Introduction
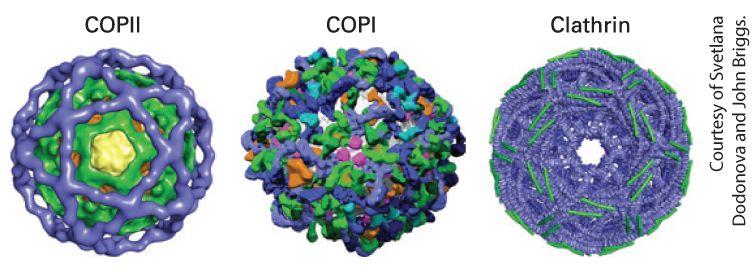
Chapter 14 Vesicular Traffic, Secretion, and Endocytosis Structural models of the coat protein complexes for three types of transport vesicles. The coat complexes are responsible for sculpting the curvature of the vesicle membrane and for selecting the types of cargo proteins incorporated into the vesicle. [Data from A. J. Noble and S. M. Stagg, 2015, Science 349(6244):142–143; https://doi.org/10.1126/science.aac6537.]
14.1 Techniques for Studying the Secretory Pathway
14.2 Molecular Mechanisms of Vesicle Budding and Fusion
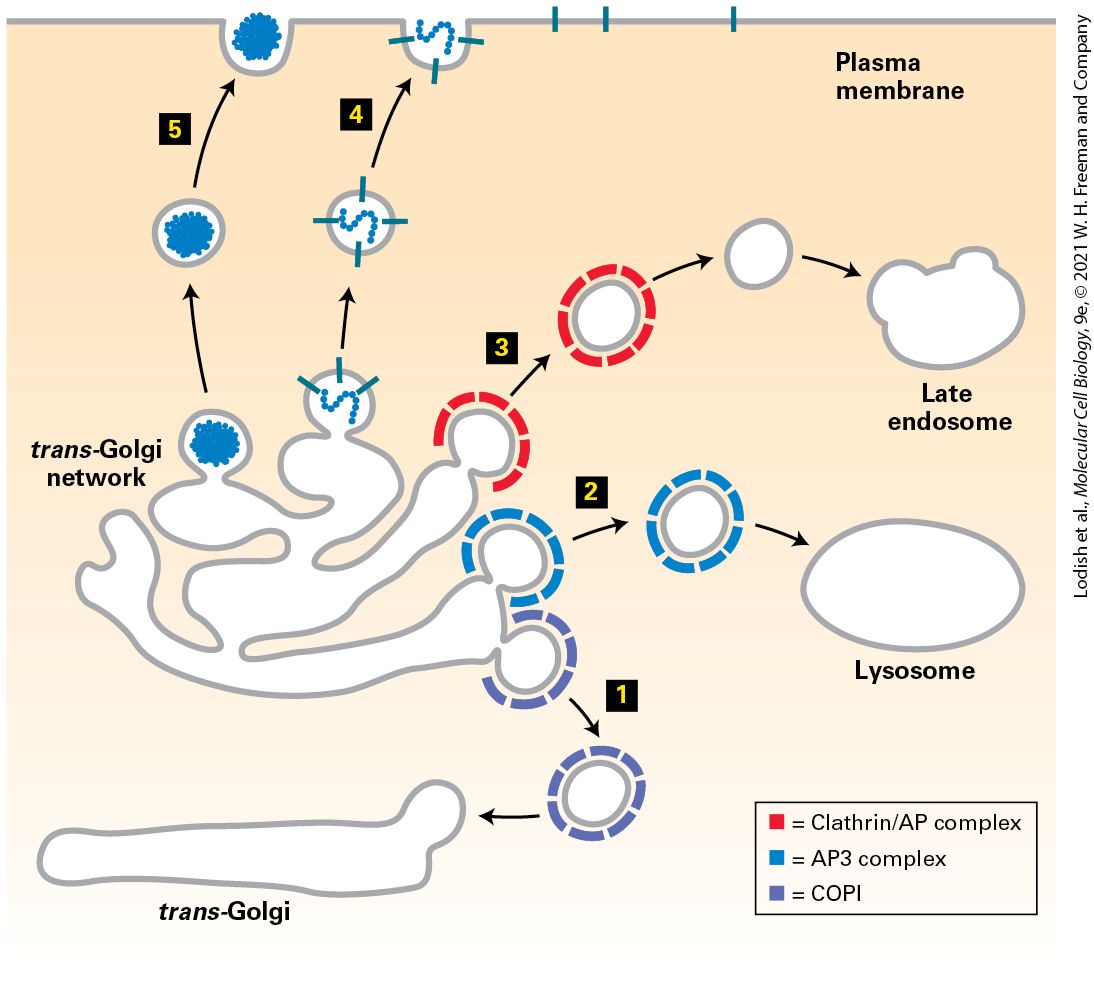
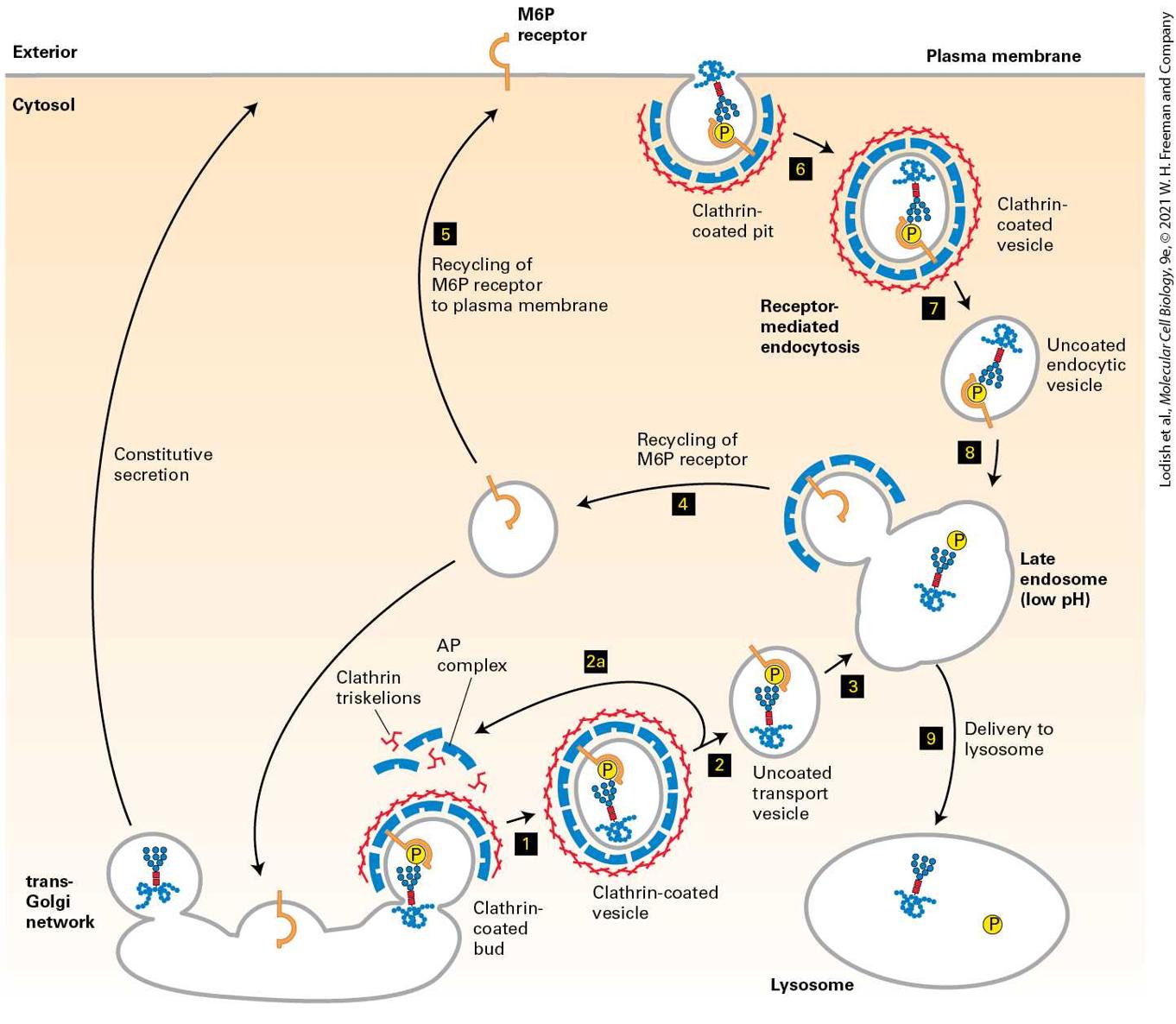
14.6 Directing Membrane Proteins and Cytosolic Materials to the Lysosome for Degradation In the previous chapter, we explored how proteins are targeted to and translocated across the membranes of different intracellular organelles, including the endoplasmic reticulum (ER), mitochondria, chloroplasts, peroxisomes, and the nucleus. In this chapter, we turn our attention to the secretory pathway and the mechanisms of vesicular traffic that allow proteins to be secreted from the cell or delivered to the plasma membrane and the lysosomes. We also discuss the related processes of endocytosis, which delivers proteins and small molecules from outside the cell, or autophagy, which delivers organelles, proteins, and small molecules from the interior of the cell to the lysosome for degradation. The secretory pathway is so named because it was initially studied in dedicated secretory cells that produce and secrete large quantities of proteins such as insulin or digestive enzymes to the outside of the cell. It was later discovered that the same pathway used for extracellular secretion of proteins delivers almost all newly synthesized membrane lipids and
membrane proteins from the endoplasmic reticulum (ER) to the cell surface. Newly synthesized membrane proteins delivered to the plasma membrane include cell-surface receptors, transporters for nutrient uptake, and ion channels that maintain the proper ionic and electrochemical balance across the plasma membrane. Soluble secreted proteins also follow the secretory pathway to the cell surface, but instead of remaining embedded in the membrane, they are released into the aqueous extracellular environment. Examples of secreted proteins include digestive enzymes, peptide hormones, serum proteins, and collagen. The secretory pathway is also responsible for the distribution of particular sets of soluble and membrane proteins to the organelles that lie along the secretory pathway including the ER itself, the Golgi, the endosome, and the lysosome. As we saw in Chapter 13, the ER is the site of synthesis and folding of all proteins that enter the secretory pathway. In this chapter, we describe the principal function of the Golgi — remodeling of carbohydrate modifications of secretory proteins. The Golgi along with the endosome are two major hubs for distributing proteins to their final destination at the plasma membrane or the lysosome. The lysosome, as described in Chapter 4, is the organelle with an acidic interior that is used for degradation of proteins and storage of small molecules such as amino acids. Accordingly, the types of proteins delivered to the lysosomal membrane include subunits of the V-class proton pump that pumps from the cytosol into the acidic lumen of the lysosome, as well as transporters that release small molecules stored in the lysosome into the cytoplasm. Soluble proteins delivered by this pathway include lysosomal digestive enzymes such as proteases, glycosidases, phosphatases, and lipases.
For all of its apparent complexity, the organization and operation of the secretory pathway can be understood as the consequence of repeated variations of just two basic mechanistic themes. The first theme is that transport of membrane and soluble proteins from one membrane-bounded compartment to another is mediated by coated transport vesicles that collect cargo proteins in buds arising from the membrane of one compartment and then deliver these cargo proteins to the next compartment by fusing with the membrane of that compartment. As we will see, different types of vesicles are responsible for each step in the secretory pathway, but the underlying mechanisms of budding of a coated vesicle from a donor membrane and the specific fusion with the appropriate target membrane are remarkably conserved, regardless of vesicle type. Importantly, as transport vesicles bud from one membrane and fuse with the next, the same face of the membrane remains oriented toward the cytosol. Therefore, once a protein has been inserted into the membrane or the lumen of the ER, that protein can be carried along the secretory pathway, moving from one organelle to the next without being translocated across another membrane or altering its orientation within the membrane. The second repeated theme is that each organelle along the secretory pathway both receives new proteins delivered by targeted transport vesicle fusion and donates proteins that are removed from the organelle by transport vesicle budding. Because transport vesicles package and deliver only specific subsets of proteins, the outcome of vesicular trafficking to and from each organelle establishes the composition and thus the identity of each organelle.
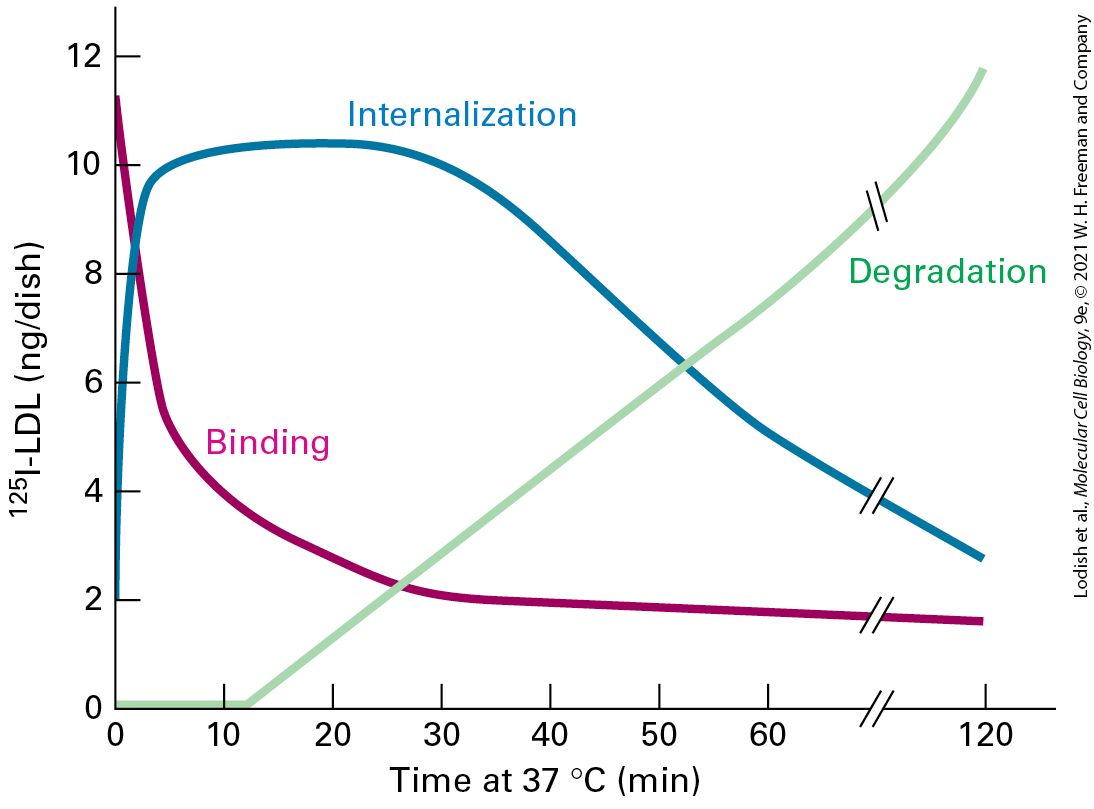
The composition of the plasma membrane is determined by two pathways: vesicle transport to the plasma membrane by the secretory pathway, which is counterbalanced by removal of proteins and membrane by the endocytic pathway (Figure 14-1). Both selective delivery by secretory vesicles and selective removal of proteins by endocytic vesicles can be regulated, allowing the protein composition of the plasma membrane to be regulated in response to environmental or developmental signals. In addition, the endocytic pathway is used to ingest certain nutrients that are too large to be transported across the plasma membrane by one of the transport mechanisms discussed in Chapter 11. For example, the endocytic pathway is used in the uptake of cholesterol carried in LDL particles. In addition, the endocytic pathway can be used to remove receptor proteins from the cell surface as a way to down-regulate their activity.
FIGURE 14-1 Basic principles of vesicle trafficking govern the composition of the plasma membrane. 1 : Membranes and proteins are delivered to the plasma membrane by exocytosis of coated vesicles that bud from the Golgi complex. The types of proteins incorporated into these vesicles are determined by interactions with vesicle coat proteins. Note that the membrane proteins preserve their orientation in the membrane such that the domain facing the vesicle interior will ultimately face the exoplasmic face of the plasma membrane. Correspondingly, soluble proteins within the vesicle interior will be released into the extracellular space. 2 : Proteins and membrane are removed from the plasma membrane by the process of endocytosis by which coated vesicles bud from the plasma membrane into the cytoplasm. In this example, a membrane receptor protein is only
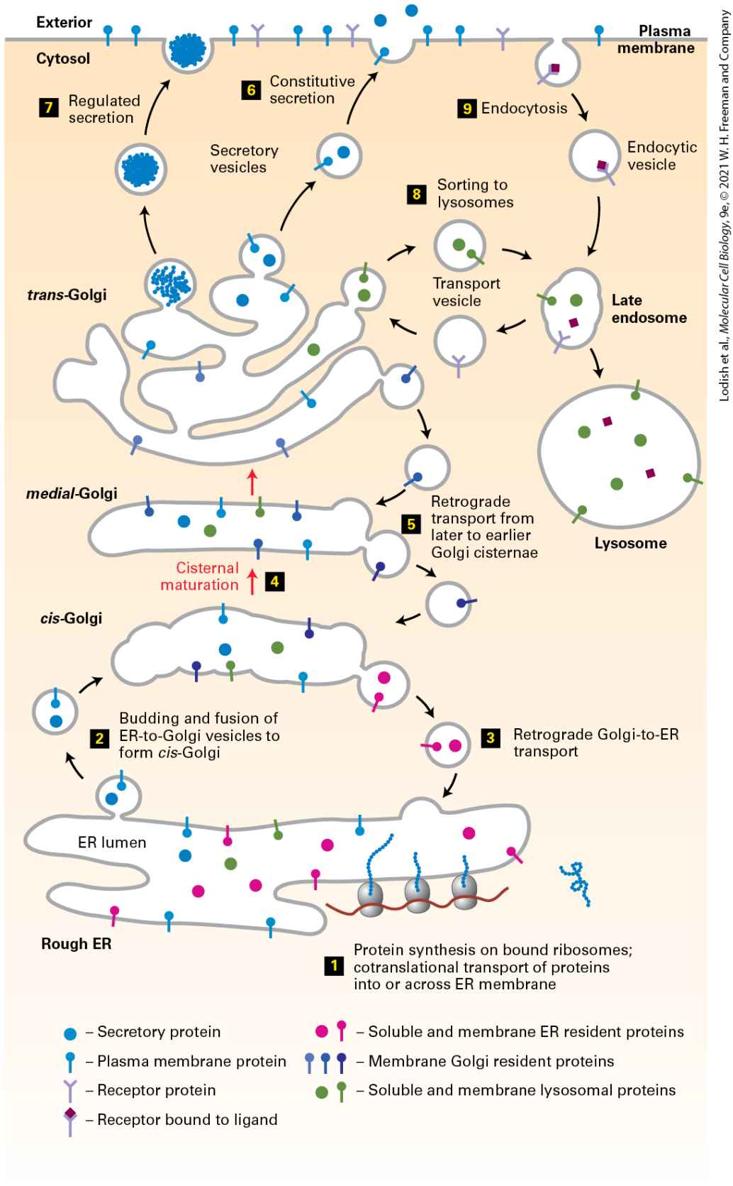
incorporated into endocytic vesicles when bound to ligand. The overall composition of the plasma membrane is controlled by a balance between the proteins that are selectively delivered by exocytic vesicles and the proteins that are removed by endocytic vesicles. Description Two numbered pathways are depicted. The information presented is as follows: 1. Exocytosis: A secretory vesicle inside the cell is circular and is made of the secretory vesicle coat. The thumbtack shaped plasma membrane proteins and y-shaped receptor proteins are attached to the insides of the secretory vesicle coat. The secretory vesicle coat also encloses secretory proteins that are represented by tiny spheres. The secretory vesicle coat disintegrates to release the secretory protein outside the cell, while the plasma membrane proteins and the receptor proteins bind to the plasma membrane of the cell. 2. Endocytosis: Endocytic vesicle coat attached to a receptor bound to a ligand in the inner membrane of the plasma membrane. Endocytosis leads to the formation of a circular endocytotic vesicle. The insides of the endocytotic vesicle are attached to two receptors bound to ligands. The overall organization of the individual vesicle trafficking steps to form the intersecting secretory and endocytic pathways is outlined in Figure 142. The first stage of the secretory pathway takes place in the rough endoplasmic reticulum (ER) (Figure 14-2, step 1 ). As described in
Chapter 13, newly synthesized soluble and membrane proteins are translocated into the ER, where they fold into their proper conformation and receive covalent modifications such as N-linked and O-linked carbohydrates and disulfide bonds. Once correctly folded and modified in the ER lumen, the newly synthesized proteins progress to the second stage of the secretory pathway: transport to and through the Golgi complex.
FIGURE 14-2 Overview of the secretory and endocytic pathways of protein sorting. Secretory pathway: Synthesis of proteins bearing an ER signal sequence is completed on the rough ER (step 1 ), and the newly made polypeptide chains are inserted into the ER membrane or cross it into the ER lumen (see Chapter 13). ER resident proteins such as BiP and PDI remain within the ER. The remainder are packaged into transport vesicles (step 2 ) that bud from the ER and fuse to form new cis-Golgi cisternae. Missorted ER-resident proteins and vesicle membrane proteins that need to be reused are retrieved to the ER by vesicles (step 3 ) that bud from the cis-Golgi and fuse with the ER. Each cis-Golgi cisterna, with its protein content, physically moves from the cis to the trans face of the Golgi complex (step 4 ) by a nonvesicular process called cisternal maturation. Retrograde transport vesicles (step 5 ) move Golgi-resident proteins to the proper Golgi compartments. In all cells, certain soluble proteins move to the cell surface in transport vesicles (step 6 ) and are secreted continuously (constitutive secretion). In certain cell types, some soluble proteins are stored in secretory vesicles (step 7 ) and are released only after the cell receives an appropriate neuronal or hormonal signal (regulated secretion). Lysosome-destined membrane and soluble proteins, which are transported in vesicles that bud from the trans-Golgi (step 8 ), first move to the late endosome and then to the lysosome. Endocytic pathway: Plasma membrane proteins such as receptor proteins bound to their ligands can be taken up in vesicles that bud from the plasma membrane (step 9 ) and move to the endosome from where they can either be recycled to the plasma membrane or be delivered to the lysosome for degradation. Description In this illustration, different proteins are represented by various shapes and a legend is given for each one. 1. Protein synthesis on bound ribosomes; and co-translational transport of proteins into or across the E R membrane. 2. Budding and fusion of E R-to-Golgi vesicles to form cis-Golgi 3. Retrograde transport from the Golgi to the E R. 4. Cisternal maturation occurs, transporting proteins from the cis-Golgi to the medialGolgi and, finally, trans-Golgi.
5. During cisternal maturation, retrograde transport of vesicles from later to earlier Golgi cisternae can occur. The following setup occurs from the trans-Golgi network: 6. Constitutive secretion. 7. Regulated secretion. 8. Sorting of proteins to lysosomes in transport vesicles. 9. In addition, proteins can be endocytosed from outside of the cell and added to late endosomes destined to become lysosomes or be transported via transport vesicles to the trans-Golgi network. The transport of cargo proteins from the ER to the Golgi occurs via anterograde (forward-moving) transport vesicles (Figure 14-2, step 2 ). These vesicles fuse with one another to form a flattened membranebounded compartment known as the cis-Golgi network or cis-Golgi cisterna (a “cistern” is a container for holding water or other liquid). Certain proteins, mainly proteins that function in the ER, can be retrieved from the cis-Golgi cisterna and returned to the ER via a different set of retrograde (backward-moving) transport vesicles (step 3 ). In a manner reminiscent of an assembly line, the new cis-Golgi cisterna, with its cargo of proteins, physically moves from the cis position (nearest the ER) to the trans position (farthest from the ER), successively becoming first a medial-Golgi cisterna and then a trans-Golgi cisterna (step 4 ). This process, known as cisternal maturation, primarily involves retrograde transport vesicles (step 5 ), which retrieve enzymes and other Golgiresident proteins from later to earlier Golgi cisternae, thereby maturing the cis-Golgi cisternae to medial-Golgi cisternae, and medial-Golgi
cisternae to trans-Golgi cisternae. As secretory proteins move through the Golgi, their linked carbohydrates may be further modified by specific glycosyltransferases that are housed in the different Golgi compartments. Proteins in the Golgi are eventually delivered to a complex network of membranes and vesicles termed the trans-Golgi network. The trans-Golgi network is a major branch point in the secretory pathway. It is at this stage that proteins are loaded into different kinds of vesicles and thereby trafficked to different destinations. Depending on which kind of vesicle the protein is loaded into, it will be transported to the plasma membrane and secreted immediately, stored for later release, or shipped to the lysosome (steps 6 – 8 ). The process by which a vesicle moves to and fuses with the plasma membrane and releases its contents is known as exocytosis. In all cell types, at least some proteins are secreted continuously (a process commonly called constitutive secretion), while others are stored inside the cell in specialized vesicles known as secretory granules until a signal for exocytosis causes them to be released (regulated secretion). Secretory proteins destined for lysosomes are first transported by vesicles from the trans-Golgi network to a compartment usually called the late endosome; the proteins are then transferred to the lysosome by direct fusion of the late endosome with the lysosomal membrane. In this chapter, we first discuss the experimental techniques that have contributed to our knowledge of the secretory pathway and endocytosis. Then we focus on the general mechanisms of membrane budding and fusion. We will see that although different kinds of transport vesicles use
distinct sets of proteins for their formation and fusion, all vesicles use the same general mechanism for budding, selection of particular sets of cargo molecules, and fusion with the appropriate target membrane. In the remaining sections of the chapter, we discuss both the early and late stages of the secretory pathway, including how specificity of targeting to different destinations is achieved. We conclude with a discussion of how proteins are transported to the lysosome by the endocytic pathway.
Transport of a Protein Through the Secretory Pathway Can Be Assayed in Live Cells
14.1 Techniques for Studying the Secretory Pathway The key to understanding how proteins are transported through the organelles of the secretory pathway has been to develop a basic description of the function of transport vesicles and the mechanism by which they incorporate particular cargo molecules. Many components required for the formation and fusion of transport vesicles have been identified by a remarkable convergence of the genetic and biochemical approaches described in this section. All studies of intracellular protein trafficking employ some method for assaying the transport of a given protein from one compartment to another. We begin by describing how intracellular protein transport can be followed in live cells and then consider genetic and in vitro systems that have proved useful in elucidating the secretory pathway. Transport of a Protein Through the Secretory Pathway Can Be Assayed in Live Cells The classic studies of George Palade and his colleagues in the 1960s first established the order in which proteins move from one organelle to the next in the secretory pathway (see Classic Experiment 14-1). These early
studies also showed that secretory proteins are not released into the cytosol — the first indication that transported proteins are always associated with some type of membrane-bounded intermediate. In these experiments, radioactively labeled amino acids were injected into the pancreas of hamsters and the newly synthesized proteins that incorporated the labeled amino acids could be tracked as they made their way to the cell surface. At different times after injection, the animals were sacrificed and the pancreatic cells were immediately fixed with glutaraldehyde, sectioned, and subjected to autoradiography to visualize the locations of the radiolabeled proteins. Because the radioactive amino acids were administered in a short pulse, only those proteins synthesized immediately after injection were labeled, forming a distinct cohort of labeled proteins whose transport could be followed. In addition, because pancreatic acinar cells are dedicated secretory cells, almost all of the labeled amino acids in these cells were incorporated into secretory proteins, facilitating the observation of transported proteins. Although autoradiography is rarely used today to localize proteins within cells, these early experiments illustrate the two basic requirements for any assay of intercompartmental transport. First, it is necessary to label a cohort of proteins in an early compartment so that their subsequent transfer to later compartments can be followed over time. Second, it is necessary to have a way to identify the compartment in which a labeled protein resides. Here we describe two modern experimental procedures for observing the intracellular trafficking of a secretory protein in almost any type of cell.
In both procedures, a gene encoding an abundant membrane glycoprotein (G protein) from vesicular stomatitis virus (VSV) is introduced into cultured mammalian cells either by transfection or simply by infecting the cells with the virus. The transfected cells efficiently produce the VSV G protein, which is then inserted into the membrane of the ER. The use of a mutant gene encoding a temperature-sensitive VSV G protein allows researchers to turn subsequent transport of this protein on and off. At the restrictive temperature of 40 °C, newly made VSV G protein is misfolded and therefore retained within the ER by the quality-control mechanisms discussed in Chapter 13, whereas at the permissive temperature of 32 °C, the protein is correctly folded and transported through the secretory pathway to the cell surface. Importantly, the misfolding of the temperature-sensitive VSV G protein is reversible; thus when cells synthesizing mutant VSV G protein are grown at 40 °C and then shifted to 32 °C, the misfolded mutant VSV G protein that had accumulated in the ER will refold and be transported normally. This clever use of a temperature-sensitive mutation in effect defines a protein cohort whose subsequent transport can be followed. In two variations of this basic procedure, transport of VSV G protein is monitored by different techniques. Studies using both of these modern trafficking assays came to the same conclusion as Palade’s early experiments: in mammalian cells, vesicle-mediated transport of a protein molecule from its site of synthesis on the rough ER to its arrival at the plasma membrane takes from 30 to 60 minutes. Microscopy of GFP-Labeled VSV G Protein
One approach for observing the transport of VSV G protein employs a hybrid gene in which the viral gene is fused to the gene encoding green fluorescent protein (GFP), a naturally occurring fluorescent protein (see
Chapter 4). The hybrid gene is transfected into cultured cells by techniques described in Chapter 6. When cells expressing the temperaturesensitive form of the hybrid protein (VSVG-GFP) are grown at the restrictive temperature, VSVG-GFP accumulates in the ER, which appears as a lacy network of membranes when the cells are observed in a fluorescent microscope. When the cells are subsequently shifted to a permissive temperature, the VSVG-GFP can be seen to move first to the membranes of the Golgi complex, which are densely concentrated at the edge of the nucleus, and then to the cell surface (Figure 14-3a). By observing the distribution of VSVG-GFP at different times after shifting cells to the permissive temperature, researchers have determined how long VSVG-GFP resides in each organelle of the secretory pathway (Figure 143b). EXPERIMENTAL FIGURE 14-3 Protein transport through the secretory pathway can be visualized by fluorescence microscopy of cells producing a GFP-tagged membrane protein. Cultured cells were transfected with a hybrid gene encoding the viral membrane glycoprotein VSV G linked to the gene for green fluorescent protein (GFP). A temperaturesensitive mutant version of the viral gene was used so that newly made hybrid protein
(VSVG-GFP) was retained in the ER at 40 °C but was released for transport at 32 °C. (a) Fluorescence micrographs of cells just before and at two times after they were shifted to the lower temperature. Movement of VSVG-GFP from the ER to the Golgi and finally to the cell surface occurred within 180 minutes. The scale bar is . (b) Plot of the amount of VSVG-GFP in the endoplasmic reticulum (ER), Golgi, and plasma membrane (PM) at different times after the shift to the permissive temperature. The kinetics of transport from one organelle to another can be reconstructed from computer analysis of these data. The decrease in total fluorescence that occurs at later times probably results from slow inactivation of GFP fluorescence. Description The three micrographs are labeled 0 minutes, 40 minutes, and 180 minutes respectively. In the first micrograph, the fluorescence is restricted to the E R. In the second micrograph, fluorescence is restricted to the Golgi, and in the third micrograph, the fluorescently labeled protein has been delivered to the cell membrane, leading to the fluorescence of the cell surface. In the graph labeled (b), the vertical axis represents the amount of V Z V G protein labeled with G F P (times 10 to the power 6) ranging from 0 to 20 in increments of 5. The horizontal axis represents time in minutes ranging from 0 to 600 in increments of 100. The red curve corresponding to E R fluorescence starts at (0, 20) and rapidly declines to (0, 100). A green curve, corresponding to fluorescence in the Golgi starts at (0, 0) increases sharply, reaches a maximum at (50, 7.5), and ends at (0,200). The fluorescence of the plasma membrane indicated by a blue curve starts at (0, 0), increases sharply, reaches a maximum at (150, 15), and ends at (6, 600). Detection of Compartment-Specific Oligosaccharide Modifications A second way to follow the transport of secretory proteins takes advantage of modifications to their carbohydrate side chains that occur at different
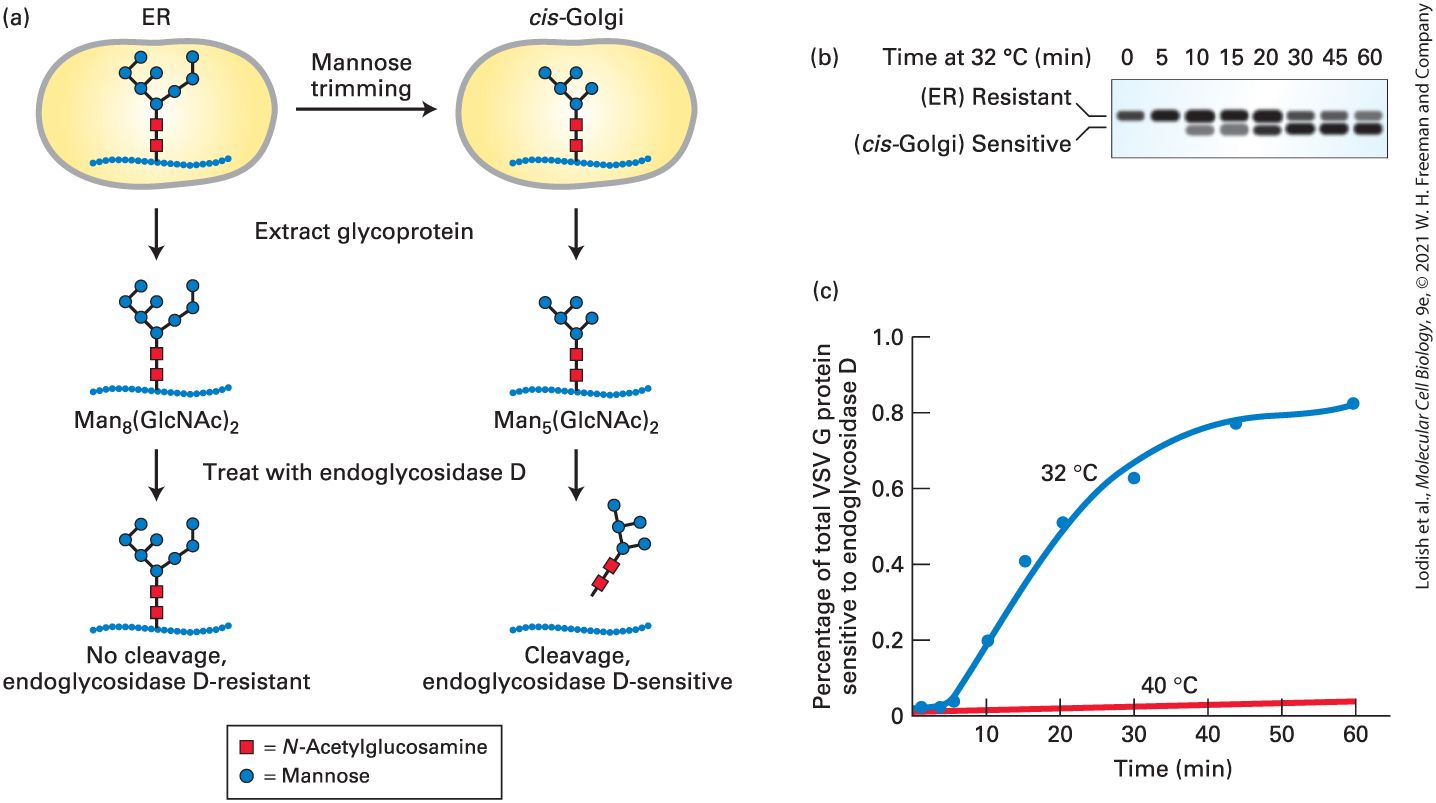
stages of the secretory pathway. To understand this approach, recall that many secretory proteins leaving the ER are carrying one or more copies of the N-linked oligosaccharide , which are synthesized and attached to secretory proteins in the ER (see Figure 13-18). As a protein moves through the Golgi complex, different enzymes localized to the cis-, medial-, and trans-Golgi cisternae catalyze an ordered series of modifications to these core chains, as discussed in a later section of this chapter. For instance, glycosidases that reside specifically in the cis-Golgi compartment sequentially trim mannose residues off the core oligosaccharide to yield a trimmed form, . By monitoring the trimming of the N-linked oligosaccharide, scientists can distinguish glycosylated VSV G protein that has entered the cis-Golgi from VSV G protein that remains in the ER. A specialized carbohydratecleaving enzyme known as endoglycosidase D enables the trimming of the N-linked oligosaccharide to be readily detected by gel electrophoresis; endoglycosidase D will cleave trimmed cis-Golgi-specific oligosaccharides from proteins, but will not cleave the core (untrimmed) oligosaccharide chains on secretory proteins within the ER (Figure 14-4a). Because a deglycosylated VSV G protein produced by endoglycosidase D digestion moves faster on an SDS gel than the corresponding glycosylated protein, these proteins can be readily distinguished (Figure 14-4b).
EXPERIMENTAL FIGURE 14-4 Transport of a membrane glycoprotein from the ER to the Golgi can be assayed based on sensitivity to cleavage by endoglycosidase D. Cells expressing a temperature-sensitive VSV G protein were labeled with a pulse of radioactive amino acids at the nonpermissive temperature so that the labeled protein was retained in the ER. At periodic times after a return to the permissive temperature of 32 °C, VSV G protein was extracted from cells and digested with endoglycosidase D. (a) As proteins move to the cis-Golgi from the ER, the core oligosaccharide is trimmed to by enzymes that reside in the cis-Golgi compartment. Endoglycosidase D cleaves the oligosaccharide chains from proteins processed in the cisGolgi but not from proteins in the ER. (b) SDS-polyacrylamide gel electrophoresis of the digestion mixtures resolves the resistant, uncleaved (slower migrating) and sensitive, cleaved (faster migrating) forms of labeled VSV G protein. Initially, as this gel shows, all of the VSV G protein was resistant to digestion, but over time, an increasing fraction was sensitive to digestion, reflecting transport of the protein from the ER to the Golgi and its processing there. In control cells kept at 40 °C, only slow-moving, digestion-resistant VSV G protein was detected after 60 minutes (not shown). (c) A plot of the percentage of VSV G protein that is sensitive to digestion, derived from electrophoretic data, reveals the time course of ER-to-Golgi transport. [Part (b) Data from C. J. Beckers et al., 1987, “Semi-Intact Cells Permeable to Macromolecules: Use in Reconstitution of Protein Transport from the Endoplasmic
Reticulum to the Golgi Complex,” Cell 50(4):523–34.] Description The illustration labeled (a) depicts the experimental assaying of transport of proteins from the E R to the Golgi: The sequence is as follows: 1. A V S V G protein labeled with M a n 8 (G l c N A c) subscript 2 oligosaccharide (made of eight mannose sugars and two N-acetylglucosamines) is present inside the E R. 2. Transport to the cis-Golgi results in mannose trimming of the oligosaccharide. 3. The glycoproteins are extracted. 4. The glycoproteins are treated with endoglycosidase D, which cleaves the oligosaccharides from proteins originating from cis-Golgi, but not those originating from the E R. The gel documentation image labeled (b) shows the increase in endoglycosidase D sensitive protein over time: The time of incubation at 32 degrees Celsius in minutes is indicated. There are eight bands, corresponding to 0, 5, 10, 15, 20, 30, 45, and 60 minutes respectively. Electrophoresis channels show dark bands. The dark bands in the first row correspond to proteins resistant (E R) to the endoglycosidase and the dark bands in the second row correspond to those sensitive (cis-Golgi) to it. With time, the sensitive band increases in darkness, indicating concentration of the protein in the cisGolgi. The graph labeled (c) plots the percentage of total V S V G protein sensitive to endoglycosidase D against time. The vertical axis of the graph represents percentage of total V S V G protein sensitive to endoglycosidase D, ranging from 0 to 1, in increments of 0.2 percent. The horizontal axis represents time in minutes, ranging from 0 to 60 in increments of 10 minutes. Two curves are plotted, a blue curve, corresponding to incubation at 32 degrees Celsius shows an almost linear increase to 0.6 percent in 30 minutes. After this time, the curve begins to plateau, reaching a value
Yeast Mutants Define Major Stages and Components of Vesicular Transport
of 0.8 percent at about 45 minutes. A red curve, corresponding to incubation at 40 degrees shows a slow, linear increase from 0 to about 0.5 percent in 60 minutes. This type of assay can be used to track movement of VSV G protein from the ER to the cis-Golgi in virus-infected cells pulse-labeled with radioactive amino acids. Immediately after labeling, all the labeled VSV G protein is still in the ER and, upon extraction, is resistant to digestion by endoglycosidase D. Over time, however, the fraction of the extracted glycoprotein that has received carbohydrate trimming in the cis-Golgi and is thus sensitive to digestion by endoglycosidase D increases. Note that transport of VSV G protein from the ER to the Golgi takes about 30 minutes, as measured either by the assay based on oligosaccharide processing or by fluorescence microscopy of VSVG-GFP (Figure 14-4c). A variety of assays based on specific carbohydrate modifications that occur in later Golgi compartments have been developed to measure progression of VSV G protein through each compartment of the Golgi complex. Yeast Mutants Define Major Stages and Components of Vesicular Transport The general organization of the secretory pathway and many of the molecular components required for vesicle trafficking are similar in all eukaryotic cells. Because of this conservation, genetic studies with yeast have been useful in identifying many of the proteins that participate in
vesicular traffic. For yeast cells, as for all cells, the secretory pathway is essential for transport and delivery of new protein and membrane to the cell surface. Thus genes encoding important components of the secretory pathway are essential for cell growth and can be studied only as conditional temperature-sensitive mutants, as described in Chapter 8. A large number of yeast mutants were initially identified by their inability to secrete proteins at a nonpermissive temperature. When these temperature-sensitive secretion (sec) mutants are transferred from the lower permissive temperature to the higher nonpermissive temperature, they accumulate secretory proteins at the point in the secretory pathway blocked by the mutation. Analysis of such mutants identified five classes (A–E) characterized by protein accumulation in the cytosol, rough ER, small vesicles taking proteins from the ER to the Golgi complex, Golgi cisternae, or constitutive secretory vesicles (Figure 14-5). Subsequent characterization of sec mutants in these various classes has helped elucidate the fundamental components and molecular mechanisms of vesicle trafficking that we discuss in later sections.
EXPERIMENTAL FIGURE 14-5 Phenotypes of yeast sec mutants identified five stages in the secretory pathway. These temperature-sensitive mutants can be grouped into five classes (A–E) based on the site where newly made secretory proteins (red dots) accumulate when cells are shifted from the permissive temperature to the higher, nonpermissive one. Analysis of double mutants permitted the sequential order of the steps to be determined. Note that organelle structures before the stage blocked by a mutant are exaggerated because the sec mutants block movement of membranes as well as cargo. See P. Novick et al., 1981, Cell 25:461; and C. A. Kaiser and R. Schekman, 1990, Cell 61:723. Description A normal yeast cell shows proteins in the E R, the Golgi, and in vesicles that are being transported in or out of the cell. The classes of mutants are listed from A to E. Class A mutants accumulated proteins in the cytosol due to defective transport into the E R. Class B mutants accumulated proteins in the rough E R due to defective budding of vesicles from the rough E R. Class C mutants accumulated vesicles used for transport from the E R to the Golgi due to defective fusion of transport vesicles with the Golgi. Class D mutants accumulate proteins in the Golgi due to defective transport from the Golgi to secretory vesicles. Class E mutants accumulate proteins in secretory vesicles due to defected transport of secretory vesicles to the cell surface. To determine the order of the steps in the pathway, researchers analyzed double sec mutants. For instance, when yeast cells contain mutations in both class B and class D functions, proteins accumulate in the rough ER, not in the Golgi cisternae. Because proteins accumulate at the earliest blocked step, this finding shows that class B mutations must act at an earlier point in the secretory pathway than class D mutations do. These studies confirmed that as a secreted protein is synthesized and processed, it moves sequentially from the cytosol to the rough ER, to ER-to-Golgi transport vesicles, to Golgi cisternae, to secretory vesicles, and finally is exocytosed.
Cell-Free Transport Assays Allow Dissection of Individual Steps in Vesicular Transport
Most importantly, the yeast sec mutants define many of the genes and encoded proteins that are required for vesicle budding and fusion at each of the major steps of the secretory pathway. Each of the individual steps in the secretory pathway is currently being studied in mechanistic detail, and biochemical assays and protein structural studies are being used to understand each of these steps in terms of the structure and function of individual protein molecules. Cell-Free Transport Assays Allow Dissection of Individual Steps in Vesicular Transport In vitro assays for intercompartmental transport are powerful complementary approaches to studies with yeast sec mutants for identifying and analyzing the cellular components responsible for vesicular trafficking. In one application of this approach, cultured mutant cells lacking one of the enzymes that modify N-linked oligosaccharide chains in the Golgi are infected with vesicular stomatitis virus, and the fate of the VSV G protein is followed. For example, if infected cells lack N-acetylglucosamine transferase I, they produce abundant amounts of VSV G protein but cannot add N-acetylglucosamine residues to the oligosaccharide chains in the medial-Golgi as wild-type cells do (Figure 14-6a). When Golgi membranes isolated from such mutant cells are mixed with Golgi membranes from wild-type, uninfected cells, the addition of N-acetylglucosamine to VSV G protein is restored (Figure 14-6b). This modification is the consequence of vesicular transport of N-
acetylglucosamine transferase I from the wild-type medial-Golgi to the cis-Golgi isolated from virally infected mutant cells. EXPERIMENTAL FIGURE 14-6 A cell-free assay demonstrates protein transport from one Golgi cisterna to another. (a) A mutant line of cultured fibroblasts is essential in this type of assay. In this example, the cells lack the enzyme N-acetylglucosamine transferase I (see step 2 in Figure 14-15). In wild-type cells, this enzyme is localized to the medialGolgi and modifies N-linked oligosaccharides by the addition of one N-acetylglucosamine. In VSV-infected wild-type cells, the oligosaccharide on the viral G protein is modified to a typical complex oligosaccharide, as shown in the trans-Golgi panel. In infected mutant cells, however, the G protein reaches the cell surface with a simpler high-mannose oligosaccharide containing only two N-acetylglucosamine and five mannose residues. (b) When Golgi cisternae isolated from infected mutant cells are incubated with Golgi cisternae from normal, uninfected cells, the VSV G protein produced in vitro contains the additional N-acetylglucosamine. This modification is carried out by transferase enzyme that is moved by transport vesicles from the wild-type medial-Golgi cisternae to the mutant cis-Golgi cisternae in the reaction mixture. See W. E. Balch et al., 1984, Cell 39:405 and 525; W. A. Braell et al., 1984, Cell 39:511; and J. E. Rothman and T. Söllner, 1997, Science 276:1212. Description The illustration labeled (a) shows the transport of protein from cis-Golgi to trans-Golgi in both V S V infected wild-type and mutant cells. The illustration shows two fibroblast cultures. One culture contains (V S V)-infected wild-type fibroblasts, while the other contains V S V-infected mutant cells. In the wild-type cells, A V S V G protein in the
cis-Golgi, labeled with a five mannose, two N-acetyl glucosamine oligosaccharide is transported to the medial Golgi. There, N-acetyl glucosamine transferase 1 adds additional N-acetyl glucosamine to the oligosaccharide. On further transport to the trans-Golgi, reactions add additional N-acetyl glucosamine, galactose, and N-acetylneuraminic acid groups the oligosaccharide. The mutant cells do not undergo an N-acetyl glucosamine transferase 1 reaction. Hence, there is no growth of the oligosaccharide chain during the transport of the protein from cis-Golgi to trans-Golgi. The illustration labeled (b) shows Golgi isolated from uninfected wild-type cells and protein in Golgi from infected mutant cells incubated to form a hybrid Golgi. The hybrid Golgi contains the glycoprotein. The oligosaccharide is found to contain an additional N-Acetylglucosamine in the oligosaccharide attached to the G-protein. Proteins that play a role in Golgi transport have been identified by fractionation methods. Under appropriate conditions, a uniform population of the transport vesicles that move N-acetylglucosamine transferase I from the medial- to cis-Golgi can be separated from the donor wild-type Golgi membranes by centrifugation. By examining the proteins that are enriched in these vesicles, scientists have been able to identify many of the integral membrane proteins and peripheral vesicle coat proteins that are the structural components of this type of vesicle. Moreover, fractionation of the cytosolic extract required for transport in cell-free reaction mixtures has permitted isolation of the various proteins required for formation of transport vesicles and of proteins required for the targeting and fusion of vesicles with appropriate acceptor membranes. In vitro assays similar in general design to the one shown in Figure 14-6 have been used to study the proteins that play a role at various transport steps in the secretory pathway. KEY CONCEPTS OF SECTION 14.1
Techniques for Studying the Secretory Pathway All assays for following the trafficking of proteins through the secretory pathway require a way to label a cohort of secretory proteins and a way to identify the compartments where the labeled proteins are subsequently located. Transport of a fluorescently labeled protein along the secretory pathway can be observed by microscopy (see Figure 14-3). A temperature-sensitive mutant protein that is retained in the ER due to misfolding at the nonpermissive temperature will be released as a cohort for transport when cells are shifted to the permissive temperature. Alternatively, pulse labeling with radioactive amino acids can specifically label a cohort of newly made proteins in the ER and the transport of a radiolabeled protein can be tracked by following compartment-specific covalent modifications to the protein. Many of the components required for intracellular protein trafficking have been identified in yeast by analysis of temperature-sensitive sec mutants defective for the secretion of proteins at the nonpermissive temperature (see Figure 14-5). Cell-free assays for intercompartmental protein transport have allowed the biochemical dissection of individual steps of the secretory pathway. Such in vitro reactions can be used to produce pure transport vesicles and to test the biochemical function of individual transport proteins.
Assembly of a Protein Coat Drives Vesicle Formation and Selection of Cargo Molecules
14.2 Molecular Mechanisms of Vesicle Budding and Fusion Small membrane-bounded vesicles that transport proteins from one organelle to another are the fundamental functional elements in the secretory and endocytic pathways (see Figure 14-1). These vesicles bud from the membrane of a particular parent (donor) organelle and fuse with the membrane of a particular target (destination) organelle. Although each step in the secretory and endocytic pathways employs a different type of vesicle, studies employing genetic and biochemical techniques have revealed that each of the different vesicular transport steps is simply a variation on a common theme. In this section, we explore the basic mechanisms underlying vesicle budding and fusion that all vesicle types have in common, before discussing the details unique to each step in the pathway. Assembly of a Protein Coat Drives Vesicle Formation and Selection of Cargo Molecules The budding of a vesicle from its parent membrane is triggered by the activation of a GTP-binding protein followed by the polymerization of soluble protein complexes on the membrane to form a proteinaceous
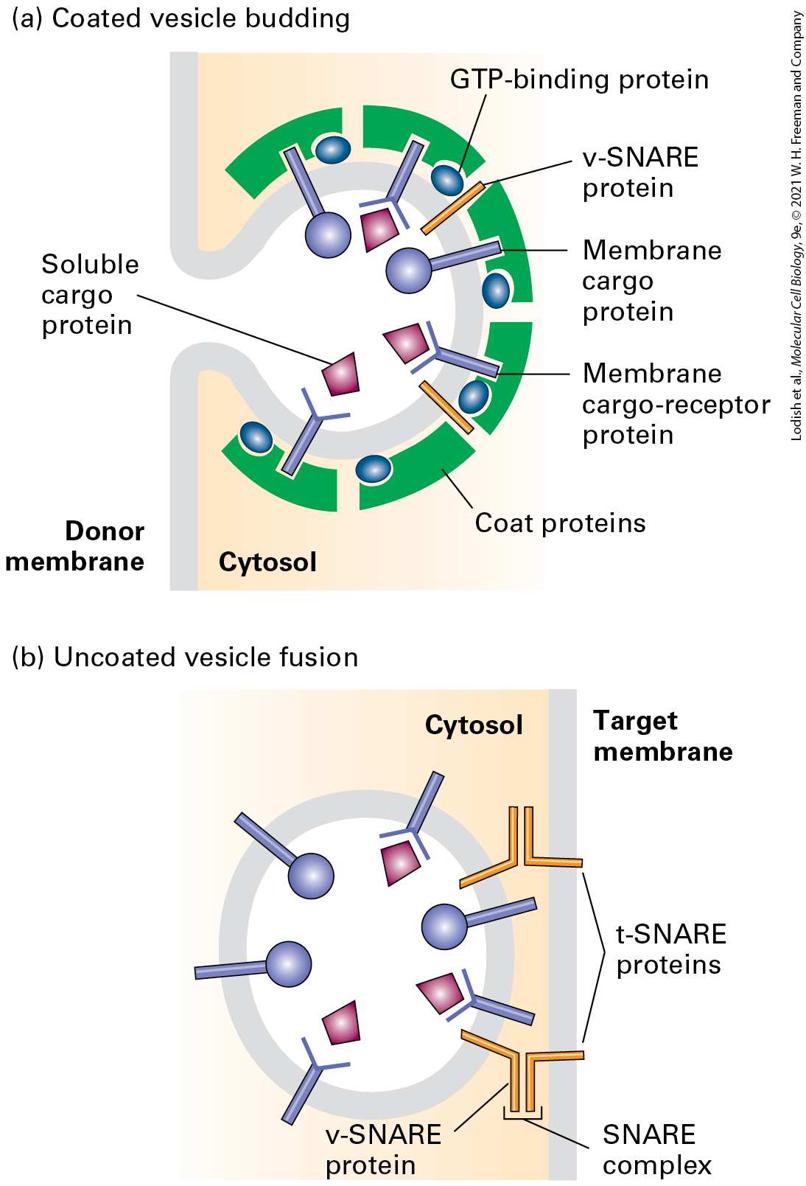
vesicle coat (Figure 14-7a). Interactions between the cytosolic portions of integral membrane proteins and the vesicle coat gather the appropriate cargo proteins into the forming vesicle. Thus the coat gives curvature to the membrane to form a vesicle and acts as the filter to determine which proteins are admitted into the vesicle.
FIGURE 14-7 Overview of vesicle budding and fusion with a target membrane. (a) Budding is initiated by recruitment of a small GTP-binding protein to a patch of donor membrane. Complexes of coat proteins in the cytosol then bind to the cytosolic domain of membrane cargo proteins, some of which also act as receptors that bind soluble proteins in the lumen, thereby recruiting luminal cargo proteins into the budding vesicle. (b) After being released and shedding its coat, a vesicle fuses with its target membrane in a process that involves interaction of cognate SNARE proteins. Description The illustration labeled (a) titled coated vesicle budding shows a vesicle buds in the cytosol from the donor membrane. On the cytosolic side of the budding membrane, coat proteins are present. The coat proteins contain G T P-binding proteins and membrane cargo receptor proteins. Several v-S N A R E proteins are embedded in the vesical membrane. The membrane cargo-receptor proteins are y-shaped and are bound to soluble cargo proteins. In addition to the membrane cargo receptors, membrane cargo proteins from the coat protein are also embedded in the vesicle. The illustration labeled (b) titled uncoated vesicle fusion shows a spherical vesicle inside the cytosol approaching the target membrane. It has no coat. The vesicle contains membrane cargo proteins and membrane cargo-receptor proteins embedded in the vesicular membrane and several v-S N A R E proteins. The target membrane contains two t-S N A R E proteins, which can interact with the v-S N A R E proteins in the membrane. Proteins responsible for the eventual fusion of a vesicle with the target membrane, known as v-SNAREs, are incorporated into the vesicle membrane during assembly of the vesicle coat. Once the coat is at least partially shed from a completed vesicle, v-SNARE proteins embedded in the vesicle membrane become accessible to join with cognate t-SNAREs in the target membrane to which the vesicle is docked. This joining brings the membranes into close apposition, allowing the two bilayers to fuse
(Figure 14-7b). Regardless of target organelle, all transport vesicles use vSNAREs and t-SNAREs to fuse. Three major types of coated vesicles have been characterized, each with a different type of protein coat and each formed by reversible polymerization of a distinct set of protein subunits (Table 14-1). Each type of vesicle, named for its primary coat proteins, transports cargo proteins from particular parent organelles to particular target organelles:
TABLE 14-1 • Coated Vesicles Involved in Protein Trafficking Vesicle Type Transport Step Mediated Coat Proteins Associated GTPase
ER to cis-Golgi Sec23/Sec24 and Sec13/Sec31 complexes, Sec16 Sar1
cis-Golgi to ER later to earlier Golgi cisternae Coatomers containing seven different COP subunits ARF Clathrin and adapter proteins trans-Golgi to endosome ARF trans-Golgi to endosome ARF Plasma membrane to endosome ARF Golgi to lysosome, melanosome, or platelet vesicles AP3 complexes ARF Each type of AP complex consists of four different subunits. It is not known whether the coat of AP3 vesicles contains clathrin. i i
COPII vesicles transport proteins from the ER to the Golgi. COPI vesicles mainly transport proteins in the retrograde direction between Golgi cisternae and from the cis-Golgi back to the ER. Clathrin-coated vesicles transport proteins from the plasma membrane (cell surface) and the trans-Golgi network to late endosomes. Every vesicle-mediated trafficking step is thought to use some kind of vesicle coat; however, a specific coat protein complex has not been identified for every type of vesicle. For example, vesicles that move proteins from the trans-Golgi to the plasma membrane during either constitutive or regulated secretion exhibit a uniform size and morphology, which suggests that their formation is driven by assembly of a regular coat structure, yet researchers have not identified specific coat proteins surrounding these vesicles. The general scheme of vesicle budding shown in Figure 14-7a applies to all three known types of coated vesicles. Experiments with isolated or artificial membranes and purified coat proteins have shown that polymerization of the coat proteins on the cytosolic face of the parent membrane is necessary to produce the high curvature of the membrane that is typical of a transport vesicle that is about 50 nm in diameter. Electron micrographs of in vitro budding reactions often reveal structures that exhibit discrete regions of the parent membrane bearing a dense coat accompanied by the curvature characteristic of a completed vesicle (Figure 14-8). Such structures, usually called vesicle buds, appear to be
intermediates that are visible after the coat has begun to polymerize but before the completed vesicle pinches off from the parent membrane. The polymerized coat proteins form a curved lattice that drives the formation of a vesicle bud by adhering to the cytosolic face of the membrane. EXPERIMENTAL FIGURE 14-8 Vesicle buds can be visualized during in vitro budding reactions. When purified COPII coat components are incubated with isolated ER vesicles or artificial phospholipid vesicles (liposomes), polymerization of the coat proteins on the vesicle surface induces emergence of highly curved buds. In this electron micrograph of an in vitro budding reaction, note the distinct membrane coat, visible as a dark protein layer, present on the vesicle buds. [Republished with permission from Elsevier, from K. Matsuoka et al., 1998, “COPII-Coated Vesicle Formation Reconstituted with Purified Coat Proteins and Chemically Defined Liposomes,” Cell 93(2):263–275; permission conveyed through Copyright Clearance Center, Inc.]
A Conserved Set of GTPase Switch Proteins Controls the Assembly of Different Vesicle Coats
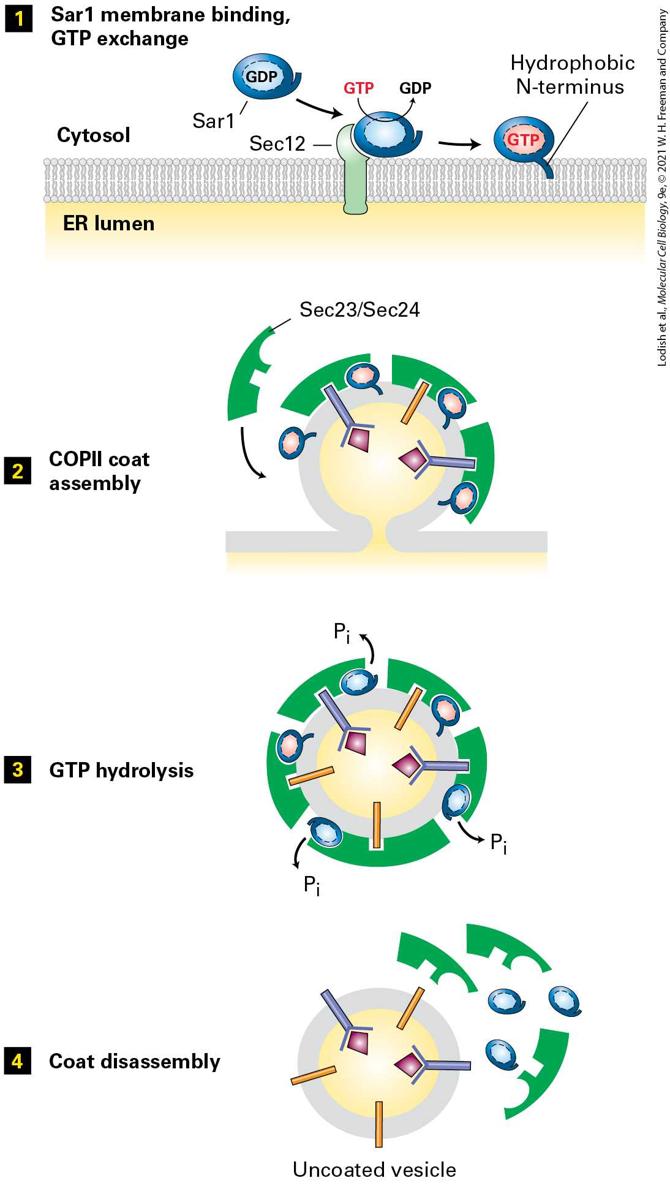
A Conserved Set of GTPase Switch Proteins Controls the Assembly of Different Vesicle Coats Using in vitro vesicle-budding reactions among isolated membranes and purified coat proteins, scientists have determined the minimum set of coat components required to form each of the three major types of vesicles. Although most of the coat proteins differ considerably from one type of vesicle to another, the coats of all three vesicles contain a small GTPbinding protein that acts as a regulatory subunit to control coat assembly (see Figure 14-7a). A GTP-binding protein known as ARF protein plays this role in COPI and clathrin-coated vesicles. A different but related GTP-binding protein known as Sar1 protein is present in the coat of COPII vesicles. Both ARF and Sar1 are monomeric proteins with a structure generally similar to that of Ras, a key intracellular signal-transducing protein (see Figure 16-12). ARF and Sar1 proteins, like Ras, belong to the GTPase superfamily of switch proteins that cycle between GDP-bound and GTP-bound forms (see Figure 3-9 to review the mechanism of GTPase switch proteins). As depicted in Figure 14-9, activation of Sar1 by binding to GTP is the initial event that triggers assembly of the COPII vesicle coat. Note that Sar1 is the only GTPase switch protein activated at the ER membrane and thus only COPII-coated vesicles bud from the ER membrane. This
initiating event begins with an ER membrane protein known as Sec12, which acts as a guanine nucleotide exchange factor (GEF) for Sar1 by catalyzing the release of GDP from cytosolic Sar1·GDP and the binding of GTP. Sec12 apparently receives and integrates multiple as yet unknown signals, probably including the presence of cargo proteins that are ready to be transported at the ER membrane. Binding of GTP causes a conformational change in Sar1; this exposes its amphipathic N-terminus, which then becomes embedded in the phospholipid bilayer and tethers Sar1·GTP to the ER membrane (Figure 14-9, step 1 ). The membraneattached Sar1·GTP drives the polymerization of cytosolic complexes of COPII subunits on the membrane, eventually leading to formation of vesicle buds (step 2 ). Once COPII vesicles are released from the donor membrane, the Sar1 GTPase activity hydrolyzes Sar1·GTP in the vesicle membrane to Sar1·GDP with the assistance of one of the coat subunits (step 3 ). This hydrolysis triggers disassembly of the COPII coat (step 4 ). Thus Sar1 couples a cycle of GTP binding and hydrolysis to the formation and then disassembly of the COPII coat.
FIGURE 14-9 Model for the role of Sar1 in the assembly and disassembly of the COPII coat. Step 1 : Interaction of soluble GDP-bound Sar1 with the GEF Sec12, an ER integral membrane protein, catalyzes exchange of GTP for GDP on Sar1. The hydrophobic N-terminus of the GTP-bound form of Sar1 extends outward from the protein’s surface and anchors Sar1 to the ER membrane. Step 2 : Once attached to the membrane, Sar1 serves as a binding site for the Sec23/Sec24 coat protein complex. Membrane cargo proteins are recruited to the forming vesicle bud by binding of specific short sequences (sorting signals) in their cytosolic regions to sites on the Sec23/Sec24 complex. Some membrane cargo proteins also act as receptors that bind soluble proteins in the lumen. The coat is completed by assembly of a second type of coat complex composed of Sec13 and Sec31 (not shown). Step 3 : After the vesicle coat is complete, the Sec23 coat subunit promotes GTP hydrolysis by Sar1. Step 4 : Release of Sar1·GDP from the vesicle membrane causes disassembly of the coat. See S. Springer et al., 1999, Cell 97:145. Description Step 1: A s e c 12 protein is embedded in the plasma membrane separating the cytosol from the E R lumen. In the cytosol, S A R 1, a G D P-containing protein binds with S E C 12. In this step G T P is converted to G D P. The hydrophobic N-terminus of S A R 1 gets embedded in the plasma membrane, binding the protein to the cytosolic face of the membrane. Step 2: S e c 23 and S e c 24 coat proteins bind with the S A R 1 proteins in the budding vesicle. During this process, membrane target receptor proteins bind with the coating proteins. Thus, C O P 2 coat assembly is complete. Step 3: The fully coated vesicle undergoes G T P hydrolysis. Three molecules of inorganic phosphates are removed from the S A R 1 proteins. Step 4: The coat disassembles along with S A R 1 proteins, leaving an uncoated vesicle behind. ARF protein undergoes a similar cycle of nucleotide exchange and hydrolysis coupled to the assembly of vesicle coats composed either of
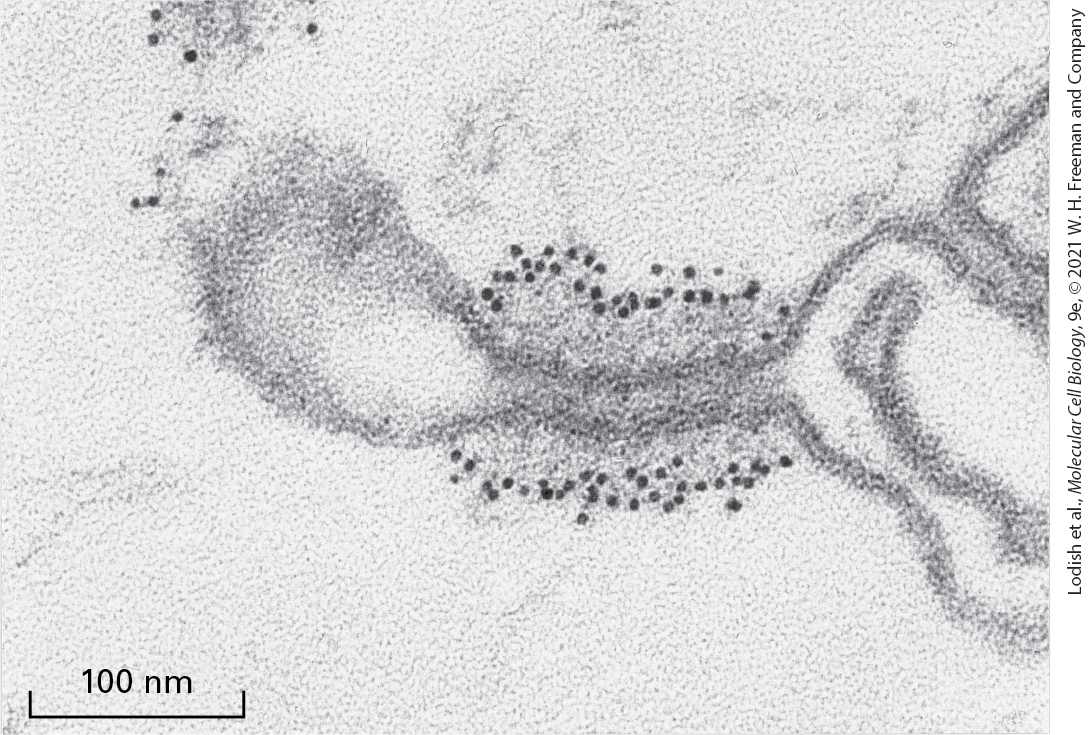
COPI or of clathrin and other coat proteins (AP complexes), discussed later. A covalent protein modification known as a myristate anchor on the N-terminus of the ARF protein weakly tethers ARF·GDP to the Golgi membrane. When GTP is exchanged for the bound GDP by a GEF attached to the Golgi membrane, the resulting conformational change in ARF allows hydrophobic residues in its N-terminal segment to insert into the membrane bilayer. The resulting tight association of ARF·GTP with the membrane serves as the foundation for further coat assembly. Drawing on the structural similarities of Sar1 and ARF to other small GTPase switch proteins, researchers have constructed genes encoding mutant versions of the two proteins that have predictable effects on vesicular traffic when transfected into cultured cells. For example, in cells expressing mutant versions of Sar1 or ARF that cannot hydrolyze GTP, vesicle coats form and vesicle buds pinch off. However, because the mutant proteins cannot trigger disassembly of the coat, all available coat subunits eventually become permanently assembled into coated vesicles that are unable to fuse with target membranes. Addition of a nonhydrolyzable GTP analog to in vitro vesicle-budding reactions causes a similar blocking of coat disassembly. The vesicles that form in such reactions have coats that never dissociate, allowing their composition and structure to be more readily analyzed. The purified COPI vesicles shown in Figure 14-10 were produced in such a budding reaction.
EXPERIMENTAL FIGURE 14-10 Coated vesicles accumulate during in vitro budding reactions in the presence of a nonhydrolyzable analog of GTP. When isolated Golgi membranes are incubated with a cytosolic extract containing COPI coat proteins, vesicles form and bud off from the membranes. Inclusion of a nonhydrolyzable analog of GTP in the budding reaction prevents disassembly of the coat after vesicle release. This micrograph shows COPI vesicles generated in such a reaction and separated from membranes by centrifugation. Coated vesicles prepared in this way can be analyzed to determine their components and properties.
Targeting Sequences on Cargo Proteins Make Specific Molecular Contacts with Coat Proteins
A second general function of small GTPases in vesicle formation is in the pinching off of a completed vesicle from the parent membrane. In vitro budding experiments show that the Sar1 GTPase that accumulates at the neck of the budding vesicle is required for the pinching off of COPII vesicles and that the ARF GTPase similarly drives the pinching off of COPI vesicles. The mechanism by which these small GTPases convert the energy from GTP hydrolysis to a mechanical force to complete the pinching off of the membrane is not understood. As we will see in Section 14.4, a large polymeric GTPase known as dynamin plays this role in clathrin-coated vesicles. Targeting Sequences on Cargo Proteins Make Specific Molecular Contacts with Coat Proteins In order for transport vesicles to move specific proteins from one compartment to the next, vesicle buds must be able to discriminate among potential membrane and soluble cargo proteins, accepting only those cargo proteins that should advance to the next compartment and excluding those that should remain as residents in the donor compartment. In addition to sculpting the curvature of a donor membrane, the vesicle coat functions in selecting specific proteins as cargo. Membrane cargo proteins and soluble cargo proteins carry two different kinds of sorting signals. The sorting signals for membrane cargo proteins usually lie in the cytosolic portion of the protein and bind to one or another of the vesicle coat proteins (see
Figure 14-7a). The polymerized coat thus acts as an affinity matrix to
cluster selected membrane cargo proteins into forming vesicle buds. Because soluble proteins within the lumen of the parent organelle cannot contact the coat directly, they require a different kind of sorting signal. Soluble luminal proteins often contain what can be thought of as luminal sorting signals, which bind to the luminal domains of certain membrane cargo proteins. The properties of some of the known sorting signals in membrane and soluble proteins are summarized in Table 14-2. We describe the role of these signals in more detail in later sections.
TABLE 14-2 • Known Sorting Signals That Direct Proteins to Specific Transport Vesicles Signal Sequence Signal-Bearing Protein Proteins with Signal Signal Receptor Cytoplasmic Sorting Signals Lys-Lys-X-X (KKXX) ER-resident membrane proteins COPI α and β subunits
Di-arginine (X-Arg-Arg- X) ER-resident membrane proteins COPI α and β subunits
Di-acidic (e.g., Asp-X- Glu) Cargo membrane proteins in ER COPII Sec24 subunit
Asn-Pro-X- Tyr (NPXY) LDL receptor in plasma membrane AP2 complex Clathrin/AP2 Tyr-X-X-Φ (YXX Φ) Membrane proteins in transGolgi AP1 ( subunit) Clathrin/AP1 Plasma membrane proteins AP2 ( subunit) Clathrin/AP2 Leu-Leu (LL) Plasma membrane proteins AP2 complexes Clathrin/AP2 ii
Rab GTPases Control Docking of Vesicles on Target Membranes
Luminal Sorting Signals Lys-Asp-Glu- Leu (KDEL) ER-resident soluble proteins KDEL receptor in cis-Golgi membrane
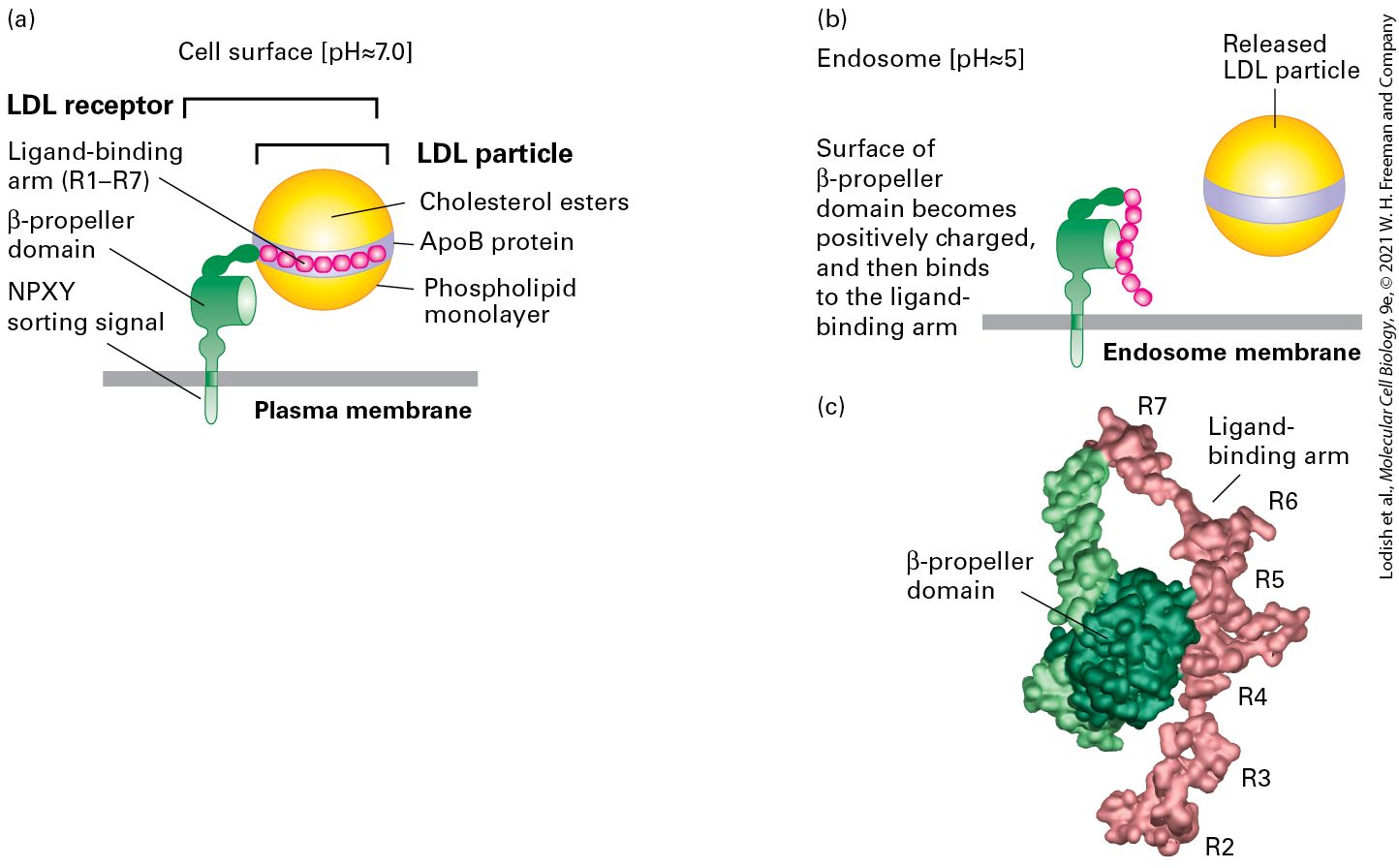
Mannose 6phosphate (M6P) Soluble lysosomal enzymes after processing in cis-Golgi M6P receptor in trans-Golgi membrane Clathrin/AP1 Secreted lysosomal enzymes M6P receptor in plasma membrane Clathrin/AP2 . Single-letter amino acid abbreviations are in parentheses. Rab GTPases Control Docking of Vesicles on Target Membranes A second set of small GTP-binding proteins, known as Rab proteins, associate with transport vesicles and act as key regulators of vesicle trafficking to and fusion with the appropriate target membrane. Like Sar1 and ARF, Rab proteins belong to the GTPase superfamily of switch proteins. Rab proteins also contain an isoprenoid anchor that allows them to become tethered to the vesicle membrane. Association of an activated Rab protein with a specific vesicle type is generally a two-step process. In the first step, cytosolic Rab·GDP is targeted to the appropriate vesicle, becoming attached there by insertion of its isoprenoid anchor into the vesicle membrane. Often this attachment step is facilitated by a protein that can associate with Rab·GDP along with its isoprenoid anchor, usually known as a guanine nucleotide dissociation inhibitor (GDI). In the second ii
step, a specific GEF located in the vesicle membrane converts membranebound Rab·GDP to Rab·GTP. Once localized and activated in this way, Rab·GTP is enabled to bind to a variety of different proteins, known as Rab effectors. Binding of Rab·GTP to a Rab effector can ultimately lead to docking of the vesicle on an appropriate target membrane (Figure 14-11a, step 1 ). After vesicle fusion occurs, the GTP bound to the Rab protein is hydrolyzed to GDP, triggering the release of Rab·GDP, which can then undergo another cycle of GDP-GTP exchange, binding, and hydrolysis.
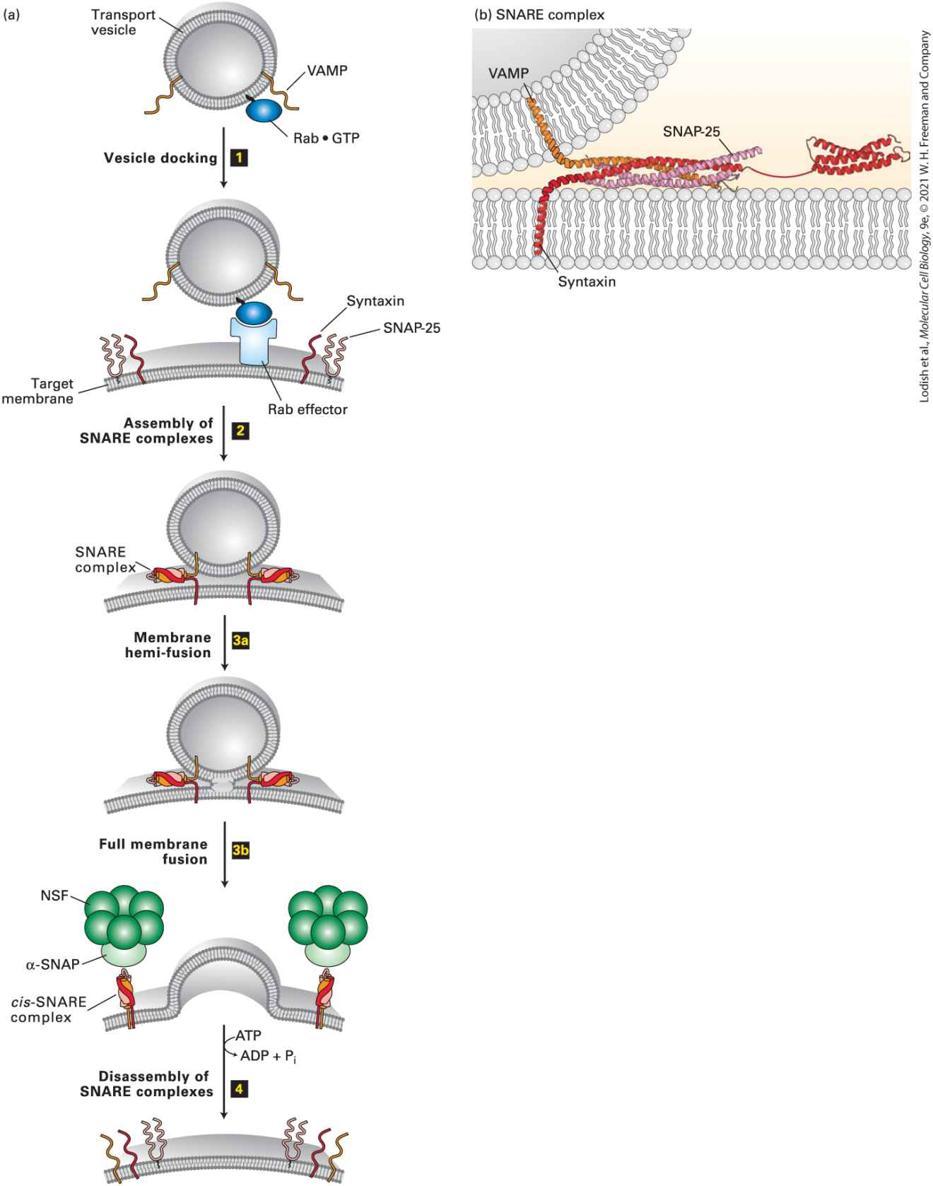
FIGURE 14-11 Model for docking and fusion of transport vesicles with their target membranes. (a) The proteins shown in this example participate in fusion of secretory vesicles with the plasma membrane, but similar proteins mediate all vesicle-fusion events.
Step 1 : A Rab protein tethered via a lipid anchor to a secretory vesicle binds to an effector protein complex on the plasma membrane, thereby docking the transport vesicle on the appropriate target membrane. Step 2 : A v-SNARE protein (in this case, VAMP) interacts with the cytosolic domains of the cognate t-SNAREs (in this case, syntaxin and SNAP-25). The very stable coiled-coil SNARE complexes that are formed hold the vesicle close to the target membrane. Step 3 : Fusion of the two membranes immediately follows formation of SNARE complexes, first by fusion of one leaflet of the bilayer to form a hemi-fused state ( 3a ), followed by fusion of the second leaflet ( 3b ). Step 4 : Following membrane fusion, NSF, in conjunction with α-SNAP, binds to the SNARE complexes. The NSFcatalyzed hydrolysis of ATP then drives dissociation of the SNARE complexes, freeing the SNARE proteins for another round of vesicle fusion. Also at this time, Rab·GTP is hydrolyzed to Rab·GDP and dissociates from the Rab effector (not shown). (b) The SNARE complex. Numerous noncovalent interactions between four long α helices, two from SNAP25 and one each from syntaxin and VAMP, stabilize the coiled-coil structure. See J. E. Rothman and T. Söllner, 1997, Science 276:1212; Y. A. Chen and R. H. Scheller, 2001, Nat. Rev. Mol. Cell Biol. 2:98; and W. Weis and R. Scheller, 1998, Nature 395:328. [Part (b) Data from I. Fernandez et al., 1998, Cell 94:841–849, PDB ID 1br0; and R. B. Sutton et al., 1998, Nature 395:347–353, PDB ID 1sfc.] Description The illustration labeled (a) depicts the docking and fusion of vesicles in four steps. A circular transport vesicle has a Rab-G T P protein embedded in the cytosolic face of the vesicle. Two V A M P proteins extend from the vesicle into the cytosol. Step 1: The target membrane contains a Rab effector, two syntaxins, and two S N A P25 proteins. Once the vesicle is close to the target membrane, the Rab- G T P protein docks with the Rab effector. Step 2: The V A M P proteins, S N A P – 25 proteins, and the syntaxin proteins bind together to form the S N A R E complex between the vesicle and target membrane. Step 3 a: The vesicle membrane and the target membrane undergo membrane hemifusion between the S N A R E complexes.
Step 3 b: The vesicle undergoes full membrane fusion with the target membrane. The now released cis-S N A R E complexes bind with the alpha S N A P domain of the N-ethylmaleimide sensitive fusion protein (NSF). Step 4: The S N A R E complexes disassemble, using a molecule of A T P in the process. A T P is converted to A D P and inorganic phosphate. The illustration labeled (b) depicts the formation of the S N A R E complex is. A vesicle is in close proximity to the target membrane. V A M P protein is embedded in the vesicle; syntaxin is embedded in the target membrane, as is S N A P-25. The alphahelices of these three proteins wrap around each other, forming a coiled-coil. A well-understood example of a Rab protein that enables vesicle fusion with the correct target membrane is the Sec4 protein of yeast, which specifically tags secretory vesicles, enabling them to fuse with the plasma membrane. In experiments, yeast cells expressing mutant Sec4 proteins accumulate secretory vesicles that are unable to fuse with the plasma membrane (class E mutants in Figure 14-5). Sec4·GDP binds to secretory vesicles, where it is activated to Sec4·GTP by its cognate GEF, which is itself located on secretory vesicles. Sec4·GTP, in turn, binds to its effector, a large tethering complex composed of eight subunits, known as the exocyst. Tethering of secretory vesicles to the exocyst by binding of Sec4·GTP ultimately leads to vesicle fusion with the plasma membrane. In mammalian cells, Rab5 protein is localized to endocytic vesicles, also known as early endosomes. These uncoated vesicles form from clathrincoated vesicles just after they bud from the plasma membrane during endocytosis (see Figure 14-2, step 9 ). The fusion of early endosomes with one another in cell-free systems requires the presence of Rab5, and
addition of Rab5 and GTP to cell-free extracts accelerates the rate at which these vesicles fuse with one another. A long coiled protein known as EEA1 (early endosome antigen 1), which resides on the membrane of the early endosome, functions as the effector for Rab5. In this case, Rab5·GTP on one endocytic vesicle is thought to bind specifically to EEA1 on the membrane of another endocytic vesicle, setting the stage for fusion of the two vesicles. Every type of transport vesicle appears to be labeled with one or more specific Rab proteins. These Rab proteins, through their specific association with effectors that are membrane tethers and molecular motors, ensure that the vesicles are directed to the correct target membrane address (Table 14-3). The tracks on which the molecular motors run to deliver the vesicles to their destinations — microfilaments — will be discussed in Chapter 17.
TABLE 14-3 • Rab Proteins Involved in Vesicle Tethering and Vesicle Movement Rab Protein Transport Step Effector Proteins Rab1/Ypt1 ER to cis-Golgi TRAPP complex and GM130 complex in cis-Golgi Sec4 trans-Golgi to plasma membrane Exocyst on plasma membrane and Type V Myosin Rab5 Plasma membrane to endosome EEA1 tether on endosome Rab6 Golgi movement along microtubules Kinesin
Paired Sets of SNARE Proteins Mediate Fusion of Vesicles with Target Membranes
Rab27 Melanosome movement to cell periphery Type V Myosin Paired Sets of SNARE Proteins Mediate Fusion of Vesicles with Target Membranes As noted previously, shortly after a vesicle buds off from the donor membrane, the vesicle coat disassembles to uncover a vesicle-specific membrane protein, a v-SNARE (see Figure 14-7b). Likewise, each type of target membrane in a cell contains t-SNARE membrane proteins, which interact specifically with v-SNAREs. After Rab-mediated docking of a vesicle on its target membrane, the interaction of cognate SNAREs brings the two membranes close enough together that they can fuse. One of the best understood examples of SNARE-mediated fusion occurs during exocytosis of secreted proteins (see Figure 14-11a, steps 1 and 3 ). In this case, the v-SNARE, known as VAMP (vesicle-associated membrane protein), is incorporated into secretory vesicles as they bud from the trans-Golgi network. The t-SNAREs are syntaxin, an integral membrane protein in the plasma membrane, and SNAP-25, which is attached to the plasma membrane by a hydrophobic lipid anchor in the middle of the protein. The cytosolic region in each of these three SNARE proteins contains a repeating heptad sequence that allows four α helices — one from VAMP, one from syntaxin, and two from SNAP-25 — to coil around one another to form a four-helix bundle (Figure 14-11b). The
unusual stability of this bundled SNARE complex is conferred by the arrangement of hydrophobic and charged amino acid residues in the heptad repeats. The hydrophobic amino acids are buried in the central core of the bundle, and amino acids of opposite charge are aligned to form favorable electrostatic interactions between helices. As multiple four-helix bundles form, the embedded transmembrane domains of VAMP and syntaxin pull the vesicle and target membranes together into very close apposition. The energetically favorable formation of four-helix bundles can overcome the electrostatic repulsion of the generally negatively charged phospholipid head groups in the vesicle and target membranes, allowing the hydrophobic interiors of the two membranes to come into contact, creating an opening between the two membranes, and ultimately causing the vesicle membrane to fuse with the target membrane. In vitro experiments have shown that when liposomes containing purified VAMP are incubated with other liposomes containing syntaxin and SNAP25, the two classes of membranes fuse, albeit slowly. This finding is strong evidence that the close apposition of membranes resulting from formation of SNARE complexes is sufficient to bring about membrane fusion. Fusion of a vesicle and target membrane occurs more rapidly and efficiently in the cell than it does in liposome experiments in which fusion is catalyzed only by SNARE proteins. The likely explanation for this difference is that in the cell, other proteins, such as Rab proteins and their effectors, are involved in targeting vesicles to the correct membrane. Yeast cells, like all eukaryotic cells, express more than 20 different related v-SNARE and t-SNARE proteins. Analyses of yeast mutants defective in
Dissociation of SNARE Complexes After Membrane Fusion Is Driven by ATP Hydrolysis
each of the SNARE genes have identified specific membrane-fusion events in which each SNARE protein participates. For all fusion events that have been examined, the SNAREs form four-helix bundled complexes similar to the VAMP/syntaxin/SNAP-25 complexes that mediate fusion of secretory vesicles with the plasma membrane. However, in other fusion events (e.g., fusion of COPII vesicles with the cis-Golgi network), each participating SNARE protein contributes only one α helix to the bundle (unlike SNAP-25, which contributes two helices); in these cases, the SNARE complexes comprise one v-SNARE and three t-SNARE molecules. Using the in vitro liposome fusion assay, researchers have tested the ability of various combinations of individual v-SNARE and t-SNARE proteins to mediate fusion of donor and target membranes. Of the very large number of different combinations tested, only a small number could efficiently mediate membrane fusion. To a remarkable degree, the functional combinations of v-SNAREs and t-SNAREs revealed in these in vitro experiments correspond to the actual SNARE protein interactions that mediate known membrane-fusion events in the yeast cell. Thus together with the specificity of interaction between Rab and Rab effector proteins, the specificity of the interaction between SNARE proteins can account for most, if not all, of the specificity of fusion between a particular vesicle type and its target membrane. Dissociation of SNARE Complexes After Membrane Fusion Is Driven by
ATP Hydrolysis After a vesicle and its target membrane have fused, the SNARE complexes must dissociate to make the individual SNARE proteins available for additional fusion events. Because of the stability of SNARE complexes, which are held together by numerous noncovalent intermolecular interactions, their dissociation depends on additional proteins and the input of energy. The first clue that dissociation of SNARE complexes required the assistance of other proteins came from in vitro transport reactions depleted of certain cytosolic proteins. The observed accumulation of vesicles in these reactions indicated that vesicles could form under these conditions, but were unable to fuse with a target membrane. Eventually two proteins, designated NSF and α-SNAP, were found to be required for ongoing vesicle fusion in the in vitro transport reaction. Yeast mutants have also contributed to our understanding of SNARE function. Among the class C yeast sec mutants are strains that lack functional Sec18 or Sec17, the yeast counterparts of mammalian NSF and α-SNAP, respectively. When these class C mutants are kept at the nonpermissive temperature, they accumulate ER-to-Golgi transport vesicles; when the cells are shifted to the lower, permissive temperature, the accumulated vesicles are able to fuse with the cis-Golgi.
Subsequent to the initial biochemical and genetic studies that identified NSF and α-SNAP, more sophisticated in vitro transport assays were developed. Using these newer assays, researchers have shown that NSF and α-SNAP proteins are not necessary for actual membrane fusion, but rather are required for regeneration of free SNARE proteins. NSF, a hexamer of identical subunits, associates with a SNARE complex with the aid of α-SNAP (soluble NSF attachment protein). The bound NSF then hydrolyzes ATP, releasing sufficient energy to dissociate the SNARE complex (see Figure 14-11a, step 4 ). Evidently, the defects in vesicle fusion observed in the earlier in vitro fusion assays and in the yeast mutants after a loss of Sec17 or Sec18 were a consequence of free SNARE proteins rapidly becoming sequestered in undissociated SNARE complexes and thus being unavailable to mediate membrane fusion. KEY CONCEPTS OF SECTION 14.2 Molecular Mechanisms of Vesicle Budding and Fusion The three well-characterized types of transport vesicles — COPI, COPII, and clathrincoated vesicles — are distinguished by the proteins that form their coats and the transport routes they mediate (see Table 14-1). All types of coated vesicles are formed by polymerization of cytosolic coat proteins on a parent (donor) membrane to form vesicle buds that eventually pinch off from the membrane to release a complete vesicle. Shortly after vesicle release, the coat is shed, exposing proteins required for fusion with the target membrane (see Figure 14-7). Small GTP-binding proteins (ARF or Sar1) belonging to the GTPase superfamily control polymerization of coat proteins, the initial step in vesicle budding (see Figure 14-9). After vesicles are released from the donor membrane, hydrolysis of GTP bound to ARF or Sar1 triggers disassembly of the vesicle coats. Specific sorting signals in membrane and luminal proteins in donor organelles interact with coat proteins during vesicle budding, thereby recruiting cargo proteins to vesicles (see Table 14-2).
A second set of GTP-binding proteins, the Rab proteins, label specific vesicle types and mediate their targeting to the appropriate membrane (see Table 14-3). Activated Rab·GTP in a vesicle can bind to a specific type of effector protein. One type of effector is a peripheral membrane protein complex that tethers the vesicle to the correct target membrane. Each v-SNARE in a vesicular membrane specifically binds to a complex of cognate tSNARE proteins in the target membrane, inducing fusion of the two membranes. After fusion is completed, the SNARE complex is disassembled in an ATP-dependent reaction mediated by other cytosolic proteins (see Figure 14-11).
14.3 Early Stages of the Secretory Pathway
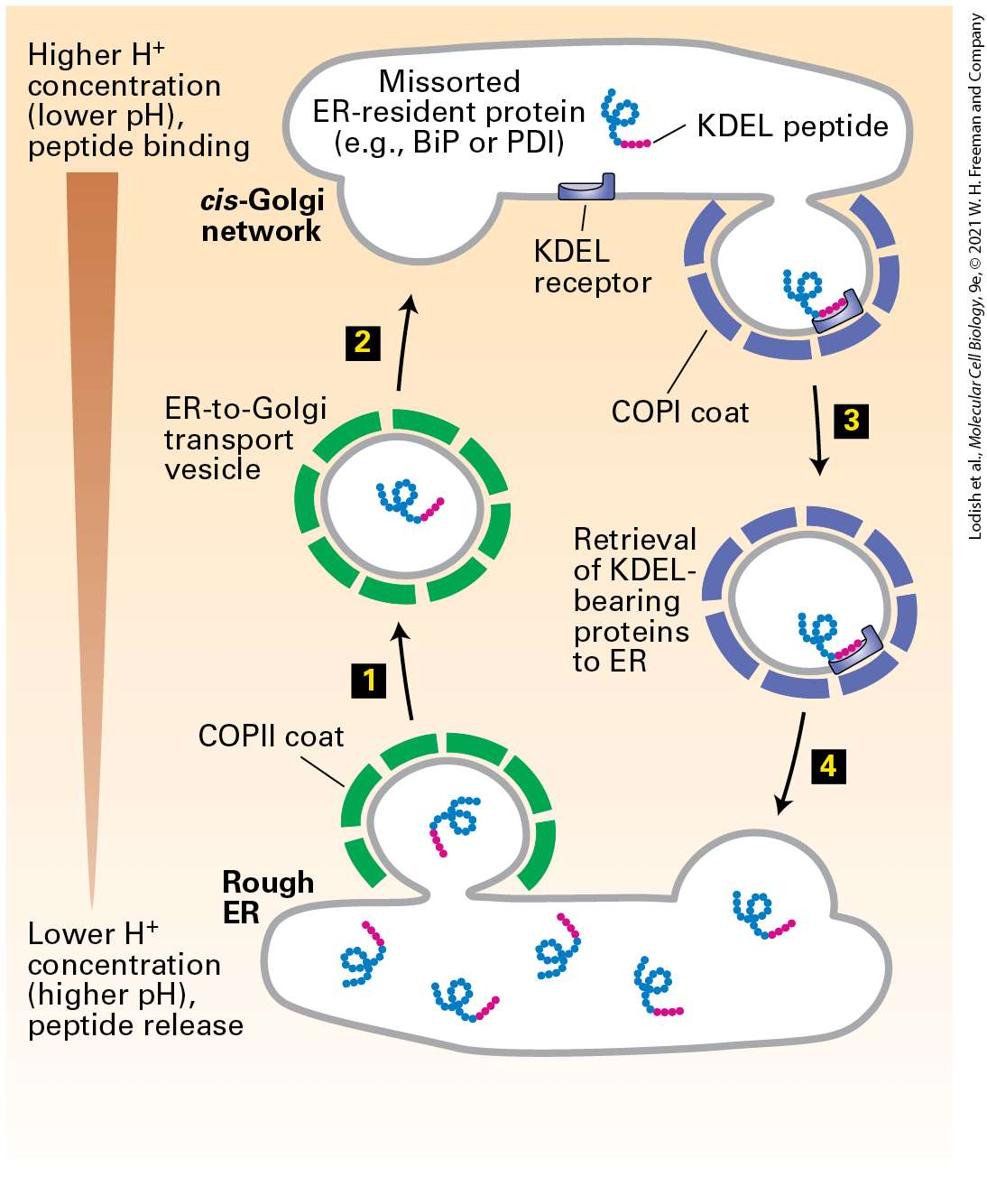
14.3 Early Stages of the Secretory Pathway In this section, we take a closer look at vesicular traffic between the ER and the Golgi and at some of the evidence supporting the general mechanisms discussed in the previous section. Recall that anterograde transport from the ER to the Golgi, the first vesicle trafficking step in the secretory pathway, is mediated by COPII vesicles. These vesicles contain newly synthesized proteins destined for the Golgi, cell surface, or lysosomes, as well as vesicle components such as v-SNAREs that are required to target vesicles to the cis-Golgi membrane. Proper sorting of proteins between the ER and Golgi also requires retrograde transport from the cis-Golgi to the ER, which is mediated by COPI vesicles (Figure 1412). This retrograde vesicle transport serves to retrieve v-SNARE proteins and components of the membrane itself to provide the necessary material for additional rounds of vesicle budding from the ER. COPI-mediated retrograde transport also retrieves missorted ER-resident proteins from the cis-Golgi.
FIGURE 14-12 Vesicle-mediated protein trafficking between the ER and cis-Golgi. Steps 1 – 3 : Forward (anterograde) transport is mediated by COPII vesicles, which are formed
by polymerization of soluble COPII coat protein complexes (green) on the ER membrane. v-SNAREs (orange) and other cargo proteins (blue) in the ER membrane are incorporated into the vesicle by interacting with coat proteins. Soluble cargo proteins (magenta) are recruited by binding to appropriate receptors in the membrane of budding vesicles. Dissociation of the coat recycles free coat complexes and exposes v-SNARE proteins on the vesicle surface. After the uncoated vesicle becomes tethered to the cis-Golgi membrane in a Rab-mediated process, pairing between the exposed v-SNAREs and cognate t-SNAREs in the Golgi membrane allows membrane fusion, releasing the contents of the vesicle into the cis-Golgi compartment (see Figure 14-10). Steps 4 – 6 : Reverse (retrograde) transport, mediated by vesicles coated with COPI proteins (purple), recycles the membrane bilayer and certain proteins, such as v-SNAREs and missorted ER-resident proteins (not shown), from the cis-Golgi to the ER. All SNARE proteins are shown in orange, although v-SNAREs and t-SNAREs are distinct proteins. Description The illustration shows the rough E R with various membrane cargo proteins, membrane receptors, and S N A R E protein. Soluble cargo proteins are present inside the rough E R. Step 1: Coated vesicles begin to bud out of the E R forming a C O P 2 coated vesicle. Step 2: The vesicle coating proteins disintegrate to be used for further budding process. Step 3: The uncoated vesicle reaches the cis-Golgi network, forming S N A R E pairs between the membrane proteins. The soluble cargo proteins are transported into the cisGolgi. Step 4: A coated vesicle buds out from the cis-Golgi network, coated by C O P 1 coat proteins. Step 5: The C O P 1 vesicle coat proteins disintegrate to be used for further budding processes from the cis-Golgi network. Step 6: The uncoated vesicles dock with the rough E R via S N A R E pairs.
COPII Vesicles Mediate Transport from the ER to the Golgi
We also discuss in this section the process by which proteins that have been correctly delivered to the Golgi advance through successive compartments of the Golgi, from the cis- to the trans-Golgi network. This process of cisternal maturation involves budding and fusion of retrograde rather than anterograde transport vesicles. COPII Vesicles Mediate Transport from the ER to the Golgi COPII vesicles were first recognized when cell-free extracts of yeast rough ER membranes were incubated with cytosol and a nonhydrolyzable analog of GTP. The vesicles that formed from the ER membranes had a distinct coat similar to that on COPI vesicles, but composed of different proteins, designated COPII proteins. The genes encoding COPII proteins were identified by the analysis of yeast cells with class B sec mutations that accumulate proteins in the rough ER (see Figure 14-5). As described previously, formation of COPII vesicles is triggered when Sec12, a GEF in the ER membrane, catalyzes the exchange of bound GDP for GTP on cytosolic Sar1. This exchange induces binding of Sar1 to the ER membrane, followed by binding of a complex of Sec23 and Sec24 proteins (see Figure 14-9). The resulting ternary complex formed between Sar1·GTP, Sec23, and Sec24 is shown in Figure 14-13. This core coat protein complex then provides binding sites for the recruitment of a second complex of Sec13 and Sec31 proteins to complete the coat structure. Since pure Sec13 and Sec31 proteins can spontaneously
assemble into cagelike lattices, it is thought that Sec13 and Sec31 form the structural scaffold for COPII vesicles (see the COPII vesicle model in the chapter opener figure). Finally, a large fibrous protein called Sec16, which is bound to the cytosolic surface of the ER, interacts with Sar1·GTP and the Sec13/31 and Sec23/24 complexes to organize the other coat proteins, increasing the efficiency of coat polymerization.
FIGURE 14-13 Three-dimensional structure of the ternary complex comprising the COPII coat proteins Sec23, Sec24, and Sar1·GTP. Early in the formation of the COPII coat, Sec23 (orange)/Sec24 (green) complexes are recruited to the ER membrane by Sar1 (red) in its GTP-bound state. A cargo protein in the ER membrane can be recruited to COPII vesicles by the interaction of a tripeptide di-acidic sorting signal (purple) in the cargo protein’s cytosolic domain with Sec24. The likely positions of the COPII vesicle membrane
and the transmembrane segment of the cargo protein are indicated. The N-terminal segment of Sar1 that tethers it to the membrane is not shown. [Republished with permission from Nature, from X. Bi et al., 2002, “Structure of the Sec23/24-Sar1 Prebudding Complex of the COPII Vesicle Coat,” Nature 419(6904):271– 277; permission conveyed through the Copyright Clearance Center, Inc.] Description The three-dimensional model of the S a r 1, S e c 23, and S e c 24 ternary protein complex is present on the cytosolic face of the vesicular membrane. The S e c 24 protein is attached to the di-acidic sorting signal attached to the transmembrane segment of a cargo protein. The transmembrane segment is represented by red wavy connected dots through the membrane. The cargo protein that follows is represented by blue connected dots that are present inside the vesicle membrane. Certain integral ER membrane proteins are specifically recruited into COPII vesicles for transport to the Golgi. The cytosolic segments of many of these proteins contain a di-acidic sorting signal (the key residues in this sequence are Asp-X-Glu, or DXE in the one-letter code) (see Table 14-2). This sorting signal, which binds to the Sec24 subunit of the COPII coat, is essential for the selective export of certain membrane proteins from the ER (see Figure 14-12). Biochemical and genetic studies have identified additional signals that help direct membrane cargo proteins into COPII vesicles. All of the known sorting signals bind to one or another site on the Sec24 subunit of COPII. Ongoing studies seek to determine how soluble cargo proteins are selectively loaded into COPII vesicles. The inherited disease cystic fibrosis is characterized by an imbalance in chloride and sodium ion transport in the epithelial cells of
COPI Vesicles Mediate Retrograde Transport Within the Golgi and from the Golgi to the ER
the lungs, leading to fluid buildup and difficulty breathing. Cystic fibrosis is caused by mutations in a protein known as CFTR, which is synthesized as an integral membrane protein in the ER and is transported to the Golgi before being transported to the plasma membranes of epithelial cells, where it functions as a chloride channel. Researchers have recently shown that the CFTR protein contains a di-acidic sorting signal that binds to the Sec24 subunit of the COPII vesicle coat and is necessary for transport of the CFTR protein out of the ER. The most common CFTR mutation is a deletion of a phenylalanine at position 508 in the protein sequence (known as ). This mutation prevents normal transport of CFTR to the plasma membrane by blocking its packaging into COPII vesicles budding from the ER. While the CFTR protein with the mutation triggers the ER quality-control machinery, a folded CFTR with this mutation can still function properly as a normal chloride channel. However, it never reaches the membrane; the disease state is therefore caused by the absence of the channel, rather than by a defective channel. COPI Vesicles Mediate Retrograde Transport Within the Golgi and from the Golgi to the ER COPI vesicles were first discovered when isolated Golgi fractions were incubated in a solution containing cytosol and a nonhydrolyzable analog of GTP (see Figure 14-10). Subsequent analysis of these vesicles showed that the coat is formed from large cytosolic complexes, called coatomers, composed of seven polypeptide subunits. Yeast cells containing
temperature-sensitive mutations in COPI proteins accumulate proteins in the rough ER at the nonpermissive temperature and thus are categorized as class B sec mutants (see Figure 14-5). Although the discovery of these mutants initially suggested that COPI vesicles mediate ER-to-Golgi transport, subsequent experiments showed that their main function is retrograde transport, both between Golgi cisternae and from the cis-Golgi to the rough ER (see Figure 14-12, right). Because COPI mutants cannot recycle key membrane proteins back to the rough ER, the ER gradually becomes depleted of ER proteins, such as v-SNAREs, that are necessary for COPII vesicle function. Eventually, vesicle formation from the rough ER grinds to a halt; secretory proteins continue to be synthesized but accumulate in the ER — the defining characteristic of class B sec mutants. The general ability of sec mutations involved in either COPI or COPII vesicle function to eventually block both anterograde and retrograde transport illustrates the fundamental interdependence of these two transport processes. As discussed in Chapter 13, the ER contains several soluble proteins dedicated to the folding and modification of newly synthesized secretory proteins. They include the chaperone BiP and the enzyme protein disulfide isomerase, which are necessary for the ER to carry out its functions. Although such ER-resident luminal proteins are not specifically selected by COPII vesicles, their sheer abundance causes them to be continuously loaded passively into vesicles destined for the cis-Golgi. The transport of these soluble proteins back to the ER, mediated by COPI vesicles, prevents their eventual depletion.
Most soluble ER-resident proteins carry a Lys-Asp-Glu-Leu (KDEL in the one-letter code) sequence at their C-terminus (see Table 14-2). Several experiments have demonstrated that this KDEL sorting signal is both necessary and sufficient to cause a protein bearing this sequence to be located in the ER. For instance, when a mutant protein disulfide isomerase lacking these four residues is synthesized in cultured fibroblasts, the protein is secreted. Moreover, if a protein that is normally secreted is altered so that it contains the KDEL sorting signal at its C-terminus, the protein is located in the ER. The KDEL sorting signal is recognized and bound by the KDEL receptor, a transmembrane protein found primarily on small transport vesicles shuttling between the ER and the cis-Golgi and on the cis-Golgi membrane. In addition, soluble ER-resident proteins that carry the KDEL signal have oligosaccharide chains bearing modifications that are catalyzed by enzymes found only in the cis-Golgi or cis-Golgi network; thus at some time these proteins must have left the ER and been transported at least as far as the cis-Golgi network. These findings indicate that the KDEL receptor acts mainly to retrieve soluble proteins containing the KDEL sorting signal that have escaped to the cis-Golgi network and return them to the ER (Figure 14-14). The KDEL receptor binds more tightly to its ligand at low pH, and it is thought that the receptor is able to bind KDEL peptides in the cis-Golgi and release these peptides in the ER because the pH of the Golgi is slightly lower than that of the ER.
FIGURE 14-14 Role of the KDEL receptor in retrieval of ER-resident luminal proteins from the Golgi. ER luminal proteins, especially those present at high concentrations, can be passively incorporated into COPII vesicles and transported to the Golgi (steps 1 and 2 ). Many such proteins bear a C-terminal KDEL (Lys-Asp-Glu-Leu) sequence (red) that allows them to be retrieved. The KDEL receptor, located mainly in the cis-Golgi network and in
both COPII and COPI vesicles, binds proteins bearing the KDEL sorting signal and returns them to the ER (steps 3 and 4 ). This retrieval system prevents depletion of ER luminal proteins such as those needed for proper folding of newly made secretory proteins. The binding affinity of the KDEL receptor is very sensitive to pH. The small difference between the pH of the ER and that of the Golgi favors binding of KDEL-bearing proteins to the receptor in Golgi-derived vesicles and their release in the ER. See J. Semenza et al., 1990, Cell 61:1349. Description The steps involved in the mechanism are as follows, Step 1: In the rough E R, which has a higher p H than the cis-Golgi network, K D E L peptides are produced. During vesicular budding, these proteins can become contained in the C O P 2 coated vesicles. Step 2: The K D E L protein in the coated transport vesicle is transported to the cisGolgi network. In the cis-Golgi, the missorted E R-resident proteins like B i P or P D I bind to K D E L receptor in the Golgi membrane and are included inside C O P 1coated budding vesicles. Step 3: The C O P 1-coated vesicles are returned to the rough E R. Step 4: The K D E L peptides are now present inside the rough E R. The KDEL receptor and other membrane proteins that are transported back to the ER from the Golgi contain a Lys-Lys-X-X sequence at the very end of their C-terminal segment, which faces the cytosol (see Table 14-2). This KKXX sorting signal, which binds to a complex of the COPI α and β subunits (two of the seven polypeptide subunits in the COPI coatomer), is both necessary and sufficient to incorporate membrane proteins into COPI vesicles for retrograde transport to the ER. Temperature-sensitive yeast mutants lacking COPIα or COPIβ are not only unable to bind the KKXX
signal, but are also unable to transport proteins bearing this signal back to the ER, indicating that COPI vesicles mediate retrograde Golgi-to-ER transport. A second sorting signal that targets proteins to COPI vesicles and thus enables recycling from the Golgi to the ER is a di-arginine sequence. Unlike the KKXX sorting signal, which must be located at the cytoplasmically oriented C-terminus of a protein, the di-arginine sorting signal can reside in any segment of a membrane protein that is on the cytoplasmic face of the membrane. The partitioning of proteins between the ER and Golgi complex is a highly dynamic process depending on both COPII (anterograde) and COPI (retrograde) vesicles, with each type of vesicle responsible for recycling the components necessary for the function of the other type of vesicle. The organization of this partitioning process raises an interesting puzzle: How do vesicles preferentially use the v-SNAREs that specify fusion with the correct target membrane instead of the v-SNAREs that are being recycled and specify fusion with the donor membrane? This basic question concerning correct membrane partitioning has recently been answered for COPII vesicles. After these vesicles form, the COPII coat proteins remain assembled long enough for the Sec23/Sec24 complex to interact with a specific tethering factor attached to the cis-Golgi membrane. Vesicle uncoating to expose the v-SNAREs is completed only after the COPII vesicle is already closely associated with the cis-Golgi membrane and the COPII v-SNAREs are in position to form complexes
Anterograde Transport Through the Golgi Occurs by Cisternal Maturation
with their cognate t-SNAREs. Although COPII vesicles also carry COPIspecific v-SNARE proteins, which are being recycled back to the cisGolgi, these COPI v-SNARE proteins never have the opportunity to form SNARE complexes with cognate ER-localized t-SNARE proteins. Anterograde Transport Through the Golgi Occurs by Cisternal Maturation The Golgi complex is organized into three compartments, often arranged in a stacked set of flattened sacs, called cisternae. The compartments of the Golgi differ from one another according to the enzymes they contain. Many of the enzymes are glycosidases and glycosyltransferases that are involved in modifying the N-linked or O-linked carbohydrates attached to secretory proteins as they transit through the Golgi complex. On the whole, the Golgi complex operates much like an assembly line, where each compartment serves as an assembly station in which the carbohydrate chains modified in the previous compartment are substrates for the modifying enzymes of the next compartment. (Figure 14-15 shows a representative sequence of modification steps.)
FIGURE 14-15 Processing of N-linked oligosaccharide chains on glycoproteins within cis-, medial-, and trans-Golgi cisternae in vertebrate cells. After removal of three mannose residues in the cis-Golgi (step 1 ), the protein moves by cisternal maturation to the medial-Golgi. Here three N-acetylglucosamine (GlcNAc) residues are added (steps 2 and 4 ), two more mannose residues are removed (step 3 ), and a single fucose is added (step 5 ). Processing is completed in the trans-Golgi by addition of three galactose residues (step 6 ) and finally by linkage of an N-acetylneuraminic acid residue to each of the galactose residues (step 7 ). Specific transferase enzymes add sugars to the oligosaccharide, one at a time, from sugar nucleotide precursors imported from the cytosol. The enzymes catalyzing each step are localized to the indicated compartments. This pathway represents the Golgi processing events for a typical mammalian glycoprotein. Variations in the structure of N-linked oligosaccharides can result from differences in processing steps in the Golgi. See R. Kornfeld and S. Kornfeld, 1985, Annu. Rev. Biochem. 45:631. Description Step: 1. In the cis-Golgi, proteins labeled with eight mannose and two N-acetyl glucosamine oligosaccharides are processed, removing three mannose units. M a n subscript 8(G l c N A c) subscript 2 is converted to M a n subscript 5(G l c N A c) subscript 2. Step 2: In the medial-Golgi, a U D P-N-acetyl glucosamine is attached to M a n subscript 5(G l c N A c) subscript 2. Step 3: Two mannose units are removed from the product formed in the previous step. Step 4 and 5: U D P-N-acetyl glucosamine and G D P- Fucose is added to the product of the previous step. Step 6: In the trans-Golgi, U D P-galactose is added to the last product formed in the previous step. Step 7: to the product from the previous step, C M P-N-acetylneuraminic acid is added. This is followed by budding and transport.
For many years, it was thought that the Golgi complex was an essentially static set of compartments with small transport vesicles carrying secretory proteins forward, from the cis- to the medial-Golgi and from the medialto the trans-Golgi. Indeed, electron microscopy reveals many small vesicles associated with the Golgi complex that appear to move proteins from one Golgi compartment to another (Figure 14-16). However, electron microscopy only provides an image of structures that is frozen in time and cannot show the direction of movement of the transport vesicles. We now know that the small vesicles in the vicinity of the Golgi complex are actually moving in the retrograde direction, retrieving ER or Golgi enzymes from a later compartment and transporting them to an earlier compartment in the secretory pathway. Thus the Golgi appears to have a highly dynamic organization, continually forming transport vesicles, though only in the retrograde direction. To see the effect of this retrograde transport on the organization of the Golgi, consider the net effect on the medial-Golgi compartment as enzymes from the trans-Golgi move to the medial-Golgi while enzymes from the medial-Golgi are transported to the cis-Golgi. As this process continues, the medial-Golgi acquires enzymes from the trans-Golgi while losing medial-Golgi enzymes to the cis-Golgi and thus progressively becomes a new trans-Golgi compartment. In this way, secretory cargo proteins acquire carbohydrate modifications in the proper sequential order without being moved from one cisterna to another via anterograde vesicle transport.
EXPERIMENTAL FIGURE 14-16 Anterograde-moving COPII transport vesicles and retrograde-moving COPI transport vesicles are difficult to distinguish in an electron micrograph of the Golgi complex in a pancreatic acinar cell. Elements of the rough ER are on the bottom and left in this micrograph. Adjacent to the rough ER are transitional elements from which smooth protrusions appear to be budding. These buds form the small COPII vesicles that transport secretory proteins in the anterograde direction from the rough ER to the Golgi complex. Interspersed among the Golgi cisternae are other small vesicles that are similar in appearance to the COPII vesicles, but these transport vesicles, which bear COPI coats, are now known to carry resident Golgi enzymes between Golgi compartments in the retrograde direction. A large anterograde secretory vesicle can be seen forming from the trans-Golgi network. Description
The parts are labeled on the right side of the electron micrograph. From top to bottom, a gray oval labeled forming secretory vesicle, a smaller oval labeled trans-Golgi network, a set of three oval areas grouped into one label: Golgi cisternae, trans-, medial-, cis-. Next is a group of small circles is labeled cis-Golgi network. Then there are groups of circles that are labeled E R-to-Golgi transport vesicles, a smooth line labeled smooth protrusion, and the last label reads, transitional elements. The scale bar reads 0.5 micrometers. The first evidence that the forward transport of cargo proteins from the cis- to the trans-Golgi occurs by this progressive mechanism of cisternal maturation came from careful microscopic analysis of the synthesis of large macromolecular complexes that follow the secretory pathway to form scales on the surface of certain types of unicellular algae. These algal scales are assembled from glycoproteins in the cis-Golgi into large dense complexes visible in the electron microscope. Like other secretory proteins, newly made scales move from the cis- to the trans-Golgi, but they were never observed in the transport vesicles that bud from Golgi cisternae. In further experiments, the observation that the movement of large cargo molecules through the Golgi can occur without entry of cargo into transport vesicles was shown also to be true of collagen synthesized in animal cells. In fibroblasts, large aggregates of the procollagen precursor often form in the lumen of the cis-Golgi. The procollagen aggregates are too large to be incorporated into small transport vesicles, and investigators could never find such aggregates in transport vesicles. These observations show that the forward movement of these, and perhaps all, secretory proteins from one Golgi compartment to another does not occur via small vesicles.
A particularly elegant demonstration of cisternal maturation in yeast takes advantage of different-colored fluorescent labels to image two different Golgi proteins simultaneously. Figure 14-17 shows how a cis-Golgi resident protein labeled with a green fluorescent protein and a trans-Golgi resident protein labeled with a red fluorescent protein behave in the same yeast cell. At any given moment, individual Golgi cisternae appear to have a distinct compartmental identity, in the sense that they contain either the cis-Golgi protein or the trans-Golgi protein, but only rarely contain both proteins. However, over time, an individual cisterna labeled with the cisGolgi protein can be seen to progressively lose this protein and acquire the trans-Golgi protein. This behavior is exactly that predicted by the cisternal maturation model, in which the composition of an individual cisterna changes as Golgi resident proteins move from later to earlier Golgi compartments. EXPERIMENTAL FIGURE 14-17 Fluorescence-tagged fusion proteins demonstrate Golgi cisternal maturation in a live yeast cell. Yeast cells expressing the early Golgi protein Vrg4 fused to GFP (green fluorescence) and the late Golgi protein Sec7 fused to DsRed (red fluorescence) were imaged by time-lapse microscopy. The top series of images, taken approximately 1 minute apart, shows a collection of Golgi cisternae, which at any one time are labeled with either Vrg4 or Sec7. The bottom series of images are digitally processed to show just the Golgi cisterna, identified by the white arrows in the top set of
images. At first, only Vrg4-GFP is located in the isolated cisterna, and later only Sec7DsRed is located in the isolated cisterna, following a brief period in which both proteins are co-localized in this compartment. This experiment is a direct demonstration of the cisternal maturation hypothesis, showing that the composition of individual cisternae follows a process of maturation characterized by loss of early Golgi proteins and gain of late Golgi proteins. [Republished with permission from Nature, from E. Losev et al., 2006, “Golgi Maturation Visualized in Living Yeast,” Nature 441:1002–1006; permission conveyed through Copyright Clearance Center, Inc.] Description Two sets, each set contains seven time-lapse fluorescence micrographs of yeast cells that show the fluorescence of early and late fluorescently labeled Golgi proteins. The first row shows a mix of red and green fluorescence. The second row shows one Golgi cisterna. The cisterna glows green, but with time, the fluorescence changes to red. The scale bar reads 1 micrometer. Although most protein traffic moves through the Golgi complex by a cisternal maturation mechanism, there is evidence that at least some cargo proteins can be detected in COPI transport vesicles that bud from Golgi membranes. This observation suggests that there may be a subset of COPI transport vesicles that move cargo in an anterograde direction. KEY CONCEPTS OF SECTION 14.3 Early Stages of the Secretory Pathway COPII vesicles transport proteins from the rough ER to the cis-Golgi; COPI vesicles transport proteins in the reverse direction (see Figure 14-12). COPII coats comprise three major components: the small GTP-binding protein Sar1, a Sec23/Sec24 complex, and a Sec13/Sec31 complex.
Components of the COPII coat bind to membrane cargo proteins containing a diacidic or other sorting signal in their cytosolic regions (see Figure 14-13). Soluble cargo proteins are probably targeted to COPII vesicles by binding to a membrane protein receptor. Many soluble ER-resident proteins contain a KDEL sorting signal. Binding of this retrieval sequence to a specific receptor protein in the cis-Golgi membrane recruits missorted ER proteins into retrograde COPI vesicles (see Figure 14-14). Membrane proteins needed to form COPII vesicles can be retrieved from the cis-Golgi by COPI vesicles. One of the sorting signals that directs membrane proteins into COPI vesicles is a KKXX sequence, which binds to subunits of the COPI coat. A distinct diarginine sorting signal operates by a similar mechanism. COPI vesicles also carry Golgi-resident proteins from later to earlier compartments in the Golgi complex. Soluble and membrane proteins advance through the Golgi complex by cisternal maturation, a process of anterograde transport that depends on resident Golgi enzymes moving by COPI vesicular transport in a retrograde direction.
14.4 Later Stages of the Secretory Pathway
14.4 Later Stages of the Secretory Pathway As cargo proteins move from the cis- to the trans-Golgi by cisternal maturation, modifications to their oligosaccharide chains are carried out by Golgi-resident enzymes. The retrograde trafficking of COPI vesicles from later to earlier Golgi compartments maintains sufficient levels of these carbohydrate-modifying enzymes in the appropriate compartments. Eventually, properly processed cargo proteins reach the trans-Golgi network, the most distal Golgi compartment. Here they are sorted into a number of different kinds of vesicles for delivery to their final destination. Each of the target destinations, such as the plasma membrane, endosomes, and lysosomes, has a unique composition of lipids and membrane proteins, and it is primarily the sorting in the trans-Golgi network that gives each of these organelles its unique identity. In this section, we discuss the different kinds of vesicles that bud from the trans-Golgi network, the mechanisms that segregate cargo proteins among them, and key processing events that occur late in the secretory pathway. The various types of vesicles that bud from the trans-Golgi and will be described in this section are summarized in Figure 14-18.
FIGURE 14-18 Vesicle-mediated protein trafficking from the trans-Golgi network. COPI (purple) vesicles mediate retrograde transport within the Golgi ( 1 ). Proteins that function in the lumen or in the membrane of the lysosome are first transported from the trans-Golgi network via AP complex and clathrin-coated (red) vesicles ( 2 ); after uncoating, these vesicles fuse with late endosomes, which deliver their contents to the lysosome. Some vesicles from the trans-Golgi carrying cargo destined for the lysosome fuse with the lysosome directly ( 3 ), bypassing the late endosome. These vesicles are coated with AP3 complex (blue); it is unknown whether these vesicles also contain clathrin. The coat proteins surrounding constitutive ( 4 ) and regulated ( 5 ) secretory vesicles have not yet been characterized; these vesicles carry secreted proteins and plasma-membrane proteins from the trans-Golgi network to the cell surface.
Vesicles Coated with Clathrin and Adapter Proteins Mediate Transport from the trans-Golgi
Description The illustration shows a trans-Golgi network out of which C O P 1 coated vesicles, A P 3 complex coated vesicles, clathrin or A P complex coated vesicles, uncoated vesicles with secreted proteins and plasma membrane proteins budding out. 1. The C O P 1 coated vesicles budding from the trans-Golgi network returns to the trans-Golgi. 2. The A P 3-complex coated vesicle moves towards the lysosome to fuse with it. 3. The clathrin-coated vesicles lose their coat to fuse with the late endosome. 4. Secreted proteins are held by receptor proteins inside the uncoated vesicles. The secreted protein is represented by small blue dotted lines. They move towards the cell surface to fuse with the plasma membrane. 5. Some secreted proteins inside uncoated vesicles are represented by a large tangle of blue lines. These vesicles move towards the cell surface to eliminate the secreted proteins. Vesicles Coated with Clathrin and Adapter Proteins Mediate Transport from the trans-Golgi The best characterized vesicles that bud from the trans-Golgi network have a two-layered coat: an outer layer composed of the fibrous protein clathrin and an inner layer composed of adapter protein (AP) complexes. Purified clathrin molecules, which have a three-limbed shape, are called triskelions, from the Greek for “three-legged” (Figure 14-19a). Each limb contains one clathrin heavy chain (180 kDa) and one clathrin light chain . Triskelions polymerize to form a polygonal lattice with an
intrinsic curvature (Figure 14-19b). When clathrin polymerizes on a donor membrane, it does so in association with AP complexes, which fill the space between the clathrin lattice and the membrane. Each AP complex (340 kDa) contains one copy each of four different adapter subunit proteins. A specific association between the globular domain at the end of each clathrin heavy chain in a triskelion and one subunit of the AP complex both promotes the co-assembly of clathrin triskelions with AP complexes and adds to the stability of the completed vesicle coat.
FIGURE 14-19 Structure of clathrin coats. (a) A clathrin molecule, called a triskelion, is composed of three heavy and three light chains. It has an intrinsic curvature due to the bend in the heavy chains. (b) Clathrin coats were formed in vitro by mixing purified clathrin heavy and light chains with AP2 complexes in the absence of membranes. Cryoelectron micrographs of more than 1000 assembled hexagonal clathrin barrel particles were analyzed by digital image processing to generate an average structural representation. The processed image shows only the clathrin heavy chains in a structure composed of 36
triskelions. Three representative triskelions are highlighted in red, yellow, and green. Some of the AP2 complexes packed into the interior of the clathrin cage are also visible in this representation. See B. Pishvaee and G. Payne, 1998, Cell 95:443. [Part (b) Republished with permission from Nature, from A. Fotin et al., 2004, “Molecular Model for a Complete Clathrin Lattice from Electron Cryomicroscopy,” Nature 432:573– 579; permission conveyed through Copyright Clearance Center, Inc.] Description The illustration labeled (a) shows the Triskelion structure of clathrin with three heavy chains and three light chains, arranged in a three-armed, right-handed propeller arrangement. At the tips of the 'propeller' are binding sites for assembly particles. The cryoelectron micrograph labeled (b) shows the sections of clathrin coat. The clathrin coat is composed of hexagonal sections and triskelion structures highlighted in red, green, and yellow. Adapter proteins determine which cargo proteins are specifically included in a budding transport vesicle by binding to the cytosolic face of membrane proteins. Thus each of the multiple adapter protein complexes that operate in the late secretory pathway can be classified based on the types of cargo proteins that they select as well as the specific transport step that they mediate (see Table 14-1). Three different AP complexes are known (AP1, AP2, and AP3), each with four subunits of different, though related, proteins. A second general type of adapter protein, known as GGA, consists of a single 70 kDa polypeptide. This monomeric adapter protein has been shown to contain both clathrin- and cargo-binding elements similar to those found in the much larger hetero-tetrameric AP complexes. All vesicles whose coats contain one of these complexes use ARF to initiate coat assembly on the donor membrane. As discussed previously,
ARF also initiates assembly of COPI coats. The additional membrane features or protein factors that determine which type of coat will assemble after ARF attachment are not well understood at this time. Vesicles that bud from the trans-Golgi network en route to the lysosome by way of the late endosome (see Figure 14-18, 3 ) have clathrin coats associated with either AP1 or GGA. One of the AP1 subunits in the vesicle coat binds to membrane proteins containing a Tyr-X-X-Φ sequence. Proteins bearing this YXXΦ sorting signal are recruited into clathrin/AP1coated vesicles budding from the trans-Golgi network. As we discuss in the next section, vesicles with clathrin/AP2 coats, which bud from the plasma membrane during endocytosis, can also recognize the YXXΦ sorting signal. Vesicles coated with GGA proteins and clathrin bind cargo molecules with different kinds of sorting sequences including Asp-X-Leu- Leu and Asp-Phe-Gly-X-Φ sequences. Some vesicles that bud from the trans-Golgi network have coats composed of the AP3 complex. Although the AP3 complex does contain a binding site for clathrin similar to those in the AP1 and AP2 complexes, it is not clear whether clathrin is necessary for the functioning of AP3-containing vesicles because mutant versions of AP3 that lack the clathrin binding site appear to be fully functional. AP3-coated vesicles mediate trafficking to the lysosome, but they appear to bypass the late endosome and fuse directly with the lysosomal membrane (see Figure 14-18, 2 ). In certain types of cells, such AP3 vesicles mediate protein transport to specialized storage compartments related to the lysosome. For example, AP3 is required for delivery of proteins to melanosomes, which contain the black
Dynamin Is Required for Pinching Off of Clathrin-Coated Vesicles
pigment melanin in skin cells, and to platelet storage vesicles in megakaryocytes, large cells that fragment into dozens of platelets. Mice with mutations in either of two different subunits of AP3 not only have abnormal skin pigmentation but also exhibit bleeding disorders. The latter occur because platelets require normal storage vesicles in order to repair tears in blood vessels. Dynamin Is Required for Pinching Off of Clathrin-Coated Vesicles A fundamental step in the formation of a transport vesicle that we have not yet considered is how a vesicle bud is pinched off from the donor membrane. In the case of clathrin-coated vesicles, a cytosolic protein called dynamin is essential for the release of complete vesicles. At the later stages of bud formation, dynamin polymerizes around the neck portion of the bud and then hydrolyzes GTP. The energy derived from GTP hydrolysis is thought to drive a conformational change in dynamin that stretches the neck until the vesicle pinches off (Figure 14-20).
FIGURE 14-20 Model for dynamin-mediated pinching off of clathrin-coated vesicles. After a vesicle bud forms, dynamin polymerizes over the neck. By a mechanism that is not well understood, dynamin-catalyzed hydrolysis of GTP leads to release of the vesicle from the donor membrane. Note that membrane proteins in the donor membrane are incorporated into vesicles by interacting with AP complexes in the coat. See K. Takel et al., 1995, Nature 374:186.
Description A vesicle starts to buds into the cytosol. The vesicle contains an integral cargo protein and an integral receptor protein bound to a soluble cargo protein. The vesicle is coated with A P complex proteins and a fibrous clathrin coat. Dynamin forms a ring around the budding vesicle just below the membrane (cytosolic face). Hydrolysis of G T P to G D P and a molecule of inorganic phosphate results in the pinching and budding of the clathrin-coated vesicle. Incubation of cell extracts with a nonhydrolyzable derivative of GTP provides dramatic evidence for the importance of dynamin in the pinching off of clathrin/AP2-coated vesicles during endocytosis. Such treatment leads to accumulation of clathrin-coated vesicle buds with excessively long necks that are surrounded by polymeric dynamin but do not pinch off (Figure 14-21). Likewise, cells expressing mutant forms of dynamin that cannot bind GTP do not form clathrin-coated vesicles and instead accumulate similar long-necked vesicle buds encased with polymerized dynamin.
EXPERIMENTAL FIGURE 14-21 GTP hydrolysis by dynamin is required for the pinching off of clathrin-coated vesicles in cell-free extracts. A preparation of nerve terminals, which undergo extensive endocytosis, was lysed by treatment with distilled water and incubated with GTP-γ-S, a nonhydrolyzable derivative of GTP. After sectioning, the preparation was treated with gold-tagged anti-dynamin antibody and viewed in the electron microscope. This image, which shows a long-necked clathrin/AP-coated bud with polymerized dynamin lining the neck, reveals that buds can form in the absence of GTP hydrolysis, but vesicles cannot pinch off. The extensive polymerization of dynamin that occurs in the presence of GTP-γ-S probably does not occur during the normal budding process. [Republished by permission from Nature, from K. Takei et al., 1995, “Tubular Membrane Invaginations Coated by Dynamin Rings Are Induced by GTP-Gamma S in Nerve Terminals,” Nature 374:(6518):186–190; permission conveyed through Copyright Clearance Center, Inc.]
Mannose 6-Phosphate Residues Target Resident Enzymes to Lysosomes
Like COPI and COPII vesicles, clathrin-coated vesicles normally lose their coat soon after their formation. Cytosolic Hsp70, a constitutive chaperone protein found in all eukaryotic cells, is thought to use energy derived from the hydrolysis of ATP to drive depolymerization of the clathrin coat into triskelions. In the case of endocytic vesicles, uncoating not only releases triskelions for reuse in the formation of additional vesicles but also exposes v-SNAREs for use in fusion with target membranes. Vesicle uncoating by cytosolic Hsp70 appears to be activated by a co-chaperone, auxillin, that contains a domain that stimulates the ATP hydrolysis by Hsp70. Conformational changes that occur when ARF switches from the GTP-bound to the GDP-bound state are thought to regulate the timing of clathrin coat depolymerization, but how the action of Hsp70 and auxillin is coupled to ARF switching is not well understood. Mannose 6-Phosphate Residues Target Resident Enzymes to Lysosomes As we have seen, many of the sorting signals that direct cargo-protein trafficking in the secretory pathway are short amino acid sequences in the targeted protein. In contrast, the sorting signal that directs soluble lysosomal enzymes from the trans-Golgi network to the late endosome is a carbohydrate residue, mannose 6-phosphate (M6P), which is formed in the cis-Golgi. The addition and initial processing of one or more preformed N-linked oligosaccharide precursors in the rough ER is the same for lysosomal enzymes as for membrane and secreted proteins,
yielding core chains (see Figure 13-18). In the cis-Golgi, the N-linked oligosaccharides present on most lysosomal enzymes undergo a two-step reaction sequence that generates M6P residues (Figure 14-22). The addition of M6P residues to the oligosaccharide chains of soluble lysosomal enzymes prevents these proteins from undergoing the further processing reactions characteristic of secreted and membrane proteins (see Figure 14-15).
FIGURE 14-22 Formation of mannose 6-phosphate (M6P) residues that target soluble enzymes to lysosomes. The M6P residues that direct proteins to lysosomes are generated in the cis-Golgi by two Golgi-resident enzymes. Step 1 : An N-acetylglucosamine (GlcNAc) phosphotransferase transfers a phosphorylated GlcNAc group to carbon atom 6 of one or more mannose residues. Because only lysosomal enzymes contain sequences (red) that are recognized and bound by this enzyme, phosphorylated GlcNAc groups are added specifically to lysosomal enzymes. Step 2 : After release of the modified protein from the phosphotransferase, a phosphodiesterase removes the GlcNAc group, leaving a phosphorylated mannose residue on the lysosomal enzyme. See A. B. Cantor et al., 1992, J. Biol. Chem. 267:23349; and S. Kornfeld, 1987, FASEB J. 1:462. Description Step 1: U D P-N-acetyl glucosamine and a lysosomal enzyme with attached oligosaccharide bind to the N-acetyl glucosamine phosphotransferase recognition sites. U M P is eliminated in this process. The N-acetyl glucosamine phosphate unit in the catalytic site and the mannose unit of the lysosomal enzyme in the recognition site of N-acetyl glucosamine phosphotransferase bind together.
Step 2. The targeting protein is released and phosphodiesterase catalyzes the removal of N-acetyl glucosamine on the lysosomal enzyme. As shown in Figure 14-23, the segregation of M6P-bearing lysosomal enzymes from secreted and membrane proteins occurs in the trans-Golgi network. Here transmembrane mannose 6-phosphate receptors bind the M6P residues on lysosome-destined proteins very tightly and specifically. Clathrin/AP1-coated vesicles containing the M6P receptor and bound lysosomal enzymes then bud from the trans-Golgi network, lose their coats, and subsequently fuse with a late endosome by mechanisms described previously (Figure 14-18). Because M6P receptors can bind M6P at the slightly acidic of the trans-Golgi network, but not at a pH of less than 6, the bound lysosomal enzymes are released within late endosomes, which have an internal pH of 5.0–5.5. Furthermore, a phosphatase within late endosomes usually removes the phosphate from M6P residues on lysosomal enzymes, preventing any rebinding to the M6P receptor that might occur in spite of the low pH there. Vesicles budding from late endosomes, coated with a protein complex known as the retromer, recycle the M6P receptor back to the trans-Golgi network. Eventually, mature late endosomes fuse with lysosomes, delivering the lysosomal enzymes to their final destination.
FIGURE 14-23 Trafficking of soluble lysosomal enzymes from the trans-Golgi network and cell surface to lysosomes. Newly synthesized lysosomal enzymes, produced in the ER, acquire mannose 6-phosphate (M6P) residues in the cis-Golgi (see Figure 14-22). For simplicity, only one phosphorylated oligosaccharide chain is depicted, although lysosomal enzymes typically have many such chains. In the trans-Golgi network, proteins that bear the M6P sorting signal interact with M6P receptors in the membrane and thereby are directed into clathrin/AP1-coated vesicles (step 1 ). The coat surrounding released vesicles is rapidly depolymerized (step 2 ), and the uncoated transport vesicles fuse with late endosomes (step 3 ). Note that disassembled coat proteins are recycled for additional rounds of vesicle assembly (step 2a ). After the phosphorylated enzymes dissociate from the M6P receptors in late endosomes, free M6P receptors are recycled by incorporation into vesicles that bud from the late endosome (step 4 ). Vesicles carrying free M6P receptors can return to the trans-Golgi or fuse with the plasma membrane delivering M6P receptors to
the cell surface (step 5 ). Phosphorylated lysosomal enzymes are also occasionally sorted from the trans-Golgi to the cell surface and secreted. These secreted enzymes can be retrieved by receptor-mediated endocytosis (steps 6 – 8 ). Ultimately, fusion of the late endosome with the lysosome delivers phosphorylated lysosomal enzymes to the interior of the lysosomes (step 9 ). See G. Griffiths et al., 1988, Cell 52:329; S. Kornfeld, 1992, Annu. Rev. Biochem. 61:307; and G. Griffiths and J. Gruenberg, 1991, Trends Cell Biol. 1:5. Description Step 1: The illustration shows a trans-Golgi network in cytosol with a clathrin-coated bud containing mannose 6 phosphate (M 6 P) receptor with associated targeted proteins. The trans-Golgi network also has another M 6 P receptor and an uncoated bud. The uncoated bud undergoes constitutive secretion. The clathrin-coated bud becomes a vesicle. Step 2: The clathrin coating of the vesicle gets removed to become the uncoated transport vesicle. Step 2 a: The disassembled clathrin triskelion and A P complex coats are reused by the trans-Golgi network. Step 3: The vesicle merges with the late endosome at a low p H, depositing the glycoprotein. Step 4 and 5: The late endosome releases the M 6 P receptor inside an empty vesicle, which either gets recycled back to the trans-Golgi complex or to the plasma membrane. Step 6: From the surface of the cell, labeled proteins bind with the mannose 6 phosphate receptor on the surface of the vesicle to form clathrin-coated pits in the cell membrane. Via receptor-mediated endocytosis, a clathrin-coated vesicle is formed. Step 7: The clathrin coating of the vesicle gets removed to become the uncoated endocytic vesicle. Step 8: The uncoated endocytic vesicle moves to bind with the late endosome. Step 9: The late endosome delivers proteins and phosphate to a lysosome.
Study of Lysosomal Storage Diseases Revealed Key Components of the Lysosomal Sorting Pathway
The sorting of soluble lysosomal enzymes in the trans-Golgi network (Figure 14-23, steps 1 – 4 ) shares many features with the trafficking of proteins between the ER and cis-Golgi compartments mediated by COPII and COPI vesicles. First, M6P acts as a sorting signal by interacting with the luminal domain of a receptor protein in the donor membrane. Second, the membrane-embedded receptors with their bound ligands are incorporated into the appropriate vesicles — in this case, either GGA- or AP1-containing clathrin-coated vesicles — by interacting with the vesicle coat. Third, these transport vesicles fuse with only one specific organelle, here the late endosome, as the result of interactions between specific vSNAREs and t-SNAREs. And finally, intracellular transport receptors dissociated from their bound ligand are recycled by retrograde vesicle trafficking. Study of Lysosomal Storage Diseases Revealed Key Components of the Lysosomal Sorting Pathway A group of genetic disorders termed lysosomal storage diseases are caused by the absence of one or more lysosomal enzymes. As a result, undigested glycolipids and extracellular components that would normally be degraded by lysosomal enzymes accumulate in lysosomes as large inclusions. Patients with lysosomal storage diseases can have a variety of developmental, physiological, and neurological abnormalities depending on the type and severity of the storage defect. I-cell disease is a
particularly severe type of lysosomal storage disease in which multiple enzymes are missing from the lysosomes. Cells from affected individuals lack the N-acetylglucosamine phosphotransferase that is required for formation of M6P residues on lysosomal enzymes in the cis-Golgi (see
Figure 14-22). Biochemical comparison of lysosomal enzymes from normal individuals with those from patients with I-cell disease led to the initial discovery of M6P as the lysosomal sorting signal. Lacking this signal, the lysosomal enzymes of affected individuals are secreted rather than being sorted to and sequestered in lysosomes. When fibroblasts from patients with I-cell disease are grown in a medium containing lysosomal enzymes bearing M6P residues, the diseased cells acquire a nearly normal intracellular content of lysosomal enzymes. This finding indicates that the plasma membrane of these cells contains M6P receptors, which can internalize extracellular phosphorylated lysosomal enzymes by receptor-mediated endocytosis. This process, used by many cell-surface receptors to bring bound proteins or particles into the cell, is discussed in detail in the next section. It is now known that even in normal cells, some M6P receptors are transported to the plasma membrane and some phosphorylated lysosomal enzymes are secreted (see Figure 14-23). The secreted enzymes can be retrieved by receptor-mediated endocytosis and directed to lysosomes. This pathway thus scavenges any lysosomal enzymes that escape the usual M6P sorting pathway.
Protein Aggregation in the trans-Golgi May Function in Sorting Proteins to Regulated Secretory Vesicles
Hepatocytes from patients with I-cell disease contain a normal complement of lysosomal enzymes and no inclusions, even in the absence of an exogeneous source of lysosomal enzymes bearing M6P residues. This finding implies that hepatocytes (the most abundant type of liver cell) employ a pathway for sorting lysosomal enzymes that does not rely on a targeting signal based on M6P. This pathway probably relies on some yet unidentified protein-based sorting signal present in lysosomal enzymes. Protein Aggregation in the trans-Golgi May Function in Sorting Proteins to Regulated Secretory Vesicles As noted in the chapter introduction, all eukaryotic cells continuously secrete certain proteins (constitutive secretion). Specialized secretory cells store other proteins in vesicles and secrete them only when triggered by a specific stimulus. One example of such regulated secretion occurs in pancreatic β cells, which store newly made insulin in specialized secretory vesicles and secrete it in response to an elevation in blood glucose (see
Figure 21-1b). These and other secretory cells simultaneously use two different types of secretory vesicles to move proteins from the trans-Golgi network to the cell surface: unregulated transport vesicles (also called constitutive secretory vesicles) and regulated transport vesicles. A common mechanism appears to sort regulated proteins as diverse as ACTH (adrenocorticotropic hormone), insulin, and trypsinogen into
Some Proteins Undergo Proteolytic Processing After Leaving the trans-Golgi
regulated secretory vesicles. Evidence for such a common mechanism comes from experiments in which recombinant DNA techniques were used to induce the synthesis of insulin and trypsinogen in pituitary tumor cells already synthesizing ACTH. In these cells, which do not normally express insulin or trypsinogen, all three proteins segregate into the same regulated secretory vesicles and are secreted together when a hormone binds to a receptor on the pituitary cells and causes a rise in cytosolic . Morphologic evidence suggests that the ability to form protein aggregates is a common feature of all three proteins that governs their packaging into regulated secretory vesicles. For instance, immature vesicles in this pathway — those that have just budded from the trans-Golgi network — contain diffuse aggregates of secretory protein that are visible in the electron microscope. These aggregates are also found in vesicles that are in the process of budding, indicating that proteins destined for regulated secretory vesicles selectively aggregate together before their incorporation into the vesicles. Some Proteins Undergo Proteolytic Processing After Leaving the transGolgi For some secretory proteins (e.g., growth hormone) and certain viral membrane proteins (e.g., VSV G protein), removal of the N-terminal ER signal sequence from the nascent chain is the only known proteolytic cleavage required to convert the polypeptide to the mature, active protein
(see Figure 13-10). However, some membrane proteins and many soluble secretory proteins are initially synthesized as relatively long-lived, inactive precursors, termed proproteins, that require further proteolytic processing to generate the mature, active proteins. Examples of proteins that undergo such processing are soluble lysosomal enzymes; many membrane proteins, such as influenza hemagglutinin (HA); and secreted proteins such as serum albumin, insulin, glucagon, and the yeast α mating factor. In general, the proteolytic conversion of a proprotein to the corresponding mature protein occurs after the proprotein has been sorted in the trans-Golgi network to appropriate vesicles. In the case of soluble lysosomal enzymes, the proproteins, called proenzymes, are sorted by the M6P receptor as catalytically inactive enzymes. Once in the late endosome or lysosome, a proenzyme undergoes a proteolytic cleavage that generates a smaller but enzymatically active polypeptide. Delaying the activation of lysosomal proenzymes until they reach the lysosome prevents them from digesting macromolecules in earlier compartments of the secretory pathway. The proproteins of most constitutively secreted proteins (e.g., albumin) are cleaved only once at a site C-terminal to a dibasic recognition sequence such as Arg-Arg or Lys-Arg (Figure 14-24a). Proteolytic processing of proteins whose secretion is regulated generally entails additional cleavages. In the case of proinsulin, multiple cleavages of the single polypeptide chain yield the N-terminal B chain and the C-terminal A chain of mature insulin, which are linked by disulfide bonds, and the
central C peptide, which is lost and subsequently degraded (Figure 1424b).
FIGURE 14-24 Proteolytic processing of proproteins in the constitutive and regulated secretory pathways. The processing of proalbumin and proinsulin is typical of the constitutive and regulated pathways, respectively. The endoproteases that function in such processing cleave at the C-terminal end of a sequence of two consecutive amino acids. (a) The endoprotease furin acts on the precursors of constitutive secreted proteins. (b) Two endoproteases, PC2 and PC3, act on the precursors of regulated secreted proteins. The final processing of many such proteins is catalyzed by a carboxypeptidase that sequentially removes two basic amino acid residues at the C-terminus of a polypeptide. See D. Steiner et al., 1992, J. Biol. Chem. 267:23435. Description The illustration labeled (a) is titled constitutive secreted proteins. Proalbumin consists of the albumin protein which has a C-terminal (C O O superscript minus) end and a double arginine sequence at the N-terminal (N H subscript 3 superscript plus) end. A furin endoprotease cleaves the terminal arginines. The illustration labeled (b) is titled regulated secreted proteins. Proinsulin contains a B chain at the N-terminal, an A chain at the c-terminal, and a C chain in the center. An arginine-arginine sequence is located after the B chain and a lysine arginine sequence is located before the A chain. The B and A chains are linked by disulfide bonds, and there is a disulfide bond present in the A chain. P C 3 and P C 2 endoproteases act to cut out the C chain at the end of the arginine- arginine sequence. Carboxypeptidase removes the arginine-arginine sequence leaving mature insulin. The processing cleavages are carried out by the members of a family of endoproteases located in the late secretory pathway, all of which cleave a protein chain on the C-terminal side of an Arg-Arg or Lys-Arg sequence. One, called furin, is found in all mammalian cells; it processes proteins such as albumin that are secreted constitutively. In contrast, the PC2 and PC3 endoproteases are found only in cells that exhibit regulated secretion;
Distinct Pathways Sort Membrane Proteins to the Apical or Basolateral Region of Polarized Cells
these enzymes are localized to regulated secretory vesicles and proteolytically cleave the precursors of many hormones at specific sites. Distinct Pathways Sort Membrane Proteins to the Apical or Basolateral Region of Polarized Cells The plasma membrane of a polarized epithelial cell is divided into two domains: apical and basolateral. Tight junctions located between the two domains prevent the movement of plasma-membrane proteins between them (see Figure 20-19). Several sorting mechanisms direct newly synthesized membrane proteins to either the apical or the basolateral domain of epithelial cells, and any one protein may be sorted by more than one mechanism. As a result of this sorting and the restriction on protein movement within the plasma membrane by tight junctions, distinct sets of proteins are found in the apical and basolateral domains. This preferential localization of certain transport proteins is critical to a variety of important physiological functions, such as absorption of nutrients from the intestinal lumen and acidification of the stomach lumen (see Figures 1130 and 11-31). In one mechanism, the sorting takes place in the trans-Golgi network. Microscopic and cell-fractionation studies indicate that proteins destined for either the apical or the basolateral membrane are initially transported together to the membranes of the trans-Golgi network. In some cases, proteins destined for the apical membrane are sorted into their own
transport vesicles that bud from the trans-Golgi network and then move to the apical region, whereas proteins destined for the basolateral membrane are sorted into other vesicles that move to the basolateral region. The different vesicle types can be distinguished by their protein constituents, including distinct Rab and v-SNARE proteins, which apparently target them to the appropriate plasma-membrane domain. In this mechanism, segregation of proteins destined for the two domains occurs as cargo proteins are incorporated into particular types of vesicles budding from the trans-Golgi network. Such direct basolateral-apical sorting has been investigated in cultured Madin-Darby canine kidney (MDCK) cells, a line of cultured polarized epithelial cells (see Figure 4-4). In MDCK cells infected with the influenza virus, progeny viruses bud only from the apical membrane, whereas in cells infected with vesicular stomatitis virus, progeny viruses bud only from the basolateral membrane. This difference occurs because the HA glycoprotein of influenza virus is transported from the Golgi complex exclusively to the apical membrane and the VSV G protein is transported only to the basolateral membrane (Figure 14-25).
FIGURE 14-25 Sorting of proteins destined for the apical and basolateral plasma membranes of polarized cells. Cells that form an epithelial cell layer contain distinct apical and basolateral domains of the plasma membrane separated by tight junctions. Distinct sorting pathways in the late secretory pathway govern sorting to either the basolateral or apical membrane. 1 : In the trans-Golgi, one type of vesicle transports proteins such as VSV G protein and endogenous proteins that carry a basolateral targeting sequence directly to the basolateral membrane. 2 : A second type of vesicle budding from the trans-Golgi carries influenza HA protein and GPI-linked proteins directly to the apical membrane. 3 : Through a process known as transcytosis, proteins can move from the basolateral to the apical membrane. Transcytosis involves endocytosis in clathrin/AP coated vesicles from the basolateral membrane, recycling from an endocytic compartment of basolateral proteins back to the basolateral membrane, and delivery of transcytosed apical proteins to the apical membrane. See K. Simons and A. Wandinger-Ness, 1990, Cell 62:207; and K. Mostov et al., 1992, J. Cell Biol. 116:577. Description The illustration has a legend at the bottom with figures that represent 5 different proteins namely G P I-anchored protein, Influenza H A glycoprotein, transcytosed protein, V S V G protein, and basolateral protein. Each protein is represented by differently shaped structures. 1. Basolateral sorting: A vesicle with V S V G and basolateral proteins buds out of the trans-Golgi to move towards the basolateral membrane to fuse with the same. 2. Direct apical sorting: From the same trans-Golgi another vesicle with G P I-anchored protein and Influenza H A glycoprotein moves towards the apical membrane at the top. The vesicle fuses with the apical membrane. 3. Endocytosis: A coated vesicle covered in clathrin-coated pits with transcytosed protein and V S V G protein moves up from the basolateral membrane through endocytosis, where V S V G protein gets eliminated for recycling. The uncoated vesicle undergoes transcytosis into the apical membrane. A section of the membrane at the right is labeled tight junction.
Mutational studies on proteins, such as the VSV G protein, that are specifically targeted to the basolateral domain have defined Tyr-X-X-Φ and Asp-X-Leu-Leu sorting signals. As we have seen, proteins bearing these motifs are recruited into clathrin/AP-coated vesicles, strongly implicating clathrin-coated vesicles in the sorting of proteins to the basolateral membrane. Among the cellular proteins that undergo similar apical-basolateral sorting in the Golgi are those with a glycosylphosphatidylinositol (GPI) membrane anchor. In MDCK cells and most other types of epithelial cells, GPIanchored proteins are targeted to the apical membrane. In membranes, GPI-anchored proteins are clustered into lipid rafts, which are rich in sphingolipids (see Chapter 10). This finding suggests that lipid rafts are localized to the apical membrane along with proteins that preferentially partition them in many cells. However, the GPI anchor is not an apical sorting signal in all polarized cells; in thyroid cells, for example, GPIanchored proteins are targeted to the basolateral membrane. Other than GPI anchors, no unique sequences have been identified that are both necessary and sufficient to target proteins to either the apical or basolateral domain. Instead, each membrane protein may contain multiple sorting signals, any one of which can target it to the appropriate plasmamembrane domain. The identities of these complex signals and of the vesicle coat proteins that recognize them are currently being pursued for a number of different proteins that are sorted to specific plasma-membrane domains of polarized epithelial cells.
Another mechanism for sorting apical and basolateral proteins, also illustrated in Figure 14-25, operates in hepatocytes. The basolateral membranes of hepatocytes face the blood (like those of intestinal epithelial cells), and the apical membranes line the small intercellular channels into which bile is secreted. In hepatocytes, newly made apical and basolateral proteins are first transported in vesicles from the transGolgi network to the basolateral region and incorporated into the plasma membrane by exocytosis (i.e., fusion of the vesicle membrane with the plasma membrane). From there both basolateral and apical proteins are endocytosed in the same vesicles, but then their paths diverge. The endocytosed basolateral proteins are sorted into transport vesicles that recycle them to the basolateral membrane. In contrast, the apically destined endocytosed proteins are sorted into transport vesicles that move across the cell and fuse with the apical membrane, a process called transcytosis. This process is also used to move extracellular materials from one side of an epithelium to another. Even in epithelial cells, such as MDCK cells, in which apical-basolateral protein sorting occurs in the Golgi, transcytosis may provide an editing function by which an apical protein sorted incorrectly to the basolateral membrane is subjected to endocytosis and then correctly delivered to the apical membrane. KEY CONCEPTS OF SECTION 14.4 Later Stages of the Secretory Pathway The trans-Golgi network is a major branch point in the secretory pathway where soluble secreted proteins, lysosomal proteins, and in some cells, membrane proteins destined for the basolateral or apical plasma membrane are segregated into different transport vesicles.
Many vesicles that bud from the trans-Golgi network as well as endocytic vesicles bear a coat composed of AP (adapter protein) complexes and clathrin (see Figure 1419). The pinching off of clathrin-coated vesicles requires dynamin, which forms a collar around the neck of the vesicle bud and hydrolyzes GTP (see Figure 14-20). Soluble enzymes destined for lysosomes are modified in the cis-Golgi by the addition of multiple mannose 6-phosphate (M6P) residues to their oligosaccharide chains. M6P receptors in the membrane of the trans-Golgi network bind proteins bearing M6P residues and direct them to late endosomes, where receptors and their ligand proteins dissociate. The receptors are then recycled to the Golgi or plasma membrane, and the lysosomal enzymes are delivered to lysosomes (see Figure 14-23). Regulated secreted proteins are concentrated and stored in secretory vesicles to await a neuronal or hormonal signal for exocytosis. Protein aggregation within the transGolgi network may play a role in sorting secreted proteins to the regulated secretory pathway. Many proproteins transported through the secretory pathway undergo post-Golgi proteolytic cleavages that yield the mature, active proteins. This proteolytic maturation can occur in vesicles carrying proteins from the trans-Golgi network to the cell surface, in the late endosome, or in the lysosome. In polarized epithelial cells, membrane proteins destined for the apical and basolateral domains of the plasma membrane are sorted in the trans-Golgi network into different transport vesicles (see Figure 14-25). Tyr-X-X-Φ and Asp-X-Leu-Leu sorting signals target proteins to the basolateral membrane, whereas a GPI anchor targets proteins to the apical membrane. In hepatocytes and some other polarized cells, all plasma-membrane proteins are directed first to the basolateral membrane. Apically destined proteins are then endocytosed and moved across the cell to the apical membrane (transcytosis).
14.5 Receptor-Mediated Endocytosis
14.5 Receptor-Mediated Endocytosis In previous sections, we have explored the main pathways of vesicle trafficking responsible for delivering soluble and membrane secretory proteins synthesized on the rough ER to the cell surface. The same basic principle of coated vesicle budding and fusion are responsible for the ability of animal cells to internalize macromolecules from their surroundings and to deliver these macromolecules to particular intracellular destinations. In this process, known as receptor-mediated endocytosis, a specific receptor on the cell surface binds tightly to an extracellular macromolecular ligand that it recognizes; then the complex of receptor and ligand are recruited to a plasma-membrane region that buds inward to form a coated transport vesicle. Plant cells and fungal cells take up small molecules by endocytosis, but generally do not take up macromolecules, which cannot readily pass through the cell wall because of their size. Among the common macromolecules that vertebrate cells internalize by receptor-mediated endocytosis are cholesterol-containing low-density lipoprotein (LDL) particles, the iron-carrying protein transferrin, many protein hormones (e.g., insulin), and certain glycoproteins. Receptormediated endocytosis of such ligands generally occurs via clathrin/AP2coated pits and vesicles in a process similar to the packaging of lysosomal
enzymes by the binding of M6P in the trans-Golgi network (see Figure 1423). As noted earlier, some M6P receptors are found on the cell surface, and these receptors participate in the receptor-mediated endocytosis of lysosomal enzymes that are mistakenly secreted. In general, the transmembrane receptor proteins that function in the uptake of extracellular ligands are internalized from the cell surface during endocytosis and are then sorted and recycled back to the cell surface, much as M6P receptors are recycled to the plasma membrane and transGolgi. The rate at which a ligand is internalized is limited by the amount of its corresponding receptor on the cell surface. Clathrin/AP2-coated pits make up about 2 percent of the surface of cells such as hepatocytes and fibroblasts. Many internalized ligands have been observed in clathrin/AP2-coated pits and vesicles, which are thought to function as intermediates in the endocytosis of most (though not all) ligands bound to cell-surface receptors (Figure 14-26). Some receptors are clustered over clathrin-coated pits even in the absence of ligand. Other receptors diffuse freely in the plane of the plasma membrane but undergo a conformational change when they bind to ligand, so that when the receptor-ligand complex diffuses into a clathrin-coated pit, it is retained there. Two or more types of receptor-bound ligands, such as LDL and transferrin, can be seen in the same coated pit or vesicle.
EXPERIMENTAL FIGURE 14-26 The initial stages of receptor-mediated endocytosis of low-density lipoprotein (LDL) particles are revealed by electron microscopy. Cultured human fibroblasts were incubated in a medium containing LDL particles covalently linked to the electron-dense, iron-containing protein ferritin; each small iron particle in ferritin is visible as a small dot under the electron microscope. Cells were initially incubated at 4 °C; at this temperature LDL can bind to its receptor, but internalization does not occur. After excess LDL not bound to the cells was washed away, the cells were warmed to 37 °C and then prepared for microscopy at periodic intervals. (a) A coated pit, showing the clathrin coat on the inner (cytosolic) surface of the pit, soon after the temperature was raised. (b) A pit containing LDL, apparently closing on itself to form a coated vesicle. (c) A coated vesicle containing ferritin-tagged LDL particles. (d) Ferritin-tagged LDL particles in a
Cells Take Up Lipids from the Blood in the Form of Large, Well-Defined Lipoprotein Complexes
smooth-surfaced early endosome, 6 minutes after internalization began. See M. S. Brown and J. Goldstein, 1986, Science 232:34. [Republished with permission from Nature, from J. Goldstein et al., 1979, “Coated Pits, Coated Vesicles, and Receptor-Mediated Endocytosis,” Nature 279:679–685; permission conveyed through the Copyright Clearance Center, Inc.] Description The micrograph labeled (a) shows a clathrin-coated U shaped pit containing L D Lferritin (tiny granules). The vesicle has a diameter of approximately 0.2 micrometers. The micrograph labeled (b) shows a pit containing L D L ferritin that has almost closed off to form an oval vesicle. The micrograph labeled (c) shows a fully formed circular vesicle containing L D L ferritin. The micrograph labeled (d) shows an early endosome full of L D L ferritin. The oval vesicle looks larger and tiny granules are within it. Cells Take Up Lipids from the Blood in the Form of Large, Well-Defined Lipoprotein Complexes Lipids absorbed from the diet in the intestines or stored in adipose tissue can be distributed to cells throughout the body. To facilitate the mass transfer of lipids between cells, animals have evolved an efficient way to package hundreds or even thousands of lipid molecules into water-soluble, macromolecular carriers, called lipoproteins, which cells can take up from the circulation as an ensemble. A lipoprotein particle has a shell composed
of proteins (apolipoproteins) overlying a cholesterol-containing phospholipid monolayer. The shell is amphipathic because its outer surface is hydrophilic, making the particle water soluble, and its inner surface is hydrophobic. Beneath the hydrophobic inner surface of the shell is a core of neutral lipids containing mostly cholesteryl esters, triglycerides, or both. Mammalian lipoproteins fall into different classes, defined by their differing buoyant densities. The class we will consider here is low-density lipoprotein (LDL). A typical LDL particle, depicted in Figure 14-27, is a sphere 20–25 nm in diameter. The amphipathic outer shell is composed of a phospholipid monolayer and a single molecule of a large protein known as apoB-100; the core of the particle is packed with cholesterol in the form of cholesteryl esters.
FIGURE 14-27 Model of low-density lipoprotein (LDL). All classes of lipoproteins have the same general structure: an amphipathic shell composed of apolipoprotein, a phospholipid monolayer (not bilayer), and cholesterol, and a hydrophobic core composed mostly of cholesteryl esters or triglycerides, or both, but with minor amounts of other neutral lipids (e.g., some vitamins). This model of LDL is based on electron microscopy and other low-resolution biophysical methods. LDL is unique in that it contains only a single molecule of one type of apolipoprotein (ApoB), which appears to wrap around the outside of the particle as a band of protein. The other lipoproteins contain multiple apolipoprotein molecules, often of different types. See M. Krieger, 1995, in E. Haber, ed., Molecular Cardiovascular Medicine, Scientific American Medicine, pp. 31–47. Description The illustration of the L D L model shows a sphere made of tiny spheres having two tail-like projections, called the phospholipids. The phospholipids make up the polar surface. The phospholipids cover the apolar core made from cholesteryl ester. Many unesterified cholesterol molecules are embedded in between the phospholipids. An apolipoprotein B molecule is embedded in the phospholipids. We have seen that a general feature of coated vesicles is the ability to select specific cargo molecules; this process of cargo selection was first studied in the context of endocytosis of labeled LDL particles added to intact cells. Two general experimental approaches have been used to study how labeled LDL particles enter cells. The first method makes use of LDL that has been labeled by the covalent attachment of radioactive to the side chains of tyrosine residues in apoB-100 on the surfaces of the LDL particles. After cultured cells are incubated for several hours with the labeled LDL, it is possible to determine how much LDL is bound to the surfaces of cells, how much is internalized, and how much of the apoB100 component of the LDL is transported to the lysosome and there
degraded by enzymatic hydrolysis to individual amino acids. The degradation of apoB-100 can be detected by the release of into the culture medium. Figure 14-28 shows the time course of these events in receptor-mediated cellular LDL processing, determined by pulse-chase experiments with a fixed concentration of LDL. These experiments clearly demonstrate the order of events: surface binding of LDL → internalization → degradation. The second approach involves tagging LDL particles with an electron-dense label that can be detected by electron microscopy. Such studies can reveal the details of how LDL particles first bind to the surface of cells at sites of nascent endocytic vesicles known as clathrin-coated pits, then remain associated with the coated pits as they invaginate and bud off to form clathrin-coated vesicles, and are finally transported to endosomes.
EXPERIMENTAL FIGURE 14-28 Pulse-chase experiment demonstrates precursorproduct relations in cellular uptake of LDL. Cultured normal human skin fibroblasts were incubated in a medium containing for 2 hours at 4 °C (the pulse). After excess not bound to the cells was washed away, the cells were incubated at 37 °C for the indicated amounts of time in the absence of external LDL (the chase). The amounts of surface-bound, internalized, and degraded (hydrolyzed) were measured. Binding, but not internalization or hydrolysis, of LDL apoB-100 occurs during the 4 °C pulse. The data show the very rapid disappearance of bound from the surface as it is internalized after the cells have been warmed to allow membrane movements. After a lag period of 15–20 minutes, lysosomal degradation of the internalized commences. See M. S. Brown and J. L. Goldstein, 1976, Cell 9:663. Description In the graph, the vertical axis represents the amount of 125-iodine labeled L D L in nanograms per dish ranging from 0 to 12 in increments of 2. The vertical axis
Receptors for Macromolecular Ligands Contain Sorting Signals That Target Them for Endocytosis
represents time at 37 degrees Celsius in minutes ranging from 0 to 60 in increments of 10. After 60 minutes, a break indicates that the experiment is continued for 120 minutes. The binding curve starts at zero minutes, 11 nanograms, and rapidly slopes downward at 5 minutes, 6 nanograms, and plateaus at 30 minutes, 2 nanograms. The internalization curve starts at 0 minutes, 2 nanograms and sharply rises in first few minutes to 10 nanograms, mirroring the binding curve. From 5 to 25 minutes, the internalization curve is flat, then slopes down and ends at 120 minutes, 3 nanograms. The degradation curve starts at 0 minutes, zero nanograms, and does not rise until 11 minutes into the experiment. After 11 minutes, the rise is linear for 70 minutes. Towards the end of the experiment, the gradient of the degradation curve increases. Receptors for Macromolecular Ligands Contain Sorting Signals That Target Them for Endocytosis The key to understanding how LDL particles bind to the cell surface and are then taken up into endocytic vesicles was the discovery of the LDL receptor. The LDL receptor is an 839-residue type I membrane glycoprotein; it has a long N-terminal exoplasmic segment that contains the LDL-binding domain, a single transmembrane segment, and a short C-terminal cytosolic segment. Seven cysteine-rich repeats form the LDLbinding domain, which interacts with the apoB-100 molecule in an LDL particle. Figure 14-29 shows how the LDL receptor facilitates internalization of LDL particles by receptor-mediated endocytosis. After internalized LDL particles reach lysosomes, lysosomal proteases hydrolyze their surface apolipoproteins and lysosomal cholesteryl esterases hydrolyze their core cholesteryl esters. The unesterified
cholesterol is then free to leave the lysosome and be used as necessary by the cell in the synthesis of membranes or various cholesterol derivatives.
FIGURE 14-29 Endocytic pathway for internalizing low-density lipoprotein (LDL). Step 1 : A cell-surface LDL receptor binds to an ApoB protein embedded in the phospholipid outer layer of an LDL particle. Interaction between the NPXY sorting signal in the cytosolic tail of the LDL receptor and the AP2 complex incorporates the receptor-ligand complex into a nascent endocytic vesicle. Step 2 : Clathrin-coated pits containing receptor-LDL complexes are pinched off by the same dynamin-mediated mechanism used to form clathrin/AP1-coated vesicles on the trans-Golgi network (see Figure 14-19). Step 3 : After the vesicle coat is shed, the uncoated endocytic vesicle (early endosome) fuses with a late endosome. The acidic pH in this compartment causes a conformational change in the LDL
receptor that leads to release of the bound LDL particle. Step 4 : The late endosome fuses with a lysosome, and the proteins and lipids of the free LDL particle are broken down into their constituent parts by enzymes in the lysosome. Step 5 : The LDL receptor is recycled to the cell surface, where at the neutral pH of the exterior medium the receptor undergoes a conformational change so that it can bind another LDL particle. See M. S. Brown and J. L. Goldstein, 1986, Science 232:34; and G. Rudenko et al., 2002, Science 298:2353. Description Step 1: L D L particle made up of a phospholipid monolayer and a band of Apo B protein binds with the L D L receptor attached to A P 2 complex present along with clathrin forming a coated pit in the plasma membrane. The endings of the coated pit come close together to internalize the L D L particle. Step 2: A coated vesicle containing L D L is formed inside the cytosol. Step 3: The coating of the vesicle disintegrates. The uncoated vesicle fuses with the late endosome, which has a pH of 5, causing a conformation change resulting in the release of L D L from its receptor. Step 4: The late endosome fuses with a lysosome and the L D L particle is broken into amino acids, fatty acids, and cholesterol. Step 5: A vesicles containing the L D L receptor move from the late endosome to the plasma membrane where the neutral p H of the exterior causes a conformational change and the receptor can bind to another L D L particle. Mutations in the human gene for the LDL receptor (LDLR) can cause a hereditary disease that is marked by elevated plasma LDL known as familial hypercholesterolemia (FH). In patients who have one normal and one defective copy of the LDLR gene (heterozygotes), LDL in the blood is increased about twofold. Those with two defective LDLR genes (homozygotes) have LDL levels that are from fourfold to sixfold higher
The Acidic pH of Late Endosomes Causes Most Receptor-Ligand Complexes to Dissociate
than normal. High levels of blood LDL can lead to excessive deposition of LDL, forming atherosclerotic plaque in coronary arteries. Without medical intervention, FH homozygotes usually die of heart attacks before reaching their late twenties. A variety of mutations in the LDLR gene can cause FH. Some mutations prevent the synthesis of the LDL receptor protein; others prevent proper folding of the receptor protein in the ER, leading to its premature degradation (see Chapter 13); and still other mutations reduce the ability of the LDL receptor to bind LDL tightly. A particularly informative group of mutant receptors are expressed on the cell surface and bind LDL normally but cannot mediate the internalization of bound LDL. In individuals with this type of defect, plasma-membrane receptors for other ligands are internalized normally, but the mutant LDL receptor is not recruited into coated pits. Analysis of this mutant receptor and other mutant LDL receptors generated experimentally and expressed in fibroblasts identified a four-residue motif in the cytosolic segment of the receptor that is crucial for its internalization: Asn-Pro-X-Tyr, where X can be any amino acid. This NPXY sorting signal binds to the AP2 complex, linking the clathrin/AP2 coat to the cytosolic segment of the LDL receptor in coated pits. A mutation in any of the conserved residues of the NPXY signal abolishes the ability of the LDL receptor to be incorporated into coated pits. The Acidic pH of Late Endosomes Causes Most Receptor-Ligand
Complexes to Dissociate The overall rate of endocytic internalization of the plasma membrane is quite high: cultured fibroblasts regularly internalize 50 percent of their cell-surface proteins and phospholipids each hour. Most cell-surface receptors that undergo endocytosis will repeatedly deposit their ligands within the cell and then recycle to the plasma membrane to mediate internalization of ligand molecules once again. The LDL receptor, for instance, makes one round trip into and out of the cell interior every 10–20 minutes, for a total of about a hundred trips in its 20-hour life span. Internalized receptor-ligand complexes commonly follow the pathway depicted for the M6P receptor in Figure 14-22 and the LDL receptor in
Figure 14-29. Endocytosed cell-surface receptors typically dissociate from their ligands within late endosomes, which appear as spherical vesicles with tubular branching membranes located a few micrometers from the cell surface. The dissociation of receptor-ligand complexes in late endosomes occurs not only in the endocytic pathway but also in the delivery of soluble lysosomal enzymes via the secretory pathway (see Figure 14-23). As discussed in Chapter 11, the membranes of late endosomes and lysosomes contain V-class proton pumps that act in concert with channels to acidify the vesicle lumen (see Figure 11-13). Most receptors, including the M6P receptor and the LDL receptor, bind their ligands tightly at neutral pH, but release their ligands if the pH is lowered to 6.0 or below. The late
endosome is the first organelle encountered by receptor-ligand complexes whose luminal pH is sufficiently acidic to promote dissociation of most endocytosed receptors from their tightly bound ligands. The mechanism by which the LDL receptor releases bound LDL particles is now understood in detail (Figure 14-30). At the endosomal pH of 5.0– 5.5, histidine residues in a region known as the β-propeller domain of the receptor become protonated, forming a site that can bind with high affinity to the negatively charged cysteine-rich repeats in the LDL-binding domain. This intramolecular interaction sequesters the repeats in a conformation like that of a closed jackknife that cannot simultaneously bind to apoB-100, thus causing release of the bound LDL particle. The LDL receptor in this relatively compact closed state is efficiently recycled back to the plasma membrane for another round of binding and uptake of LDL.
FIGURE 14-30 Model for pH-dependent binding of LDL particles by the LDL receptor. Schematic depiction of an LDL receptor at the neutral pH found at the cell surface and at the acidic pH found in the interior of the late endosome. (a) At the cell surface, apoB-100 on the surface of an LDL particle binds tightly to the receptor. Of the seven cysteine-rich repeats (R1–R7) in the ligand-binding arm, R4 and R5 appear to be most critical for LDL binding. (b) Within the endosome, histidine residues in the β-propeller domain of the LDL receptor become protonated. The positively charged propeller can bind with high affinity to the ligand-binding arm, which contains negatively charged residues, causing release of the LDL particle. (c) Experimental electron density and backbone trace model of the extracellular region of the LDL receptor at pH 5.3 based on x-ray crystallographic analysis. In this conformation, extensive hydrophobic and ionic interactions occur between the β-propeller and the R4 and R5 repeats. [Part (b) data from G. Rudenko et al., 2002, Science 298:2353; PDB ID 1n7d.] Description The illustration labeled (a) shows an L D L receptor attached to the plasma membrane of a cell. The receptor consists of an N P X Y sorting signal embedded in the plasma membrane, a beta-propeller domain, and a ligand-binding arm with a seven residue-
Receptor-Mediated Endocytosis Can Down-Regulate Signaling Receptors
binding region (R 1 to R 7). The ligand-binding arm is bound to the Apo B band on the L D L particle. The particle has the labels for phospholipid monolayer and cholesterol esters. The cell surface has a p H of 7. The illustration labeled (b) shows an endosome membrane, which has a p H of 5. The L D L receptor is attached to the endosome membrane, while the L D L particle is released. A label reads, surface of beta-propeller domain becomes positively charged, and then binds to the ligand-binding arm. A space-filling three-dimensional model labeled (c) shows the L D L receptor with a beta-propeller domain and six residue ligand-binding arms. The six residues are labeled, from top to bottom: R 7, R 6, R 5, R 4, R 3, and R 2. As described in Chapter 6, human mutations that cause a loss of function of the gene PCSK9 lead to a decrease in blood LDL. We now know that PCSK9 encodes a secreted protein whose normal function is to negatively regulate the LDL receptor. Biochemical and structural studies show that PCSK9 binds to the LDL-binding domain of the LDL receptor, thereby preventing the receptor from closing into a compact state. This more open conformation of the receptor cannot be recycled to the plasma membrane and is transported to the lysosome where it is degraded. Inhibitors of PCSK9 are a promising new class of LDL-lowering drugs. Receptor-Mediated Endocytosis Can Down-Regulate Signaling Receptors Signaling receptors are an important class of plasma membrane proteins that can undergo receptor-mediated endocytosis. These receptors bind to extracellular ligands and respond by generating an appropriate
intracellular signal. As described in Chapter 16, the epidermal growth factor receptor (EGFR) activates a signal transduction pathway in response to the extracellular growth factor protein EGF. Treatment of cells with EGF both activates EGFR and stimulates receptor-mediated endocytosis of EGFR. Removal of EGFR from the plasma membrane by endocytosis leads to an attenuation of the signaling response in an adaptive process, known as down-regulation, which extends the dynamic range of EGF concentrations over which the system can respond. The mechanism of EGFR down-regulation is one of the best understood examples of regulated endocytosis. EGFR is a Type I cell surface glycoprotein with a large extracellular domain that binds the ligand EGF, a single transmembrane domain, and a cytoplasmic signaling domain. Normally, EGFR resides in the plasma membrane as a monomer, but when EGF is present, two copies of EGF bind to two copies of EGFR, forming a dimer that brings the cytoplasmic signaling domains of EFGR together and activates signal transduction (see
Figure 16-8). Activated EGFR enters the endocytic pathway because the dimeric form of the receptor binds to clathrin/AP2-coated pits. Mutational studies of the cytoplasmic domain of EGFR have identified a di-leucine (Leu-Leu) sorting signal that is necessary for rapid endocytosis. Other proteins employ a cytoplasmic di-leucine sorting signal for entry into the endocytic pathway, but EGFR is unusual in that the cytoplasmic di-leucine signal only interacts with clathrin/AP2 complexes during ligand-induced endocytosis when the cytoplasmic domain is dimeric.
Endocytosis delivers EGFR to the late endosome, and from this location EGFR is available to recycle to the plasma membrane, in a pathway similar to that for the LDL receptor (see Figure 14-29). Complete downregulation depends on further transport of EGFR to the lysosome where it is degraded. In addition to enabling interaction with clathrin/AP2 complexes, ligand-induced activation of EGFR also triggers covalent modification of the cytoplasmic domain of EGFR by the small protein ubiquitin. As we will see in the next section, monoubiquitinylation marks EGFR for entry into multivesicular bodies that are ultimately degraded in lysosomes (see Figure 14-32). KEY CONCEPTS OF SECTION 14.5 Receptor-Mediated Endocytosis Some extracellular ligands that bind to specific cell-surface receptors are internalized, along with their receptors, in clathrin-coated vesicles whose coats also contain AP2 complexes (see Figure 14-26). Sorting signals in the cytosolic domain of cell-surface receptors target them into clathrin/AP2-coated pits for internalization. Known signals include the Asn-Pro-X- Tyr, Tyr-X-X-Φ, and Leu-Leu sequences (see Table 14-2). The endocytic pathway delivers some ligands (e.g., LDL particles) to lysosomes, where they are degraded. Transport vesicles from the cell surface first fuse with late endosomes, which subsequently fuse with lysosomes. Most receptor-ligand complexes dissociate in the acidic milieu of the late endosome; the receptors are recycled to the plasma membrane, while the ligands are sorted to lysosomes (see Figure 14-29). Signaling receptors can be down-regulated by endocytosis. In these cases, the cytoplasmic domain of the receptors only interact with clathrin/AP2-coated pits when the receptor is in a ligand-activated state.
14.6 Directing Membrane Proteins and Cytosolic Materials to the Lysosome for Degradation
14.6 Directing Membrane Proteins and Cytosolic Materials to the Lysosome for Degradation The major function of lysosomes is to degrade extracellular materials taken up by the cell and to degrade intracellular components under specific conditions. Materials to be degraded must be delivered to the lumen of the lysosome, where the various degradative enzymes reside. As we have just seen, endocytosed ligands (e.g., LDL particles) that dissociate from their receptors in the late endosome subsequently enter the lysosomal lumen when the membrane of the late endosome fuses with the membrane of the lysosome (see Figure 14-29). It is apparent how the general vesicular trafficking mechanism discussed in this chapter can be used to deliver the luminal contents of an endosomal organelle to the lumen of the lysosome for degradation. In this section, we will discuss two further pathways. Membrane proteins delivered to the lysosome by the typical vesicular trafficking process we have discussed the previous section should ultimately be delivered to the membrane of the lysosome. How, then, are membrane proteins delivered to the interior of the lysosome for degradation? The first pathway discussed in this section, used to degrade endocytosed membrane proteins, utilizes an unusual type of vesicle that buds into the lumen of the endosome to produce a multivesicular endosome. The second pathway that will be discussed is
Multivesicular Endosomes Segregate Membrane Proteins Destined for the Lysosomal Membrane from Proteins Destined for Lysosomal Degradation
used by the cell for delivery of cytosolic materials to the lysosomal lumen for degradation. This second pathway, known as autophagy, involves the de novo formation of a double-membrane organelle known as an autophagosome that envelops cytosolic material, such as soluble cytosolic proteins, or sometimes organelles, such as peroxisomes or mitochondria. Both pathways lead to fusion of either the multivesicular endosome or autophagosome with the lysosome, depositing the contents of these organelles into the lysosomal lumen for degradation. Multivesicular Endosomes Segregate Membrane Proteins Destined for the Lysosomal Membrane from Proteins Destined for Lysosomal Degradation Resident lysosomal membrane proteins, such as V-class proton pumps and amino acid transporters, can carry out their functions and remain in the lysosomal membrane, where they are protected from degradation by the soluble hydrolytic enzymes in the lumen. Such proteins are delivered to the lysosomal membrane by transport vesicles that bud from either the trans-Golgi network or the endosome by the same basic mechanisms described in earlier sections. In contrast, endocytosed membrane proteins that are to be degraded are transferred in their entirety to the interior of the lysosome by a specialized delivery mechanism. Lysosomal degradation of cell-surface receptors for extracellular signaling molecules is a common mechanism for controlling the sensitivity of cells to extracellular signals
(see Chapter 15). Receptors that become damaged are also targeted for lysosomal degradation. Early evidence that membranes can be delivered to the lumen of a membrane-bounded compartment came from electron micrographs showing membrane vesicles and fragments of membranes within endosomes and lysosomes. Parallel experiments in yeast revealed that endocytosed receptor proteins targeted to the vacuole (the yeast organelle equivalent to the lysosome) were primarily associated with membrane fragments and small vesicles within the interior of the vacuole rather than with the vacuole surface membrane. These observations suggest that endocytosed membrane proteins can be incorporated into specialized vesicles that form at the endosomal membrane (Figure 14-31). Although these vesicles are similar in size and appearance to transport vesicles, they differ topologically. Transport vesicles bud outward from the surface of a donor organelle into the cytosol, whereas vesicles within the endosome bud inward from the surface into the lumen (away from the cytosol). Mature endosomes containing numerous vesicles in their interior are usually called multivesicular endosomes (or bodies). The surface membrane of a multivesicular endosome then fuses with the membrane of a lysosome, thereby delivering its internal vesicles and the membrane proteins they contain into the lysosome interior for degradation. Thus the sorting of proteins in the endosomal membrane determines which ones will remain on the lysosome surface (e.g., pumps and transporters) and which ones
will be incorporated into internal vesicles and ultimately degraded in lysosomes.
FIGURE 14-31 Delivery of plasma-membrane proteins to the lysosome interior for degradation. Early endosomes carrying endocytosed plasma-membrane proteins (blue) and vesicles carrying lysosomal membrane proteins (green) from the trans-Golgi network fuse with the late endosome, transferring their membrane proteins to the endosomal membrane (steps 1 and 2 ). Proteins to be degraded, such as those from the early endosome, are incorporated into vesicles that bud into the interior of the late endosome, eventually forming a multivesicular endosome containing many such internal vesicles (step 3 ). Fusion of a multivesicular endosome directly with a lysosome releases the internal vesicles into the lumen of the lysosome, where they can be degraded (step 4 ). Because proton pumps and other lysosomal membrane proteins normally are not incorporated into internal endosomal vesicles, they are delivered to the lysosomal membrane and are protected from degradation. See F. Reggiori and D. J. Klionsky, 2002, Eukaryot. Cell 1:11; and D. J. Katzmann et al., 2002, Nat. Rev. Mol. Cell Biol. 3:893. Description Step 1: A vesicle for degradation buds from the Golgi. The coat proteins cover the bud formed in the Golgi. The coated vesicle formed moves towards the late endosome and becomes an uncoated vesicle. The uncoated vesicle fuses with the late endosome membrane.
Step 2: Proteins from the plasma membrane are endocytosed into the cytosol. These vesicles containing proteins for degradation merge with the late endosome, becoming embedded in the membrane of the endosome. Step 3: The proteins bound to the late endosome membrane bud into the endosome, forming a multivesicular body. Step 4: Merging with a lysosome, the contents of the multivesicular body, including the membrane proteins, are digested. Many of the proteins required for inward budding of the endosomal membrane were first identified by mutations in yeast that blocked the delivery of membrane proteins to the interior of the vacuole. The current model of endosomal budding to form multivesicular endosomes is based primarily on studies in yeast, but mammalian cells appear to have essentially the same mechanism (Figure 14-32). Most cargo proteins that enter a multivesicular endosome are tagged with ubiquitin. Cargo proteins destined to enter a multivesicular endosome usually receive their ubiquitin tags at the plasma membrane, the trans-Golgi network, or the endosomal membrane. We have already seen how ubiquitin tagging can serve as a signal for degradation of cytosolic or misfolded ER proteins by proteasomes (see Chapters 3 and 13). When used as a signal for proteasomal degradation, the ubiquitin tag usually consists of a chain of covalently linked ubiquitin molecules (polyubiquitin), whereas ubiquitin used to tag proteins for entry into the multivesicular endosome usually takes the form of a single (monoubiquitin) molecule. In the membrane of the endosome, a ubiquitin-tagged peripheral membrane protein, known as Hrs, facilitates recruitment of a set of at least three different protein complexes to the membrane. These ESCRT (endosomal sorting
complexes required for transport) proteins include the ubiquitin-binding protein Tsg101. The membrane-associated ESCRT proteins act to drive vesicle budding directed into the interior of the endosome as well as the loading of specific monoubiquitinylated membrane cargo proteins into the vesicle buds. Finally, the ESCRT proteins pinch off the vesicle by forming a filamentous spiral inside the neck of a vesicle bud, releasing it and the specific membrane cargo proteins it carries into the interior of the endosome. An ATPase, known as Vps4, uses the energy from ATP hydrolysis to disassemble the ESCRT proteins, releasing them into the cytosol for another round of budding. In the fusion event that pinches off a completed endosomal vesicle, the ESCRT proteins and Vps4 may function like SNAREs and NSF, respectively, in the typical membrane-fusion process discussed previously (see Figure 14-11).
Retroviruses Bud from the Plasma Membrane by a Process Similar to Formation of Multivesicular Endosomes
FIGURE 14-32 Model of the mechanism for formation of multivesicular endosomes. In endosomal budding, ubiquitinylated Hrs on the endosomal membrane directs the loading of specific membrane cargo proteins (blue) into vesicle buds and then recruits cytosolic ESCRT protein complexes to the membrane (step 1 ). Note that both Hrs and the recruited cargo proteins are tagged with ubiquitin. After the set of bound ESCRT complexes mediates the completion and pinching off of the inwardly budding vesicles (step 2 ), these complexes are disassembled by the ATPase Vps4 and returned to the cytosol (step 3 ). See text for discussion. See O. Pornillos et al., 2002, Trends Cell Biol. 12:569. Description Step 1: An illustration shows an endosome. In the endosomal membrane, many cargo proteins and H r s proteins are embedded. These proteins are labeled with ubiquitin. E S C R T complexes are bound to the ubiquitin-bound h r s proteins. A vesicle starts to bud from the plasma membrane into the lumen of the endosome with the cargo proteins embedded in them. Step 2: An endosomal vesicle containing cargo proteins is formed from the bud. Step 3: On the surface of the endosome, the E S C R T complex is disassembled via V P S 4 using an A T P molecule. Retroviruses Bud from the Plasma Membrane by a Process Similar to Formation of Multivesicular Endosomes The vesicles that bud into the interior of endosomes have a topology similar to that of enveloped virus particles that bud from the plasma membrane of virus-infected cells. Moreover, recent experiments have
demonstrated that a common set of proteins is required for both types of membrane-budding events. In fact, the two processes so closely parallel each other in mechanistic detail as to suggest that enveloped viruses have evolved mechanisms to recruit the cellular proteins used in inward endosomal budding for their own purposes. The human immunodeficiency virus (HIV) is an enveloped retrovirus that buds from the plasma membrane of infected cells in a process driven by the viral Gag protein, the major structural component of completed virus particles. Gag protein binds to the plasma membrane of an infected cell, and some 4000 Gag molecules polymerize into a spherical shell, producing a structure that looks like a vesicle bud protruding outward from the plasma membrane. Mutational studies with HIV have revealed that the N-terminal segment of Gag protein is required for association with the plasma membrane, whereas the C-terminal segment is required for pinching off of complete HIV particles. For instance, if the portion of the viral genome encoding the C-terminus of Gag is removed, HIV buds will form in infected cells, but pinching off does not occur, and thus no free virus particles are released. The first indication that HIV budding employs the same molecular machinery as vesicle budding into endosomes came from the observation that Tsg101, an ESCRT protein, binds to the C-terminus of Gag protein. Subsequent findings have clearly established the mechanistic parallels between the two processes. For example, Gag is ubiquitinylated as part of the process of virus budding, and in cells with mutations in Tsg101 or Vps4, HIV virus buds accumulate but cannot pinch off from the membrane
(Figure 14-33). Moreover, when a segment from the cellular Hrs protein is added to a truncated Gag protein by construction of the appropriate hybrid gene, proper budding and release of virus particles is restored. Taken together, these results indicate that Gag protein mimics the function of Hrs, redirecting ESCRT proteins to the plasma membrane, where they can function in the budding of virus particles.
FIGURE 14-33 Mechanism for budding of HIV from the plasma membrane. Proteins required for the formation of multivesicular endosomes are exploited by HIV for virus budding from the plasma membrane. (a) Budding of HIV particles from HIV-infected cells occurs by a mechanism similar to that shown in Figure 14-32, using the virally encoded Gag protein and cellular ESCRT and Vps4 proteins (steps 1 – 3 ). Ubiquitinylated Gag located near a budding particle functions like Hrs. (b) In wild-type cells infected with HIV, virus particles bud from the plasma membrane and are rapidly released into the extracellular space. (c) In cells that lack the functional ESCRT protein Tsg101, the viral Gag protein
The Autophagic Pathway Delivers Cytosolic Proteins or Entire Organelles to Lysosomes
forms dense viruslike structures, but budding of these structures from the plasma membrane cannot be completed, and chains of incomplete viral buds still attached to the plasma membrane accumulate. Description The illustration labeled (a) shows H I V budding from the plasma membrane of a cell. On the cytosolic side of the membrane, many H I V G A G proteins are embedded. The H I V particle also contains H I V envelope proteins. In the cytosol, the G A G proteins are ubiquitinated. The E S C R T assembly leads to pinching off of the vesicle containing the core particle. The H I V buds and the E S C R T complex in the cytosol disassemble by the action of V p s 4 which uses a molecule of A T P. The electron micrograph labeled (b) shows wild-type H I V budding from a cell membrane into the extracellular space. It is dark and circular. The electron micrograph labeled (c) shows a clump of buds with dark outlines. These are H I V buds attached to the plasma membrane, unable to leave the plasma membrane. Other enveloped retroviruses, such as murine leukemia virus and Rous sarcoma virus, have also been shown to require ESCRT complexes for their budding, although each virus appears to have evolved a somewhat different mechanism to recruit ESCRT complexes to the site of virus budding. The Autophagic Pathway Delivers Cytosolic Proteins or Entire Organelles to Lysosomes
When cells are placed under stressful conditions, such as starvation, they have the capacity to recycle macromolecules for use as nutrients in a process of lysosomal degradation known as autophagy (“eating oneself”). The autophagic pathway begins with the formation of a flattened doublemembrane cup-shaped structure that envelops a region of the cytosol or an entire organelle (e.g., a mitochondrion), forming an autophagosome, or autophagic vesicle (Figure 14-34). The outer membrane of an autophagosome can fuse with a lysosome, delivering a large vesicle, bounded by a single membrane bilayer, to the interior of the lysosome. Lipases and proteases within the lysosome degrade the autophagosome and its contents into their molecular components, just as they do when the contents of multivesicular endosomes are delivered to the lysosome. Amino acid permeases in the lysosomal membrane then allow for the transport of free amino acids back into the cytosol for use in synthesis of new proteins.
FIGURE 14-34 The autophagic pathway. The autophagic pathway allows cytosolic proteins and organelles to be delivered to the lysosome interior for degradation. In the
autophagic pathway, a cup-shaped structure forms around a portion of the cytosol (right) or an organelle such as a mitochondrion (left). Continued addition of membrane eventually leads to the formation of an autophagosome that envelops its contents in two complete membranes (steps 1a and 1b ). Fusion of the outer membrane with the membrane of a lysosome releases a single-membrane vesicle and its contents into the lysosome interior (steps 2a and 2b ). After degradation of the protein and lipid components by hydrolases in the lysosome interior (step 3 ), the released amino acids are transported across the lysosomal membrane into the cytosol (step 4 ). Proteins known to participate in the autophagic pathway include Atg8, which forms a coat structure around the autophagosome. Description This illustration has a beginning pathway coming in from the top left and labeled with steps 1 a and 2 a, and a second pathway coming down from the top right and labeled 1 b and 2 b. 1 a: In the pathway from top left, the mitochondrion is represented by a pink circle being enclosed by an autophagic vesicle. 2 a: The vesicle fully encloses the mitochondrion to enter into the lysosome. 1 b. A C-shaped autophagic vesicle with tiny A t g 8 spheres attached to it is present in the cytosol. 2 b: The autophagic vesicle encloses cytosol inside it. 3: Inside the lysosome, both vesicles from 2 a and 2 b and the mitochondrion dissolve. 4: Amino acids exit the lysosome. By studying mutants with defects in the autophagic pathway, scientists have identified processes other than recycling of cellular components during starvation that also depend on autophagy. Experiments carried out principally in Drosophila and mice have shown that autophagy participates in a type of quality-control mechanism that removes
organelles that have ceased to function properly. In particular, the autophagic pathway can target dysfunctional mitochondria that have lost their integrity and no longer have an electrochemical gradient across their inner membrane. In certain cell types, pathogenic bacteria and viruses that are multiplying in the cytosol of host cells can be targeted to the autophagic pathway for destruction in the lysosome as part of a host defense mechanism against infection. In each of these processes, and in all eukaryotic organisms, the autophagic pathway takes place in three basic steps. Although the mechanisms underlying each of these steps are relatively poorly understood, they are thought to be related to the basic mechanisms for vesicular trafficking discussed in this chapter. Autophagosome Nucleation The autophagosome is thought to originate from a fragment of a membrane-bounded organelle. The origin of this membrane has been difficult to trace because no known unique integral membrane proteins, which might serve to identify the source of this membrane, are known to be required for the formation of the autophagosome. Studies in yeast have shown that some mutants defective in Golgi trafficking are also defective in autophagy, suggesting that the autophagosome is initially derived from a fragment of the Golgi. Autophagy that is induced by starvation appears to be a nonspecific process in which a random portion of the cytoplasm, including organelles, becomes enveloped by an autophagosome. In these cases, the site of nucleation is probably random. In cases in which
organelles are enveloped by the autophagosome in response to nutrient starvation, a signal based on ubiquitin modification on the surface of the organelle targets nucleation of the autophagosome. Autophagosome Growth and Completion New membrane must be delivered to the autophagosome membrane in order for this cup-shaped organelle to grow. This growth probably occurs by the fusion of transport vesicles with the membrane of the autophagosome. About 30 proteins that participate in the formation of autophagosomes have been identified in genetic screens for yeast mutants that are defective in autophagy. One of these proteins is Atg8, shown in
Figure 14-34, which is covalently linked to the lipid phosphatidylethanolamine and thus becomes attached to the cytoplasmic face of the autophagosome. Association of Atg8 with a membrane vesicle appears to be the key step in enabling a vesicle to fuse with the growing autophagosome. Fusion of Atg8-containing vesicles with the autophagosome involves the formation of a cytosolic assembly of Atg12, Atg5, and Atg16. Atg12 is similar in structure to ubiquitin, and a set of proteins related to ubiquitinconjugating enzymes are responsible for covalently joining Atg12 to Atg5 by a process similar to that used for covalently joining ubiquitin to a target protein (see Figure 3-32). The covalently linked Atg12-Atg5 dimer then co-assembles with Atg16 to form a polymeric complex localized to the site of a growing autophagosome. By an unknown mechanism, this
cytosolic complex is thought to bring about the fusion of Atg8-containing vesicles into a cup-shaped autophagosome. Autophagosome Targeting and Fusion The outer membrane of the completed autophagosome is thought to contain a set of proteins that target it for fusion with the membrane of a lysosome. Two vesicle-tethering proteins have been found to be required for autophagosome fusion with a lysosome, but the corresponding SNARE proteins have not been identified. Fusion of the autophagosome with the lysosome occurs after Atg8 has been released from the autophagosome membrane by proteolytic cleavage; this proteolysis step occurs only after the autophagosome has completely formed a sealed double-membrane system. Thus Atg8 protein appears to mask fusion proteins and to prevent premature fusion of the autophagosome with the lysosome. KEY CONCEPTS OF SECTION 14.6 Directing Membrane Proteins and Cytosolic Materials to the Lysosome for Degradation Endocytosed membrane proteins destined for degradation in the lysosome are incorporated into vesicles that bud into the interior of the endosome. Multivesicular endosomes, which contain many of these internal vesicles, can fuse with the lysosome to deliver the vesicles to the interior of the lysosome (see Figure 14-31). Some of the cellular components (e.g., ESCRT) that mediate inward budding of endosomal membranes are used in the budding and pinching off of enveloped viruses, such as HIV, from the plasma membrane of virus-infected cells (see Figures 14-32 and 14-33). In the process of autophagy, a portion of the cytoplasm or an entire organelle (e.g., a mitochondrion) can be enveloped in a flattened membrane and eventually incorporated into a double-membrane autophagosome. Fusion of the outer vesicle
membrane with the lysosome delivers the enveloped contents to the interior of the lysosome for degradation (see Figure 14-34).
Key Terms
End of Chapter Visit Achieve to access study tools and to learn more about the content in this chapter: Perspectives for the Future Analyze the Data Classic Experiment 14-1: Following a Protein out of the Cell Chapter References Additional study tools, including videos, animations, and quizzes Key Terms adapter protein (AP) complexes anterograde ARF protein autophagy cisterna cisternal maturation clathrin constitutive secretion
Review the Concepts
exocytosis late endosome low-density lipoprotein (LDL) mannose 6-phosphate (M6P) multivesicular endosomes Rab proteins receptor-mediated endocytosis regulated secretion retrograde retromer secretion (sec) mutants secretory pathway sorting signals transcytosis trans-Golgi network transport vesicles t-SNAREs v-SNAREs Review the Concepts 1. What two modern methods allow the process of intercompartmental transport to be followed? What basic features do these experimental approaches to study protein transport have in common? 2. Vesicle budding is associated with coat proteins. What is the role of coat proteins in vesicle budding? How are coat proteins
recruited to membranes? What kinds of molecules are likely to be included or excluded from newly formed vesicles? What is the best-known example of a protein likely to be involved in the pinching off of vesicles? 3. Treatment of cells with the drug brefeldin A (BFA) has the effect of causing Golgi membranes in the process of budding COPI vesicles to shed their vesicle coats. The result of BFA treatment is to cause the vast majority of Golgi proteins to be relocated in the ER. What inferences can be made from this observation regarding roles of coat proteins, other than promoting vesicle formation? Predict what type of mutation in ARF might have the same effect as treating cells with BFA. 4. Microinjection of an antibody known as EAGE, which reacts with the “hinge” region of the β subunit of COPI, causes accumulation of Golgi enzymes in transport vesicles and inhibits anterograde transport of newly synthesized vesicles from the ER to the plasma membrane. What effect does the antibody have on COPI activity? Explain the results. 5. Specificity in fusion between vesicles involves two discrete and sequential processes. Describe the first of the two processes and its regulation by GTPase switch proteins. What effect on the size of early endosomes might result from overexpression of a mutant form of Rab5 that is stuck in the GTP-bound state? 6. Sec18 is a yeast gene that encodes NSF. Mutations of this gene produce class C mutants. What is the mechanistic role of NSF in membrane trafficking? And, as indicated by its class C phenotype, why does an NSF mutation produce accumulation of
vesicles at what appears to be only one stage of the secretory pathway? 7. What feature of procollagen synthesis provided early evidence for the Golgi cisternal maturation model? 8. Sorting signals that cause retrograde transport of a protein in the secretory pathway are sometimes known as retrieval sequences. List the two known examples of retrieval sequences for soluble and membrane proteins of the ER. How does the presence of a retrieval sequence on a soluble ER protein result in its retrieval from the cis-Golgi complex? Describe how the concept of a retrieval sequence is essential to the cisternal-maturation model. 9. Clathrin adapter protein (AP) complexes bind directly to the cytosolic face of membrane proteins and also interact with clathrin. What are the four known adapter protein complexes? What observation regarding AP3 suggests that clathrin is an accessory protein to a core coat composed of adapter proteins? 10. I-cell disease is a classic example of an inherited human defect in protein targeting that affects an entire class of proteins: the soluble enzymes of the lysosome. What is the molecular defect in I-cell disease? Why does it affect the targeting of an entire class of proteins? What other types of mutations might produce the same phenotype? 11. The trans-Golgi network is the site of multiple sorting processes as proteins and lipids exit the Golgi complex. Compare and contrast the sorting of proteins to lysosomes with the packaging of proteins into regulated secretory vesicles such as those containing insulin. Compare and contrast the sorting of proteins
to the basolateral versus apical cell surfaces in MDCK cells and in hepatocytes. 12. What does the budding of influenza virus and vesicular stomatitis virus (VSV) from polarized MDCK cells reveal about the sorting of newly synthesized plasma membrane proteins to the apical or basolateral domains? Now consider the following result: a peptide with a sequence identical to that of the VSV G protein cytoplasmic domain inhibits targeting of the G protein to the basolateral surface and has no effect on HA targeting to the apical membrane, but a peptide in which the single tyrosine residue is mutated to an alanine has no effect on G protein basolateral targeting. What does this tell you about the sorting process? 13. Describe the role of pH in regulating the interaction between mannose 6-phosphate and the M6P receptor. Why does a rise in endosomal pH lead to the secretion of newly synthesized lysosomal enzymes into the extracellular medium? 14. What mechanistic features are shared by (a) the formation of multivesicular endosomes by budding into the interior of an endosome and (b) the outward budding of HIV virus at the cell surface? You wish to design a peptide inhibitor/competitor of HIV budding and decide to mimic a portion of the HIV Gag protein in a synthetic peptide. Which portion of the HIV Gag protein would be a logical choice? What normal cellular process might this inhibitor block? 15. The endocytic and autophagic pathways serve two fundamentally different roles, but both deliver their vesicles to the lysosome. What are the fundamental differences between the two
pathways? Describe the three basic steps in the formation and fusion of autophagosomes. 16. Describe the location and pH sensitivity of the receptor-ligand interaction in the LDL receptor-mediated endocytosis pathway. 17. What do mutations in the cytoplasmic domain of the LDL receptor that cause familial hypercholesterolemia reveal about the receptor-mediated endocytosis pathway?