potently block certain transporters. By altering the normal recycling process of various transmitters, the drugs lead to chemical imbalances in the brain that can have profound effects on mood and behavior. It is also possible that defects in transporters can lead to psychiatric or neurological disease; certainly, some of the drugs that are therapeutically useful in psychiatry work by blocking transporters. The numerous links between transmitters, drugs, disease, and treatment are tantalizing but complex, and will be discussed further in Chapters 15 and 22.
ChAT transfers an acetyl group from acetyl CoA to choline (Figure 6.11a). The source of choline is the extracellular fluid, where it exists in low micromolar concentrations. Choline is taken up by the cholinergic axon terminals via a specific transporter that requires the cotransport of Na+ to power the movement of choline (see Box 6.2). Because the availability of choline limits how much ACh can be synthesized in the axon terminal, the transport of choline into the neuron is said to be the rate-limiting step in ACh synthesis. For certain diseases in which a deficit in cholinergic synaptic transmission has been noted, dietary supplements of choline are sometimes prescribed to boost ACh levels in the brain.

FIGURE 6.11 Acetylcholine. (a) ACh synthesis. (b) ACh degradation. Description
Cholinergic neurons also manufacture the ACh degradative enzyme acetylcholinesterase (AChE). AChE is secreted into the synaptic cleft and is associated with cholinergic axon terminal membranes. However, AChE is also manufactured by some noncholinergic neurons, so this enzyme is not as useful a marker for cholinergic synapses as ChAT.
AChE degrades ACh into choline and acetic acid (Figure 6.11b). This happens very quickly because AChE has one of the fastest catalytic rates among all known enzymes. Much of the resulting choline is taken up by the cholinergic axon terminal via a choline transporter and reused for ACh synthesis (see the red arrow in Figure 6.10). In Chapter 5, we mentioned that AChE is the target of many nerve gases and insecticides. Inhibition of AChE prevents the breakdown of ACh, disrupting transmission at cholinergic synapses on skeletal muscle and heart muscle. Acute effects include marked decreases in heart rate and blood pressure; however, death from the irreversible inhibition of AChE typically results from respiratory paralysis.
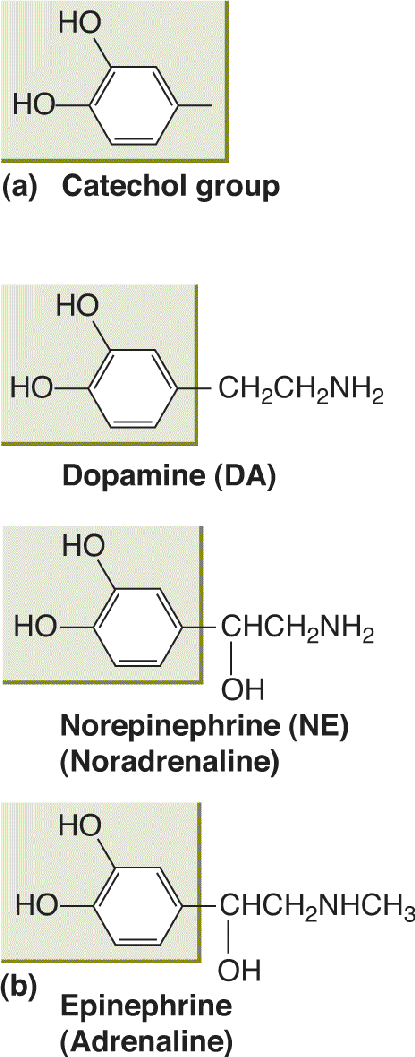
The amino acid tyrosine is the precursor for three different amine neurotransmitters that contain a chemical structure called a catechol (Figure 6.12a). These neurotransmitters are collectively called catecholamines. The catecholamine neurotransmitters are dopamine (DA), norepinephrine (NE), and epinephrine, also called adrenaline (Figure 6.12b). Catecholaminergic neurons are found in regions of the nervous system involved in the regulation of movement, mood, attention, and visceral function (discussed further in Chapter 15).

FIGURE 6.12 The catecholamines. (a) A catechol group. (b) The catecholamine neurotransmitters. Description
All catecholaminergic neurons contain the enzyme tyrosine hydroxylase (TH), which catalyzes the first step in catecholamine synthesis, the conversion of tyrosine to a compound called dopa (L-dihydroxyphenylalanine) (Figure 6.13a). The activity of TH is rate limiting for catecholamine synthesis. The enzyme’s activity is regulated by various signals in the cytosol of the axon terminal. For example, decreased catecholamine release by the axon terminal causes the catecholamine concentration in the cytosol to rise, thereby inhibiting TH. This type of regulation is called end-product inhibition. On the other hand, during periods when catecholamines are released at a high rate, the elevation in [Ca2+]i that accompanies neurotransmitter release triggers an increase in the activity of TH, so transmitter supply keeps up with demand. In addition, prolonged periods of stimulation actually cause the synthesis of more mRNA that codes for the enzyme.

FIGURE 6.13 The synthesis of catecholamines from tyrosine. The catecholamine neurotransmitters are in boldface type. Description
Dopa is converted into the neurotransmitter DA by the enzyme dopa decarboxylase (Figure 6.13b). Dopa decarboxylase is abundant in catecholaminergic neurons, so the amount of DA synthesized depends primarily on the amount of dopa available. In the movement disorder known as Parkinson’s disease, dopaminergic neurons in the brain slowly degenerate and eventually die. One strategy for treating Parkinson’s disease is the administration of dopa, which causes an increase in DA synthesis in the surviving neurons, increasing the amount of DA available for release. (We will learn more about dopamine and movement in Chapter 14.)
Neurons that use NE as a neurotransmitter contain, in addition to TH and dopa decarboxylase, the enzyme dopamine β-hydroxylase (DBH), which converts DA to NE (Figure 6.13c). Interestingly, DBH is not found in the cytosol but instead is located within the synaptic vesicles. Thus, in noradrenergic axon terminals, DA is transported from the cytosol to the synaptic vesicles, and there it is made into NE.
The last in the line of catecholamine neurotransmitters is epinephrine (adrenaline). Adrenergic neurons contain the enzyme phentolamine N-methyltransferase (PNMT), which converts NE to epinephrine (Figure 6.13d). Curiously, PNMT is in the cytosol of adrenergic axon terminals. Thus, NE must first be synthesized in the vesicles and released into the cytosol for conversion into epinephrine, and then the epinephrine must again be transported into vesicles for release. In addition to serving as a neurotransmitter in the brain, epinephrine acts as a hormone when it is released by the adrenal gland into the bloodstream. As we shall see in Chapter 15, circulating epinephrine acts at receptors throughout the body to produce a coordinated visceral response.
The catecholamine systems have no fast extracellular degradative enzyme analogous to AChE. Instead, the actions of catecholamines in the synaptic cleft are terminated by selective uptake of the neurotransmitters back into the axon terminal via Na+-dependent transporters. This step is sensitive to a number of different drugs. For example, amphetamine and cocaine block catecholamine uptake and therefore prolong the actions of the neurotransmitter in the cleft. Once inside the axon terminal, the catecholamines may be reloaded into synaptic vesicles for reuse, or they may be enzymatically destroyed by the action of monoamine oxidase (MAO), an enzyme found on the outer membrane of mitochondria.
The amine neurotransmitter serotonin, also called 5-hydroxytryptamine and abbreviated 5-HT, is derived from the amino acid tryptophan. Serotonergic neurons are relatively few in number, but, as we shall see in Part III, they appear to play an important role in the brain systems that regulate mood, emotional behavior, and sleep.
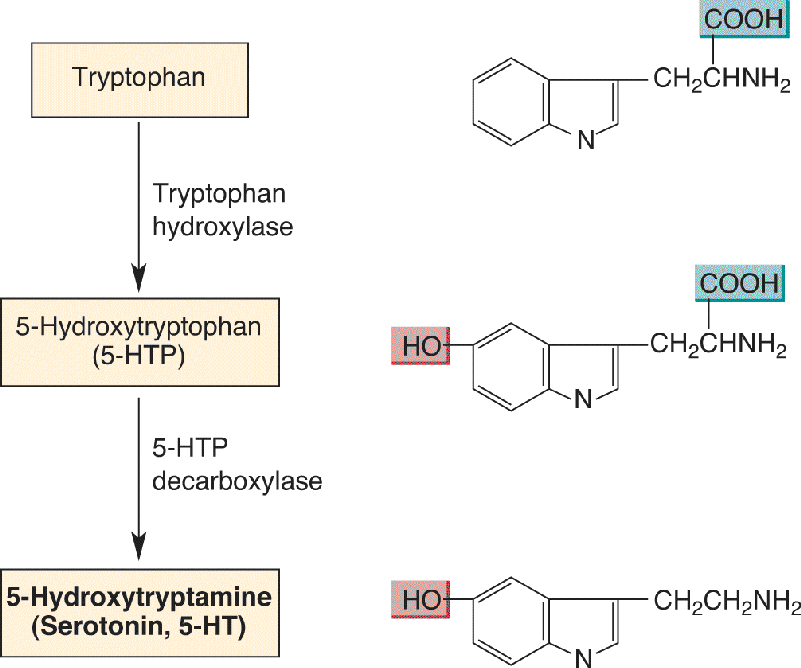
The synthesis of serotonin occurs in two steps, just like the synthesis of DA (Figure 6.14). Tryptophan is converted first into an intermediary called 5-hydroxytryptophan (5-HTP) by the enzyme tryptophan hydroxylase. The 5-HTP is then converted to 5-HT by the enzyme 5-HTP decarboxylase. Serotonin synthesis appears to be limited by the availability of tryptophan in the extracellular fluid bathing neurons. The source of brain tryptophan is the blood, and the source of blood tryptophan is the diet (grains, meat, dairy products, and chocolate are particularly rich in tryptophan).

FIGURE 6.14 The synthesis of serotonin from tryptophan. Description
Following release from the axon terminal, 5-HT is removed from the synaptic cleft by the action of a specific transporter. The process of serotonin reuptake, like catecholamine reuptake, is sensitive to a number of different drugs. For example, numerous clinically useful antidepressant and antianxiety drugs, including fluoxetine (trade name Prozac), are selective inhibitors of serotonin reuptake. Once it is back in the cytosol of the serotonergic axon terminal, the transmitter is either reloaded into synaptic vesicles or degraded by MAO.
The amino acids glutamate (Glu), glycine (Gly), and gamma-aminobutyric acid (GABA) serve as neurotransmitters at most CNS synapses (Figure 6.15). Of these, only GABA is unique to those neurons that use it as a neurotransmitter; the others are among the 20 amino acids that make up proteins.

FIGURE 6.15 The amino acid neurotransmitters. Description
Glutamate and glycine are synthesized from glucose and other precursors by the action of enzymes that exist in all cells. Differences among neurons in the synthesis of these amino acids are therefore quantitative rather than qualitative. For example, the average glutamate concentration in the cytosol of glutamatergic axon terminals has been estimated to be about 20 mM, two or three times higher than that in nonglutamatergic cells. The more important distinction between glutamatergic and nonglutamatergic neurons, however, is the transporter that loads the synaptic vesicles. In glutamatergic axon terminals, but not in other types, glutamate transporters concentrate glutamate until it reaches a value of about 50 mM in the synaptic vesicles.
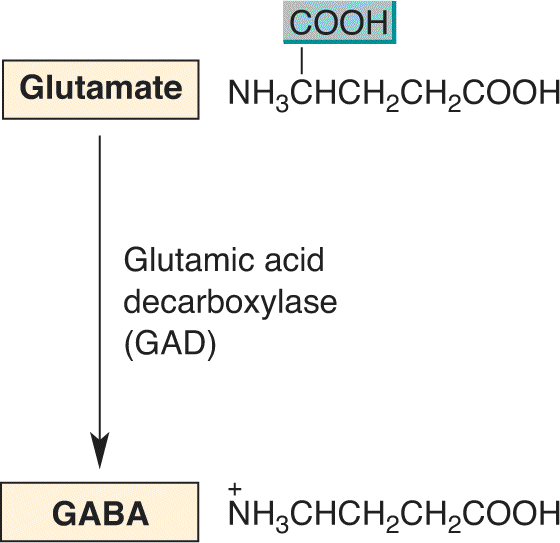
Because GABA is not one of the 20 amino acids used to construct proteins, it is synthesized in large quantities only by the neurons that use it as a neurotransmitter. The precursor for GABA is glutamate, and the key synthesizing enzyme is glutamic acid decarboxylase (GAD) (Figure 6.16). GAD, therefore, is a good marker for GABAergic neurons. Immunocytochemical studies have shown that GABAergic neurons are distributed widely in the brain. GABAergic neurons are the major source of synaptic inhibition in the nervous system. Therefore, remarkably, in one chemical step, the major excitatory neurotransmitter in the brain is converted into the major inhibitory neurotransmitter in the brain!

FIGURE 6.16 The synthesis of GABA from glutamate. Description
The synaptic actions of the amino acid neurotransmitters are terminated by selective uptake into the presynaptic terminals and glia, once again via specific Na+-dependent transporters. Inside the terminal or glial cell, GABA is metabolized by the enzyme GABA transaminase.
Other Neurotransmitter Candidates and Intercellular Messengers
In addition to the amines and amino acids, a few other small molecules serve as chemical messengers between neurons. One of the most common is adenosine triphosphate (ATP), a key molecule in cellular metabolism (see Figure 2.13). ATP is also a neurotransmitter. It is concentrated in all synaptic vesicles in the CNS and PNS, and it is released into the cleft by presynaptic spikes in a Ca2+-dependent manner, just as the classic transmitters are. ATP is often packaged in vesicles along with another classic transmitter. For example, catecholamine-containing vesicles may have 100 mM of ATP, an enormous quantity, in addition to 400 mM of the catecholamine itself. In this case, the catecholamine and ATP are co-transmitters. ATP also occurs as a co-transmitter with GABA, glutamate, ACh, DA, and peptide transmitters in various specialized types of neurons.
ATP directly excites some neurons by gating cation channels. In this sense, some of the neurotransmitter functions of ATP are similar to those of glutamate and ACh. ATP binds to purinergic receptors, some of which are transmitter-gated ion channels. There is also a large class of G-protein-coupled purinergic receptors. Following its release from synapses, ATP is degraded by extracellular enzymes, yielding adenosine. Adenosine itself does not meet the standard definition of a neurotransmitter because it is not packaged in vesicles, but it does activate several adenosine-selective receptors.
The most interesting discovery about neurotransmitters in the past few years is that small lipid molecules, called endocannabinoids (endogenous cannabinoids), can be released from postsynaptic neurons and act on presynaptic terminals (Box 6.3). Communication in this direction, from “post” to “pre,” is called retrograde signaling; thus, endocannabinoids are retrograde messengers. Retrograde messengers serve as a kind of feedback system to regulate the conventional forms of synaptic transmission, which of course go from “pre” to “post.” The details about endocannabinoid signaling are still emerging, but one basic mechanism is now clear (Figure 6.17). Vigorous firing of action potentials in the postsynaptic neuron causes voltage-gated calcium channels to open, Ca2+ enters the cell in large quantities, and intracellular [Ca2+] rises. The elevated [Ca2+] then stimulates the synthesis of endocannabinoid molecules from membrane lipids by somehow activating endocannabinoid-synthesizing enzymes. There are several unusual qualities about endocannabinoids:

FIGURE 6.17 Retrograde signaling with endocannabinoids.
Most neurotransmitters were discovered long before their receptors, but modern techniques have tended to reverse this tradition. In this story, the receptors were discovered before their transmitters.
Cannabis sativa is the botanical name for hemp, a fibrous plant used through the ages for making rope and cloth. These days, cannabis is much more popular as dope than rope. It is widely, and usually illegally, sold as marijuana or hashish, although medical uses of cannabis-related compounds are slowly being recognized, and medical or recreational use is being legalized in some states and other parts of the world. The Chinese first recognized the potent psychoactive properties of cannabis 4000 years ago, but Western society learned of its intoxicating properties only in the nineteenth century, when Napoleon III’s troops returned to France with Egyptian hashish. As a member of Napoleon’s Commission of Sciences and Arts reported in 1810, “For the Egyptians, hemp is the plant par excellence, not for the uses they make of it in Europe and many other countries, but for its peculiar effects. The hemp cultivated in Egypt is indeed intoxicating and narcotic” (cited in Piomelli, 2003, p. 873).
At low doses, the effects of cannabis can be euphoria, feelings of calm and relaxation, altered sensations, reduced pain, increased laughter, talkativeness, hunger, and lightheadedness, as well as decreased problem-solving ability, short-term memory, and psychomotor performance (i.e., the skills necessary for driving). High doses of cannabis can cause profound personality changes and even hallucinations. In recent years, forms of cannabis have been approved for limited medicinal use in the United States, primarily to treat nausea and vomiting in cancer patients undergoing chemotherapy and to stimulate appetite in some AIDS patients.
The active ingredient in cannabis is an oily chemical called Δ9-tetrahydrocannabinol, or THC. During the late 1980s, it became apparent that THC can bind to specific G-protein-coupled “cannabinoid” receptors in the brain, particularly in motor control areas, the cerebral cortex, and pain pathways. At about the same time, a group at the National Institute of Mental Health cloned the gene for an unknown (or “orphan”) G-protein-coupled receptor. Further work showed that the mystery receptor was a cannabinoid (CB) receptor. Two types of cannabinoid receptors are known: CB1 receptors are in the brain, and CB2 receptors are mainly in immune tissues elsewhere in the body.
Remarkably, the brain has more CB1 receptors than any other G-protein-coupled receptor. What are they doing there? We are quite certain they did not evolve to bind the THC from hemp. The natural ligand for a receptor is never the synthetic drug, plant toxin, or snake venom that might have helped us identify that receptor in the first place. It is much more likely that the cannabinoid receptors exist to bind some signaling molecule made naturally by the brain: THC-like neurotransmitters called endocannabinoids. Research has identified two major endocannabinoids: anandamide (from ananda, the Sanskrit word for “internal bliss”) and arachidonoylglycerol (2-AG). Anandamide and 2-AG are both small lipid molecules (Figure A), quite different from any other known neurotransmitter.

As the search for new transmitters continues, the hunt is also on for more selective compounds that bind to the CB receptors. Cannabinoids are potentially useful for relieving nausea, suppressing pain, relaxing muscles, treating seizures, and decreasing the intraocular pressure of glaucoma. Antagonists of CB1 receptors have recently been tested as appetite suppressants, but they cause unfortunate side effects. Cannabinoid therapies might be more practical if new drugs can be developed that retain the therapeutic benefits without causing psychoactive and other side effects.
- They are not packaged in vesicles like most other neurotransmitters; instead, they are manufactured rapidly and on demand.
- They are small and membrane permeable; once synthesized, they can diffuse rapidly across the membrane of their cell of origin to contact neighboring cells.
- They bind selectively to the CB1 type of cannabinoid receptor, which is mainly located on certain presynaptic terminals.
CB1 receptors are G-protein-coupled receptors, and their main effect is often to reduce the opening of presynaptic calcium channels. With its calcium channels inhibited, the ability of the presynaptic terminal to release its neurotransmitter (usually GABA or glutamate) is impaired. Thus, when a postsynaptic neuron is very active, it releases endocannabinoids, which suppress either the inhibitory or excitatory drive onto the neuron (depending on which presynaptic terminals have the CB1 receptors). This general endocannabinoid mechanism is used throughout the CNS, for a wide range of functions that we don’t completely understand.
One of the more exotic chemical messengers to be proposed for intercellular communication is actually a gaseous molecule, nitric oxide (NO). The gases carbon monoxide (CO) and hydrogen sulfide (H2S) have also been suggested to be messengers in the brain, although evidence for “gasotransmitter” functions is still sparse. These are the same NO, carbon monoxide, and hydrogen sulfide that are often major air pollutants. NO is synthesized from the amino acid arginine by many cells of the body and has powerful biological effects, particularly in the regulation of blood flow. In the nervous system, NO may be another example of a retrograde messenger. Because NO is small and membrane permeable, similar to endocannabinoids, it can diffuse much more freely than most other transmitter molecules, even penetrating through one cell to affect another beyond it. Its influence may spread throughout a small region of local tissue, rather than being confined to the site of the cells that released it. On the other hand, NO is evanescent and breaks down very rapidly. The functions of gaseous transmitters are being extensively studied and hotly debated.
Before leaving the topic of neurotransmitter chemistry, we point out, once again, that many of the chemicals we call neurotransmitters may also be present in high concentrations in non-neural parts of the body. A chemical may serve dual purposes, mediating communication in the nervous system but doing something entirely different elsewhere. Amino acids, of course, are used to make proteins throughout the body. ATP is the energy source for all cells. NO is released from endothelial cells and causes the smooth muscle of blood vessels to relax. (One consequence in males is penile erection.) The cells with the highest levels of ACh are not in the brain but in the cornea of the eye, where there are no ACh receptors. Likewise, the highest serotonin levels are not in neurons but in blood platelets. These observations underscore the importance of rigorous analysis before a chemical is assigned a neurotransmitter role.
The operation of a neurotransmitter system is like a play with two acts. Act I is presynaptic and culminates in the transient elevation of neurotransmitter concentration in the synaptic cleft. We are now ready to move on to Act II, the generation of electrical and biochemical signals in the postsynaptic neuron. The main players are transmitter-gated channels and G-protein-coupled receptors.
In Chapter 5, we learned that ACh and the amino acid neurotransmitters mediate fast synaptic transmission by acting on transmitter-gated ion channels. These channels are magnificent minuscule machines. A single channel can be a sensitive detector of chemicals and voltage, it can regulate the flow of surprisingly large currents with great precision, it can sift and select between very similar ions, and it can be regulated by other receptor systems. Yet each channel is only about 11 nm long, just barely visible with the best computer-enhanced electron microscopic methods.
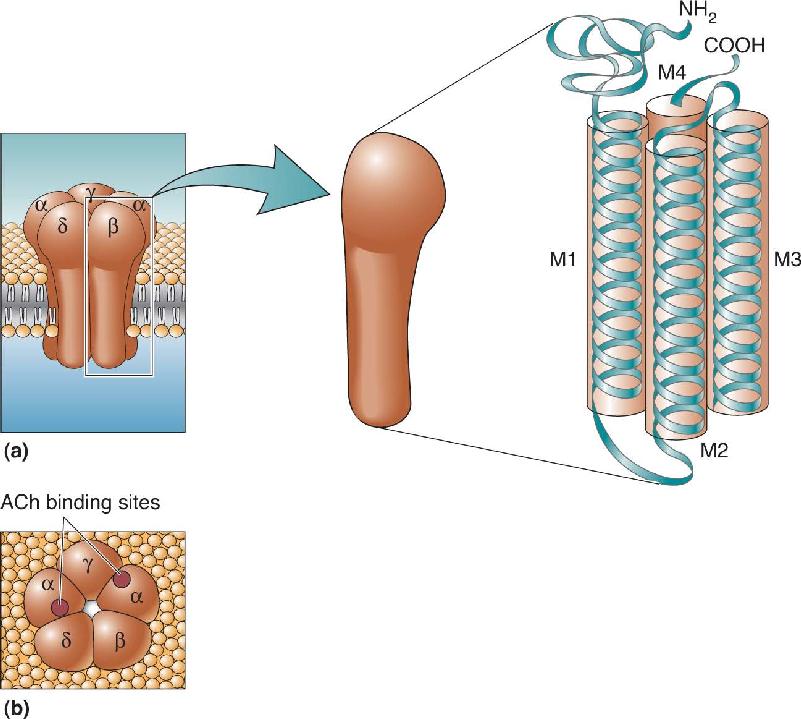
The most thoroughly studied transmitter-gated ion channel is the nicotinic ACh receptor at the neuromuscular junction in skeletal muscle. It is a pentamer, an amalgam of five protein subunits arranged like the staves of a barrel to form a single pore through the membrane (Figure 6.18a). Four different types of polypeptides are used as subunits for the nicotinic receptor, designated α, β, γ, and δ. A complete mature channel is made from two α subunits, and one each of β, γ, and δ (abbreviated α2βγδ). There is one ACh binding site on each of the α subunits; the simultaneous binding of ACh to both sites is required for the channel to open (Figure 6.18b). The nicotinic ACh receptor on neurons is also a pentamer, but, unlike the muscle receptor, most of these receptors are composed of α and β subunits only (in a ratio of α3β2).

FIGURE 6.18 The subunit arrangement of the nicotinic ACh receptor. (a) Side view, with an enlargement showing how the four alpha helices of each subunit are packed together. (b) Top view, showing the location of the two ACh binding sites. Description
Although each type of receptor subunit has a different primary structure, there are stretches where the different polypeptide chains have a similar sequence of amino acids. For example, each subunit polypeptide has four separate segments that will coil into alpha helices (see Figure 6.18a). Because the amino acid residues of these segments are hydrophobic, the four alpha helices are believed to be where the polypeptide is threaded back and forth across the membrane, similar to the pore loops of potassium and sodium channels (see Chapters 3 and 4).
The primary structures of the subunits of other transmitter-gated channels in the brain are also known, and there are obvious similarities (Figure 6.19). Most contain the four hydrophobic segments that span the membrane in subunits of the nicotinic ACh receptor, the GABAA receptor, and the glycine receptor. These three neurotransmitter receptors are all pentameric complexes of subunits (Figure 6.19b). The glutamate-gated channels are slightly different. Glutamate receptors are tetramers, having four subunits that comprise a functional channel. The M2 region of the glutamate subunits does not span the membrane but instead forms a hairpin that both enters and exits from the inside of the membrane (Figure 6.19c). The structure of the glutamate receptors resembles that of some potassium channels (see Figure 3.17), and this has inspired the surprising hypothesis that glutamate receptors and potassium channels evolved from a common ancestral ion channel. The purinergic (ATP) receptors also have an unusual structure. Each subunit has only two membrane-spanning segments, and three subunits make up a complete receptor.

FIGURE 6.19 Similarities in the structure of subunits for different transmitter-gated ion channels. (a) If the polypeptides for various channel subunits were stretched out in a line, this is how they would compare. They have in common the four regions called M1 to M4, which are segments where the polypeptides will coil into alpha helices to span the membrane. Kainate receptors are subtypes of glutamate receptors. (b) M1–M4 regions of the ACh α subunit as they are threaded through the membrane. (c) M1–M4 regions of the glutamate receptor subunits; M1, M3, and M4 span the entire thickness of the membrane, whereas M2 penetrates only part way. Description
The most interesting variations among channel structures are the ones that account for their differences. Different transmitter binding sites let one channel respond to Glu while another responds to GABA; certain amino acids around the narrow ion pore allow only Na+ and K+ to flow through some channels, Ca2+ through others, and only Cl– through yet others.
Amino acid-gated channels mediate most of the fast synaptic transmission in the CNS. Let’s take a closer look at their functions because they are central to topics as diverse as sensory systems, memory, and disease. Several properties of these channels distinguish them from one another and define their functions within the brain.
- The pharmacology of their binding sites describes which transmitters affect them and how drugs interact with them.
- The kinetics of the transmitter binding process and channel gating determine the duration of their effect.
- The selectivity of the ion channels determines whether they produce excitation or inhibition and whether Ca2+ enters the cell in significant amounts.
- The conductance of open channels helps determine the magnitude of their effects.
All of these properties are a direct result of the molecular structure of the channels.
Glutamate-Gated Channels. As we discussed previously, three glutamate receptor subtypes bear the names of their selective agonists: AMPA, NMDA, and kainate. Each of these is a glutamate-gated ion channel. The AMPA-gated and NMDA-gated channels mediate the bulk of fast excitatory synaptic transmission in the brain. Kainate receptors also exist throughout the brain, on both presynaptic and postsynaptic membranes, but their functions are not clearly understood.
AMPA-gated channels are permeable to both Na+ and K+, and most of them are not permeable to Ca2+. The net effect of activating them at normal, negative membrane potentials is to admit an excess of cations into the cell (i.e., more Na+ enters than K+ leaves), causing a rapid and large depolarization. Thus, AMPA receptors at CNS synapses mediate excitatory transmission in much the same way as nicotinic receptors mediate synaptic excitation at neuromuscular junctions.
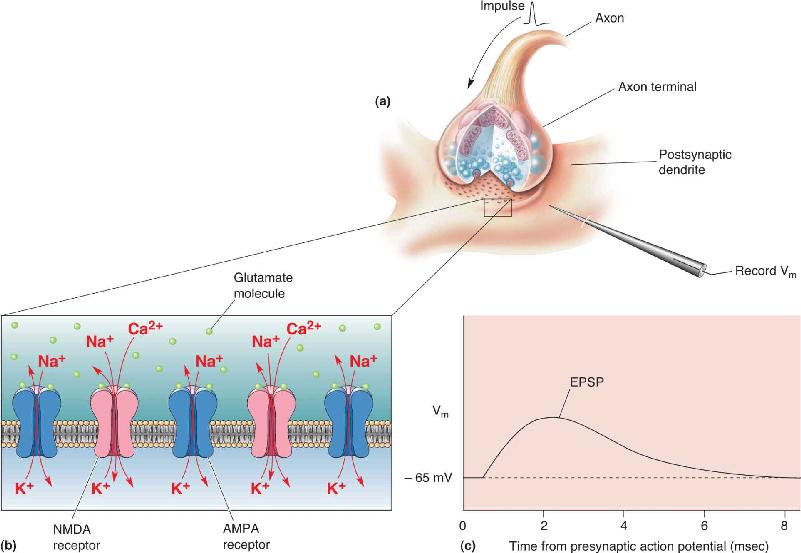
AMPA receptors coexist with NMDA receptors at many synapses in the brain, so most glutamate-mediated excitatory postsynaptic potentials (EPSPs) have components contributed by both (Figure 6.20). NMDA-gated channels also cause excitation of a cell by admitting an excess of Na+, but they differ from AMPA receptors in two very important ways: (1) NMDA-gated channels are permeable to Ca2+, and (2) inward ionic current through NMDA-gated channels is voltage dependent. We’ll discuss each of these properties in turn.

FIGURE 6.20 The coexistence of NMDA and AMPA receptors in the postsynaptic membrane of a CNS synapse. (a) An action potential arriving in the presynaptic terminal causes the release of glutamate. (b) Glutamate binds to AMPA receptor channels and NMDA receptor channels in the postsynaptic membrane. (c) The entry of Na+ through the AMPA channels, and Na+ and Ca2+ through the NMDA channels, causes an EPSP. Description
It is hard to overstate the importance of intracellular Ca2+ to cell functions. We have already seen that Ca2+ can trigger presynaptic neurotransmitter release. Postsynaptically, Ca2+ can also activate many enzymes, regulate the opening of a variety of channels, and affect gene expression; in excessive amounts, Ca2+ can even trigger the death of a cell (Box 6.4). Thus, activation of NMDA receptors can, in principle, cause widespread and lasting changes in the postsynaptic neuron. Indeed, as we will see in Chapter 25, Ca2+ entry through NMDA-gated channels may cause the changes that lead to long-term memory.
Neurons of the mammalian brain almost never regenerate, so each dead neuron is one less we have for thinking. One of the fascinating ironies of neuronal life and death is that glutamate, the most essential neurotransmitter in the brain, is also a common killer of neurons. A large percentage of the brain’s synapses release glutamate, which is stored in large quantities. Even the cytosol of nonglutamatergic neurons has a very high glutamate concentration, greater than 3 mM. An ominous observation is that when you apply this same amount of glutamate to the outside of isolated neurons, they die within minutes. Mae West once said, “Too much of a good thing can be wonderful,” but apparently she wasn’t talking about glutamate.
The voracious metabolic rate of the brain demands a continuous supply of oxygen and glucose. If blood flow ceases, as in cardiac arrest, neural activity will stop within seconds, and permanent damage will result within a few minutes. Disease states such as cardiac arrest, stroke, brain trauma, seizures, and oxygen deficiency can initiate a vicious cycle of excess glutamate release. Whenever neurons cannot generate enough ATP to keep their ion pumps working hard, membranes depolarize, and Ca2+ leaks into cells. The entry of Ca2+ triggers the synaptic release of glutamate. Glutamate further depolarizes neurons, which further raises intracellular Ca2+ and causes still more glutamate to be released. At this point, there may even be a reversal of the glutamate transporter, further contributing to the cellular leakage of glutamate.
When glutamate reaches high concentrations, it kills neurons by overexciting them, a process called excitotoxicity. Glutamate simply activates its several types of receptors, which allow excessive amounts of Na+, K+, and Ca2+ to flow across the membrane. The NMDA subtype of the glutamate-gated channel is a critical player in excitotoxicity because it is the main route for Ca2+ entry. Neuron damage or death occurs because of swelling resulting from water uptake and stimulation by Ca2+ of intracellular enzymes that degrade proteins, lipids, and nucleic acids. Neurons literally digest themselves.
Excitotoxicity has been implicated in several progressive neurodegenerative human diseases such as amyotrophic lateral sclerosis (ALS, also known as Lou Gehrig disease), in which spinal motor neurons slowly die, and Alzheimer’s disease, in which brain neurons slowly die. The effects of various environmental toxins mimic aspects of these diseases. Eating large quantities of a certain type of chickpea can cause lathyrism, a degeneration of motor neurons. The pea contains an excitotoxin called β-oxalylaminoalanine, which activates glutamate receptors. A toxin called domoic acid, found in contaminated shellfish, is also a glutamate receptor agonist. Ingesting small amounts of domoic acid causes seizures and brain damage. And another plant excitotoxin, β-methylaminoalanine, may cause a hideous condition that combines signs of ALS, Alzheimer’s disease, and Parkinson’s disease in individual patients on the island of Guam.
As researchers sort out the tangled web of excitotoxins, receptors, enzymes, and neurological disease, new strategies for treatment emerge. Glutamate receptor antagonists that can obstruct these excitotoxic cascades and minimize neuronal suicide have shown some clinical promise. Genetic manipulations may eventually thwart neurodegenerative conditions in susceptible people.
When the NMDA-gated channel opens, Ca2+ and Na+ enter the cell (and K+ leaves), but the magnitude of this inward ionic current depends on the postsynaptic membrane potential in an unusual way, for an unusual reason. When glutamate binds to the NMDA receptor, the pore opens as usual. However, at normal negative resting membrane potentials, the channel becomes clogged by Mg2+ ions, and this “magnesium block” prevents other ions from passing freely through the NMDA channel. Mg2+ pops out of the pore only when the membrane is depolarized, which usually follows the activation of AMPA channels at the same and neighboring synapses. Thus, inward ionic current through the NMDA channel is voltage dependent, in addition to being transmitter gated. Both glutamate and depolarization must coincide before the channel will pass current (Figure 6.21). This property has a significant impact on synaptic integration at many locations in the CNS.

FIGURE 6.21 Inward ionic current through the NMDA-gated channel. (a) Glutamate alone causes the channel to open, but at the resting membrane potential, the pore becomes blocked by Mg2+. (b) Depolarization of the membrane relieves the Mg2+ block and allows Na+ and Ca2+ to enter. Description
GABA-Gated and Glycine-Gated Channels. GABA mediates most synaptic inhibition in the CNS, and glycine mediates most of the rest. Both the GABAA receptor and the glycine receptor gate a chloride channel. Surprisingly, inhibitory GABAA and glycine receptors have a structure very similar to that of excitatory nicotinic ACh receptors, despite the fact that the first two are selective for anions while the last is selective for cations. Each receptor has α subunits that bind the transmitter and β subunits that do not.
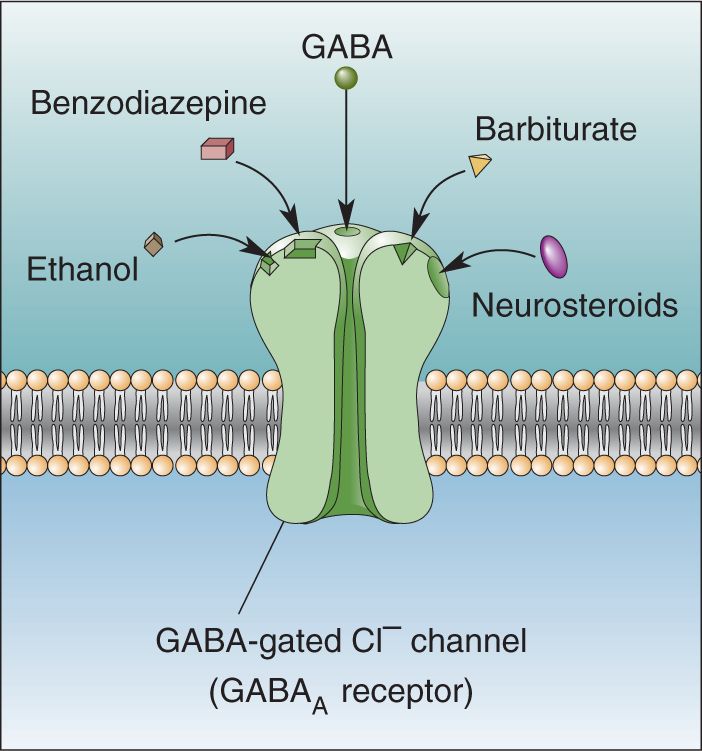
Synaptic inhibition must be tightly regulated in the brain. Too much causes a loss of consciousness and coma; too little leads to a seizure. The need to control inhibition may explain why the GABAA receptor has, in addition to its GABA binding site, several other sites where chemicals can dramatically modulate its function. For example, two classes of drugs, benzodiazepines (such as the tranquilizer diazepam, with the trade name Valium) and barbiturates (including phenobarbital and other sedatives and anticonvulsants), each bind to their own distinct site on the outside face of the GABAA channel (Figure 6.22). By themselves, these drugs do very little to the channel. But when GABA is present, benzodiazepines increase the frequency of channel openings, while barbiturates increase the duration of channel openings. The result in each case is more inhibitory Cl– current, stronger inhibitory postsynaptic potentials (IPSPs), and the behavioral consequences of enhanced inhibition. The actions of benzodiazepines and barbiturates are selective for the GABAA receptor, and the drugs have no effect on glycine receptor function. Some of this selectivity can be understood in molecular terms; only receptors with the γ type of GABAA subunit, in addition to α and β subunits, respond to benzodiazepines.

FIGURE 6.22 The binding of drugs to the GABAA receptor. The drugs by themselves do not open the channel, but they change the effect that GABA has when it binds to the channel at the same time as the drug. Description
Another popular drug that strongly enhances the function of the GABAA receptor is ethanol, the form of alcohol imbibed in beverages. Ethanol has complex actions that include effects on NMDA, glycine, nicotinic ACh, and serotonin receptors. Its effects on GABAA channels depend on their specific structure. Evidence indicates that particular α, β, and γ subunits are necessary for constructing an ethanol-sensitive GABAA receptor, similar to the structure that is benzodiazepine sensitive. This explains why ethanol enhances inhibition in some brain areas but not others. By understanding this molecular and anatomical specificity, we can begin to appreciate how drugs like ethanol lead to such powerful, and addictive, effects on behavior.
These myriad drug effects present an interesting paradox. Surely, the GABAA receptor did not evolve modulatory binding sites just for the benefit of our modern drugs. The paradox has motivated researchers to search for endogenous ligands, natural chemicals that might bind to benzodiazepine and barbiturate sites and serve as regulators of inhibition. Substantial evidence indicates that natural benzodiazepine-like ligands exist, although identifying them and understanding their functions are proving difficult. Other good candidates as natural modulators of GABAA receptors are the neurosteroids, natural metabolites of steroid hormones that are synthesized from cholesterol primarily in the gonads and adrenal glands, but also in glial cells of the brain. Some neurosteroids enhance inhibitory function while others suppress it, and they seem to do both by binding to their own sites on the GABAA receptor (see Figure 6.22), distinct from those of the other drugs we’ve mentioned. The functions of natural neurosteroids are also obscure, but they suggest a means by which brain and body physiology could be regulated in parallel by the same chemicals.
There are multiple subtypes of G-protein-coupled receptors in every known neurotransmitter system. In Chapter 5, we learned that transmission at these receptors involves three steps: (1) binding of the neurotransmitter to the receptor protein, (2) activation of G-proteins, and (3) activation of effector systems. Let’s focus on each of these steps.
The Basic Structure of G-Protein-Coupled Receptors
Most G-protein-coupled receptors are simple variations on a common plan, consisting of a single polypeptide containing seven membrane-spanning alpha helices (Figure 6.23). Two of the extracellular loops of the polypeptide form the transmitter binding sites. Structural variations in this region determine which neurotransmitters, agonists, and antagonists bind to the receptor. Two of the intracellular loops can bind to and activate G-proteins. Structural variations here determine which G-proteins and, consequently, which effector systems are activated in response to transmitter binding.

FIGURE 6.23 The basic structure of a G-protein-coupled receptor. Most metabotropic receptors have seven membrane-spanning alpha helices, a transmitter binding site on the extracellular side, and a G-protein binding site on the intracellular side. Description
A very partial list of G-protein-coupled receptors appears in Table 6.2. The human genome has genes coding for about 800 different G-protein-coupled receptors, which are organized into five major families with similar structures. Most of these receptors were unknown before the powerful methods of molecular biology were applied to their discovery. It is also important to recall that G-protein-coupled receptors are important in all of the body’s cell types, not just neurons.
5-HT1A, 5-HT1B, 5-HT1D, 5-HT1E, 5-HT2A, 5-HT2B, 5-HT4, 5-HT5A
G-proteins are the common link in most signaling pathways that start with a neurotransmitter receptor and end with effector proteins. G-protein is short for guanosine triphosphate (GTP) binding protein, which is actually a diverse family of about 20 types. There are many more transmitter receptors than G-proteins, so some types of G-proteins can be activated by many receptors.
Most G-proteins have the same basic mode of operation (Figure 6.24):

FIGURE 6.24 The basic mode of operation of G-proteins. (a) In its inactive state, the α subunit of the G-protein binds GDP. (b) When activated by a G-protein-coupled receptor, the GDP is exchanged for GTP. (c) The activated G-protein splits, and both the Gα (GTP) subunit and the Gαγ subunit become available to activate effector proteins. (d) The Gα subunit slowly removes phosphate (PO4) from GTP, converting GTP to GDP and terminating its own activity. Description
- Each G-protein has three subunits, termed α, β, and γ. In the resting state, a guanosine diphosphate (GDP) molecule is bound to the Gα subunit, and the whole complex floats around on the inner surface of the membrane.
- If this GDP-bound G-protein bumps into the proper type of receptor and if that receptor has a transmitter molecule bound to it, then the G-protein releases its GDP and exchanges it for a GTP that it picks up from the cytosol.
- The activated GTP-bound G-protein splits into two parts: the Gα subunit plus GTP and the Gβγ complex. Both can then move on to influence various effector proteins.
- The Gα subunit is itself an enzyme that eventually breaks down GTP into GDP. Therefore, Gα eventually terminates its own activity by converting the bound GTP to GDP.
- The Gα and Gβγ subunits come back together, allowing the cycle to begin again.
The first G-proteins that were discovered had the effect of stimulating effector proteins. Subsequently, it was found that other G-proteins could inhibit these same effectors. Thus, the simplest scheme for subdividing the G-proteins is GS, designating that the G-protein is stimulatory, and Gi, designating that the G-protein is inhibitory.
In Chapter 5, we learned that activated G-proteins exert their effects by binding to either of two types of effector proteins: G-protein-gated ion channels and G-protein-activated enzymes. Because the effects do not involve any other chemical intermediaries, the first route is sometimes called the shortcut pathway.
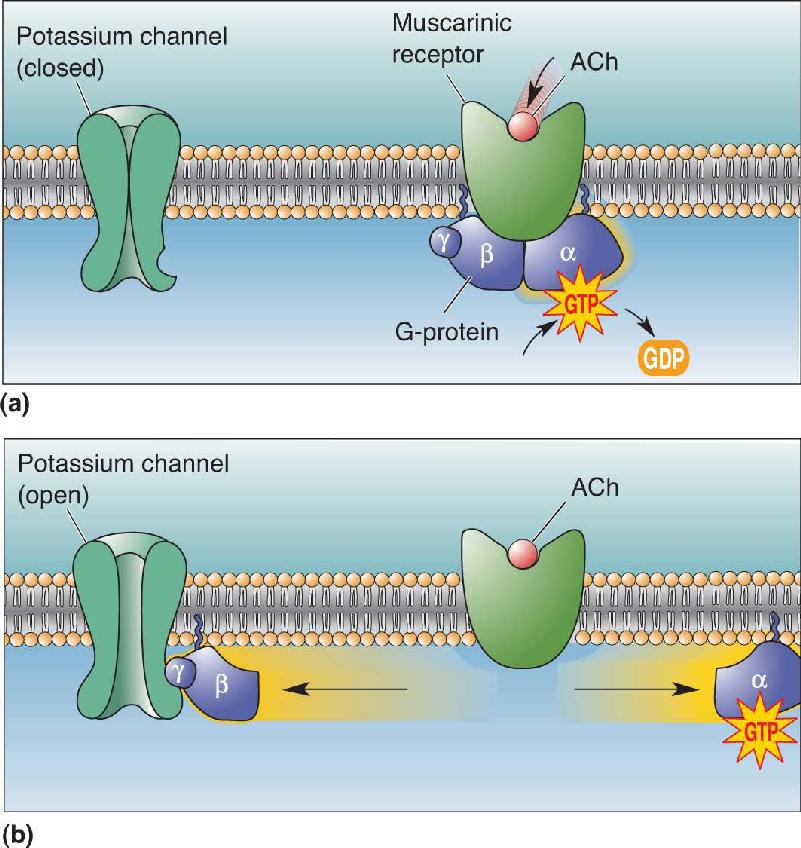
The Shortcut Pathway. A variety of neurotransmitters use the shortcut pathway, from receptor to G-protein to ion channel. One example is the muscarinic receptors in the heart. These ACh receptors are coupled via G-proteins to particular types of potassium channels, explaining why ACh slows the heart rate (Figure 6.25). In this case, the βγ subunits migrate laterally along the membrane until they bind to the right type of potassium channel and induce it to open. Another example is neuronal GABAB receptors, also coupled by the shortcut pathway to potassium channels.

FIGURE 6.25 The shortcut pathway. (a) G-proteins in heart muscle are activated by ACh binding to muscarinic receptors. (b) The activated Gβγ subunit directly induces a potassium channel to open. Description
Shortcut pathways are the fastest of the G-protein-coupled systems, having responses beginning within 30–100 msec of neurotransmitter binding. Although not quite as fast as a transmitter-gated channel, which uses no intermediary between receptor and channel, this is faster than the second messenger cascades we describe next. The shortcut pathway is also very localized compared with other effector systems. As the G-protein diffuses within the membrane, it apparently cannot move very far, so only channels nearby can be affected. Because all the action in the shortcut pathway occurs within the membrane, it is sometimes called the membrane-delimited pathway.
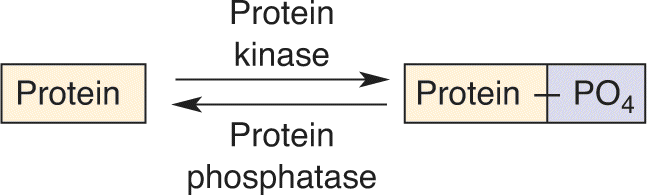
Second Messenger Cascades. G-proteins can also exert their effects by directly activating certain enzymes. Activation of these enzymes can trigger an elaborate series of biochemical reactions, a cascade that often ends in the activation of other “downstream” enzymes that alter neuronal function. Between the first enzyme and the last are several second messengers. The whole process that couples the neurotransmitter, via multiple steps, to activation of a downstream enzyme is called a second messenger cascade (Figure 6.26).

FIGURE 6.26 The components of a second messenger cascade. Description
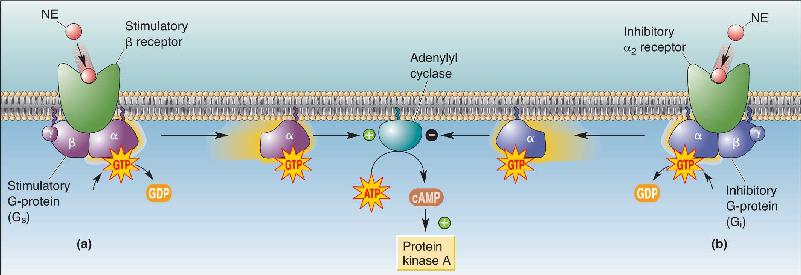
In Chapter 5, we introduced the cAMP second messenger cascade initiated by the activation of the NE β receptor (Figure 6.27a). It begins with the β receptor activating the stimulatory G-protein, GS, which proceeds to stimulate the membrane-bound enzyme adenylyl cyclase. Adenylyl cyclase converts ATP to cAMP. The subsequent rise of cAMP in the cytosol activates a specific downstream enzyme called protein kinase A (PKA).

FIGURE 6.27 The stimulation and inhibition of adenylyl cyclase by different G-proteins. (a) Binding of NE to the β receptor activates Gs, which in turn activates adenylyl cyclase. Adenylyl cyclase generates cAMP, which activates the downstream enzyme protein kinase A. (b) Binding of NE to the α2 receptor activates Gi, which inhibits adenylyl cyclase. Description
Many biochemical processes are regulated with a push–pull method, one to stimulate them and one to inhibit them, and cAMP production is no exception. The activation of a second type of NE receptor, called the α2 receptor, leads to the activation of Gi (the inhibitory G-protein). Gi suppresses the activity of adenylyl cyclase, and this effect can take precedence over the stimulatory system (Figure 6.27b).
Some messenger cascades can branch. Figure 6.28 shows how the activation of various G-proteins can stimulate phospholipase C (PLC), an enzyme that floats in the membrane-like adenylyl cyclase. PLC acts on a membrane phospholipid (PIP2, or phosphatidylinositol-4, 5-bisphosphate), splitting it to form two molecules that serve as second messengers: diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3). DAG, which is lipid-soluble, stays within the plane of the membrane where it activates a downstream enzyme, protein kinase C (PKC). At the same time, the water-soluble IP3 diffuses away in the cytosol and binds to specific receptors on the smooth ER and other membrane-enclosed organelles in the cell. These receptors are IP3-gated calcium channels, so IP3 causes the organelles to discharge their stores of Ca2+. As we have said, elevations in cytosolic Ca2+ can trigger widespread and long-lasting effects. One effect is activation of the enzyme calcium-calmodulin-dependent protein kinase, or CaMK. CaMK is an enzyme implicated in, among other things, the molecular mechanisms of memory, as we’ll see in Chapter 25.

FIGURE 6.28 Second messengers generated by the breakdown of PIP2, a membrane phospholipid. ① Activated G-proteins stimulate the enzyme phospholipase C (PLC). ② PLC splits PIP2 into DAG and IP3. ③ DAG stimulates the downstream enzyme protein kinase C (PKC). ④ IP3 stimulates the release of Ca2+ from intracellular stores. The Ca2+ can go on to stimulate various downstream enzymes. Description
Phosphorylation and Dephosphorylation. The preceding examples show that key downstream enzymes in many of the second messenger cascades are protein kinases (PKA, PKC, CaMK). As mentioned in Chapter 5, protein kinases transfer phosphate from ATP floating in the cytosol to proteins, a reaction called phosphorylation. The addition of phosphate groups to a protein changes its conformation slightly, thereby changing its biological activity. The phosphorylation of ion channels, for example, can strongly influence the probability that they will open or close.
Consider the consequence of activating the β type of NE receptors on cardiac muscle cells. The subsequent rise in cAMP activates PKA, which phosphorylates the cell’s voltage-gated calcium channels, and this enhances their activity. More Ca2+ flows, and the heart beats more strongly. By contrast, the stimulation of β-adrenergic receptors in many neurons seems to have no effect on calcium channels, but instead causes inhibition of certain potassium channels. Reduced K+ conductance causes a slight depolarization, increases the length constant, and makes the neuron more excitable (see Chapter 5).
If transmitter-stimulated kinases were allowed to phosphorylate without some method of reversing the process, all proteins would quickly become saturated with phosphates, and further regulation would become impossible. Enzymes called protein phosphatases save the day because they act rapidly to remove phosphate groups. The degree of channel phosphorylation at any moment therefore depends on the dynamic balance of phosphorylation by kinases and dephosphorylation by phosphatases (Figure 6.29).

FIGURE 6.29 Protein phosphorylation and dephosphorylation. Description
The Function of Signal Cascades. Synaptic transmission using transmitter-gated channels is simple and fast. Transmission involving G-protein-coupled receptors is complex and slow. What are the advantages of having such long chains of command? One important advantage is signal amplification: The activation of one G-protein-coupled receptor can lead to the activation of not one, but many, ion channels (Figure 6.30).

FIGURE 6.30 Signal amplification by G-protein-coupled second messenger cascades. When a transmitter activates a G-protein-coupled receptor, there can be amplification of the messengers at several stages of the cascade, so that ultimately, many channels are affected. Description
Signal amplification can occur at several places in the cascade. A single neurotransmitter molecule, bound to one receptor, can activate perhaps 10–20 G-proteins; each G-protein can activate an adenylyl cyclase, which can make many cAMP molecules that can spread to activate many kinases. Each kinase can then phosphorylate many channels. If all cascade components were tied together in a clump, signaling would be severely limited. The use of small messengers that can diffuse quickly (such as cAMP) also allows signaling at a distance, over a wide stretch of cell membrane. Signal cascades also provide many sites for further regulation, as well as interaction between cascades. Finally, signal cascades can generate very long-lasting chemical changes in cells, which may form the basis for, among other things, a lifetime of memories.
DIVERGENCE AND CONVERGENCE IN NEUROTRANSMITTER SYSTEMS
Glutamate is the most common excitatory neurotransmitter in the brain, while GABA is the pervasive inhibitory neurotransmitter. But this is only part of the story because any single neurotransmitter can have many different effects. A molecule of glutamate can bind to any of several kinds of glutamate receptors, and each of these can mediate a different response. The ability of one transmitter to activate more than one subtype of receptor, and cause more than one type of postsynaptic response, is called divergence.
Divergence is the rule among neurotransmitter systems. Every known neurotransmitter can activate multiple receptor subtypes (see Table 6.2), and evidence indicates that the number of known receptors will continue to escalate as the powerful methods of molecular neurobiology are applied to each system. Because of the multiple receptor subtypes, one transmitter can affect different neurons (or even different parts of the same neuron) in very different ways. Divergence also occurs at points beyond the receptor level, depending on which G-proteins and which effector systems are activated. Divergence may occur at any stage in the cascade of transmitter effects (Figure 6.31a).

FIGURE 6.31 Divergence and convergence in neurotransmitter signaling systems. (a) Divergence. (b) Convergence. (c) Integrated divergence and convergence. Description
Neurotransmitters can also exhibit convergence of effects. Multiple transmitters, each activating their own receptor type, can converge to influence the same effector system (Figure 6.31b). Convergence in a single cell can occur at the level of the G-protein, the second messenger cascade, or the type of ion channel. Neurons integrate divergent and convergent signaling systems, resulting in a complex map of chemical effects (Figure 6.31c). The wonder is that it ever works; the challenge is to understand how it does.
Neurotransmitters are the essential links between neurons, and between neurons and other effector cells, such as muscle cells and glandular cells. But it is important to think of transmitters as one link in a chain of events, inciting chemical changes both fast and slow, divergent and convergent. You can envision the many signaling pathways onto and within a single neuron as a kind of information network. This network is in delicate balance, shifting its effects dynamically as the demands on a neuron vary with changes in the organism’s behavior.
The signaling network within a single neuron resembles in some ways the neural networks of the brain itself. It receives a variety of inputs, in the form of transmitters bombarding it at different times and places. These inputs cause an increased drive through some signal pathways and a decreased drive through others, and the information is recombined to yield a particular output that is more than a simple summation of the inputs. Signals regulate signals, chemical changes can leave lasting traces of their history, drugs can shift the balance of signaling power—and, in a literal sense, the brain and its chemicals are one.
calcium-calmodulin-dependent protein kinase (CaMK) (p. 174)
1. List the criteria that are used to determine whether a chemical serves as a neurotransmitter. What are the various experimental strategies you could use to show that ACh fulfills the criteria of a neurotransmitter at the neuromuscular junction?
2. What are three methods that could be used to show that a neurotransmitter receptor is synthesized or localized in a particular neuron?
3. Compare and contrast the properties of (a) AMPA and NMDA receptors and (b) GABAA and GABAB receptors.
4. Synaptic inhibition is an important feature of the circuitry in the cerebral cortex. How would you determine whether GABA or Gly, or both, or neither, is the inhibitory neurotransmitter of the cortex?
5. Glutamate activates a number of different metabotropic receptors. The consequence of activating one subtype is the inhibition of cAMP formation. A consequence of activating a second subtype is activation of PKC. Propose mechanisms for these different effects.
6. Do convergence and divergence of neurotransmitter effects occur in single neurons?
7. Ca2+ are considered to be second messengers. Why?
Cooper JR, Bloom FE, Roth RH. 2009. Introduction to Neuropsychopharmacology. New York: Oxford University Press.
Cowan WM, Südhof TC, Stevens CF. 2001. Synapses. Baltimore: Johns Hopkins University Press.
Katritch V, Cherezov V, Stevens RC. 2012. Diversity and modularity of G protein-coupled receptor structures. Trends in Pharmacological Sciences 33:17–27.
Mustafa AK, Gadalla MM, Snyder SH. 2009. Signaling by gasotransmitters. Science Signaling 2(68):re2.
Nestler EJ, Hyman SE, Malenka RC. 2008. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience, 2nd ed. New York: McGraw-Hill Professional.
Piomelli D. 2003. The molecular logic of endocannabinoid signalling. Nature Reviews Neuroscience 4:873–884.
Regehr WG, Carey MR, Best AR. 2009. Activity-dependent regulation of synapses by retrograde messengers. Neuron 63:154–170.
Additional figures