Introduction
Chapter 16 Growth Factor and Cytokine Signaling Pathways That Control Gene Expression A planarian with multiple heads. Several hormones and their receptors regulate the regeneration of complex body parts in wounded planaria; the extracellular signaling protein Wnt promotes tail regeneration and inhibits head regeneration. In an experiment, the gene encoding beta-catenin-1, an essential protein in the Wnt signal transduction pathway, was inhibited by feeding animals an inhibitory double-stranded RNA. During the subsequent month, normal uninjured animals developed heads around their periphery in a process induced by small wounds caused by defects in stem-cell mediated tissue turnover and repair; the complete absence of a Wnt signal at these wounds promotes formation of a head.
16.1 Growth Factors and Their Receptor Tyrosine Kinases
16.2 The Ras/MAP Kinase Signal Transduction Pathway
16.3 Phosphoinositide Signal Transduction Pathways
16.4 Cytokines, Cytokine Receptors, and the JAK/STAT Signaling Pathway
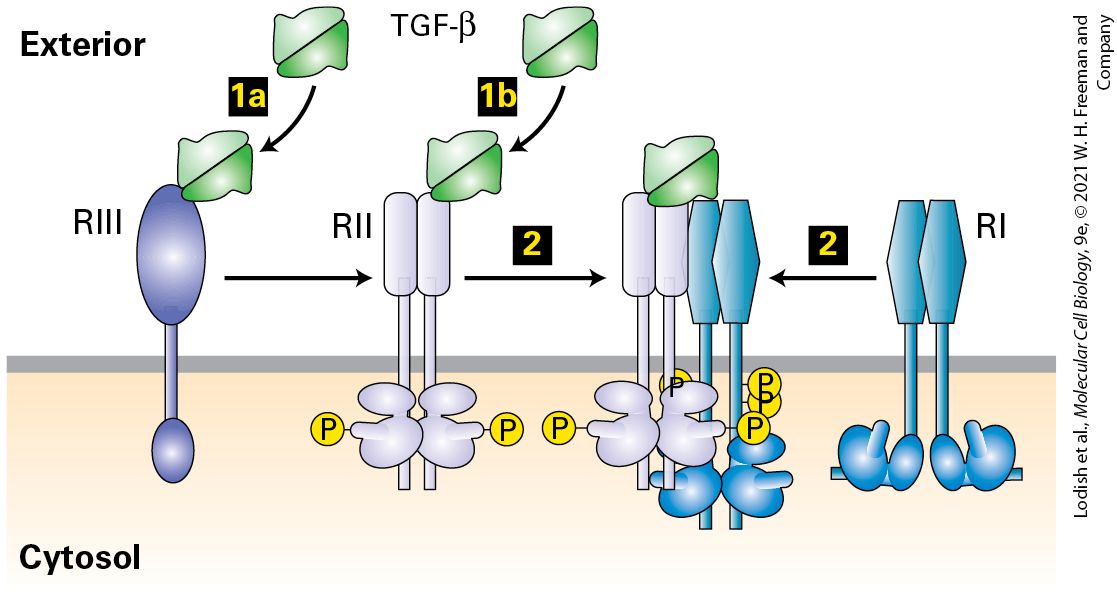
16.5 The TGF-β Family of Growth Factors, Their Receptor Serine Kinases, and the Smad Transcription Factors They Activate
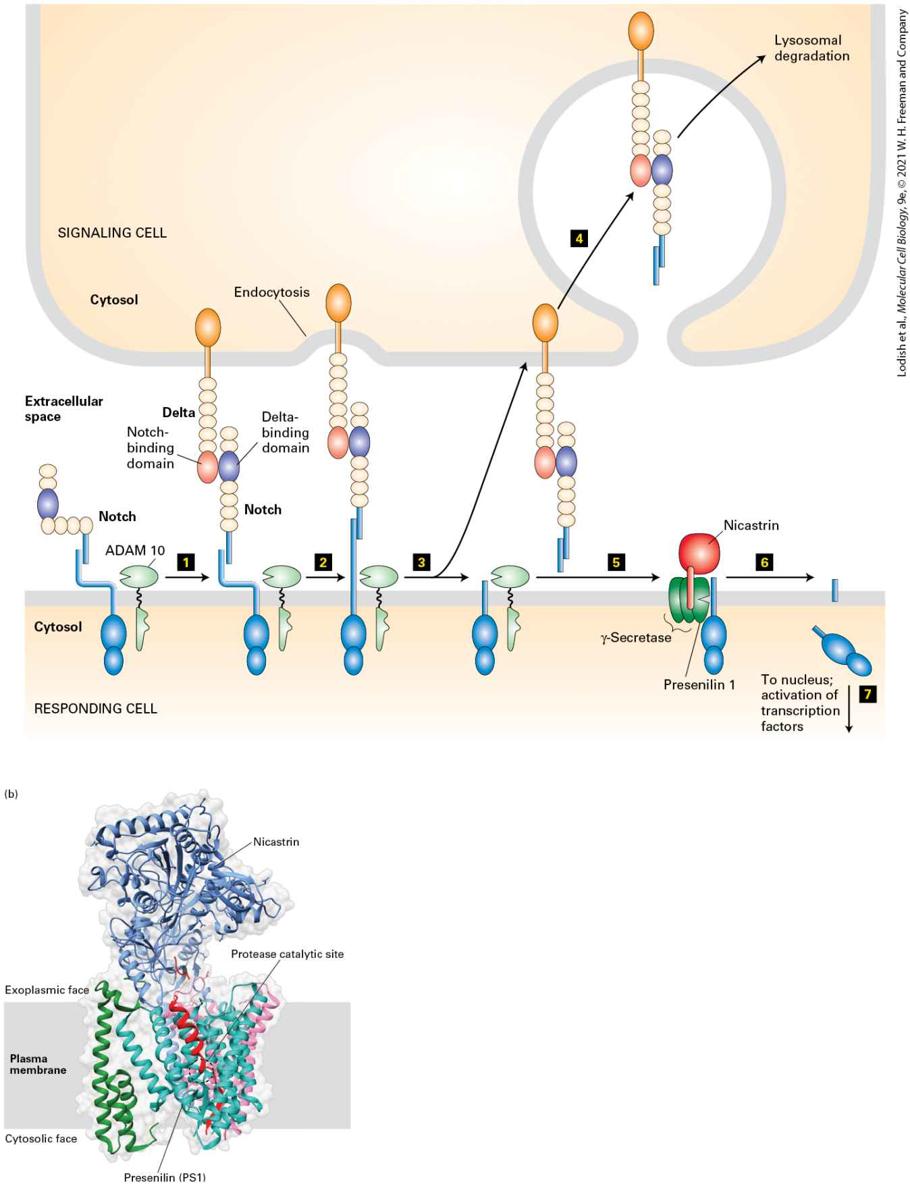
16.6 Signal Transduction Pathways That Utilize Regulated, Site-Specific Protein Cleavage: Notch/Delta and EGF Precursors
16.7 Signal Transduction Pathways That Utilize Proteasomal Degradation of Signaling Components: Wnt, Hedgehog, and the Many Hormones That Activate NF-κB Hundreds of extracellular signaling molecules regulate the growth and fate of cells and in this way control the development of all metazoans; these signaling molecules can have both short- and long-term effects on cells. Short-term effects are usually triggered by modification of existing enzymes or other proteins, as we saw in Chapter 15. Long-term changes in cell function, however, require changes in gene expression. Thus many extracellular signaling molecules alter the expression of genes and in doing so can alter cell division and cell differentiation. For example, signaling molecules called cytokines induce the body’s production of red blood cells, white blood cells, and platelets. Once differentiated, cells respond to their environment by changing their shape, metabolism, or movement. Here again, extracellular signaling molecules may lead to changes in gene expression. In response to infection, for example, several hormones activate or repress the expression of several hundred genes in
immune system cells that carry out multiple responses to infection. Given the extensive role of gene transcription in mediating critical aspects of development, metabolism, and movement, it is not surprising that mutations in such signaling pathways cause many human diseases, including cancer, diabetes, and immune-system disorders. In this chapter, we explore the main groups of hormones, receptors, and their associated intracellular signal transduction pathways that cells use mainly to influence gene expression. In eukaryotes, there are about a dozen classes of highly conserved cell-surface receptors, and these receptors activate several types of highly conserved and often complex intracellular signal transduction pathways, leading to both short-term and long-term effects. These pathways have been conserved throughout evolution and operate in much the same manner in flies, worms, planaria, mice, and humans; their commonalities have enabled researchers to study them in a variety of experimental systems. For instance, the signaling protein Hedgehog (Hh) and its receptor were first identified in fruit fly (Drosophila) mutants that had impaired development. Subsequently, the human and mouse homologs of these proteins were cloned and shown to participate in a number of important signaling events during cell differentiation, resulting in the discovery that abnormal activation of the Hh pathway occurs in several human tumors. Despite the complexity of many of these signal transduction pathways, there are features common to many of them. We learned about some in our study of GPCR signaling, and we will encounter others in the following sections. Signal transduction pathways that regulate gene expression act
through transcription factors that switch genes on or off. In non-stimulated cells, most regulated transcription factors are sequestered in the cytosol, unable to move into the nucleus, bind to DNA, or affect gene expression. Activation of the relevant signal transduction pathway triggers transport of the transcription factor into the nucleus, in some cases by releasing it from an inhibitory complex in the cytosol, in others by unmasking a nuclear localization sequence. Alternatively, a kinase in the signal transduction pathway may phosphorylate the transcription factor, converting it into an active form that is able to bind to DNA. Another common theme is negative feedback regulation through changes in gene expression; a regulated transcription factor may induce synthesis of proteins that repress or dampen a signal transduction pathway — preventing overstimulation of the cell. We begin this chapter with a large class of receptors — the Receptor Tyrosine Kinases (RTKs; Section 16.1) — that are activated by a large group of protein growth factors. Ligand binding to the extracellular domain induces two monomers to bind together as a dimer, activating a protein kinase contained in its cytosolic domain (Figure 16-1a). One important RTK signaling pathway includes the activation of the small GTP-binding protein termed Ras; Ras in turn activates a cascade of protein kinases in which one kinase activates another — the Ras/MAP Kinase Pathway (Figure 16-1a, Pathway A; Section 16.2). The final kinase phosphorylates and activates one or more transcription factors (Tfs).
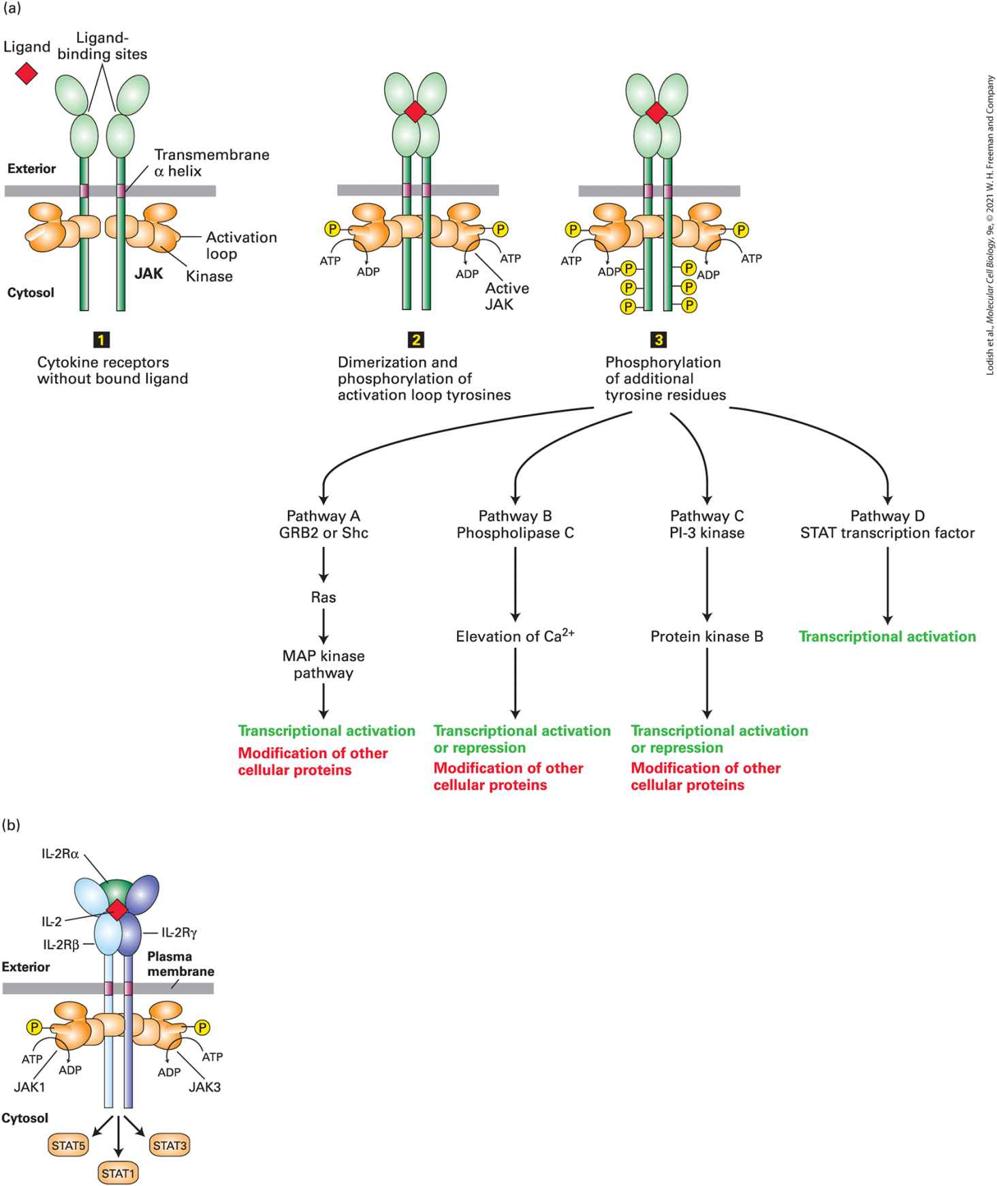
FIGURE 16-1 Common types of cell-surface receptors and signal transduction pathways that regulate gene expression. (a) Signal transduction pathways activated by receptor tyrosine kinases (RTKs) and cytokine receptors. The cytosolic domain of RTKs contains an intrinsic protein tyrosine kinase domain; the cytosolic domain of cytokine receptors bind a separate protein tyrosine kinase. In the absence of ligand these receptors generally exist as monomers that have poor kinase activity. Binding of ligands to the extracellular domains of these receptors triggers dimerization of two monomers and activation of the tyrosine kinase and phosphorylation of several tyrosine residues in the receptor’s cytosolic domain. The resulting phosphotyrosines function as docking sites for binding and activating several downstream signal-transducing proteins. Pathway A: Binding of one type of adapter protein (e.g., GRB2) to an activated receptor leads to activation of the small GTP-binding “switch” protein Ras (see Section 16.2). Signal transduction pathways downstream of Ras involve several kinases; in these MAP kinase pathways, one kinase phosphorylates and thus activates the activity of another. This leads to phosphorylation and activation of transcription factors, which are often different in different cells. Pathways B and C: Two signal transduction pathways that involve phosphoinositides are triggered by recruitment of phospholipase C and PI-3 kinase, respectively, to the plasma membrane (see Section 16.3). Elevated levels of and activated protein kinase B modulate the activity of transcription factors as well as of cytosolic proteins that are involved in metabolic pathways or cell movement or shape change. Pathway D: In a pathway mainly employed by cytokine receptors, a STAT transcription factor binds to the activated receptor, becomes phosphorylated, dimerizes, moves to the nucleus, and directly activates transcription. (b) Other common types of cell-surface receptors and signal transduction pathways. Pathway A: Many GPCRs activate the heterotrimeric GTP-binding protein, ultimately leading to activation of protein kinase A, and phosphorylation and activation of transcription factors such as CREB. Pathway B: The cytosolic domains of receptors for the TGF-β family of signaling proteins contain a serine-threonine kinase, which directly phosphorylates and activates a member of the Smad class of transcription factors, unmasking a nuclear localization signal. Pathway C: Binding of a Delta ligand to the extracellular domain of a member of the Notch family of receptors triggers proteolytic cleavage of the receptor, releasing its cytosolic domain, which moves into the nucleus and regulates gene expression. Pathway D: Signal transduction pathways activated by binding of members of the Wnt, Hedgehog, or Interleukin 1 (IL-1) families of ligands to their respective receptors lead to ubiquitination and degradation of components of multiprotein
complexes in the cytosol, releasing a transcription factor that then translocates into the nucleus. Description The illustration labeled (a) shows a common cell surface receptor at the top in the plasma membrane. From here, 4 arrows points to different pathways. The left list is titled pathway A, G R B 2 or S h c. Downward arrow to R a s then to M A P kinase pathway, lastly transcriptional activation, modification of other cellular proteins. The second list starts with the title Pathway B, Phospholipase C, downward arrows to Elevation of C a superscript 2 plus and transcriptional activation or repression, modifications of other cellular proteins. The third list is titled Pathway C, P 1-3 kinase, downward arrow to protein kinase B, then transcriptional activation or repression, modification of other cellular proteins. The final list is titled pathway D, S T A T transcription factor, downward arrow to transcriptional activation. The illustration labeled (b) shows the exterior, membrane, cytosol, nuclear membrane, and nucleus. In the cell membrane at the top is pathway A shows a chain of cylinders with a G T Pbinding protein and G T P. This is moved with downward arrows to the nucleus where it combines to G P C R. Pathway B also starts in the cell membrane, moves into the cytosol with a pink structure labeled transcription factor, then into the nucleus where the structure is labeled T G F-beta. Pathway C starts in the cell membrane, adds a transcription factor of a different shape than pathway B, and into the nucleus where the label is notch/delta. Pathway D starts with the cylinder formation in the cell membrane, adds another different transcription factor, and goes to the nucleus where it is labeled W n t hedgehog I L-1 T N F alpha. Alternatively, RTKs can initiate signal transduction pathways containing phosphorylated inositols that often although not always lead to changes in gene expression. In Chapter 15 we saw one that leads to an elevation in the concentration of in the cytosol (Figure 16-1a, Pathway B). Depending on the type of cell, this pathway triggers processes as diverse as protein secretion and cell migration. RTKs also induce other signaling
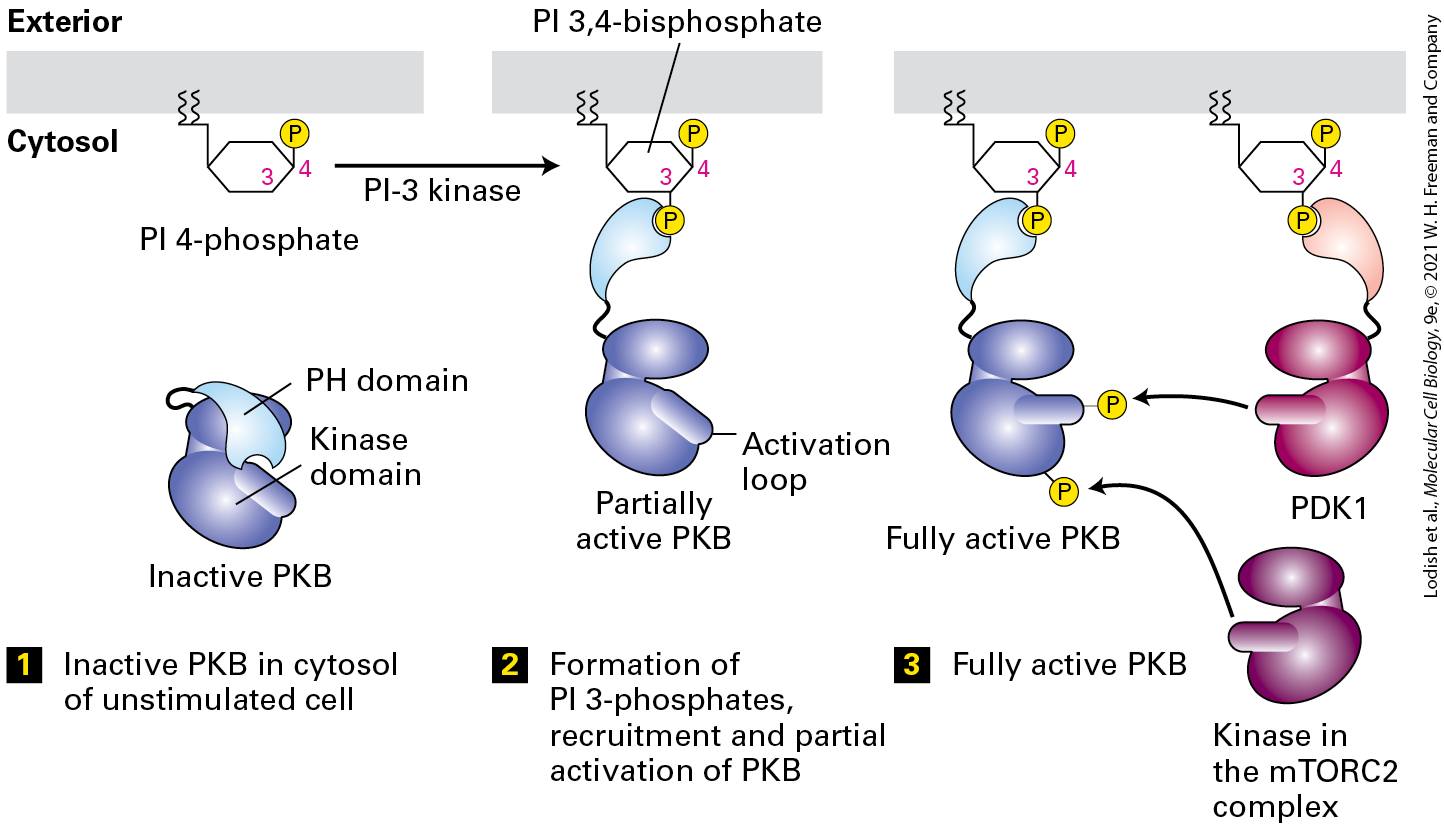
pathways involving phosphorylated inositols that activate yet other protein kinases, including Protein Kinase B, that in turn alter gene expression to regulate cellular metabolism and cell death (Figure 16-1a, Pathway C; Section 16-3). Cytokines activate a distinct class of signaling receptors. The cytokines form a large family of protein hormones that regulate, for example, the formation and function of all types of blood cells, including all of the many kinds of cells that compose our immune system (Chapter 24). Cytokine receptor proteins are similar to RTKs in that ligand binding leads to the activation of a protein tyrosine kinase (JAK), although in this case the kinase is a separate protein tightly associated with the receptor’s cytosolic domain. Cytokine receptors and RTKs activate many of the same signal transduction pathways (see Figure 16-1a), and in Section 16-4 we learn how a JAK kinase directly phosphorylates a STAT transcription factor (Figure 16-1a, Pathway D). Phosphorylation leads to the exposure of a nuclear localization signal that guides the transcription factor to the nucleus. The large TGF-β family of extracellular signaling proteins acts to regulate many developmental pathways. Like RTKs, these structurally distinct receptors employ activation of a kinase that is part of their cytosolic domain. The activated kinase directly phosphorylates transcription factors in the Smad family of DNA-binding proteins (Figure 16-1b, Pathway B, and Section 16.5). In this case, phosphorylation unmasks a nuclear localization signal that allows the transcription factor to move directly to the nucleus.
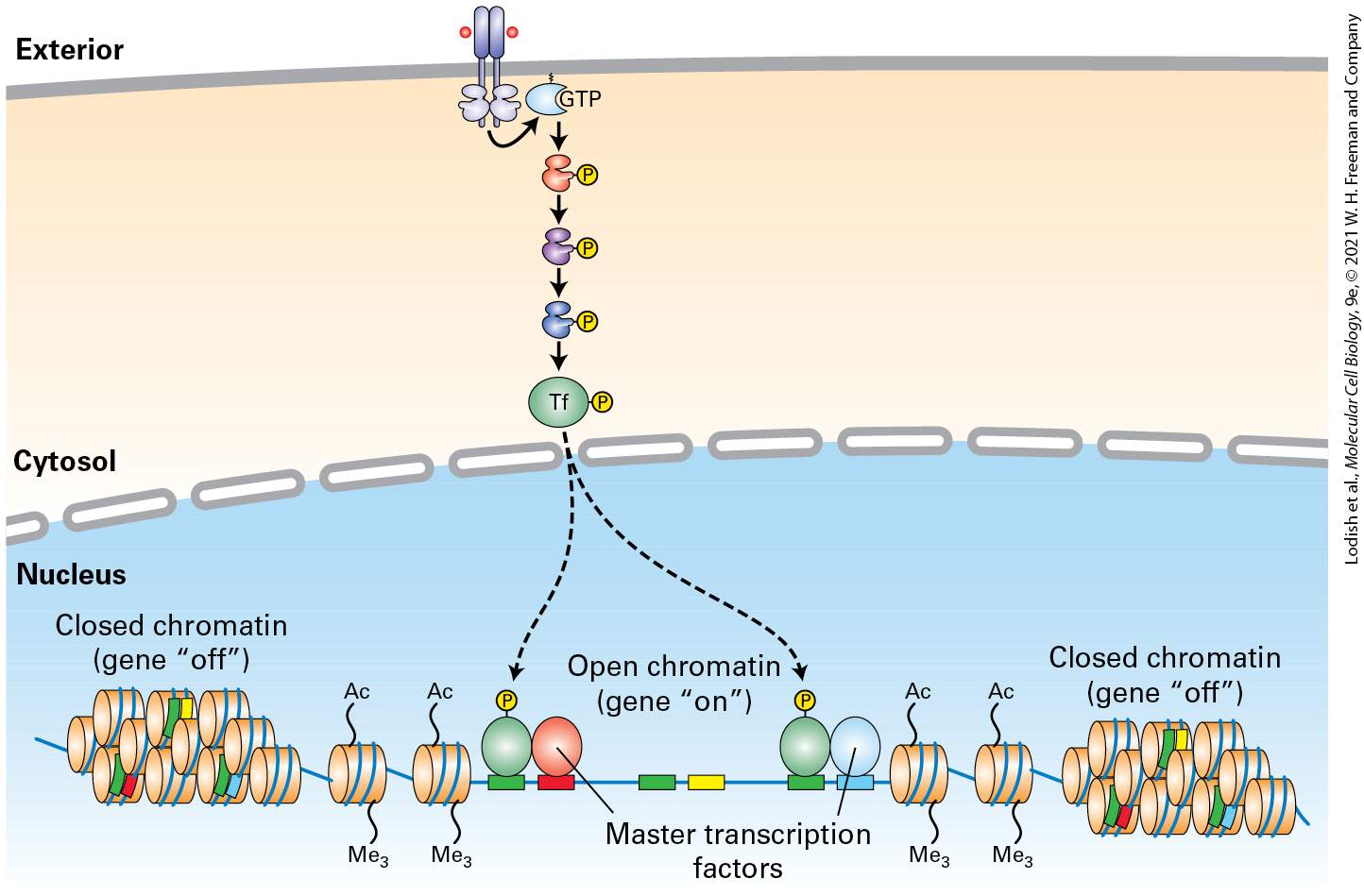
Not all of the signaling receptors described in this chapter lead to the activation of protein kinases or GTP-binding proteins in the earliest steps of their signal transduction pathways. For example, binding of a ligand to the extracellular domain of certain receptors, such as Notch, triggers proteolytic cleavage of the receptor itself, releasing the cytosolic domain. The released domain is a transcription factor that travels into the nucleus and interacts with other transcription factors to alter gene expression (Figure 16-1b, Pathway C, and Section 16-6). Finally, we discuss several different pathways in which binding of the ligand to its receptor ultimately leads to ubiquitination and degradation of one or more proteins in a multiprotein cytosolic complex, consequently releasing a transcription factor that is part of the complex and that then translocates into the nucleus and affects gene expression (Figure 16-1b, Pathway D). These include signaling pathways activated by the Wnt and Hedgehog signaling proteins, and others downstream of several types of receptors that all lead to activation of the NF-κB transcription factor. As with GPCRs, many receptors discussed in this chapter are expressed in multiple types of body cells, but activation of these receptors by the same hormone triggers induction (or repression) of very different sets of genes in different cell types. As we learned in Chapter 8, the epigenetic state of the cell is determined by its developmental history. Whether a transcription factor activated by a cell-surface receptor induces (or represses) a particular gene in a particular cell depends, first (Figure 162), on the epigenetic state of the gene: whether the gene is in an active “open” chromatin conformation, and therefore accessible to binding by the
transcription factor, or in a silenced “closed” state that is not accessible by the transcription factor. In other words, a given transcription factor can potentially bind to multiple gene regulatory sites in chromosomal DNA, but in any given cell type only a fraction of these sites will be accessible for binding.
FIGURE 16-2 Induction of a particular gene by a transcription factor depends on DNAbinding sites for the factor, on the gene’s epigenetic state, and on the presence of master transcription factors and other nuclear proteins. Any given activated transcription factor has multiple sites on the chromosomal DNA to which it can potentially bind (green), but in any given cell it will bind only to those sites that are in an “open” chromatin conformation and in which specific master transcription factors or other cell-specific proteins (blue and red, respectively) are bound to adjacent sites on the DNA; together these proteins activate (or repress) expression of the adjacent gene. Other potential transcription-factor binding sites are adjacent to binding sites for other master transcription factors (yellow) that are not expressed in this cell type, and thus the activated transcription factor will not bind to those sites.
Description The illustration shows an open section of chromatin in the nucleus. The cell membrane shows the start of a series of transcription factors going to the nuclear membrane. Several transcription factors bound to D N A at several sites. Activation of a cell surface receptor leads to the activation of a transcription factor via phosphorylation, which further leads to binding of the activated transcription factor to sites on the D N A next to the master transcription factor. Possible binding sites for the transcription factor that do not have an adjacent master transcription factor are not bound. Additionally, many cell types express one or more master transcription factors that determine the identity and developmental fate of the cell; a recent finding is that many transcription factors activated by cell-surface receptors bind to chromosomal DNA at regulatory sites — mainly enhancers — adjacent to these master factors, and together these transcription factors induce (or repress) cell-specific genes (see Figure 162). No cell-surface receptor, and no signal transduction pathway, works in isolation. Expression of many if not most genes in metazoans are regulated by multiple transcription factors, and these in turn are activated or repressed by different intracellular signaling pathways, each of which can be regulated by multiple extracellular signals. An emerging subfield of molecular cell biology — systems analysis — uses a combination of experimental and computational analyses to understand how cells of different types integrate these signals over time. For example, in Chapter 21 we consider how multiple hormones and multiple signal transduction
pathways in different types of cells regulate the body’s needs for the key metabolites glucose and fatty acids.
Binding of Ligand to the Extracellular Domain of an RTK Leads to Dimerization and Activation of Its Intrinsic Cytosolic Tyrosine Kinase
16.1 Growth Factors and Their Receptor Tyrosine Kinases The extracellular signaling molecules that activate receptor tyrosine kinases (RTKs) constitute a large class of soluble and membrane-bound protein hormones, including many that were initially identified because they stimulate the growth of specific types of cells (hence the name growth factor). Those RTK ligands, such as nerve growth factor (NGF), platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), and epidermal growth factor (EGF), trigger proliferation of cells that bear a cognate receptor. Others, such as insulin, regulate expression of multiple genes that control sugar and lipid metabolism in liver, muscle, and adipose (fat) cells. The large family of ephrins play critical roles in regulating the migration of many nerve cell axons to their target destinations (see
Chapter 23). Many RTKs and their ligands were first identified in studies of human cancers, in which mutant forms of growth-factor receptors stimulate cell proliferation even in the absence of the growth-factor ligand. The mutation locks the receptor conformation in a state such that the kinase is permanently active, even if the ligand is not present (constitutively active; see Chapter 25). Other RTKs were uncovered during analysis of developmental mutations that lead to blocks in differentiation of specific cell types in Caenorhabditis elegans, Drosophila, and mice.
Binding of Ligand to the Extracellular Domain of an RTK Leads to Dimerization and Activation of Its Intrinsic Cytosolic Tyrosine Kinase All RTK proteins have four essential components: an extracellular domain containing a ligand-binding site, a single hydrophobic transmembrane α helix, a cytosolic segment that includes a domain with protein tyrosine kinase activity, and a C-terminal segment that contains tyrosine residues that become phosphorylated by the receptor’s own kinase (Figure 16-3). Most RTKs are monomeric, and ligand binding to the extracellular domain induces two monomers to join together as a dimer.
FIGURE 16-3 General structure and activation of receptor tyrosine kinases (RTKs). The cytosolic domain of RTKs contains an intrinsic protein tyrosine kinase domain. In the absence of ligand (step 1 ), RTKs generally exist as monomers with poor kinase activity. Binding of two ligands to the extracellular domains of two RTKs forms or stabilizes an activated dimeric receptor. This brings together two poorly active kinases such that each one phosphorylates the other on a tyrosine residue in the activation loop (step 2 ).
Phosphorylation increases the activity of the kinase by causing the loop to move out of the kinase catalytic site, thus increasing the ability of ATP and/or the protein substrate to bind (see Chapter 3). The activated kinase then phosphorylates several tyrosine residues in the receptor’s cytosolic domain (step 3 ). The resulting phosphotyrosines function as docking sites for SH2 and other binding domains on downstream signal-transducing proteins, as summarized in Figure 16-1a. Description The illustration shows tyrosine kinase receptor monomers embedded in the plasma membrane. The ligand-binding regions are present on the cell exterior, held in place by transmembrane alpha helices. Step 1: In the cytosol, the activation loop of the receptor is present, and the protein tyrosine kinase is poorly active when no ligand is bound to the receptors in the exoplasm. Step 2: On binding, the two dimers are brought together, and the protein kinase in the cytosol is activated by activation of the loop tyrosines, resulting in phosphorylation of the additional tyrosine residues. Step 3: Phosphorylation of additional tyrosine residues results in the addition of six more phosphates in the cytosolic domain. RTK activation and signaling can be summarized as follows: in the resting, unstimulated (no ligand bound) state, the intrinsic kinase activity of an RTK is very low (Figure 16-3, step 1 ). Like most other kinases, the cytosolic kinase domain of an RTK contains a flexible domain termed the activation loop. In the resting state, the activation loop is not phosphorylated and assumes a conformation that blocks kinase activity. In some receptors (e.g., the insulin receptor), this loop prevents binding of ATP. In others (e.g., the FGF receptor), it prevents binding of substrate. Binding of ligand causes a conformational change that promotes the dimerization of the RTK. The extracellular domains of two RTK monomers join, which brings their transmembrane segments — and therefore their cytosolic domains — close together and results in
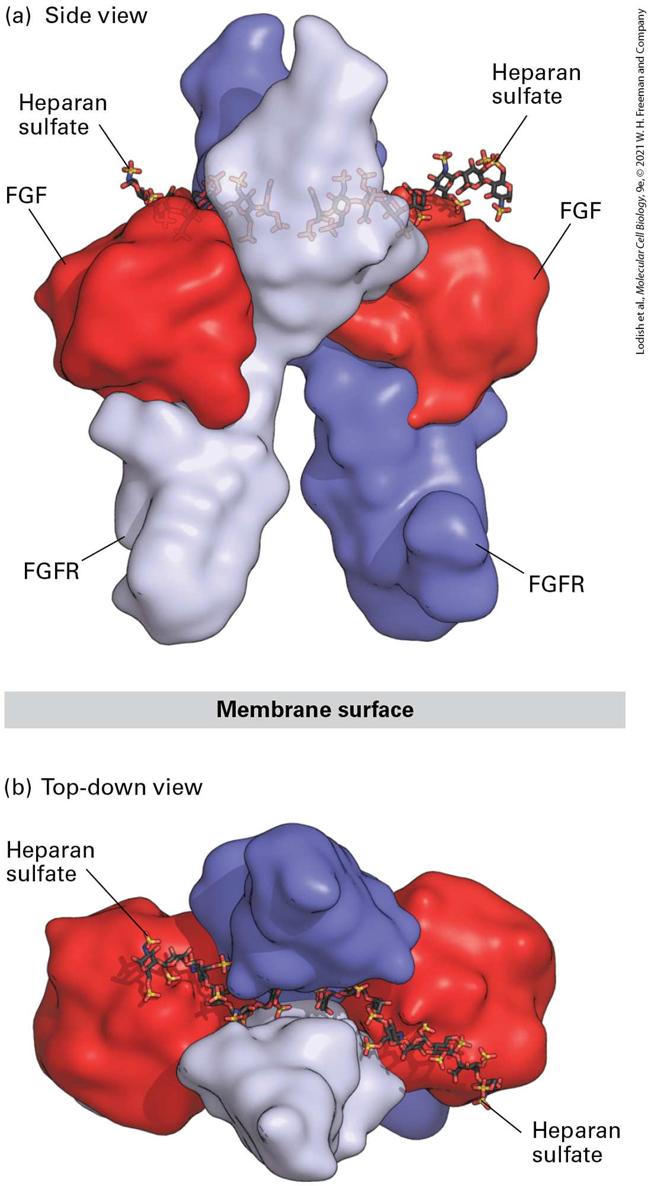
activation of the tyrosine kinases. For most, but not all, RTKs, the weakly active kinase in each subunit is thought to phosphorylate a key tyrosine residue in the activation loop of the other subunit (Figure 16-3, step 2 ). This phosphorylation leads to a conformational change in the activation loop that unblocks the kinase active site, reducing the for either ATP or the substrate to be phosphorylated and greatly enhances kinase activity (see Chapter 3). The strongly active kinase then phosphorylates additional tyrosine residues in the cytosolic domain of the receptor (Figure 16-3, step 3 ). These phosphotyrosine residues serve as binding sites for proteins that then activate several signal transduction pathways (see Figure 16-1a), each of which will be detailed in subsequent sections of this chapter. Although dimerization is a necessary step in the activation of all RTKs, functional dimers can be formed in multiple ways, as we shall see. For example, many receptors are dimerized in a manner similar to the fibroblast growth factor (FGF) receptor (Figure 16-4), in which each of two FGF ligands binds simultaneously to the extracellular domains of both subunits of the dimerized receptor, stabilizing the dimer. An additional feature is that FGF binding, and thus FGF receptor dimerization, is enhanced by the additional binding to both the ligand FGF and its receptor of the negatively charged polysaccharide heparan sulfate (see Figure 164), which is a component of some cell-surface proteins and of the extracellular matrix (see Chapter 20). The participation of the heparan sulfate is essential for efficient receptor activation; we see later (Section 16.5) that binding of other hormones to components of the extracellular matrix is also essential for their signaling functions.
FIGURE 16-4 Structure of the extracellular domains of the active dimeric fibroblast growth factor (FGF) receptor, stabilized by binding of two ligands and by heparan sulfate. Shown here are side and top-down views of the complex comprising the extracellular domains of two FGF receptor (FGFR) monomers (purple and violet), two bound FGF molecules (red), and two short heparan sulfate chains that are bound tightly to FGF. (a) In the side view, the upper domain of one receptor monomer (purple) is seen situated behind that of the other (violet); the plane of the plasma membrane is at the bottom. A small segment of the receptor’s extracellular domain connects to the membrane-spanning α-helical segment of each of the two receptor monomers (not shown) that protrude downward into the membrane. (b) In the top view, the heparan sulfate chains are seen threading between and making numerous contacts with the upper domains of both receptor monomers. These interactions enhance binding of the FGF ligand to the receptor and receptor dimerization. [Data from J. Schlessinger et al., 2000, Mol. Cell 6:743, PDB ID 1fq9.] Description The illustration labeled (a) titled side view shows a space-filling three-dimensional model of a dimeric fibroblast. The top right and left of the ribbon model has the ball and stick structures of heparan sulfate. The top sections and bottom sections of the ribbon model are labeled F G F and F G F R, respectively. The illustration labeled (b) titled top-down view shows two heparan sulfate molecules on the top of the membrane surface. Yet other RTKs, such as the insulin receptor, form disulfide-linked dimers even in the absence of hormone; binding of ligand to this type of predimerized but inactive RTK alters the conformation of the dimer in such a way that the receptor’s kinase becomes activated (Figure 16-5). In the absence of insulin, the structure of the dimeric receptor’s extracellular domain forces the weakly active cytosolic kinase domains to be far apart, unable to phosphorylate and thus activate each other. Upon insulin
binding, the receptor’s extracellular domain converts from an inverted Ushape into a T-shaped conformation that brings the transmembrane domains, and thus the kinase domains, together, presumably facilitating phosphorylation of the activation loop in the tyrosine kinase domain of one receptor by the kinase in the other.
FIGURE 16-5 Insulin-induced activation of the insulin receptor. (a) Schematic cartoon showing the extracellular domain (ECD), transmembrane domain (TMD), and tyrosine kinase domain (TKD) as well as a structural model of the full-length human insulin receptor in the absence of insulin. The receptor is produced as a single polypeptide chain that undergoes cleavage in the Golgi complex; the two resulting subunits, α and β, are linked together by a disulfide bond, as are the two α subunits in a dimer. (b) Electron microscopic views of single insulin receptors reconstituted in tiny lipid discs, both in the absence and presence of insulin. (c) Cartoon illustrating the ligand (green) binding–induced conformational change in the extracellular domain of the insulin receptor and its coupling to the transmembrane domains (TMDs) with concomitant activation of the tyrosine kinase domains by phosphorylation. In the absence of insulin, the extracellular domain adopts a symmetric inverted U-shaped conformation; upon insulin binding, the extracellular domain is converted into a T-shaped conformation that brings the transmembrane domains together, presumably facilitating phosphorylation of the activation loop of one kinase by the poorly active kinase of the other (trans-phosphorylation), leading to activation of both tyrosine kinase domains.
Homo- and Hetero-Oligomers of Epidermal Growth Factor Receptors Bind Members of the Epidermal Growth Factor Family
This last example highlights the fact that simply having two identical receptor monomers in close contact is not sufficient for receptor activation — the proper conformational changes must accompany receptor dimerization. Once an RTK converts into a functional state in a dimer, its associated tyrosine kinase becomes activated. Homo- and Hetero-Oligomers of Epidermal Growth Factor Receptors Bind Members of the Epidermal Growth Factor Family During evolution, the genes encoding some growth factors and their receptors underwent duplication and diverged to encode similar, but distinct, proteins with distinct functions. For example, in humans there are 22 FGF-related and 4 FGF receptor-related genes with many different functions. There are also multiple members of the epidermal growth factor (EGF) and EGF receptor families that have been studied intensively because of their involvement in many human diseases. Drugs targeting EGF receptors that are overproduced in tumors or that have undergone mutations that cause their kinase domains to be active even in the absence of a hormone signal are used to treat many cancers, as we learn in Chapter 25. The first known member of this family of hormones, epidermal growth factor, was originally identified by Stanley Cohen as a small protein that stimulated the early opening of eyelids in a baby mouse; it is now known
EGF Receptor Homodimers
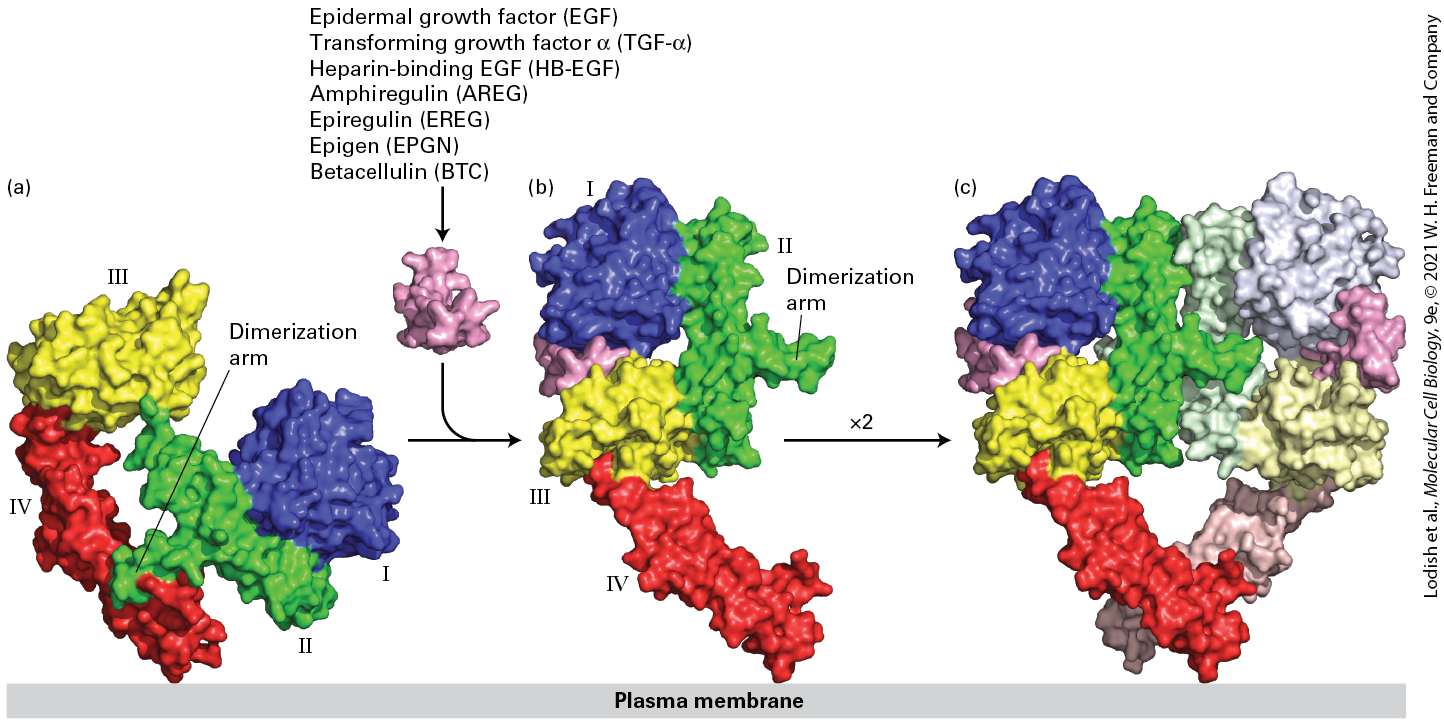
to stimulate the proliferation of many types of epithelial cells. For this research and the parallel discovery of nerve growth factor, Cohen and Rita Levi-Montalcini were awarded Nobel Prizes. Four related EFG receptors participate in signaling by the many members of the epidermal growth factor family of signaling molecules. In the mouse these receptors are termed Erb-B1, 2, 3, and 4. In humans these receptors are called HER (human epidermal growth factor receptor) 1, 2, 3, and 4, respectively. EGF Receptor Homodimers In the resting state, the majority of EGF receptor molecules are monomeric. We first consider the activation of the EGF receptor HER1 (Erb-B1). HER1 directly binds EGF as well as six other members of the EGF family; binding of any of these ligands leads to formation of a homodimer (homodimerization) of the HER1 extracellular domain (Figure 16-6). Binding of EGF binding triggers a dramatic conformational change in the extracellular domain of HER1, exposing a portion of the receptor that is inaccessible in the inactive monomer, termed the dimerization arm. The dimerization arms from two receptor monomers then bind tightly together, forming a receptor homodimer that is further stabilized by interactions between another extracellular domain (domain IV) in each of the two receptors. Several members of the EGF family — neuregulins 1, 2, 3, and 4 — bind to HER4 that, like HER1, can also form homodimers. As
detailed below, formation of a receptor dimer triggers activation of the kinase domain.
FIGURE 16-6 Ligand-induced dimerization of HER1, a human receptor for epidermal growth factor (EGF). (a) The extracellular region of all EGF receptors contains four domains: domains I (blue) and III (yellow) are closely related in sequence, as are domains II (green) and IV (red). In the absence of bound EGF, the receptor is mostly monomeric and the intracellular kinase is inactive. The extracellular region adopts a configuration in which the β-hairpin from domain II that forms the dimerization arm binds to domain IV of the same receptor molecule. (b) EGF, as well as the six other listed members of the EGF family, binds simultaneously to domains I and III; binding induces a major conformational change in the extracellular domain, exposing the dimerization arm of domain II. (c) Dimerization of two identical ligand-bound receptor monomers in the plane of the membrane occurs primarily through interactions between the dimerization arms of the two receptors. [Data from H. Ogiso et al., 2002, Cell 110:775, PDB ID 1ivo.] Description The illustration labeled (a) shows three-dimensional surface models of four domains. 1 (blue), 2 (green), 3 (yellow), and 4 (red). The domains 2 and 4 are connected and labeled dimerization arm. In the illustration labeled (b), the addition of Epidermal
EGF Receptor Heterodimers with HER2
growth factor (E G F), Transforming growth factor-alpha (TGF-alpha), Heparin-binding EGF (H B-E G F), Amphiregulin (A R E G), Epiregulin (E R E G), Epigen (E P G N), Betacellulin (B T C) induces the conformational change which affects the interaction between domains 2 and 4 and the monomer opens out. The illustration labeled (c) shows the formation of a dimer in the shape of the heart. EGF Receptor Heterodimers with
Remarkably, although one of the EGF receptors, HER2 (mouse Erb-B2), cannot bind any ligand, it participates in signaling by forming heterodimers with HER1, HER3, or HER4 when they are bound to their respective ligands. HER2 can form heterodimers with these other EGF receptors because it adopts an active conformation with its dimerization arm always accessible for binding to the dimerization arms of ligandbound receptors. Thus, by HER2 forming heterodimers, its kinase domain becomes activated and in so doing it facilitates signaling by all EGF family members that bind to one of the other three EGF receptors (Figure 16-7).
FIGURE 16-7 The HER family of receptors and their ligands. Humans and mice express a family of four receptor tyrosine kinases, denoted HER1, 2, 3, and 4 in humans and ErbB1, 2, 3, and 4 in mice and other animals. Only the extracellular domains of these receptors are depicted here. These receptors bind epidermal growth factor (EGF) and the other EGF family members: heparin-binding EGF (HB-EGF), transforming growth factor α (TGF-α),
amphiregulin (AREG), epiregulin (EREG), epigen (EPGN), betacellulin (BTC), and the four neuregulins (NRG1–4). Notice that Erb-B2 (HER2), which does not directly bind a ligand, exists in a conformation that is very similar to that of the activated Erb-B1 with a bound EGF. Erb-B2 can form a heterodimer with ligand-activated Erb-B1, 3, or 4; thus Erb-B2 facilitates signaling by all EGF family members. Erb-B3 (HER3) has a very poorly active kinase domain and can signal only when complexed with Erb-B2. [Erb-B1 data from K. M. Ferguson et al., 2003, Mol. Cell 11:507, PDB ID 1nql. Erb-B2 data from H.-S. Cho et al., 2003, Nature 421:756, PDB ID 1n8z. Erb-B3 data from H. S. Cho and D. J. Leahy, 2002, Science 297:1330, PDB ID 1m6b. Erb-B4 data from S. Bouyain et al., 2005, Proc. Nat’l. Acad. Sci. USA 102:15024, PDB ID 1ahx. EFG and Erb-B1 data from H. Ogiso et al., 2002, Cell 110:775, PDB ID 1ivo.] Description Molecules that bind E R B-B 1 include E G F, T G F-alpha, H B-E G F, amphiregulin (A R E G), Epiregulin (E R E G), Epigen (E P G N), and betacellulin (B T C). Molecules that bind E R B-B 3 include N R G 1 and N R G 2. Molecules that bind E R B-B 4 are H B-E G F, and N R G 1 to 4. E G F binds to E R B- B 1, which turn binds to a dimer of E R B - B 2. HER3 is an EGF receptor that can bind ligands but lacks a functional kinase domain. HER3 can still participate in signaling; after binding a ligand, it heterodimerizes with HER2 and activates the HER2 kinase. Later we describe how the active kinases in the receptor cytoplasmic domains mediate downstream signal transduction pathways. An increase in the numbers of HER2 molecules on the cell surface makes a cell more sensitive to signaling by many EGF family members, because the greater number of HER2 receptors enhances the rate and extent of formation of signaling heterodimers following binding of a
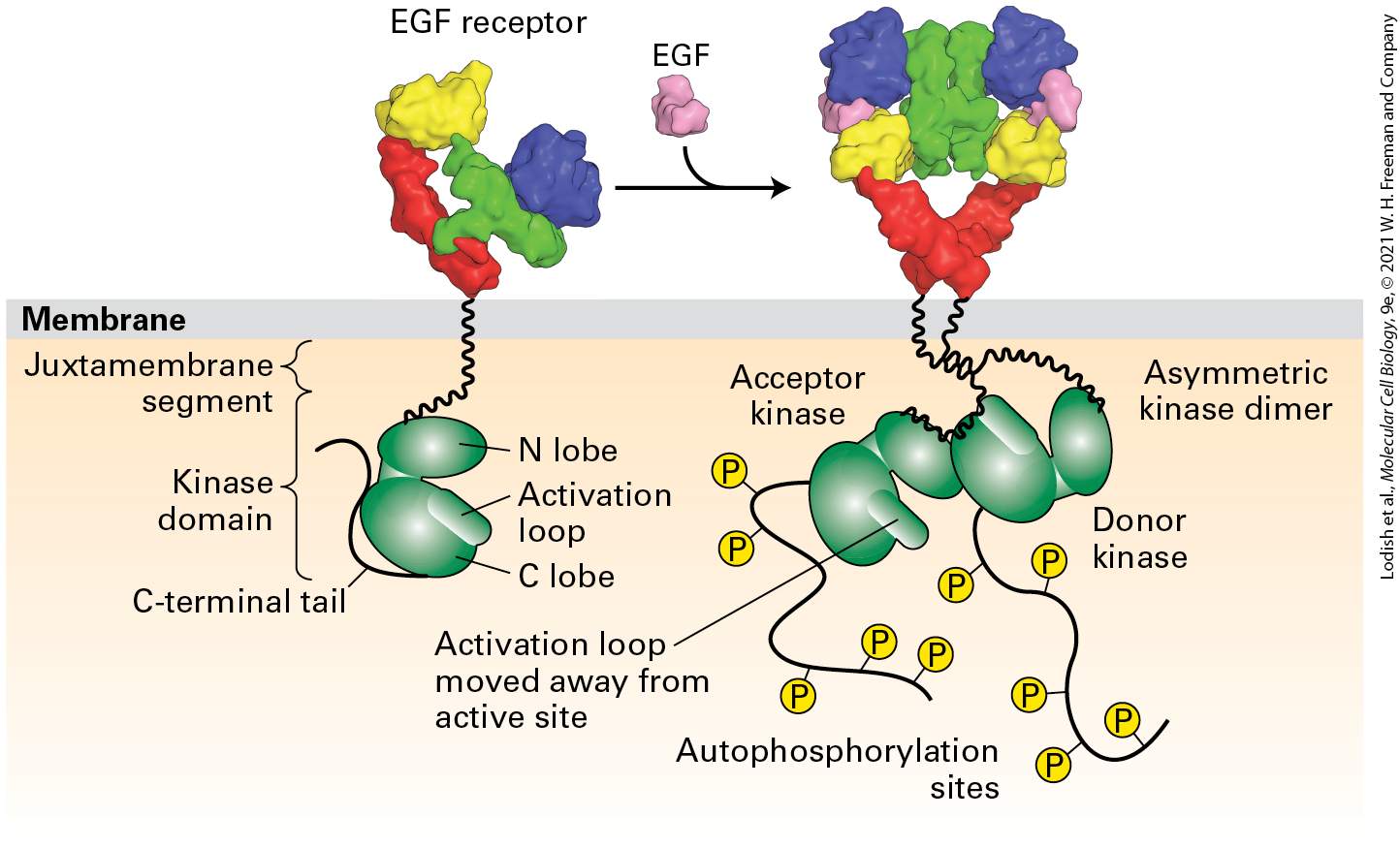
Ligand Binding to the EGF Receptor and Receptor Dimerization Results in the Formation of an Active Asymmetric Kinase Domain Dimer
member of the EGF family to HER1, HER3, or HER4. As we learn in
Chapter 25, in about 25 percent of breast cancers the tumor cells have multiple copies of the HER2 gene and thus elevated levels of HER2. Cells that overproduce HER2 are hypersensitive to ambient levels of all members of the EGF family of hormones, levels that normally would not lead to activation of many receptors. As a consequence, at even low levels EGF family members can stimulate growth of these tumor cells inappropriately. Monoclonal antibodies that bind HER2 and thereby block signaling by EGF have proved useful in treatment of breast and other tumors that overexpress HER2. Ligand Binding to the EGF Receptor and Receptor Dimerization Results in the Formation of an Active Asymmetric Kinase Domain Dimer In the case of most receptor tyrosine kinases, the kinase domain is activated by phosphorylation of a tyrosine in the activation loop following formation of a receptor dimer. In contrast, loop phosphorylation is not necessary for initial activation of the kinase domains of the EGF receptors, though it is important for full activation. The mechanism of activation was uncovered through structural studies of the receptor cytosolic domain in both the active and inactive states.
The kinase domains are separated from the transmembrane segment by a so-called juxtamembrane segment (Figure 16-8). As we learned in Chapters 3 and 15, kinase domains typically contain two regions termed the N and C lobes. In the inactive, monomeric state of the EGF receptor, the activation loop is situated in the active site of the kinase near the interface of the N and C lobes, blocking the site’s activity; in this way the kinase is maintained in the inactive or “off” state (Figure 16-8, left). Receptor dimerization brings the kinase domains of the two subunits close together, so that the C lobe of one kinase domain (called the donor) binds to the N lobe of the other kinase domain (the acceptor), producing what is called an asymmetric kinase dimer (Figure 16-8, right). The binding of the two lobes changes the conformation of the N lobe of the acceptor kinase, displacing the activation loop and activating the kinase activity of the acceptor domain, thereby initiating intracellular signal transduction.
FIGURE 16-8 Activation of the EGF receptor by EGF results in the formation of an active asymmetric kinase domain dimer. In the inactive, monomeric state, the activation loop is positioned in the kinase active site, blocking substrate binding and thus inhibiting kinase function. Receptor dimerization generates an asymmetric kinase dimer such that the C lobe of the donor kinase binds to the N lobe of the acceptor kinase in the opposite receptor; the dimer is stabilized by interactions between the juxtamembrane segments of the two receptors. These interactions cause a conformational change that pushes out the activation loop from the kinase site of the acceptor kinase, activating its kinase activity. The active acceptor kinase then phosphorylates tyrosine residues in the C-terminal segments of both receptor cytosolic domains. [EGF receptor data from H. Ogiso et al., 2002, Cell 110:775, PDB ID 1ivo; and K. M. Ferguson et al., 2003, Mol. Cell. 11:507, PDB ID 1nql. Asymmetric kinase dimer data from E. Kovacs et al., 2015, Annu. Rev. Biochem. 84:739.] Description The illustration shows the E G F receptor on the exoplasmic side of the plasma membrane. A side arrow points toward the right and shows the addition of E G F, which changes the shape of the E G F receptor to be almost heart-shaped. Through the membrane is the schematic model, first of the E G F receptor, is with two parts of green structures. Labels top to bottom on the left side of it are Juxtamembrane segment, Kinase domain, and C-terminal tail. On the right side of this structure are labeled N lobe, Activation loop, and C lobe. The schematic under the heart-shaped receptor shows the acceptor kinase (green structure) added to an asymmetric kinase dimer. Phosphates are added to autophosphorylation sites. The activation loop is labeled and noted that it is moved away from the active site. The lobe of the green structure on the right is labeled donor kinase. Thus evolution has produced many variations on the theme of ligandinduced RTK signaling: RTKs are activated by dimerization, but different receptors use different mechanisms to accomplish this. Similarly, kinases become activated by movement of the activation loop away from the
Signal Transduction After Activation of RTKs: Phosphotyrosine Residues on the Receptor Are Binding Surfaces for Multiple Proteins with SH2 Domains
kinase catalytic site, but, again, different receptors use different mechanisms to accomplish this. Signal Transduction After Activation of RTKs: Phosphotyrosine Residues on the Receptor Are Binding Surfaces for Multiple Proteins with SH2 Domains Once the RTK kinases become activated, they phosphorylate several tyrosine residues on their cytosolic domains (see Figures 16-3 and 16-8). Each of these phosphotyrosine residues, together with a few adjacent residues, serve as binding sites for signal-transducing proteins that have conserved phosphotyrosine-binding domains (see Figure 16-1a). The recruitment of each signal-transducing protein to the activated receptor sets off a specific subsequent signal transduction pathway. There are several classes of phosphotyrosine-binding domains; one is called the SH2 domain. An SH2 domain is able to bind to a short target sequence of polypeptide that contains a phosphotyrosine. The SH2 domains in different signal-transducing proteins have very similar threedimensional structures, but each binds to a distinct, short target sequence of amino acids adjacent to a specific phosphotyrosine residue (often abbreviated “pY”). The unique amino acid sequence of each SH2 domain determines the specific target sequence to which it binds (Figure 16-9).
Each SH2 domain contains a site for binding the phosphotyrosine and sites for binding the side chains of the adjacent residues in that SH2’s target sequence.
FIGURE 16-9 Surface model of an SH2 domain bound to a phosphotyrosine-containing peptide. The peptide bound by this SH2 domain, part of the Src tyrosine kinase, is shown in stick form (blue backbone with red oxygen atoms). This SH2 domain binds strongly to short target peptides containing a critical four-residue core sequence: phosphotyrosine ( and )–glutamic acid –glutamic acid –isoleucine . Binding resembles the insertion of a two-pronged plug — the phosphotyrosine and isoleucine side chains of the peptide — into a two-pronged socket in the SH2 domain. The two glutamate residues are bound to sites on the surface of the SH2 domain between the two sockets. Other SH2 domains bind phosphotyrosine residues surrounded by other sequences of amino acids. [Data from G. Waksman et al., 1993, Cell 72:779, PDB ID 1sps.]
Receptor-Mediated Endocytosis and Lysosomal Degradation Squelch Signaling from RTKs
As an example, consider the protein in which SH2 domains were first identified, the tyrosine kinase called Src (Src is an acronym for a sarcoma tumor). A mutant form of the src gene, which inappropriately activates a downstream signal transduction pathway, was originally found incorporated in the Rous sarcoma virus that in chickens causes sarcomas (a muscle tumor, as Chapter 25 details). The SH2 domain of the Src tyrosine kinase binds strongly to any peptide containing the four-residue sequence: phosphotyrosine–glutamic acid–glutamic acid–isoleucine (single letter code: pY-E-E-I, see Figure 16-9). These four amino acids make intimate contact with the peptide-binding site in the Src SH2 domain. Binding resembles the insertion of a two-pronged plug — the phosphotyrosine and isoleucine side chains of the peptide — into a twopronged socket in the SH2 domain. The two glutamic acids fit snugly onto the surface of the SH2 domain between the phosphotyrosine binding socket and the hydrophobic socket that binds the isoleucine residue. The specificity of SH2 domains for their target sequence plays an important role in determining which SH2-domain-containing signal-transducing proteins bind to which receptors and thus which signal transduction pathways are activated. Receptor-Mediated Endocytosis and Lysosomal Degradation Squelch Signaling from RTKs In Chapter 15 we emphasized that, to prevent overstimulation, cells must be able to suppress or even turn off their signal transduction pathways. In
addition, once the cell has adequately responded to the signal, or when the signal has been removed, it is important to prevent any further cellular response. Suppression of signaling from RTKs is common, and different mechanisms have evolved to accomplish this task. For example, phosphotyrosine phosphatases continuously hydrolyze the bonds linking phosphates to tyrosine residues on the receptor, reducing the ability of activated RTKs to bind proteins containing phosphotyrosinebinding domains and thus diminishing the activation of signal transduction pathways. A second mechanism — receptor-mediated endocytosis followed by receptor degradation in lysosomes — is another common method for dampening RTK signal transduction. Treatment of cells with ligand often reduces the number of available cellsurface receptors, so that the cells have a less robust response to continuous exposure to a given concentration of ligand than they did before ligand addition. This desensitization response helps prevent inappropriately prolonged receptor activity. In the absence of EGF, for instance, cell-surface HER1 receptors are relatively long-lived, with an average half-life of 10–15 hours. Unbound receptors are internalized via clathrin-coated pits into endosomes at a relatively slow rate, on average once every 30 minutes, and are often recycled rapidly to the plasma membrane so that in the absence of ligands there is little reduction in the total number of surface receptors. Following binding of an EGF ligand, the rate of endocytosis of HER1 is increased about tenfold, and only a fraction of the internalized receptors return to the plasma membrane; the rest are degraded in lysosomes. Each time a HER1–EGF complex is internalized
through endocytosis (see Figure 14-29), the receptor has about a 20–80 percent chance of being degraded, depending on the cell type. In epithelial cells exposed to high levels of EGF, for example, almost all cell-surface EGF receptor molecules are internalized and degraded, markedly reducing the cell’s sensitivity to EGF. In this way, prolonged treatment with EGF desensitizes the cell to that hormone. If the EGF is removed, newly synthesized receptors will eventually replenish the cell surface and allow the cell to respond to a second treatment of EGF. Several processes influence whether surface receptors internalized in endosomes are recycled to the plasma membrane or transported to lysosomes for degradation. One is covalent modification by the small protein ubiquitin (see Chapter 3). The enzyme c-Cbl adds a single ubiquitin to a given lysine of a protein, a process called monoubiquitinylation. There is a strong correlation between the extent of monoubiquitinylation of the HER1 cytosolic domain and HER1 degradation; such monoubiquitinylation also occurs to other ligandactivated RTKs. The enzyme c-Cbl, which is an E3 ubiquitin ligase (see
Figure 3-32), contains a domain that binds directly to phosphorylated EGF receptors, and a RING finger domain, which recruits ubiquitin-conjugating enzymes and mediates transfer of ubiquitin to the receptor. The ubiquitin functions as a tag on the receptor that stimulates its incorporation from endosomes into multivesicular bodies (see Figure 14-32) that are ultimately engulfed by and degraded in lysosomes. A role for c-Cbl in EGF receptor trafficking emerged from genetic studies in C. elegans, which established that c-Cbl negatively regulates the function of the nematode EGF receptor (Let-23), probably by inducing its degradation.
Similarly, knockout mice lacking c-Cbl show hyperproliferation of mammary gland epithelia, consistent with a role of c-Cbl as a negative regulator of EGF signaling. Interestingly, the other EGF family receptors — HER2, HER3, and HER4 — do not undergo internalization after exposure to ligand, an observation that emphasizes how each receptor evolved to be regulated in its own appropriate manner. KEY CONCEPTS OF SECTION 16.1 Growth Factors and Their Receptor Tyrosine Kinases Receptor tyrosine kinases, which bind to peptides and signaling proteins such as growth factors and insulin, may exist as preformed dimers or dimerize when ligands bind. Ligand binding triggers conformational changes leading to formation of functional dimeric receptors, a necessary step in activating the receptor’s kinase domain. Different receptors accomplish this function in different ways (see Figures 16-3 to 16-7). Activation of many RTKs leads to phosphorylation of the activation loop in the protein tyrosine kinase that is an intrinsic part of the cytoplasmic domain, enhancing its catalytic activity (see Figure 16-3). The activated kinase then phosphorylates tyrosine residues in the receptor’s cytosolic domain and in other protein substrates. Humans express many RTKs, four of which (HER1–HER4) define the family of epidermal growth factor receptors that mediates signaling from the many members of the epidermal growth factor family of signaling molecules (see Figure 16-7). One of these receptors, HER2, does not bind ligand; it forms active heterodimers with ligandbound monomers of the other three HER proteins. Overexpression of HER2 is implicated in many breast cancers. In RTKs, short amino acid sequences containing a phosphotyrosine residue are bound by signal-transducing proteins with conserved SH2 domains. The sequence of amino acids surrounding the phosphorylated tyrosine determines which SH2 domain will bind to it (see Figure 16-9). Such protein-protein interactions are important in determining specificity in many signaling pathways (see Figure 16-3).
Endocytosis of receptor-ligand complexes and their degradation in lysosomes is a principal way of reducing the number of receptor tyrosine kinases on the cell surface and thus decreasing the sensitivity of cells to many peptide and protein hormones. Other receptors, such as cytokine receptors, are also regulated by endocytosis and degradation.
16.2 The Ras/MAP Kinase Signal Transduction Pathway
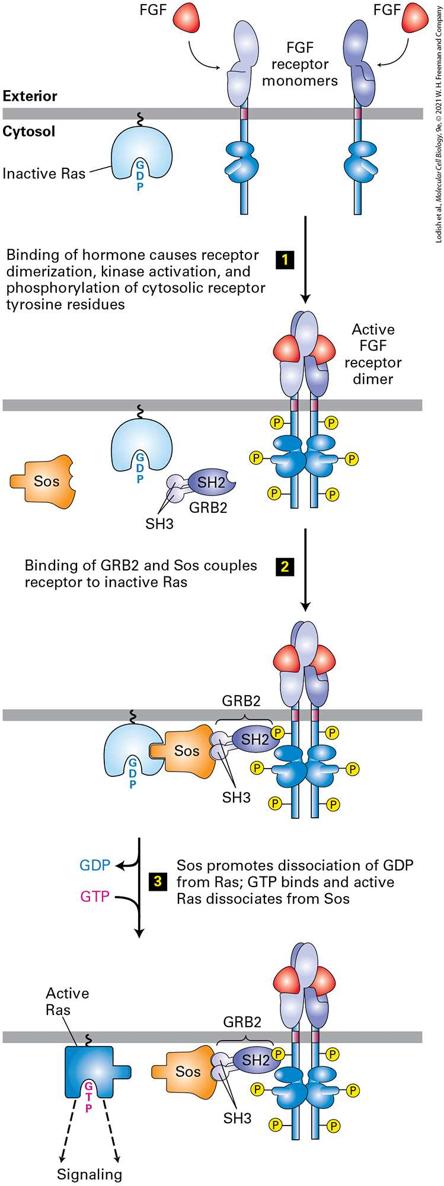
16.2 The Ras/MAP Kinase Signal Transduction Pathway Signal transduction by RTKs begins with the binding of a protein with an SH2 domain to a phosphorylated tyrosine of the receptor’s cytosolic domain. After one or two additional steps that we will see shortly, virtually all receptor tyrosine kinases activate the Ras/MAP kinase signal transduction pathway (Figure 16-10; also see Figure 16-1a, Pathway A). This pathway is also activated by most cytokine receptors. It is conserved in evolution, as versions are found in vertebrates, invertebrates, and even in yeasts, and it plays a key role in many developmental pathways and regulates the growth and differentiation of many types of cells.
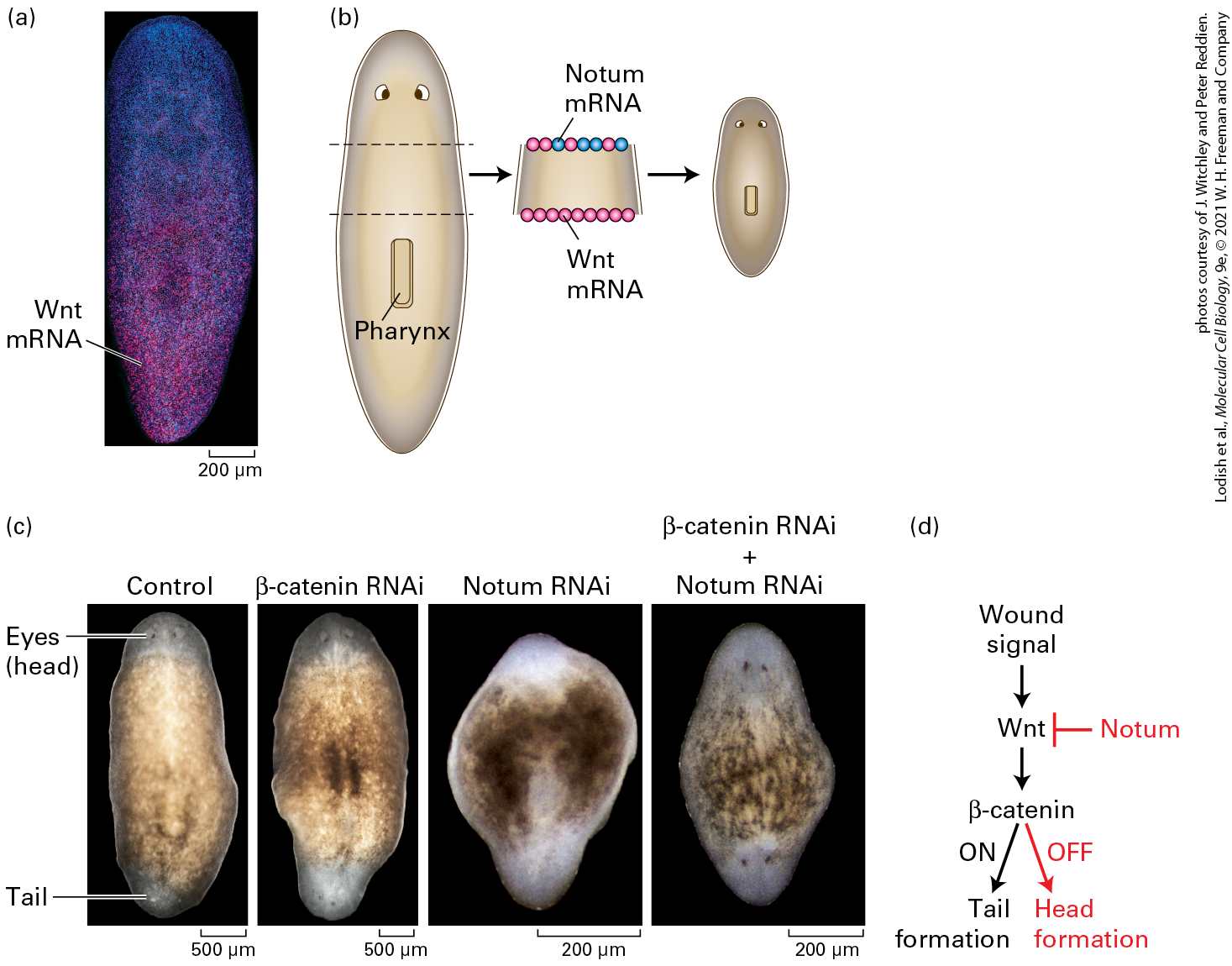
FIGURE 16-10 Activation of Ras following ligand binding to receptor tyrosine kinases (RTKs) or cytokine receptors. The receptor for fibroblast growth factor (FGF) exemplifies the activation of this signaling pathway. The SH2 domain of the cytosolic adapter protein GRB2 binds to a specific phosphotyrosine on an activated, ligand-bound receptor, and its SH3 domains bind to a cytosolic Sos protein, bringing it near the cytosolic surface of the plasma membrane and close to its substrate, the inactive Ras·GDP. The guanine nucleotide exchange factor (GEF) activity of Sos then promotes formation of active Ras·GTP from Ras·GDP. Note that Ras is tethered to the cytosolic surface of the plasma membrane by a hydrophobic farnesyl anchor (see Figure 10-19). See J. Schlessinger, 2000, Cell 103:211; and M. A. Simon, 2000, Cell 103:13. Description The illustration shows the following sequence. Step 1. Two F G F receptor molecules are on the membrane. On the cytosolic side, inactive R A S is bound. On binding of F G F hormone, the receptor dimerizes, kinase activation occurs, and the cytosolic receptor tyrosine residues are phosphorylated. Step 2. G R B 2 (consisting of S H 2 and S H 3) and S O S bind to inactive R A S. Step 3. S O S promotes dissociation of G D P from R A S, G T P binds, and the active form of R A S dissociates from S O S. The active R A S then results in signaling. The monomeric Ras protein belongs to the GTPase superfamily of intracellular switch proteins (see Figure 15-7). Activated Ras promotes the formation of a signal transduction complex, containing three sequentially acting protein kinases, at the cytosolic surface of the plasma membrane. This kinase cascade culminates in activation of certain members of the MAP kinase family, which can translocate into the nucleus and phosphorylate many different proteins. Among the target proteins for MAP kinase are transcription factors that regulate the expression of proteins with important roles in the cell cycle and in cell differentiation. Importantly, different hormones and their cell-surface receptors often
Ras, a GTPase Switch Protein, Operates Downstream of Most RTKs and Cytokine Receptors
differ in the strength or duration of the activation of a MAP kinase and thus in their effects on the cell. An activating mutation in an RTK, Ras, or a protein in the MAP kinase pathway that induces inappropriate cell division is found in almost all types of human tumors. Thus the RTK/Ras/MAP kinase signal transduction pathway has been studied extensively, and a great deal is known about its component proteins and their regulation (see Chapter 25). We begin our discussion of the Ras/MAP kinase signal transduction pathway by reviewing how Ras cycles between the active GTP-bound and inactive GDP-bound states. We then describe how activated Ras passes a signal to the MAP kinase. Finally, we examine recent studies indicating that both yeasts and cells of higher eukaryotes contain multiple MAP kinase pathways, and we consider the ways in which cells keep different MAP kinase pathways separate from one another through the use of scaffold proteins. Ras, a GTPase Switch Protein, Operates Downstream of Most RTKs and Cytokine Receptors Like the other G proteins, including the subunits in trimeric G proteins that were discussed in Chapter 15, monomeric Ras proteins are G protein switches that alternate between an inactive “off” state with a bound GDP and a receptor-activated “on” state with a bound GTP (see Figure 15-7). Trimeric G proteins are activated upon binding directly to a ligand-bound
GPCR that acts as a guanine nucleotide exchange factor (GEF) that triggers the release of the bound GDP. GTP then binds spontaneously to the nucleotide-free G protein. Activated RTKs and cytokine receptors are not GEFs; rather, the activated RTK or cytokine receptor recruits several adaptor proteins to the cytosolic surface of the plasma membrane, and one of these acts as the GEF that activates Ras. Ras (∼170 amino acids) is smaller than proteins (∼300 amino acids), but the GTP-binding domains of the two protein types have similar structures (see Figure 15-7 to review the structure of Ras). As with proteins, hydrolysis of the bound GTP to GDP deactivates Ras. proteins contain a GTPase-activating protein (GAP) domain that increases the intrinsic rate of GTP hydrolysis, but this domain is not present in Ras. As a result, Ras has an intrinsically slower rate of GTP hydrolysis. Thus the average lifetime of a GTP bound to Ras is about 1 minute, much longer than the average lifetime of a complex. Because of its low intrinsic GTPase activity, Ras⋅GTP requires the assistance of another protein, a GTPase-activating protein (GAP), to become deactivated. Binding of a GAP to Ras⋅GTP accelerates the intrinsic GTPase activity of Ras by more than a hundredfold; the actual hydrolysis of GTP to GDP and is catalyzed by amino acids from both Ras and the GAP. The GAP for RAS (called a RAS-GAP) works by inserting one of its arginine side chains into the Ras active site, thus stabilizing an intermediate in the hydrolysis reaction.
Receptor Tyrosine Kinases Are Linked to Ras by Adapter Proteins
As noted, mammalian Ras proteins have been studied in great detail because mutant Ras proteins are associated with many types of human cancer. These mutant proteins, which bind but cannot hydrolyze GTP, are permanently in the “on” state and contribute to the cellular transformation that leads to cancer (see Chapter 25). Determination of the threedimensional structure of the Ras-GAP complex and tests of mutant forms of Ras explained the puzzling observation that most oncogenic, constitutively active Ras proteins contain a mutation at position 12. Replacement of the normal glycine 12 with any other amino acid (except proline) blocks the functional binding of GAP proteins, vastly decreases the rate of GTP hydrolysis, and in essence locks Ras in the active GTP-bound state. The first indication that Ras functions downstream from RTKs in a common signaling pathway came from experiments on cultured fibroblast cells. These cells were induced to proliferate by treatment with a mixture of two protein hormones that activate RTKs: PDGF and EGF. Microinjection of anti-Ras antibodies into these cells blocked cell proliferation. Conversely, injection of , a constitutively active mutant Ras protein, caused the cells to proliferate in the absence of the growth factors. These findings are consistent with studies using the pull-down assay method detailed in Figure 15-11 in which addition of FGF to fibroblasts led to a rapid increase in the proportion of Ras present in the GTP-bound active form.
Receptor Tyrosine Kinases Are Linked to Ras by Adapter Proteins In order for activated RTKs and cytokine receptors to activate Ras, two cytosolic proteins — GRB2 and Sos — must first be recruited to provide a link between the receptor and Ras (see Figure 16-10). GRB2 is an adapter protein, meaning that it has no enzyme activity and serves as a link, or scaffold, between two other proteins — in this case, between the activated receptor and Sos. Sos is a GEF that catalyzes conversion of inactive GDPbound Ras to the active GTP-bound form. (The name Sos is an acronym for Son of Sevenless, a gene defined by analysis of Drosophila mutants with defects in eye development, which is critically dependent on a Rasmediated signaling pathway.) GRB2 is able to serve as an adapter protein because it has an SH2 domain, which binds to specific phosphotyrosine residues in the cytosolic domains of many activated RTKs. In addition to its SH2 domain, the GRB2 adapter protein contains two SH3 domains, which bind to Sos (see Figure 16-10). Like phosphotyrosine-binding SH2 domains, SH3 domains are present in a large number of proteins involved in intracellular signaling. Although the three-dimensional structures of various SH3 domains are similar, their amino acid sequences differ. The SH3 domains in GRB2 selectively bind to proline-rich sequences in Sos; different SH3 domains in other proteins bind to proline-rich target sequences distinct from those in Sos. Proline residues play two roles in the interaction between an SH3 domain in an adapter protein (e.g., GRB2) and a proline-rich sequence in its
corresponding target protein (e.g., Sos). First, the proline-rich sequence assumes an extended conformation that permits extensive contacts with the SH3 domain, thereby facilitating interactions. Second, a subset of the prolines fit into binding pockets on the surface of the SH3 domain (Figure 16-11). Several nonproline residues also interact with the SH3 domain and are responsible for determining which SH3 domain binds to which target protein (binding specificity). Hence the binding of proteins to SH3 and to SH2 domains follows a similar strategy: certain residues provide the key structural motif necessary for binding (phosphotyrosine for binding to SH2 domains, prolines for binding to SH3 domains), and neighboring residues confer specificity to the binding.
Binding of Sos to Inactive Ras Causes a Conformational Change That Triggers an Exchange of GTP for GDP
FIGURE 16-11 Surface model of an SH3 domain bound to a target peptide. The short, proline-rich target peptide is shown as a stick model and the SH3 domain as a space-filling model. In this target peptide, two prolines (Pro4 and Pro7, dark blue) fit into binding pockets on the surface of the SH3 domain. Interactions involving an arginine (Arg1, red), two other prolines (gray), and other residues in the target peptide (green) determine the specificity of binding of the target peptide to an SH3 domain. [Data from S. Feng et al., 1995, Proc. Nat’l. Acad. Sci. USA 92:12408, PDB ID 1qwf.]
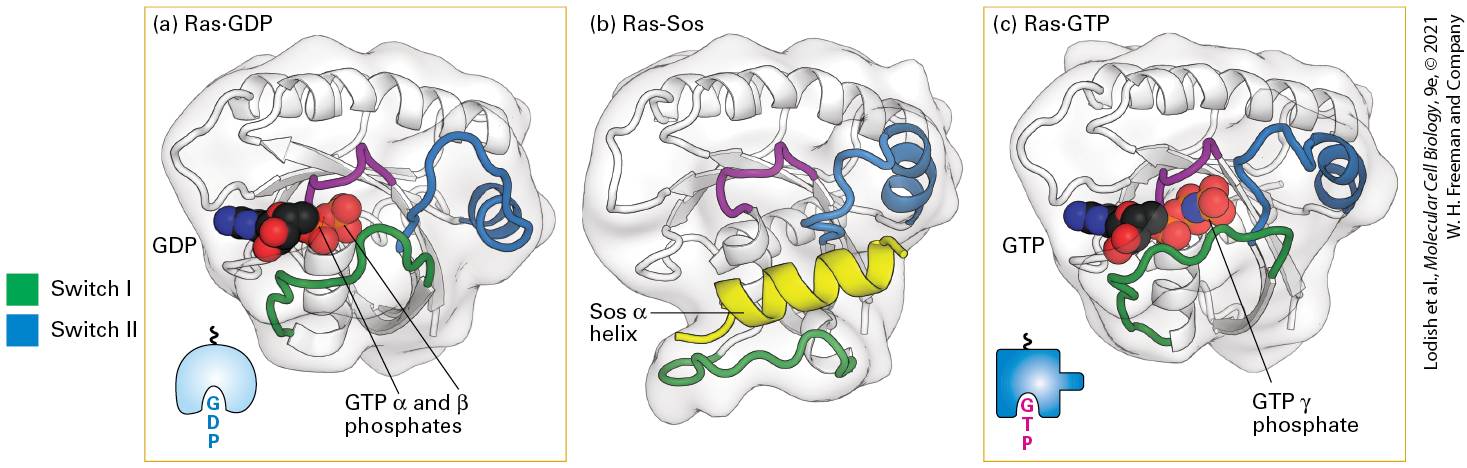
Binding of Sos to Inactive Ras Causes a Conformational Change That Triggers an Exchange of GTP for GDP Following activation and autophosphorylation of an RTK (e.g., the FGF receptor) and binding of GRB2, Sos binds to GRB2, forming a complex on the cytosolic face of the plasma membrane (see Figure 16-10). The formation of this complex depends on the ability of GRB2 to bind simultaneously to the activated receptor and to Sos. Thus on receptor activation, Sos relocates from the cytosol to the membrane, bringing Sos close to its substrate, Ras⋅GDP, which is already bound to the cytosolic surface of the plasma membrane by means of a covalently attached lipid. Binding of Sos to Ras⋅GDP activates Ras⋅GDP by releasing its GDP so that GTP can replace it. The binding of Sos leads to conformational changes in the switch I and switch II segments of Ras, thereby permitting GDP to diffuse away (Figure 16-12). In other words, Sos functions as a GEF for Ras. Binding of GTP to Ras, driven by the high GTP concentration in the cytosol relative to that of GDP, induces a specific conformation of switch I and switch II that allows Ras⋅GTP to activate Raf, the next protein in the Ras/MAP kinase pathway.
FIGURE 16-12 Structures of Ras bound to GDP, Sos protein, and GTP. (a) As with other G proteins bound to GDP, in Ras⋅GDP, the switch I (green) and switch II (blue) segments do not directly interact with GDP. (b) One α helix (yellow) in Sos binds to both switch segments of Ras⋅GDP, leading to a massive conformational change in Ras. In effect, Sos pries Ras open by displacing the switch I region, thereby allowing GDP to diffuse out. (c) GTP is thought to bind to the Ras-Sos complex first through its base (guanine); subsequent binding of the GTP phosphates completes the interaction. The resulting conformational change in the switch I and switch II segments of Ras, allowing both to bind to the GTP γ phosphate, displaces Sos and promotes interaction of Ras⋅GTP with its effectors (discussed later). The P loop (purple) is a sequence motif found in many ATP- and GTP-binding proteins that binds the β phosphate of the nucleotide. See Figure 15-7 for another depiction of Ras⋅GDP and Ras⋅GTP. [Part (a) Data from M. V. Milburn et al., 1990, Science 247:939, PDB ID 4q21. Part (b) Data from J. Sejbal et al., 1996, J. Med. Chem. 39:1281, PDB ID 1bdk. Part (c) Data from M. E. Pacold et al., 2000, Cell 103:931, PDB ID 1he8.] Description The illustration labeled (a) shows the ribbon model of titled R a s - G D P. Highlighted areas are G T P alpha and beta phosphates. The G D P is highlighted to the left-center of the model. The illustration labeled (b) shows the ribbon model of R a s - S O S. In this model, the blue ribbon that goes through the top center is labeled switch 2, and a green ribbon at the bottom is labeled Switch 1. Between these ribbons is a yellow ribbon labeled S O S alpha helix. The illustration labeled (c) shows the ribbon model of R a s - G T P. The Switch 1 green ribbon has moved up to the G T P area at the leftcenter. G T P gamma phosphate is highlighted. The Switch 2 ribbon is beside the G T P complex.
Signals Pass from Activated Ras to a Cascade of Protein Kinases Ending with MAP Kinase
Signals Pass from Activated Ras to a Cascade of Protein Kinases Ending with MAP Kinase Biochemical and genetic studies in yeast, C. elegans, Drosophila, and mammals have revealed that downstream of Ras is a highly conserved cascade of three protein kinases, culminating in MAP kinase. Although activation of the kinase cascade does not yield the same biological results in all cells, all Ras/MAP kinase pathways have a common set of three sequentially acting kinases, as outlined in Figure 16-13: kinase.
FIGURE 16-13 Ras/MAP kinase pathway. In unstimulated cells, most Ras is tethered to the cytosolic surface of the plasma membrane in the inactive form with bound GDP. Binding of a ligand to its RTK or cytokine receptor leads to formation of the active Ras⋅GTP complex (step 1 ; see also Figure 16-12). Activated Ras triggers the activation of the Raf kinase, activating the kinase cascade depicted in steps 2 – 6 and culminating in activation of MAP kinase (MAPK). In unstimulated cells, binding of a dimer of the 14-3-3 protein to Raf stabilizes its kinase domain in an inactive conformation. Each 14-3-3 monomer binds to a phosphoserine residue in Raf, one to phosphoserine-259 in the N-terminal domain and the other to phosphoserine-621 in the kinase domain; binding to 14-33 maintains the kinase in a closed, inactive state in the cytosol. Step 2 : Binding of the Raf N-terminal regulatory domain to Ras⋅GTP results in dephosphorylation by a cytosolic enzyme of one of the serines that bind Raf to 14-3-3, leading to the consequent loss of 143-3 binding and partial activation of Raf kinase activity. Maximum activation of Raf kinase activity occurs in part by formation of Raf dimers (not depicted), in which the kinase domain of one phosphorylates serine or threonine residues in the activation loop of the other. Step 3 : After the GTP bound to Ras is hydrolyzed to GDP, a process accelerated by a Ras-GAP, the inactive Ras⋅GDP dissociates from Raf. The Ras⋅GDP can be reactivated by signals from activated receptors, thereby recruiting additional Raf molecules to the membrane. As detailed in the text, in step 4 Raf phosphorylates and thus activates a MEK kinase, and in step 5 MEK phosphorylates and thus activates a MAP kinase. See E. Kerkhoff and U. Rapp, 2001, Adv. Enzyme Regul. 41:261; J. Avruch et al., 2001, Recent Prog. Horm. Res. 56:127; and D. Matallanas et al., 2011, Genes Cancer 2:232. Description The illustration shows the steps involved in the pathway. Step 1. R a s is activated by the exchange of G D P for G T P. Step 2. The active R A S binds and activates R A F, which is composed of an N-terminal regulatory domain and a C-terminal kinase domain held together by a 14-3-3 domain. R A F is activated by loss of the 14-3-3 domain. Step 3. G T P hydrolysis leads to dissociation of R A S from R A F. Step 4. R A F activates M E K by phosphorylation. Step 5. M E K activates M A P K by phosphorylation. Step 6. Activated M A P kinase translocates to the nucleus where it activates many transcription factors.
In unstimulated cells, Raf is phosphorylated by several cytosolic kinases on two sites, one in its serine/threonine kinase domain and a second in its N-terminal autoinhibitory domain. These phosphate groups then bind to a dimer of the protein 14-3-3, which locks the autoinhibitory domain in a position where it blocks and inhibits the kinase active site — a molecular handcuff. Binding to 14-3-3 also retains the inactive Raf protein in the cytosol. As noted, Ras is activated by the exchange of GDP for GTP (Figure 16-13, step 1 ). Active Ras⋅GTP (but not Ras⋅GDP) binds to the N-terminal autoinhibitory domain of Raf, altering its conformation and allowing the phosphate attached to this domain to be removed by cytosolic phosphatases, thereby causing loss of 14-3-3 binding. This unblocks the kinase active site and partially activates its kinase activity (step 2 ). Hydrolysis of Ras⋅GTP to Ras⋅GDP releases Raf from Ras (step 3 ), and the released Raf then forms dimers in which the kinase activity of one phosphorylates serine or threonine residues on the activation loop of the other, further increasing Raf kinase activity. In summary, binding of Ras⋅GTP to Raf, followed by hydrolysis of Ras⋅GTP to Ras⋅GDP, releases active Raf from its inhibitory complex with 14-3-3 (step 3 ) and leads to activation of Raf kinase activity. The now active Raf subsequently phosphorylates MEK on one or two serine residues in its activation loop, thereby activating MEK kinase activity (step 4 ). MEK is a kinase that phosphorylates its target proteins on both tyrosine and serine/threonine residues; hence it is called a dualspecificity protein kinase. Active MEK mainly phosphorylates MAP
kinase in its activation loop (step 5 ). Active MAP kinase phosphorylates many different proteins, including nuclear transcription factors that mediate cellular responses that are discussed later (step 6 ). Several types of experiments have established the kinase pathway. For example, cultured mammalian cells that express a mutant, nonfunctional Raf protein cannot be stimulated to proliferate uncontrollably by a constitutively active protein, one that has a permanently bound GTP. This finding established a link between the Raf and Ras proteins and showed that Raf lies downstream of Ras in the signaling pathway. In vitro binding studies further showed that the purified Ras⋅GTP protein binds directly to the N-terminal regulatory domain of Raf and activates its catalytic activity. That MAP kinase is activated in response to Ras activation was demonstrated in quiescent (nondividing) cultured cells expressing a constitutively active protein. In these cells, activated MAP kinase is generated in the absence of stimulation by growth-promoting hormones. Biochemical studies showed, however, that Raf cannot directly phosphorylate MAP kinase or otherwise stimulate its activity. The final link in the kinase cascade activated by Ras⋅GTP emerged from studies in which scientists fractionated extracts of cultured cells to search for a kinase activity that could phosphorylate MAP kinase and that was active only in cells stimulated with growth factors, not in unstimulated cells. This work led to the identification of MEK. Phosphorylation promotes not only the catalytic activity of MAP kinase, but also its dimerization. The dimeric form of MAP kinase is translocated to the nucleus, where it
regulates the activity of many nuclear transcription factors. Later studies showed that MEK binds to the C-terminal catalytic domain of Raf and is phosphorylated on one or two serine residues in its activation loop by the Raf serine/threonine kinase (see Figure 16-13, step 4 ); this phosphorylation activates the catalytic activity of MEK. Given that the Ras/MAP kinase pathway is used in many cells and activated by many different receptors, it is not surprising that there are multiple isoforms of each of its components. In humans, there are three RAS, three Raf, two MEK, and two MAP proteins, and the isoforms have overlapping but also nonredundant functions. Because Raf phosphorylates MEK, Raf and analogous MEK phosphorylating kinases in cells are often called MEK kinases, or MEKK. All eukaryotes including mammals possess multiple highly conserved Ras/MAP kinase pathways that are activated by different extracellular signals and that activate different MAP kinase proteins that phosphorylate different transcription factors; these transcription factors, in turn, trigger different changes in cell division, differentiation, or function. Mammalian MAP kinases include Jun N-terminal kinases (JNKs) and p38 kinases, which are activated by signaling pathways in response to various types of stresses and which phosphorylate various transcription factors and other types of signaling proteins that affect cell division. Mutations in the Raf gene that lock its kinase domain in a permanently activated conformation occur in many types of tumors, in particular in about 50 percent of melanomas, skin cancers that are often
MAP Kinase Regulates the Activity of Many Transcription Factors Controlling Early Response Genes
caused by exposure to the ultraviolet radiation in sunlight. One such mutation, a glutamic acid substitution for the valine at position 600, occurs in over 90 percent of melanomas caused by exposure to UV radiation. This mutant constitutively active Raf kinase stimulates MAPkinase signaling in cells in the absence of growth factors, promoting cell proliferation and preventing apoptosis (programmed cell death; see
Chapter 22). Selective inhibitors of this mutant form of Raf have recently entered the clinic and generally produce short-term remission of the cancer. A combination of drugs that inhibit Raf and MEK appear more promising and have recently entered clinical trials in patients with melanomas triggered by this mutant Raf. MAP Kinase Regulates the Activity of Many Transcription Factors Controlling Early Response Genes Addition of a growth factor such as EGF or PDGF to quiescent, nondividing cultured mammalian cells causes a rapid increase in the expression of as many as a hundred different genes. These genes are called early response genes because they are induced well before cells, in the or phase of the cell cycle, enter the S phase and replicate their DNA (see Chapter 19); many are turned on downstream of activated (phosphorylated and dimeric) MAP kinase (Figure 16-14). Consider the master transcription factor c-Fos. Together with other transcription factors, c-Fos induces the expression of many genes that encode proteins necessary for cells to progress through the cell cycle. The enhancer that
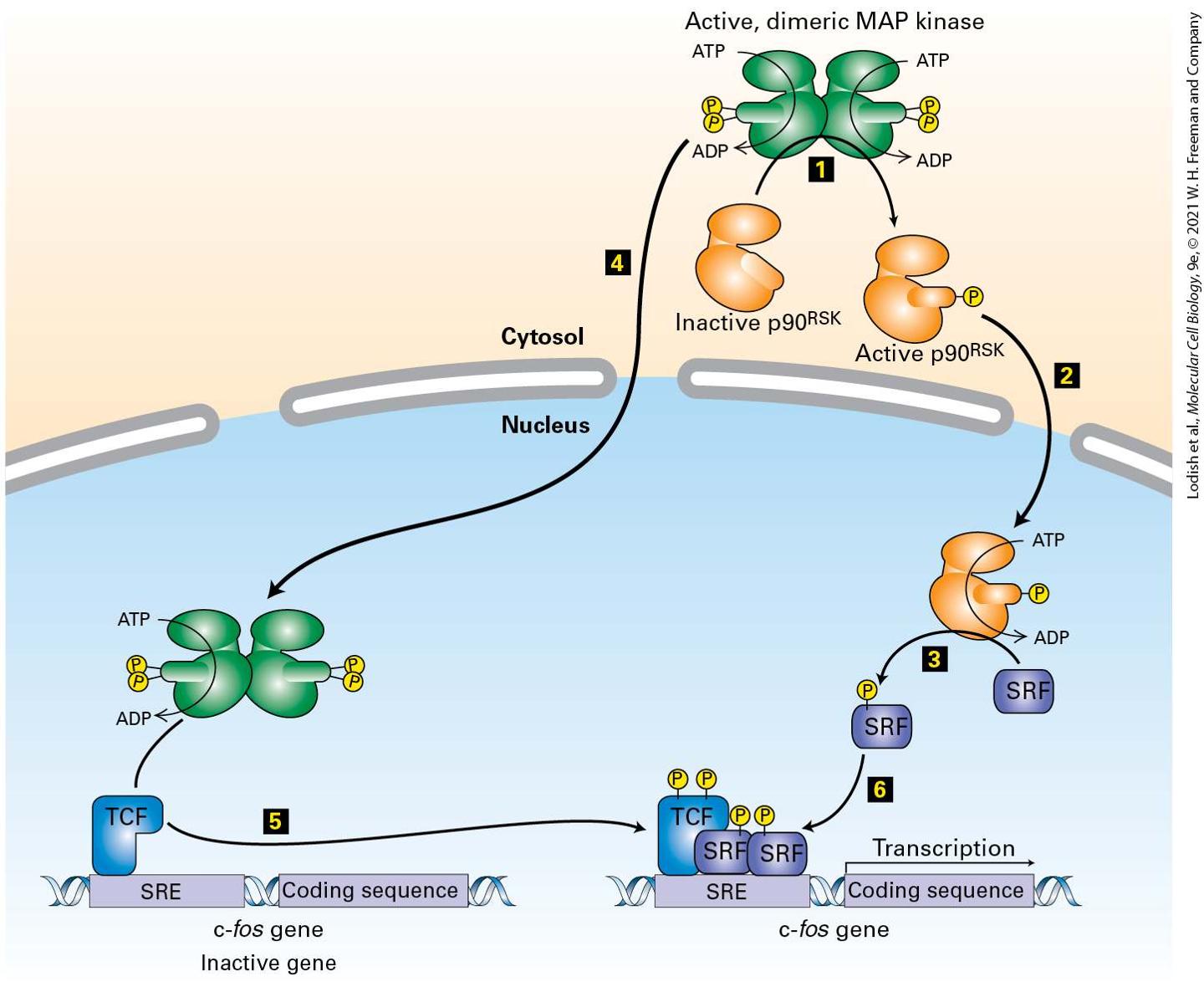
regulates c-fos gene expression contains a serum response element (SRE), so named because it is activated by many growth factors in serum. Activated MAP kinase induces transcription of the c-fos gene by directly phosphorylating and thus activating one transcription factor, ternary complex factor (TCF). MAP kinase also phosphorylates and activates yet another kinase, termed , that in turn phosphorylates another transcription factor termed serum response factor (SRF). Association of phosphorylated TCF with two molecules of phosphorylated SRF forms an active transcription factor complex that activates c-fos gene transcription.
FIGURE 16-14 Induction of gene transcription by MAP kinase. Steps 1 – 3 : In the cytosol, MAP kinase phosphorylates and activates the kinase , which then moves into the nucleus and phosphorylates a specific serine in the SRF transcription factor. Steps 4 and 5 : After translocating into the nucleus, MAP kinase directly phosphorylates specific serines in the transcription factor TCF, which is already bound to the promoter of the c-fos gene. Step 6 : Phosphorylated TCF and SRF act together to stimulate transcription of c-fos and other genes that contain an SRE sequence in their promoter. See the text for details. See R. Marais et al., 1993, Cell 73:381; and V. M. Rivera et al., 1993, Mol. Cell Biol. 13:6260. Description The illustration shows cytosol, membrane, and nucleus. The steps involved in the process are as follows. At step 1. Active dimeric M A P kinase in the cytosol is represented in green. The orange structure below is labeled inactive p 90 R S K that moves through the active dimeric M A P kinase and becomes active. At this point, the active p 90 R S K moves separately into the nucleus at step 2. The active p 90 R S K uses A T P and adds S R F (blue rectangle) and phosphate (yellow circle). The schematic moves to the cytosol for step 4, in which the active dimeric M A P kinase moves into the nucleus. In the nucleus, at the bottom, kinase adds phosphates to T C F, a blue structure attached to a c-f o s gene (an inactive gene with S R E and coding sequence). At step 5, the c-f o s gene moves toward the S R F with phosphate from step 3, and joins with it and initiates transcription. Most RTKs that bind growth factors use the Ras/MAP kinase pathway to activate genes encoding proteins such as c-Fos, which in turn propel the cell through the cell cycle. Unlike the Raf and MEK kinases, which phosphorylate one or only a few target kinases in the Ras/MAP kinase cascade, MAP kinases are known to phosphorylate more than 200 proteins in the nucleus and cytosol. Many MAP kinase targets are regulators of gene expression, and different MAP kinase targets are expressed in
Multiple Feedback Mechanisms Restrain MAP Kinase Activation
different mammalian cell types. As with other transcription factors activated directly or indirectly by cell-surface receptors, the precise proteins induced by MAP kinase depend on the particular target proteins expressed in the cell, on epigenetic markers on DNA and chromatin proteins, and on the presence of other transcription factors (see Figure 162). Multiple Feedback Mechanisms Restrain MAP Kinase Activation Prolonged activation of the RTK–Ras/MAP kinase cascade may lead to inappropriate cell proliferation that would be deleterious to the organism. Accordingly, to maintain proper normal cell regulation, MAP kinase activation is tightly regulated through several feedback mechanisms. As with feedback control of other cellular pathways, these fall into two main categories: post-translational modification of pathway components and induction of synthesis of proteins that inhibit specific steps in the pathway. Inhibition of the Ras/MAP kinase pathway by direct post-translational modification is nearly instantaneous, while many minutes are required to produce inhibitory protein(s) by de novo gene expression and protein synthesis and for these inhibitory proteins to influence their targets. At the post-translational level, activated MAP kinase phosphorylates multiple proteins that act earlier in the pathway, reducing their ability to activate the MAP kinase pathway. For example, activated MAP kinase phosphorylates the HER1 EGF receptor on a threonine residue near the
Scaffold Proteins Isolate Distinct MAP Kinase Pathways in the Same Cell from One Another
juxtamembrane domain (see Figure 16-8). Phosphorylation of this threonine residue decreases the ability of the receptor kinase to become fully active. Active MAP kinase also phosphorylates Raf on multiple sites, reducing its association with Ras and thus its ability to activate Ras. Such regulation effectively limits both the intensity and duration of MAP kinase activation. Activation of the Ras/MAP kinase pathway also promotes increased transcription of regulators that lower the intensity and duration of MAP kinase signaling, a classic example of regulation by negative feedback. For example, activated MAP kinase triggers an increase in the expression of dual-specificity phosphatases (DUSPs), particularly DUSP6. Their name is derived from their ability to dephosphorylate both the threonine and tyrosine phosphorylation sites in the MAP kinase activation loop, causing the loop to block the MAP kinase active site. DUSP6, along with several other negative regulators of the RTK–Ras/MAP kinase signaling cascade, are among the early response genes whose transcription is increased rapidly — within the first 15 minutes — following MAP kinase activation. Increased expression of these phosphatases typically leads to transient inhibition of MAP kinase and can also suppress the response to a subsequent stimulation. Scaffold Proteins Isolate Distinct MAP Kinase Pathways in the Same Cell from One Another
Two distinct signal transduction pathways within the same cell, initiated by two distinct receptors and having two distinct results, may share the identical protein kinase at some step. Yet this kinase will phosphorylate a different substrate depending on the pathway, even though both potential substrates are present in the cell. The principal mechanism that ensures that the correct substrate is phosphorylated is the formation of a complex by a scaffold protein. An example from yeast illustrates how the action of a scaffold protein guides the kinase to the correct substrate. Yeast cells, like mammalian cells, utilize several MAP kinase pathways (Figure 16-15), but they are not activated by Ras-like proteins (which do not exist in yeasts). One pathway is activated by placing the cells in medium of high osmotic strength. The MAP kinase in this pathway, Hog1, phosphorylates transcription factors that in turn activate genes whose proteins protect the cell against this osmotic challenge (Figure 16-15b). A second and distinct pathway is activated during mating of yeast cells of opposite mating type. Haploid yeast cells are either of the a or α mating type. An a haploid cell secretes the a mating factor and has cell-surface GPCRs that bind the α factor; an α cell secretes the α factor and has GPCRs that bind the a factor (see Figure 1-24). GPCRs that bind these factors initiate a MAP kinase pathway; the MAP kinase in this pathway, Fus3, translocates into the nucleus. There it phosphorylates and activates several transcription factors that turn on genes whose encoded proteins inhibit progression of the cell cycle and other genes that enable cells of opposite mating type to fuse together and ultimately form a diploid cell (Figure 16-15a).
FIGURE 16-15 Scaffold proteins separate yeast MAP kinase cascades in the mating and osmoregulatory pathways. In the budding yeast Saccharomyces cerevisiae, different cellsurface receptors activate different MAP kinase pathways, two of which are outlined here. (a) Mating pathway: The GPCR receptors for yeast α and a mating factors are coupled to the same trimeric protein. Following ligand binding and dissociation of the G protein subunits, the released membrane-tethered subunit binds the Ste5 scaffold to the plasma membrane. then activates a signal transduction pathway (arrows) that culminates with the phosphorylation and thus activation of Ste11, which is analogous to Raf and other mammalian MEK kinase (MEKK) proteins. Ste11 initiates a MAP kinase cascade in which the final component, Fus3, is functionally equivalent to the MAP kinases (MAPK) in higher eukaryotes. Like other MAP kinases, activated Fus3 then translocates into the nucleus where it leads to phosphorylation and activation of several transcription factors — in this case ones that initiate transcription of genes that enable cells of opposite mating type to fuse together and ultimately form a diploid cell. (b) Osmoregulatory pathway: Two plasma membrane proteins, Sho1 and Msb1, are activated by exposure of yeast cells to media of high osmotic strength. Activated Sho1 recruits the Pbs2 scaffold protein, which contains a MEK domain, to the plasma membrane, and activates a signal transduction
pathway (arrows) that also results in phosphorylation and activation of Ste11. This initiates a different MAP kinase cascade that activates Hog1, a MAP kinase. After translocating to the nucleus, Hog1 phosphorylates several transcription factors and chromatin-modifying enzymes that induce transcription of genes that support survival in high-osmotic-strength media. Note that the MEK in this pathway is an integral domain of the Ste5 scaffold. See N. Dard and M. Peter, 2006, BioEssays 28:146; and R. Chen and J. Thorner, 2007, Biochim. Biophys. Acta 1773:1311. Description The illustration labeled (a) is titled mating pathway. It begins in the exterior areas with a mating factor, represented by a purple circle, entering a receptor. Inside the cytosol, a structure with G D P labeled is attached. A sideways arrow to the right is labeled activation of G protein. This G protein is then attached to a blue structure labeled gamma at the top, and beta at the bottom, and attached to a long rectangle labeled S t e 5 scaffold protein. Also attached to the scaffold are structures labeled M E K K, M E K, M A P K. The M A P K (F u s 3) enters into the nucleus. Inside the nucleus, two structures labeled transcription factors are attached to ribbons labeled mating-type genes initiates transcription. The illustration labeled (b) is titled osmoregulatory pathway. It begins with two structures within the plasma membrane, labeled S h o 1 and M s b 2. A right arrow is labeled activation by high osmotic strength. The activation leads to the attachment of these two structures with a long rectangle labeled P b s 2 scaffold protein. Along with this rectangle from top to bottom are M E K K, M E K, M A P K proteins. The M A P K (H o g 1) enters into the nucleus. Inside the nucleus, a structure labeled transcription factor is attached to ribbons labeled osmoresponsive genes, initiates transcription. There is a potential source of signaling confusion because, as Figure 16-15 shows, the same MEKK protein kinase, Ste11, is found in both pathways but phosphorylates different MEK kinases that are unique to each pathway. Scaffold proteins prevent Ste11 from phosphorylating the wrong MEK kinase. The scaffold protein Ste5, employed in the mating pathway, has no obvious catalytic function but stabilizes a large complex that includes the
MEKK kinase Ste11, the MEK kinase Ste7, and the MAP kinase Fus3 in the mating signal transduction pathway. By binding to the protein that is released following activation of the mating factor GPCR, Ste5 ensures that the Ste11 bound to it is activated by the mating pathway. In contrast, the receptors that sense changes in extracellular osmotic strength, Sho1 and Msb2, cannot activate the MEKK Ste11 in the mating pathway complex. Similarly, the Pbs2 scaffold protein is employed in the kinase cascade in the osmoregulatory pathway (see Figure 16-15) and binds to Sho1, an activator of the osmolarity pathway. This ensures that the Ste11 bound to it is activated only by the osmotic pathway; the protein released following activation of the mating factor GPCR cannot activate the MEKK Ste11 in this osmo-sensing pathway complex. Thus signaling downstream from Ste11 is restricted to the complex in which it is localized. As a result, exposure of yeast cells to mating factors induces activation of a single MAP kinase, Fus3, whereas exposure to a high osmolarity induces activation of a different MAP kinase, Hog1. As emphasized in Figure 3-31, scaffolds also increase the rate of enzyme– catalyzed reactions by bringing the enzyme (e.g., a MEKK) close together to its substrate (a MEK), reducing the time needed for an enzyme to diffuse through an aqueous solution and find a substrate molecule. Scaffolds for MAP kinase pathways are well documented in yeast, fly, worm, and mammalian cells. In addition to their ability to function as
insulators for specific pathways, as described above for yeast Fus3 and Hog1, scaffold proteins can also regulate pathway activity. IQGAP1 is a mammalian scaffold protein that binds to RAF, MEK, and ERK kinases, as well as to a number of other proteins. Expression of IQGAP1 is increased in multiple different cancer types, and when its expression increases, so does the activity of the Ras–MAP kinase signaling cascade. Presumably IQGAP1 acts by bringing Raf, MEK, and ERK together into a single complex and thus facilitating their activation. Intriguingly, expressing in cells just the domain of IQGAP that binds ERK is sufficient to inhibit the growth of melanoma cells that express a mutant, activated Raf protein. This small protein blocks the binding of ERK to the full-length IQGAP1 scaffold, thus preventing ERK’s incorporation into the functional scaffold complex. This function of IQGAP suggests an alternate strategy for reducing MAP kinase activity in cancers. The signal specificity of the different MAP kinases in mammalian cells may arise from their association with different scaffold-like proteins, but much additional research is needed to test this possibility. KEY CONCEPTS OF SECTION 16.2 The Ras/MAP Kinase Signal Transduction Pathway Ras is an intracellular GTPase switch protein that acts downstream from most RTKs and cytokine receptors. Like , Ras cycles between an inactive GDP-bound form and an active GTP-bound form. Ras cycling requires the assistance of two proteins: a guanine nucleotide exchange factor (GEF) and a GTPase-activating protein (GAP). RTKs are linked indirectly to Ras via two proteins: GRB2, an adapter protein, and Sos, which has GEF activity (see Figure 16-10). The SH2 domain in GRB2 binds to a phosphotyrosine in activated RTKs, while its two SH3 domains bind Sos, thereby bringing Sos, a GEF, close to membrane-bound Ras·GDP and activating it.
Binding of Sos to inactive Ras causes a large conformational change that permits release of GDP and binding of GTP, forming active Ras (see Figure 16-12). Activated Ras triggers a kinase cascade in which Raf (a MEK kinase or MEKK), MEK, and MAP kinase are sequentially phosphorylated and thus activated. Activated MAP kinase then translocates to the nucleus (see Figure 16-13). Activation of MAP kinase following stimulation of a growth-factor receptor leads to phosphorylation and activation of two transcription factors that promote transcription of various early response genes (see Figure 16-14). Different extracellular signals induce activation of different MAP kinase pathways, which regulate diverse cellular processes by phosphorylating different sets of transcription factors. Multiple negative feedback mechanisms restrain MAP kinase activation — some depending on proteins whose gene transcription is induced by activated MEK. The kinase components of MAP kinase cascades can assemble into a large pathwayspecific complex often stabilized by a scaffold protein (see Figure 16-15). This ensures that activation of one MAP kinase pathway by a particular extracellular signal does not lead to inappropriate activation of other pathways containing shared components.
16.3 Phosphoinositide Signal Transduction Pathways
16.3 Phosphoinositide Signal Transduction Pathways In previous sections, we have seen how activated receptor tyrosine kinases (RTKs) initiate the Ras/MAP kinase pathway (see Figures 16-10 and 1613). Here we discuss how these same receptors initiate some very different signaling pathways. These pathways have as intermediates certain phosphorylated phospholipids derived from phosphatidylinositol. As discussed in Chapter 15, these membrane-bound lipids are collectively referred to as phosphoinositides. These signaling pathways include several enzymes that synthesize different phosphoinositides on the cytosolic face of the plasma membrane as well as cytosolic proteins with domains that can bind to these molecules. These proteins can thus be recruited to the cytosolic surface of the plasma membrane when the corresponding phosphoinositide is generated. Several of these phosphoinositide pathways have long-term effects on patterns of gene expression as well as short-term effects. We will see that different phosphoinositide pathways end with activation of specific kinases, including protein kinase C (PKC) and protein kinase B (PKB), that play key roles in cell growth and metabolism. As an example, we will
Phospholipase Cγ Is Activated by Many RTKs and Cytokine Receptors
see in Chapter 21 how insulin activation of PKB plays a key role in rapidly stimulating glucose import into muscle and fat cells. Phospholipase Is Activated by Many RTKs and Cytokine Receptors In Chapter 15 we learned that hormonal stimulation of certain G protein– coupled receptors leads to the activation of phospholipase C (PLC). This membrane-associated enzyme then cleaves phosphatidylinositol 4,5bisphosphate to generate two important second messengers: 1,2-diacylglycerol (DAG) and inositol 1,4,5-trisphosphate . One of the resulting products, , triggers an increase in the concentration of in the cytosol with its many and diverse effects in different types of cells. A rise in the cytosolic level causes protein kinase C to translocate to the cytosolic surface of the plasma membrane, where it binds to DAG and becomes activated (see Figure 15-28); activation of PKC in different cells results in a varied array of cellular responses, indicating that it plays a key role in many aspects of cellular growth and metabolism. As in the case for many signal transduction proteins, there are multiple isoforms of PLC. Via trimeric G proteins, GPCRs specifically activate the β isoform of this enzyme . Many RTKs (and the cytokine receptors discussed in the following section) activate a different isoform of phospholipase C, the γ isoform , which contains an SH2 domain. This SH2 domain binds to specific phosphotyrosines on the cytosolic
Recruitment of PI-3 Kinase to Activated Receptors Leads to Accumulation of Phosphatidylinositol-3-Phosphates in the Plasma Membrane and to Activation of Several Downstream Kinases
domains of these activated receptors, thus recruiting the enzyme from the cytosol to the cytosolic face of the plasma membrane, close to its substrate, . In addition, the receptor phosphorylates tyrosine residues on the bound , enhancing its hydrolase activity. Thus activated RTKs and cytokine receptors promote activity in two ways: by localizing the enzyme to the membrane and by phosphorylating it. As detailed in Chapter 15, the pathway initiated by PLC has multiple physiological effects. Recruitment of PI-3 Kinase to Activated Receptors Leads to Accumulation of Phosphatidylinositol3-Phosphates in the Plasma Membrane and to Activation of Several Downstream Kinases In addition to the pathway, many activated RTKs and cytokine receptors initiate another phosphoinositide pathway by recruiting the enzyme phosphatidylinositol-3 kinase (PI-3 kinase) to the cytosolic surface of the plasma membrane. PI-3 kinase is recruited to the plasma membrane by binding of its SH2 domain to phosphotyrosines on the cytosolic domain of many activated RTKs and cytokine receptors. This recruitment positions the catalytic domain of PI-3 kinase near its phosphoinositide substrates on the cytosolic face of the plasma membrane.
Unlike kinases we have encountered earlier, which phosphorylate proteins, PI-3 kinase adds a phosphate to the hydroxyl group on the carbon in one of two separate phosphatidylinositol substrates, leading to formation of either phosphatidylinositol 3,4-bisphosphate or phosphatidylinositol 3,4,5-trisphosphate — collectively called PI 3-phosphates (Figure 16-16). These membrane-bound PI 3phosphate products of the PI-3 kinase act as docking sites for multiple signal-transducing proteins. Once bound to these phosphoinositides, these proteins transduce signals in several important pathways.
FIGURE 16-16 Generation of phosphatidylinositol 3-phosphates. The enzyme phosphatidylinositol-3 kinase (PI-3 kinase) is recruited to the cytosolic face of the plasma membrane by many activated receptor tyrosine kinases (RTKs) and cytokine receptors. The 3-phosphate added by this enzyme, to yield or , is a binding site for various signal-transducing proteins, such as the PH domain of protein kinase B. See L. Rameh and L. C. Cantley, 1999, J. Biol. Chem. 274:8347. Description The vertical flow chart shows the generation of phosphatidylinositol 3-phosphates using chemical models. The structure of P I 4- phosphate (P I P) contains a zig-zag line labeled cytosolic leaflet attaches to Inositol. Phosphate molecule attaches to the fourth carbon. By the action of enzyme P I-5 kinase A T P is dephosphorylated to A D P and forms P I 4,5-bisphosphate (P I P subscript 2). Phosphate molecules attach to the fourth and fifth carbon. P I 4- phosphate (P I P) is converted to P I 3,4-bisphosphate by the action of enzyme P I-3 kinase. A T P is dephosphorylated to A D P. Phosphate molecules attach to the third and fourth carbon. P I 4,5-bisphosphate (P I P subscript 2) is converted to P I 3,4,5-trisphosphate by the action of enzyme P I-3 kinase. A T P is dephosphorylated to A D P. Phosphate molecules attach to the third, fourth, and fifth carbon. P I 3,4-bisphosphate is converted to P I 3,4,5-trisphosphate by the action of enzyme P I-5 kinase. A T P is dephosphorylated to A D P. Phosphate molecules attach to the third, fourth, and fifth carbon. In some cells, this PI-3 kinase pathway triggers cell division and prevents apoptosis, thus ensuring cell survival. In other cells, this pathway induces specific changes in cell metabolism. An important signal transduction protein that binds to PI 3-phosphates is protein kinase B (PKB), a serine/threonine kinase that is also called Akt. In addition to its kinase domain, PKB contains a PH domain, a conserved protein domain present in a wide variety of signaling proteins that binds
with high affinity to the 3-phosphate in both and . In unstimulated, resting cells, the concentration of these phosphoinositides is low; PKB is present in the cytosol in an inactive form because its PH domain physically blocks the kinase active site (Figure 16-17). Following ligand-induced receptor activation, the concentration of PI 3-phosphates rises. The PH domain of PKB binds to these PI 3-phosphates, so it no longer blocks the PKB kinase active site. PKB is thus recruited to the cytosolic face of the plasma membrane, and its unblocked kinase domain becomes partially activated. Maximal activation of PKB depends on recruitment to the cytosolic surface of the plasma membrane of two other kinases, named PDK1 and PDK2.
FIGURE 16-17 Recruitment and activation of protein kinase B (PKB) in PI-3 kinase pathways. In unstimulated cells (step 1 ), PKB is in the cytosol with its PH domain bound to its catalytic kinase domain, inhibiting its activity. Hormone stimulation leads to activation of PI-3 kinase and subsequent formation of or (see Figure 1616). The 3-phosphate group on both serves as a docking site on the plasma membrane for
Activated Protein Kinase B Induces Many Cellular Responses
the PH domain of PKB (step 2 ) and another kinase, PDK1. Full activation of PKB requires phosphorylation both in the activation loop by PDK1 and at the C-terminus by a second kinase, PDK2, that is part of the mTORC2 complex described in Chapter 21 (step 3 ). See A. Toker and A. Newton, 2000, Cell 103:185; and S. Sarbassov et al., 2005, Curr. Opin. Cell Biol. 17:596. Description The illustration shows the membrane, represented by gray bars, with P I 4-Phosphate attached to it. Outside the membrane is labeled exterior and inside is labeled cytosol. The steps involved in the activation of protein kinase B are as follows. Step 1, labeled inactive P K B in cytosol of unstimulated cell. The inactive P K B has a P H domain and kinase domain. Step 2, labeled Formation of P I 3-phosphates, recruitment, and partial activation of P K B. The P H domain of P K B attaches to the phosphate group in the third carbon of P I 3,4-bisphosphate. The partially activated P K B is labeled with activation loop. Step 3, labeled fully active P K B shows P D K 1 and kinase in the m T O R C 2 complex phosphorylates the activation loop and the C-terminus of P K B. PDK1 is recruited to the plasma membrane via binding of its own PH domain to PI 3-phosphates. Anchored to distinct PI 3-phosphates, both PKB and PDK1 diffuse randomly in the plane of the plasma membrane, eventually coming close enough together so that PDK1 can phosphorylate PKB on a critical threonine residue in its activation loop — yet another example of kinase activation by phosphorylation. Phosphorylation by the mTORC2 kinase complex, described in Chapter 21, of a second serine on PKB that is not in the activation loop is necessary for maximal PKB activity (see Figure 16-17). As in the regulation of Raf activity (see Figure 16-13), release of an inhibitory domain and phosphorylation by other kinases regulate the activity of PKB.
Activated Protein Kinase B Induces Many Cellular Responses Once fully activated, PKB can dissociate from the plasma membrane and phosphorylate its many target proteins throughout the cell. These targets have a wide range of effects on cell behavior. In many cells, activated PKB directly phosphorylates and inactivates a protein termed Bad, which as its name suggests is not good for a cell’s survival — it will induce programmed cell death (apoptosis; see Figure 22-42). Phosphorylation of Bad by PKB protects against apoptosis. Activated PKB also promotes survival of many cultured cells by phosphorylating the Forkhead transcription factor FOXO3a on multiple serine and threonine residues. When FOXO3a is not phosphorylated, it mainly localizes to the nucleus, where it activates transcription of several genes encoding proteins that induce apoptosis. When growth factors are added to the cells, PI-3 kinase is activated, and consequently so is PKB. Activated PKB phosphorylates FOXO3a. The cytosolic phosphoserinebinding protein 14-3-3 then binds to phosphorylated FOXO3a and sequesters it in the cytosol. (Recall that many other phosphorylated proteins, including Raf, that bind to 14-3-3 are similarly retained in an inactive state in the cytosol; see Figure 16-13.) A FOXO3a mutant in which the three serine residues that are targets for PKB are mutated to alanines is constitutively active as a transcription factor and initiates apoptosis even in the presence of activated PKB. This finding
The PI-3 Kinase Pathway Is Negatively Regulated by PTEN Phosphatase
demonstrates the importance of phosphorylation of FOXO3a by PKB in preventing induction of apoptosis. Misregulation of PKB is implicated in the pathogenesis of both cancer and diabetes, and in Chapter 21 we will see how PKB, activated downstream of the insulin RTK, promotes glucose uptake and storage in muscle and liver. This is another example of one signaling pathway controlling different cellular functions in different cells. The PI-3 Kinase Pathway Is Negatively Regulated by PTEN Phosphatase Like most intracellular signaling events, phosphorylation by PI-3 kinase is reversible. The phosphatase that removes the phosphates added by PI-3kinase, termed PTEN, has an unusually broad specificity. Although PTEN can remove phosphate groups attached to serine, threonine, and tyrosine residues in proteins, its major function in cells is thought to be its ability to remove the 3-phosphate from and and thus suppress the PI-3 kinase signaling pathway. Overexpression of PTEN in cultured mammalian cells promotes apoptosis by reducing the level of and , and hence reducing the activation of PKB and limiting its anti-apoptotic effect.
The PTEN gene is deleted in multiple types of advanced human cancers, and its loss contributes to the uncontrolled growth of cells. Indeed, cells lacking PTEN have elevated levels and PKB activity. Because PKB inactivates a protein that induces apoptosis, loss of PTEN reduces the programmed cell death that is the normal fate of many cells including many tumors. In certain cells, such as neuronal stem cells, absence of PTEN not only prevents apoptosis but also stimulates cell cycle progression and enhances the rate of cell proliferation. Knockout mice lacking PTEN have big brains with an excess number of neurons, attesting to PTEN’s importance in the control of normal brain development. KEY CONCEPTS OF SECTION 16.3 Phosphoinositide Signal Transduction Pathways Many RTKs and cytokine receptors initiate the signaling pathway by activating phospholipase , a different PLC isoform than the one activated by G protein–coupled receptors. Activated RTKs and cytokine receptors also can initiate another phosphoinositide pathway by binding a PI-3 kinase, thereby allowing the enzyme access to its membrane-bound phosphoinositide substrates, which then become phosphorylated at the 3 position (PI 3-phosphates and ; see Figure 16-16). The PH domains in various proteins bind to PI 3-phosphates, forming signaling complexes associated with the cytosolic face of the plasma membrane. Protein kinase B (PKB) becomes partially activated by binding with its PH domain to PI 3-phosphates. Full activation of PKB requires phosphorylation by the kinase PDK1, which is also recruited to the membrane by binding to PI 3-phosphates, and by a second kinase, PDK2 (see Figure 16-17). Activated PKB promotes survival of many cells by directly phosphorylating and inactivating several pro-apoptotic proteins and by phosphorylating and inactivating the FOXO3a transcription factor, which otherwise induces synthesis of pro-apoptotic proteins.
Signaling via the PI-3 kinase pathway is terminated by the PTEN phosphatase, which hydrolyzes the 3-phosphate in PI 3-phosphates. Loss of PTEN, a common occurrence in human tumors, promotes cell survival and proliferation.
16.4 Cytokines, Cytokine Receptors, and the JAK/STAT Signaling Pathway
16.4 Cytokines, Cytokine Receptors, and the JAK/STAT Signaling Pathway The cytokines form a family of relatively small, secreted signaling molecules, generally containing about 160–200 amino acids, that control growth and differentiation of many types of cells. All cytokines have a similar three-dimensional structure — four long, conserved α helices folded into a conserved three-dimensional structure. All cytokine receptors also have similar three-dimensional structures; they function similarly to RTKs and activate similar signal transduction pathways. Whereas in RTKs the protein tyrosine kinase is part of the receptor cytosolic domain, cytokine receptors and the kinase enzyme are separate polypeptides, encoded by different genes, yet are bound tightly together. The tightly bound kinase is known as a JAK kinase. (JAK is an acronym for “Just Another Kinase” because when the first member of the family was cloned researchers knew it was a kinase from its sequence, but had no idea of its function.) In this section, we focus here on a pathway — the JAK/STAT pathway — mainly employed by cytokine receptors: the JAK kinase phosphorylates and activates a STAT transcription factor that moves directly to the nucleus. More specifically, the SH2 domain of a STAT transcription factor binds to a phosphotyrosine in the cytosolic domain of an activated cytokine receptor; the STAT transcription factor
Cytokines Regulate the Development and Function of Many Cell Types
then becomes phosphorylated by the JAK kinase, moves to the nucleus, and directly activates gene expression. Cytokines Regulate the Development and Function of Many Cell Types Growth hormone (GH), as its name implies, is a 191-amino-acid protein that stimulates proliferation of many types of body cells; it is made and secreted by cells in the anterior pituitary gland in response to another hormone, growth hormone–releasing hormone, that is made by the part of the brain termed the hypothalamus. GH was one of the first protein drugs to be made by recombinant DNA; it is used clinically to treat short stature caused by GH deficiency in children and adults. The bovine version is used to increase milk production in dairy cows. During pregnancy, a related hormone, the cytokine prolactin, induces epithelial cells lining the immature ductules of the mammary gland to differentiate into the acinar cells that produce milk proteins and secrete them into the ducts. GH and prolactin have three-dimensional structures very similar to those of the cytokines that induce the formation of important types of blood cells. All blood cells are derived from hematopoietic stem cells, which form a series of progenitor cells that then differentiate into the mature blood cells (see Figure 22-18). For instance, the cytokine granulocyte colony– stimulating factor (G-CSF) induces a granulocyte progenitor cell in the bone marrow to divide several times and then differentiate into granulocytes, a type of white blood cell that inactivates invading bacteria
and other pathogens. A related cytokine, thrombopoietin, stimulates a different progenitor cell to divide and differentiate into megakaryocytes, huge cells that fragment into the platelets that are essential for blood clotting. A structurally related cytokine, erythropoietin (Epo), triggers production of erythrocytes (red blood cells) by inducing the proliferation and differentiation of erythroid progenitor cells in the bone marrow (Figure 16-18a). Erythropoietin is synthesized by certain kidney cells. The major function of erythrocytes is to transport oxygen complexed to hemoglobin. Thus a drop in blood oxygen, such as that caused by loss of blood from a large wound, signifies a lower than optimal level of red blood cells. As we learn in Chapter 21, the oxygen-sensing transcription factor HIF-1α becomes activated and induces the transcription of the erythropoietin gene; the kidney cells synthesize more of this cytokine and secrete it into the blood. As the level of erythropoietin rises, more and more erythroid progenitors are induced to divide and differentiate; each progenitor produces 30 to 50 erythrocytes in only a few days. In this way, the body can respond to the loss of blood or reduction in available atmospheric oxygen at high elevations by accelerating the production of erythrocytes. GH, prolactin, G-CSF, thrombopoietin, and Epo undoubtedly evolved from a common ancestral protein, since all of these cytokines have a similar tertiary structure.
FIGURE 16-18 Erythropoietin and formation of red blood cells (erythrocytes). (a) Differentiation of erythroid progenitors to erythrocytes. Erythroid progenitor cells, called colony-forming units–erythroid (CFU-E), are derived from hematopoietic stem cells, which also give rise to progenitors of other blood cell types (see Figure 22-18). In the absence of erythropoietin, CFU-E cells undergo apoptosis (programmed cell death). Binding of Epo to its receptors on a CFU-E cell induces transcription of several genes whose encoded proteins prevent apoptosis, allowing the cell to survive. Other Epo-induced proteins trigger a developmental program of three to six terminal cell divisions, induction of hemoglobin and many other erythroid-important genes, reduction in cell and nuclear size, and finally, loss of the cell nucleus. If CFU-E cells are cultured with Epo in a semisolid medium (e.g., containing methylcellulose), daughter cells cannot move away, and thus each CFU-E cell produces a colony of 30–100 erythroid cells; hence its name. (b) Structure of erythropoietin bound to an erythropoietin receptor. Like other cytokines, erythropoietin (Epo) contains four conserved, long α helices that are folded into a conserved structural arrangement. The activated erythropoietin receptor (EpoR) is a dimer of identical subunits. Like the extracellular domains of all cytokine receptors, the extracellular domain of each monomer of the EpoR is constructed of two subdomains, each containing seven conserved β strands folded together in a characteristic and conserved structure. Side chains of residues on two of the α helices in Epo, termed site 1, contact loops on one EpoR monomer, while residues on the two other Epo α helices, termed site 2, bind to the same loop segments in a second receptor monomer, thereby stabilizing the dimeric receptor in a specific active conformation. [Part (a) Data from M. Socolovsky et al., 2001, Blood 98:3261. Part (b) Data from R. S. Syed et al., 1998, Nature 395:511, PDB ID 1eer.] Description The illustration labeled (a) starts from hematopoietic stem cells, and leads to progenitors of other types of blood cells, and erythroid progenitor with E P O receptors on its surface. In the absence of E P O, apoptosis, cell death, occurs. In the presence of E P O, cell division and maturation occurs in 4 generations. The mature red blood cells are labeled at the bottom. The three-dimensional ribbon diagram labeled (b) shows E P O receptors bound to the plasma membrane at the C-terminal, and erythropoietin bound in between the two
Binding of a Cytokine to Its Receptor Activates One or More Tightly Bound JAK Protein Tyrosine Kinases
identical receptor units. Both Epo and G-CSF are produced commercially by recombinant expression in cultured mammalian cells. Patients with kidney disease, especially those undergoing dialysis, frequently are anemic (have a low red blood cell count) and therefore are treated with recombinant Epo to boost red cell levels. Epo and G-CSF are used as adjuncts to certain cancer therapies because many cancer treatments reduce the production of red cells and granulocytes in the bone marrow. Epo can also be misused to increase red blood cell levels in endurance athletes (blood “doping”). Cytokines include the interferons, which are produced and secreted by several cell types following viral infection. Interferons act on nearby cells to induce enzymes that render those cells more resistant to viral infection. Other cytokines, exemplified by Interleukin 2 (IL-2), mainly regulate cells of the immune system. IL-2 promotes the proliferation, differentiation, and survival of key cells of the immune system, including antibodyforming B cells and also several types of T cells (Chapter 24). Binding of a Cytokine to Its Receptor Activates One or More Tightly Bound JAK Protein Tyrosine Kinases GH, prolactin, G-CSF, thrombopoietin, and Epo all have similar structures, and they activate receptors of similar structure by forming dimers of two
identical cytokine receptor proteins, a process termed receptor homodimerization. The extracellular domains of these cytokine receptors are constructed of two subdomains, each of which contains seven conserved β strands folded together in a characteristic fashion (Figure 1618b). Cytokines bind their receptors very tightly, as a consequence of molecular complementarity and multiple weak, noncovalent forces — that is, ionic, van der Waals, and hydrophobic interactions [see Figure 15-4; dissociation constants of about ]. Thus very low concentrations of GH and most other cytokines — about 1 microgram per liter (roughly one part in a billion) — are sufficient to activate cytokine receptors. Other cytokines, including the interleukins and the interferons, bind simultaneously to two or more different cytokine receptors, a process called receptor hetero-dimerization or hetero-oligomerization (Figure 1619b). IL-2 can bind first to the alpha chain (IL-2Rα), which does not directly interact with downstream signal transduction molecules, but rather enhances the binding of IL-2 to the two signaling subunits, the beta and gamma chains IL-2Rβ and IL-2Rγ. The same gamma chain is also an essential subunit of the receptors for several other interleukins, including IL-4, IL-7, IL-9, IL-15, and IL-21, cytokines that are essential for formation of B cells and other types of immune-system cells. Severe combined immunodeficiency (SCID) is a genetic disease in which neither T nor B cells are produced. People with SCID cannot cope with any bacterial or viral infection, so they must be kept in a sterile
environment (like the famous “bubble boy” who was kept in a plastic bubble and eventually succumbed to an infection). Many cases of SCID are due to a deficiency in the IL-2 receptor gamma chain, whose gene is located on the X chromosome. These children can now be cured by gene therapy: a lentiviral vector (see Figure 6-37) is used to introduce a functional gamma chain gene into the hematopoietic stem cells that generate all immune-system cells (see Chapter 23), and the children can live normal lives. The JAK family of kinases contain four members. An identical JAK kinase of one of these types is bound to the cytosolic domain of each of the two polypeptides in homodimeric receptors (Figure 16-19a, step 1 ). Two distinct JAK kinase isoforms bind to heterodimeric receptors (Figure 1619b). Despite this difference, the signaling pathways activated by all cytokine receptors are broadly similar; here we focus on the simpler case of receptors that homodimerize. Our example is the interaction of one erythropoietin molecule with two identical erythropoietin receptor (EpoR) proteins (Figures 16-19a and 16-20a).
FIGURE 16-19 Structure and activation of cytokine receptors. (a) Homodimeric receptors. The cytosolic domains of both cytokine receptor subunits bind tightly and irreversibly to the same JAK protein tyrosine kinase. In the absence of ligand (step 1 ), the receptors are largely monomers and the JAK kinases are poorly active. Ligand binding causes a conformational change in the receptors that brings together the JAK kinase
domains, which then phosphorylate each other on a tyrosine residue in the activation loop, activating kinase activity (step 2 ). The active JAK kinases then phosphorylate multiple tyrosine residues in the receptor cytosolic domain (step 3 ). As in RTKs (see Figure 161a), the resulting phosphotyrosines function as docking sites for SH2 domains of multiple signal-transducing proteins, including STAT proteins. (b) Heteromeric receptors, as exemplified by the binding of Interleukin 2 (IL-2) to the extracellular domains of the three subunits of the IL-2 receptor. Both the beta and gamma subunits of the IL-2 receptor (IL2Rβ and IL-2Rγ) have structures similar to that of the Epo receptor. The alpha subunit (IL2Rα) is expressed in some but not all responding cells and does not activate a signal transduction pathway. Its function is to bind IL-2 and facilitate the binding of IL-2 to the signaling IL-2Rβ and IL-2Rγ subunits. Each of the two signaling receptors, IL-2Rβ and IL2Rγ, is bound to a different JAK kinase and activates different STAT signal-transducing proteins but otherwise the receptor complex activates similar same downstream signaling pathways as depicted in (a). Description The illustration labeled (a) shows the steps involved in the activation of homodimeric receptors. Step 1 is labeled cytokine receptors without bound ligands. In this step, the illustration shows two green structures (with ligand binding sites labeled) above the membrane and several orange structures below. The orange structure is labeled J A K and has an activation loop and kinase labeled. Step 2 is labeled dimerization and phosphorylation of activation loop tyrosines. Here, the orange structures are attached to phosphates (yellow circles) and the conversion of A T P to A D P takes place in this step. The right side of the structure is labeled active J A K. Step 3 is labeled phosphorylation of additional tyrosine residues and shows multiple phosphates. Also, four downward arrows indicate pathways A, B, C, D with lists of their proteins. The left list is titled pathway A, G R B 2 or S h c. Downward arrow to R a s then to M A P kinase pathway, lastly transcriptional activation, modification of other cellular proteins. The second list starts with the title Pathway B, Phospholipase C, downward arrows to Elevation of C a superscript 2 plus and transcriptional activation or repression, modifications of other cellular proteins. The third list is titled Pathway C, P 1-3 kinase, downward arrow to protein kinase B, then transcriptional activation or repression, modification of other cellular proteins. The final list is titled pathway D, S T A T transcription factor, downward arrow to transcriptional activation.
The illustration labeled (b) shows the structure of a heterodimeric receptor. Above the plasma membrane is a structure labeled I L-2 R alpha, I L-2, I L-2 R beta, and I L-2 R gamma. Below the membrane are the orange structures with phosphates and the labels J A K 1 on the left and J A K 3 on the right, interacting with two cylinders that come down from the exterior. At the end of these cylinders are 3 arrows labeled S T A T 5, S T A T 1, and S T A T 3. Each of the four members of the JAK family of kinases contains an N-terminal domain that binds to the receptor, a C-terminal kinase domain that has low catalytic activity when no ligand is bound, and a middle pseudokinase domain that regulates kinase activity by an as yet unknown mechanism. These kinases become activated after ligand binding and receptor dimerization (Figure 16-19a, step 1 ); as with many RTKs, ligand binding triggers a conformational change in the JAKs such that they phosphorylate each other on a critical tyrosine in the activation loop (Figure 16-19a, step 2 ; see Figure 15-6), which greatly enhances kinase activity. Also as with RTKs, the active JAK kinase phosphorylates several tyrosine residues in the receptor’s cytosolic domain (Figure 16-19a, step 3 ), creating binding sites for SH2 domains of many signal transduction proteins, including those that activate the Ras/MAP kinase and other signaling pathways discussed in Sections 16.2 and 16.3. Here we focus on a pathway — the JAK/STAT pathway — mainly employed by cytokine receptors: the JAK kinase phosphorylates and activates a STAT transcription factor that moves directly to the nucleus (Figure 16-20a).
FIGURE 16-20 Activation and structure of STAT proteins. (a) Phosphorylation and dimerization of STAT proteins. Step 1 : Following dimerization and activation of a JAK kinase bound to a cytokine receptor (see Figure 16-19a), the SH2 domain of an inactive monomeric STAT transcription factor binds to a phosphotyrosine in the receptor cytosolic domain, bringing the STAT close to the active JAK associated with the receptor. The JAK then phosphorylates a specific C-terminal tyrosine in the STAT. Steps 2 and 3 :
JAK Kinases Phosphorylate and Activate STAT Transcription Factors
Phosphorylated STATs spontaneously dissociate from the receptor and spontaneously homodimerize. Because the STAT homodimer has two phosphotyrosine-SH2 domain interactions, whereas the receptor-STAT complex is stabilized by only one such interaction, phosphorylated dimeric STATs tend not to dissociate and rebind to the receptor. Step 4 : The STAT dimer moves into the nucleus, where it binds to promoter sequences and activates transcription of target genes. (b) Ribbon diagram of the STAT1 dimer bound to DNA (black). The STAT1 dimer forms a C-shaped clamp around DNA that is stabilized by reciprocal and highly specific interactions between the SH2 domain (purple) of one monomer and the phosphorylated tyrosine residue (yellow with red oxygens) on the C-terminal segment of the other. The phosphotyrosine-binding site of the SH2 domain in each monomer is coupled structurally to the DNA-binding domain (magenta), suggesting a potential role for the SH2-phosphotyrosine interaction in the stabilization of DNA interacting elements. [Part (b) Data from X. Chen et al., 1998, Cell 93:827, PDB ID 1bf5.] Description The illustration labeled (a) is a schematic of a receptor with a S T A T protein on it. The E p o receptor is above the membrane, and the active J A K is below. Below the J A K and attached to the receptor is S T A T. The structure of S T A T has three parts: a yellow line with a phosphate, an S H 2 domain, and a D N A-binding domain. The illustration labeled (b) is a ribbon model of S T A T 1 protein. A circle of D N A is in the center with D N A binding domains (blue and purple ribbons) on each side. Above the D N A area is the S H 2 domain, tyrosine, and phosphate. An inset shows a magnified view of the D N A, S H 2 domain, tyrosine, and phosphate. JAK Kinases Phosphorylate and Activate STAT Transcription Factors All STAT proteins contain an N-terminal DNA-binding domain, an SH2 domain, and a C-terminal domain with a critical tyrosine residue. Through
its SH2 domain, a monomeric STAT binds to a specific phosphotyrosine in the cytosolic domain of an activated receptor (Figure 16-20a, step 1 ). The C-terminal tyrosine then becomes phosphorylated by the adjacent JAK kinase (step 2 ). This arrangement ensures that in a particular cell, only those STAT proteins with an SH2 domain that can bind to a particular receptor will be activated when that receptor is activated by cytokine binding. For example, the erythropoietin receptor, as well as the receptors for GH, prolactin, G-CSF, and several other cytokines, activates STAT5, but not STATs 1, 2, 3, or 4. In contrast the IL-2 receptor complex activates STATs 1, 3, and 5 but not STATs 2 or 4 (see Figure 16-19b). A phosphorylated STAT dissociates spontaneously from the receptor, and two phosphorylated STAT proteins form a homodimer in which the SH2 domain on each molecule in the dimer binds to the phosphotyrosine in the other molecule (Figure 16-20b). Conformational changes take place on dimerization that expose the nuclear-localization signal (NLS), which had been inaccessible in the unphosphorylated STAT. NLSs are present in virtually all transcription factors found in the cytosol and are required for their transport into the nucleus (see Chapter 13). Targeted by the NLS, the STAT dimers move into the nucleus, where they bind to specific enhancers or promoters (DNA regulatory sequences) controlling target genes and thus alter gene expression. As noted above (see Figure 16-2), because different cell types have unique complements of transcription factors and unique epigenetic modifications of their chromatin, the genes that are available to be activated by any STAT are different in different cell types. For example, in mammary gland
Phosphotyrosine Phosphatases
cells, STAT5 becomes activated following prolactin binding to the prolactin receptor and induces transcription of genes encoding milk proteins. In contrast, when STAT5 becomes activated in erythroid progenitor cells following binding of Epo to the EpoR, it induces expression of the protein prevents the apoptosis of these progenitors (see Chapter 22), allowing them to proliferate and differentiate into red blood cells. More generally, it is thought that in each type of cell, activated STAT proteins, like the other transcription factors discussed earlier, bind only to DNA sites in open chromatin and mainly to sites that have master transcription factors or other cell-specific gene regulatory proteins bound at adjacent sites. This strategy of activating sets of genes specific to specific cell types by a relatively limited set of receptors, JAK kinases, and STAT proteins, allows the control, via combinatorial diversity, of a vast array of cellular activities. Multiple Mechanisms Suppress Signaling from Cytokine Receptors In Chapter 15 and in this one, we saw several ways in which signaling from G protein–coupled receptors and RTKs is terminated to prevent cells from overresponding to external stimuli. Here we discuss two mechanisms by which cytokine receptor signaling is regulated. Phosphotyrosine Phosphatases
SOCS Proteins
Phosphotyrosine phosphatases are enzymes that remove (hydrolyze) phosphates from specific phosphotyrosines on specific target proteins. An excellent example of how phosphotyrosine phosphatase enzymes function to suppress the activity of protein tyrosine kinases is provided by SHP1, a phosphatase that negatively regulates signaling by several types of cytokine receptors. Its role was first identified by analysis of mutant mice (termed motheaten) that lacked this protein; they died because of excess production of several types of blood cells, including erythrocytes. SHP1 dampens cytokine signaling by binding to a cytokine receptor and inactivating the associated JAK protein, as depicted in Figure 16-21a. In addition to a phosphatase catalytic domain, SHP1 has two SH2 domains. When cells are in the resting state, unstimulated by a cytokine, SHP1 is free in the cytosol and one of its SH2 domains physically binds to and masks the catalytic site in the enzyme’s phosphatase domain. In the stimulated state, however, this blocking SH2 domain preferentially binds to a specific phosphotyrosine residue in the activated receptor and unblocks the catalytic site. The accompanying conformational change brings the SHP1 catalytic site adjacent to the phosphotyrosine residue in the activation loop of the JAK associated with the receptor. By removing this phosphate, SHP1 inactivates the JAK, so that it can no longer phosphorylate the receptor or other substrates including the STATs. SOCS Proteins
In another example of negative feedback, among the genes whose transcription is induced by STAT proteins are those encoding a class of small proteins termed suppressors of cytokine signaling (SOCS), which terminate signaling by cytokine receptors. All SOCS proteins contain an SH2 domain that binds to specific phosphotyrosines on an activated receptor (Figure 16-21b). Binding of SOCS to an activated receptor results in the degradation of both the receptor and its associated JAK kinase.
FIGURE 16-21 Two mechanisms for terminating cytokine signal transduction, as exemplified by the erythropoietin receptor (EpoR). (a) Short-term regulation: SHP1, a phosphotyrosine phosphatase, is present in an inactive form in the cytosol of unstimulated cells. Binding of an SH2 domain in SHP1 to a particular phosphotyrosine in the activated receptor unmasks its phosphatase catalytic site and positions it near the phosphorylated tyrosine in the activation loop region of JAK2. Removal of the phosphate from this tyrosine inactivates the JAK kinase. See S. Constantinescu et al., 1999, Trends Endocrin. Met. 10:18. (b) Long-term regulation: SOCS proteins, whose expression is induced by the STAT5 protein in erythropoietin-stimulated erythroid progenitor cells, inhibit or permanently terminate signaling over longer periods. Binding of SOCS to specific phosphotyrosine residues on EpoR or JAK2 blocks binding of other signaling proteins (left). The SOCS box also targets the receptor as well as JAK2 for degradation by the ubiquitin-proteasome pathway (right). Similar mechanisms regulate signaling from other cytokine receptors. See B. T. Kile and W. S. Alexander, 2001, Cell. Mol. Life Sci. 58:1627. Description The illustration labeled (a) titled short-term regulation: J A K 2 deactivation by S H P 1 phosphate shows the E p o receptor above the membrane. Below the membrane are the active and inactive J A K 2 kinase represented in orange to the left and right of the receptor, respectively. Active S H P 1 is attached to the active J A K 2. Below this is an inactive S H P 1 with S H 2 domains and phosphatase domain labeled. The illustration labeled (b) titled long-term regulation: signal blocking and protein degradation by S O C 5 protein shows the E p o receptor above the membrane. The area below the membrane shows 6 S H 2 proteins with phosphates attached. Arrows moving to the right from 3 of the domains points to a label: recruitment of E 3 ubiquitin ligase, proteolytic degradation of J A K 2 and receptor. Below is the S O C 5 protein with S H 2 domain labeled on the left, and S O C 5 box labeled on the right. SOCS acts by targeting these two proteins to be degraded by a proteasome. SOCS proteins contain a domain, called the SOCS box, that recruits components of E3 ubiquitin ligases (see Figure 3-32) that polyubiquitinylate the receptor and the JAK kinase (a polymer of
ubiquitins is covalently attached). The polyubiquitin tail marks the proteins for destruction by a proteasome (see Chapter 3), thereby permanently turning off JAK2-mediated signaling pathways until new receptors and JAK2 proteins can be made. The observation that proteasome inhibitors prolong JAK2 signal transduction supports this mechanism. One SOCS protein, SOCS-1, also binds to the critical phosphotyrosine in the activation loop of an activated JAK2 kinase, thereby inhibiting its catalytic activity. Studies with cultured mammalian cells have shown that the cytokine receptor for growth hormone is suppressed by another SOCS protein, SOCS-2. Strikingly, mice deficient in SOCS-2 grow significantly larger than their wild-type counterparts; these mice have long bones and proportionate enlargement of most organs. Thus SOCS proteins play an essential, negative role in regulating intracellular signaling from the receptors for erythropoietin, growth hormone, and other cytokines. KEY CONCEPTS OF SECTION 16.4 Cytokines, Cytokine Receptors, and the JAK/STAT Signaling Pathway Cytokines play numerous roles in development. Erythropoietin, a cytokine secreted by kidney cells, promotes proliferation and differentiation of erythroid progenitor cells in the bone marrow to increase the number of mature red cells in the blood (see
Figure 16-18a). Cytokines such as GH, prolactin, Epo, and G-CSF have very similar tertiary structures, as do their receptors. These and related cytokines form a cytokine: receptor homodimer complex on the cell surface (see Figures 16-18b and 16-19a). Other cytokines, exemplified by Interleukin 2, interact with two or more different receptor subunits, forming cytokine: receptor heteromultimeric complexes (see Figure 16-19b).
The cytosolic domains of cytokine receptors are tightly bound to a JAK protein tyrosine kinase, which becomes activated after cytokine binding and receptor dimerization or heteromerization and phosphorylates tyrosine residues on the cytosolic domain of the receptor (see Figure 16-19a). The JAK/STAT pathway operates downstream from all cytokine receptors and some RTKs. STAT monomers bound to phosphotyrosines on receptors are phosphorylated by receptor-associated JAKs, then dissociate from the receptor, dimerize and move to the nucleus, where they activate transcription (see Figure 16-20). Signaling from cytokine receptors is terminated by the phosphotyrosine phosphatase SHP1 and several SOCS proteins (see Figure 16-21).
16.5 The TGF-β Family of Growth Factors, Their Receptor Serine Kinases, and the Smad Transcription Factors They Activate
16.5 The TGF-β Family of Growth Factors, Their Receptor Serine Kinases, and the Smad Transcription Factors They Activate In this section, we discuss an evolutionarily conserved large family of signaling molecules, called the transforming growth factor β (TGF-β) family, as well as a conserved family of cell-surface receptors, called the TGF-β receptor family. Members of these two families are present in invertebrate organisms in phyla such as Porifera (sponges), and Cnidaria (coral and hydra) as well as in all vertebrates. Kinase domains in TGF-β receptors phosphorylate and thus trigger the activation of a conserved class of transcription factors (the Smads) that regulate many growth and differentiation pathways. Like the STATs discussed in the previous section, Smads are located in the cytosol in unstimulated cells, but when phosphorylated they move into the nucleus to regulate transcription. As with cytokine signaling, the TGF-β pathways have widely diverse effects in different types of cells because different members of the TGF-β family activate different members of the TGF-β receptor family, which activate different members of the Smad group of transcription factors. In addition, as with other receptor-activated
transcription factors, we will see that the same activated Smad protein partners with different transcription factors and thus activates different sets of genes in different types of cells. The mammalian TGF-β family contains 33 members and includes a number of related extracellular signaling molecules that play widespread roles in regulating development. The founding member of the TGF-β family, TGF-β1, was identified on the basis of its ability to induce a malignant phenotype in several cultured early-stage mammalian cancer cell lines (hence the name “transforming growth factor”); in this case, TGF-β1 promoted metastases, the spreading and invasion of primary tumors, as discussed in Chapter 25. Ironically, the principal function of all three human TGF-β isoforms, TGF-β1, 2, and 3, in most normal (noncancerous) mammalian cells is to prevent their proliferation by inducing the synthesis of proteins, including , which inhibit the cyclindependent kinases (CDKs) that are essential for progression into the DNA synthesis (S) phase of the cell cycle (see Figures 1-22 and 19-12). TGF-β is secreted by many cells in the body and inhibits the growth of both the secreting cell (autocrine signaling) and neighboring cells (paracrine signaling). Loss of TGF-β receptors, or of any of several proteins in the TGF-β signal transduction pathway, releases cells from this growth inhibition and is seen frequently in the early development of human tumors. TGF-β proteins also promote expression of cell-adhesion molecules and extracellular-matrix molecules, which play important roles in tissue organization (see Chapter 20).
TGF-β Proteins Are Stored in an Inactive Form in the Extracellular Matrix
Other members of the TGF-β family, bone morphogenetic proteins (BMP), were initially identified by their ability to induce bone formation when implanted in mice. One of these, BMP2, is used clinically to strengthen bone after severe fractures. Of the numerous BMP proteins subsequently recognized, many induce key steps in development, including formation of mesoderm and the earliest blood-forming cells; some are important for maintaining the undifferentiated state of cultured embryonic and adult stem cells (see Chapter 22). Many have nothing to do with bones; for example, the BMP4/BMP7 heterodimer is important in dorsal-ventral (back-to-front) patterning in vertebrate embryos. Similarly, a Drosophila homolog of TGF-β, called Dpp, participates in dorsal-ventral patterning in fly embryos. In the early human male embryo another member of the human TGF-β family, Müllerian-inhibiting hormone (MIH), inhibits the development of the female reproductive tract, and thus is critical to sex differentiation at a specific time during the fetal development of human males. TGF-β Proteins Are Stored in an Inactive Form in the Extracellular Matrix In mammals, the monomeric active forms of all members of the TGF-β family contain a conserved cysteine residue that forms a disulfide bond linking two monomers into a dimer (Figure 16-22a), and the precursor proteins of all TGF-β family members dimerize in the endoplasmic reticulum immediately after synthesis. The genes for all members of the
family encode a precursor polypeptide comprising a secretion signal peptide (Chapter 13), a ∼250-residue prodomain, and the C-terminal ∼110-residue TGF-β domain. The amino-terminal prodomain is required for proper folding and dimerization of the C-terminal TGF-β domain. As these dimeric precursor proteins pass through the Golgi complex, a protease severs the covalent connection between the prodomain and the TGF-β domain, but the prodomain and the mature TGF-β protein remain noncovalently attached.
FIGURE 16-22 Model for TGF-β latency and activation. (a) Ribbon diagram structure of a mature TGF-β homodimer. Three intrachain disulfide linkages (yellow) in each monomer stabilize a cystine-knot domain; another disulfide bond (red) links the two monomers. (b, c) TGF-β latency. TGF-β is synthesized with a 250-residue amino-terminal prodomain that in
the Golgi is proteolytically cleaved from the mature growth factor but remains noncovalently attached even after secretion. Both the mature growth factor and the prodomain form homodimers. The prodomain, its “arm” and “straitjacket” segments remaining tightly wrapped around the TGF-β growth factor, prevents the latent TGF-β from binding to a cell-surface receptor. Disulfide bonds on the prodomain link it covalently to LTBP or to other proteins in the extracellular matrix (b), or to the GARP protein on the surface of the secreting cell (c), localizing the latent TGF-β near the cell that produced it. (d) TGF-β activation. Certain integrins, perhaps on the surface of a nearby cell, bind to the RGD motifs on the arm segment of the prodomain. Because these integrins are also linked to the rigid actin cytoskeleton on the cytosolic side of the plasma membrane (Chapter 20), movements of the cell exert a traction force against the RGD motif, pulling and elongating the straitjacket segment of the prodomain. Consequently, the TGF-β dimer is released from the complex and is now able to bind to a cell-surface receptor and initiate signaling. [Part (a) Data from S. Daopin et al., 1992, Science 257:369, PDBID 2tgi. Parts (c, d) From A. Hinck et al., 2016, Cold Spring Harb. Perspect. Biol. 8:a022103.] Description The illustration labeled (a) shows a space-filling three-dimensional model of mature dimeric T G F-beta. The illustration labeled (b) is titled latent T G F-beta. At the top are two blue ovals labeled arm domain, below them is the green structure T G F-beta. This structure is attached to light blue ovals labeled L T B P, by a blue line labeled straight jacket. A series of loose lines are labeled extracellular matrix. The illustration labeled (c) is titled latent T G F-beta. The diagram is the same as that of illustration (b), except it is attached to a pink structure labeled G A R P, which has a protein tail going through the plasma membrane. The illustration labeled (d) titled T G F-beta activation by cytoskeletal force transmitted by integrins shows the same latent T G F-beta structure with label R G D above it with arrows pointing upward labeled integrin. A sideward arrow shows the active T G F-beta being removed with tensile force, leading to the arm domain structures attached to L T B P or G A R P. After TGF-β is secreted from the cell, the presence of the prodomain prevents the mature TGF-β protein from binding to its cell-surface
receptors; the TGF-β growth factor is thus said to be latent. In addition, the prodomain forms linkages that localize the latent TGF-β to the surface of the secreting cell, to a nearby cell, or to the extracellular matrix. Specifically, the side chain of a cysteine residue near the N-terminus of the prodomain becomes disulfide linked to one of several types of proteins in the extracellular matrix, such as latent TGF-β binding protein (LTBP;
Figure 16-22b) or to a protein called GARP on the surface of the secreting cell (Figure 16-22c). Several mechanisms can release the mature TGF-β dimer from the inhibitory prodomain, enabling the active TGF-β dimer to rapidly bind to nearby TGF-β receptors and initiate highly localized autocrine or paracrine signaling (Figure 16-22d). These mechanisms include cleavage of the prodomain by extracellular proteases and mechanical stress that pulls the prodomain away. Mechanical stress may be generated by the binding of the prodomains to a protein on the surface of a moving cell. The prodomains of some TGF-β family members (e.g., TGF-β1 and TGF-β3) contain a three-amino-acid sequence motif, RGD (Arg-Gly-Asp), that allows them to bind to certain members of the class of membrane proteins termed integrins on the surface of a nearby cell (discussed in Chapter 20); integrins bind to RGD motifs in many extracellular-matrix proteins. During cell migration, for example, a tensile force is generated due to movement of the cell containing the integrin relative to the extracellular matrix containing LTBP and the prodomain/TGF-β dimer. The force physically distorts the prodomain and frees the active TGF-β dimer, initiating TGF-β signaling.
Three Separate TGF-β Receptor Proteins Participate in Binding TGF-β and Activating Signal Transduction
Three Separate TGF-β Receptor Proteins Participate in Binding TGF-β and Activating Signal Transduction The approach taken to identify the receptors for TGF-β1 is representative of typical biochemical approaches to identifying receptors (see Section 15.2). Investigators first labeled the purified TGF-β1 protein with the radioisotope iodine-125 ( ; see Chapter 3) using conditions that covalently link to exposed tyrosine residues. The -labeled TGF-β1 was then incubated at 4 °C with cultured fibroblast cells that express the receptor. At 4 °C ligands bind to receptors, but endocytosis is inhibited. At the end of the incubation, unbound in the extracellular fluid was washed away and the cells with receptorbound were treated with a chemical that covalently crosslinks proteins that are physically close to one another, including ligands bound to their receptors. The cells were then disrupted and the investigators purified any proteins (and cross-linked protein complexes) that were radioactively labeled. Analysis of the purified proteins showed that was cross-linked primarily to three different polypeptides with molecular weights of 55, 85, and 280 kDa, referred to as TGF-β receptors RI, RII, and RIII, respectively. Many additional experiments established that RI and RII are each homodimeric proteins; each polypeptide chain has both a single transmembrane segment and a serine/threonine kinase as part of its cytosolic domain. RI and RII together are sufficient to serve as a
functional heterotetrameric TGF-β receptor. RIII is a transmembrane cellsurface protein expressed in some but not all cell types. Because RIII contains large, covalently bound oligosaccharide chains, it is a member of the class of glyco (sugar containing) proteins called proteoglycans (see
Figure 20-33). The function of RIII for TGF-β receptors is similar to that of the α subunit of the IL-2 receptor described above (Figure 16-19b); RIII binds and concentrates TGF-β molecules near the cell surface, facilitating their subsequent binding to the signaling receptors RII and RI (Figure 1623, step 1a ). As most cells lack RIII, TGF-β binds directly to the RII dimer (Figure 16-23, step 1b ). This step takes place before the RII dimer and RI bind each other (Figure 16-23, step 2 ). The complex is inactive until RI binds.
FIGURE 16-23 Activation of the TGF-β receptor kinases. Step 1a : In cells that express the type III TGF-β receptor (RIII), TGF-β binds first to this receptor, which concentrates TGF-β near the cell surface and thus facilitates the binding of TGF-β to one of the two type II TGF-β signaling receptors (RII) in a homodimeric complex. Step 1b : In most cells, RIII
is not expressed and TGF-β binds directly to one molecule of RII, a constitutively active kinase. Step 2 : Ligand-bound RII recruits a type I TGF-β receptor (RI), such that one monomer of the TGF-β dimer binds simultaneously to one RII and one RI protein. The constitutively active RII protein kinase then phosphorylates serine and threonine residues in the juxtamembrane segment of the RI receptor, releasing the inhibition of RI kinase activity. The now active type I receptor kinase then phosphorylates a Smad transcription factor. Description The illustration shows the membrane as a gray line with cytosol labeled below and exterior above. Step 1 a, the R 3 protein is represented by a blue oval with a large section above and a small section below the membrane. T G F-beta attaches to R 3. In step 1 b, T G F-beta attaches to R 2. In step 2, a right arrow shows the attachment of T G F-beta with R 2 and R 1. Step 3 shows the movement of R 1 toward R 2 after the attachment of T G F-beta to R 2. In the tyrosine kinase and cytokine receptors we have already discussed, ligand binding induces a substantial increase in kinase activity, which is required for activating intracellular signal transduction pathways. In contrast, the dimeric RII subunits of the TGF-β receptor exhibit constitutive kinase activity; that is, their kinase domains are enzymatically active even when there is no TGF-β bound. Active RII kinase activity is not sufficient to generate an intracellular signal. However, the binding of TGF-β to RII generates a new molecular surface at the TGF-β–RII interface that binds to a dimeric RI receptor (Figure 16-23, step 2 ), generating a complex that contains one dimeric TGF-β molecule and one each of the RI and RII receptor homodimers — another example of ligandinduced receptor hetero-oligomerization. RI will not bind to RII unless RII is bound to TGF-β. An RII subunit in the complex then phosphorylates serine and threonine residues in a highly conserved sequence of the
Activated RI TGF-β Receptors Phosphorylate Smad Transcription Factors
attached RI subunit adjacent to the cytosolic face of the plasma membrane, thereby activating RI kinase activity. The RI kinase then sets in motion the downstream intracellular signaling pathway. Activated RI TGF-β Receptors Phosphorylate Smad Transcription Factors The transcription factors downstream of the TGF-β receptors are called Smads. Three types of Smad proteins function in the TGF-β signaling pathway: R-Smads (receptor-regulated Smads; Smad2 and Smad3), coSmads (Smad4), and I-Smads (inhibitory Smads; Smad7). Smad2 and/or Smad3 become phosphorylated by active RI receptors and together with Smad4 move into the nucleus to activate gene transcription; in contrast, ISmads block the TGF-β signaling pathway, as discussed at the end of this section. As illustrated in Figure 16-24a, all R-Smads share a similar domain architecture comprising an N-terminal MH1 domain, a central proline-rich linker, and a C-terminal MH2 domain. The MH1 domain contains a DNAbinding segment as well as a nuclear-localization signal (NLS). However, when R-Smads are in their inactive, nonphosphorylated state, the NLS is masked so that it cannot direct the Smad into the nucleus (see Figure 1335), and the MH1 and MH2 domains associate in such a way that they could not bind to DNA or to a co-Smad. Phosphorylation of two serine residues in a common Ser-X-Ser motif at the extreme C-terminus of the
MH2 domain by the activated RI subunits of the TGF-β receptor separates the two domains, exposing the NLS for binding to importins and nuclear import (see Figure 13-36). Simultaneously, the two serines in each Smad2 or Smad3 that were phosphorylated by the TGF-β RI receptor kinase bind to phosphoserine-binding sites in the MH2 domains of a Smad4 or Smad2/3 protein, forming a stable complex containing two molecules of phosphorylated Smad3 (or Smad2) and one molecule of the co-Smad (Smad4). (Smad4 does not get phosphorylated but is essential to form a functional heterotrimeric Smad complex.) The bound importin then mediates translocation of the R-Smad/co-Smad complex into the nucleus. After importin dissociates inside the nucleus, the Smad3/Smad4 (or Smad2/Smad4) complex binds to other transcription factors to activate transcription of specific target genes, for example, the protease inhibitor gene PAI-1 in Figure 16-24a.
FIGURE 16-24 Activation of the TGF-β/Smad signaling pathway and its inhibition. (a) Activation. Step 1 : The activated type RI receptor kinase (see Figure 16-23) phosphorylates Smad2 or Smad3 (shown here as Smad2/3), causing a conformational change that unmasks its nuclear-localization signal (NLS). Step 2 : Two phosphorylated molecules of Smad2/3 bind to one co-Smad (Smad4) molecule, which is not phosphorylated, and to an importin, forming a multiprotein cytosolic complex. Steps 3 and 4 : After the entire complex translocates into the nucleus, Ran·GTP causes dissociation of the importin, as discussed in Chapter 13. Step 5 : One or more nuclear transcription factors (e.g., TFE3) then associates with the Smad2/3/Smad4 complex, forming an activation complex that cooperatively binds to a regulatory sequence of a target gene. Step 6 : This complex then recruits transcriptional co-activators and induces gene transcription (see Chapter 8). Smad2/3 is eventually dephosphorylated by a nuclear phosphatase (step 7 ) and escorted by an exportin (not shown) returns to the cytosol through a nuclear pore
(step 8 ), where it can be reactivated by another TGF-β receptor complex. Shown is the activation complex for the gene encoding plasminogen activator inhibitor (PAI-1); similar transcription complexes activate expression of genes encoding other extracellular-matrix proteins such as fibronectin. (b) Ski-mediated inhibition of transcription mediated by the Smad complex. Ski, whose synthesis is induced by the Smad2/3/Smad4 complex formed after TGF-β addition, represses Smad function by binding directly to Smad4. Since the Skibinding domain on Smad4 significantly overlaps with the Smad4 MH2 domain required for binding the phosphorylated C-terminus of Smad3, binding of Ski disrupts the normal interactions between Smad3 and Smad4 necessary for transcriptional activation. In addition, Ski recruits the protein N-CoR, which binds directly to mSin3A; in turn, mSin3A interacts with a histone deacetylase (HDAC), an enzyme that promotes histone deacetylation on nearby promoters and enhancers, repressing gene expression (see Chapter 8). As a result of both processes, transcription activation induced by TGF-β and mediated by Smad complexes is shut down. The related protein SnoN functions similarly to Ski in repressing TGF-β signaling. [Part (a) Data from S. Daopin et al., 1992, Science 257:369, PDB ID 2tgi; A. Moustakas and C.-H. Heldin, 2009, Development 136:3699; and D. Clarke and X. Liu, 2008, Trends Cell Biol. 18:430. Part (b) Data from J. Deheuninck and K. Luo, 2009, Cell Res. 19:47.] Description The illustration labeled (a) shows the membrane as a gray line with cytosol labeled below and exterior above. Above the membrane is the group of R 3, R 2, R 1 receptors with T G F-beta. Below the membrane, these receptors have phosphates attached. At step 1, S m a d 2 slash 3-P comes from the receptor. At step 2, S m a d 4 is added along with M H 2-M H 1 and I m p-beta along a downward arrow. These items are joined and enter the nucleus at step 3. At step 4, a green oval labeled R a n - G T P goes in, and R a n comes out. At step 5, T F E 3 comes in and the S m a d 4 continues to join with S m a d 2 slash 3-P at step 6. The D N A is produced and at step 7 the phosphatase dephosphorylates S m a d 2 slash 3. Step 8 shows S m a d 2 slash 3 and M H 1 returning to the cytosol and to the membrane. The illustration labeled (b) shows a D N A helix from top left, to the bottom left corner, then to the right, and changes into basepair notation. A multicolor structure is attached to the D N A on the left and at the bottom. The labels on these shapes are, from top to bottom, H D A C, m S i n 3 A, N-C o R, S k i, and S m a d 4. At the top of this structure, a red line from the H D A C
The R-Smad/co-Smad Complex Activates Expression of Different Genes in Different Cell Types
portion is labeled Histone deacetylation. This line moves down to the D N A where the label states: transcription and the rectangle below it, in the DNA, is labeled P A I - 1. An R-Smad’s time in the nucleus is limited to prevent overstimulation of the cell. Within the nucleus, R-Smads are further modified by phosphorylation of their linker domains, acetylation of their MH1 domains, and dephosphorylation of the C-terminal serines by nuclear phosphatases. Collectively, these many modifications result in diminution of transcriptional activity and ultimately in dissociation of the R-Smad/coSmad complex and export of the Smads from the nucleus via exportin proteins. Thus the concentration of active Smads within the nucleus closely reflects the levels of activated TGF-β receptors on the cell surface, allowing the regulation of gene transcription to closely follow the level of active TGF-β in the environment. BMP proteins, which also belong to the TGF-β family, bind to and activate a different set of receptors that are similar to the TGF-β RI and RII proteins but phosphorylate other R-Smads, Smad1, Smad5, and Smad8. Two of these phosphorylated Smads then form a trimeric complex with Smad4, and this Smad complex activates different transcriptional responses from those induced by the TGF-β receptors. The R-Smad/co-Smad Complex Activates Expression of Different Genes in Different Cell Types
Virtually all mammalian cells secrete at least one TGF-β isoform, and most have TGF-β receptors on their surface. However, the cellular responses induced by TGF-β vary among cell types, depending on the transcription factors that can partner with the R-Smad/co-Smad complex. In epithelial cells and fibroblasts, for example, TGF-β induces expression of extracellular-matrix proteins (e.g., fibronectins and collagens; see
Chapter 20); it also induces expression of proteins that inhibit serum proteases, which otherwise would degrade these extracellular-matrix proteins. The inhibition of serum proteases stabilizes the matrix, allowing cells to form stable tissues. In many cells the R-Smad/co-Smad complex promotes expression of genes encoding other proteins such as , which arrests the cell cycle at the stage and thus blocks cell proliferation (see Chapter 19). More generally, binding of the R-Smad/coSmad complex to DNA requires other transcription factors to bind at adjacent sites in the DNA; often these transcription factors are master transcription factors that determine the identity of a cell during its development. Equally important, in order for the R-Smad/co-Smad complex to bind to a DNA regulatory region and activate a given gene, the DNA binding segment must be in an active “open” chromatin conformation (see Figure 16-2). Loss of TGF-β signaling plays a key role in the early development of many cancers. Many human tumors contain inactivating mutations in either TGF-β receptors or Smad proteins and thus are resistant to growth inhibition by TGF-β (see Figure 25-18). Most human pancreatic cancers, for instance, contain a deletion in the gene encoding Smad4 and thus
Negative Feedback Loops Restrain TGF-β/Smad Signaling
cannot induce cell-cycle inhibitors such as in response to TGF-β. In fact, Smad4 was originally called DPC (deleted in pancreatic cancer). Retinoblastoma, colon and gastric cancer, hepatomas, and some T- and Bcell malignancies are also unresponsive to TGF-β growth inhibition. This loss of responsiveness correlates with loss of RI or RII; responsiveness to TGF-β can be experimentally restored by recombinant expression of the “missing” protein. Loss-of-function mutations in Smad2 also commonly occur in several types of human tumors. Negative Feedback Loops Restrain TGF-β/Smad Signaling As discussed previously in the context of other signaling pathways, the response to most growth factors and other signaling molecules decreases with time, a phenomenon called desensitization. This response is adaptive because it prevents overreaction and makes fine-tuned control of cellular responses possible. Two cytosolic proteins called SnoN and Ski (Ski stands for “Sloan-Kettering Cancer Institute”) are induced by TGF-β signaling in virtually all body cells and serve to suppress the TGF-β/Smad signaling pathway. These proteins were originally identified as cancer-causing oncoproteins because their expression is elevated in many cancers, including melanomas and certain breast cancers; consequently, growthinhibitory proteins normally induced by the TGF-β signal transduction pathway are not produced. Indeed, when overexpressed in cultured primary fibroblast cells, Ski or SnoN can cause abnormal cell
proliferation, and suppression of Ski in pancreatic cancers reduces tumor growth. SnoN and Ski trigger abnormal cell proliferation by binding to both the co-Smad (Smad4) and phosphorylated R-Smads (Smad3) after stimulation by TGF-β. SnoN and Ski do not prevent an R-Smad/co-Smad complex from forming or a Smad complex from binding to DNA regulatory regions. Rather, they block a Smad complex bound to DNA from activating transcription, in part by inducing deacetylation of histones in adjacent chromatin segments. With Smad blocked from activating transcription, the growth-inhibitory effects of TGF-β are diminished (Figure 16-24b). The increased levels of SnoN and Ski proteins induced by TGF-β are thought to dampen the long-term effects of signaling due to continued exposure to TGF-β; this is another example of negative feedback, in which a gene induced by TGF-β signaling, in this case SnoN, inhibits further signaling by TGF-β. Among other proteins induced after TGF-β stimulation are the I-Smads, especially Smad7. Smad7 binds to the activated RI subunit of TGF-β receptors and blocks its ability to phosphorylate Smad2 or Smad3. Like SOCS proteins in the cytokine signaling pathway (see Figure 16-21), Smad7 also recruits an E3 ubiquitin ligase that targets TGF-β receptors for degradation. In these ways, Smad7, like Ski and SnoN, participates in a negative feedback loop: its induction inhibits intracellular signaling stimulated by long-term exposure to the stimulating TGF-β hormone. KEY CONCEPTS OF SECTION 16.5
The TGF-β Family of Growth Factors, Their Receptor Serine Kinases, and the Smad Transcription Factors They Activate The transforming growth factor β (TGF-β) family includes a number of related extracellular signaling molecules that play widespread roles in regulating development. TGF-β dimers are stored in an inactive form bound to the inhibitory prodomain on a cell surface or in the extracellular matrix; release of active dimers — by mechanical stretching or protease digestion of the prodomain — initiates TGF-β signaling (see
Figure 16-22). TGF-β receptors consist of three types of subunits (RI, RII, RIII). Binding of members of the TGF-β family to the RII subunit, a constitutively active kinase, allows recruitment of the RI subunit to the RII subunit. RII phosphorylates the cytosolic domain of the RI subunit, activating RI’s serine/threonine kinase domain. RI then phosphorylates an R-Smad, exposing a nuclear-localization signal (see Figures 16-23 and 16-24). After phosphorylated R-Smads bind a co-Smad, the resulting complex translocates into the nucleus, where it interacts with various transcription factors to induce expression of target genes (see Figure 16-24). The Smad3/Smad4 complex often induces different genes in different cells by binding to regulatory DNA sequences at sites adjacent to those occupied by cell-specific master transcription factors (see Figure 16-24a). TGF-β signaling generally inhibits cell proliferation. Loss of any of several components of this signaling pathway can contribute to abnormal cell proliferation and malignancy. Oncoproteins (e.g., Ski and SnoN) and I-Smads (e.g., Smad7) suppress the TGF-β signaling pathway by inhibiting transcription mediated by the Smad2/3/Smad4 complex (see Figure 16-24b).
On Binding Delta, the Notch Receptor Is Cleaved, Releasing a Component Transcription Factor
16.6 Signal Transduction Pathways That Utilize Regulated, SiteSpecific Protein Cleavage: Notch/Delta and EGF Precursors All of the signaling pathways we have discussed so far are reversible and so can be suppressed or turned off relatively quickly if the extracellular signal is removed. In this section and the next, we discuss several pathways that are irreversible or only slowly reversible. Many of these pathways trigger key steps in metazoan differentiation, and the irreversibility of a signal transduction pathway can lock the cell into undergoing a specific series of developmental events. In these pathways, a critical protein — a receptor, a transcription factor, or a protein in a signaling cascade that regulates a transcription factor — undergoes regulated cleavage. On Binding Delta, the Notch Receptor Is Cleaved, Releasing a Component Transcription Factor We begin by looking at a pathway in which cleavage of a protein releases a domain that affects the transcription of multiple target genes. In the
Notch/Delta pathway (Figure 16-25a), the signaling molecule is Delta, an integral membrane protein bound to the surface of the signaling cell. Delta binds to the extracellular domain of the receptor, Notch, on a neighboring responding cell. The binding of Delta triggers two sequential proteolytic cleavages of Notch, releasing the Notch intracellular domain. That cleavage product then moves into the nucleus and functions as a transcriptional activator.
FIGURE 16-25 Notch/Delta signaling pathway. (a) In the absence of Delta, the transmembrane subunit of Notch on the surface of a responding cell is noncovalently
associated with its extracellular subunit; the extracellular domain is folded so that it cannot be cleaved by the cell-surface matrix metalloprotease ADAM 10. Binding of Notch to its ligand Delta on an adjacent signaling cell (step 1 ) is followed by endocytosis of Delta by the signaling cell (step 2 ), stretching the Notch extracellular domain so that ADAM 10 is able to cleave it (step 3 ). The released fragment of the Notch extracellular domain remains bound to Delta and is endocytosed by the signaling cell and degraded in lysosomes (step 4 ). Next the four-protein γ-secretase complex binds to the stump generated by ADAM 10 (step 5 ), and then the protease in the complex, presenilin 1, catalyzes cleavage of the Notch transmembrane alpha helix near the cytosolic face of the membrane, releasing the cytosolic segment of Notch (step 6 ). Following translocation to the nucleus, this Notch segment does not directly bind to DNA but rather interacts with several transcription factors to affect expression of genes that in turn influence the determination of cell fate during development (step 7 ). See R. Kovall et al., 2017, Dev. Cell 41:228; and S. Bray, 2016, Nat. Rev. Mol. Cell Biol. 17:772. (b) High resolution structure of the four-protein γ-secretase complex determined by cryoelectron microscopy. A bound Notch transmembrane peptide that contains the intermembrane cleavage site is colored red and the presenilin (PS1) subunit is colored cyan. The protease catalytic site in PS1 is located near the cytosolic surface. [Part (b) Data from G. Yang et al., 2019, Nature 565:192.] Description The illustration labeled (a) shows a signaling cell with bound delta protein. A responding cell is with notch bound to the transmembrane notch receptor. Nearby, an A D A M 10 protease is also embedded in the membrane. Binding of the notch and delta binding domains together results in cleavage of the notch protein and subsequent cleavage of the cytosolic portion of notch be a nicastrin-containing gamma-secretase complex, resulting in activation of nuclear transcription factors. Both Notch and Delta are cell-surface, single membrane-spanning proteins. Notch also has other ligands, but the molecular mechanisms of Notch activation are the same with each. Notch is synthesized as a monomeric membrane protein in the endoplasmic reticulum. In the Golgi
complex, it undergoes a proteolytic cleavage that generates an extracellular subunit and a transmembrane subunit that remain noncovalently associated with each other on the cell surface. The first of two cleavages of the Notch extracellular domain is performed by ADAM 10, a metalloprotease and member of a class of metalcontaining enzymes localized to the plasma membrane that cleave the extracellular segments of target proteins at a site near the plasma membrane. ADAM is an abbreviation for a disintegrin and metalloprotease; a disintegrin is a conserved protein domain that binds integrins and disrupts cell-matrix interactions (see Chapter 20). ADAM 10 cannot cleave Notch in the absence of Delta on an adjacent cell, because then the Notch extracellular domain is folded such that ADAM 10 cannot access the protease cleavage site (see Figure 16-25a). After Delta binds to Notch (Figure 16-25a, step 1 ), the Delta in the signaling cell undergoes endocytosis (step 2 ). The force accompanying the movement of Delta into the signaling cell stretches the Notch protein on the responding cell, changing its conformation and allowing access by ADAM 10, which cleaves the Notch extracellular domain (step 3 ). The Notch extracellular domain remains bound to Delta, is internalized by the signaling cell, and is probably degraded in lysosomes (step 4 ). A second cleavage of Notch follows quickly but this time it occurs within the hydrophobic membrane-spanning region of Notch; it is catalyzed by a transmembrane complex termed γ-secretase that contains presenilin 1
Metalloproteases Catalyze Cleavage of Many Signaling Proteins from the Cell Surface
(PS1), the protease enzyme, and three other essential subunits (Figure 1625a, step 5 , and Figure 16-25b). Cleavage by the γ-secretase complex releases the Notch cytosolic segment, which immediately translocates to the nucleus (Figure 16-25a, step 6 ). This Notch intracellular domain does not bind directly to DNA. Rather it interacts with a single DNA-binding transcription factor, CSL (RBPJ in vertebrates) whereby it affects transcription of different target genes depending on the cell type. Its effect, like those of other transcription factors activated downstream of other cell-surface receptors, depends on the constellation of epigenetic chromatin marks and the presence of cell-specific transcription factors; it can both increase and decrease gene expression. Metalloproteases Catalyze Cleavage of Many Signaling Proteins from the Cell Surface We next look at a pathway in which the cleavage of a cell surface protein releases a signaling molecule rather than a transcription factor. Many growth factors and other signaling molecules are synthesized as transmembrane proteins whose signal domain extends into the extracellular space. Some such proteins, like Delta described above, are often biologically active but can signal only by binding to receptors on adjacent cells. In other cases, proteolytic cleavage of such proteins can release the soluble, active signaling molecule into the extracellular space;
from there the signaling molecule can bind to receptors on nearby cells. The cleavage producing these signaling molecules is often carried out by ADAMs. The human genome encodes 21 metalloproteases in the ADAM family, but only 12 are known to be catalytically active; many are involved in cleaving the transmembrane precursors of signaling proteins just outside their transmembrane segment, releasing the soluble signaling protein into the extracellular space. Medically important examples of signaling proteins released from the cleavage of precursors are members of the EGF family, including EGF, HB-EGF, TGF-α, NRG1, and NRG2 discussed earlier (see Figures 16-6 and 16-7), which affect the proliferation and differentiation of many types of body cells. The activity of one or more ADAMs is increased in many cancers, leading to high levels of extracellular EGF-family growth factors. These stimulate the secreting cells (autocrine signaling) or adjacent cells (paracrine signaling) to proliferate inappropriately. ADAM proteases also are an important factor in heart disease. As we learned in the last chapter, stimulation of β-adrenergic receptors in heart muscle by epinephrine (adrenaline) causes glycogenolysis and an increase in the rate of muscle contraction. Prolonged treatment of heart muscle cells with epinephrine, however, leads to activation of an ADAM by an unknown mechanism. This ADAM protease cleaves the transmembrane precursor of HB-EGF, and the released HB-EGF then binds to EGF receptors on heart muscle cells and stimulates their inappropriate growth. The excessive proliferation of heart muscle cells can lead to an enlarged
but weakened heart — a condition known as cardiac hypertrophy, which may cause early death. How cleavage of these proteins by ADAM metalloproteases is regulated and how specificity of substrate cleavage on the cell surface is achieved are still not completely understood. KEY CONCEPTS OF SECTION 16.6 Signal Transduction Pathways That Utilize Regulated, Site-Specific Protein Cleavage: Notch/Delta and EGF Precursors On binding to its ligand, Delta, on the surface of an adjacent cell, the receptor Notch protein undergoes two proteolytic cleavages (see Figure 16-25), first by an ADAM protease and then within the plane of the plasma membrane by γ-secretase. The released Notch cytosolic segment then translocates into the nucleus and modulates transcription of target genes critical in determining cell fate during development. Many important growth factors and other signaling proteins such as EGFs are synthesized as transmembrane proteins; regulated cleavage of the precursor near the plasma membrane by members of the ADAM protease family releases the active molecule into the extracellular space to signal distant cells. Inappropriate cleavage of EGF-family precursors can result in abnormal cell proliferation, potentially leading to cancer, cardiac hypertrophy, and other diseases.
16.7 Signal Transduction Pathways That Utilize Proteasomal Degradation of Signaling Components: Wnt, Hedgehog, and the Many Hormones That Activate NF-κB
16.7 Signal Transduction Pathways That Utilize Proteasomal Degradation of Signaling Components: Wnt, Hedgehog, and the Many Hormones That Activate NF-κB Next we discuss signal transduction pathways in which either a transcription factor or an inhibitor of a transcription factor becomes ubiquitinated and then is either cut or totally degraded by the proteasome. We begin with signal transduction pathways induced by the Wnt and Hedgehog (Hh) proteins, two evolutionarily conserved families of signaling proteins that play key roles in many developmental pathways and often induce expression of genes required for a cell to acquire a new identity or fate. Although Wnt and Hedgehog signaling pathways use different sets of receptors and signaling proteins, they do share similarities, which is why we group them together: In the unstimulated state, key transcription factors in both pathways are initially localized in large cytosolic multiprotein complexes; enzymes in the complex add ubiquitin chains to the transcription factors. In the Wnt pathway the transcription factor becomes totally degraded by the proteasome, whereas in the Hh pathway the
Wnt Signaling Prevents Destruction of a Transcription Factor by a Cytosolic Protein Complex
transcription factor is cleaved by the proteasome, generating a repressor of transcription. Activation of both pathways involves disassembly of these protein complexes, inhibition of cleavage or degradation of the transcription factor, and movement of the full-length, active transcription factor into the nucleus. Both pathways regulate cell fate decisions in animal development. Next we examine the NF-κB signal transduction pathway that is activated downstream of many important hormones that affect the immune system. In this case, an inhibitor of a transcription factor, rather than a transcription factor itself, is degraded following ubiquitinylation. In the resting state, the transcription factor termed NF-κB is sequestered in the cytosol and bound to an inhibitor. Several signal transduction pathways, activated by different hormone receptors, converge on an enzyme that ubiquitinates and thus causes rapid degradation of the inhibitor. Freed of its inhibitor, NF-κB immediately and vigorously moves to the nucleus and activates transcription of multiple genes. In learning how the NF-κB pathway is activated by one class of surface receptors, we also see a very different function of polyubiquitinylation: the formation of a scaffold to assemble a key signal transduction complex. Wnt Signaling Prevents Destruction of a Transcription Factor by a Cytosolic Protein Complex
The components of the Wnt signaling pathways have been conserved throughout the evolution of metazoan organisms and were elucidated mainly through genetic analysis of developmental mutants in Drosophila. In vertebrates, mutations in these pathways are thought to trigger several types of cancers. In fact, the first vertebrate Wnt gene to be discovered, the mouse Wnt-1 gene, attracted notice because it was overexpressed in certain mouse mammary cancers. When a mouse retroviral DNA, the mammary tumor virus (MMTV) genome, became inserted near the Wnt-1 gene, the retrovirus LTR promoter (see Figure 7-13) activated inappropriate expression of the Wnt-1 gene. The human genome encodes 19 different Wnt proteins, and many Wnts are essential for critical developmental events, including bone formation, the segmented development of the musculoskeletal system in vertebrates, and the formation and maintenance of skin and hair. Yet all Wnt proteins are thought to use the same set of cell-surface receptors and signal transduction proteins. Wnt proteins are signaling molecules secreted from the cell and are modified by linkage of a monounsaturated fatty acid, palmitoleic acid, to a serine in the middle of the protein. Like other secreted signaling proteins, Wnt proteins interact with several extracellular and cell-surface proteins and activate multiple downstream signal transduction pathways. The principal signaling receptor for Wnt proteins is Frizzled (Fz), which contains seven transmembrane α helices. Like the glucagon receptor (see
Figure 15-15), Fz has a large extracellular domain that is connected to the first membrane-spanning α helix and contains the major Wnt binding site.
Unlike the glucagon receptor, however, as far as is known Fz does not activate a G protein. The palmitoleate attached to the Wnt protein binds to a specific site on the Fz extracellular domain and stabilizes the Wnt-Fz complex. Thus this lipid is central to receptor engagement by Wnt proteins and is one of the few known examples of post-translational modification by a lipid that mediates a ligand-receptor interaction. At least three different signal transduction pathways can be activated by the binding of different Wnt proteins to particular Fz proteins. The most widespread, canonical Wnt signaling pathway has a second transmembrane receptor, LRP (called Arrow in Drosophila), that associates with Fz such that Wnt binds simultaneously to both (Figure 1626). Inactivating mutations in the genes encoding Wnt proteins, Fz, or LRP all have similar effects on the development of mouse embryos, indicating that all three proteins are essential for Wnt signaling.
FIGURE 16-26 Canonical Wnt signaling pathway. (a) In the absence of Wnt, the transcription factor TCF is bound to promoters or enhancers of Wnt target genes. Its activator β-catenin is absent from the nucleus, and TCF is associated with transcriptional repressor proteins such as Groucho (Gro) that inhibit gene transcription. The key signaling molecule in the canonical Wnt pathway is the cytoplasmic protein β-catenin, whose stability is controlled by a large multiprotein β-catenin destruction complex. The tumor suppressor protein Axin is the scaffold of the complex, and binds to β-catenin, the tumor suppressor APC, the Dishevelled (Dvl) protein, and two constitutively active protein kinases, glycogen synthase kinase 3β (GSK 3β) and casein kinase 1 (CK1). CK1 and GSK3 sequentially phosphorylate β-catenin at multiple serine and threonine residues. The E3 βTrCP ubiquitin ligase then binds to two phosphorylated residues on β-catenin, leading to ubiquitinylation of β-catenin and its degradation in proteasomes. (b) Wnt proteins bind to a complex of two receptor proteins on the cell surface, Frizzled (Fz) and LRP. Fz proteins have seven transmembrane segments and an extracellular N-terminal domain that binds Wnt. Binding
of Wnt to these receptors induces phosphorylation of the LRP cytosolic domain by several kinases, followed by binding of Axin to phosphorylated LRP and Dvl to Fz. This causes part of the Axin–APC–CK1–GSK3–β-catenin complex to fall apart, preventing phosphorylation of β-catenin by CK1 and GSK3, inhibiting ubiquitination of β-catenin, and allowing β-catenin to accumulate in the cytosol. After translocation to the nucleus, β-catenin binds to TCF, displaces the Gro repressor, and recruits co-activator proteins to activate expression of Wnt target genes. See R. Nusse and H. Clevers, 2017, Cell 169:985; and also the Wnt home page, http://web.stanford.edu/group/nusselab/cgi-bin/wnt. Description The illustration labeled (a) is titled W n t OFF. It shows a cell membrane, cytosol, nuclear membrane, and nucleus. Starting at the top, there is a set of 7 cylinder shaped structures together in the membrane, with one end at the left in the cytosol, and the other end at the right in the exterior. The label Frizzled is on this structure. Next to this at the right is a purple rectangle labeled L R P, which goes through the membrane. In the cytosol, a multicolored diagram is labeled beta-catenin destruction complex. At the left end of this is a row of phosphates (yellow circles) connects to U b circles and a beta-cat (light yellow oval). An arrow from this group points to a rectangle labeled proteasome. An arrow from proteasome points to small pieces representing the degradation of proteasome. Within the nucleus below, a separate structure is labeled W n t target gene and shows a D N A ribbon with a pink rectangle labeled T C F and a brown oval labeled Groucho. A cross symbol denotes no activation of W n t target gene The illustration labeled (b) is a similar diagram but is titled W n t ON. In this diagram, the seven cylinders are in the membrane and labeled Frizzled. The end of the receptor thread in the exterior is attached to a red oval labeled W n t, and also it is attached to the L R P rectangle. The action then moves through the membrane to the beta-catenin destruction complex and adds only 2 phosphates. From the complex at the left, downward arrows show several yellow ovals labeled beta-cat moves into the nucleus. In the nucleus, the beta-cat attaches to the T C F oval and D N A ribbon, and activates the expression of W n t target gene.
The central player in the canonical Wnt intracellular signal transduction pathway is a protein called β-catenin in vertebrates and Armadillo in Drosophila. This multi-talented protein functions both as a transcription factor in the Wnt signaling pathway and as a linker protein that connects cell adhesion proteins in the plasma membrane to the actin cytoskeleton (see Figure 20-14). In the absence of a Wnt signal, the β-catenin molecules that are not involved in membrane-cytoskeleton interactions (“free” β-catenin) are targeted for degradation by a cytosolic destruction complex that is held together by the scaffold protein Axin. This complex contains the tumor suppressor adenomatous polyposis coli (APC) protein, so named because its loss may result in colorectal cancer. The large size of the destruction complex observed by fluorescence microscopy suggests that it contains many copies of each component. In the resting state, two constitutively active kinases in the destruction complex, casein kinase 1 (CK1) and GSK3, sequentially phosphorylate β-catenin on multiple serine and threonine residues; some of these phosphorylated residues serve as binding sites for an E3 ubiquitin-ligase protein named βTrCP. β-Catenin is then ubiquitinylated by βTrCP and rapidly degraded by the 26S proteasome (Figure 16-26a; for more on ubiquitinylation, see Figure 3-32). Degradation prevents free β-catenin from entering the nucleus and activating transcription of Wnt target genes. The complete mechanism by which Wnt signaling blocks the degradation of β-catenin, enabling it to act as a transcription factor, has not yet been fully elucidated. The formation of the Wnt/Fz/LRP complex at the cell surface causes the adaptor protein Dishevelled (Dvl), a component of the
Concentration Gradients of Wnt Protein Are Essential for Many Steps in Development
destruction complex, to bind to Fz and Axin to bind to LRP. This results in the destabilization and inactivation of the destruction complex — no phosphorylation or subsequent polyubiquitinylation of β-catenin. Thus free β-catenin accumulates; it transfers into the nucleus and activates transcription of Wnt target genes (Figure 16-26b). Aberrant hyperactive Wnt signaling and abnormally high levels of free β-catenin is implicated in the progression of many cancers; the Wnt signaling pathway is hyperactive in more than 90 percent of human colon cancers (see Chapter 25). This observation provided one of the earliest clues that β-catenin can activate many growth-promoting genes. Inactivating mutations in genes encoding the tumor suppressors APC and Axin, components of the destruction complex, are found in multiple types of human cancers, as are mutations in β-catenin phosphorylation sites for the destruction complex kinases GSK3 or CK1; these mutations either reduce the formation of the destruction complex or reduce the phosphorylations on β-catenin (see Figure 16-26a). Thus they reduce degradation of β-catenin and allow free β-catenin to activate gene expression even in the absence of the normal Wnt signal. Among the Wnt target genes are many that also control Wnt signaling, indicating a high degree of feedback regulation. The importance of β-catenin stability and localization means that Wnt signals affect a critical balance between the three pools of β-catenin in the cell: at the membranecytoskeleton interface, in the cytosol, and in the nucleus.
Concentration Gradients of Wnt Protein Are Essential for Many Steps in Development Wnts are proteins secreted from the cell; however, in part because of the hydrophobic lipid that is covalently linked to these proteins, these proteins diffuse only a short distance from a signaling cell and generally have only localized effects. As Wnt diffuses farther and farther away from secreting cells, its concentration decreases. Different Wnt concentrations induce different fates in target cells: cells that detect a large amount of Wnt turn on certain genes and form certain structures; cells that detect a smaller amount turn on different genes and so form different structures. Signals that induce different cell fates depending on their concentration at their target cells are referred to as morphogens. In the development of the vertebrate nervous system, for instance, high levels of Wnt signaling specify that a certain progenitor cell becomes a posterior neuron while low levels of Wnt signaling specify that the progenitor cell becomes an anterior neuron. A striking example of a Wnt gradient regulating tissue pattern occurs in the adult planarian Schmidtea mediterranea (see Figure 1-23e); Wnt mRNA and protein are expressed in a posterior-to-anterior gradient (Figure 16-27a). If the planarian’s head is cut off, a new one regenerates within 14 days; a normal, albeit smaller, worm is regenerated. Similarly, after removal of the tail, a new one regenerates.
EXPERIMENTAL FIGURE 16-27 Gradients of Wnt are essential for normal regeneration of a head and a tail by planaria. (a) As shown by in situ hybridization, the WntP-2 gene (pink dots) is expressed in a posterior-to-anterior gradient in adult planaria. J. Witchley et al., 2013, Cell Rep. 4:633. (b) As indicated in the diagram, a small piece excised from the middle of the body of the planarian S. mediterranea was placed in culture. In situ mRNA hybridization performed after 4 days indicated that Wnt mRNA (pink dots) was expressed in cells at both the anterior and posterior wound sites, but that the Wnt inhibitor Notum (blue dots) was expressed only at the anterior wound site. Thus a posteriorto-anterior gradient of Wnt protein is formed. (c) After 14 days in culture, the excised body piece has regenerated a normal, albeit smaller, worm, with a head, easily visualized by the two eyes, from the anterior wound site and a tail from the posterior. Treatment of the excised body piece with an inhibitory RNA specific for β-catenin results in regeneration of a two-headed planarian, whereas treatment with an inhibitory RNA specific for Notum results in regeneration of a two-tailed planarian. Treatment of the excised body piece with two inhibitory RNAs, one specific for β-catenin and the other for Notum, results in a
phenotype similar to that caused by the loss of β-catenin alone: a two-headed planarian is regenerated. (d) These experiments give rise to a model in which β-catenin, stabilized by addition of Wnt to cells, causes expression of genes that promote tail formation; inhibition of Wnt/β-catenin signaling by Notum causes a head to be formed. [Part (a) photo: Jessica Witchley and Peter Reddien. Part (c, left two photos) Republished with permission from AAAS, from C. P. Petersen and P. W. Reddien, 2008, “Smed-β-catenin-1 Is Required for Anteroposterior Blastema Polarity in Planarian Regeneration,” Science 319(5861):327–330; photos courtesy of J. Witchley and Peter Reddien. Part (c, right two photos) Republished with permission from AAAS, from C. P. Petersen and P. W. Reddien, 2011, “Polarized notum Activation at Wounds Inhibits Wnt Function to Promote Planarian Head Regeneration,” Science 332(6031):852–855; permission conveyed through Copyright Clearance Center, Inc.; photos courtesy of J. Witchley and Peter Reddien.] Description The micrograph labeled (a) shows W n t m R N A in adult Planaria. The illustration labeled (b) shows the effect of W n t gradient on head and tail regeneration in Planaria. A schematic of a planarian is marked with dotted lines between the head and pharynx. The anterior portion contains more notum m R N A than W n t m R N A and the posterior section only has W n t m R N A. The micrographs labeled (c) show dissected planarians treated with inhibitory R N A. The illustration labeled (d) shows the messenger pathway for regeneration. In the pathway, a wound signal causes the generation of W n t, which is blocked by N O T U M. W n t activates beta-catenin, which when present leads to the tail formation, and when absent, leads to head formation. Strikingly, a small body piece taken from the middle of the animal regenerates a normal head from the anterior (head-facing) wound and a tail from the posterior wound (Figure 16-27b). Wnt signaling is required for the choice of head versus tail made at the wounds in these fragments. The gene Wnt-1 is expressed at all planarian wounds. By contrast, the gene encoding the secreted extracellular enzyme Notum is expressed only at
anterior-facing wounds. Notum inhibits Wnt signaling by cleaving off palmitoleic acid, the fatty acid that is covalently attached to Wnt and, as we learned above, is essential for Wnt signaling. RNA interference (RNAi) can be used to inhibit either the β-catenin gene, eliminating Wnt signaling, or the notum gene, or both genes. As shown in
Figure 16-27c, in the absence of all Wnt signaling, a two-headed planarian is regenerated. In contrast, in the absence of Notum, a two-tailed planarian is regenerated; in this case, Wnt is present and active at both the anterior and posterior wounds, and thus two tails are formed. In the absence of both Wnt signaling and Notum, the phenotype is similar to that caused by the loss of Wnt alone: a two-headed planarian is regenerated. Wnt signaling is absent at both the anterior and posterior wounds, inducing head regeneration at both sites, and the absence of Notum is irrelevant in the absence of Wnt. These experiments show that Wnt/β-catenin signaling promotes tail regeneration and inhibits head regeneration; the absence of Wnt signaling induces head formation at wounds (Figure 16-27d). In Chapter 22, we will learn that mature planarians contain pluripotent stem cells termed neoblasts that can differentiate into any body cell type. Gradients of Wnt protein play a major role in instructing neoblasts to differentiate into the appropriate cell types of tissues that constitute the planarian head or tail. As evidence, RNA interference (RNAi) has been used to specifically inhibit β-catenin throughout an uninjured animal. Even in these uninjured animals, inhibition of Wnt signaling by this means causes heads to appear around the body. These are consequences of defects
Hedgehog Signaling Relieves Repression of Target Gene Expression
in the pattern of tissue replacement and repair mediated by stem cells (see the chapter opening figure). Hedgehog Signaling Relieves Repression of Target Gene Expression The Hedgehog (Hh) signaling pathway is similar to the Wnt pathway in that two membrane proteins, one with seven membrane-spanning segments, are required to receive and transduce a signal. The Hh pathway also involves the disassembly of an intracellular complex containing a transcription factor. Hh signaling differs from Wnt signaling in that its two membrane receptors move between the plasma membrane and intracellular vesicles, and that in mammalian cells, the Hh signaling pathway is restricted to the single primary cilium that protrudes from the cell surface of most vertebrate cells. Like Wnt proteins, Hh proteins contain a covalently attached lipid and often act as morphogens and signal to influence the fate of nearby cells. Hh signaling plays essential roles in the development of numerous organ systems in animals. For instance, Hh signaling specifies the identities of neurons made along the ventral (belly) to dorsal (back) axis of the embryonic neural tube, which makes the spinal cord and brain. Hh signaling also regulates lung morphogenesis and hair follicle formation. One of the three mammalian Hh proteins, Sonic Hedgehog (Shh), is essential for normal patterning of the limbs; abnormal expression of Shh
Processing of Hh Precursor Protein
in an anterior region of the developing limb, in addition to its normal expression in the posterior domain, leads to polydactyly (extra digits). Processing of Hh Precursor Protein Hedgehog proteins are formed from a precursor protein with autoproteolytic activity that enables the protein to cut itself in half while still in the endoplasmic reticulum. The cleavage produces an N-terminal fragment, which is subsequently secreted to signal other cells, and a C-terminal fragment, which is degraded. As shown in Figure 16-28, cleavage of the precursor is accompanied by covalent addition of the lipid cholesterol to the new carboxyl terminus of the N-terminal fragment. A second modification to Hedgehog, the addition of a palmitoyl group to the N-terminus, makes the protein even more hydrophobic.
FIGURE 16-28 Processing of Hedgehog (Hh) precursor protein. Cells synthesize a 45 kDa Hh precursor, which in the endoplasmic reticulum undergoes a nucleophilic attack by the thiol side chain of cysteine 258 (Cys-258) on the carbonyl carbon of the adjacent residue glycine 257 (Gly-257), forming a high-energy thioester intermediate. Enzyme
The Hh Receptors Patched and Smoothened and the Downstream Signaling Pathway Were Initially Elucidated by Genetic Studies of Drosophila Development
activity in the C-terminal domain then catalyzes the formation of an ester bond between the hydroxyl group of cholesterol and glycine 257, cleaving the precursor into two fragments. The N-terminal signaling fragment (blue) retains the cholesterol group and is also modified by the addition of a palmitoyl group to the N-terminus. Once Hh has been secreted, the two hydrophobic anchors may tether the processed Hh protein to the plasma membrane. See P. Thérond, 2012, Curr. Opin. Cell Biol. 24:173. Description The hedgehog (H h) precursor protein is represented schematically by a 20-kilo Dalton portion at the N-terminal (colored in blue) and a 25-kilo Dalton portion (colored in green) at the C-terminal end. At the connection between these portions, two residues are present, a glycine residue and a cysteine residue. The Glycine-257 portion is attached to the Cysteine-258 by a sulfur molecule, forming a thioester. The thioester is then cleaved by nucleophilic attachment of the cholesterol hydroxyl group, forming a membrane-tethered H h. A palmitoyl group is tethered to the N-terminal, resulting in the membrane-tethered hedgehog. Despite its hydrophobicity Hh proteins can travel relatively long distances — up to 300 μm in the developing vertebrate limb. Both Hh and Wnt are often anchored to the phospholipid monolayers of extracellular lipoprotein particles (see Figure 14-27 for the structure of a typical lipoprotein) via their attached lipid groups, and while anchored to these particles they can diffuse in extracellular spaces. However, the majority of the Hh proteins produced by a cell can remain bound to its plasma membrane; in such cases, Hh signals mainly by cell-cell contact. As with Wnt proteins, spatial restriction plays a crucial role in constraining the effects of Hh proteins. The Hh Receptors Patched and Smoothened and the Downstream
Signaling Pathway Were Initially Elucidated by Genetic Studies of Drosophila Development Two membrane proteins, Smoothened (Smo) and Patched (Ptc), are required to receive a Hedgehog signal and transduce it to the cell nucleus. Smo has seven membrane-spanning α helices, is a member of the GPCR superfamily of proteins, and is related in sequence to the Wnt receptor Fz. Ptc contains 12 transmembrane α helices and is most similar structurally to the Niemann-Pick C1 (NPC1) protein, a cholesterol-transporting member of the ABC superfamily of membrane transport proteins (see
Table 11-3). Ptc is the receptor to which Hh binds, but Smo is the membrane protein that initiates the signal to the nucleus. One can think of the pathway as two series of steps: The binding of Hh to Ptc initiates a series of steps that brings Smo to the plasma membrane, and from there Smo initiates another series of steps that frees a transcription factor from a cytosolic complex.
Figure 16-29 depicts the current model of the Hedgehog pathway in Drosophila, where the pathway is known in the most detail. Early evidence supporting this model came from the study of fly embryos with loss-offunction mutations in the hedgehog (Hh) or smoothened (Smo) genes. Both types of mutant embryos have very similar developmental phenotypes; the name hedgehog came from the appearance of Hh mutant embryos, which were covered by an array of disorganized hairlike bristles that resembled
the spines on hedgehogs, as a result of a defect in the patterning of body segments. Moreover, both the Hh and Smo genes are required to activate transcription of the same target genes (e.g., patched and wingless) during embryonic development. In contrast, loss-of-function mutations in the patched (Ptc) gene produce a quite different phenotype, one similar to the effect of flooding the embryo with Hedgehog protein. These findings suggested that, in the absence of Hh, Ptc represses target genes by inhibiting a signaling pathway that requires Hh and Smo and is needed for gene activation. The additional observation that Smo is required for the transcription of Hh target genes in mutants lacking patched function places Smo downstream of Ptc in the Hh pathway. Together with biochemical experiments showing that Hh binds directly to Ptc, this genetic evidence indicates that, in the absence of Hh, Ptc blocks Smomediated activation of the transcription of Hh target genes. Binding of Hh to Ptc relieves this inhibition and permits the transcription of target genes.
FIGURE 16-29 Hedgehog signaling in Drosophila. (a) In the absence of Hedgehog (Hh), the Hh receptor protein Patched (Ptc) inhibits Smoothened (Smo), which is present largely in the membranes of internal vesicles. Smo becomes ubiquitinated and then is degraded in proteasomes. A cytosolic complex containing the kinase Fused (Fu); other kinases including protein kinase A (PKA), glycogen synthase kinase 3β (GSK3β), and casein kinase 1 (CK1), the zinc-finger transcription factor Cubitis interruptus (Ci), and SUFU (Suppressor of Fu), a protein that binds to and inhibits Ci, is bound to a microtubule by the kinesin-related motor protein Costal-2 (Cos2). In this complex, Ci becomes phosphorylated in a series of steps catalyzed by PKA, GSK3β, and CK1. The phosphorylated Ci is then proteolytically cleaved by the ubiquitin-proteasome pathway, generating an N-terminal fragment Ci75, which is transported into the nucleus and functions as a transcriptional repressor of Hh target genes. (b) Hh binds to Ptc, causing Ptc to be endocytosed from the cell surface and degraded, thereby relieving the inhibition of Smo. Smo then moves to the plasma membrane and its cytosolic C-terminus is phosphorylated by PKA, CK1, and other kinases. Then the C-terminal segment of Smo binds Cos2, and Smo is stabilized from degradation. Both Fu and Cos2 become extensively phosphorylated, and most importantly, the Fu-Cos2-Ci complex dissociates. This leads to the stabilization of the full-length Ci, which moves into the nucleus, displaces the repressor Ci75 from the promoter of target genes, recruits the CREBbinding activator protein (CBP), and induces expression of target genes. The exact membrane compartments in which Ptc and Smo respond to Hh and function are unknown. See S. Goetz and K. Anderson, 2010, Nat. Rev. Genet. 11:331; and R. Heck et al., 2016, Development 143:367. Description The illustration labeled is titled minus H h. The background is the cell membrane with exterior and cytosol label, plus the nucleus and nuclear membrane toward the bottom of the diagram. At the top left, in the membrane is a cylinder structure labeled P t c. Below is a gray oval labeled vesicle with a second cylinder structure labeled S m o. Below the S m o is a downward arrow leading to the word degradation. To the right of the vesicle, is a multicolored structure with a light blue rectangle labeled S U F U, blue circle labeled C o s 2, a blue structure labeled F u, and a light purple one labeled Kinases C K 1, P K A, G S K-3. A dark purple circle labeled C i is attached. Microtubules are thin green structures attached below this structure. An arrow moves
from the C i circle toward the nucleus and changes to C i 75 as it passes the nuclear membrane. This C i 75 attaches to the target gene represented in ribbon form. The illustration labeled (b) is titled plus H h. In this diagram, the cylinder structure at the left now has a red rectangle labeled H h attached. The vesicle is empty, but the S m o structures went up into the cell membrane next to the P t c structure. The light purple kinases are shown as a separated structure, and the multicolored structure is now attached to a second S m o cylinder structure in the membrane. From this area at the right, a purple C i oval is attached to an S U F U rectangle and the C i moves into the nucleus and attaches to the target genes along with a gray oval labeled C B P. Subsequent immunostaining and subcellular fractionation studies showed that, in the absence of Hh, Ptc is enriched in the plasma membrane but Smo is found in membranes of internal vesicles (Figure 16-29a). Precisely how Ptc inhibits Smo function is poorly understood. In the absence of Hh, a microtubule-bound complex containing several different proteins including Fu and Cos2 sequesters in the cytosol the zincfinger transcription factor called, in Drosophila, Cubitis interruptus (Ci). The protein complex contains at least four kinases, including the same two that phosphorylate β-catenin in the Wnt pathway. Phosphorylation of Ci in this complex by these kinases triggers binding of a component of an E3 ubiquitin ligase complex, which in turn directs ubiquitinylation of Ci and its targeting to proteasomes. There Ci undergoes a specific proteolytic cleavage but not complete degradation to amino acids; the resulting Ci fragment, designated Ci75, translocates to the nucleus and represses expression of Hh target genes.
Feedback Regulation of Hh Signaling
Following binding of Hh to the receptor Ptc, the Hh-Ptc complex, like many other receptor-ligand complexes, is endocytosed from the cell surface into internal vesicles and eventually degraded; the binding of Hh to Ptc also inhibits the ability of Ptc to inhibit Smo (Figure 16-29b). The now active Smo moves from internal vesicles to the plasma membrane, where it initiates the signaling pathway that sends the full-length transcription factor Ci to the nucleus. The C-terminal cytosol-facing domain of Smo becomes phosphorylated through the coordinated activities of several kinases. Then Cos2 binds to the phosphorylated C-terminal tail of Smo and itself becomes phosphorylated. This leads to a disruption of the cytosolic complex of Fu, Cos2, and Ci, and its dissociation from microtubules, triggering a reduction in both phosphorylation and proteolytic cleavage of Ci. As a consequence, full-length Ci is released from the dissociated complex and translocates to the nucleus, where it binds to the transcriptional co-activator CREB-binding protein (CBP), activating rather than repressing Hh target genes. Feedback Regulation of Hh Signaling As in other signaling pathways, feedback control of the Hh pathway is important because unrestrained Hh signaling can cause cancerous overgrowth or formation of the wrong cell types. In Drosophila, one of the genes induced by the Hh signal is patched. The subsequent increase in expression of Ptc dampens the Hh signal in large measure by reducing the pool of active Smo protein inside the cell and by sequestering Hh protein on the cell surface. Thus the system is buffered: if during development too
Hedgehog Signaling in Vertebrates Requires Primary Cilia
much Hh signal is made, a consequent increase in Ptc will compensate; if too little Hh signal is made, the amount of Ptc produced is decreased. Hedgehog Signaling in Vertebrates Requires Primary Cilia The Hedgehog signaling pathway in vertebrates shares many conserved features with the Drosophila pathway, but there are also some striking differences. First, mammalian genomes contain three Hh genes and two Ptc genes, which are expressed differentially among various tissues. Second, mammals express three Gli transcription factors, which collectively perform the roles of the single Ci transcription factor in Drosophila. All other components of the Hh pathway in Drosophila also are conserved in mammals. The most fascinating aspect of the mammalian Hh pathway is the involvement of the primary cilium. Cilia are long, thin structures enveloped by a plasma membrane that protrude from the cell surface. The roles of cilia and flagella in specialized cell types — such as moving materials along the airway surface formed by tracheal cells and powering the locomotion of sperm — are well known (see Figure 1-15 and Chapter 18). Most vertebrate cells have a single cilium called the primary cilium, a slim, nonmotile structure that projects from the surface, but primary cilia are conspicuously absent in almost all invertebrate cell types that have been examined.
As we will learn in Chapter 18, a cilium is extended and maintained by the transport of proteins and particles along a bundle of microtubules in its center; different intraflagellar transport (IFT) motor proteins move proteins and particles along the microtubule bundle from the base of the cilium to the tip and in the opposite direction. Some of the first evidence for a role of primary cilia in Hh signaling came from a screen for mutations in mice that altered early development in a manner similar to that seen in embryos with altered Hh signaling: the mutant phenotypes included losses of certain types of cells in the neural tube that require high concentrations of a particular Hh protein to develop. Many of these mutations were in genes encoding IFT proteins, indicating a role for cilia (or flagella) in Hh signaling. Further studies showed that in the absence of Hh signaling, Ptc is localized to the membrane of the primary cilium and Smo is located in internal vesicles near the base of the cilium. As in Drosophila, in the absence of Hh, kinases in a cytosolic complex phosphorylate Gli, inducing cleavage of Gli by the proteasome and translocation of the Gli fragment (termed ) into the nucleus, where it binds to regulatory regions of Gliresponsive genes and represses their expression. As a result of Hh binding to Ptc, Smo moves into the membrane of the primary cilium. Gli then accumulates at the tip of the cilium, a process requiring a kinesin motor protein (Chapter 18) to move material along microtubules to the tip of the cilium. At the cilia tip, Gli becomes activated by proteins attached to Smo by a mechanism not yet known in detail, but involving dissociation of Gli from Sufu, an inhibitor of Gli that
is a component of the cytosolic complex. Another motor protein, in this case the retrograde motor dynein (Chapter 18), is required to move the full-length and activated Gli, (termed ) to the base of the cilium. As in Drosophila, this active transcription factor then moves into the nucleus, where it activates, rather than represses, expression of multiple target genes. The detailed mechanisms of Hh signaling and Gli activation remain unclear. Ptc is predicted to transport cholesterol or a structurally related sterol that could either activate or inhibit Smo. The vertebrate Smo protein can bind cholesterol as well as several kinds of oxysterol molecules, both by a site in its N-terminal extracellular segment and one within its membranespanning domain. As cholesterol is sufficient to activate vertebrate Smo, in vertebrates cholesterol or a related sterol, perhaps transported into the cytosol by Ptc, is likely to be the activator of Smo. In contrast, in Drosophila, cholesterol is not sufficient to activate Smo, and the identity of the activator for Drosophila Smo is completely unknown. Thus much remains to be learned about the mechanism of Smo activation both in flies and man! Inappropriate activation of Hh signaling is the cause of several types of human tumors, including medulloblastomas (cerebellum tumors) and rhabdomyosarcomas (muscle tumors). Primary cilia are essential for this abnormal Hh signaling, and drugs that inhibit the function of primary cilia are being tested on animal models of these cancers. For instance, expression of a mutant activated form of Smo in the postnatal mouse brain
Degradation of an Inhibitor Protein Activates the NF-κB Transcription Factor
causes medulloblastomas, but these tumors do not form if, simultaneously, a gene encoding an essential ciliary protein is inactivated. Degradation of an Inhibitor Protein Activates the NF-κB Transcription Factor As we have seen, in the non-stimulated state of the Wnt and Hh signal transduction pathways, a key transcription factor is ubiquitinylated and subjected to limited proteolytic cleavage or complete degradation; activation of the signaling pathway blocks ubiquitinylation and releases the transcription factor in its active state. The NF-κB pathway works in the opposite manner: in the resting state, the NF-κB transcription factor is retained in the cytosol bound to an inhibitor; activation of the signaling pathway results in the inhibitor becoming ubiquitinylated and then degraded, triggering release of the active transcription factor. This mechanism allows cells to respond to a variety of stress signals by quickly activating gene transcription. The steps in the NF-κB pathway were revealed in studies with both mammalian cells and Drosophila. The NF-κB signaling pathway is named for its key transcription factor NFκB (an acronym for the somewhat unwieldy descriptor “nuclear-factor kappa-light-chain enhancer of activated B cells”). This transcription factor is rapidly activated in many mammalian immune-system cells in response to bacterial and viral infection, inflammation, and a number of other stressful situations, such as ionizing radiation. In certain cells of the
immune system, for example, the NF-κB pathway is activated when components of bacterial or fungal cell walls or segments of bacterial DNA bind to certain Toll-like receptors on the cell surface or in endosomal membranes (see Figure 24-37). In several types of cells, this pathway is activated by so-called inflammatory cytokines, such as tumor necrosis factor alpha (TNFα) and Interleukin 1 (IL-1), which are released by nearby immune cells in response to infection. In all of these cases, binding of ligand to its receptor induces a signaling pathway that activates the NF-κB transcription factor. NF-κB was originally discovered on the basis of its transcriptional activation of the gene encoding the light chains of antibodies (immunoglobulins) in B cells. It is now thought to be a master transcriptional regulator of the immune system in mammals. Although flies do not make antibodies, NF-κB homologs in Drosophila induce synthesis of a large number of secreted antimicrobial peptides in response to bacterial and viral infection. The existence of these homologs indicates that the NF-κB regulatory system has been conserved during evolution and is more than half a billion years old. Biochemical studies in mammalian cells and genetic studies in flies have provided important insights into the operation of the NF-κB pathway. The NF-κB transcription factor is a heterodimer formed of two subunits, p65 and p50. These two subunits share a region of homology at their N-termini that is required both for their dimerization and binding to DNA. In cells that are not undergoing a stress or responding to signs of an infection, NFκB binds directly to an inhibitor called I-κBα and by this linkage is
sequestered in an inactive state in the cytosol. A single molecule of I-κBα binds to the paired N-terminal domains of the p50–p65 subunits, thereby masking their nuclear-localization signals (Figure 16-30).
FIGURE 16-30 Activation of the NF-κB signaling pathway. In resting cells, the heterodimeric transcription factor NF-κB, composed of p50 and p65 subunits, is sequestered in the cytosol, bound to the inhibitor protein I-κBα. Step 1 : As indicated by the arrows, the three-subunit I-κB kinase is activated downstream of signal transduction pathways stimulated by diverse agents, including activation of any of several Toll-like receptors (Figure 24-37), and by components of invading viruses, bacteria, or fungi. Step 2 : The β subunit of I-κB kinase phosphorylates the inhibitor protein I-κBα, creating a binding site on I-κBα for an E3 ubiquitin ligase. Steps 3 and 4 : Subsequent lysine 48– linked polyubiquitinylation of I-κBα targets it for degradation by proteasomes. Step 5 : The removal of I-κBα unmasks the nuclear-localization signals in both subunits of NF-κB, allowing their translocation to the nucleus. Step 6 : In the nucleus, NF-κB activates transcription of numerous target genes, including the gene encoding I-κBα, which acts to
terminate signaling, and, depending on the cell, activates genes encoding various inflammatory cytokines and other proteins. See R. Khush et al., 2001, Trends Immunol. 22:260; and J. L. Luo et al., 2005, J. Clin. Invest. 115:2625. A three-protein complex termed I-κB kinase operates immediately upstream of NF-κB and is responsible for releasing it from sequestration. Activation of the kinase activity of the β subunit of I-κB kinase, abbreviated IKKβ, is the point of convergence of all of the extracellular signals noted above that activate NF-κB. Within minutes of stimulation of the cell by an infectious agent or inflammatory cytokine, the β subunit of I-κB kinase becomes activated by phosphorylation and then it phosphorylates two N-terminal serine residues on I-κBα (Figure 16-30, steps 1 and 2 ). An E3 ubiquitin ligase then binds to these phosphoserines and ubiquitinylates I-κBα. This E3 ubiquitin ligase links the carboxyl terminus of one ubiquitin to lysine 48 (K48) on another, forming poly-K48-linked ubiquitin chains that target the attached I-κBα protein to the proteasome and its immediate degradation (steps 3 and 4 ; also see Chapter 3). In cells expressing mutant forms of I-κBα in which these two serines have been changed to alanine and so cannot be phosphorylated, NF-κB is permanently inactive, demonstrating that phosphorylation of I-κBα is essential for pathway activation. The degradation of I-κBα exposes the nuclear-localization signals on the two NF-κB subunits, which then translocate into the nucleus and activate transcription of a multitude of target genes (Figure 16-30, steps 5 and 6 ). NF-κB signaling is eventually turned off by negative feedback regulation because one of the genes whose transcription is immediately
Enormous Signalsomes with Polyubiquitin Chain Scaffolds Link Many Cell Surface Receptors to Downstream Proteins in the NF-κB Pathway
induced by NF-κB encodes I-κBα. As its levels increase, the I-κBα protein binds active NF-κB in the nucleus and returns it to the cytosol. In many immune-system cells, NF-κB stimulates transcription of more than 150 genes, including those encoding cytokines and chemokines; the latter attract other immune-system cells and fibroblasts to sites of infection using the signal transduction pathways we described earlier. NFκB also promotes expression of receptor proteins that enable neutrophils (a type of white blood cell) to migrate from the blood into the underlying tissue (see Figure 20-42). In certain immune cells, NF-κB stimulates expression of iNOS, the inducible isoform of the enzyme that produces nitric oxide, which is toxic to bacterial cells, as well as expression of several anti-apoptotic proteins, which prevent cell death. Thus this single transcription factor coordinates and activates the body’s defense, either directly by responding to pathogens and stress or indirectly by responding to signaling molecules released from other infected or wounded tissues and cells. Enormous Signalsomes with Polyubiquitin Chain Scaffolds Link Many Cell Surface Receptors to Downstream Proteins in the NF-κB Pathway
As we have just seen, activation of the β subunit of I-κB kinase is the point of convergence for extracellular signals transmitted through multiple receptors, including Toll-like and IL-1 receptors. Since the cytosolic domains of the Toll-like and IL-1 receptors have no enzyme activity, it was a mystery for many years how activation of these receptors led to phosphorylation and activation of the I-κB kinase β subunit. Recent work showed that huge multiprotein complexes termed signalsomes are formed by activation of these receptors and that several kinases incorporated into these complexes phosphorylate and thus activate I-κB kinase. Oligomerization occurs at multiple levels of the signal transduction cascade, as exemplified by the IL-1 signalsome (Figure 16-31). An MyD88 adapter protein binds to the cytosolic domain of the ligandactivated IL-1 receptor, triggering formation of long helical filaments formed by interactions between MyD88 proteins and the kinases IRAK 2 (Interleukin 1 receptor-associated kinase 2) and IRAK4. TRAF6 proteins form homotrimers each of which then bind to three molecules of IRAK 2, the proteins at the ends of these filaments, and the tips of adjacent TRAF6 trimers bind together forming large two-dimensional lattices containing thousands of copies of TRAF6 and the other signalsome proteins.
FIGURE 16-31 The giant interleukin 1 (Il-1) signalsome and activation of NF-κB. Like the cytosolic domains of the Toll-like receptors (Figure 24-37), the cytosolic domains of both subunits of the interleukin-1β (IL-1β) receptors contain a globular TIR domain; binding of IL-1β to interleukin 1 receptors triggers dimerization. MyD88 (red) proteins then bind to the receptors through interactions with the TIRs, and this in turn facilitates the successive binding of many copies of the kinases IRAK4 (green) and IRAK1 or IRAK2
(orange), leading to phosphorylation and activation of their kinase activities. Activated IRAK1 or IRAK2, in turn, binds TRAF6, an E3 ubiquitin ligase, to the signalsome, and promotes the formation of an extended two-dimensional lattice formed by TRAF6-TRAF6 interactions. TRAF6 also catalyzes synthesis of long lysine-63-linked polyubiquitin chains that are covalently linked to TRAF6 itself. Several proteins including the protein kinase TAK1 and the protein NEMO bind to these K-63 linked polyubiquitin chains. NEMO (a component of the I-κB kinase complex depicted in Figure 16-30), in turn, recruits IKK kinases α and β to the complex, forming the three subunit I-κB kinase complex. IKK α and β kinases then become activated by phosphorylation by TAK1 or other active kinases in the supramolecular complex. As depicted in Figure 16-30, the β subunit of I-κB kinase then phosphorylates the inhibitor I-κB, leading to proteasome destruction of I-κBα and activation of the NF-κB signaling pathway. Only a small portion of an actual signalsome is depicted in this figure as the free ends of the TRAF6 proteins will bind to other TRAF6 molecules, expanding the area of the signalsome just inside the plasma membrane. [Data from R. Ferrao et al., 2012, Sci. Signal. 5:re3; B. Skaug et al., 2009, Annu. Rev. Biochem. 78:769; and J. Napetschnig and H. Wu, 2013, Annu. Rev. Biophys. 42:443.] TRAF6 is an E3 ubiquitin ligase that synthesizes polyubiquitin chains. At the time TRAF6 was discovered all polyubiquitinylation was thought to signal degradation by proteasomes, so researchers looked for ubiquitinylated target proteins that were quickly destroyed. Not finding these, scientists looked for other possible roles for polyubiquitin and soon found that, depending on the specific E3 ubiquitin ligase, multiple types of polyubiquitin chains are formed that have different structures and biological functions. The E3 ligase TRAF6 links the carboxyl terminus of one ubiquitin to lysine 63 (K63) on another ubiquitin (see Figure 3-39), forming a linear poly-K63 ubiquitin chain. Rather than targeting the protein to which it is attached for degradation, these ubiquitin chains act as scaffolds that bind proteins that have a poly-K63 ubiquitin-binding domain. One of these proteins is the protein kinase TAK1, which becomes
activated by binding to the polyubiquitin chain; another is the NEMO subunit of the I-κB kinase. Binding to adjacent sites on the poly-K63 ubiquitin brings the TAK1 kinase and its target, the β subunit of I-κB kinase, into proximity so that TAK1 can phosphorylate and thereby activate this downstream kinase (see Figure 16-31). I-κB kinases can also become activated by phosphorylation by other already active I-κB kinases in the complex. As noted above, I-κB kinase then phosphorylates I-κBα, leading to activation of the NF-κB factor. Thus two different types of polyubiquitin chains participate in very different ways in transmitting the IL-1 signal to activation of the NF-κB transcription factors. What is the advantage of such huge signalsome complexes? Why did they evolve as part of signal transduction pathways downstream of Toll-like receptors and receptors for many inflammatory hormones, including IL-1 and TNFα? One hypothesis is that many hormone receptors must cooperate to form signalsomes, leading to a threshold all-or-none response: a low concentration of an extracellular signal would not trigger assembly of a stable functional signalsome whereas just a slightly higher concentration would trigger a maximum response. This prevents the immune system from mounting a potentially damaging immune response in the absence of significant danger. Depending on the TNFα concentration, formation of the signalsome by the TNFα receptor can take between 20 and 50 minutes, and this delay allows formation of a signalsome to be reversed if the initial stimulation does not persist. Such a fail-safe mechanism might have evolved to allow a response only when there is sufficient danger, as signaled by a persistent and high dose of stimulation.
KEY CONCEPTS OF SECTION 16.7 Signal Transduction Pathways ThatUtilize Proteasomal Degradation of Signaling Components: Wnt, Hedgehog, and the Many Hormones That Activate NF-κB Many signaling pathways involve ubiquitinylation and proteolysis of target proteins and so are irreversible or only slowly reversible. These target proteins can be either a transcription factor or an inhibitor of a transcription factor. Wnt signaling controls numerous critical developmental events, such as brain development, limb patterning, and organogenesis. Like Wnt, Hedgehog (Hh) also functions as a morphogen during development. Activating mutations in both pathways can cause cancer. Both Hh and Wnt are secreted proteins that contain lipid anchors that reduce their signaling ranges. The fatty acid covalently attached to Wnt is essential for binding to its Frizzled (Fz) receptor. Wnt signals act through two cell-surface proteins, the receptor Fz and co-receptor LRP, and an intracellular complex containing β-catenin (see Figure 16-26). Binding of Wnt promotes the stability and nuclear localization of β-catenin, which either directly or indirectly promotes activation of the TCF transcription factor. Gradients of Wnt protein concentration are essential for many steps in development, including regeneration of a head and tail in planaria (see Figure 16-27). The Hh signal also acts through two cell-surface proteins, Smoothened (Smo) and Patched (Ptc), and an intracellular complex containing the Cubitis interruptus (Ci) transcription factor (see Figure 16-29). An activating form of Ci is generated in the presence of Hh; a repressing Ci fragment is generated in the absence of Hh. Both Ptc and Smo change their subcellular location in response to Hh binding to Ptc. Hh signaling in vertebrates requires primary cilia and intraflagellar transport proteins. Ptc localizes to the ciliary membrane in the absence of Hh, and Smo moves from internal membranes to the cilium membrane when Hh is present. The NF-κB transcription factor regulates many genes that permit immune system cells to respond to infection and inflammation. In unstimulated cells, NF-κB is localized to the cytosol, bound to the inhibitor protein I-κBα. In response to many types of extracellular signals, I-κBα is phosphorylated and then ubiquitinylated, targeting it for degradation by proteasomes. The destruction of I-κBα releases active NF-κB, which translocates to the nucleus (see Figure 16-30). Signal transduction by the interleukin 1 and Toll-like receptors involves formation of huge multiprotein complexes termed signalsomes. Polyubiquitin chains formed by the TRAF6 protein in this complex form a scaffold that brings the TAK1 kinase near its
substrate, the β subunit of the I-κB kinase, activating the I-κB kinase that phosphorylates the inhibitor I-κBα. The resultant destruction of I-κBα allows signals to be transmitted from the receptor to downstream components of the NF-κB pathway (see Figure 16-31).
Key Terms
End of Chapter Visit Achieve to access study tools and to learn more about the content in this chapter: Perspectives for the Future Analyze the Data Chapter References Additional study tools, including videos, animations, and quizzes Key Terms activation loop Akt cytokine receptors cytokines enhancer erythropoietin (Epo) granulocyte colony–stimulating factor (G-CSF) Hedgehog (Hh) HER MAP kinase morphogen NF-κB phosphoinositides prolactin
Review the Concepts
promoter protein kinase B (PKB) scaffold proteins Smads transforming growthfactor β (TGF-β) Wnt Review the Concepts 1. Even though GRB2 lacks intrinsic enzyme activity, it is an essential component of the epidermal growth factor (EGF) signaling pathway that activates MAP kinase. What is the function of GRB2? What roles do the SH2 and SH3 domains play in the function of GRB2? Many other signaling proteins possess SH2 domains. What determines the specificity of SH2 interactions with other molecules? 2. Once an activated signaling pathway has elicited the proper changes in target-gene expression, the pathway must be inactivated. Otherwise, pathological consequences may result, as exemplified by persistent growth factor–initiated signaling in many cancers. Many signaling pathways possess intrinsic negative feedback by which a downstream event in a pathway turns off an upstream event. Describe the negative feedback that down-regulates signals induced by (a) erythropoietin and (b) TGF-β. 3. A mutation in the Ras protein renders Ras constitutively active . What is constitutive activation? How does constitutively
active Ras promote cancer? What type of mutation might render the following proteins constitutively active: (a) Smad3, (b) MAP kinase, and (c) NF-κB? 4. The enzyme Ste11 participates in several distinct MAP kinase signaling pathways in the budding yeast Saccharomyces cerevisiae. What is the substrate for Ste11 in the mating factor signaling pathway? When a yeast cell is stimulated by mating factor, what prevents the induction of osmolytes required for survival in high-osmotic-strength media, given that Ste11 also participates in the MAP kinase pathway initiated by high osmolarity? 5. Describe the events required for full activation of protein kinase B. 6. Describe the function of the PTEN phosphatase in the PI-3 kinase signaling pathway. Why does a loss-of-function mutation in PTEN promote cancer? Predict the effect of constitutively active PTEN on cell growth and survival. 7. Name three features common to the activation of cytokine receptors and receptor tyrosine kinases. Name one difference with respect to the enzyme activity of these receptors. 8. Erythropoietin (Epo) is a hormone that is produced naturally in the body in response to low levels in the blood. The intracellular events that occur in response to Epo binding to its cell-surface receptor are well characterized. What molecule translocates from the cytosol to the nucleus after (a) JAK2 activates STAT5 and (b) GRB2 binds to the Epo receptor? Why did some endurance athletes use Epo to improve their
performance (“blood doping”) until it was banned by most sports? 9. Explain how expression of a dominant-negative mutant of JAK blocks the erythropoietin (Epo)-cytokine signaling pathway. 10. Binding of TGF-β to its receptors can elicit a variety of responses in different cell types. For example, TGF-β induces plasminogen activator inhibitor 1 in epithelial cells and specific immunoglobulins in B cells. In both cell types, Smad3 is activated. Given the conservation of the signaling pathway, what accounts for the diversity of the response to TGF-β in various cell types? 11. How is the signal generated by binding of TGF-β to cell-surface receptors transmitted to the nucleus, where changes in targetgene expression occur? What activity in the nucleus ensures that the concentration of active Smads closely reflects the level of activated TGF-β receptors on the cell surface? 12. What feature of Delta ensures that only neighboring cells are signaled? 13. What biochemical reaction is catalyzed by γ-secretase? 14. The extracellular signaling protein Hedgehog can remain anchored to cell membranes. What modifications to Hedgehog enable it to be membrane bound? Why is this property useful? 15. Explain why loss-of-function hedgehog and smoothened mutations yield the same phenotype in flies, but a loss-offunction patched mutation yields the opposite phenotype. 16. Why is the signaling pathway that activates NF-κB considered to be relatively irreversible compared with cytokine or RTK signaling pathways? Nonetheless, NF-κB signaling must be
down-regulated eventually. How is the NF-κB signaling pathway turned off? 17. Describe two roles for polyubiquitinylation in the NF-κB signaling pathway.