Motivation
THE HYPOTHALAMUS, HOMEOSTASIS, AND MOTIVATED BEHAVIOR
Hormonal and Hypothalamic Regulation of Body Fat and Feeding
BOX 16.1 OF SPECIAL INTEREST: The Starving Brains of the Obese
The Effects of Elevated Leptin Levels on the Hypothalamus
The Effects of Decreased Leptin Levels on the Hypothalamus
The Control of Feeding by Lateral Hypothalamic Peptides
BOX 16.2 OF SPECIAL INTEREST: Marijuana and the Munchies
BOX 16.3 OF SPECIAL INTEREST: Diabetes Mellitus and Insulin Shock
BOX 16.4 OF SPECIAL INTEREST: Self-Stimulation of the Human Brain
BOX 16.5 OF SPECIAL INTEREST: Dopamine and Addiction
BOX 16.6 PATH OF DISCOVERY: Learning to Crave, by Julie Kauer
Behavior happens. But why? In Part II of this book, we discussed various types of motor responses. At the lowest level are unconscious reflexes initiated by sensory stimulation—dilation of the pupils when the lights go out, sudden removal of the foot from a thumbtack, and so on. At the highest level are conscious movements initiated by the neurons of the frontal lobe—for example, the finger movements that tap this text into the computer. Voluntary movements are incited to occur, or motivated, in order to satisfy a need. The motivation can be very abstract (the “need” to go sailing on a warm and breezy summer afternoon), but it can also be quite concrete (the need to go to the bathroom when your bladder is full).
Motivation can be thought of as a driving force on behavior. By analogy, consider the driving force on sodium ions to cross the neuronal membrane (an odd analogy, perhaps, but not for a neuroscience text). As we learned back in Chapters 3 and 4, ionic driving force depends on a number of factors, including the concentration of the ion on both sides of the membrane and the electrical membrane potential. Variations in driving force make transmembrane ionic current in a particular direction more or less likely. But the driving force alone does not determine whether the current flows; the transmembrane movement of ions also requires the appropriate gated ion channels to be opened and capable of conducting the current.
Of course, human behavior will never be described by anything as simple as Ohm’s law. Still, it is useful to consider that the probability and direction of a behavior will vary with the level of driving force to perform that behavior. And while motivation may be required for a certain behavior, it does not guarantee that behavior. The membrane analogy also allows us to highlight the fact that a crucial part of the control of behavior is to appropriately gate the expression of different motivated actions that have conflicting goals—tapping on the computer keyboard versus spending the afternoon sailing, for example.
Despite tangible progress in recent years, neuroscience cannot yet provide a detailed explanation for why the sailing expedition was abandoned in favor of writing this chapter. Nonetheless, much has been learned about what motivates certain behaviors that are basic to survival.
THE HYPOTHALAMUS, HOMEOSTASIS, AND MOTIVATED BEHAVIOR
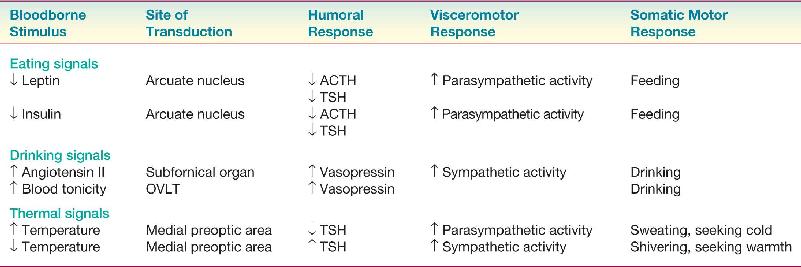
The hypothalamus and homeostasis were introduced in Chapter 15. Recall that homeostasis refers to the processes that maintain the internal environment of the body within a narrow physiological range. Although homeostatic reflexes occur at many levels of the nervous system, the hypothalamus plays a key role in the regulation of body temperature, fluid balance, and energy balance.
The hypothalamic regulation of homeostasis starts with sensory transduction. A regulated parameter (e.g., temperature) is measured by specialized sensory neurons, and deviations from the optimal range are detected by neurons concentrated in the periventricular zone of the hypothalamus. These neurons then orchestrate an integrated response to bring the parameter back to its optimal value. The response generally has three components:
- Humoral response: Hypothalamic neurons respond to sensory signals by stimulating or inhibiting the release of pituitary hormones into the bloodstream.
- Visceromotor response: Neurons in the hypothalamus respond to sensory signals by adjusting the balance of sympathetic and parasympathetic outputs of the autonomic nervous system (ANS).
- Somatic motor response: Hypothalamic neurons (particularly within the lateral hypothalamus) respond to sensory signals by inciting an appropriate somatic motor behavioral response.
You are cold, dehydrated, and depleted of energy. The appropriate humoral and visceromotor responses kick in automatically. You shiver, blood is shunted away from the body surface, urine production is inhibited, body fat reserves are mobilized, and so on. But the fastest and most effective way to correct these disturbances of brain homeostasis is to actively seek or generate warmth by moving, to drink water, and to eat. These are examples of motivated behaviors generated by the somatic motor system, and they are incited to occur by the activity of the lateral hypothalamus. Our goal in this chapter is to explore the neural basis for this type of motivation. To illustrate, we will concentrate on a subject dear to our hearts: eating.
As you know, even a brief interruption in a person’s oxygen supply can lead to serious brain damage and death. You may be surprised to learn that the brain’s requirement for food, in the form of glucose, is no less urgent. Only a few minutes of glucose deprivation will lead to a loss of consciousness, eventually followed by death if glucose is not restored. While the external environment normally provides a constant source of oxygen, the availability of food is less assured. Thus, complex internal regulatory mechanisms have evolved to store energy in the body so that it is available when needed. One primary reason we are motivated to eat is to keep these reserves at a level sufficient to ensure that there will not be an energy shortfall.
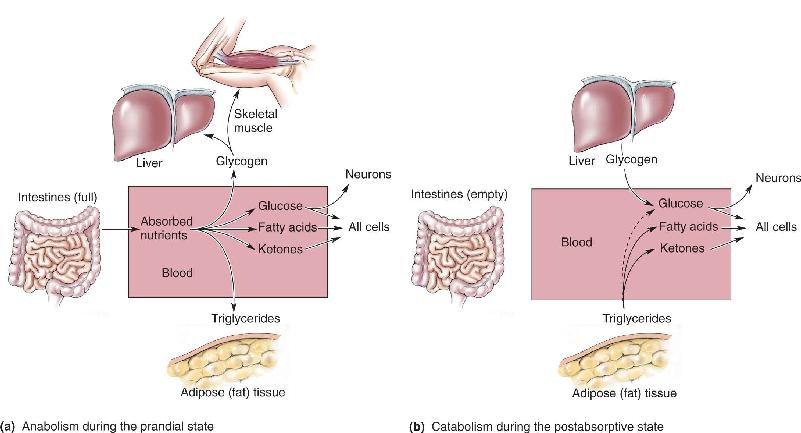
The body’s energy stores are replenished during and immediately after consuming a meal. This condition, in which the blood is filled with nutrients, is called the prandial state (from the Latin word for “breakfast”). During this time, energy is stored in two forms: glycogen and triglycerides (Figure 16.1). Glycogen reserves have a finite capacity, and they are found mainly in the liver and skeletal muscle. Triglyceride reserves are found in adipose (fat) tissue, and they have a virtually unlimited capacity. The assembly of macromolecules such as glycogen and triglycerides from simple precursors is called anabolism, or anabolic metabolism.

FIGURE 16.1 Loading and emptying the body’s energy reserves. (a) After a meal, when we are in the prandial state, excess energy is stored as glycogen or as triglycerides. (b) During the time between meals, when we are in the postabsorptive state, the glycogen and triglycerides are broken down (catabolized) into smaller molecules that can be used as fuel by the cells of the body. Description
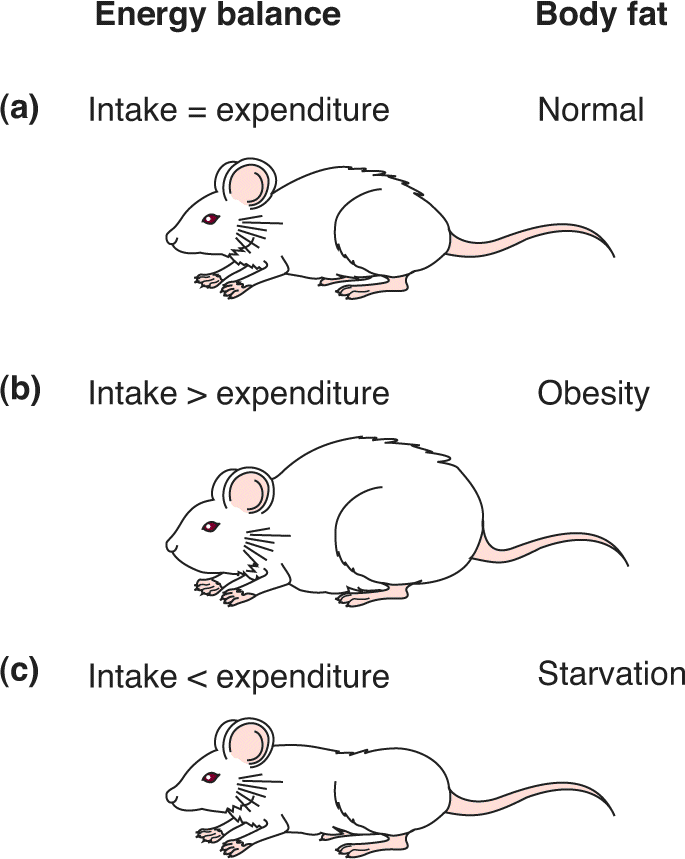
During the fasting condition between meals, called the postabsorptive state, stored glycogen and triglycerides are broken down to provide the body with a continuous supply of the molecules used as fuel for cellular metabolism (glucose for all cells, and fatty acids and ketones for all cells other than neurons). The process of breaking down complex macromolecules is called catabolism, or catabolic metabolism; it is the opposite of anabolism. The system is in proper balance when energy reserves are replenished at the same average rate that they are expended. If the intake and storage of energy consistently exceed the usage, the amount of body fat, or adiposity, increases, eventually resulting in obesity. (The word obese is derived from the Latin word for “fat.”) If the intake of energy consistently fails to meet the body’s demands, loss of fat tissue occurs, eventually resulting in starvation. Figure 16.2 summarizes the concept of energy balance and body fat.

FIGURE 16.2 Energy balance and body fat. (a) Normal energy balance leads to normal adiposity. (b) Prolonged positive energy balance leads to obesity. (c) Prolonged negative energy balance leads to starvation. Description
For the system to stay in balance, there must be some means of regulating feeding behavior, based on the size of the energy reserves and their rate of replenishment. In recent decades, research has made substantial progress in understanding the various means by which this regulation occurs—and none too soon, because eating disorders and obesity are widespread health problems. It is now apparent that there are multiple regulatory mechanisms, some acting over a long period of time to maintain the body’s fat reserves, and others acting over a shorter time period to regulate meal size and frequency. We begin our investigation by looking at long-term regulation.
Hormonal and Hypothalamic Regulation of Body Fat and Feeding
The study of the homeostatic regulation of feeding behavior has a long history, but the pieces of the puzzle have only recently fallen into place. As we will see, feeding is stimulated when neurons in the hypothalamus detect a drop in the level of a hormone released by fat cells. These hypothalamic cells are concentrated in the periventricular zone; those neurons that incite feeding behavior are in the lateral hypothalamus.
Body Fat and Food Consumption. If you’ve ever dieted, you don’t need to be told that the body works hard to frustrate any efforts to alter adiposity. Consider Figure 16.3, showing that a rat can be induced to lose body fat by severely restricting its caloric intake. However, once free access to food is restored, the animal overeats until the original level of body fat has fully returned. It also works the other way around. Animals force fed in order to gain fat mass, once given the chance to regulate their own diet, eat less until their fat levels return to normal. The heavy rat’s behavioral response is obviously not a reflection of vanity; it is a mechanism for maintaining energy homeostasis. The idea that the brain monitors the amount of body fat and acts to “defend” this energy store against perturbations, first proposed in 1953 by British scientist Gordon Kennedy, is called the lipostatic hypothesis.

FIGURE 16.3 The maintenance of body weight around a set value. Body weight is normally very stable. Weight lost during a period of starvation is rapidly gained when food is freely available. Similarly, if an animal is force fed, it will gain weight, but the weight is lost as soon as the animal can regulate its own food intake. Description
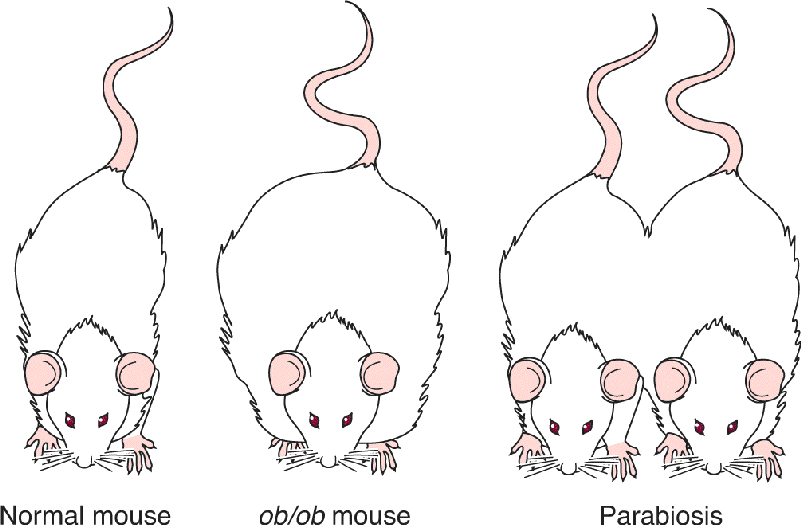
The connection between body fat and feeding behavior suggests that there must be communication from adipose tissue to the brain. A bloodborne hormonal signal was immediately suspected, and this suspicion was confirmed in the 1960s by Douglas Coleman and his colleagues at the Jackson Laboratories in Bar Harbor, Maine, working with genetically obese mice. The DNA of one strain of an obese mouse lacked both copies of a gene called ob (these mice are therefore called ob/ob mice). Coleman hypothesized that the protein encoded by the ob gene is the hormone telling the brain that fat reserves are normal. Thus, in the ob/ob mice that lack this hormone, the brain is fooled into thinking that the fat reserves are low, and the animals are abnormally motivated to eat. To test this idea, a parabiosis experiment was performed. Parabiosis is the long-term anatomical and physiological union of two animals, as in Siamese twins. Fusion can also be achieved surgically, thereby resulting in parabiosed animals sharing a common blood supply. Coleman and his colleagues found that when ob/ob animals were parabiosed with normal mice, their feeding behavior and obesity were greatly reduced, as if the missing hormone had been replaced (Figure 16.4).

FIGURE 16.4 The regulation of body fat by a circulating hormone. If a genetically obese ob/ob mouse is surgically fused with a normal mouse so that bloodborne signals are shared between the animals, the obesity of the ob/ob mouse is greatly moderated.
Then the search was on for the protein encoded by the ob gene. In 1994, a group of scientists led by Jeffrey Friedman at Rockefeller University finally isolated the protein, which they called leptin (from the Greek for “slender”). Treating ob/ob mice with leptin completely reverses the obesity and the eating disorder (Figure 16.5). The hormone leptin, released by adipocytes (fat cells), regulates body mass by acting directly on neurons of the hypothalamus that decrease appetite and increase energy expenditure.

FIGURE 16.5 The reversal of obesity in ob/ob mice by leptin. Both of these mice have a defect in the ob gene that encodes the fat hormone leptin. The animal on the right received daily hormone replacement treatment, which prevented the obesity that is apparent in the animal on the left. (Courtesy of John Sholtis, Rockefeller University.)
Well-fed humans tend to focus on how increased leptin can fight obesity (Box 16.1). However, more significant for survival is how leptin depletion fights starvation. Leptin deficiency stimulates hunger and feeding, suppresses energy expenditure, and inhibits reproductive competence—adaptive responses when food is scarce and energy reserves are low.
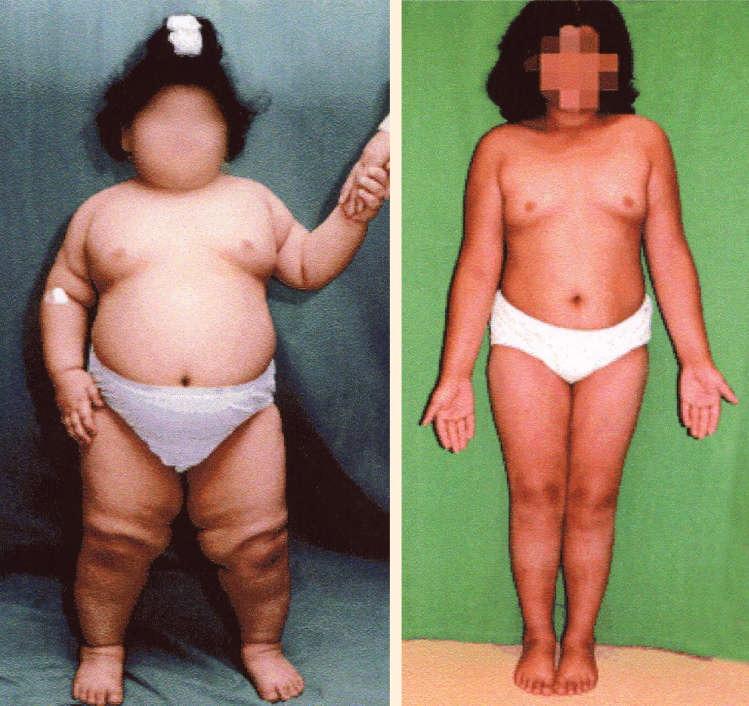
Like the ob/ob mouse, humans lacking leptin crave food, have slowed metabolism, and become morbidly obese. For these individuals, leptin replacement therapy can be a “miracle cure” (Figure A). While mutations affecting the leptin gene are rare, there is evidence for a genetic basis for many forms of human obesity. The hereditability of obesity is equivalent to that of height and greater than that of many other conditions, including heart disease and breast cancer. Many genes are involved, and the search is on to discover them.

Figure A The effect of hormone replacement in a leptin-deficient human. Daily leptin treatment begun at age 5 (left) brought this girl’s weight down to a near-normal level, shown here at age 9 (right). (Source: Gibson, et al., 2004, p. 4823.)
Obesity is a major human health problem. In the United States, two-thirds of the population is overweight, and millions are morbidly obese. Many obese people experience intense cravings for food but at the same time have slowed metabolism. In the case of leptin deficiency, the brain and body respond as if the person is starving, despite massive obesity.
Leptin offered tremendous promise as a treatment for obesity. By supplementing leptin, the logic went, the brain could be fooled into decreasing appetite and increasing metabolism. Unfortunately, other than the rare individuals who congenitally lack the hormone, most obese patients have failed to respond to leptin therapy. Indeed, many have been found to have abnormally elevated blood levels of leptin. It appears that the problem for these patients stems from decreased sensitivity of brain neurons to the leptin circulating in the blood. The problem can arise from decreased penetration of the leptin through the blood-brain barrier, reduced expression of the leptin receptor in the neurons of the periventricular hypothalamus, or altered CNS responses to changes in hypothalamic activity. Intensive efforts are now underway to identify drug targets in the feeding circuits of the brain that are downstream of leptin.
The Hypothalamus and Feeding. A.W. Hetherington and S.W. Ranson of Northwestern University made the seminal discovery, published in 1940, that small lesions made on both sides of a rat’s hypothalamus can have large effects on subsequent feeding behavior and adiposity. Bilateral lesions of the lateral hypothalamus caused anorexia, a severely diminished appetite for food. In contrast, bilateral lesions of the ventromedial hypothalamus caused the animals to overeat and become obese (Figure 16.6). This basic scenario applies to humans as well. Anorexia caused by damage to the lateral hypothalamus is commonly referred to as the lateral hypothalamic syndrome; overeating and obesity caused by lesions to the ventromedial hypothalamus is called the ventromedial hypothalamic syndrome.

FIGURE 16.6 Altered feeding behavior and body weight resulting from bilateral lesions of the rat hypothalamus. (a) The lateral hypothalamic syndrome, characterized by anorexia, is caused by lesions of the lateral hypothalamus. (b) The ventromedial hypothalamic syndrome, characterized by obesity, is caused by lesions of the ventromedial hypothalamus. Description
For a time, it was a popular idea that the lateral hypothalamus is a “hunger center” acting in opposition to the ventromedial hypothalamus “satiety center” and that lesions of the medial or lateral hypothalamus bring the system out of balance. Destruction of the lateral hypothalamus leaves the animals inappropriately satiated, so they do not eat; destruction of the ventromedial hypothalamus leaves the animals insatiable, so they overeat. However, this “dual center” model has proven to be overly simplistic. We now have a better idea why hypothalamic lesions affect body fat and feeding behavior; it has much to do with leptin signaling.
The Effects of Elevated Leptin Levels on the Hypothalamus. Although still sketchy in places, a picture is beginning to emerge of how the hypothalamus participates in body fat homeostasis. First, let’s consider the response when leptin levels are high, as they are right after several days of “forced” holiday feasting.
Circulating leptin molecules, released into the bloodstream by fat cells, activate leptin receptors on neurons of the arcuate nucleus of the hypothalamus, which lies near the base of the third ventricle (Figure 16.7). The arcuate neurons that are activated by a rise in blood leptin levels contain peptide neurotransmitters called αMSH and CART, and the levels of these peptides in the brain vary in proportion to the level of leptin in the blood. (To explain the alphabet soup: Peptides are often named by their first discovered function, and these names can lead to confusion when other roles are recognized. Therefore, neuropeptides are usually referred to simply by their abbreviations. For the record, αMSH stands for alpha-melanocyte-stimulating hormone, and CART stands for cocaine- and amphetamine-regulated transcript. Like other neurotransmitters, the functional role of these molecules depends on the circuits in which they participate.)

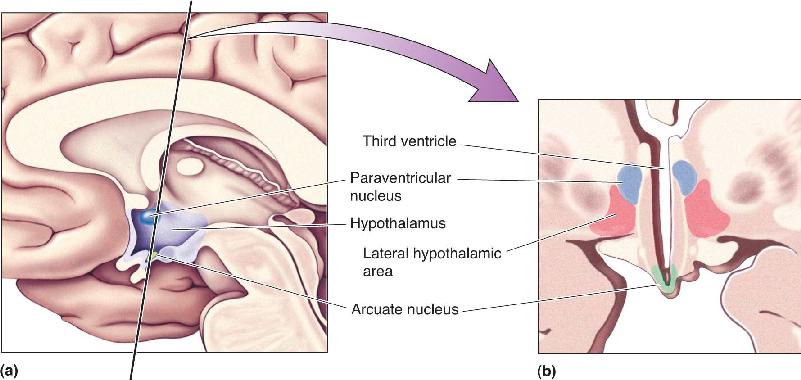
FIGURE 16.7 Hypothalamic nuclei important for the control of feeding. (a) A midsagittal view of the human brain, showing the location of the hypothalamus. (b) A coronal section, taken in the plane indicated in part a, showing three important nuclei for the control of feeding: the arcuate nucleus, the paraventricular nucleus, and the lateral hypothalamic area. Description
Before going any further, let’s take a moment to consider the body’s integrated response to excessive adiposity, high leptin levels, and activation of the αMSH/CART neurons of the arcuate nucleus. The humoral response consists of increased secretion of TSH (thyroid-stimulating hormone) and ACTH (adrenocorticotropic hormone) (see Table 15.1 in Chapter 15). These pituitary hormones act on the thyroid and adrenal glands and have the effect of raising the metabolic rate of cells throughout the body. The visceromotor response increases the tone of the sympathetic division of the ANS, which also raises metabolic rate, in part by raising body temperature. The somatic motor response decreases feeding behavior. The αMSH/CART neurons of the arcuate nucleus project their axons directly to the regions of the nervous system that orchestrate this coordinated response (Figure 16.8).

FIGURE 16.8 The response to elevated leptin levels. A rise in leptin levels in the blood is detected by neurons in the arcuate nucleus that contain the peptides αMSH and CART. These neurons project axons to the lower brain stem and spinal cord, the paraventricular nuclei of the hypothalamus, and the lateral hypothalamic area. Each of these connections contributes to the coordinated humoral, visceromotor, and somatic motor responses to increased leptin levels. (Source: Adapted from Sawchenko, 1998, p. 437.) Description
αMSH/CART neurons trigger the humoral response by the activating neurons in the paraventricular nucleus of the hypothalamus, which in turn causes the release of the hypophysiotropic hormones that regulate the secretion of TSH and ACTH from the anterior pituitary (see Chapter 15). The paraventricular nucleus also controls the activity of the sympathetic division of the ANS with direct axonal projections to neurons in the lower brain stem and to preganglionic neurons in the spinal cord. Additionally, there is also a direct path for arcuate control of the sympathetic response: The αMSH and CART neurons themselves project axons directly down to the intermediolateral gray matter of the spinal cord. Finally, feeding behavior is inhibited via connections of the arcuate nucleus neurons with cells in the lateral hypothalamus. We will take a closer look at the lateral hypothalamus in a moment.
The injection of αMSH or CART into the brain mimics the response to elevated leptin levels. Thus, these are said to be anorectic peptides; they diminish appetite. The injection of drugs that block the actions of these peptides increases feeding behavior. These findings suggest that αMSH and CART normally participate in the regulation of energy balance, in part by acting as the brain’s own appetite suppressants.
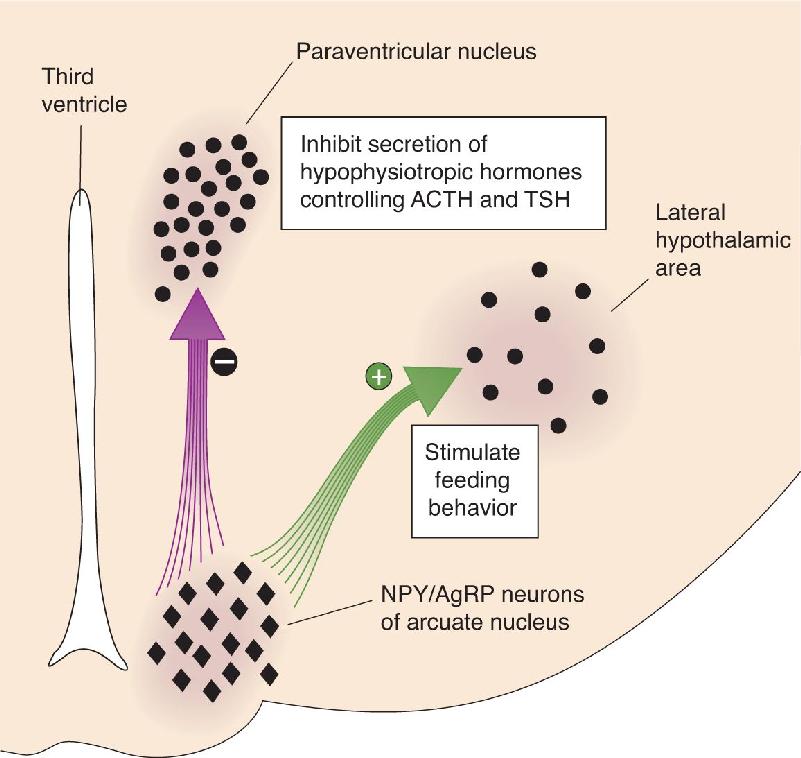
The Effects of Decreased Leptin Levels on the Hypothalamus. In addition to turning off the responses mediated by αMSH/CART neurons, a fall in leptin levels actually stimulates another type of arcuate nucleus neuron. These neurons contain their own mix of peptides: NPY (neuropeptide Y) and AgRP (agouti-related peptide). The NPY/AgRP neurons of the arcuate nucleus also have connections with the paraventricular nucleus and the lateral hypothalamus (Figure 16.9), and the effects of these neuropeptides on energy balance are the opposite of the effects caused by the αMSH/CART neurons. NPY and AgRP inhibit the secretion of TSH and ACTH, they activate the parasympathetic division of the ANS, and they stimulate feeding behavior. They are therefore referred to as orexigenic peptides (from the Greek for “appetite”). The brain’s coordinated response to changing leptin levels is summarized in Figure 16.10.

FIGURE 16.9 The response to decreased leptin levels. A reduction in blood levels of leptin is detected by neurons in the arcuate nucleus that contain the peptides NPY and AgRP. These arcuate nucleus neurons inhibit the neurons in the paraventricular nuclei that control the release of TSH and ACTH from the pituitary. In addition, they activate the neurons in the lateral hypothalamus that stimulate feeding behavior. Some of the activated lateral hypothalamic neurons contain the peptide MCH (melanin-concentrating hormone). Description

FIGURE 16.10 Summary of the responses to increased and decreased adiposity (fat). The arcuate nucleus senses changes in blood levels of leptin. A rise in leptin increases activity in the aMSH/CART neurons, and a fall in leptin increases activity in the NPY/AgRP neurons. These two populations of arcuate nucleus neurons orchestrate the humoral, visceromotor, and somatic motor responses to increased or decreased adiposity, respectively. Description
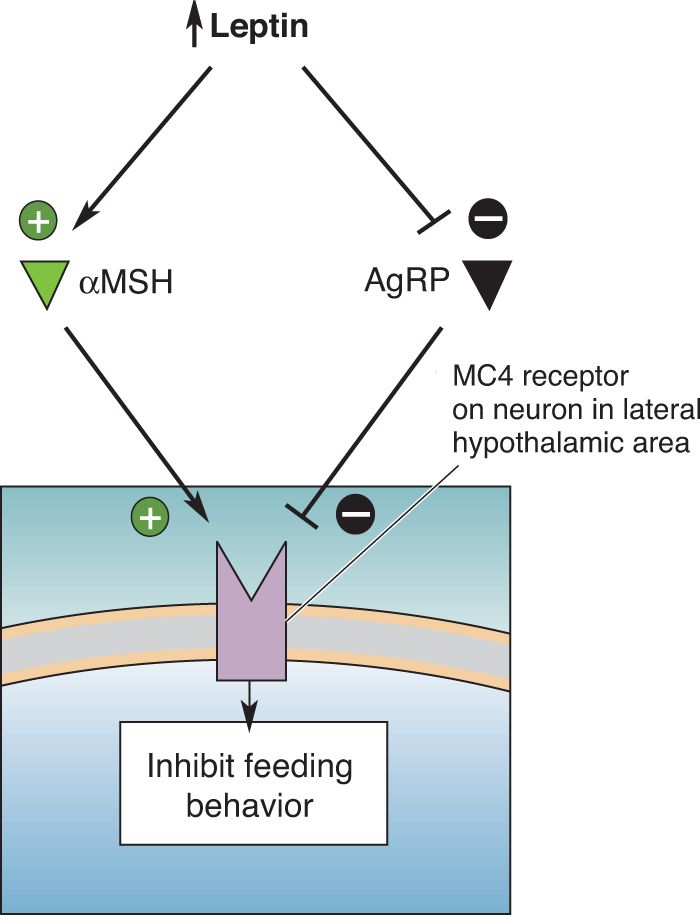
AgRP and αMSH are literally antagonistic neurotransmitters. Both peptides bind to the MC4 receptor on postsynaptic neurons in the hypothalamus. αMSH activates the receptor and AgRP inhibits it. Activation of MC4 receptors on lateral hypothalamic neurons inhibits feeding; inhibiting the receptors stimulates feeding (Figure 16.11).

FIGURE 16.11 Competition for activation of the MC4 receptor. One way that αMSH, an anorectic peptide, and AgRP, an orexigenic peptide, exert opposite effects on metabolism and feeding behavior is via an interaction with the MC4 receptor on some hypothalamic neurons. While αMSH stimulates the MC4 receptor, AgRP inhibits it. Description
The Control of Feeding by Lateral Hypothalamic Peptides. We now come to the mysterious lateral hypothalamus, which appears to have a special role in motivating us to eat. Because this region of the brain is not organized into well-defined nuclei, it has the nondistinctive name lateral hypothalamic area (see Figure 16.7). As mentioned earlier, the first indication that the lateral hypothalamus is involved in motivating feeding behavior was that a lesion here causes animals to stop eating. Moreover, electrical stimulation of this area triggers feeding behavior, even in satiated animals. These basic findings apply to all mammals that have been examined, including humans. However, crude lesions and electrical stimulation not only affect the neurons with cell bodies in this region but also affect many different axonal pathways passing through the lateral hypothalamus. Modern experiments using optogenetic methods to stimulate and silence specific types of neurons (see Chapter 4) reveal that both neurons intrinsic to the lateral hypothalamus and axons passing through the lateral hypothalamus contribute to the motivation of feeding behavior. Let’s first concentrate on the role of the neurons within the lateral hypothalamic area.
One group of neurons in the lateral hypothalamus that receives direct input from the leptin-sensitive cells of the arcuate nucleus has yet another peptide neurotransmitter called MCH (melanin-concentrating hormone). These cells have extremely widespread connections in the brain, including direct monosynaptic innervation of most of the cerebral cortex. The cortex is involved in organizing and initiating goal-directed behaviors, such as raiding the refrigerator. The MCH system is in a strategic position to inform the cortex of leptin levels in the blood and therefore could contribute significantly to motivating the search for food. Supporting this idea, the injection of MCH into the brain stimulates feeding behavior. Moreover, mutant mice that lack this peptide exhibit reduced feeding behavior, have an elevated metabolic rate, and are lean.
A second population of lateral hypothalamic neurons with widespread cortical connections has been identified, containing another peptide called orexin. These cells also receive direct inputs from the arcuate nucleus. As is the case for MCH, and as the name suggests, orexin is an orexigenic peptide (i.e., it stimulates feeding behavior). The levels of both MCH and orexin rise in the brain when leptin levels in the blood fall. These two peptides are complementary, not redundant. For example, orexin promotes meal initiation, whereas MCH prolongs consumption. Additionally, orexin, also called hypocretin, plays a very important role in the regulation of wakefulness. As we will learn in Chapter 19, gene mutations that disable orexin (hypocretin) signaling not only lead to weight loss but also to excessive daytime sleepiness. Perhaps it is obvious that sleep inhibits feeding behavior; after all, it is difficult to eat when you are asleep. However, you might be surprised to learn that insomnia and obesity also often go together. Orexin (hypocretin) provides an interesting link between these conditions.
To conclude this section, let’s briefly summarize hypothalamic responses to blood leptin levels. Remember, leptin levels rise when body fat is increased, and they fall when body fat is decreased:
- A rise in leptin levels stimulates the release of αMSH and CART from arcuate nucleus neurons. These anorectic peptides act on the brain, in part by activating the MC4 receptor, to inhibit feeding behavior and increase metabolism.
- A fall in leptin levels stimulates the release of NPY and AgRP from arcuate nucleus neurons, and the release of MCH and orexin from neurons in the lateral hypothalamic area. These orexigenic peptides act on the brain to stimulate feeding behavior and decrease metabolism.
Regulation of the tendency to seek and consume food by the body’s levels of leptin is very important, but it is not the whole story. Setting aside social and cultural factors (such as a mother’s command: “Eat!”), the motivation to eat depends on how long it has been since the last meal and how much we ate at that time. Moreover, the motivation to continue eating once a meal starts depends on how much food (and what type) has already been eaten. These are examples of what we are calling the short-term regulation of feeding behavior.
A useful way to think about this regulatory process is to imagine that the drive to eat, which may vary rather slowly with the rise and fall of leptin, is increased by orexigenic signals generated in response to a period of fasting, and inhibited by satiety signals that occur when we eat and begin the process of digestion (i.e., the prandial period). These satiety signals both terminate the meal and inhibit feeding for some time afterward. During this postabsorptive (fasting) period, the satiety signals slowly dissipate, and the orexigenic signals build, until the drive to eat again takes over (Figure 16.12). We will use this model to explore the biological basis for the short-term regulation of feeding behavior.

FIGURE 16.12 A hypothetical model for the short-term regulation of feeding behavior. This graph shows a possible means of regulating food consumption by satiety signals. Satiety signals rise in response to feeding. When satiety signals are high, food consumption is inhibited. When the satiety signals fall to zero, the inhibition is eliminated, and food consumption ensues. Description
You have awakened in the morning after a long night’s slumber. You come to the kitchen to find pancakes cooking on the stove; when they are ready, you enthusiastically eat them until you’re satiated. Your body’s reactions during this process can be divided into three phases: cephalic, gastric, and substrate (also called the intestinal phase):
- Cephalic phase. The sight and smell of the pancakes trigger a number of physiological processes that anticipate the arrival of breakfast. The parasympathetic and enteric divisions of the ANS are activated, causing the secretion of saliva into your mouth and digestive juices into your stomach.
- Gastric phase. These responses grow much more intense, when you start chewing, swallowing, and filling your stomach with food.
- Substrate phase. As your stomach fills and the partially digested pancakes move into your intestines, nutrients begin to be absorbed into your bloodstream.
As you pass through these phases, signals that motivate consumption of the pancakes are replaced by those that terminate your meal. Let’s look at some of the orexigenic and satiety signals that shape eating behavior during a meal (Box 16.2).
A well-known consequence of marijuana intoxication is stimulation of appetite, an effect known by users as “the munchies.” The active ingredient in marijuana is D9-tetrahydrocannabinol (THC), which alters neuronal functions by stimulating a receptor called cannabinoid receptor 1 (CB1). CB1 receptors are abundant throughout the brain, so it is overly simplistic to view these receptors as serving only appetite regulation. Nevertheless, “medical marijuana” is often prescribed (where legal) as a means to stimulate appetite in patients with chronic diseases, such as cancer and AIDS. A compound that inhibits CB1 receptors, rimonabant, was also developed as an appetite suppressant. However, human drug trials had to be discontinued because of psychiatric side effects. Although this finding underscores the fact that these receptors do much more than mediate the munchies, it is still of interest to know where in the brain CB1 receptors act to stimulate appetite. Not surprisingly, the CB1 receptors are associated with neurons in many regions of the brain that control feeding, such as the hypothalamus, and some of the orexigenic effects of THC are related to changing the activity of these neurons. However, neuroscientists were surprised to learn in 2014 that much of the appetite stimulation comes from enhancing the sense of smell, at least in mice. Collaborative research conducted by neuroscientists in France and Spain, countries incidentally known for their appreciation of good tastes and smells, revealed that activation of CB1 receptors in the olfactory bulb increases odor detection and is necessary for the increase in food intake stimulated in hungry mice by cannabinoids.
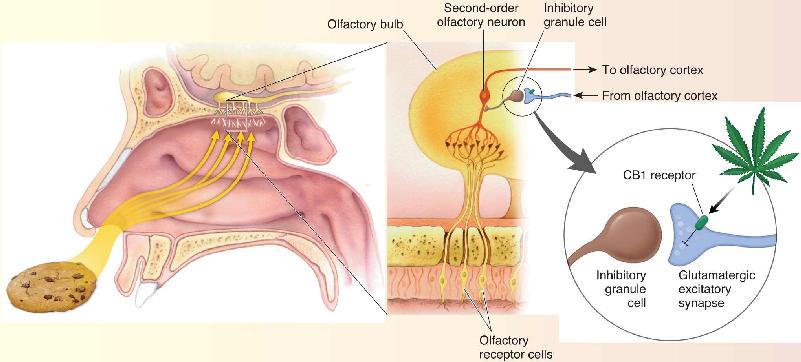
In Chapter 8, we discussed how smells activate neurons in the olfactory bulb which, in turn, relay information to the olfactory cortex. The cortex also sends feedback projections to the bulb that synapse on inhibitory interneurons called granule cells. By activating the inhibitory granule cells, this feedback from the cortex dampens ascending olfactory activity. These corticofugal synapses use glutamate as a neurotransmitter. The brain’s own endocannabinoids (anandamide and 2-arachidonoylglycerol) are synthesized under fasting conditions, and they inhibit glutamate release by acting on CB1 receptors on the corticofugal axon terminals. Reducing granule cell activation by glutamate in the bulb has the net effect of enhancing the sense of smell (Figure A). It remains to be determined if the munchies arise from enhanced olfaction in marijuana users, but a simple experiment, such as holding your nose while eating, confirms that much of the hedonic value of food derives from the sense of smell.

Figure A Activation of CB1 receptors by THC, the psychoactive ingredient in marijuana, enhances olfaction by suppressing the release of glutamate from corticofugal inputs to inhibitory granule cells in the olfactory bulb. (Source: Adapted from Soria-Gomez et al., 2014.) Description
Ghrelin. You don’t need to be told that the meal begins because you are hungry. Until recently, scientists believed that hunger was merely the absence of satiety. This view changed in 1999 with the discovery of a peptide called ghrelin. Ghrelin was isolated originally as a factor that stimulates growth hormone release. However, researchers quickly found that the peptide is highly concentrated in the stomach and is released into the bloodstream when the stomach is empty. Your “growling” stomach releases ghrelin (“ghrrrrrrrelin”). Intravenous administration of ghrelin strongly stimulates appetite and food consumption by activating the NPY/AgRP-containing neurons of the arcuate nucleus (the same neurons activated by a drop in leptin in the bloodstream).
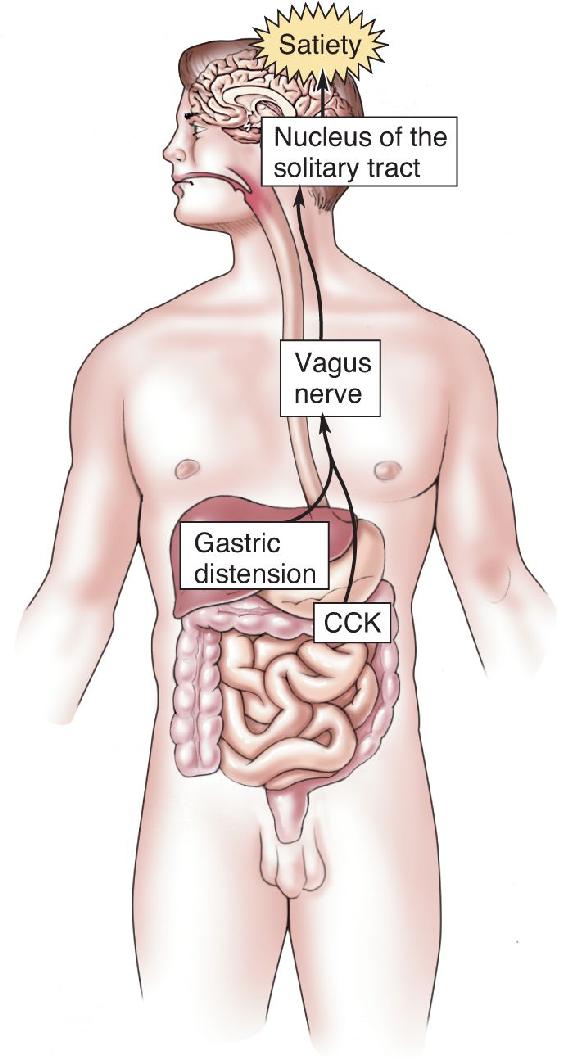
Gastric Distension. We all know what it is like to feel “full” after a big meal; as you might expect, the stretching of the stomach wall is a powerful satiety signal. The stomach wall is richly innervated by mechanosensory axons, and most of these ascend to the brain via the vagus nerve. Recall from the appendix to Chapter 7 that the vagus nerve (cranial nerve X) contains a mixture of sensory and motor axons, originates in the medulla, and meanders through much of the body cavity (vagus is from the Latin for “wandering”). The vagal sensory axons activate neurons in the nucleus of the solitary tract in the medulla. These signals inhibit feeding behavior.
You may recall that the nucleus of the solitary tract has been mentioned several times in different contexts. The gustatory nucleus, which receives direct sensory input from the taste buds (see Chapter 8), is actually a subdivision of the nucleus of the solitary tract. The nucleus of the solitary tract is also an important center in the control of the ANS (see Chapter 15). Now we find that the same nucleus receives visceral sensory input from the vagus nerve. It is easy to see how a nucleus with such widespread connections could serve as an important integration center in the control of feeding and metabolism. As you know, satiety induced by a full stomach can be delayed quite a while if what you are eating is tasty enough.
Cholecystokinin. In the 1970s, researchers discovered that the administration of the peptide cholecystokinin (CCK) inhibits meal frequency and size. CCK is present in some of the cells that line the intestines and some of the neurons of the enteric nervous system. It is released in response to stimulation of the intestines by certain types of food, especially fatty ones. The major action of CCK as a satiety peptide is exerted on the vagal sensory axons. CCK acts synergistically with gastric distension to inhibit feeding behavior (Figure 16.13). Curiously, CCK, like many other gastrointestinal peptides, is also contained within selected populations of neurons within the central nervous system (CNS).

FIGURE 16.13 The synergistic action of gastric distension and CCK on feeding behavior. Both signals converge on axons in the vagus nerve that trigger satiety. Description
Insulin. Released into the bloodstream by the β cells of the pancreas, insulin is a vitally important hormone (Box 16.3). Although glucose is always readily transported into neurons, glucose transport into the other cells of the body requires insulin. This means that insulin is important for anabolic metabolism when glucose is transported into liver, skeletal muscle, and adipose cells for storage as well as for catabolic metabolism when the glucose liberated from storage sites is taken up as fuel by the other cells of the body. Thus, the level of glucose in the blood is tightly regulated by the level of insulin: Blood glucose levels are elevated when insulin levels are reduced; blood glucose levels fall when insulin levels rise.
Insulin, released by the β cells of the pancreas, plays a pivotal role in maintaining energy balance. After a meal, glucose levels in the blood rise. To be used by the cells of the body, glucose must be shuttled across the plasma membrane by specialized proteins called glucose transporters. In all cells other than neurons, the insertion of glucose transporters into the membrane occurs when insulin binds to cell surface insulin receptors. Thus, for the glucose to be utilized or stored by these cells, a rise in blood insulin levels must accompany the rise in blood glucose levels. In the clinical condition known as diabetes mellitus, defects in insulin production and release, or in the cellular response to insulin, prevent the normal reaction to elevated glucose. The consequence is elevated blood sugar levels (hyperglycemia) because the glucose absorbed from the intestines cannot be taken up by the cells of the body (other than neurons). The excess glucose passes to the urine, making it sweet. Indeed, the disorder’s name is from the Latin for “siphoning honey.”
An effective treatment for some types of diabetes mellitus is hypodermic injections of insulin. However, this treatment has risks. An overdose of insulin causes blood glucose levels to plummet (hypoglycemia), starving the neurons of the brain. The resulting condition is called insulin shock, characterized by sweating, tremor, anxiety, dizziness, and double vision. If it is not corrected promptly, these early signs are followed by delirium, convulsions, and loss of consciousness. The sudden neurological response to hypoglycemia illustrates how vital energy balance is for the normal functioning of the brain (Figure A).

Figure A A PET image superimposed on an MRI image of the human body. The hot colors (red to yellow) show regions with high glucose utilization. Note that the brain, even at rest, has a very high demand for fuel. When glucose levels in the blood fall, as they do during insulin shock, brain functions are very rapidly lost. (Source: Siemens Healthcare and Professor Marcus Raichle, Washington University, St. Louis.)
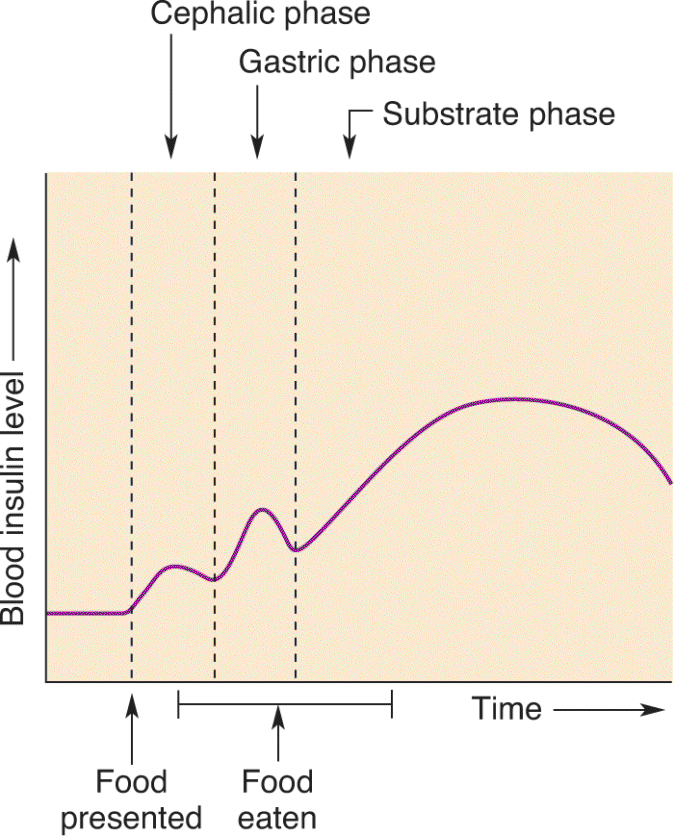
Insulin release by the pancreas is controlled in a number of ways (Figure 16.14). Consider the example of your pancake breakfast. During the cephalic phase, when you are anticipating food, the parasympathetic innervation of the pancreas (delivered by the vagus nerve) stimulates the β cells to release insulin. In response, blood glucose levels fall slightly, and this change, detected by the neurons of the brain, increases your drive to eat (in part, by activation of the NPY/AgRP neurons of the arcuate nucleus). During the gastric phase, when food enters your stomach, insulin secretion is stimulated further by gastrointestinal hormones, such as CCK. Insulin release is maximal when the food is finally absorbed in the intestines and blood glucose levels rise, during the substrate phase. Indeed, the primary stimulus for insulin release is increased blood glucose levels. This rise in insulin, coupled with the elevated blood glucose levels, is a satiety signal and causes you to stop eating.

FIGURE 16.14 Changes in blood insulin levels before, during, and after a meal. (Source: Adapted from Woods and Stricker, 1999, p. 1094.) Description
In contrast to the other satiety signals we’ve discussed, which communicate with the brain mainly via the vagus nerve, bloodborne insulin acts to inhibit feeding behavior by acting directly on the arcuate and ventromedial nuclei of the hypothalamus. It appears that insulin acts in much the same way as leptin to regulate feeding behavior.
We have talked about the signals that motivate feeding behavior, but we still have not discussed what that really means in psychological terms. Obviously, we eat because we like food. This aspect of motivation is hedonic: It feels good, so we do it. We derive pleasure from the taste, smell, sight, and feel of food and from the act of eating. However, we also eat because we are hungry and we want food. This aspect of motivation can be considered as a drive reduction: satisfying a craving. A reasonable assumption is that “liking” and “wanting” are two aspects of a unified process; after all, we typically crave food that we like. However, research on humans and animals suggests that liking and wanting are mediated by separate circuits in the brain.
In experiments performed in the early 1950s, James Olds and Peter Milner at McGill University in Montreal, Canada, implanted electrodes in the brains of rats to investigate the effect of electrical brain stimulation on the animals’ behavior. The animals were allowed to freely explore a box about 3 ft2. Every time the rats wandered into one corner of the box, the researchers delivered brain stimulation. They observed that when the electrodes were lodged in certain parts of the brain, the stimulation appeared to cause the animals to spend all their time in the corner that led to stimulation. In a brilliant twist on this experiment, Olds and Milner set up a new box with a lever on one side that, when depressed, caused the brain to be stimulated (Figure 16.15). At first, the rats wandered about the box, stepping on the lever occasionally by accident. But before long, the rats were pressing the lever repeatedly to receive the electrical stimulation. This behavior is called electrical self-stimulation. Sometimes the rats would become so involved in pressing the lever that they would shun food and water, stopping only after collapsing from exhaustion (Box 16.4).

FIGURE 16.15 Electrical self-stimulation by a rat. When the rat presses the lever, it receives a brief electrical current to an electrode in its brain. Description
To determine the sensations evoked by brain stimulation, it would be desirable to stimulate a person’s brain by inserting electrodes and ask how it feels. Obviously, this is not normally feasible or ethical. However, as treatments of last resort for debilitating medical conditions, humans have occasionally been fitted with intracranial electrodes they can self-stimulate. Let’s consider two patients studied by Robert Heath at the Tulane University School of Medicine in the 1960s.
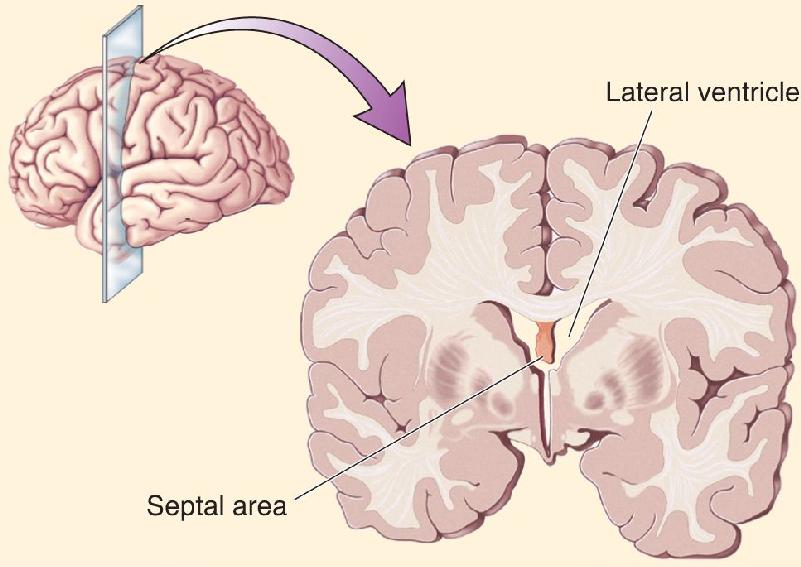
The first patient had severe narcolepsy; he would abruptly go from being awake into a deep sleep. (Narcolepsy and sleep will be discussed in Chapter 19.) The condition significantly interfered with his life and obviously made it difficult to hold a job. He was implanted with 14 electrodes in different areas of the brain in the hope of finding a self-stimulation site that might keep him alert. When he stimulated his hippocampus, he reported feeling mild pleasure. Stimulation of his midbrain tegmentum made him feel alert but unpleasant. The site he chose to frequently self-stimulate was the septal area of the forebrain (Figure A). Stimulating this area made him more alert and gave him a good feeling, which he described as building toward orgasm. He reported that he would sometimes push the button over and over, trying unsuccessfully to achieve orgasm, ultimately ending in frustration.

Figure A The septal area, a site of electrical self-stimulation in humans, is in the rostral forebrain below the lateral ventricle.
The second patient’s case is a bit more complex. This person had electrodes implanted at 17 brain sites in the hope of learning something about the location of his severe epilepsy. He reported pleasurable feelings with stimulation of the septal area and the midbrain tegmentum. Consistent with the first case above, septal stimulation was associated with sexual feelings. The midbrain stimulation gave him a “happy drunk” feeling. Other mildly positive feelings were produced by stimulation of the amygdala and caudate nucleus. Interestingly, the site he most frequently stimulated was in the medial thalamus, even though stimulation here induced an irritable feeling, one that was less pleasurable than stimulation at other locations. The patient stated that he stimulated this area the most because it gave him the feeling he was about to recall a memory. He repeated the stimulation in a futile attempt to fully bring the memory into his mind, even though, in the end, this process proved to be frustrating.
These two specific cases and many others suggest that self-stimulation is not synonymous with pleasure. Often some reward or anticipated reward is associated with the stimulation, but the experience is not always pleasant.
Electrical self-stimulation appeared to provide a reward that reinforced the habit to press the lever. By systematically moving the stimulating electrode to different regions of the brain, researchers were able to identify specific sites that were reinforcing. It became apparent that the most effective sites for self-stimulation fell along the trajectory of dopaminergic axons arising in the ventral tegmental area, projecting through the lateral hypothalamus to several forebrain regions (Figure 16.16). Drugs that block dopamine receptors reduced self-stimulation, suggesting that the animals were working to stimulate the release of dopamine in the brain. This idea was further supported when researchers discovered that animals will press a lever to receive an injection of amphetamine, a drug that releases dopamine in the brain. Although there is more to electrical self-stimulation than dopamine, there is little question that dopamine release in the brain will reinforce the behavior that causes it. These experiments suggested a mechanism by which natural rewards (food, water, sex) reinforce particular behaviors. Indeed, a hungry rat will press a lever to receive a morsel of food, and this response is also greatly reduced by dopamine receptor blockers.

FIGURE 16.16 The mesocorticolimbic dopamine system. Animals are motivated to behave in ways that stimulate the release of dopamine in the basal forebrain area. Description
For many years, this dopamine projection, from the ventral tegmental area to the forebrain, was believed to serve hedonic reward—in other words, pleasure. In the case of feeding, it was believed that dopamine was released in response to palatable foods, making the sensation pleasurable. Animals were motivated to seek palatable food for the hedonic reward: a squirt of dopamine in the forebrain.
However, this simple idea has been challenged in recent years. Destruction of the dopamine axons passing through the lateral hypothalamus fails to reduce the hedonic responses to food, even though animals stop eating. If a tasty morsel is placed on the tongue of a rat that has sustained such a lesion, the animal will still behave as if the food evokes a pleasurable sensation (the rat equivalent of lip smacking), and the morsel will be consumed. The dopamine-depleted animal behaves as though it likes food but does not want food. The animal apparently lacks the motivation to seek food, even though it seems to enjoy it when it is available. Conversely, stimulation of the dopamine axons in the lateral hypothalamus of normal rats appears to produce a craving for food without increasing the food’s hedonic impact. Not surprisingly, recent research on the cravings associated with addiction (to drugs and alcohol, as well as to chocolate) has focused on the role of this dopaminergic pathway (Box 16.5). It is no coincidence that some of the most highly addictive drugs (cocaine and amphetamine, for example) act directly on dopamine synapses in the brain.
What do the drugs heroin, nicotine, and cocaine have in common? They act on different neurotransmitter systems in the brain—heroin on the opiate system, nicotine on the cholinergic system, and cocaine on the dopaminergic and noradrenergic systems—and they produce different psychoactive effects. However, all three drugs are highly addictive. This common quality is explained by the fact that they all act on the brain circuitry that motivates behavior—in this case, drug-seeking behavior. We can learn much about the brain mechanisms of motivation by studying drug addiction and vice versa.
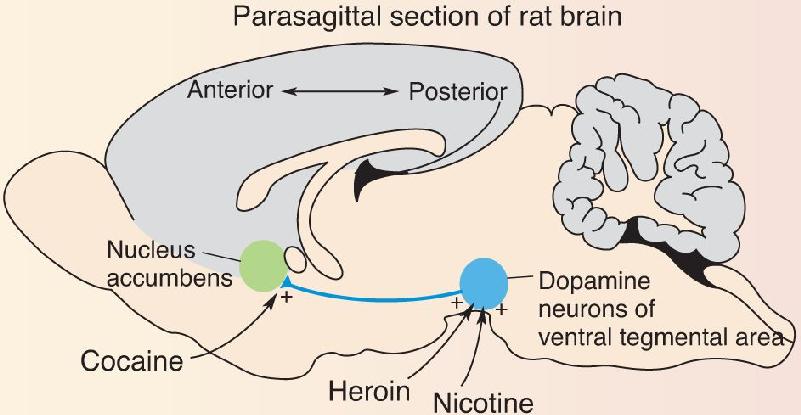
Rats, like humans, will self-administer drugs and will develop clear signs of drug dependence. Studies using microinfusions of drugs directly into the brain have mapped out the sites where the drugs cause addiction. In the case of heroin and nicotine, the key site of action is the ventral tegmental area (VTA), home of the dopamine neurons that project axons through the lateral hypothalamus to the forebrain. These dopaminergic neurons have both opiate and nicotinic acetylcholine receptors. In the case of cocaine, a key site of action is the nucleus accumbens, one of the major targets of the ascending dopaminergic axons in the forebrain (Figure A). Recall from Chapter 15 that cocaine prolongs the actions of dopamine at its receptors. Thus, these three drugs either stimulate dopamine release (heroin, nicotine) or enhance dopamine actions (cocaine) in the nucleus accumbens.

Figure A Addictive drugs act on the dopaminergic pathway from the ventral tegmental area to the nucleus accumbens. (Source: Adapted from Wise, 1996, p. 248, Fig. 1.) Description
The exact role of dopamine in motivating behavior continues to be explored. However, much evidence suggests that animals are motivated to perform behaviors that stimulate dopamine release in the nucleus accumbens and related structures. Behaviors associated with the delivery of drugs that act to stimulate dopamine release are therefore strongly reinforced. However, chronic overstimulation of this pathway causes a homeostatic response: The dopamine “reward” system is downregulated. This adaptation leads to the phenomenon of drug tolerance; it takes more and more of the drug to get the desired (or required) effect. Indeed, drug discontinuation in addicted animals is accompanied by a marked decrease in dopamine release and function in the nucleus accumbens. And, of course, one withdrawal symptom is the powerful craving for the discontinued drug.
Clues into how dopamine signaling influences behavior have come from animal studies in which the activity of dopamine neurons in the ventral tegmental area of the midbrain is monitored with microelectrodes. In one important study, Wolfram Schultz and colleagues at the University of Cambridge, England, explored what happens to dopamine neurons when a sip of juice is given to a monkey shortly after a light was turned on. Initially, before the monkey learned that the light predicts the delivery of juice, Schultz found that the dopamine neurons had no response to light but became briefly active when the juice was delivered. This is what one might expect if the dopamine neurons were simply registering the occurrence of a pleasurable experience. After the light and the juice were repeatedly paired, however, the dopamine neurons had changed firing patterns. They now responded briefly when the light came on but had no response when the juice was delivered. Furthermore, if Schultz and colleagues tricked the trained monkey and failed to deliver juice after the light, they found that the dopamine neuron firing decreased at the time of anticipated reward (Figure 16.17). These findings have led to the concept that activity of dopamine neurons signals errors in reward prediction: Events that are “better than expected” cause dopamine neurons to come to life, those that are “worse than expected” cause them to be inhibited, and those that occur “as expected” cause no change in firing, even if these events still provide hedonic reward (the juice still tastes good even if you have come to expect it). Behaviors that cause expected or better-than-expected outcomes are repeated; those with outcomes that are worse than expected are not.

FIGURE 16.17 Dopamine neurons in the VTA fire when reward is unexpected. Description
Just as the monkey learned that the light predicted delivery of juice, you have learned that the smell or sight of pancakes and coffee predict the delivery of breakfast. This type of learning is integral to the body’s “cephalic” preparation for ingestion of a meal. Dopamine is intimately involved in the mechanism behind this learning. Synaptic connections that are active during and shortly before a rise in dopamine are persistently changed to store this memory. While this type of learning is clearly beneficial under normal circumstances, it is hijacked during exposure to addictive drugs, often with devastating consequences. As earlier mentioned, addictive drugs have in common the fact that they act on the central dopaminergic system in the brain. By studying how synapses are modified by drug exposure, researchers have gained insight not only into the neurobiology of addiction and its possible treatments but also into how the brain creates memories (Box 16.6). We will take a closer look at the mechanisms of memory formation in Chapter 25.
After college, I was fortunate enough to work as a lab technician in Anne Bekoff’s lab at the University of Colorado. Anne studied motor pattern generators, the simple circuits in the spinal cord that allow coordinated muscle movements to take place. Anne and I researched what happens to the hatching pattern generator in chicks after the bird hatches and has no apparent further use for it. When a chick is ready to hatch from the egg, it is tightly curled with its head under the wing pointing up toward the shell. Every 20 seconds or so, it executes two strong leg movements that propel the body slightly within the egg. The beak gradually makes a circular hole, and when this is large enough, the strong leg movements allow the chick to hatch out. To test the fate of the hatching pattern generator, my job was to place recording electrodes in the leg muscles and then carefully fold an already-hatched chick back into the hatching position, this time in a glass egg. Remarkably, the chick became quiet and soon began making leg movements indistinguishable from normal hatching movements. More amazing, we found that chicks even up to 2 months old can be induced to “hatch”; the hatching pattern generator appeared to remain available even weeks after the last time it was needed. While I had a ball putting weeks-old chickens back in glass eggs, I was simultaneously hatching my own scientific approach. I developed a great appreciation for Anne’s strategy of asking simple questions that could generate thorough answers, and breaking a complex problem into smaller parts that could be understood clearly. This approach has remained a driving principle in my scientific life ever since.
How does the nervous system store information? This question has been the focus of my work since graduate school, where I first investigated the cellular basis of persistent changes in the nervous system of Aplysia, a giant sea slug [will be discussed in Chapter 25]. My fascination with long-lasting changes in neuronal excitability led me to post-doctoral work on long-term potentiation (LTP) of synaptic transmission, a recently discovered phenomenon—and I was hooked forever! Excitatory synapses, when stimulated only for a second or two, increase their strength persistently, for many hours. The opportunity to study how individual synapses are persistently modified was just what I was looking for.
To store information, the brain needs to change in response to environmental stimuli, so it makes sense that many circuits would have the capability of synaptic modification. When I began my own lab in 1991, this idea became more and more interesting to me, and led directly to our ongoing work at Brown University on circuits that underlie motivation. My best friend from graduate school, Marina Wolf, had been studying addiction-related brain alterations and suggested that drugs of abuse might alter synaptic plasticity in the motivational circuit that includes the ventral tegmental area (VTA) and nucleus accumbens. Her hunch launched our lab and others on a quest for the synaptic basis of addictive behaviors.
Animals will self-administer the same drugs that humans abuse, and their drug-seeking behavior closely resembles that of human substance abusers. Rodents will press a lever to receive cocaine, for example, and will do work or even suffer painful shocks in order to press the lever for the drug, much as substance abusers will suffer tremendous personal loss to acquire the drug. A critical idea in the field has been that drugs of abuse hijack the midbrain dopamine neurons, part of the motivational control system, and by doing so produce an overwhelming craving for the drug, analogous perhaps to the craving for water if one is deprived for a long period. Intriguingly, we found that inhibitory, GABAergic synapses on dopamine cells lost their normal ability to exhibit LTP after a single drug exposure. It had been known for some time that all drugs of abuse increase dopamine release from VTA neurons, and the loss of LTP at inhibitory synapses (and net loss of inhibition) on dopamine neurons is likely a contributing factor.
We next made two key findings. First, multiple different drugs of abuse all erased the GABAergic synapse LTP. Secondly, a brief stressor (5-minute exposure to cold water) had exactly the same effect. What could this mean, when the rewarding effects of drugs seem so different from the aversive effects of stress? Previous work had shown that in rats that had “recovered” from cocaine self-administration (they had learned that the lever press no longer caused drug delivery), either a small dose of the drug or a stressful experience restores powerful drug-seeking behavior, a process known as reinstatement. Human patients also report that minimal drug exposure or stress can trigger relapse and drug craving. It has been suggested that by activating the motivational circuitry, either drugs or stress promote drug seeking.
How could our reductionist approach of studying the details of synaptic function tell us anything about a complex disorder like drug addiction? We did many experiments to tease out which molecules and pathways are needed for stress to block LTP at the inhibitory VTA synapses. We found one molecule that was clearly required: the kappa opioid receptor. If we used an inhibitor to block kappa receptors prior to stress, we found that LTP was unaffected by the stressful experience. Thus, we had found a pharmacological tool that prevents this brain alteration triggered by acute stress. Might the kappa receptor blocker affect relapse behavior as well? Our colleagues at the University of Pennsylvania, Chris Pierce and Lisa Briand, taught rats to self-administer cocaine in response to a lever press; then they no longer provided cocaine when the lever was pressed. Over several days, the rats pressed the lever less and less, and as expected, a brief stressful experience at this point restored robust lever pressing, even when no cocaine was forthcoming. If the kappa receptor inhibitor was administered before the stress, however, we saw no such reinstatement! These exciting findings support the idea that kappa opiate receptors are normally activated during a stressful experience and contribute directly to the initiation of drug-seeking behavior in animals, and perhaps to relapse in humans. Kappa receptor inhibitors may therefore have clinical utility in treating stress-induced drug relapse. Despite the brain’s complexity, the approach of understanding component parts and processes proved to be powerful in unpredictable and surprising ways.
Working with this team of outstanding scientists for many years has been tremendous fun. Together we have shared ups and downs and dry periods as well as periods of exciting discovery. Our project demonstrates how understanding the building blocks of a complex system not only help us understand how the brain works but can also suggest ways to control brain plasticity. In our case, a reductionist approach gave insight into a possible therapeutic strategy for addicted individuals.
Mood and food are connected. Consider how grouchy you are when you’re on a restricted diet or how good you feel with a whiff and a bite of a freshly baked chocolate chip cookie. As mentioned in Chapter 15, one system in the brain involved in the control of mood uses serotonin as a neurotransmitter. Serotonin provides one of the links between food and mood.
Measurements of serotonin in the hypothalamus reveal that levels are low during the postabsorptive period, rise in anticipation of food, and spike during a meal, especially in response to carbohydrates (Figure 16.18). Serotonin is derived from the dietary amino acid tryptophan, and tryptophan levels in the blood vary with the amount of carbohydrate in the diet (see Box 15.2 in Chapter 15). The rise in blood tryptophan and brain serotonin is one likely explanation for the mood-elevating effects of a chocolate chip cookie. This effect of “carbs” on mood is particularly evident during periods of stress, possibly explaining the food-seeking behavior and subsequent weight gain of many first-year college students.

FIGURE 16.18 Changes in hypothalamic serotonin levels before and during a meal. The mood-elevating effects of eating are believed to be related to the release of serotonin in the brain. (Source: Adapted from Schwartz et al., 1990.) Description
It is interesting to note that drugs that elevate serotonin levels in the brain are powerful appetite suppressants. One of these drugs is dexfenfluramine (trade name Redux), which was used successfully as a treatment for human obesity. Unfortunately, the drug had toxic side effects, leading to its withdrawal from the market in 1997.
Abnormalities in brain serotonin regulation are believed to be one factor that contributes to eating disorders. The defining characteristic of anorexia nervosa is a compulsion to maintain body weight at an abnormally low level, while bulimia nervosa is characterized by frequent eating binges, often compensated for by forced vomiting. These disorders are also commonly accompanied by depression, a severe disturbance of mood that has been linked to lowered brain serotonin levels (we will discuss mood disorders in Chapter 22). The serotonin connection is clearest in the case of bulimia. In addition to depressing mood, low serotonin levels reduce satiety. Indeed, antidepressant drugs that act to elevate brain serotonin levels (e.g., fluoxetine, or Prozac) are also an effective treatment for most bulimia nervosa patients.
We have used eating and the regulation of energy balance to give you a fairly detailed picture of the brain mechanisms that incite behavior. The systems involved in motivating several other behaviors that are basic for survival have also been intensively studied. Although we will not cover these other systems in depth, a quick overview will show that the basic principles are the same as those for eating. We will see that the transduction of physiological stimuli in the blood occurs in specialized regions of the hypothalamus, that humoral and visceromotor responses are initiated by activation of the periventricular and medial hypothalamus, and that behavioral action depends on the lateral hypothalamus.
Two different physiological signals stimulate drinking behavior. As mentioned in Chapter 15, one of these is a decrease in blood volume, or hypovolemia. The other is an increase in the concentration of dissolved substances (solutes) in the blood, or hypertonicity. These two stimuli trigger thirst by different mechanisms.
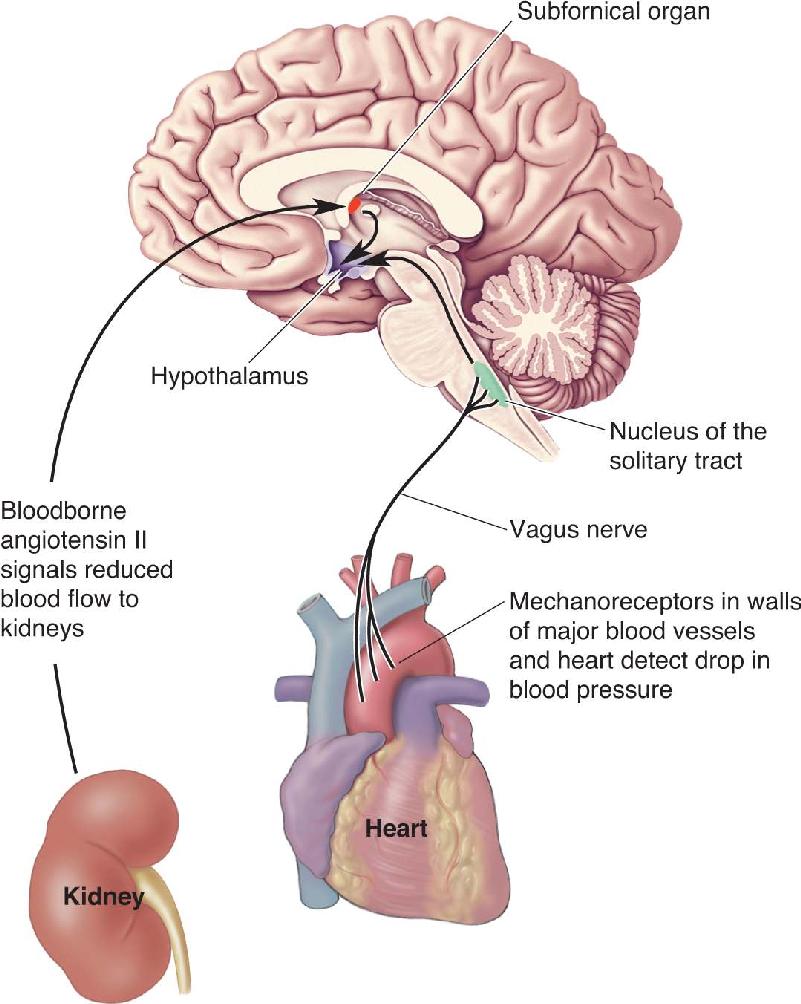
Thirst triggered by hypovolemia is called volumetric thirst. In Chapter 15, we used the example of decreased blood volume to illustrate when and how vasopressin is released in the posterior pituitary by the magnocellular neurosecretory cells. Vasopressin (also called antidiuretic hormone, or ADH) acts directly on the kidneys to increase water retention and inhibit urine production. The release of vasopressin associated with volumetric thirst is triggered by two types of stimuli (Figure 16.19). First, a rise in blood levels of angiotensin II occurs in response to reduced blood flow to the kidneys (see Figure 15.5 in Chapter 15). The circulating angiotensin II acts on the neurons of the subfornical organ in the telencephalon, which in turn directly stimulate the magnocellular neurosecretory cells of the hypothalamus to release vasopressin. Second, mechanoreceptors in the walls of the major blood vessels and heart signal the loss of blood pressure that accompanies a loss of blood volume. These signals make their way to the hypothalamus via the vagus nerve and the nucleus of the solitary tract.

FIGURE 16.19 Pathways triggering volumetric thirst. Hypovolemia is detected in two ways. First, angiotensin II, released into the bloodstream in response to decreased blood flow to the kidneys, activates neurons in the subfornical organ. Second, mechanosensory axons in the vagus nerve, detecting a drop in blood pressure, activate neurons in the nucleus of the solitary tract. The subfornical organ and nucleus of the solitary tract relay this information to the hypothalamus, which orchestrates the coordinated response to reduced blood volume. Description
In addition to this humoral response, reduced blood volume (1) stimulates the sympathetic division of the ANS, which helps correct the drop in blood pressure by constricting arterioles, and (2) powerfully motivates animals to seek and consume water. Not surprisingly, the lateral hypothalamus has been implicated in inciting the behavioral response, although the details of this process are still poorly understood.
The other stimulus for thirst, hypertonicity of the blood, is sensed by neurons in yet another specialized region of the telencephalon lacking a blood–brain barrier, the vascular organ of the lamina terminalis (OVLT). When the blood becomes hypertonic, water leaves cells by the process of osmosis. This loss of water is transduced by the OVLT neurons into a change in action potential firing frequency. The OVLT neurons (1) directly excite the magnocellular neurosecretory cells that secrete vasopressin, and (2) stimulate osmometric thirst, the motivation to drink water when dehydrated (Figure 16.20). Lesions of the OVLT completely prevent the behavioral and humoral responses to dehydration (but not the responses to loss of blood volume).

FIGURE 16.20 Osmometric thirst: the hypothalamic response to dehydration. Blood becomes hypertonic when it loses water. Blood hypertonicity is sensed by neurons of the vascular organ of the lamina terminalis (OVLT). The OVLT activates magnocellular neurosecretory cells and cells in the lateral hypothalamus. The neurosecretory cells secrete vasopressin into the blood, and the neurons of the lateral hypothalamus trigger osmometric thirst. Description
The motivation to drink and the secretion of vasopressin from the hypothalamus (and the retention of water by the kidneys) normally go hand in hand. However, selective loss of the vasopressin-secreting neurons of the hypothalamus produces a curious condition called diabetes insipidus, in which the body works against the brain. As a consequence of the loss of vasopressin, the kidneys pass too much water from the blood to the urine. The resulting dehydration stimulates the strong motivation to drink water; however, the water absorbed from the intestines passes quickly through the kidneys into the urine. Thus, diabetes insipidus is characterized by extreme thirst coupled with frequent excretion of a large amount of pale, watery urine. This condition can be treated by replacing the missing vasopressin.
You are hot; you seek a cool place. You are cold; you seek warmth. We are all motivated to interact with our environment to keep our bodies within a narrow range of temperatures. The need for such regulation is clear: The cells of the body are fine-tuned for a constant temperature, 37 °C (98.6°F), and deviations from this temperature interfere with cellular functions.
Neurons that change their firing rate in response to small changes in temperature are found throughout the brain and spinal cord. However, the most important neurons for temperature homeostasis are found clustered in the anterior hypothalamus. These cells transduce small changes in blood temperature into changes in their firing rate. Humoral and visceromotor responses are subsequently initiated by neurons in the medial preoptic area of the hypothalamus; somatic motor (behavioral) responses are initiated by the neurons of the lateral hypothalamic area. Lesions in these different regions can selectively abolish different components of the integrated response.
A fall in temperature is detected by cold-sensitive neurons of the anterior hypothalamus. In response, TSH is released by the anterior pituitary. TSH stimulates the release of the hormone thyroxin from the thyroid gland, which causes a widespread increase in cellular metabolism. The visceromotor response is constricted blood vessels in the skin and piloerection (goose bumps). An involuntary somatic motor response is shivering (to generate heat in the muscles), and, of course, the other somatic response is to seek warmth.
A rise in temperature is detected by warm-sensitive neurons of the anterior hypothalamus. In response, metabolism is slowed by reducing TSH release, blood is shunted toward the body periphery to dissipate heat, and behavior is initiated to seek shade. In some mammals, an involuntary motor response is panting—in humans, it is sweating—which helps cool the body.
The strong parallels between the hypothalamic control of energy balance, water balance, and temperature should now be clear. In each case, specialized neurons detect variations in the regulated parameter. The hypothalamus orchestrates responses to these challenges, which always include adjustments in physiology and the stimulation of different types of behavior. Table 16.1 summarizes some of the hypothalamic responses we have discussed in this chapter.
Hypothalamic Responses to Stimuli That Motivate Behavior

In the motor system chapters of Part II, we addressed “how” questions related to behavior. How do muscles contract? How is movement initiated? How are the actions of our different muscles coordinated? The discussion of motivation, however, asks a different question: Why? Why do we eat when our energy reserves become depleted? Why do we drink when we are dehydrated? Why do we seek warmth when our blood temperature falls?
Neuroscientists have found concrete answers to both the “how” and the “why” of behavior in the body’s periphery. We move because of the release of acetylcholine at the neuromuscular junction. We drink because we are thirsty, and we are thirsty when angiotensin II levels rise in response to decreased blood flow to the kidneys. However, we remain largely ignorant about the convergence of “how” and “why” in the brain. In this chapter, we chose to focus on feeding behavior, in part because the trail leads farthest into the brain. The discovery of orexigenic peptide neurons in the lateral hypothalamus that respond to changes in leptin levels was a major breakthrough. We are now able to frame the question of how these neurons act elsewhere in the brain to initiate feeding behavior. Advances in this research will have a significant impact on how we interpret our own behavior and the behavior of those around us.
After reading about the bloodborne signals that motivate eating and drinking, you might begin to feel that, indeed, we are ruled by our hormones. However, while bloodborne signals do have a strong effect on the probability of specific types of behavior, we are not their slaves. Clearly, one of the great triumphs of human evolution is the ability to exert cognitive, cortical control over our more primitive instincts. This is not to say that we humans make decisions solely on rational thought, however (Box 16.7). In addition to the powerful forces of self-preservation and heredity, our behaviors are molded by many factors that include our personal fears, ambitions, incentives, and history. In the coming chapters, we will explore additional influences on behavior, including how past experiences leave their mark on the brain.
The field of economics was born in 1776 with the publication of Adam Smith’s The Wealth of Nations. Among other endeavors, economists attempt to understand how choices are made about the allocation of resources. Economics was called “the dismal science” in the nineteenth century, initially because of dire predictions by economists that humanity was doomed to unending poverty because the food supply could not keep up with population growth. However, that phrase might also apply to how difficult it has been to understand and predict how humans make choices, economic and otherwise (Figure A).

What if we could get under the hood and find out what goes on in the brain during a decision? Advances in the technology of neuroscience, particularly the ability to measure and influence brain activity in awake behaving animals, including humans, make this an attainable goal. In the past decade, economists have increasingly looked to studies of the brain to test the validity of their theoretical assumptions, and neurophysiologists and psychologists have embraced economic theories to interpret their data on the neural basis of choice. The mutual attraction of these disciplines spawned a new field, called neuroeconomics. The central goal of neuroeconomics is to combine the tools and insights from economics, neuroscience, and psychology to determine how individuals make economic decisions. The history of science shows that great advances often occur when traditional disciplines come together to solve a common problem. There is perhaps no more urgent scientific challenge than understanding human behavior. More than any other factor, our individual and collective behaviors will determine the destiny of our species and our planet. Although success in this endeavor is by no means assured, it is certain that the understanding of behavior will require the understanding of neuroscience.
Glimcher PW, Fehr E. 2014. Neuroeconomics: Decision Making and the Brain, 2nd ed. San Diego, CA: Academic Press.
The Hypothalamus, Homeostasis, and Motivated Behavior
vascular organ of the lamina terminalis (OVLT) (p. 574)
1. A surgical approach to reducing excessive body fat is liposuction—the removal of adipose tissue. Over time, however, body adiposity usually returns to precisely the same value as before surgery. Why does liposuction not work permanently? Contrast this with the effect of gastric surgery to treat obesity.
2. Bilateral lesions of the lateral hypothalamus lead to reduced feeding behavior. Name three types of neurons, distinguished by their neurotransmitter molecules, which contribute to this syndrome.
3. What neurotransmitter agonists and antagonists would you design to treat obesity? Consider drugs that could act on the neurons of the brain as well as drugs that could act on the peripheral nervous system.
4. Name one way the axons of the vagus nerve might stimulate feeding behavior and one way they inhibit it.
5. What does it mean, in neural terms, to be addicted to chocolate? How could chocolate elevate mood?
6. Compare and contrast the functions of these three regions of the hypothalamus: the arcuate nucleus, the subfornical organ, and the vascular organ of the lamina terminalis.
Berridge KC. 2009. ‘Liking’ and ‘wanting’ food rewards: brain substrates and roles in eating disorders. Physiology and Behavior 97: 537–550.
Flier JS. 2004. Obesity wars: molecular progress confronts an expanding epidemic. Cell 116:337–350.
Friedman JM. 2004. Modern science versus the stigma of obesity. Nature Medicine 10: 563–569.
Gao Q, Hovath TL. 2007. Neurobiology of feeding and energy expenditure. Annual Review of Neuroscience 30:367–398.
Kauer JA, Malenka RC. 2007. Synaptic plasticity and addiction. Nature Reviews Neuroscience 8:844–858.
Schultz W. 2002. Getting formal with dopamine and reward. Neuron 36:241–263.
Wise RA. 2004. Dopamine, learning, and motivation. Nature Reviews Neuroscience 5:483–494.
Additional figures