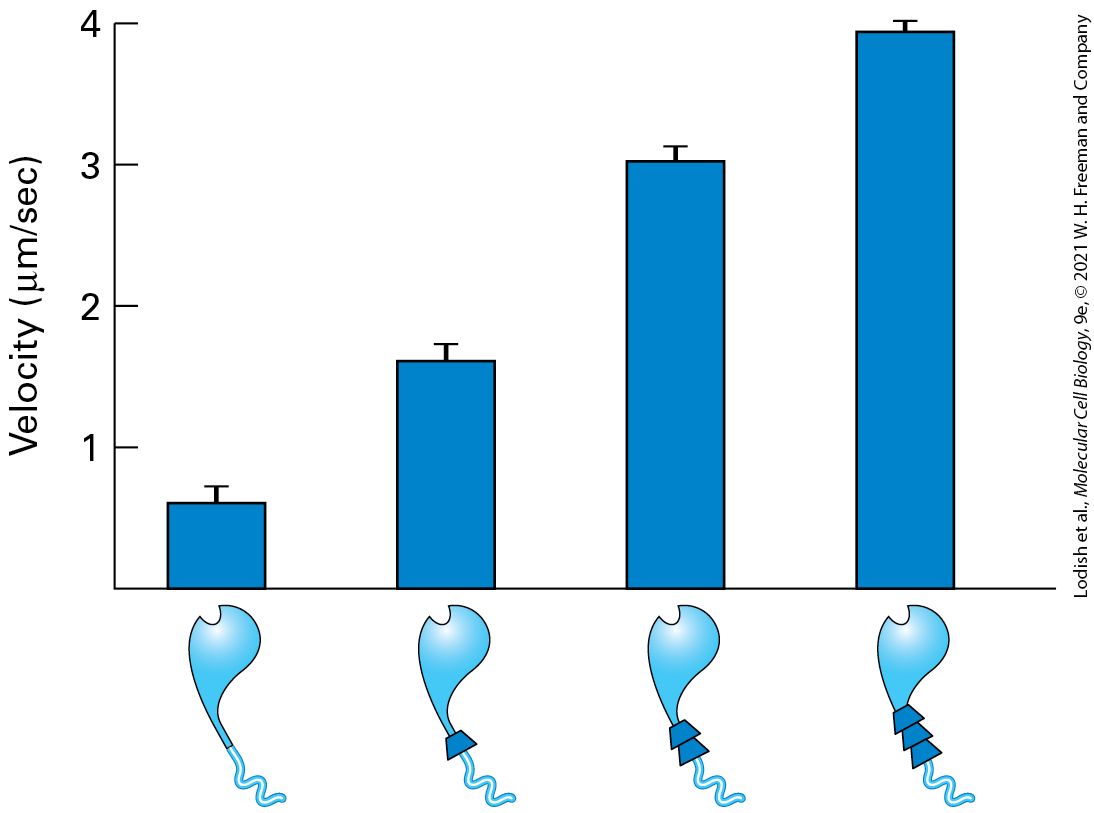
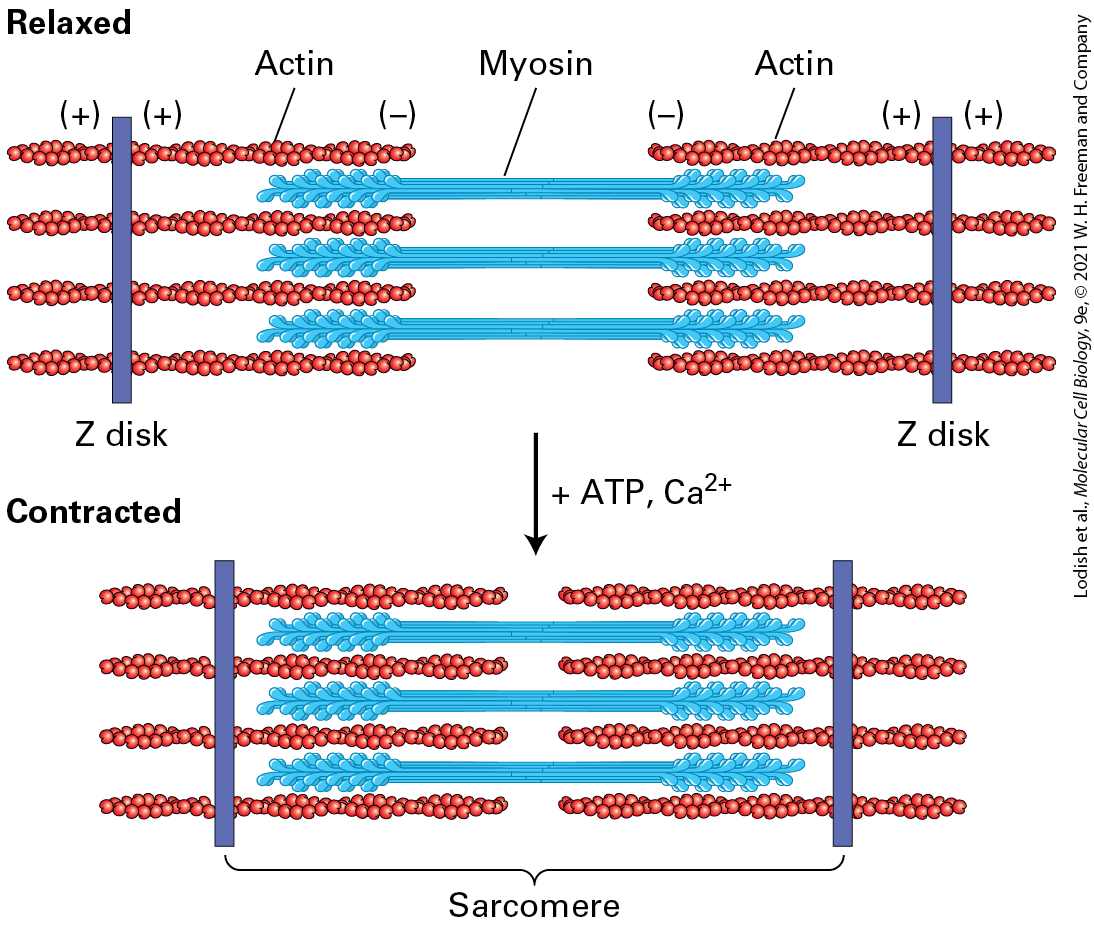
Introduction
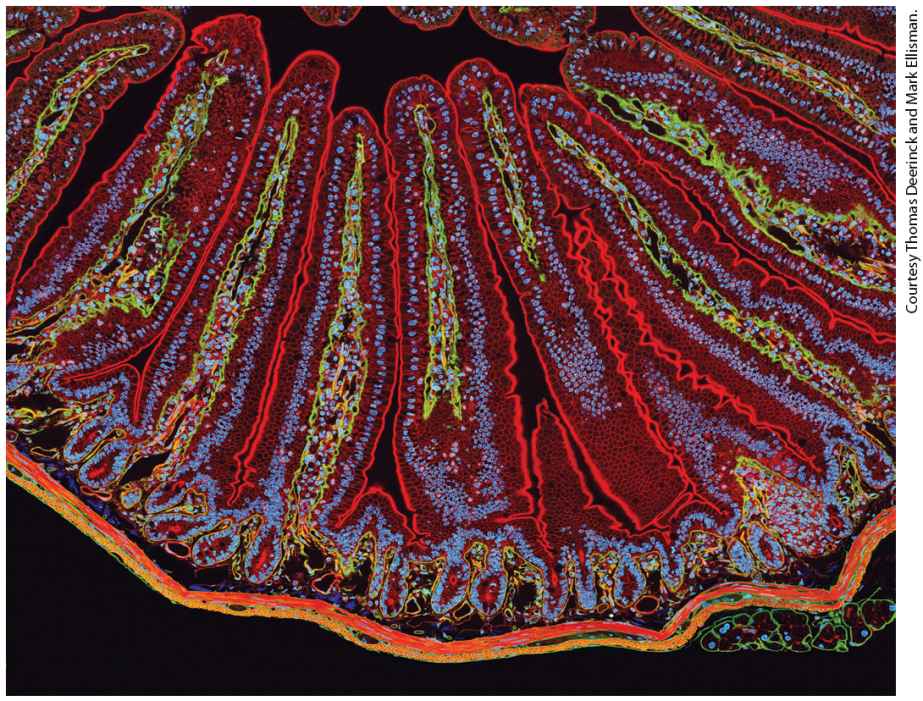
Chapter 17 Cell Organization and Movement I: Microfilaments A section of mouse intestine stained for actin (red), the extracellular matrix protein laminin (green), and DNA (blue). Each blue dot of DNA indicates the presence of a cell. Actin in the microvilli on the apical end of the epithelial cells can be seen lining the surface facing the lumen (top). Actin can also be seen prominently in the smooth muscle that surrounds the intestine (bottom).
17.4 Organization of Actin-Based Cellular Structures
17.7 Cell Migration: Mechanism, Signaling, and Chemotaxis When we look through a microscope at the diversity of cells in nature, the variety of cell shapes and movements we see is astonishing. We may notice that some cells, such as vertebrate sperm, ciliates such as Tetrahymena, or flagellates such as Chlamydomonas, swim rapidly, propelled by cilia and flagella. Other cells, such as amoebae and human macrophages, move more sedately, propelled not by external appendages, but by coordinated movement of the cell itself. If we examine tissues under the microscope, we might notice that some cells connect to form a pavement-like sheet, whereas other cells — neurons, for example — have processes up to 3 feet in length that make selective contacts with other cells. When we look at the internal organization of cells, we see that organelles have characteristic locations. For example, the Golgi complex is generally near the nucleus, which itself is found in the center of the cell. How is this diversity of shape, cellular organization, and motility achieved? Why is it important for cells to have a distinct shape and well-
defined internal organization? To begin to answer these questions, let us consider two very different cells: epithelial cells and macrophages. The epithelial cells that line the intestine form a tight, pavement-like layer of brick-shaped cells, known as an epithelium (Figure 17-1a, b). The epithelium imports nutrients from the intestinal lumen across the apical (top) domain of the plasma membrane and exports the nutrients across the basolateral (bottom-side) domain of the plasma membrane toward the bloodstream. To perform this directional transport, the apical and basolateral domains of the plasma membranes of epithelial cells contain different transport proteins.
FIGURE 17-1 Overview of the cytoskeletons of an epithelial cell and a migrating cell. (a) Transmission electron micrograph of a thin section of an epithelial cell from the small intestine, showing the core bundles of microfilaments that provide support to the microvilli. (b) Epithelial cells are highly polarized, with distinct apical and basolateral domains. An intestinal epithelial cell transports nutrients into the cell through the apical domain and out of the cell across the basolateral domain. (c) Transmission electron micrograph of part of the leading edge of a migrating cell. The cell was treated with a mild detergent to dissolve the membranes, which also allows solubilization of most cytoplasmic components. The remaining cytoskeleton was shadowed with platinum and visualized in the electron microscope. Note the network of actin filaments visible in this micrograph. (d) A migrating cell, such as a fibroblast or a macrophage, has morphologically distinct domains, with a leading edge at the front. Microfilaments are indicated in red, microtubules in green, and intermediate filaments in dark blue. The position of the nucleus (light blue oval) is also shown. [Part (a) M. S. Mooseker and L. G. Tilney, 1975, J. Cell Biol. 67(3):725–743; https://doi.org/10.1083/jcb.67.3.725. Part (c) T. M. Svitkina and G. G. Borisy, 1999, J. Cell Biol. 145(5):1009–1026; https://doi.org/10.1083/jcb.145.5.1009.] Description The micrograph labeled (a) shows microvilli. The microvilli are finger-like projections from cells and bundles of filaments are visible within the microvilli as darker stained regions. The illustration labeled shows epithelial cells with structural components and microvilli. Epithelial cells have microvilli projecting from their cell membrane. The microvilli are full of structural microfilaments (apical domain). Around the border of the main cell body, microfilaments, microtubules, and intermediate filaments are also present (basolateral domain). The nucleus consists of intermediate filaments. The extracellular matrix below also has microfilaments. The micrograph labeled (c) shows the leading edge of a migrating cell, with many crisscross filaments at the leading edge. The illustration labeled (d) shows a migratory cell shaped almost like a foot. A layer beneath the cell membrane makes up the microfilaments, which enclose more
microfilaments, microtubules, and intermediate filaments. A layer made of intermediate filaments lies beneath the nuclear membrane. The direction of the migration is labeled from left to right. The leading edge and the filopodium are labeled. Epithelial cells are sealed together by cell junctions (discussed in Chapter 20), which create a physical barrier between the apical and basolateral domains of the membrane. This separation allows the cell to place the proper transport proteins in the appropriate places: either the apical or the basolateral membrane. In addition, the apical membrane domain has a unique morphology, with numerous fingerlike projections called microvilli that dramatically increase the area of the plasma membrane available for nutrient absorption. So we see that the distinct functions of different regions of the intestinal epithelium are due to specific morphological features. To achieve these features (such as membrane domains, microvilli, and cell junctions), epithelial cells must have an internal structure that gives the cells shape, promotes cell-to-cell adhesion, and aids in delivering the right proteins to the right membrane domain. Now consider the macrophage, a type of white blood cell that seeks out infectious agents and destroys them by an engulfing process called phagocytosis. For example, bacteria release chemicals that attract the macrophage and guide it to the site of infection. As the macrophage follows the chemical gradient, twisting and turning to get to the bacteria and phagocytose them, it must constantly reorganize its cell locomotion machinery. As we will see, the internal machinery of macrophages that allows them to crawl across a substrate is always oriented in the direction in which they crawl (Figure 17-1c, d).
These are just two examples of cell polarity, the ability of a cell to generate functionally distinct regions within itself. In fact, as you think about various types of cells, you might realize that most of them have some form of cell polarity. A universal example of cell polarity is the ability of cells to divide — they must first select an axis for cell division and then set up the machinery to segregate their organelles along that axis. The shape, internal organization, and functional polarity of a cell are due to a network of filamentous proteins called the cytoskeleton. We can visualize the cytoskeleton in different ways. For example, the cytoskeleton can be visualized by electron microscopy after treating cells with gentle detergents that solubilize the plasma membrane and internal organelles, releasing most of the cytoplasm (see Figure 17-1c). The cytoskeleton can also be visualized by immunofluorescence microscopy, as shown in Figure 17-2. We know from various studies that the cytoskeleton extends throughout the cell and is attached to the plasma membrane and to internal organelles. Thus the cytoskeleton provides a framework for organizing cellular components and processes. The term cytoskeleton may imply a fixed structure like a bony skeleton in vertebrates. In fact, the cytoskeleton can be very dynamic, with components reorganizing in less than a minute under some conditions. Under different conditions, it can be stable for hours. As we will see throughout this chapter, the lengths of cytoskeletal filaments and the dynamics of filament assembly vary both in different regions of the cell and depending on the needs of the cell. The result is that cytoskeletal filaments are assembled into many types of structures and are regulated within different regions of the cell.
FIGURE 17-2 The components of the cytoskeleton. Each filament type is assembled from specific subunits in a reversible process so that cells can assemble and disassemble filaments as needed. Bottom panels show localization of the three filament systems in cultured cells as seen by immunofluorescence microscopy of actin, tubulin, and an intermediate filament protein, respectively. The image of microtubules shows their distribution in an interphase cell. In mitosis, they make up the machinery for chromosome segregation, as we discuss in Chapter 18. Description On the left, titles for each row, from top to bottom are subunit, structure, and localization. The first column is labeled microfilaments, the subunit name is actin. A diagram of actin shows a helical structure with a measurement of 7-9 nanometers, and a fluorescence image shows many red straight lines in the web. The second column is labeled microtubules. The subunit is alpha beta-tubulin dimer. The dimer has a cylindrical structure with rows alternating in green and white spheres. It is 25 nanometers in width. The fluorescence image shows a series of green lines forming a circle at the center. The last column is labeled intermediate filaments. It has various subunits. The structure has several threads wrapped around each other into a rope-like structure with a width of 10 nanometers. The fluorescence image shows many curved blue lines forming a web. The cytoskeleton is composed of three different filament systems, shown in Figures 17-1b, 17-1d, and 17-2. The filaments are polymers whose
assembly and disassembly are regulated. This gives the cell flexibility to assemble and disassemble different types of cytoskeletal structures as needed. The three filament systems are: Microfilaments are polymers of the protein actin that are organized into functional bundles and networks by actin-binding proteins. Microfilaments are especially important in organizing the plasma membrane, giving shape to surface structures such as microvilli. Microfilaments can provide mechanical support on their own or can serve as tracks for ATP-powered myosin motor proteins. Myosin can ferry cargo along microfilaments, and myosin and microfilaments working together provide a contractile function (as in muscle). Microtubules are polymeric tubes formed from the protein tubulin and organized by microtubule-associated proteins. Microtubules make up the mitotic spindle, the machine for segregating duplicated chromosomes at mitosis. In interphase cells, as shown in Figure 17-2, microtubules often extend throughout the cell, providing a framework for the organization of organelles and structural support to cilia and flagella. Motor proteins called kinesins and dyneins transport cargo along microtubules, and like myosins, they are powered by ATP hydrolysis. Intermediate filaments are also polymers, but assembled from tissue-specific subunits that lend structural support to the nuclear membrane, provide structural integrity to cells in tissues, and serve structural and barrier functions in skin, hair, and nails. Unlike microfilaments and microtubules, intermediate filaments are not used as tracks by motor proteins.
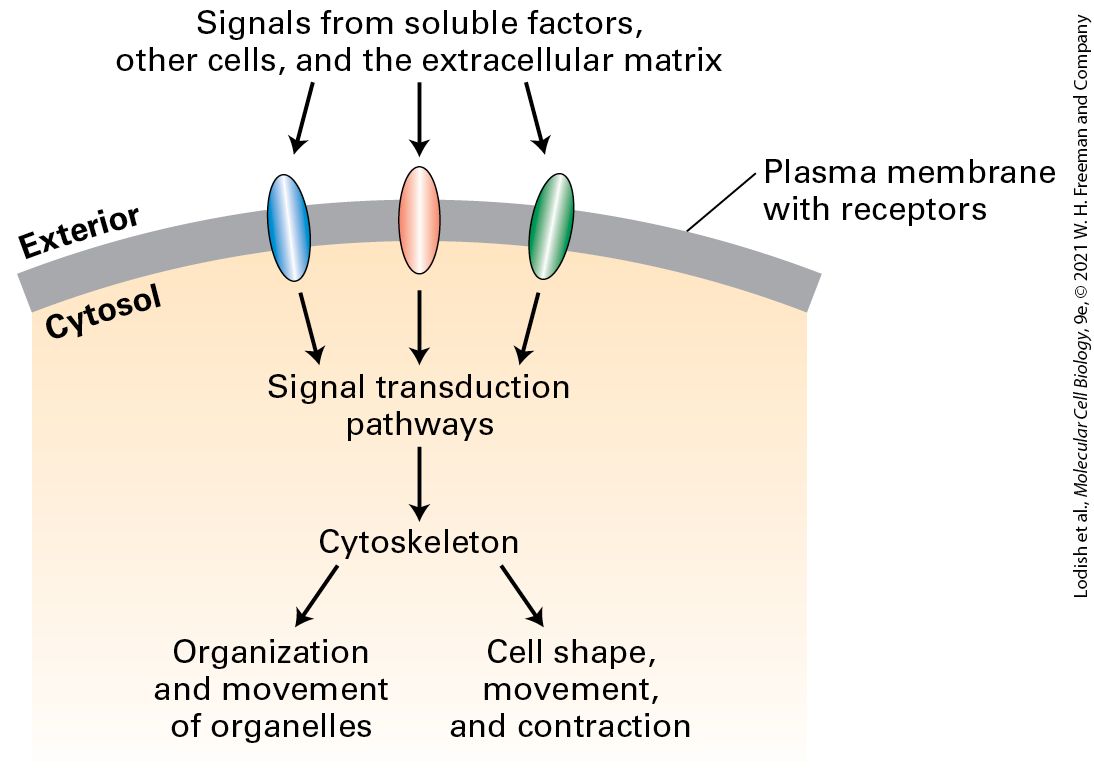
As we can see in Figure 17-1, cells can organize their cytoskeletons in distinct ways. How cells organize the cytoskeleton depends on the cells’ ability to sense signals — from soluble factors bathing the cell, from adjacent cells, or from the extracellular matrix — and interpret them (Figure 17-3). These signals are detected by cell-surface receptors that activate signal transduction pathways, which ultimately converge on factors that regulate cytoskeletal organization.
FIGURE 17-3 Regulation of cytoskeleton function by cell signaling. Cells use cell-surface receptors to sense external signals from the extracellular matrix, other cells, or soluble factors. These signals are transmitted across the plasma membrane and activate specific cytosolic signaling pathways. Signals — often integrated from more than one receptor — lead to the organization of the cytoskeleton so as to provide cells with their shape as well as
to determine organelle distribution and movement. In the absence of external signals, cells still organize their internal structure, but not in a polarized manner. Description The illustration shows a part of the plasma membrane of a cell with three receptors. The exterior and the cytosol are labeled. The flow chart has the following sequence: 1. Signals from soluble factors, other cells, and the extracellular matrix act on receptors in the plasma membrane. 2. Signal transduction pathways are activated. 3. The cytoskeleton is organized, resulting in organization and movement of organelles or cell shape, movement, and contraction. The importance of the cytoskeleton for normal cell function and motility is evident when a defect in a cytoskeletal component — or in cytoskeletal regulation — causes disease. For example, about 1 in 500 people has a cytoskeletal defect that affects the contractile apparatus of the heart resulting in various cardiomyopathies (diseases of the heart muscle). Many defects in red blood cytoskeletal components that support these cells’ plasma membrane are known to cause disease. Metastatic cancer cells break away from their tissue of origin and migrate to new locations because of misregulation of the cytoskeleton. In this and the following chapter, we discuss the structure, function, and regulation of the cytoskeleton. We look at how a cell arranges its cytoskeleton to generate cell shape and cell polarity to provide organization and motility to its organelles and to be the structural framework for such processes as cell swimming and cell crawling. We discuss how cells assemble the three different filament systems and how
signal transduction pathways regulate these structures. Regulation of the cytoskeleton during the cell cycle is discussed in Chapter 19. In Chapter 20, we examine how the cytoskeleton participates in the functional organization of tissues. Our focus in this current chapter is on microfilaments and actin-based structures. Although we initially examine the three cytoskeletal systems separately, in the next chapter we will see that microfilaments cooperate with microtubules and intermediate filaments in the normal functioning of cells.
17.1 Microfilaments and Actin Structures
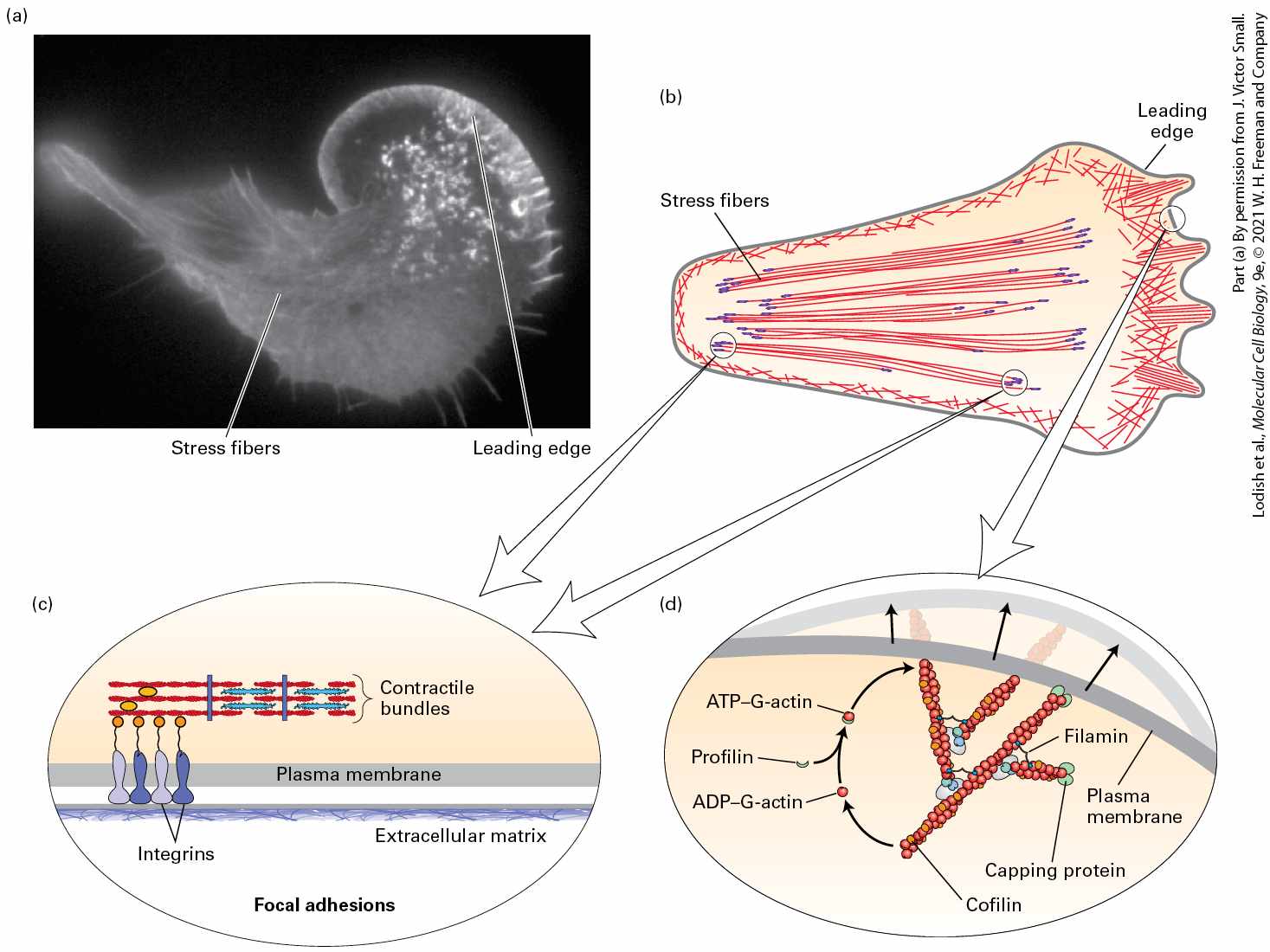
17.1 Microfilaments and Actin Structures Microfilaments can assemble into a wide variety of structures within a cell (Figure 17-4a). Each of these structures is critical for particular cellular functions. For example, microfilaments can form a tight bundle of filaments making up the core of the fingerlike microvilli, but they can also be found in a less ordered network just beneath the plasma membrane, in a region of cytoplasm known as the cell cortex, where they provide support and organization to membrane proteins. In epithelial cells, microfilaments form a contractile band around the cell, the adherens belt, that provides strength to the epithelium. In migrating cells, a network of microfilaments is found at the front of the cell in the leading edge, or lamellipodium, from which bundles of filaments called filopodia may protrude. Many cells have contractile microfilaments called stress fibers, which attach to the external substratum as cells migrate (discussed in
Chapter 20). Specialized cells such as macrophages use contractile microfilaments to engulf and internalize pathogens though phagocytosis. Highly dynamic, short bursts of actin filament assembly can power the movement of endocytic vesicles away from the plasma membrane. At a late stage of cell division in animals, after all the organelles have been duplicated and segregated, a contractile ring forms and constricts to generate two daughter cells in the process of cytokinesis. Thus cells use actin filaments in many ways: in structural roles, as contractile
mechanisms, by harnessing actin polymerization and depolymerization to do work, and to move vesicles around within a cell. Multiple arrangements of microfilaments often coexist within a single cell, as shown in a migrating fibroblast (Figure 17-4b).
FIGURE 17-4 Examples of microfilament-based structures. (a) In each panel, microfilaments are depicted in red. (b) A cell moving toward the top of the page, stained for actin with fluorescent phalloidin, a drug that specifically binds F-actin. Note how many different organizations of microfilaments can exist in one cell. Description The illustration labeled (a) has three rows. Each row has a set of three similar cells within which microfilaments in different areas are highlighted. In the first row, the microfilaments in the microvilli, the cell cortex (lining of the cell membrane), and the adherens belt (present below the microvilli) are highlighted. In the second row, the cells are shaped like an animal track with three claws to the right. In the first cell labeled filopodium, the microfilaments present inside the claw-like structure are highlighted. In the second cell labeled lamellipodium or leading-edge, the microfilaments in the area behind the claw-like structure are highlighted. In the third cell labeled stress fibers, the microfilaments present in the center of the foot like structure are highlighted. In the
Actin Is Ancient, Abundant, and Highly Conserved
third row, in the first cell, the microfilaments lining a part of a cell performing phagocytosis is highlighted. In the second cell, the microfilaments moving endocytic vesicles are highlighted. In the third cell, the microfilaments squeezing the middle of a phagocyte are highlighted. It is labeled as the contractile ring. The micrograph labeled (b) shows a cell with three different areas labeled: the leading edge, stress fibers, and filopodia. The fibers around the leading edge, stress fibers, and filopodia are all visible. The basic building block of microfilaments is actin, a protein that has the remarkable capacity to reversibly assemble into a polarized filament with functionally distinct ends. These filaments are molded into the various structures described in the previous paragraph by actin-binding proteins. The name microfilament, which arose from the very thin filaments seen by electron microscopists in thin-section preparations of cells, refers to actin in its polymerized form, with its associated proteins. In this section, we look at the actin protein itself and the filaments into which it assembles. Actin Is Ancient, Abundant, and Highly Conserved Actin is an abundant intracellular protein in eukaryotic cells. In muscle cells, for example, actin constitutes 10 percent by weight of the total cellular protein; even in nonmuscle cells, actin makes up 1–5 percent of the cellular protein. The cytosolic concentration of actin in nonmuscle cells ranges from 0.1 to 0.4 mM. In structures such as microvilli, however, the local actin concentration can be as high as 5 mM. To grasp how much actin is present in cells, consider a typical liver cell, which has
G-Actin Monomers Assemble into Long, Helical F-Actin Polymers
insulin receptor molecules but approximately , or half a billion, actin molecules. Because they form structures that extend across large parts of the cell interior, cytoskeletal proteins are among the most abundant proteins in a cell. Actin is encoded by a gene family that gives rise to some of the most conserved proteins within and across species. The protein sequences of actins from amoebae and from animals are identical at 80 percent of their amino acid positions, despite about a billion years of evolution. The multiple actin genes found in modern eukaryotes are related to a bacterial gene, MreB, whose product forms filaments that are important in bacterial cell-wall synthesis. Some single-celled eukaryotes, such as yeasts and amoebae, have one or two actin genes, whereas multicellular organisms often contain multiple actin genes. For instance, humans have 6 actin genes, while Arabidopsis has 8 to 10, and maize has 21 actin genes. Each functional actin gene encodes a different isoform of the protein. In vertebrates, four actin isoforms are present in specific types of muscle cells, and two other isoforms are found in nonmuscle cells. These six isoforms differ at only about 25 of the 375 residues in the complete protein or show about 93 percent identity. Actin isoforms have historically been classified into three groups based on their overall charge: the α-actins, β-actins, and γ-actins. Alpha-actins are associated with various contractile structures, β-actins are enriched in the cell cortex and leading edge of motile cells, and γ-actin is found in stress fibers.
G-Actin Monomers Assemble into Long, Helical F-Actin Polymers Actin exists as a globular monomer called G-actin and as a filamentous polymer called F-actin, which is a linear chain of actin subunits. Each actin molecule contains a ion complexed with either ATP or ADP. In fact, actin is an ATPase, which hydrolyzes ATP to ADP and . The importance of the interconversion between the ATP and the ADP forms of actin is discussed below. X-ray crystallographic analysis reveals that the G-actin monomer is separated into two lobes by a deep cleft (Figure 17-5a). At the base of the cleft is the ATPase fold, which is the site where ATP and are bound. The ATPase fold has structural similarity to the GTP-binding cleft of the GTPase molecular switches (see Figure 15-7). The floor of the cleft in Gactin acts as a hinge that allows the lobes to flex relative to each other. When ATP or ADP is bound to G-actin, the nucleotide affects the conformation of the molecule (in fact, without a bound nucleotide, G-actin denatures very quickly).
FIGURE 17-5 Structures of monomeric G-actin and F-actin filaments. (a) Structure of the actin monomer , which is divided by a central cleft into two approximately equal-sized lobes and four subdomains, numbered I–IV. ATP (yellow) binds at the bottom of the cleft and contacts both lobes (the green ball represents ). The N- and C-termini lie in subdomain I. (b) An actin filament appears as two strands of subunits. One repeating unit consists of 28 subunits (14 in each strand, indicated by * for one strand), covering a distance of 72 nm. The ATP-binding cleft of every actin subunit is oriented toward the same end of the filament. The end of a filament with an exposed binding cleft is the end; the opposite end is the end. (c) In the electron microscope, negatively stained actin filaments appear as long, flexible, and twisted strands of beaded subunits. Because of the twist, the filament appears alternately thinner (7-nm diameter) and thicker (9-nm diameter) (arrows). (The microfilaments visualized in a cell by electron microscopy are F-actin filaments plus any bound proteins.) [Part (a) Data from C. E. Schutt et al., 1993, Nature 365:810, PDB ID 2btf, courtesy of M. Rozycki.] Description The illustration labeled (a) shows an actin monomer with four subdomains labeled. The ribbon is enclosed in a semi-transparent three-dimensional model. A space-filling
model made up of A T P and magnesium cation is attached to the ribbon at the center. The A T P-binding cleft is labeled. The illustration labeled (b) shows the structure of a single actin filament. An actin filament has two rope-like structures made up of spheres twisted around each other. The negative end is labeled at the top and the positive end at the bottom. The top half and the bottom half of the structure are each 36 nanometers in length. The electron micrograph labeled (c) shows actin filaments in a cell. It shows two long tube-like structures with dark outlines. Four arrows point at a part of one of the tubelike structures. Dotted lines from a part of the actin show the actin filament in illustration b. G-actin can polymerize, in a reversible reaction, into F-actin. The F-actin filaments that form in vitro are indistinguishable from microfilaments seen in cells, indicating that F-actin is the major component of microfilaments. From the results of x-ray diffraction studies of actin filaments and from the actin monomer structure shown in Figure 17-5a, scientists have determined that the subunits in an actin filament are arranged in a helical structure (Figure 17-5b). In this arrangement, the filament can be considered as two helical strands wound around each other. Each subunit in the structure contacts one subunit above it and one below it in its own strand as well as two subunits in the other strand. The subunits in a single strand wind around the back of the other strand and repeat after 72 nm, or 14 actin subunits. Since there are two strands, the actin filament appears to repeat every 36 nm (see Figure 17-5b). When F-actin is viewed by electron microscopy after negative staining with uranyl acetate, it appears as a twisted string whose diameter varies between 7 and 9 nm (Figure 17-5c).
F-Actin Has Structural and Functional Polarity
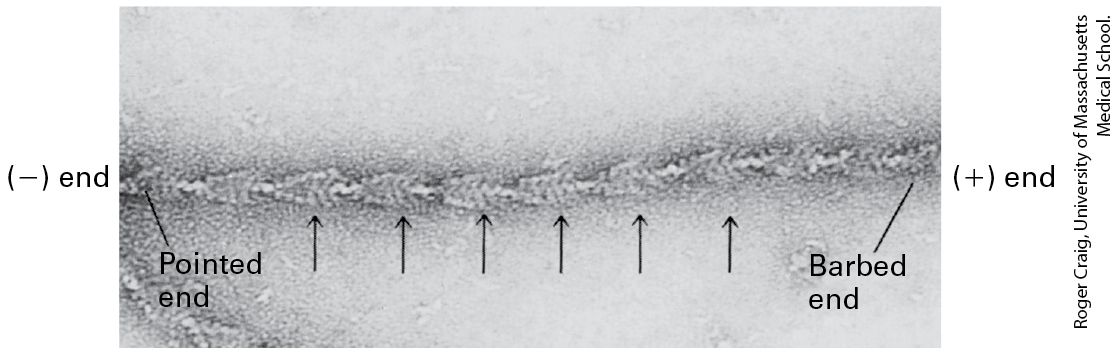
F-Actin Has Structural and Functional Polarity All subunits in an actin filament are oriented the same way. A consequence of this subunit orientation is that the filament as a whole exhibits polarity; that is, one end of the filament differs from the other. As we will see, a result of this subunit orientation is that one end of the filament is favored for the addition of actin subunits and is designated the end, whereas the other end is favored for removal of subunits and is designated the end. At the end, the ATP-binding cleft of the terminal actin subunit contacts the neighboring subunit, whereas on the end, the cleft is exposed to the surrounding solution (see Figure 17-5b). The cleft in an actin subunit, and therefore the polarity of a filament, is not detectable without the atomic resolution afforded by x-ray crystallography. However, the polarity of actin filaments can be demonstrated by electron microscopy in so-called decoration experiments, which exploit the ability of the motor protein myosin (discussed in Section 17.5) to bind specifically to actin filaments. In this type of experiment, an excess of myosin S1, a proteolytic cleavage product of myosin containing the actinbinding head domain (see Figure 17-22), is mixed with actin filaments under conditions where binding takes place. Myosin attaches to the sides of a filament and binds with a slight tilt. When all the actin subunits are bound by myosin, the filament appears as if decorated with arrowheads that all point toward the end of a filament (Figure 17-6). Thus the end is often called the pointed end of an actin filament. The end is
known as the barbed end. Because myosin binds to actin filaments and not to microtubules or intermediate filaments, arrowhead decoration is one criterion by which actin filaments can be definitively identified among the other cytoskeletal fibers in electron micrographs of cells. EXPERIMENTAL FIGURE 17-6 Myosin S1 decoration demonstrates the identity and polarity of an actin filament. Myosin S1 head domains bind to actin subunits in a particular orientation. When bound to all the subunits in a filament, S1 appears to spiral around the filament. This coating of myosin heads produces a series of arrowhead-like decorations (arrows). The polarity in decoration defines a pointed end and a barbed end. KEY CONCEPTS OF SECTION 17.1 Microfilaments and Actin Structures Microfilaments can be assembled into diverse structures, many of which are associated with the plasma membrane (see Figure 17-4a). Actin, the basic building block of microfilaments, is an abundant protein of eukaryotic cells and is highly conserved among species. Actin can reversibly assemble into filaments that consist of two helices of actin subunits. The actin subunits in a filament are all oriented in the same direction, with the nucleotide-binding site exposed on the end (see Figure 17-5).
17.2 Dynamics of Actin Filaments
17.2 Dynamics of Actin Filaments The actin cytoskeleton is a dynamic structure, in which the assembly and disassembly of actin filaments, and their association into functional structures, are exquisitely regulated. In some cytoskeletal structures, microfilaments are stable for hours, whereas in others they grow or shrink in seconds. These changes in the organization of actin filaments can generate forces that cause large changes in the shape of a cell or drive movement of intracellular structures. In this section, we first consider actin’s intrinsic ability to polymerize and depolymerize and then look at how these processes are modulated by accessory proteins. We will see how actin-binding proteins make important contributions to the stability and regulated disassembly of filaments. In subsequent sections, we turn to the mechanisms that cells use to assemble and organize actin filaments, how the location of assembly within a cell is regulated by signaling pathways, and how together with myosins they can drive motility processes. A summary of the actin-binding proteins discussed in this chapter is presented in Table 17-1.
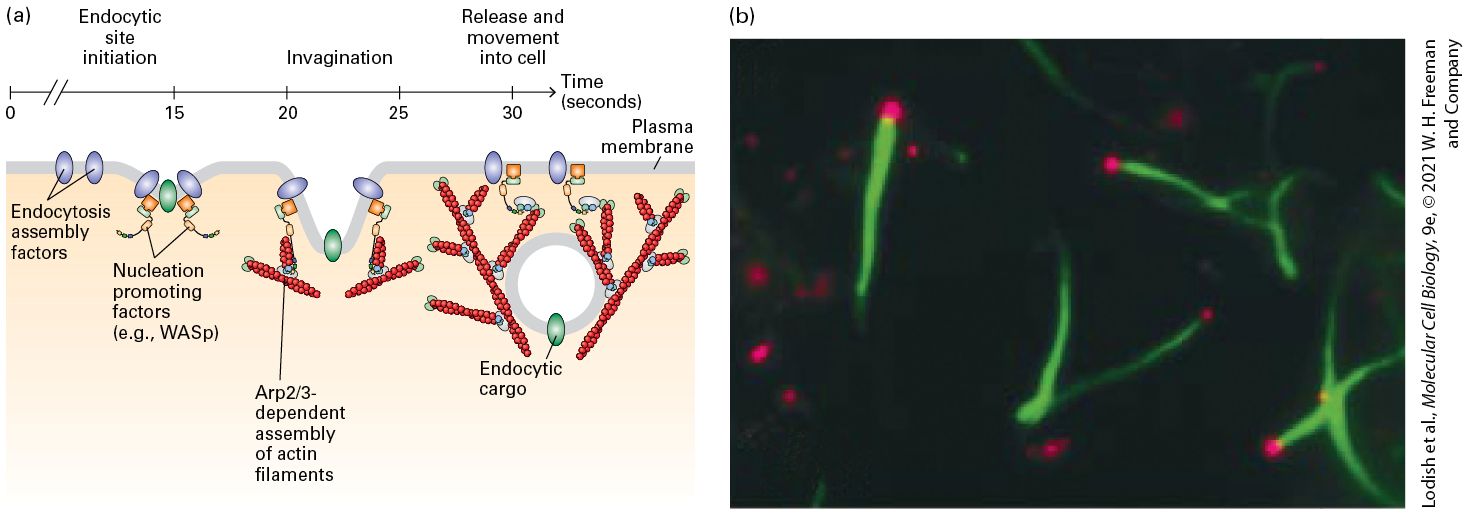
Table 17-1 • Summary of Actin-Binding Proteins Discussed in This Chapter Name Primary Function Profilin G-actin nucleotide exchange Cofilin F-actin disassembly
Actin Polymerization In Vitro Proceeds in Three Steps
ThymosinG-actin monomer sequestering CapZ and Tropomodulin F-actin end-binding Gelsolin F-actin severing Formins, Actin nucleating Fimbrin, α-actinin, Filamin, Spectrin F-actin cross-linking Ankyrin, Band 4.1, ERM proteins, Dystrophin F-actin-membrane linking Myosins F-actin-based molecular motors Nebulin F-actin length regulation Tropomyosin F-actin stabilizing Actin Polymerization In Vitro Proceeds in Three Steps The in vitro polymerization of G-actin monomers to form F-actin filaments can be monitored by viscometry, sedimentation, fluorescence spectroscopy, or fluorescence microscopy (see Chapter 4). When actin filaments grow long enough to become entangled, the viscosity of the solution, which is measured as a decrease in its flow rate in a viscometer, increases. The basis of the sedimentation assay is the ability of ultracentrifugation (100,000g for 30 minutes) to sediment F-actin, but not G-actin. The third assay makes use of G-actin covalently labeled with a fluorescent dye; the fluorescence spectrum of the labeled G-actin
monomer changes when it polymerizes into F-actin. Finally, growth of the fluorescently labeled filaments can be imaged in real time with fluorescence microscopy. These four assays are useful for kinetic studies of actin polymerization and for characterizing actin-binding proteins to determine how they affect actin dynamics or how they cross-link actin filaments. The mechanism of actin assembly has been studied extensively. Remarkably, one can purify G-actin at a high protein concentration without it forming filaments — provided it is maintained in a buffer with ATP and low levels of cations. The addition of cations (e.g., to 100 mM and 2 mM ) to a solution of G-actin will induce polymerization into F-actin filaments, with the kinetics of the reaction depending on the starting concentration of G-actin. The process is reversible: F-actin depolymerizes into G-actin when the ionic strength of the solution is lowered. The polymerization of pure G-actin in vitro proceeds in three sequential phases (Figure 17-7a).
FIGURE 17-7 The three phases of in vitro G-actin polymerization. (a) In the initial nucleation phase, ATP–G-actin monomers (red) slowly form stable complexes of actin (purple). These nuclei are rapidly elongated in the second phase by the addition of subunits to both ends of the filament. In the third phase, the ends of actin filaments are in equilibrium with monomeric G-actin. (b) Time course of the in vitro polymerization reaction reveals the initial lag period associated with nucleation, the elongation phase, and the steady state. (c) If some short, stable actin filament fragments are added at the start of the reaction to act as nuclei, elongation proceeds immediately, without any lag period. Description In illustration labeled (a) the formation of a microfilament occurs in several steps. 1. Nucleation: Free molecules of A T P-G-actin form a stable nucleus of actin. 2. Elongation: Addition of subunits leads to growth of the helix in both directions from the nucleus. 3. Steady state: A steady state is formed when the rate of loss and gain of actin molecules is equal. The negative end, the positive end, and the nucleus of F-actin are labeled. In the graph labeled (b), the vertical axis plots mass of filaments, and the horizontal axis plots time. No units are given. A sigmoidal curve starts slowly during nucleation, then rises sharply during elongation, and then plateaus off at the steady state.
In the graph labeled (c), the vertical axis plots mass of filaments, and the horizontal axis plots time. The curve starts at elongation with a label that reads nuclei added at t equals 0. The curve is logarithmic, rising sharply and then plateauing off, corresponding to the elongation and steady state parts of growth, respectively. 1. The nucleation phase is marked by a lag period in which G-actin subunits combine into an oligomer of two or three subunits. When the oligomer reaches three subunits in length, it can act as a seed, or nucleus, for the next phase. 2. During the elongation phase, the short oligomer rapidly increases in length by the addition of actin monomers to both of its ends. As Factin filaments grow, the concentration of G-actin monomers decreases until equilibrium is reached between the filament ends and monomers, and a steady state is reached. 3. In the steady-state phase, G-actin monomers exchange with subunits at the filament ends, but there is no net change in the total length of filaments. The kinetic curves in Figure 17-7b, c show the filament mass during each phase of polymerization. We can show experimentally that the lag period is due to nucleation, because the lag period can be eliminated by the addition of a small number of F-actin nuclei — consisting of very short filaments — to a solution of G-actin (Figure 17-7c). How much G-actin is required for nucleation to occur? Scientists have placed various concentrations of G-actin under polymerizing conditions and found that, below a certain concentration, filaments cannot assemble (Figure 17-8). Above this concentration, filaments begin to form; when
steady state is reached, the incorporation of more free subunits is balanced by the dissociation of subunits from filament ends to yield a mixture of filaments and monomers. The minimal concentration of monomers at which filaments form is known as the critical concentration . Below , filaments will not form; above , filaments form. At steady state, the concentration of monomeric actin remains at the (Figure 17-8).
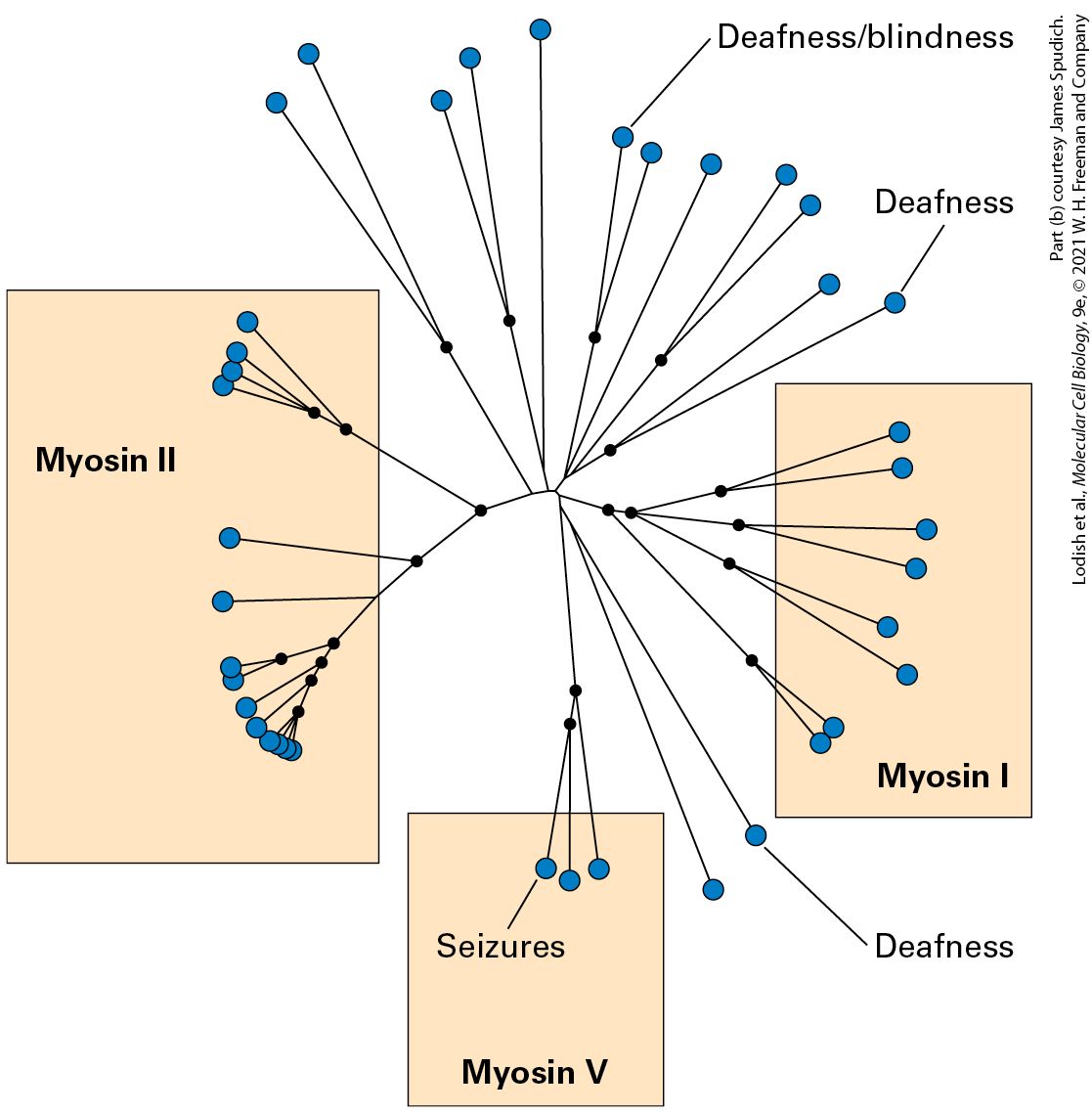
FIGURE 17-8 Determination of the critical concentration of G-actin above which filaments are formed. When different concentrations of actin are placed under polymerizing conditions and the reaction is allowed to come to steady state, the is the concentration at which actin filaments begin to form. At initial monomer concentrations below the , no polymerization takes place. At monomer concentrations above the , filaments assemble until steady state is reached with the monomer concentration remaining at . Description In the line graph, the vertical axis plots mass, and the horizontal axis plots total actin concentration (the sum of monomer and filaments). No units are given. Two curves are
Actin Filaments Grow Faster at (+) Ends than at (−) Ends
plotted. The first curve is labeled monomer. The monomer curve rises and then plateaus at the value of big C subscript small c. The second curve is labeled filament. The filament curve rises from big C subscript small c, increasing in a linear pattern. Actin Filaments Grow Faster at Ends than at Ends Using myosin S1 decoration experiments, we saw earlier that F-actin has an inherent structural polarity that is due to the uniform orientation of subunits in the filament (see Figures 17-5 and 17-6). As we described above, G-actin can bind ATP or ADP. When ATP–G-actin assembles into a filament, the ATP is rapidly hydrolyzed to , and then the is more slowly released, generating a filament mostly composed of and ADP-actin. Moreover, this gives rise to two different ends: the end is composed of ATP-actin or , whereas the end has . If free ATP–G-actin is added to such a preexisting myosin-decorated filament, the two ends grow at very different rates (Figure 17-9). In fact, the rate of addition of ATP–G-actin is nearly 10 times faster at the end than at the end. Kinetic experiments have shown that the rate of addition is about at the end and about at the end (Figure 17-10a). This means that if 1 of free ATP–G-actin is added to preformed filaments, 12 subunits, on average, will be added to the end every second, whereas only 1.3 will be added to the end every second. Note that the rate of addition (the association rate) is a function of the concentration of free ATP–G-actin. What about the rate of subunit loss from each end? By
contrast with the rates of addition, the rates of dissociation of G-actin subunits from the two ends are quite similar: about from the end and from the end. Since this dissociation is simply the rate at which subunits leave the filament ends, it does not depend on the concentration of free ATP–G-actin. EXPERIMENTAL FIGURE 17-9 The two ends of a myosin-decorated actin filament grow at different rates. When short actin filaments are decorated with myosin S1 heads to reveal the orientation of the filaments and then used to nucleate actin polymerization, Gactin monomers are added much more efficiently to the ends than to the ends of the nucleating filaments.
FIGURE 17-10 Actin treadmilling. ATP-actin subunits are added faster at the end than at the end of an actin filament, resulting in a lower critical concentration at the end and treadmilling at steady state. (a) The rate of addition of ATP–G-actin is much faster at the end than at the end, whereas the rate of dissociation of actin is similar at the two ends. This difference results in a lower critical concentration at the end. At steady state, ATP-actin is added preferentially at the end, giving rise to a short region of the filament containing ATP-actin and regions containing and ADPactin toward the end. (b) At steady state, ATP–G-actin subunits add preferentially to the end, while ADP–G-actin subunits disassemble from the end, giving rise to treadmilling of subunits. For reference, two actin subunits are colored blue, to highlight that over time subunits appear to move from the plus end toward the minus end within the filament. Description The illustration labeled (a) shows an actin filament. It has two rope-like structures made up of spheres twisted around each other. The filament is divided into three sections. From the negative end, these are composed of A D P-actin, A D P-phosphate actin, and A T P actin. The rate of loss of monomers at the negative end is 0.8 per second and 1.4 per second at the positive end. The critical concentration at the negative end is 0.6 micro molars and that at the positive end is 0.12 micro molars. The rate of addition at
the negative end is 1.3 per micromole per second and 12 per micromole per second at the positive end. The illustration labeled (b) shows three actin filaments one below the other. An A T P – G- actin molecule is added at its positive end while one of the molecules disassociates at the negative end. The nucleus represented by two differently colored spheres is present near the positive end. As these additions and deletions of A T P- G- actin molecules occur; the nucleus moves from the positive end towards the negative in the steady state. How do these association and dissociation rates impact actin dynamics? As we noted above, the association rate depends on the free ATP–G-actin concentration, whereas the dissociation rate does not. Thus when the free ATP–G-actin concentration is high, subunits will be added to the filament ends more quickly than subunits will be lost, so the filament will grow. As the concentration is lowered, a point will be reached where the rate of addition is balanced by the rate of loss, and no net filament growth occurs. Consider just the end for the moment. The free G-actin concentration at which rates of addition and loss are equal is called the critical concentration for the end, We can calculate by setting the rate of assembly equal to the rate of disassembly. Thus at the critical concentration the rate of assembly is times the measured rate of addition of , whereas the rate of disassembly is independent of the free actin concentration, namely, . Setting these two rates equal to each other yields , or , for the end. Above this free ATP–G-actin concentration, subunits are added to the end and net growth occurs, whereas below it, there is a net loss of subunits, and shrinkage occurs.
Actin Filament Treadmilling Is Accelerated by Profilin and Cofilin
Now let’s consider the end. Because the rate of addition is much lower, , yet the rate of dissociation is about the same, , we expect the critical concentration at the end to be higher than . Indeed, as we just did for the end, we can calculate to be about , or . Thus at less than free ATP–G-actin — say, end will lose subunits. Notice that at this concentration the end will grow, because is above . Because the critical concentrations for the two ends are different, at steady state the free ATP–G-actin will be intermediate between and , so the end will grow and the end will lose subunits. This phenomenon is known as treadmilling because particular subunits, such as those shown in blue in Figure 17-10b, appear to move through the filament. The treadmilling of actin filaments is powered by hydrolysis of ATP. When ATP–G-actin binds to the end of a filament, ATP is hydrolyzed to ADP and . Since the is slowly released from the subunits in the filament, the filament becomes organized into three regions: a short region of ATP-actin subunits at the end, ADP- –actin in the middle of the filament, and, following release, ADP-actin subunits toward the end (Figure 17-10a). During release of from subunits in a filament, actin undergoes a conformational change that is responsible for the different association and dissociation rates at the two ends. Our analysis relies on a plentiful supply of ATP–G-actin, which, as we will see, turns out to be the case in vivo. Thus actin can use the power generated by hydrolysis of ATP to treadmill, and treadmilling filaments can do work in vivo, as we will see later.
Actin Filament Treadmilling Is Accelerated by Profilin and Cofilin Actin treadmilling in vivo can occur at rates several times higher than happens with pure actin in vitro under physiological conditions. Why is treadmilling faster in vivo, and how does the cell recharge the ADP-actin dissociating from the end into ATP-actin that adds at the end? The answers involve two actin-binding proteins. The first of these proteins is profilin, a small protein that binds G-actin on the side opposite the nucleotide-binding cleft (see the profilin cycle in
Figure 17-11). When profilin binds ADP-actin, it opens the cleft and greatly enhances the loss of ADP. Because [ATP] in the cell is much greater than [ADP], ATP readily binds to G-actin, forming a profilin–ATPactin complex. This complex cannot bind to the end because profilin blocks the sites on G-actin for end assembly. However, the profilin– ATP-actin complex can bind efficiently to the end. Once the new actin subunit is bound to the filament, profilin dissociates. Profilin activity does not enhance the treadmilling rate, but it does ensure a constant supply of ATP-actin formed from released ADP-actin. As a consequence, essentially all of the free G-actin in a cell has bound ATP.
FIGURE 17-11 Regulation of filament turnover by actin-binding proteins. Actin-binding proteins regulate the rate of assembly and disassembly of actin filaments as well as the availability of G-actin for polymerization. In the profilin cycle (step 1 ), profilin binds
ADP–G-actin and catalyzes the exchange of ADP for ATP. The profilin–ATP–G-actin complex can deliver actin to the end of a filament with dissociation and recycling of profilin. In the cofilin cycle (step 2 ), cofilin binds preferentially to regions of filaments containing ADP-actin, inducing them to fragment and thus enhancing depolymerization by making more filament ends. In the thymosincycle (step 3 ), ATP–G-actin made available by the profilin cycle is bound by , which sequesters it from polymerization. As the free G-actin concentration is lowered by polymerization, dissociates to make free G-actin available for association with profilin and further polymerization. Description The illustration shows an actin microfilament on the top with its negative end at the left and the positive end at the right. It has two rope-like structures made up of spheres twisted around each other. Its three parts, from left to right actin-A D P, actin-A D P-P I, and actin-A T P are labeled. The actin–A D P part of the filament is embedded with cofilin molecules. Below, the structures representing A D P- actin, A T P-actin, profiling, cofilin, and thymosin-beta subscript 4 are listed. 1. Profilin cycle: a profilin molecule binds to A D P-actin. A T P gets converted to A D P to yield A T P actin, which then binds to the positive end of the actin filament. 2. Cofilin cycle: the actin–A D P part of the filament detaches from the filament to release an A D P-actin and cofilin complex. Cofilin is released from this complex to once again bind to the negative end of the filament. The released A D P – actin further undergoes the profiling cycle. 3. Thymosin-beta subscript 4 cycle: the A T P actin produced in the Profilin cycle binds to a thymosin-beta subscript 4 molecule to then be released again to be used in the profilin cycle. Profilin has another important property: it can bind proteins with sequences rich in proline residues at the same time it is bound to actin. We
Thymosin-β4 Provides a Reservoir of Actin for Polymerization
will see later how this property can enhance the rate of actin filament assembly. The second of the actin-binding proteins is cofilin, a small protein that binds specifically to ADP-actin subunits within F-actin. Recall from
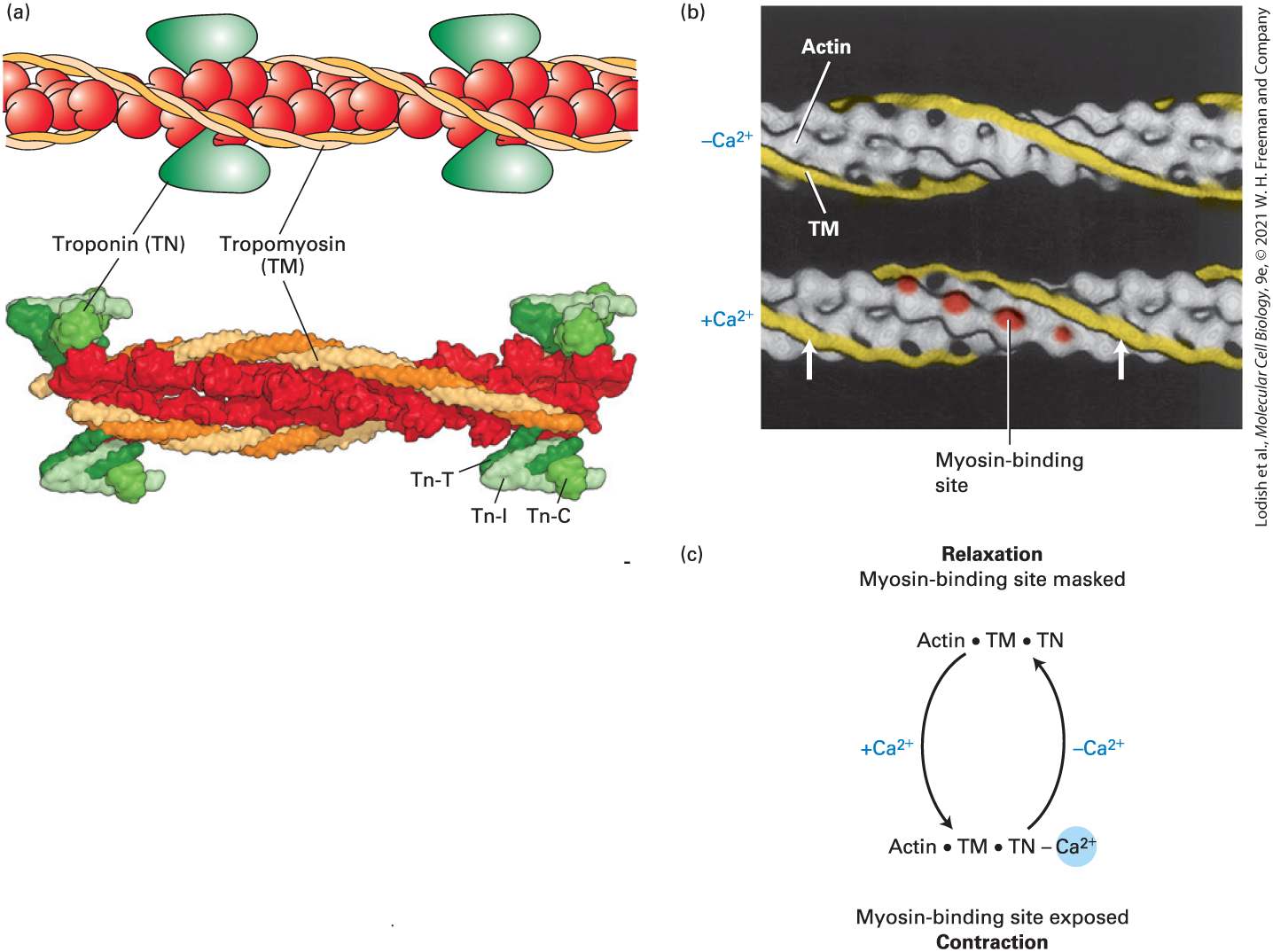
Figure 17-10a, these are the subunits found toward the end of F-actin. Cofilin binds by bridging two actin monomers and inducing a small change in the twist of the filament. This small twist destabilizes the filament between regions with and without cofilin, breaking it into short pieces. By breaking the filament in this way, cofilin generates many more free ends and therefore greatly enhances the net disassembly of the filament (see the cofilin cycle in Figure 17-11). The released ADP-actin subunits are then recharged by profilin and added to the end as described above. When purified profilin and cofilin are mixed with purified G-actin under conditions where filaments will form, rates of treadmilling in vitro increase more than tenfold compared to treadmilling in the absence of these two actin-binding proteins. These enhanced levels of in vitro treadmilling are similar to what is seen in vivo. Thymosin- Provides a Reservoir of Actin for Polymerization It has long been known that cells often have a very large pool of G-actin, sometimes constituting as much as half the actin in the cell. Since cellular
Capping Proteins Block Assembly and Disassembly at Actin Filament Ends
actin levels can be as high as , this means that there can be as much as unpolymerized actin in cells. Since the critical concentration in vitro is about , why doesn’t all this actin polymerize? The answer is, at least in part, due to G-actin sequestering proteins. One of these proteins is thymosin- . When thymosinbinds to ATP–G-actin, it inhibits addition of the actin subunit to either end of a filament. As an example, let’s consider human blood platelets. These diskshaped cell fragments are very abundant, and when they are activated during blood clotting, they undergo a burst of actin assembly. Platelets are rich in actin; they are estimated to have a total concentration of about actin, of which about is in the unpolymerized form. They also contain about thymosin- , which sequesters much of the free actin. However, as in any protein-protein interaction, free actin and free thymosinare in a dynamic equilibrium with actin–thymosin- . When some free actin is incorporated into actin filaments, then more actin– thymosinwill dissociate, thus maintaining a steady supply of actin subunits available for polymerization (see the thymosincycle in Figure 17-11). Thus thymosinbehaves as a buffer of unpolymerized actin, making ATP-actin subunits available as they are needed. Capping Proteins Block Assembly and Disassembly at Actin Filament Ends Cells further regulate treadmilling and actin filament dynamics with capping proteins that bind to filament ends. If this were not the case, actin filaments would continue to grow and disassemble in an uncontrolled
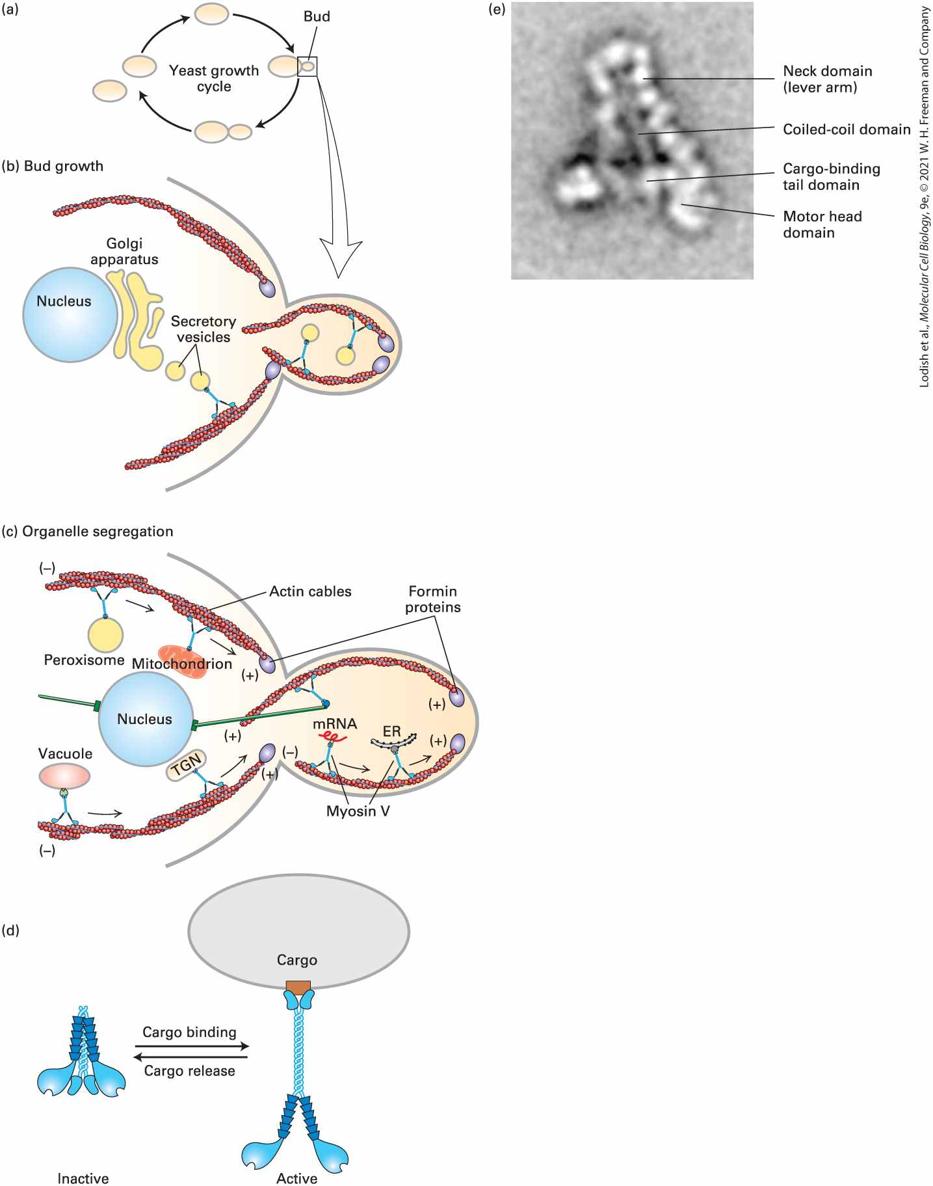
manner. As one might expect, two classes of proteins have been discovered: ones that bind the end and ones that bind the end (Figure 17-12).
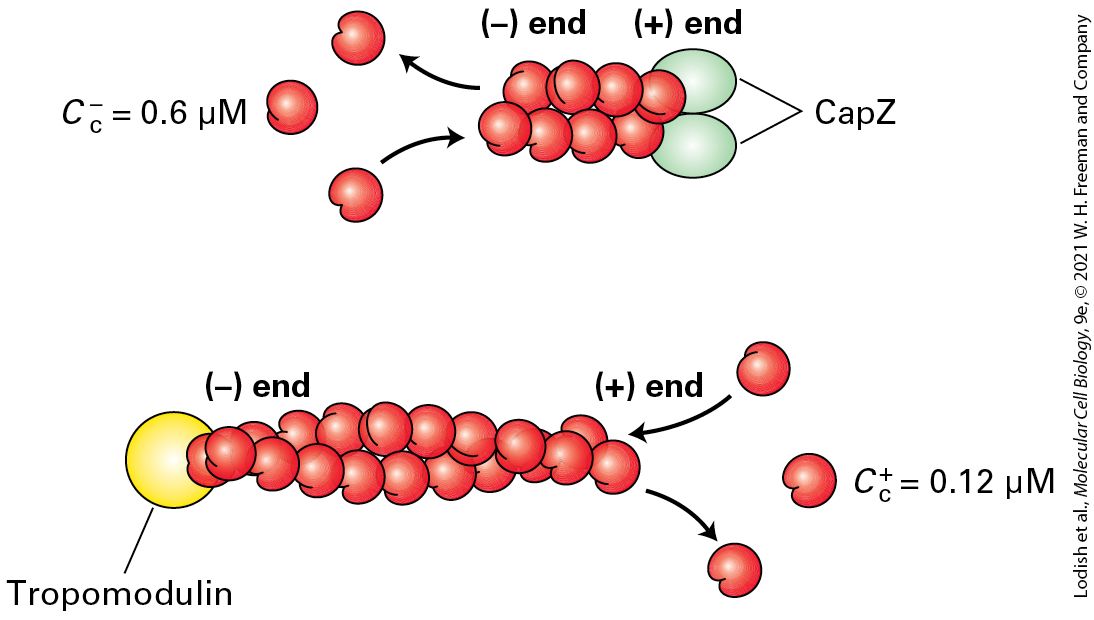
FIGURE 17-12 Filament capping proteins. Capping proteins block assembly and disassembly at filament ends. CapZ blocks the end, which is where filaments normally grow, so its function is to limit actin dynamics to the end. The capping protein tropomodulin blocks ends, where filament disassembly normally occurs; thus the major function of tropomodulin is to stabilize filaments. Description The illustration shows an actin filament where a capping protein called cap Z is bound to the positive end of the filament. This binding allows disassembly of molecules from the negative end, where the critical concentration is 0.6 micromoles. The illustration also shows another actin filament where tropomodulin is bound to the negative end of the filament. This binding allows the growth of the filament at the positive end, where the critical concentration is 0.12 micromoles.
A protein known as CapZ, consisting of two closely related subunits, binds with a very high affinity to the end of an actin filament, thereby inhibiting subunit addition or loss. The concentration of CapZ in cells is generally sufficient to rapidly cap any newly formed ends. So how can filaments grow at their ends? At least two mechanisms regulate the activity of CapZ. First, the capping activity of CapZ is inhibited by the regulatory phospholipid phosphatidylinositol 4,5bisphosphate , found in the plasma membrane (see Chapter 16). Second, recent work has shown that certain regulatory proteins are able to bind the end and protect it from CapZ while still allowing assembly there. Thus cells have evolved an elaborate mechanism to block assembly of actin filaments at their ends except when and where assembly is needed. Tropomodulin is one of the regulatory proteins that inhibit filament assembly and disassembly by binding to the end of an actin filament. This protein is found predominantly in cells in which actin filaments need to be stabilized for long periods of time, such as the short actin filaments in the cortex of red blood cells and the actin filaments in muscle. In both of these cells, tropomodulin works with the actin-binding protein tropomyosin to stabilize actin filaments. We will look at these protein interactions in more detail later in this chapter. In addition to CapZ, another class of proteins can cap the ends of actin filaments. These proteins can also sever actin filaments. One member of this family, gelsolin, is regulated by ion concentration. On binding , gelsolin undergoes a conformational change that allows it to bind to
the side of an actin filament and then insert itself between subunits of the helix, thereby breaking the filament. It then remains bound to and caps the end, generating a new end that can disassemble. As we discuss in a later section, actin cross-linking proteins can provide linkages between individual actin filaments to turn a solution of F-actin into a gel. If gelsolin is added to such a gel and the level of is elevated, gelsolin will sever the actin filaments and turn the gel back into a liquid solution. This ability to turn a gel into a sol is why the protein was named gelsolin. KEY CONCEPTS OF SECTION 17.2 Dynamics of Actin Filaments The rate-limiting step in actin filament assembly is the formation of a short actin oligomer (nucleus) that can then be elongated into filaments. The critical concentration is the concentration of G-actin below which actin filaments will not form. When the concentration of G-actin is above the , the filament end will grow; when it is less than the , the filament will shrink (see Figure 17-8). ATP–G-actin is added much faster at the end than at the end, resulting in a lower critical concentration at the end than at the end. At steady state, actin subunits treadmill through a filament. Treadmilling means ATPactin is added at the end, ATP is hydrolyzed to ADP and , is lost slowly, and ADP-actin dissociates from the end. The length and rate of turnover of actin filaments is regulated by specialized actinbinding proteins (see Figure 17-11). Profilin enhances the exchange of ADP for ATP on G-actin; cofilin binds regions of ADP–F-actin to break the filament and generate new ends to enhance the rate of disassembly of ADP-actin from filament ends, and thymosinbinds G-actin to provide reserve G-actin when it is needed. Capping proteins bind to filament ends, blocking assembly and disassembly.
Formins Assemble Unbranched Filaments
17.3 Mechanisms of Actin Filament Assembly The rate-limiting step of actin polymerization is the formation of an initial actin nucleus from which a filament can grow (see Figure 17-7a). In cells, this step is used as a control point to determine where actin filaments are assembled and what types of actin structures are generated (see Figures 17-1 and 17-4). Two major classes of actin-nucleating proteins, the formin protein family and the Arp2/3 complex, nucleate actin assembly under the control of signal transduction pathways. Moreover, they nucleate the assembly of different actin structures: formins lead to the assembly of long actin filaments, whereas the Arp2/3 complex leads to branched networks of filaments. Here we discuss each separately and see how the power of actin polymerization can drive motile processes in a cell. We then touch on other specialized actin-nucleating factors. Formins Assemble Unbranched Filaments Formins are found in nearly all eukaryotic cells as a diverse family of proteins; seven different classes are present in vertebrates. Although they are diverse, all formin family members have two adjacent domains in common, the so-called FH1 and FH2 domains (formin-homology domains 1 and 2). Two FH2 domains from two individual formin monomers
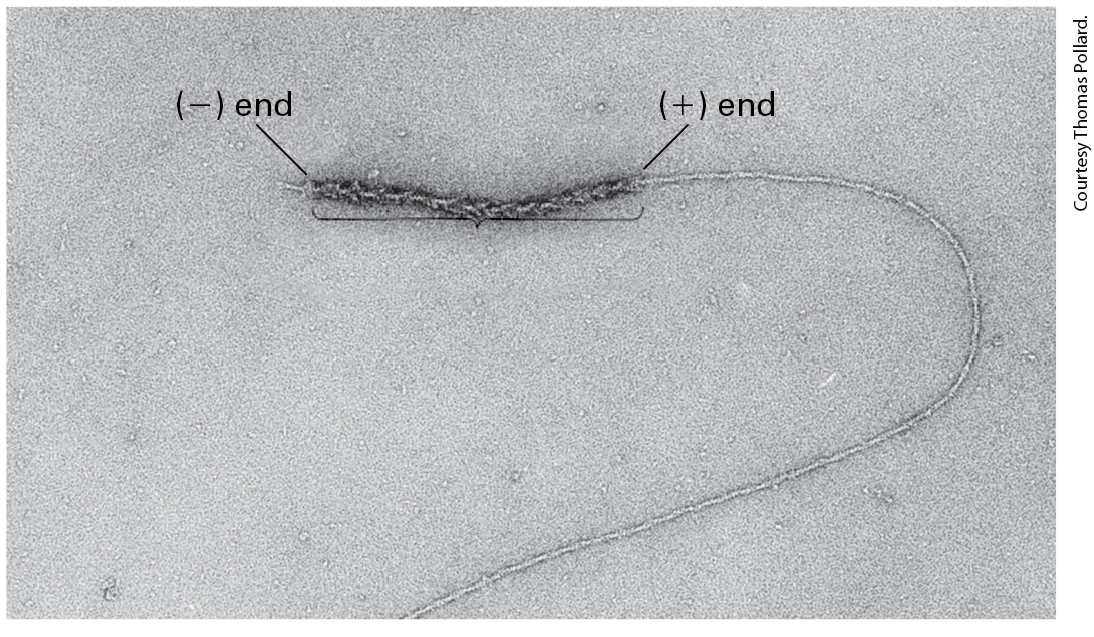
associate to form a doughnut-shaped complex (Figure 17-13a). This complex has the ability to nucleate actin assembly by binding two actin subunits, holding them so that the end of the nascent filament is toward the FH2 domains. The filament can now grow at the end while the FH2 domain dimer remains attached to it. How is this possible? As we saw earlier, an actin filament can be thought of as two intertwined strands of subunits. The FH2 dimer can bind to the two terminal subunits. It then probably rocks between the two end subunits, letting go of one to allow addition of a new subunit and then binding the newly added subunit and freeing up space for the addition of another subunit to the other strand. In this way, rocking between the two subunits on the end, it can remain attached while simultaneously allowing growth at the end (Figure 1713a).
FIGURE 17-13 Actin nucleation by the formin FH2 domain. (a) Formins have a domain called FH2 that can form a dimer and nucleate filament assembly. The dimer binds two actin subunits (step 1 ) and, by rocking back and forth (steps 2 – 4 ), can allow insertion of additional subunits between the FH2 domain and the end of the growing filament. The FH2 domain protects the end from being capped by capping proteins. (b) The FH2 domain of a formin was labeled with colloidal gold (black dot) and used to nucleate assembly of an actin filament. The resulting filament was visualized by electron microscopy after staining with uranyl acetate. Formins assemble long unbranched filaments. [Part (b) Republished with permission from AAAS, from D. Pruyne et al., 2002, “Role of Formins in Actin Assembly: Nucleation and Barbed-End Association,” Science 297(5581): 612–615; permission conveyed through Copyright Clearance Center, Inc.] Description The illustration labeled (a) shows four steps. Step 1: the dimer of formin F H 2 domain binds to two actin subunits. Step 2: a third subunit binds to the complex from step 1. Step 3: a fourth subunit binds to the complex from step 2. Step 4: many subunits are now bound to each other as the F H 2 domain keeps assembling subunits for the actin filament to grow. The electron micrograph labeled (b) shows a gold stained F H 2 domain on an actin filament. The filament is visible with a black spot at the tip, showing the location of the F H 2 domain. The FH1 domain adjacent to the FH2 domain also makes an important contribution to actin filament growth (Figure 17-14). This domain is rich in proline residues, which serve as sites for the binding of several profilin molecules. We discussed earlier how profilin can exchange the ADP nucleotide on G-actin for ATP to generate profilin–ATP-actin. The FH1 domain behaves as a landing site for profilin–ATP–G-actin to increase the local concentration of these complexes. The actin from these profilin-actin
complexes is then fed into the FH2 domain to add actin to the end of the filament, and the profilin is released (Figure 17-14).
FIGURE 17-14 Regulation of formin activity by an intramolecular interaction. Some of the formin classes found in vertebrates are regulated by an intramolecular interaction. In its inactive state, the formin folds back on itself to inhibit the activity of the FH2 domain. The inactive formin is activated when its Rho-binding domain (RBD) binds to membrane-bound active Rho-GTP, resulting in exposure of the formin’s FH2 domain, which can then nucleate the assembly of a new actin filament. All formins have an FH1 domain adjacent to the FH2 domain; the proline-rich FH1 domain is a site for recruitment of profilin–ATP–G-actin complexes that can then be fed into the growing end. For simplicity of representation, a single formin protein is shown, but as shown in Figure 17-13, the FH2 domain functions as a dimer to nucleate actin assembly. Regulation of the Rho family of small GTPases is detailed in Figures 17-40 and 17-42. Description The illustration shows a plasma membrane with the exterior and the cytoplasm labeled. An inactive formin protein made of a rho binding domain, F H 1, and F H 2 is present in the cytosol. The inner membrane of the plasma membrane is bound to a Rho- G T P
The Arp2/3 Complex Nucleates Branched Filament Assembly
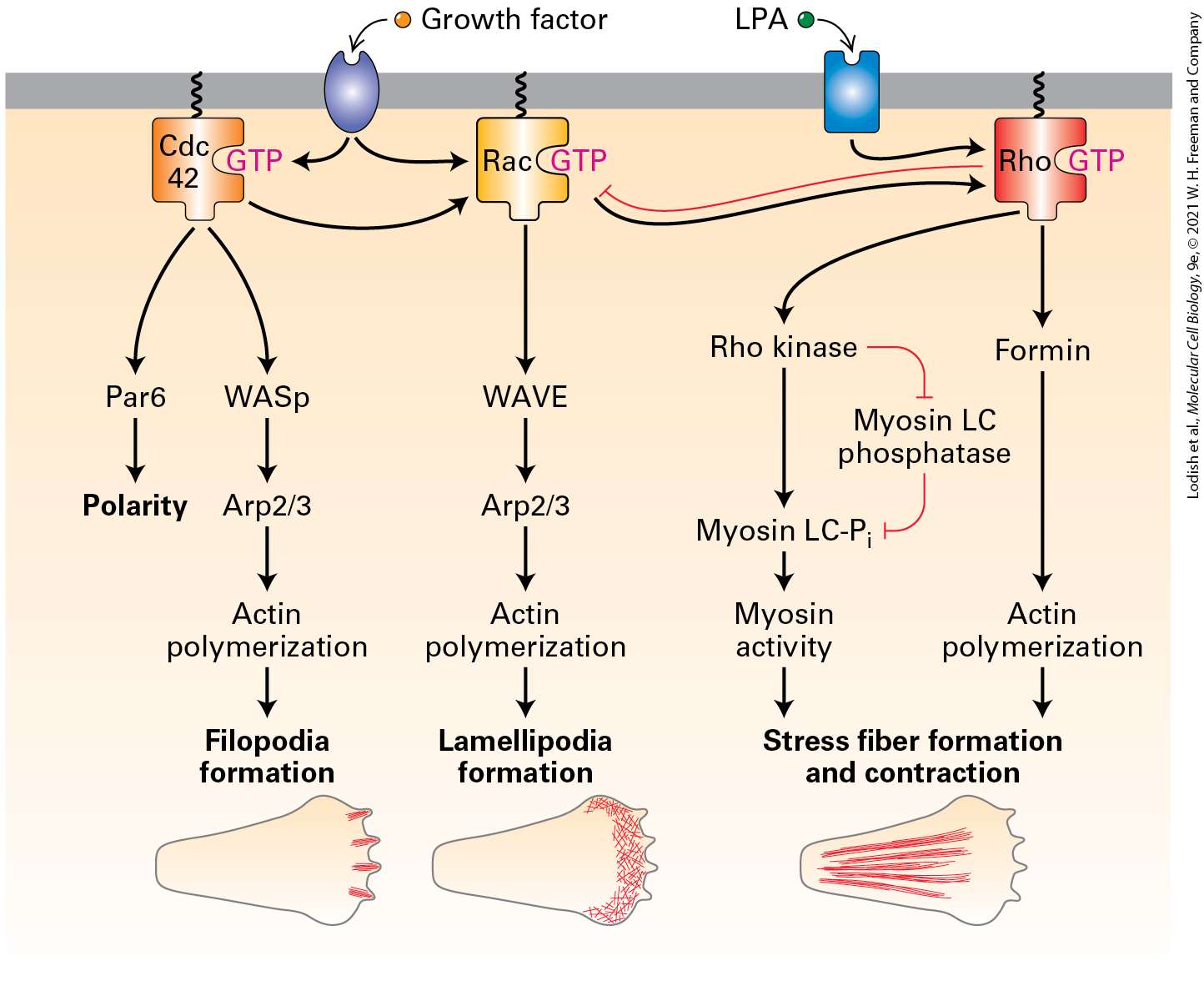
protein. The Rho binding domain of the inactive formin protein binds to the Rho- G T P protein to become active. The profilin A T P-actin monomers bind to F H 1. The monomers move to F H 2 to become a filament with a positive and a negative end. The profilin molecule is released at F H 2. Since formin allows rapid addition of actin subunits to the end, long filaments with a formin at their end are generated (Figure 17-13b). Thus formins not only nucleate actin assembly, but they remain bound to the end and, in conjunction with profilin, they facilitate rapid filament assembly. To ensure continued growth of a filament, formins bind to the end in such a way that precludes binding of a end capping protein such as CapZ, which would terminate assembly. To be useful to a cell, formin activity must be regulated. Many formins exist in a folded, inactive conformation as a result of an interaction between the first half of the protein and its C-terminal tail. These formins are activated by membrane-bound Rho-GTP, a Ras-related small GTPase (discussed in Section 17.7). When Rho switches from its inactive RhoGDP form to its activated membrane-bound Rho-GTP state, it can bind and activate these formins (see Figure 17-14). Formins are responsible for the assembly of long actin filaments such as those found in muscle cells, stress fibers, filopodia, and the contractile ring that forms during cytokinesis (see Figure 17-4). The different formin classes in animals differ in their ability to nucleate actin assembly, and some formins can also bundle actin filaments.
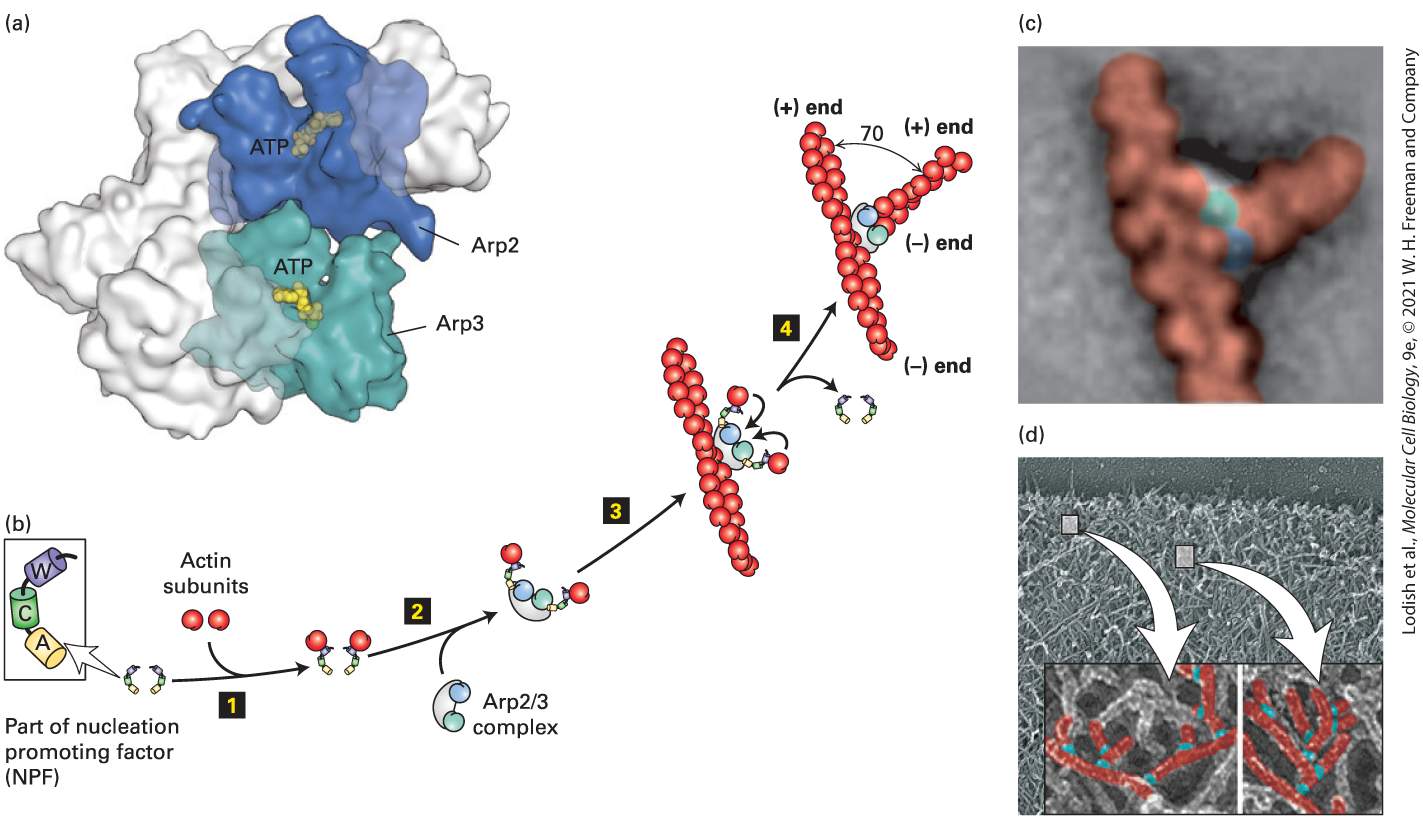
The Complex Nucleates Branched Filament Assembly The complex is a protein machine consisting of seven subunits, two of which are actin-related proteins (Arp; Figure 17-15a). It is found in essentially all eukaryotes, including plants, yeasts, and animal cells. The complex alone is a very poor nucleator. To nucleate the assembly of branched actin filaments, needs to be activated by a nucleation promoting factor (NPF). The major NPF family is characterized by the presence of a region called WCA (WH2, connector, acidic for the acidic amino acid residues it contains). Experiments have shown that if the WCA domain is added to an actin assembly assay together with preformed actin filaments, becomes a potent nucleator of further actin assembly.
FIGURE 17-15 Actin nucleation by the complex. (a) X-ray crystallographic structure of the complex, with five of the subunits in gray and the Arp2 and Arp3 subunits in green and blue. (b) To nucleate actin assembly efficiently, must interact with the activating part of an NPF, shown here with its W (WH2), C (connector), and A (acidic) domains. Step 1 involves binding of an actin subunit to the W domain of each NPF. Two NPF-actin complexes then bind the complex (step 2 ). This interaction induces a conformational change in the complex. After binding of the complex to the side of an actin filament, the actin subunits delivered by the W domains bind to the complex (step 3 ), which then initiates the assembly of an actin filament at the available end (step 4 ). The branch makes a characteristic 70° angle between the old and new filaments. (c) Averaged image compiled from several electron micrographs of at an actin branch. (d) Image of actin filaments in the leading edge of a motile cell, with a magnification and coloring of individual branched filaments. [Part (a) Data from B. J. Nolen and T. D. Pollard, 2007, Mol. Cell 26:449–457, PDB ID 2p9l. Part (c) C. Egile et al., 2005, “Mechanism of Filament Nucleation and Branch Stability Revealed by the Structure of the Arp2/3 Complex at Actin Branch Junctions.” PLoS Biol. 3(11):e383; https://doi.org/10.1371/journal.pbio.0030383. Part (d) T. M. Svitkina and G. G. Borisy, 1999, J. Cell Biol. 145:1009–1026; https://doi.org/10.1083/jcb.145.5.1009.] Description The illustration labeled (a) shows the three-dimensional X-ray structure of A r p 2 slash 3 protein complex. The space-filling structure of an A T P is bound to A r p 2 and A r p 3 subunits. The illustration (b) depicts a sequence of induction of actin nucleation by A r p 2 slash 3 and nucleation promoting factors (N P F). The sequence has 4 steps. Step 1: actin subunits bind to the N P F. Step 2: A r p 2 slash 3 complex binds to the complex formed in the previous step. Step 3: the complex formed in the previous step binds to the center of an actin filament. Step 4: the N P F gets removed. A vertical actin filament now is bound to a horizontal second filament. There is a 70-degree angle between the two filaments. Their positive and negative ends are labeled.
The electron micrograph labeled (c) shows an r p complex at a branch in actin microfilaments. The actin filaments are colored red, and the A R P 2 and A R P 3 subunits are colored green and blue. The electron micrograph labeled (d) shows branched actin filaments highlighted with arrows. How do the complex and NPFs nucleate the assembly of actin filaments? Two NPFs each bind an actin subunit at their WH2 domains, and together, they activate the complex through its interaction with their connector and acidic domains. In the inactive complex, the two actin-related polypeptides and are in the wrong configuration to nucleate filament assembly (see Figure 17-15b, step 2 ). When activated by the NPFs, Arp2 moves into a new configuration permitting the complex to bind to the side of a preexisting actin filament. The actin subunits brought in by the WH2 domains of the NPFs bind to the template to nucleate filament assembly at the end (Figure 1715b, step 3 ). The NPFs are released, and the new end then grows as long as ATP–G-actin is available or until it is capped by a end capping protein such as CapZ. The angle between the old filament and the new one is 70° (Figure 17-15b, c). This is also the angle observed experimentally in branched filaments at the leading edges of motile cells, which are believed to be formed by the action of the activated complex (Figure 1715d). As we discuss in subsequent sections, the complex can be used to drive actin polymerization that powers intracellular motility. Actin nucleation by the complex is finely controlled, and NPFs are part of that regulatory process. One NPF is called WASp, as it is defective
in patients with Wiskott-Aldrich syndrome, an X-linked disease characterized by eczema, low platelet count, and immune deficiency. WASp exists in a folded inactive conformation that makes the WCA domain unavailable (Figure 17-16). To ensure that WASp is activated only at the plasma membrane, its activation requires two signals. One signal is binding between WASp and the regulatory phospholipid ,which is characteristically enriched in the plasma membrane (Chapter 16). WASp binds through its basic domain. The second signal is binding of the activated form of the small GTP-binding protein Cdc42 to the RBD region of WASp. GTP-Cdc42 is itself activated in response to certain signaling pathways (discussed in Section 17.7). This type of two-signal input, called coincidence detection, ensures that the protein is activated only at the right place — at the plasma membrane — and by the right signaling pathway. Once bound to the two input signals, the conformation of WASp opens up, and the WCA domain becomes accessible.
FIGURE 17-16 Regulation of the complex by WASp and PI(4,5)P2. The NPF WASp is inactive due to an intramolecular interaction that masks the WCA domain. It is activated by a coincidence detection mechanism: it must bind both the regulatory phospholipid PI(4,5)P2 though its basic domain (B) and the membrane-bound active small G protein Cdc42-GTP (a member of the Rho family) through its Rho-binding domain (RBD). When activated in this way, the intramolecular interaction in WASp is relieved, allowing the W domain to bind actin and the acidic A domain to activate the complex. For simplicity, only a single interaction is shown. Regulation of the Rho family of small GTPases is detailed in Figures 17-40 and 17-42. Description The illustration shows a plasma membrane with its exterior and cytosol labeled. The cytosol has nucleation promoting factors W A S p which has the B, R B D, A, C, and W subunits. The plasma membrane is bound to a structure labeled P I (4,5) P 2. Next to it is a G-protein labeled c d c 42- G T P. The W a s P protein gets activated by binding to P I (4,5) P 2 and c d c 42- G T P. the subunit B is bound to P I (4,5) P 2. R B D is bound
Intracellular Movements Can Be Powered by Actin Polymerization
to c d c 42- G T P. The nucleation promoting factors is bound to an actin filament which has its negative and positive ends labeled. A second NPF is a large protein complex called WAVE, which also has a WCA domain that can activate the complex. WAVE is also activated by coincidence detection, specifically by binding to acidic phospholipids and to the active form of another small GTP-binding protein, Rac1. As we discuss in Section 17.7, activation of the complex by Cdc42 through WASp and by Rac1 through WAVE induces the formation of different microfilament-based structures. Although formins and the complex are found in fungi, plants, and animals, additional actin nucleators have been discovered in animal cells. One of these, called Spire, has four tandem WH2 domains, so it can bind four actin monomers. It does this in a manner that allows the actins to assemble into a filament, although the detailed mechanism remains to be understood. Given that actin filaments perform so many functions in cells, it is not surprising that additional NPF proteins and actin nucleators have recently been described. Intracellular Movements Can Be Powered by Actin Polymerization How can actin polymerization be harnessed to do work? As we have seen, actin polymerization involves the hydrolysis of ATP–G-actin to ADP–Gactin, which allows actin filaments to grow preferentially at the end
and disassemble at the end. If a collection of actin filaments were to become immobilized in the network of the cytoskeleton and you could bind to and ride on the assembling ends, you would be transported across the cell. This is just what the intracellular bacterial parasite Listeria monocytogenes does to get around the cell. The study of Listeria motility was, in fact, the way the nucleating activity of the protein was discovered. As we will see shortly, Listeria has hijacked a normal cell motility process for its own purposes. Listeria is a food-borne bacterial pathogen that causes mild gastrointestinal symptoms in most adults but can be fatal to elderly or immunocompromised individuals. It enters animal cells and divides in the cytoplasm. Its rapid movement around the cell is powered by massive local polymerization of actin to resemble a comet tail (Figure 17-17a), and when it runs into the plasma membrane, it continues to use the power of actin polymerization to push its way into the adjacent cell. Since the comet tail is embedded in the stationary cytoskeletal matrix of the cell, the bacterium is pushed forward, ahead of the polymerizing actin. Cytosolic factors then disassemble the tail so that actin subunits can be recycled for further polymerization. To do this, Listeria needs to direct the assembly of host-cell actin at one end of the bacterium and at the same time confine assembly there so that it efficiently pushes the bacterium forward. How does it do it? Listeria has on its surface a protein called ActA, which mimics an NPF by having an actin-binding site and an acidic region that efficiently activates the complex. Researchers have reconstituted Listeria motility in the test tube using purified proteins to see what are the minimal requirements for Listeria motility. Remarkably, the bacteria will
move when just four proteins are added: ATP–G-actin, the complex, CapZ, and cofilin (see Figure 17-17b, c). We have discussed the role of actin and , but why are CapZ and cofilin needed? As we have seen earlier, CapZ rapidly caps the free end of actin filaments, so when a growing filament no longer contributes to bacterial movement, it is rapidly capped and inhibited from further elongation. In this way, assembly occurs mostly adjacent to the bacterium, where ActA is stimulating the complex. Cofilin is necessary to accelerate the disassembly of the end region of the actin filament, regenerating free actin to keep the polymerization cycle going (see Figure 17-11). This minimal rate of motility can be increased by the inclusion of VASP and profilin. The ActA protein binds the host protein VASP, which has three important properties. First, VASP has a proline-rich region that can bind profilin–ATP-actin and thus enhance ATP-actin assembly into the newly formed ends generated by the complex. Second, it can hold onto the end of the newly formed filament. Third, it can protect the end of the growing filament from capping by CapZ. These properties allow VASP to enhance actin assembly and confine it to the rear of the bacterium.
EXPERIMENTAL FIGURE 17-17 Listeria uses the power of actin polymerization for intracellular movement. (a) Fluorescence microscopy of a cultured cell stained with an antibody to a bacterial surface protein (red) and fluorescent phalloidin to localize F-actin (green). Behind each Listeria bacterium is an actin “comet tail” that becomes embedded in the stationary cytoskeletal matrix and so propels the bacterium forward by actin polymerization. When the bacterium runs into the plasma membrane, it pushes the membrane out into a structure like a filopodium, which protrudes into a neighboring cell. (b) Listeria motility can be reconstituted in vitro with bacteria and just four proteins: ATP– G-actin, complex, CapZ, and cofilin. This phase-contrast micrograph shows bacteria (black), behind which are the phase-dense actin tails. (c) A model of how Listeria moves using just four proteins. The ActA protein on the bacterial cell surface activates the complex to nucleate new filament assembly from preexisting filaments. Filaments grow at their end until capped by CapZ. Actin is recycled through the action of cofilin, which enhances depolymerization at the end of the filaments. In this way, polymerization is confined to the back of the bacterium and propels it forward. [Part (a) Courtesy Julie Theriot and Timothy Michison. Part (b) Reprinted with permission from Nature Publishing Group, from T. P. Loisel et al., 1999, “Reconstitution of Actin-Based
Motility of Listeria and Shigella Using Pure Proteins,” Nature 401:613–616; permission conveyed through Copyright Clearance Center, Inc.] Description The fluorescence micrograph labeled (a) shows listeria with actin tails behind them. The bacterium is tiny and red in color, whereas the actin tail is longer and green in color. The plasma membrane of the cell is outlined in bright green color. The phase-contrast micrograph labeled (b) shows listeria with actin tails. The dark spot is the bacterium and the faded curved line behind it is the actin tail. The illustration labeled (c) shows Listeria either in vitro or within the host cell cytoplasm. The bacterial cell surface has three Act A proteins in it, represented by three ovals. The Act A proteins drive the A r p 2 slash 3 complex to make new actin filaments. The newly forming filaments attach to Act A proteins. The three filaments present have multiple branches, with each end covered with an oval capping protein. The A r p 2 slash 3 complex is present at the points where branches attach to the main filament and at the end of the main filament is a section labeled Cofilin. At the bottom right is the label actin disassembly negative end that shows A r p 2 slash 3 and actin subunits disassembling. The positive end of a branch is labeled actin assembly positive end at the top right of the illustration. To move inside cells, the Listeria bacterium hijacks a normal, regulated cellular process involved in cell locomotion. As we discuss in more detail later (Section 17.7), moving cells have a thin sheet of cytoplasm at the front of the cell called the leading edge (see Figures 17-1c, 17-4, and 17- 15d). This thin sheet of cytoplasm consists of a dense network of actin filaments that are continually elongating at the front of the cell to push the membrane forward. Factors in the leading-edge membrane activate the complex to nucleate these filaments. Thus the power of actin assembly pushes the membrane forward to contribute to cell locomotion.
Microfilaments Function in Endocytosis
It is the leading-edge machinery that is co-opted by Listeria as it moves within and between host cells. Microfilaments Function in Endocytosis As we saw in Chapter 14, endocytosis is the process that cells use to take up particles, molecules, or fluid from the external medium by enclosing them in plasma membrane and then internalizing them. The uptake of molecules or liquid is called receptor-mediated or fluid-phase endocytosis, and the uptake of large particles is called phagocytosis (“cell eating”). Microfilaments participate in both of these processes. Fluid-phase endocytosis is a multistep process, involving collection of cargo proteins in a clathrin-coated pit, deformation and invagination of the membrane by the clathrin coat, pinching off the membrane by the dynamin GTPase, and movement of the endosome into the cell interior (see Figures 14-20 and 14-29). The power of actin assembly contributes to this process. Endocytosis assembly factors recruit NPFs so that as the endocytic vesicles invaginate and pinch off from the membrane, they are driven into the cytoplasm, powered by a rapid and short-lived burst (a few seconds in duration) of actin polymerization driven by the complex (Figure 17-18a). Although the physical mechanism that drives the endocytic vesicle into the cytoplasm is not fully resolved, the actin-based machinery is similar to that used in leading-edge formation and Listeria motility.
This actin-based movement of endocytic vesicles can be reconstituted in vitro (Figure 17-18b).
Figure 17-18
-dependent actin assembly during endocytosis. (a) Clathrin- mediated endocytosis is a rapid and ordered process. It has been best studied in yeast, in which the temporal order of specific steps has been delineated. In vivo imaging has shown that endocytosis assembly factors recruit NPFs that activate the complex. A burst of -dependent actin assembly drives internalization of endocytic vesicles away from the plasma membrane. For clarity, other factors involved in endocytosis, such as clathrin, the adaptin AP2 and dynamin are not shown (see Figure 14-20). (b) Endosome movement can be reconstituted in vitro. Endosomes isolated from cells that had taken up fluorescently labeled transferrin (red) were added to a cell extract containing fluorescently labeled actin (green). The endosomes bind WASp, which then activates the complex to assemble actin tails that propel them through the cytoplasm. [Part (b) J. Taunton et al., 2000, J. Cell Biol. 148(3):519–530; https://doi.org/10.1083/jcb.148.3.519.] Description The illustration labeled (a) shows a time scale across the top starting at 0 seconds and going to 30 seconds. The following sequence is depicted. 1. Endocytosis assembly factors are embedded in the plasma membrane. 2. Within 15 seconds nucleation promoting factors such as W A S p associated with the assembly factors. 3. After 20 seconds, actin filaments assemble by the action of A R P 2 slash 3, allowing
invagination of the membrane to occur. 4. After 30 seconds, the endocytic vesicle containing its cargo is formed. The endocytic vesicle is surrounded by actin filaments. The fluorescence electron micrograph labeled (b) shows endosomes with attached actin filaments. The vesicles appear as red dots with long trailing green actin filaments. Phagocytosis by leukocytes (white blood cells) is a vital process in the recognition and removal of pathogens, such as bacteria. The immune system identifies a bacterium as foreign and makes antibodies that recognize surface features of the bacterium. As we discussed in Chapter 3, each antibody has a region called the Fab domain that binds specifically to its antigen, in this case a component on the bacterial cell surface. As antibodies coat the bacterium through interaction between their Fab domains and the cell-surface antigen, a second antibody domain, known as the Fc domain, is exposed. This process is known as opsonization (Figure 17-19, step 1 ; see Chapter 24). Leukocytes have a receptor on their cell surface, called the Fc receptor, that binds the Fc portion of the bacteriumbound antibody (step 2 ). Initial binding between the leukocyte and opsonized bacterium signals the leukocyte to engulf the bacterium (steps 3 and 4 ) inducing assembly of microfilaments at the site of interaction with the bacterium. The assembled microfilaments, together with myosin motor proteins, provide the force necessary to draw the bacterium into the cell, ultimately fully enclosing the pathogen in plasma membrane (step 4 ). Once internalized, the newly formed phagosome fuses with lysosomes, and the pathogen is killed and degraded by lysosomal enzymes.
FIGURE 17-19 Phagocytosis and actin dynamics. Actin assembly and contraction drives the phagocytic internalization of particles. Shown here is the phagocytosis and degradation of a bacterium by a leukocyte. An invading bacterium is coated by specific antibodies to a cell-surface protein in a process known as opsonization (step 1 ). The Fc region of the bound antibodies is displayed on the bacterial surface and recognized by a specific receptor, the Fc receptor, on the leukocyte surface (step 2 ). This interaction signals the cell to assemble a contractile actin structure that results in the internalization and engulfment of the bacterium (step 3 ). Once it has been internalized into a phagosome, the bacterium is killed and degraded by enzymes delivered from lysosomes (step 4 ). Description An illustration shows a Y-shaped antibody to component on bacterial surface and a bacterium, which is oval in shape. The bacterium contains surface proteins. The F c and F a b portions of the antibody are labeled. Step 1: several antibodies bind to the bacterial surface proteins to make an opsonized bacterium. Step 2: the opsonized bacterium binds to the F c receptors present in the membrane of a leukocyte. Step 3: F-actin filaments form around the activated F c receptors on the cytosolic side, causing the bacterium to be endocytosed. Step 4: A phagosome is formed around the bacterium. Two lysosomes are ready to merge with the phagosome. Actin assembly has been implicated in other steps of the endocytic pathway as well as in the secretory pathway. For example, an NPF called WASH has been implicated in the -dependent nucleation of actin filament on endosomes, regulating their shape and contributing to transport events. Another NPF, called WHAMM, localizes to the Golgi complex and is believed to direct the -dependent assembly of actin participating in membrane traffic from the endoplasmic reticulum to the
Toxins That Perturb the Pool of Actin Monomers Are Useful for Studying Actin Dynamics
Golgi. The emerging view is that actin assembly facilitates transport between different compartments of the secretory and endocytic pathways. Toxins That Perturb the Pool of Actin Monomers Are Useful for Studying Actin Dynamics Certain fungi and sponges produce toxins that target the polymerization cycle of actin and are therefore toxic to animal cells but useful for the study of actin dynamics. Two types of these toxins have been characterized. The first class is represented by two unrelated toxins, cytochalasin D and latrunculin, that promote the depolymerization of filaments, though by different mechanisms. Cytochalasin D, a fungal alkaloid, depolymerizes actin filaments by binding to the end of Factin, where it blocks further addition of subunits. Latrunculin, a toxin secreted by sponges, binds and sequesters G-actin, inhibiting it from adding to a filament end. Exposure to either toxin thus increases the monomer pool. When cytochalasin D or latrunculin is added to live cells, the actin cytoskeleton disassembles and cell movements such as locomotion and cytokinesis are inhibited. These observations were among the first to implicate actin filaments in cell motility. Latrunculin is especially useful because it binds actin monomers and prevents any new actin assembly. Thus if latrunculin is added to a cell, the rate at which actin-based structures disappear reflects their normal rate of turnover. This method has revealed that the stability of actin structures varies over a wide range. For example, experiments with latrunculin show that the leading
edges of motile cells turn over every 30–180 seconds, and that stress fibers turn over every 5–10 minutes. In the other class of toxins, the monomer-polymer equilibrium is shifted in the direction of filaments. Examples from this class of toxins are jasplakinolide, another sponge toxin, and phalloidin, which is isolated from Amanita phalloides (the “angel of death” mushroom). Jasplakinolide enhances nucleation by binding and stabilizing actin dimers and thereby lowering the critical concentration. Phalloidin binds at the interface between subunits in F-actin, locking adjacent subunits together and preventing actin filaments from depolymerizing. Even when actin is diluted below its critical concentration, phalloidin-stabilized filaments will not depolymerize. Because many actin-based processes depend on actin filament turnover, the introduction of phalloidin into a cell paralyzes these systems, and the cell dies. Since phalloidin binds only to F-actin, fluorescently labeled phalloidin is commonly used to stain actin filaments for light microscopy (see Figure 17-4). KEY CONCEPTS OF SECTION 17.3 Mechanisms of Actin Filament Assembly Actin assembly is nucleated by two classes of proteins: formins nucleate the assembly of unbranched filaments (see Figure 17-13), whereas the complex nucleates the assembly of branched actin networks (see Figure 17-15). The activities of formins and are regulated by signal transduction pathways. Functionally different actin-based structures are assembled by formins and by nucleators. Formins drive the assembly of stress fibers and the contractile ring, whereas nucleates the assembly of the branched actin filaments found in the leading edge of motile cells.
The power of actin polymerization can be harnessed to do work, as is seen in the -dependent intracellular movement of pathogenic bacteria (see Figure 1717) and the inward movement of endocytic vesicles (see Figures 17-18 and 17-19). Several toxins affect the dynamics of actin polymerization; some, such as latrunculin, bind and sequester actin monomers, whereas others, such as phalloidin, stabilize filamentous actin. Fluorescently labeled phalloidin is useful for staining actin filaments for their visualization by microscopy.
Cross-Linking Proteins Organize Actin Filaments into Bundles or Networks
17.4 Organization of Actin-Based Cellular Structures We have seen that actin filaments are assembled into a wide variety of different arrangements and that many actin-associated proteins nucleate actin assembly and regulate filament turnover. Dozens of proteins in a vertebrate cell organize these filaments into diverse structures, which the cell uses for an array of functions. In this section, we discuss a few of these proteins, including typical types of actin cross-linking proteins and adapter proteins involved in making links between actin and membrane proteins. One fascinating problem, about which very little is known, is how cells assemble different actin-based structures within the cytoplasm of the same cell. Some of this organization is due to local regulation of actin assembly, a topic we come to at the end of the chapter. Cross-Linking Proteins Organize Actin Filaments into Bundles or Networks When actin filaments are assembled in a test tube, they form a tangled network. In cells, however, actin filaments are found in organized structures, such as the highly ordered filament bundles in microvilli or the network characteristic of the leading edge of a motile cell (see Figure 17-
4a). These structures are erected with the help of the filament assembly mechanisms discussed previously and by the presence of actin crosslinking proteins. To be able to organize actin filaments, an actin cross-linking protein must have two F-actin–binding sites (Figure 17-20a). Actin cross-linking proteins can have two actin-binding sites within a single polypeptide, as with fimbrin, which builds bundles of filaments all having the same polarity, as seen in the stereocilium (a giant microvillus) in Figure 17-20b. Like fimbrin, filamin has dual actin-binding sites in a single polypeptide, but unlike fimbrin, which is short and stiff, filamin has a flexible region between the binding sites that functions like a molecular leaf spring. This enables filamin to form stabilizing cross-links between filaments in a network, such as the ones found in the leading edge of a motile cell.
FIGURE 17-20 Actin cross-linking proteins. Actin cross-linking proteins mold F-actin filaments into diverse structures. (a) Examples of four F-actin cross-linking proteins, all of which have two domains (blue) that bind F-actin. Some have a -binding site (purple) that inhibits their activity at high levels of free . (b) Transmission electron micrograph of a thin section of a stereocilium (an unfortunate name, since it is really a giant microvillus) on a sensory hair cell in the inner ear. This structure contains a bundle of actin filaments cross-linked by fimbrin, a small cross-linking protein that allows for close and regular interaction of actin filaments. (c) Super-resolution image of the repeating structure of actin
(green) and spectrin (magenta) that form a cagelike network to support the plasma membrane of an axon. [Part (b) L. G. Tilney et al., 1983, J. Cell Biol. 96(3):822–834; https://doi.org/10.1083/jcb.96.3.822. Part (c) Courtesy of Xiaowei Zhuang and Ke Xu.] Description The illustration labeled (a) shows the following proteins and their locations. Fimbrin located in microvilli, filopodia, and focal adhesions. The filament is represented by a double-wide ladder with actin sides and fimbrin rungs represented by two ovals with two tiny spheres attached at one end. Alpha-actinin is located in stress fibers, filopodia, and muscle Z-line. The filament is represented by a double-wide ladder with actin sides and alpha-actinin rungs represented by an oval structure with a rod-shaped structure attached to two tiny spheres at one end. Spectrin located in the cell cortex. Spectrin has two long rod-like structures parallel to each other made of beta and alpha units. These units are attached to two tiny spheres at one end and an oval structure at opposite ends. Six spectrins are attached at one end to a central actin filament. Each spectrin is attached to an actin filament on the other end. Filamin is located on the leading edge, stress fibers, and filopodia. It is represented by a white segmented rod-shaped structure with a v-shaped curve in the center and a blue oval attached to each end. These filamins are attached at various places to actin filaments, joining one filament to another one. The transmission electron micrograph labeled (b) shows a bundle of actin filaments represented a group of straight dark vertical lines lined by a thicker dark curvy layer. The super resolution image labeled (c) shows an axon where spectrin, in purple, is alternating with actin, in green to form a cage-like pattern. A measurement scale of 500 nanometers is labeled. Other actin cross-linking proteins possess a single actin-binding site per polypeptide chain, but the functional protein is a dimer. These dimeric cross-linking proteins assemble to generate a rigid rod with a binding site at either end, as happens with α-actinin. Like fimbrin, α-actinin bundles
Adapter Proteins Link Actin Filaments to Membranes
parallel actin filaments, but keeps them farther apart than fimbrin. Another protein, called spectrin, is a tetramer with two polypeptide chains containing actin-binding sites and two chains that extend the length of the cross-linking protein. Spectrin spans a much greater distance between actin filaments than does fimbrin or α-actinin. Spectrin assists in forming microfilament networks beneath the plasma membrane (shown in Figure 17-20c and discussed in the next section). The complex (which we discussed in terms of its ability to nucleate actin filament assembly) is also an important cross-linking protein, attaching the end of one filament to the side of another filament (see Figure 17-15). Adapter Proteins Link Actin Filaments to Membranes Actin-based structures are not useful to a cell unless they are attached to specific components by adapter proteins. In this section, we describe how adapter proteins attach microfilaments to membranes to provide mechanical support, or attach a contractile apparatus to the membrane. While we discuss some specific examples, essentially all eukaryotic cells have actin-based filaments associated with the plasma membrane as part of a structure known as the cell cortex. In these various roles, actin filaments can interact with membranes either laterally or at their ends. Our first example of actin filaments attached to a membrane involves the human erythrocyte — the red blood cell. The erythrocyte consists essentially of a plasma membrane enclosing a high concentration of the
protein hemoglobin. It transports oxygen from the lungs to tissues and brings carbon dioxide from tissues back to the lungs. Erythrocytes must be able to survive the raging torrents of blood flow in the heart, then flow down arteries and survive squeezing through narrow capillaries before being cycled past the lungs and back to the heart. To survive this grueling journey for thousands of cycles, an erythrocyte has an actin-based network underlying the plasma membrane that gives the cell both the tensile strength and the flexibility necessary for its journey. This network is made of short actin filaments about 14 subunits in length, stabilized on their sides by tropomyosin (discussed in more detail in Section 17.6) and by the capping protein tropomodulin on the end. These short filaments serve as hubs for binding about four to six flexible spectrin molecules, generating a fishnet-like structure (Figure 17-21a) that provides strength and flexibility to the cell cortex. Spectrin molecules are attached to membrane proteins in two ways. One is via the protein ankyrin, which binds about halfway along a spectrin molecule and also binds to the transmembrane bicarbonate transporter (also known as band 3). The other adapter is band 4.1, which binds F-actin, the actin-binding domain of spectrin, and glycophorin C, another transmembrane protein (Figure 1721b). Spectrin-based networks, similar to the one found in erythrocytes, occur in many types of cells. For example, an ankyrin-spectrin attachment links the ATPase to the actin cytoskeleton on the basolateral membrane of epithelial cells, and an ordered repeating actin/spectrin network supports the axonal plasma membrane of neurons (Figure 1720c).
FIGURE 17-21 Lateral attachment of microfilaments to membranes. (a) Electron micrograph of the erythrocyte membrane showing the spoke-and-hub organization of the cortical cytoskeleton supporting the plasma membrane in human erythrocytes. The long spokes are composed mainly of spectrin and can be seen to intersect at the hubs, or membrane-attachment sites. The darker spots along the spokes are ankyrin molecules, which link spectrin to integral membrane proteins. (b) Diagram of the erythrocyte cytoskeleton, showing the two main types of membrane attachments: step 1 through ankyrin to band 3 and step 2 through band 4.1 to glycophorin C. (c) Actin is incorporated into the tips of stereocilia (giant microvilli). Cells with stereocilia were transfected to express GFP-labeled actin for a short period of time and then counterstained with rhodaminephalloidin to stain all the F-actin. The experiment shows that new actin is incorporated at the tips of the stereocilia. (d) Ezrin, a member of the ezrin-radixin-moesin (ERM) family, links actin filaments laterally to the plasma membrane in surface structures such as microvilli;
attachment can be direct or indirect. Ezrin exists in an inactive, closed cytosolic form. Activation to an open form is mediated by phosphorylation and requires both the presence of the plasma membrane lipid PI(4,5)P2 and a specific ezrin kinase. Ezrin, activated by phosphorylation (P), links directly to the cytoplasmic region of transmembrane proteins (right) or indirectly through a scaffolding protein such as EBP50 (left). (e) Dystrophin, which has an actin-binding site on its N-terminal end and a C-terminal domain, binds the membrane protein dystroglycan, which itself associates with the extracellular matrix. See
Figure 20-41 for more detail. [Part (a) From T. J. Byers and D. Branton, 1985, Proc. Nat’l. Acad Sci. USA 82:6153; courtesy Daniel Branton. Part (c) A. K. Rzadzinska et al., 2004, J. Cell Biol. 164(6):887– 897; https://doi.org/10.1083/jcb.200310055.] Description The electron micrograph labeled (a) shows actin filaments below a cell membrane. Several lines extended from a few dotted areas. The illustration labeled (b) depicts the electron micrograph with actin filaments attached to oval structures labeled glycophorin C in the cell membrane. Actin filaments are attached to square particles on both sides labeled band 4.1. Two bands made of four tiny rod-shaped structures are labeled band 3. Thin tube-like structures connect various filaments and are labeled spectrin. A sphere at the center of each spectrin is labeled ankyrin. The electron micrograph labeled (c) shows stereocilia with actin attached at the top of each strand. It looks like a collection of tubules. The illustration labeled (d) shows a group of four bundles of actin with one section on the second bundle highlighted and an arrow pointing to an enlargement of that area. The enlargement shows the plasma membrane at top with two structures in the membrane. The left structure is attached to E B P 5 0 in the cytosol, which is attached to an actin filament in the filament center. A triangle labeled Ezrin is attached to the end of E B P 5 0 and also attached to the membrane structure at the right. The ezrin is attached at the right to another actin filament. The illustration labeled (d) shows muscle. The top layer is a feathery set of thin lines labeled extracellular matrix. The top layer encloses the plasma membrane which has a
structure called dystroglycan complex embedded in it. Dystroglycan complex is attached to a rod-like structure called the dystrophin which has a bulb-like ending further attached to an actin filament made up of spheres. Genetic defects in proteins of the red blood cell cytoskeleton can result in cells that rupture easily, giving rise to diseases known as hereditary spherocytic anemias (spherocytic because the cells are rounder, anemias because there is a shortage of red blood cells) and hence a shorter life span. In human patients, mutations in spectrin, band 4.1, and ankyrin can cause these diseases. Microfilaments also support cell-surface structures such as microvilli and membrane ruffles. If we look at a microvillus, it is clear that its actin filaments must have an end-on attachment at the tip and lateral attachments down its length. What is the orientation of actin filaments in microvilli? Decoration experiments show that the end is at the tip. Moreover, when fluorescent actin is added to a cell, it is incorporated at the tip of a microvillus, showing not only that the end is there, but also that actin filament assembly occurs there (Figure 17-21c). It is not yet known how actin filaments are attached at the microvillus tip or how assembly is regulated there. This end orientation of actin filaments with respect to the plasma membrane is found almost universally — not just in microvilli, but also, for example, in the leading edges of motile cells. The lateral attachments to the plasma membrane are provided, at least in part, by the ERM (ezrin-radixin-moesin) family of proteins. This family consists of regulated proteins that exist in a folded, inactive form.
They are activated after being phosphorylated by a specific kinase in a reaction requiring the plasma membrane-localized regulatory phospholipid phosphatidylinositol 4,5-bisphosphate . The Factin–binding and membrane protein–binding sites of the ERM protein are exposed to provide a lateral linkage between membrane and actin filaments (Figure 17-21d). This type of coincident-detection mechanism — requiring both a regulatory lipid and localized kinase — ensures that ERM proteins are only activated at appropriate locations. ERM proteins can link actin filaments to the cytoplasmic domain of membrane proteins directly or indirectly through scaffolding proteins (see Figure 17-21d). The types of actin-membrane linkages we have discussed so far do not involve areas of the plasma membrane attached directly to other cells in a tissue or to the extracellular matrix, but such linkages do exist. Contact between epithelial cells is mediated by highly specialized regions of the plasma membrane called adherens junctions (see Figure 17-1b). Other specialized regions of association called focal adhesions mediate attachment of cells to the extracellular matrix. In turn, these specialized attachments connect to the cytoskeleton, as will be described in more detail when we discuss cell migration (Section 17.7) and cells in the context of tissues (see Chapter 20). Muscular dystrophies are genetic diseases that are often characterized by the progressive weakening of skeletal muscle. One of these genetic diseases, Duchenne muscular dystrophy, affects the protein dystrophin, whose gene is located on the X chromosome, so the disease is
much more prevalent in males than in females. Dystrophin is a modular protein whose function is to link the cortical actin network of muscle cells to a complex of membrane proteins that link to the extracellular matrix. Thus dystrophin has an N-terminal actin-binding domain, followed by a series of spectrin-like repeats, and terminates in a domain that binds the transmembrane dystroglycan complex to the extracellular matrix protein laminin (see Figure 17-21d, e and for more detail Figure 20-41). In the absence of dystrophin, the plasma membrane of muscle cells becomes weakened by cycles of muscle contraction and eventually ruptures, resulting in death of the muscle fibers. KEY CONCEPTS OF SECTION 17.4 Organization of Actin-Based Cellular Structures Actin filaments are organized by cross-linking proteins that have two F-actin–binding sites. Actin cross-linking proteins can be long or short, rigid or flexible (see Figure 17-20). Actin filaments are attached to the plasma membrane by adapter proteins, as seen in erythrocytes and in cell-surface structures such as microvilli (see Figure 17-21). Attachment to the plasma membrane can be regulated, as seen in the activation of ERM proteins by phosphorylation. Hereditary diseases, such as spherocytic anemias and muscular dystrophy, have been traced to defects in the attachment of the microfilament-based cortical cytoskeleton to the plasma membrane.
17.5 Myosins: Actin-Based Motor Proteins
17.5 Myosins: Actin-Based Motor Proteins In Section 17.3, we discussed how actin polymerization nucleated by the complex can be harnessed to do work, such as moving vesicles during endocytosis, moving cells along a substrate at their leading edges, and propelling a Listeria bacterium through the cytoplasm of a host cell. In addition to this actin polymerization–based motility, cells have a large family of motor proteins called myosins that can perform work by moving along actin filaments. The first myosin discovered, myosin II, was isolated from skeletal muscle. For a long time, biologists thought that this was the only type of myosin found in nature. However, they then discovered other types of myosins and began to ask how many functional classes might exist. Today we know that there are multiple families of myosins, in addition to the myosin II of skeletal muscle, that move along actin. Indeed, with the discovery and analysis of all these microfilament-based motors and the corresponding microtubule-based motors described in the next chapter, our former relatively static view of cells has been replaced with the realization that the cytoplasm is incredibly dynamic — like a busy freeway system with motors busily ferrying components around.
Myosins Have Head, Neck, and Tail Domains with Distinct Functions
Myosins have the amazing ability to convert the energy released by ATP hydrolysis into mechanical work (movement along actin). While all myosins convert the chemical energy of ATP hydrolysis into mechanical work, different myosins perform very different cellular functions. For example, many molecules of myosin II pull together on actin filaments to bring about muscle contraction, whereas myosin V binds to cargo-filled vesicles and transports them along actin filaments. Other types of myosin move organelles around cells or aid in cell motility. To begin to understand myosins, we first discuss their general structure. Next we explore the diversity of myosin classes and describe in more detail some of those that are common in eukaryotes. We investigate the mechanism that converts the energy released by ATP hydrolysis into work and then see how this mechanism is modified to tailor the properties of specific myosin classes to their specific functions. Myosins Have Head, Neck, and Tail Domains with Distinct Functions Much of what we know about myosins comes from studies of myosin II isolated from skeletal muscle. In skeletal muscle, hundreds of individual myosin II molecules are assembled into bipolar bundles called thick filaments (Figure 17-22a). In a later section, we will discuss how these myosin filaments interdigitate with actin filaments to bring about muscle contraction. Here we investigate the properties of the myosin molecule itself.
FIGURE 17-22 Structure of myosin II. (a) Organization of myosin II in filaments isolated from skeletal muscle. Myosin II assembles into bipolar filaments in which the tails form the shaft of the filament and the heads are exposed at the ends. Treatment of bipolar filaments with high salt concentrations and ATP disassembles the filament into individual myosin II molecules. (b) A myosin II molecule consists of two identical heavy chains (light blue) and four light chains (green and dark blue). The tails of the heavy chains form a coiled coil to dimerize; the neck region of each heavy chain has two light chains associated with it. Limited proteolytic cleavage of myosin II generates tail fragments — LMM and S2 — and the S1 motor domain. (c) Three-dimensional model of a single S1 head domain shows that it has a curved, elongated shape and is bisected by a cleft. The nucleotide-binding pocket lies on one side of this cleft, and the actin-binding site lies on the other side near the tip of the head. Wrapped around the shaft of the α-helical neck are two light chains. These chains stiffen the neck so that it can act as a lever arm for the head. Shown here is the ADP-bound conformation. [Part (c) Data from S. Gourinath et al., 2003, Structure 11:1621–1627, PDB ID 1qvi.]
Description The illustration labeled (a) shows the structure of myosin 2. The central portion is composed of a bunch of tube-like structures that have bulb-like endings on both sides. The central portion measures 160 nanometers in length, while the whole molecule measures 325 nanometers. The illustration labeled (b) shows the structure of a myosin 2 molecule. Head portion: It has two bulb-shaped endings with U shaped actin-binding sites. Neck portion: it is made of two subunits attached to both bulb-like structures. The subunits are composed of essential light chains and regulatory light chains. Tail portion: the tail is made of two coiled heavy chains attached to the neck. Chymotrypsin Cleavage: chymotrypsin cleaves myosin 2 into a head fragment (H M M) and tail fragment (L M M). Subsequent cleavage of the H M M fragment with papain after the regulatory light chains yields S 1 and S 2 fragments. The illustration labeled (c) shows a ribbon structure enclosed by a semi-transparent three-dimensional structure of the head and neck of domain myosin. The following parts are labeled the heavy chain, essential light chain, and regulatory light chain. The actin-binding site to the left of the heavy chain portion is labeled. A space-filling model called the nucleotide-binding site is embedded in the ribbon structure of the heavy chain. Dissolving the myosin thick filament in a solution of ATP and a high concentration of salt generates a pool of individual myosin II molecules. The soluble myosin II molecule is actually a protein complex consisting of six polypeptide subunits. Two of the subunits are identical high-molecularweight polypeptides known as myosin heavy chains. Each consists of a globular head domain and a long tail domain, connected by a flexible neck domain. The tails of the two myosin heavy chains intertwine, so that the head regions are in close proximity. The remaining four subunits of the myosin complex are smaller in size and are known as the light chains.
There are two types of light chains: the essential light chain and the regulatory light chain. One light chain of each type associates with the neck region of each heavy chain (Figure 17-22b, top). The myosin heavy chain and the two types of light chains are encoded by three different genes. The soluble myosin II molecule has ATPase activity, reflecting its ability to power movements by hydrolysis of ATP, but which part of the myosin complex is responsible for this activity? A standard approach to identifying functional domains in a protein is to cleave the protein into fragments with specific proteases and then ask which fragments have the activity. Soluble myosin II can be cleaved by gentle treatment with the protease chymotrypsin to yield two fragments, one called heavy meromyosin (HMM; mero means “part of”) and the other, light meromyosin (LMM) (Figure 17-22b, middle). The heavy meromyosin can be further cleaved by the protease papain to yield subfragment 1 (S1) and subfragment 2 (S2) (Figure 17-22b, bottom). By analyzing the properties of the various fragments — S1, S2, and LMM — it was found that the intrinsic ATPase activity of myosin resides in the S1 fragment, as does its F-actin–binding site. Moreover, it was found that the ATPase activity of the S1 fragment is greatly enhanced by the presence of filamentous actin, so that fragment is said to have an actin-activated ATPase activity, which is a hallmark of all myosins. The S1 fragment of myosin II consists of the head and neck domains with associated light chains, whereas the S2 and LMM regions make up the tail domain.
X-ray crystallographic analysis of the head and neck domains has revealed the shapes of the subunits, the positions of the light chains, and the locations of the ATP-binding and actin-binding sites (Figure 17-22c). At the base of the myosin head is the α-helical neck, where two light-chain molecules wrap around the heavy chain like C-clamps. In this position, the light chains stiffen the neck region. The actin-binding site is an exposed region at the tip of the head domain. The ATP-binding site is also in the head domain, within a cleft opposite the actin-binding site. How much of the myosin II molecule is necessary and sufficient for its motor activity? To answer this question, one needs a simple in vitro motility assay. In one such assay, the sliding-filament assay, myosin molecules are tethered to a coverslip to which is added stabilized, fluorescently labeled actin filaments and ATP. Because the myosin molecules are tethered, they cannot slide; thus any force generated by the interaction of myosin heads with actin filaments forces the filaments to move relative to the myosin. The tethered myosin molecules or fragments pull toward the end of a filament, so the force that myosin generates causes the filaments to move with the end leading (Figure 17-23). Using the sliding-filament assay, one can show that the S1 fragment of myosin II is sufficient to move actin filaments (Figure 17-23a). The rate at which myosin moves an actin filament can be determined from video recordings of sliding-filament assays (Figure 17-23b).
EXPERIMENTAL FIGURE 17-23 The sliding-filament assay is used to detect myosinpowered movement. (a) After myosin S1 fragments are adsorbed onto the surface of a glass coverslip, excess unbound myosin is removed; the coverslip is then placed myosinside-down on a glass slide to form a chamber through which solutions can flow. A solution of actin filaments, made visible and stable by staining with rhodamine-labeled phalloidin, is allowed to flow into the chamber. In the presence of ATP, the myosin heads pulls toward the ends of the actin filaments by the mechanism discussed later and illustrated in Figure 17-26. Because the myosin tails are immobilized, movement of the heads toward the ends causes sliding of the filaments, which appear to be moving with their ends leading the way. (b) Movement of individual filaments can be observed in a fluorescence light microscope. These photographs show the positions of three actin filaments (numbered 1, 2, 3) at 30-second intervals recorded by video microscopy. The rate of filament movement can be determined from such recordings.
Myosins Make Up a Large Family of Mechanochemical Motor Proteins
Description The illustration labeled (a) of a sliding filament assay shows a glass coverslip on which there are seven S 1 heads of myosin attached. An actin filament has two rope-like structures made up of spheres twisted around each other. Two actin filaments bind to three S 1 myosin heads each. Their positive and negative ends are labeled. Three fluorescence micrographs labeled (b) show fluorescent actin filaments moving by the action of myosin. In the first image, three red structures are labeled from left to right as 3, 1, 2. In the second image, the structure labeled 1 has moved between 3 and 2. In the third image, the structure named 1 has moved below 2 and 3 with 3 now above 2 and 2 below the left of 3. All myosins have a domain related to the S1 domain of myosin II, which in all cases is responsible for the motor activity. As we will see in a later section, the length of the neck domain and the numbers and types of light chains associated with it vary among myosin classes. The tail domain does not contribute to motility, but rather defines what is moved by the S1related domain. Thus as might be expected, tail domains differ among myosins and are tailored to bind specific cargoes. Myosins Make Up a Large Family of Mechanochemical Motor Proteins Myosin S1 domains share considerable similarity in their primary amino acid sequence, so it is possible to determine how many myosin genes and how many different classes of myosins exist in the sequenced genome of an organism. For example, there are about 40 myosin genes in the human
genome (Figure 17-24), 9 in Drosophila, and 5 in budding yeast. Bioinformatic analysis of the sequence relationships among myosin head domains indicates that about 20 distinct classes of myosins have evolved in eukaryotes. As shown in Figure 17-24, the genetic bases for some conditions have been traced to mutations in genes encoding myosins.
FIGURE 17-24 The myosin superfamily in humans. Results of a computer analysis of the relatedness of the S1 head domains of all of the approximately 40 myosins encoded by the
human genome. Each myosin is indicated by a blue dot. The lengths of the black lines indicate phylogenetic distance relationships: myosins connected by short lines are closely related, whereas those separated by longer lines are more distantly related. Among these myosins, three classes — myosins I, II, and are widely represented among eukaryotes; others have more specialized functions. Indicated are examples in which loss of a specific myosin causes a disease. [Data from R. E. Cheney, 2001, Mol. Biol. Cell 12:780.] Description A superfamily tree of myosin is illustrated as a circular pattern, with myosin 2 highlighted on left, Myosin 5 highlighted at the bottom and myosin 1 highlighted on the right. Single blue dots among the tree lines are labeled, clockwise, Deafness/blindness, Deafness, Deafness, and Seizures. All myosin head domains use the same mechanism to convert ATP hydrolysis into mechanical work. However, as we will see, subtle variations in this mechanism can have profound effects on the functional properties of different myosin classes. How do these different classes relate with respect to their tail domains? If one uses the protein sequences of myosin tail domains to place the myosins into classes, the myosins fall into the same groupings found when comparing the motor domains. This implies that head domains with specific properties have coevolved with specific classes of tail domains, which makes a lot of sense, and further suggests that each class of myosin has evolved to carry out a specific function. Among the myosins are three especially well-studied families, commonly found in animals and fungi: the myosin I, myosin II, and myosin V families
(Figure 17-25). In humans, 8 genes encode heavy chains for the myosin I family, 14 for the myosin II family, and 3 for the myosin V family (see
FIGURE 17-25 Three common classes of myosins. Myosin I molecules consist of a head domain and a neck domain; a variable number of light chains is associated with the neck domain. Members of the myosin I class are the only myosins to have a single head domain. Some of these myosins are believed to associate directly with membranes through lipid interactions. Myosin II molecules have two head domains and two light chains per neck and are the only class that can assemble into bipolar filaments. Myosin V molecules have two head domains and six light chains per neck. They bind specific receptors (brown box) on organelles, which they transport. All myosins in these three classes move toward the end of actin filaments. Description Class one myosins have a step size of 10 to 14 nanometers and have membrane association and endocytic functions. Myosin one has a bulb-like structure with a U shaped binding site connected to three small subunits that are connected to an oval structure. This myosin attached to actin filament on one end and the inner membrane of the cell membrane on the other end. An actin filament has two rope-like structures made up of spheres twisted around each other. The positive and negative ends of the actin filament are labeled. Class two myosins have a step size of 8 nanometers and are
involved in contraction. The structure of a myosin 2 molecule has two bulb-shaped endings with U shaped binding sites. The bulb-like structures are attached to four subunits further attached to a tail made of two coiled heavy chains. A bundle of myosins is present between two rows of actin filaments. Class five myosins have a step size of 36 nanometers and are involved in organelle transport. Myosin five has a structure similar to that of myosin 2 except that it has several subunits attached to the tail which is further attached to two oval structures. The tail end is attached to a vesicle at one end, and a filament of actin on the other. The large myosin I family has a variable number of light chains associated with the neck region and is the only type of myosin that has a single heavy chain and so functions with a single head. Only a few of the functions of myosin I molecules have been determined. These include connecting actin filaments to membranes and assisting in endocytosis. Since these myosin molecules have only one head each, several of them must work together to generate movement, and at least one must always be attached to an actin filament during the work cycle. All myosin II members have a relatively short neck domain and have two light chains per heavy chain. The myosin II class is the only one that assembles into bipolar filaments. Myosin II molecules form bipolar filaments in which the tail domains assemble together into thick bundles with a cluster of head domains at each end. This organization is important for its involvement in contraction; indeed, this is the only class of myosins involved in contractile functions. The large number of members in this class reflects the need for myosin II filaments with slightly different contractile properties seen in different muscles (e.g., skeletal, cardiac, and various types of smooth muscle) as well as in nonmuscle cells.
Conformational Changes in the Myosin Head Couple ATP Hydrolysis to Movement
Members of the myosin V class have two heavy chains, resulting in a motor with two heads, long neck regions with six light chains each, and tail regions that dimerize and terminate in domains that bind to specific organelles to be transported. In every case that has been tested so far, myosins move toward the end of an actin filament — with one exception: myosin VI found in animals. This unusual myosin has an inserted amino acid sequence in its head domain that causes myosin and its cargo to move toward the end of the actin filament. Myosin VI is believed to move endocytic vesicles along actin filaments away from the plasma membrane. Recall that membraneassociated actin filaments have their ends toward the membrane, so a motor directed toward the end would take carry vesicles away from the membrane toward the center of the cell. Conformational Changes in the Myosin Head Couple ATP Hydrolysis to Movement Studies of muscle contraction provided the first evidence that myosin heads slide or walk along actin filaments. Unraveling the mechanism of muscle contraction was greatly aided by the development of in vitro motility assays and single-molecule force measurements. On the basis of information obtained with these two techniques plus the three-dimensional structure of the myosin head (see Figure 17-22c), researchers have developed a model for how myosin harnesses the energy released by ATP
hydrolysis to move along an actin filament (Figure 17-26). Because all myosins are thought to use the same basic mechanism to generate movement, we will ignore for the moment whether the myosin tail is bound to a vesicle or is part of a thick filament, as it is in muscle. The most important aspect of this model is that the hydrolysis of a single ATP molecule is coupled to each step taken by a myosin molecule along an actin filament.
FIGURE 17-26 ATP-driven myosin movement along actin filaments. (a) When ADP is bound to the myosin head, it is firmly attached to the actin filament. Step 1 : On exchanging ATP for ADP, the myosin head releases from the actin filament. Step 2 : The head hydrolyzes the ATP to ADP and , which induces a rotation in the head with respect to the neck. This “cocked” state stores the energy released by ATP hydrolysis as elastic energy, like a stretched spring. Step 3 : Myosin in the cocked state is stable until it binds actin. Step 4 : When it encounters actin, the myosin head couples release of with release of the elastic energy to move the actin filament. This movement is known as the
power stroke, as it involves moving the actin filament with respect to the end of the myosin neck domain. In the presence of bound ADP, the head remains tightly bound to the actin filament in a state known as rigor and is the basis for the unmistakable sign of death, rigor mortis. Step 5 : When ADP is replaced by fresh ATP the head releases from the filament. See R. D. Vale and R. A. Milligan, 2002, Science 288:88. (b) Molecular models of the conformational changes in the myosin head involved in “cocking” the head (upper panel) and after the power stroke (lower panel). The myosin light chains are shown in dark blue and green; the rest of the myosin head and neck are colored light blue, and actin is red. See S. Fischer et al., 2005, Proc. Nat'l Acad. Sci. USA 102:6873–6876. [Part (b) Data from Ken Holmes.] Description The illustration labeled (a) shows a thick filament made of many coiled tubules; one coiled-rod from the thick filaments has a two myosin head as endings. The U shaped binding site of the myosin is bound to one of the actin filament units. One of the myosin heads is bound to an A D P molecule. An actin filament has two rope-like structures made up of spheres twisted around each other. The positive and negative ends of the actin filament are labeled. The following sequence is depicted. 1. Binds ATP, head released from actin: The myosin head binds to A T P and the head is released from actin. A molecule of A D P is released. 2. Hydrolysis of A T P to A D P and inorganic phosphate causes the myosin head to rotate and form the cocked state. 3. The myosin head binds to the actin filament. 4. The power stroke occurs, where the release of inorganic phosphate and elastic energy straightens the myosin molecule, moving the actin filament to the left. 5. A D P is released and A T P is bound, and the head is released from actin. The illustration labeled (b) shows the structure of actin filament with its positive and negative ends labeled and of the myosin. The A D P- P i head is highlighted whereas the A T P state head is not. An arrow pointing to the right labeled head cocking points at the A D P- P i head of the myosin. The second 3- D model shows the structure of A D P state myosin head highlighted bound to an actin subunit. An arrow labeled power stroke points at the myosin head.
How does myosin convert the chemical energy released by ATP hydrolysis into mechanical work? The S1 head of myosin is an ATPase. When it has ADP bound, the head of myosin binds very tightly to F-actin (Figure 1726a). When the ADP in the head is exchanged for ATP, its affinity for Factin is greatly reduced, and it releases from actin (Figure 17-26a, step 1 ). Next the myosin head hydrolyzes the ATP, and the hydrolysis products, ADP and , remain bound to it (step 2 ). The energy from the hydrolysis of ATP induces a conformational change in the head that results in the head domain rotating with respect to the neck domain, assuming what is known as the “cocked” position (Figure 17-26b, top). Next the head binds F-actin tightly (step 3 ), inducing both release of and rotation of the head back to its original position, thus moving the actin filament relative to the neck domain (Figure 17-26a, step 4 , and 17-26b, bottom). In this way, binding to F-actin induces the movement of the head and the release of , thereby coupling the two processes. This step is known as the power stroke. The head remains bound until the ADP leaves and a fresh ATP binds the head, releasing it from the actin filament (step 5 ). The cycle repeats. How is hydrolysis of ATP in the nucleotide-binding pocket of the head converted into a mechanical force? Hydrolysis of ATP induces a small conformational change in the head domain. This small movement is amplified by a converter region at the base of the head, which acts like a fulcrum causing the rodlike neck (also known as the lever arm) to rotate. The rotation is amplified by the neck domain, so the actin filament moves by a few nanometers (see Figure 17-26b).
This model makes a strong prediction: the distance a myosin moves along actin during hydrolysis of one ATP — the myosin step size — should be proportional to the length of the neck domain. To test this prediction, mutant myosin molecules were constructed with neck domains of different lengths, and the rate they moved down an actin filament was determined. As the myosins with different neck domains step at the same rate, the rate of movement reflects the step size. As predicted, there was an excellent correlation between the length of the neck domain and the rate of movement (Figure 17-27). EXPERIMENTAL FIGURE 17-27 The length of the myosin II neck domain determines the rate of movement. To test the lever-arm model of myosin movement, investigators used recombinant DNA techniques to make myosin heads attached to neck domains with
Myosin Heads Take Discrete Steps Along Actin Filaments
increasing numbers of light-chain-binding sites and hence lengths. The rate at which these myosins moved on actin filaments was determined, which reflects the myosin step size. The longer the lever arm, the faster the myosin moved, supporting the proposed mechanism. [Data from K. A. Ruppel and J. A. Spudich, 1996, Ann. Rev. Cell Dev. Biol. 12:543–573.] Description In the column graph, the vertical axis plots the velocity in micrometers per second and ranges from 1 to 4 in increments of 1. The horizontal axis plots the neck length in neck units. No neck units yield a velocity of 0.7 micrometers per second; one neck unit, 1.7 micrometers per second; 2 neck units, 3 micrometers per second; and 3 neck units, 5 micrometers per second. Myosin Heads Take Discrete Steps Along Actin Filaments The defining property of a myosin is its ability to generate a force when it contacts an actin filament, but the details of this interaction are tailored to the function of the myosin. Researchers have measured the distance a single myosin II head moves a filament and the force generated. The results show that myosin II takes discrete steps, which average out to about 8 nm, and generates 3–5 piconewtons (pN) of force, approximately the same force as that exerted by gravity on a single bacterium. Moreover, myosin II does not interact with the actin filament continuously but rather binds, moves, and releases it. In fact, myosin II spends on average only about 10 percent of each ATPase cycle in contact with F-actin — it is said to have a duty ratio of 10 percent. This observation will be important later when we consider that in contracting muscle, hundreds of myosin heads
pull on actin filaments, so that at any one time, 10 percent of the heads are engaged to provide a smooth contraction. Now let us examine how myosin V moves. Recall that myosin V has a much longer neck region than does myosin II (see Figure 17-25). In one set of experiments, scientists added a fluorescent label to one of the two neck domains in myosin V molecules (see Section 4.2 for how this can be achieved). Mixing labeled myosin V molecules with F-actin and ATP, the movement of myosin V was observed using fluorescence microscopy (Figure 17-28a). The labeled head took many 72-nm steps without releasing from the actin — so it is said to move processively (Figure 1728b). In a filament, actin subunits are oriented to present a myosin-binding site every 36 nm (see Figures 17-5b and 17-28a), so each head binds to every other site along a filament. As the myosin has two heads, each alternately takes 72-nm steps — like someone walking on stepping stones across a river and placing each foot on every other stone. When the label was attached to the tail instead of the neck, the myosin V motor as a whole moved in 36-nm steps. Thus while the step size for each individual head is 72 nm, cargo attached to the myosin V tail region moves in 36-nm steps as a head moves from behind the cargo to in front of it (Figure 17-28a).
EXPERIMENTAL FIGURE 17-28 Myosin V has a step size of 36 nm, with each head stepping hand-over-hand in 72-nm steps. (a) Diagram of how the myosin V heads interact with binding sites spaced 36 nm along an actin filament, with a fluorescent label on the neck domains of one myosin head. While each myosin V head steps “hand-over-hand”
down an actin filament in 72-nm steps, the cargo, and hence the motor as a whole, moves in 36-nm steps. (b) A trace of the label on one myosin V neck domain as the molecule walks down an actin filament. The labeled myosin head takes successive 72-nm steps. As can be seen from the trace, myosin V takes many successive steps along a filament, so it is said to be processive. As shown in panel (a), the step size corresponds to equivalent sites on the helical structure of the actin filament. [Data from A. Yildiz et al., 2003, Science 300:2061.] Description The illustration labeled (a) shows an actin filament bound to myosin. An actin filament has two rope-like structures made up of spheres twisted around each other. The positive and negative ends of the actin filament are labeled. The structure of a myosin molecule has two bulb-shaped endings with U shaped binding sites. The bulb-like structures are attached to several subunits further attached to a tail made of two coiled heavy chains. The ends of the heavy chains are attached to two oval structures. Myosin has a fluorescent label on the neck, and the separation between the heavy head chains of myosin on the actin filament is about 36 nanometers. The single-step size is thus indicated to be double that value, 72 nanometers. The motor step size of myosin is also 36 nanometers. In the graph labeled (b), the vertical axis represents time in seconds ranging from 0 to 60 in increments of 10. The horizontal axis represents the position in nanometers ranging from 0 to 1000 in increments of 500. The data points form a stepped curve with an increment of 72 nanometers. The time between the increments varies so the steps look uneven. Presumably, myosin V has evolved a long neck domain to take steps that match the distance between myosin-binding sites. Moreover, its ATPase cycle has a high duty ratio of . (Compare this with the 10 percent duty ratio of myosin II.) Myosin V has a high duty ratio because its rate of ADP release is quite slow; thus the head remains in contact with the actin filament for a much larger percentage of the cycle. Since a single
myosin V molecule has two heads, a duty ratio greater than 50 percent ensures that at least one head is always in contact with actin as it moves down an actin filament. This keeps myosin V and its cargo from falling off. These are exactly the properties one would expect for a motor designed to transport cargo along an actin filament. KEY CONCEPTS OF SECTION 17.5 Myosins: Actin-Based Motor Proteins Myosins are actin-based motors powered by ATP hydrolysis. Myosins have a motor head domain, a lever-arm neck domain, and a cargo-binding tail domain (see Figure 17-22). There are many classes of myosins, three of which are present in many eukaryotes: myosin I, which has a single head domain; myosin II, which has two heads and assembles into bipolar filaments; and myosin V, which has two heads but does not assemble into filaments (see Figure 17-25). Myosins convert ATP hydrolysis to mechanical work by amplifying a small conformational change in the head through the neck domain when the head is bound to F-actin (see Figure 17-26). Myosin heads take discrete steps along an actin filament, which can be small (8 nm) and nonprocessive in the case of myosin II or large (36 nm for the motor overall, 72 nm for each head) and processive for myosin V.
Myosin Thick Filaments and Actin Thin Filaments in Skeletal Muscle Slide Past Each Other During Contraction
17.6 Myosin-Powered Movements In the previous section, we discussed how the head and neck domains of myosins function as a motor that converts chemical energy into mechanical force. In this section, we discuss how the whole myosin motor molecules work and are regulated. Since the functions of many of the families of myosins found in metazoans are not yet known, we look at three examples for which we have a good idea of specific myosin function. Our first example is skeletal muscle. In muscle, many myosin II heads, each with a low duty ratio, are bundled into bipolar filaments that work together to bring about muscle contraction. Contractile machinery also functions in the contraction of smooth muscle, in stress fibers, and in the contractile ring during cytokinesis. In our second example, we will look at how contraction operates in nonskeletal muscle. In the third example, we turn to the myosin V class, whose high duty ratio allows cargo to be transported over relatively long distances. Myosin Thick Filaments and Actin Thin Filaments in Skeletal Muscle Slide Past Each Other During Contraction
Muscle cells have evolved to carry out one highly specialized function: contraction. Muscle contractions must occur quickly and repetitively, and they must occur over long distances and with enough force to move large loads. A typical skeletal muscle cell is cylindrical, large (1–40 mm in length and in width), and multinucleated (containing as many as 100 nuclei) (Figure 17-29a). Within each muscle cell are many myofibrils consisting of a repeating array of a specialized structure called a sarcomere (Figure 17-29b). A sarcomere, which is about long in resting muscle, shortens by about 70 percent of its length during contraction. Electron microscopy and biochemical analysis have shown that each sarcomere contains two major types of filaments: thick filaments, composed of myosin II, and thin filaments, containing actin and associated proteins (Figure 17-29c).
FIGURE 17-29 Structure of the skeletal muscle sarcomere. (a) Skeletal muscles consist of muscle fibers made of bundles of multinucleated cells. Each cell contains a bundle of myofibrils, which consist of thousands of repeating contractile structures called sarcomeres. (b) Electron micrograph of mouse skeletal muscle in longitudinal section, showing one sarcomere. On either side of the Z disks are the lightly stained I bands, composed entirely of actin thin filaments. These thin filaments extend from both sides of the Z disk to interdigitate with the dark-stained myosin thick filaments in the A band. (c) Diagram of the arrangement of myosin and actin filaments in a sarcomere. Description The illustration labeled (a) shows muscles in the shoulders to be composed of bundles of muscle fibers, which are composed of multinucleated muscle cells, which are composed of myofibrils, which are composed of sarcomeres, muscle cells. The electron micrograph labeled (b) shows a section through a sarcomere with linear bands of actin and myosin, and perpendicular to these are indicated the z-disk bands composed mainly of actin, and the A band composed mainly of myosin. The illustration labeled (c) shows sarcomere where myosins are present in between two columns of actin filaments held by the Z- disks on both sides. The thick filaments are made of many myosin II bipolar filaments, in which the heads on each half of the filament are in opposite orientations (see Figure 17-22a). The thin actin filaments are assembled with their ends embedded in a densely staining structure known as the Z disk, so that the two sets of actin filaments in a sarcomere have opposite orientations (Figure 17-30). To understand how a muscle contracts, consider the interactions between one myosin head (among the hundreds in a thick filament) and one thin (actin) filament, as diagrammed in Figure 17-26. During these cyclical interactions, also called the cross-bridge cycle, the hydrolysis of ATP is coupled to the movement of a myosin head toward
the Z disk, which corresponds to the end of the actin thin filament. Because the thick filament is bipolar, the action of the myosin heads at opposite ends of the thick filament draws the thin filaments toward the center of the thick filament and therefore toward the center of the sarcomere (see Figure 17-30). This movement shortens the sarcomere until the ends of the thick filaments abut the Z disk. Contraction of an intact muscle results from the activity of hundreds of myosin heads on a single thick filament, amplified by the hundreds of thick and thin filaments in a sarcomere and thousands of sarcomeres in a muscle fiber. We can now see why myosin II is both nonprocessive and needs to have a low duty ratio: each head pulls a short distance on the actin filament and then lets go to allow other heads to pull, and so many heads working together allow the smooth contraction of the sarcomere. The first experimental basis for the sliding-filament model of muscle contraction is highlighted in Classic Experiment 17-1.
FIGURE 17-30 The sliding-filament model of contraction in skeletal muscle. The arrangement of thick myosin and thin actin filaments in the relaxed state is shown in the top diagram. In the presence of ATP and , the myosin heads extending from the thick filaments walk toward the ends of the thin filaments. Because the thin filaments are anchored at the Z disks (purple), movement of myosin pulls the actin filaments toward the center of the sarcomere, shortening its length in the contracted state, as shown in the bottom diagram. Description In the relaxed state, the Z disks are far apart, and the actin fibers overlap the head groups. In the contracted state, the Z disks are closer together, and the actin fibers overlap almost the whole myosin filament. The part between the Z disks is labeled sarcomere.
Skeletal Muscle Is Structured by Stabilizing and Scaffolding Proteins
The human heart is an amazing contractile organ — it contracts without interruption about 3 million times a year, or a fifth of a billion times in a lifetime. The muscle cells of the heart contain contractile machinery very similar to that of skeletal muscle, except that they are mono- and bi-nucleated cells. In each cell, the end sarcomeres insert into structures at the plasma membrane called intercalated disks, which link the cells into a contractile chain. Heart muscle cells are generated only early in life, so they cannot be replaced in response to damage, such as occurs during a heart attack. Many different mutations in proteins of the heart contractile machinery give rise to hypertrophic cardiomyopathies — thickening of the heart wall muscle, which compromises its function. For example, many mutations have been documented in the cardiac myosin heavy-chain gene that compromise the protein’s contractile function even in heterozygous individuals. In such individuals, the heart tries to compensate by hypertrophy (enlargement), which often results in fatal heart arrhythmia (irregular beating). In addition to myosin heavy-chain defects, defects that result in cardiomyopathies have been traced to mutations in other components of the contractile machinery, including actin, myosin light chains, tropomyosin and troponin, and structural components such as titin (discussed below). Skeletal Muscle Is Structured by Stabilizing and Scaffolding Proteins
The structure of the sarcomere is maintained by a number of accessory proteins (Figure 17-31). The actin filaments are stabilized on their ends by CapZ and on their ends by tropomodulin (see Section 17.2). A giant protein known as nebulin extends along the thin filament all the way from the Z disk to tropomodulin, to which it binds. Nebulin consists of repeating domains that bind to actin subunits in the filament, and it is believed that the number of actin-binding repeats, and therefore the length of nebulin, determines the length of the thin filaments. Another giant protein, called titin, has its head associated with the Z disk and extends to the M band in the middle of the thick filament, where another titin molecule extends to the subsequent Z disk. Titin is believed to be an elastic molecule that holds the thick filaments in the middle of the sarcomere and also prevents overstretching to ensure that the thick filaments remain interdigitated between the thin filaments.
FIGURE 17-31 Accessory proteins found in skeletal muscle. To stabilize the actin filaments, CapZ caps the end of the thin filaments at the Z disk, whereas tropomodulin caps the end. The giant protein titin extends from the M band through the thick filaments and attaches to the Z disk. Nebulin binds actin subunits and determines the length of the thin filament. Description The illustration shows bundles of actin with myosin in between. The Z disks are labeled, one at left and one at right holding the actin filaments. The myosin bands have a vertical M band between them in the center. At the negative ends of each actin filament is a sphere labeled tropomodulin. Against the Z disks at both sides are oval structures in groups of 2 labeled Cap Z. The myosin bundles contain thin structures through them extending outside labeled titin. Thin thread-like structure coils around the actin filaments and is labeled nebulin.
Contraction of Skeletal Muscle Is Regulated by Ca2+ and Actin-Binding Proteins
Contraction of Skeletal Muscle Is Regulated by and Actin-Binding Proteins Like many cellular processes, skeletal muscle contraction is initiated by an increase in the cytosolic concentration. As described in Chapter 11, the concentration of the cytosol is normally kept below . In skeletal muscle cells, a low cytosolic level is maintained primarily by a unique ATPase that continually pumps ions from the cytosol containing the myofibrils into the sarcoplasmic reticulum (SR), a specialized endoplasmic reticulum of muscle cells (Figure 17-32). This activity establishes a reservoir of in the SR.
FIGURE 17-32 The sarcoplasmic reticulum regulates the level of free in myofibrils. (a) When a nerve impulse stimulates a muscle cell, the action potential is transmitted down a transverse tubule (yellow), which is continuous with the plasma membrane (sarcolemma), leading to release of from the adjacent sarcoplasmic reticulum (blue) into the myofibrils. (b) Thin-section electron micrograph of skeletal muscle, showing the intimate relationship of the sarcoplasmic reticulum to the muscle fibers.
Description The illustration labeled (a) shows a muscle cell. It is cylindrical in structure, filled with bundles of myofibrils. The myofibrils are tube-like structures. The Z disk around one of the bundles is labeled. The nucleus of the cell is labeled. Around the myofibril bundles are centipede-like structures labeled sarcoplasmic reticulum. The tube at the center of this reticulum is labeled transverse tubule. Sarcolemma covers the myofibrils. The electron micrograph labeled (b) shows a tube-like muscle cell with the dark regions called Z disks labeled, and a horizontal area labeled sarcoplasmic reticulum with a transverse tubule in it. A scale bar reads 0.5 nanometers. The arrival of a nerve impulse (or action potential; see Chapter 23) at a neuromuscular junction triggers an action potential in the muscle-cell plasma membrane (also known as the sarcolemma). The action potential travels down invaginations of the plasma membrane known as transverse tubules, which pass into the cell and lie around each myofibril. The arrival of the action potential in the transverse tubules stimulates the opening of voltage-gated channels in the SR membrane, and the ensuing release of from the SR raises the cytosolic concentration in the myofibrils. This elevated concentration induces changes in two accessory proteins, tropomyosin and troponin, which are bound to the actin thin filaments and normally block myosin binding. Changes in the positions of these proteins on the actin thin filaments in turn permit the myosin-actin interactions and hence contraction. This type of regulation is very rapid and is known as thin-filament regulation. Tropomyosin (TM) is a ropelike molecule, about 40 nm in length, that binds to seven actin subunits in an actin filament. TM molecules are
strung together head to tail, forming a continuous chain along each side of the actin thin filament (Figure 17-33a, b). Associated with each TM molecule is troponin (TN), a complex of three subunits, TN-T, TN-I, and TN-C. TN-C is the calcium-binding subunit of troponin; it controls the position of TM on the surface of an actin filament through the TN-I and TN-T subunits.
Figure 17-33
dependent thin-filament regulation of skeletal muscle contraction. (a) Model and the corresponding structure of the tropomyosin-troponin regulatory complex on a thin filament. Troponin is a protein complex that is bound to the long α-helical tropomyosin molecule. (b) Three-dimensional electron-microscope reconstructions of the tropomyosin helix (yellow) on a muscle thin filament. Tropomyosin in the relaxed state (top) shifts to a new position (arrows) in the state inducing contraction (bottom) when the concentration in the sarcoplasm increases. This movement
exposes myosin-binding sites (red) on actin. (Troponin is not shown in this representation, but it remains bound to tropomyosin in both states.) (c) Summary of the regulation of skeletal muscle contraction by binding to troponin. [Part (a) Data from S. Wu et al., 2012, PLOS One 7:39422, PDB ID 2w4u. Part (b) P. Vibert, R. Craig, and W. Lehman, 1993, J. Cell Biol. 123(2):313–321; https://doi.org/10.1083/jcb.123.2.313.] Description The illustration labeled (a) shows an actin filament. An actin filament has two rope-like structures made up of spheres twisted around each other. Two thread-like structures coiled around each other wrap around the actin filaments. This structure is labeled tropomyosin (T M). Four oval structures are also attached to the actin filament. These structures are labeled troponin (T N). A matching three-dimensional model is present below. Troponin is made of three subunits called T n-T, T n-I, and T n-C. A reconstructed three-dimensional electron micrograph labeled (b) shows two actin filaments as grey bumpy ropes with tubes attached that are labeled T M. The actin filament, labeled negative C a superscript 2 plus, has no markings, whereas the one labeled positive C a superscript 2 plus, which has red areas between the actin labeled myosin binding site. The illustration labeled (c) shows a cycle titled "Relaxation Myosin-binding site masked" at the top, and "Myosin-binding site exposed Contraction" at the bottom. Actin-T M -T N with the addition of C a superscript 2 plus becomes actin-T M -T N-C a superscript 2 plus causing contraction. When the C a superscript 2 plus is removed it goes back to actin-T M -T N, a relaxed state. Under the control of and TN, TM can occupy either of two positions on a thin filament. In the absence of , TM blocks myosin’s interaction with F-actin, and the muscle is relaxed. Binding of ions to TN-C triggers movement of TM to a new position on the filament, thereby exposing the myosin-binding sites on actin (Figure 17-33b). Thus at
Actin and Myosin II Form Contractile Bundles in Nonmuscle Cells
concentrations greater than the inhibition exerted by the TM-TN complex is relieved, and contraction occurs. The -dependent cycling between relaxation and contraction states in skeletal muscle is summarized in Figure 17-33c. Heart muscle, like skeletal muscle, is subject to thin-filament regulation, using cardiac-specific tropomyosin and troponin. During a heart attack (myocardial infarction), heart cells are deprived of sufficient oxygen (cardiac ischemia) and may subsequently die. The plasma membrane of the dead cells ruptures and releases cellular components into the bloodstream. Blood tests that specifically measure the level of cardiacspecific troponins are used by physicians to determine the severity of a heart attack. Actin and Myosin II Form Contractile Bundles in Nonmuscle Cells In skeletal muscle, as we have seen, actin thin filaments and myosin II thick filaments assemble into highly organized contractile structures. Nonmuscle cells contain several types of contractile bundles composed of actin and myosin II filaments, which are similar to skeletal muscle fibers but much less organized. Moreover, these contractile bundles lack the troponin regulatory system and are instead regulated by myosin phosphorylation, as we discuss below. Let’s look at a few examples.
In epithelial cells, contractile bundles are most commonly found as an adherens belt, also known as the circumferential belt, that encircles the inner surface of the cell at the level of the adherens junction (see Figure 17-4a). These bundles are important in maintaining the integrity of the epithelium (discussed in Chapter 20). A second type of contractile bundle are stress fibers, which are seen along the lower surfaces of cells cultured on artificial (glass or plastic) surfaces or on extracellular matrices (see
Figure 17-4a, b) and are important in cell adhesion. The ends of stress fibers terminate at integrin-containing focal adhesions, special structures that attach a cell to the underlying substratum (see Figure 17-39b, c and
Chapter 20). Circumferential belts and stress fibers contain several proteins found in the contractile apparatus of smooth muscle and exhibit some organizational features resembling those of muscle sarcomeres. A third type of contractile bundle is the contractile ring, a transient structure in animal cells that assembles at the equator of a dividing cell, encircling the cell midway between the poles of the mitotic spindle (Figure 17-34a). As the ring contracts, pulling the plasma membrane in, the cytoplasm is divided and eventually pinched into two parts in a process known as cytokinesis, giving rise to two daughter cells. In a dividing cell expressing GFP-myosin-II, myosin II is localized to the contractile ring (Figure 1734b). As evidence for the role of myosin II in forming the cleavage furrow, when the gene for the heavy chain of myosin II has been deleted, cells are unable to undergo cytokinesis. Instead, these cells form a multinucleated syncytium because cytokinesis, but not nuclear division, is inhibited.
Myosin-Dependent Mechanisms Regulate Contraction in Smooth Muscle and Nonmuscle Cells
EXPERIMENTAL FIGURE 17-34 Localization of myosin II during cytokinesis. (a) Diagram of a cell going through cytokinesis, showing the mitotic spindle (microtubules green, chromosomes blue) and the contractile ring with actin filaments (red). (b) Fluorescence micrograph of a Dictyostelium amoeba expressing GFP-myosin-II reveals myosin-II enrichment in the cleavage furrow cortex, as the cell pinches into two during cytokinesis. [Part (b) Republished with permission from Elsevier, from J. C. Effler et al., 2006, Curr. Biol. 16(9):1962–1967, Fig. 1A (149); permission conveyed through Copyright Clearance Center, Inc.; photo courtesy Douglas Robinson.] Description The illustration labeled (a) shows a peanut-shaped cell with microtubules pulling the chromosomes to opposite ends while a contractile ring is formed around the central section of this dividing cell. The fluorescence micrograph labeled (b) shows Dictyostelium amoeba having a U shaped cleavage furrow. Myosin-Dependent Mechanisms Regulate Contraction in Smooth Muscle and Nonmuscle Cells
Like skeletal muscle, smooth muscle is composed of contractile cells. Smooth muscle is found in the walls of hollow organs and the walls of passageways. For example, smooth muscle surrounds blood vessels to regulate blood pressure, surrounds the intestine to move food through the gut, and restricts airway passages in the lung. The contractile apparatus of smooth muscle and its regulation are a valuable model for studying these processes because contraction in smooth muscle is regulated in a manner similar to that in nonmuscle cells. As we have just seen, in skeletal muscle the activity of the myosin II is not directly regulated, but its accessibility to F-actin is regulated by the tropomyosin-troponin complex bound to the actin thin filament that switches between the contraction-inducing state in the presence of and the relaxed state in its absence. In contrast, smooth muscle contraction is regulated by the cycling of myosin II between on and off states. Myosin II cycling, and thus contraction of smooth muscle and nonmuscle cells, is regulated in response to many extracellular signaling molecules. Contraction of vertebrate smooth muscle is regulated primarily by a pathway in which the myosin regulatory light chain (LC) associated with the myosin II neck domain (see Figure 17-22b) undergoes phosphorylation and dephosphorylation. When the regulatory LC is not phosphorylated, smooth muscle myosin II folds into a self-inhibited state where its ATPase cycle is inactive. When the regulatory LC is phosphorylated by the enzyme myosin light-chain kinase (MLC kinase), myosin II opens up by unfolding, assembles into filaments, and becomes able to induce contraction (Figure 17-35). The activity of MLC kinase is regulated by the level of free in the cytoplasm. The -dependent regulation of
MLC kinase activity is mediated by the -binding protein calmodulin (see Figure 3-34). When an extracellular signal leads to a rise in intracellular free concentration, binds to calmodulin inducing a conformational change in the protein. The calmodulin complex then binds to MLC kinase and activates it. This enables MLC kinase to phosphorylate the myosin regulatory LC. When the cytosolic concentration returns to its resting level, MLC kinase becomes inactive, and myosin light-chain phosphatase removes the phosphates from myosin, which returns the system to its relaxed state. This mode of regulation relies on the action of a protein kinase and on the diffusion of over greater distances than in sarcomeres, so contraction is slower in smooth muscle than in skeletal muscle. Because this regulation involves myosin, it is known as thick-filament regulation.
FIGURE 17-35 Myosin light-chain phosphorylation regulates smooth muscle contraction. In vertebrate smooth muscle, phosphorylation of the myosin regulatory light chain (LC) activates contraction. At concentrations of less than , the regulatory light chain is not phosphorylated, and the myosin adopts a folded conformation. When the level rises, binds calmodulin (CaM), which undergoes a conformational change (CaM). The CaM- complex binds and activates myosin light-chain kinase (MLC kinase), which then phosphorylates the myosin LC. This
phosphorylation event opens myosin II, which assembles into side-polar filaments that participate in contraction. Note that the organization of side-polar filaments is distinct from that of the bipolar filaments seen in skeletal muscle. When levels drop, the myosin LC is dephosphorylated by myosin light-chain (MLC) phosphatase, causing muscle relaxation. Description An illustration shows relaxation, where myosin is folded and inactive. The structure of myosin as a chain structure with its large ends folded down along the chain. This structure is labeled myosin L C. Moving counterclockwise, there is an equation that reads C a M and M L C kinase (inactive) yields C a M superscript asterisk minus C a superscript 2 plus M L C Kinase (active). The next part is labeled myosin L C with a circle labeled P. The myosin has a head portion: It has two bulb-shaped endings with U shaped actin-binding sites. Neck portion: it is made of two subunits attached to both bulb-like structures. The subunits are composed of essential light chains and regulatory light chains. The subunits are bound to phosphorus. Tail portion: the tail is made of two coiled heavy chains attached to the neck. A label at the left reads, myosin unfolded and active. Below, a myosin chain labeled side-polar filament assembly leading to contraction is illustrated. The cycle leads back to start with a label reading M L C phosphatase. Unlike skeletal muscle, which is stimulated to contract solely by nerve impulses, smooth muscle cells and nonmuscle cells are regulated by many types of external signals. For example, norepinephrine, angiotensin, endothelin, histamine, and other signaling molecules modulate or induce the contraction of smooth muscle or elicit changes in the shape and adhesion of nonmuscle cells by triggering various signal transduction pathways. Some of these pathways lead to an increase in the cytosolic concentration, which can stimulate myosin activity by activating MLC kinase (see Figure 17-35). As we will see in Section 17.7, other signaling
Myosin V Carries Vesicles Along Actin Filaments
pathways activate Rho kinase, which also activates myosin activity by phosphorylating the regulatory light chain, but in a -independent manner. Myosin V Carries Vesicles Along Actin Filaments The myosin V family of proteins transport cargo along actin filaments and are the most processive myosin motors known. In the next chapter we discuss how they work in conjunction with microtubule motors to transport organelles. Here we look at how these motors carry endocytic and secretory vesicles around the cell. Much is known about myosin V motors in experimentally accessible systems such as the budding yeast Saccharomyces cerevisiae. This wellstudied organism grows by budding, which requires its secretory machinery to target newly synthesized materials to the growing bud (Figure 17-36a). Myosin V transports secretory vesicles along actin filaments at to grow the bud (Figure 17-36b). Myosin V proteins are also important at a later stage of the yeast cell cycle, when the organelles have to be distributed between the mother and daughter cells. Myosin V molecules in yeast transport some of the mother cell’s peroxisomes, mitochondria, vacuoles, endoplasmic reticulum, the transGolgi network, and even the ends of microtubules and some specific messenger RNAs into the bud (Figure 17-36c). These organelles each have a distinct receptor that the tail of myosin V binds to. The motor makes
many delivery cycles, so it has to have a way to pick up, transport, and deliver its organelle cargo. Recent work has shown that myosin V can exist in two states: an inactive folded state in which the tail domain binds and inhibits the activity of the motor head domain, and an active state in which binding to cargo unfolds the tail, thus releasing the head from the tail, which allows the myosin motor to operate (Figure 17-36d, e). How the motor releases its cargo upon delivery is poorly understood — although in one case, it is known that the receptor on the organelle that binds myosin becomes degraded upon delivery of the organelle to its destination. Whereas budding yeast use myosin V and polarized actin filaments to transport many organelles, animal cells employ microtubules and their motors to transport many of the same organelles over the longer distances inside an animal cell. We discuss those transport mechanisms in the next chapter.
FIGURE 17-36 Cargo movement by myosin V. (a) The yeast Saccharomyces cerevisiae (used in making bread, beer, and wine) grows by budding. (b) Secretory vesicles are transported by myosin V along actin cables, nucleated by formins, into the bud, which
grows to swell to about the size of the mother cell. (c) Before cell division can occur, all the organelles must be segregated between the mother and the bud, including the vacuole (the yeast equivalent of a lysosome), peroxisomes, mitochondria, endoplasmic reticulum (ER), trans-Golgi network (TGN), and even selected mRNAs into the bud, which are all mediated by myosin V. Myosin V also binds the ends of cytoplasmic microtubules (green) to orient the nucleus in preparation for mitosis. See D. Pruyne et al., 2004, Ann. Rev. Cell Dev. Biol. 20:559. (d) Myosin V is regulated by binding cargo. In the inactive folded state, the tails of myosin V bind and inactivate the motor head domains. Upon cargo binding, the head-to-tail interaction is alleviated, and the motor is now active to transport cargo. (e) Electron micrograph of inactive myosin V. Myosin V molecules were negatively stained and viewed in a transmission electron microscope. The image shown is the composite average from several individual images. [Part (e) Reprinted by permission from Macmillan Publishers Ltd, from K. Thirumurugan et al., 2006, “The Cargo-Binding Domain Regulates Structure and Activity of Myosin 5,” Nature 442:212–215; permission conveyed through Copyright Clearance Center, Inc.] Description The illustration labeled (a) shows the yeast life cycle where budding occurs. An oval structure represents a yeast cell. The oval structure is bound to a smaller oval structure labeled the bud. The bud grows in size. The bud grows into a new yeast cell. The illustration labeled (b) titled bud growth shows a close up of a budding area. Within the bud, two secretory vesicles, represented by spheres, are attached to actin filaments. One filament extends into the parent cell. Formin proteins are attached at the ends of the actin filaments. At the center of the parent cell is a nucleus with the Golgi apparatus and secretory vesicles. The illustration labeled (c) titled organelle segregation shows a bud, now larger and containing structures labeled m R N A and E R that are attached to actin filaments by myosin 5. In the parent cell, the nucleus has filaments attached to it. Below the nucleus is a vacuole attaching to actin with arrows showing that it will go into the bud. Peroxisome, mitochondrion, vacuole, and T G N is attached to actin which moves towards the bud.
The illustration labeled (d) shows inactive myosin with the heads together, and then open and active with an oval structure called cargo attached. The electron micrograph labeled (e) shows myosin, with labels from top to bottom in this order: Neck domain (lever arm), Coiled-coil domain, Cargo-binding tail domain, and Motor head domain. Perhaps the most dramatic example of movement by myosin V is seen in giant green algae, such as Nitella and Chara. These algae can be found in ponds during the summer and their movement easily observed using a simple microscope. In their large cells, which can be as much as 2 cm in length, cytosol flows rapidly, at a rate approaching 4.5 mm per minute, in an endless loop around the inner circumference of the cell (Figure 17-37). This cytoplasmic streaming is a principal mechanism for distributing cellular metabolites and is also seen in other large cells such as plant cells and amoebae. The algal cells have bundles of actin filaments aligned along the length of the cell, lying just above the stationary chloroplasts located adjacent to the membrane. The bulk cytosol is propelled by myosin V (also known as myosin XI in plants) attached to parts of the ER adjacent to the actin filaments. The flow rate of the cytosol in Nitella is about 15 times faster than movement produced by any other myosins.
FIGURE 17-37 Cytoplasmic streaming in cylindrical giant algae. (a) Cells of Nitella, a freshwater alga commonly found in ponds in the summer. The cytoplasmic movement, described below, can be readily observed with a simple microscope, so go find some Nitella (or related algae) and watch this amazing phenomenon! (b) The center of a Nitella cell is filled with a single large water-filled vacuole, which is surrounded by a layer of moving cytoplasm (blue arrows). A nonmoving layer of cortical cytoplasm filled with chloroplasts lies just under the plasma membrane (enlarged in bottom figure). On the inner side of this layer are bundles of stationary actin filaments (red), all oriented with the same polarity. A motor protein (blue), a plant myosin V, carries parts of the endoplasmic reticulum (ER) along the actin filaments. The movement of the ER network propels the entire viscous cytoplasm, including organelles that are enmeshed in the ER network. (c) Electron micrograph of the cortical cytoplasm showing a large vesicle connected to an underlying bundle of actin filaments. Description
The micrograph labeled (a) shows algae with one cell highlighted and an arrow points toward the illustration (b). The illustration labeled (b) shows a Nitella cell which is oval in shape. It has a huge oval-shaped vacuole inside. Below the cell membrane is a layer labeled nonmoving cytoplasm. The nonmoving cytoplasm encloses the moving cytoplasm which covers the vacuole. The moving cytoplasm has arrows indicating that it is moving counterclockwise. A section of the cell along the bottom is highlighted and an arrow points to it. In the moving cytoplasm, the E R is labeled. The E R is attached to actin filaments via myosin fibers present above the nonmoving cortical cytoplasm. The plasma membrane is labeled as well as the cell wall. The nonmoving cortical cytoplasm has oval structures labeled chloroplasts. The electron micrograph labeled (c) shows a large circular vesicle connected to actin filaments. KEY CONCEPTS OF SECTION 17.6 Myosin-Powered Movements In skeletal muscle, contractile myofibrils are composed of thousands of repeating units called sarcomeres. Each sarcomere consists of two interdigitating filament types: myosin thick filaments and actin thin filaments (see Figure 17-29). Skeletal muscle contraction involves the ATP-dependent sliding of myosin thick filaments along actin thin filaments to shorten the sarcomere and hence the myofibril (see Figure 17-30). The ends of the actin thin filaments in skeletal muscle are stabilized by CapZ at the end and by tropomodulin at the end. Two large proteins, nebulin associated with the thin filaments and titin with the thick filaments, also contribute to skeletal muscle organization. Skeletal muscle contraction is subject to thin-filament regulation. At low levels of free , tropomyosin blocks the interaction of myosin and F-actin, and the muscle is relaxed. At elevated levels of free , the troponin complex associated with tropomyosin binds and moves the tropomyosin to uncover the myosin-binding sites on actin, allowing contraction (see Figure 17-33). Smooth muscle and nonmuscle cells have contractile bundles of actin and myosin filaments with an organization similar to that in skeletal muscle but less well ordered.
Contractile bundles are subject to thick-filament regulation. A myosin light chain is phosphorylated by myosin light-chain kinase, which activates myosin and hence induces contraction. The MLC kinase is activated by binding to -calmodulin when the free intracellular concentration rises (see Figure 17-35). Inactive myosin V is activated by binding cargo, which it transports processively along actin filaments.
17.7 Cell Migration: Mechanism, Signaling, and Chemotaxis
17.7 Cell Migration: Mechanism, Signaling, and Chemotaxis So far in this chapter, we have examined some of the mechanisms cells use to create movement using actin, myosin, and other actin-associated proteins. Some of these mechanisms generate the forces used by cells to migrate. Cell migration results from the coordination of actin-based motions generated in different parts of a cell, which are integrated with an endocytic cycle. The study of cell migration is important to many fields of biology and medicine. For example, an essential feature of animal development is the migration of specific cells along predetermined paths. Epithelial cells in an adult animal migrate to heal a wound, and white blood cells migrate to sites of infection. Less obvious are the continuous slow migration of intestinal epithelial cells along the villi in the intestine and of endothelial cells that line the blood vessels. The inappropriate migration of cancer cells after breaking away from their normal tissue results in metastasis. Cell migration is initiated by the formation of a large, broad membrane protrusion at the leading edge of a cell. Video microscopy reveals that a major feature of this movement is the polymerization of actin at the membrane. Actin filaments at the leading edge are rapidly cross-linked into bundles and networks in a protruding region, called a lamellipodium
Cell Migration Coordinates Force Generation with Cell Adhesion and Membrane Recycling
in vertebrate cells. In some cases, slender, fingerlike membrane projections, called filopodia, also extend from the leading edge. These structures form stable contacts with the underlying surface (e.g., the extracellular matrix) that the cell moves across. In this section, we take a closer look at how cells coordinate various microfilament-based processes with endocytosis to move across a surface. We also consider the role signaling pathways play in coordinating and integrating the actions of the cytoskeleton, a major focus of current research. Cell Migration Coordinates Force Generation with Cell Adhesion and Membrane Recycling If you watch a moving fibroblast (connective tissue cell), it displays a characteristic sequence of events as it migrates over a surface: initial extension of a membrane protrusion, attachment to the substratum, forward flow of cytosol, and retraction of the rear of the cell (Figure 1738). These events occur in an ordered pattern in a slowly moving cell such as a fibroblast, but in rapidly moving cells, such as macrophages, all four steps occur simultaneously in a coordinated manner. In this section, we first consider the role the actin cytoskeleton plays in assembly at the leading edge as well as attachment to (and detachment from) the substratum (Figure 17-39a, b). Second, we discuss the role of the endocytic cycle in cell migration.
FIGURE 17-38 Steps in cell locomotion. Movement begins with the extension of one or more lamellipodia from the leading edge of the cell (step 1 ); some lamellipodia adhere to the substratum by focal adhesions (step 2 ). Then the bulk of the cytoplasm in the cell body flows forward due to contraction at the rear of the cell (step 3 ). The trailing edge of the cell remains attached to the substratum until the tail eventually detaches and retracts into
the cell body. During this cytoskeleton-based cycle, the endocytic cycle internalizes membrane and integrins at the rear of the cell and transports them to the front of the cell (arrows) for reuse in making new adhesions (step 4 ). Description An illustration shows a cell with a nucleus on a surface. There are several points of adhesion. Movement of the cell in the direction of motion occurs in the following steps. 1. Extension of the lamellipodium. 2. Adhesion of the extended portion. 3. Translocation, leading to cell body movement. 4. De-adhesion and endocytic recycling.
FIGURE 17-39 Actin-based structures involved in cell locomotion. (a) Localization of actin in a fibroblast expressing GFP-actin. (b) Diagram of the classes of microfilaments involved in cell migration. The network of actin filaments in the leading edge advances the cell forward. Contractile fibers in the cell cortex squeeze the cell body forward, and stress
fibers terminating in focal adhesions also pull the bulk of the cell body up as the rear adhesions are released. (c) The structure of focal adhesions involves the attachment of the ends of stress fibers through integrins to the underlying extracellular matrix. Focal adhesions also contain many signaling molecules important for cell locomotion. (d) The dynamic actin network in the leading edge is nucleated by the complex and employs the same set of factors that control assembly and disassembly of actin filaments in the Listeria tail (see Figure 17-17). Description The electron micrograph labeled (a) shows a curved oval structure having stress fibers and leading edge. Some spots glow, which is the G F P marked actin filaments. The illustration labeled (b) shows an animal footprint like a cell with stress fibers along its length. The stress fibers have blue dots on the ends. More fibers are present lining the cell. The leading edge is labeled. The illustration labeled (c) titled focal adhesions depicts the area around the blue dot from the illustration (b). Contractile bundles are labeled at the top, and at the left side, they are attached to the plasma membrane via four structures labeled integrins. The bundles are inside the cell. The integrins are attached to the extracellular matrix. The illustration labeled (d) shows the close up of the actin filaments in the claw part of the footprint shaped cell in the illustration (b). There are four actin filaments with more being added to the inner membrane of the cell membrane. The actin filament has filamin, capping protein, cofilin labeled. Profilin binds to A D P- G actin to become A T P-G actin to elongate actin filament. Step 1: Membrane Extension The network of actin filaments at the leading edge is a type of cellular engine that pushes the membrane forward in a manner very similar to the propulsion of Listeria by actin polymerization (Figure 17-39d; for Listeria, see Figure 17-17c). At the membrane of the leading edge, actin is
nucleated by the activated Arp2/3 complex (see Figure 17-15), and filaments are elongated by assembly onto ends adjacent to the plasma membrane. Because the actin network is fixed with respect to the substratum, the front membrane is pushed out as the filaments elongate. Actin turnover, and thus treadmilling, is mediated, as it is in the comet tails of Listeria, by the action of profilin and cofilin (Figure 17-39d). Step 2: Cell-Substratum Adhesions When the membrane has been extended and the actin network has been assembled, the plasma membrane becomes firmly attached to the substratum. Time-lapse microscopy shows that actin bundles in the leading edge become anchored to structures known as focal adhesions (Figure 17-39c). The membrane proteins within a focal adhesion that connect actin bundles to the extracellular matrix are called integrins. Integrins have an external domain that binds to specific components of the extracellular matrix, such as fibronectin and collagen, and a cytoplasmic domain that links them to the actin cytoskeleton (see Figure 17-39c and
Chapter 20). The attachment of cell to substrate serves two purposes: it prevents the lamellipodium from retracting, and it allows the cell to move forward. Given the importance of focal adhesions and their regulation during cell locomotion, it is not surprising that they are very rich in molecules involved in signal transduction pathways. Focal adhesions are discussed in Chapter 20, where we discuss in detail the interactions that occur between a cell and the extracellular matrix. Step 3: Cell-Body Translocation
After the forward adhesions at the leading edge have been made, the bulk contents of the cell body are translocated forward (see Figure 17-38, step 3 ). The nucleus and the other organelles embedded in the cytoskeleton are moved forward by myosin II–dependent cortical contraction in the rear part of the cell, like toothpaste when the lower half of the tube is squeezed. Consistent with this model, myosin II is localized to the rear cell cortex. As the cell migrates forward, the adhesions effectively move toward the rear of the cell. Step 4a: Breaking Cell Attachments Finally, in the last step of movement, the focal adhesions at the rear of the cell are broken (de-adhesion), the integrins are recycled, and the freed tail of the cell is brought forward. In the light microscope, the tail is often seen to snap loose from its connections — perhaps by the contraction of stress fibers in the tail or by elastic tension — and it sometimes leaves a little bit of its membrane behind, still firmly attached to the substratum. Cells cannot move as just described if they are either too strongly attached or too weakly attached to a surface. The ability of a cell to move corresponds to a balance between the mechanical forces generated by the cytoskeleton and the resisting forces generated by cell adhesions. This relationship can be demonstrated by measuring the rate of movement in cells that express varying levels of integrins. Such measurements show that the fastest migration occurs at an intermediate level of adhesion, with the rate of movement falling off at high and low levels of adhesion. Cell
The Small GTP-Binding Proteins Cdc42, Rac, and Rho Control Actin Organization
locomotion thus results from traction forces exerted by the cell on the underlying substratum. Step 4b: Recycling Membrane and Integrins by Endocytosis The dynamic changes in the actin cytoskeleton alone are not sufficient to drive cell migration. It is also dependent on endocytic recycling of plasma membrane. The membrane needed during lamellipodium extension is provided by internal endosomes as the membrane recycles back to the cell surface. Adhesion molecules in focal adhesions at the rear of the cell are internalized as those adhesions are disassembled and are then transported by an endocytic cycle to the front of the cell to make new substratum attachments at the leading edge (see Figure 17-38, step 4 ). This cycling of adhesion molecules in a migrating cell resembles the way a tank uses its treads to move forward. The movement of membrane internally through the cell also generates a rearward membrane flow across the surface of the cell. Indeed, this type of flow may contribute to the mechanics of cell locomotion, as it has been found that white blood cells can move in a liquid (i.e., “swim”) in the absence of attachment to a substratum, presumably as surface structures operating like paddles move backward across the cell surface. The Small GTP-Binding Proteins Cdc42, Rac, and Rho Control Actin
Organization A striking feature of a moving cell is its polarity: it has a front and a back. In addition, when a cell makes a turn, a new leading edge forms in the new direction of movement. To sustain movement in a particular direction, a cell requires signals to coordinate events at the front of the cell with events at the back and, indeed, signals to tell the cell where its front is. Our understanding of how this coordination occurs has emerged from studies of growth factors. Growth factors, such as epidermal growth factor (EGF) and plateletderived growth factor (PDGF), bind to specific cell-surface receptors (see
Chapter 16) and consequently stimulate cells to move or to divide. For example, in a wound, activated blood platelets secrete PDGF to attract fibroblasts and epithelial cells to enter the wound and repair it. It is possible to watch part of this process in vitro. If you grow cells in a culture dish and, after starving them of growth factors, you add some fresh growth factor, within a minute or two the cells respond by forming membrane ruffles. The growth factor activates cellular components that control recycling of membrane from endosomes coupled with actin assembly. In this regard, membrane ruffles are very similar to the lamellipodia of migrating cells. Growth factors bind to specific receptors on the cell surface and induce a signal transduction pathway on the inner surface of the plasma membrane (see Chapter 15). The signal transduction pathway activates Rac, a
member of the small GTPase superfamily of Ras-related proteins. Rac is one member of a GTPase superfamily that regulates microfilament organization; two others are Cdc42 and Rho. (Due to the history of their discovery, the protein family of which Cdc42, Rac, and Rho are members has been collectively named “Rho proteins.”) To understand how these proteins work, we first have to recall how small GTP-binding proteins function (see Chapter 15). Like all small GTPases of the Ras superfamily, Cdc42, Rac, and Rho act as molecular switches: inactive in the GDP-bound state and active in the GTP-bound state (Figure 17-40). In their GDP-bound state, they exist free in the cytoplasm in an inactive form bound to a protein known as guanine nucleotide dissociation inhibitor (GDI). Growth factors can bind and activate their receptors to turn on specific membrane-bound regulatory proteins, called guanine nucleotide exchange factors (GEFs), which activate Rho proteins at the membrane by releasing them from GDI and catalyzing the exchange of GDP for GTP. The GTP-bound active Rho protein associates with the plasma membrane, where it binds effector proteins to transmit the biological response. The small GTPase remains active until the GTP is hydrolyzed to GDP, a process that is stimulated by specific GTPase-activating proteins (GAPs).
FIGURE 17-40 Regulation of the Rho family of small GTPases. The small GTPases of the Rho family are molecular switches regulated by accessory proteins. Rho proteins exist in the Rho-GDP bound form complexed with a protein known as GDI (guanine nucleotide dissociation inhibitor), which keeps them in an inactive state in the cytosol. Membranebound signaling pathways bring Rho proteins to the membrane and, through the action of a GEF (guanine nucleotide exchange factor), exchange the GDP for GTP, thus activating them. Membrane-bound activated Rho-GTP can then bind effector proteins that cause changes in the actin cytoskeleton. The Rho protein remains in the active Rho-GTP state until acted on by a GAP (GTPase-activating protein), which allows it to interact with the GDI and be returned to the cytoplasm. See S. Etienne-Manneville and A. Hall, 2002, Nature 420:629. Description At the top of this illustration, there is a tiny sphere labeled extracellular signal moving towards an oval receptor which is in the plasma membrane. Exterior and cytosol are labeled. To the left of the receptor, a Rho structure with G D P is attached to the inner membrane of the plasma membrane, and below it is a reversible set of two arrows to the same Rho with G D I added. An arrow to the right of the Rho in the membrane shows that the signal from the exterior is now a structure labeled G E F. The G T P is added and returned as G D P. Then the arrow continues right to show the Rho structures with G T P having released two effector proteins.
To unravel the functions of the Rho proteins in cell migration, cells were modified to express mutant proteins that were locked either in the active Rho-GTP state or in the inactive Rho-GDP state. In general, a small GTPase that is locked in the active state is called a dominant-active protein. This always active GTPase will constitutively bind effector proteins (Figure 17-40), and one can then assess the biological outcomes. Alternatively, a dominant-negative protein can be introduced into a cell, such as a protein that binds to and inhibits the GEF protein. Introduction of a dominant-negative protein interferes with the signal transduction pathway, enabling one to assess what processes are blocked. When experiments like these were performed in fibroblasts, Cdc42, Rac, and Rho were implicated in the regulation of microfilament organization because introduction of dominant-active mutant proteins had dramatic effects on the actin cytoskeleton, even in the absence of growth factors. Dominant-active Cdc42 resulted in the appearance of filopodia, dominantactive Rac resulted in the appearance of membrane ruffles, and dominantactive Rho resulted in the formation of stress fibers, which were seen to contract (Figure 17-41). How can one tell if dominant-active Rac and growth factor stimulation, both of which stimulate membrane ruffle formation, use the same signal transduction pathway? If growth factor stimulation leads to Rac activation, then introduction of a dominantnegative Rac protein into a cell should block the ability of a growth factor to induce membrane ruffling. This is precisely what was found. Using this
and many other biochemical strategies, scientists have identified the signaling pathways involving Cdc42, Rac, and Rho (Figure 17-42). EXPERIMENTAL FIGURE 17-41 Dominant-active Rac, Rho, and Cdc42 induce different actin-containing structures. To look at the effects of constitutively active Rac, Rho, and Cdc42, growth-factor-starved fibroblasts were microinjected with plasmids to
express dominant-active versions of the three proteins. The cells were then treated with fluorescent phalloidin, which stains filamentous actin. Dominant-active Rac induces the formation of peripheral membrane ruffles, whereas dominant-active Rho induces abundant contractile stress fibers, and dominant-active Cdc42 induces filopodia. [Republished with permission from AAAS, from A. Hall, 1998, “Rho GTPases and the Actin Cytoskeleton,” Science 279(5350):509–514; permission conveyed through Copyright Clearance Center, Inc.] Description The first electron micrograph labeled control and shows a bat-shaped web-like structure. The second electron micrograph labeled dominant-active Rho shows many white thread-like structures in an irregular bundle. The third electron micrograph labeled dominant-active R a c shows a bright ring with a light web inside. The fourth electron micrograph labeled dominant-active C d c 4 2 shows a spherical structure with many thin spikes around it.
FIGURE 17-42 Summary of signal-induced changes in the actin cytoskeleton. Specific signals, such as growth factors and lysophosphatidic acid (LPA), are detected by cellsurface receptors. Detection leads to the activation of the small GTP-binding proteins, which then interact with effectors to bring about cytoskeletal changes as indicated. Description The illustration shows the following signaling pathways that induce changes in the microfilament structure of cells. Growth factor activates C d c 4 2. G T P, which can activate P a r 6, leading to cell polarization, or W A S p, which via A r p 2 slash 3 results in actin polymerization and filopodia formation. Alternatively, growth factors activate R a c . G T P, which via W A V E, A r p 2 slash 3, and actin polymerization, lead to formation of lamellipodia. In addition, Growth factors, via R a c or L P A, can activate Rho, leading to two pathways to stress fiber formation and contraction. Path 1: Rho kinase, myosin L C phosphatase, and myosin L C-P subscript i, myosin activity, or
Cell Migration Involves the Coordinate Regulation of Cdc42, Rac, and Rho
path 2: formin, followed by actin polymerization. Filopodia formation shows filaments only in the claw-shaped structures of the cell. Lamellipodia formation shows filaments below the claw-shaped structures of the cell. Stress fiber formation and contraction show linear filaments in the center of the cell. Some of the pathways that these proteins regulate contain proteins we are familiar with. Activation of Cdc42 stimulates actin assembly by through activation of WASp, a nucleation promoting factor (NPF) (see
Figure 17-16), resulting in the formation of filopodia. Activation of Rac also induces , mediated by the WAVE complex, leading to the assembly of branched actin filaments in the leading edge. Activation of Rho has at least two effects. First, it can activate a formin for unbranched actin filament assembly. Second, through activation of Rho kinase, it can phosphorylate the myosin light chain to activate nonmuscle myosin II and can also inhibit light-chain dephosphorylation by phosphorylating myosin light-chain phosphatase to inhibit its activity. Both actions of Rho kinase lead to a higher level of myosin light-chain phosphorylation and therefore higher myosin activity and contraction. Cdc42, Rac, and Rho are also linked by activation and inhibition pathways, as shown in Figure 17-42. Cell Migration Involves the Coordinate Regulation of Cdc42, Rac, and Rho How does each of these small GTP-binding proteins contribute to cell migration? To answer this question, researchers developed the wounded-
cell monolayer assay (Figure 17-43a). Cells in culture are grown in a petri dish with growth factors until they are confluent and form a tight monolayer, at which point they stop dividing. The cell monolayer is then scratched with a needle to remove a swath of cells, generating a wound containing a free edge of cells. The cells on the edge sense the loss of their neighbors and, in response to components of the extracellular matrix now exposed on the dish surface, move to fill up the empty wound area (Figure 17-43b). To do this, they orient themselves toward the empty area, first extending a lamellipodium and then moving in that direction. In this way, one can study the induction of directed cell migration in vitro.
EXPERIMENTAL FIGURE 17-43 The wounded-cell monolayer assay can be used to dissect signaling pathways in directed cell movement. (a) A confluent layer of cells is scratched to remove a swath about three cells wide to generate a free cell border. The remaining cells detect the free space and newly exposed extracellular matrix and, over a period of hours, fill in the area. (b) Localization of actin in a wounded monolayer 5 minutes and 3 hours after scratching; after 3 hours, cells have migrated into the wounded area. (c) Effect of introducing dominant-negative Cdc42, Rac, and Rho into cells at the wound edge; all affect wound closure. [Part (b, c) C. D. Nobes and A. Hall, 1999, J. Cell Biol. 144(6):1235–1244; https://doi.org/10.1083/jcb.144.6.1235.] Description The illustration labeled (a) shows three Petri dishes. The first petri dish has confluent cells. The second petri dish also has confluent cells with the middle portion scratched off to create a border. The third petri dish has confluent cells. The electron micrograph labeled (b) shows the scraped region after five minutes and after three hours. After three hours, the scraped region has been filled. In the graph labeled (c), the horizontal axis represents wound closure in percentage and the vertical axis represents the effects of control, dominant-negative Rho, c d c 42, and r a c wound closure. Wound closure is 100 percent for the control, 5 percent for dominant-negative Rho, 30 percent for dominant-negative c d c 4 2, and 20 percent for dominant-negative r a c. All values are approximate. Using this wounded-cell monolayer assay, researchers have introduced dominant-negative Rac into cells on the wound edge to see how it affects the ability of the cells to migrate and fill the wound. Since Rac is needed for activation of the complex to form the lamellipodium, it is not surprising that cells containing dominant-negative Rac fail to form this structure and do not migrate, and so the wound does not close (Figure 17-
43c). A very interesting result is obtained when dominant-negative Cdc42 is introduced into the cells at the wound edge: they can form a lamellipodium, but they do not orient in the correct direction — in fact, they try to migrate in random directions. This observation suggests that Cdc42 is critical for regulating the overall polarity of the cell. Studies from yeast (in which Cdc42 was first described), wounded-cell monolayers, epithelial cells, and neurons reveal that Cdc42 is a master regulator of polarity in many different systems. Part of this regulation in animals involves the binding of Cdc42 to its effector, Par6, a polarity protein that functions in nematodes (in which it was first discovered), neurons, and epithelial cells. We explore these polarity pathways in more detail in Chapter 22. Studies such as these suggest a general model of how cell migration is controlled (Figure 17-44). Signals from the environment are transmitted to Cdc42, which orients the cell. The oriented cell has high Rac activity at the front, which induces the formation of the leading edge; Rho activity is high in the rear, inducing the assembly of contractile structures and activating the myosin-II-based contractile machinery. It is important to notice that different regions of the cell have different levels of active Cdc42, Rac, or Rho, so the amount of these regulators are controlled locally within the cell. Part of this spatial regulation occurs because some small G proteins can work antagonistically. For example, active Rho can stimulate pathways that lead to the inactivation of Rac. This process might help ensure that no leading-edge structures form at the rear of the cell.
FIGURE 17-44 Contributions of Cdc42, Rac, and Rho to cell movement. The overall polarity of a migrating cell is controlled by Cdc42, which is activated at the front of a cell. Cdc42 activation leads to active Rac in the front of the cell, which generates the leading edge, and active Rho at the back of the cell, which causes myosin II activation and contraction. Active Rho inhibits Rac activation, ensuring the asymmetry of the two active G-proteins. Description The illustration shows a migratory cell shaped almost like a foot. A layer beneath the cell membrane makes up the microfilaments, which enclose more microfilaments, microtubules, and intermediate filaments. The finger-like projections are labeled actin filament assembly and treadmilling in the leading edge. The actin filaments in the center and around the inner membrane are labeled contraction of myosin 2 filaments in both stress fibers and the cell cortex. C d c 42 activation at the front of the cell leads to activation of r a c. At the front of the cell, r a c activation leads to A r p 2 slash 3 activation. At the back of the cell, r h o activation leads to myosin 2 activation.
Migrating Cells Are Steered by Chemotactic Molecules
Migrating Cells Are Steered by Chemotactic Molecules Under certain conditions, extracellular chemical cues guide the migration of a cell in a particular direction. In some cases, the movement is guided by insoluble molecules in the underlying substratum, as in the woundedcell monolayer assay described above. In other cases, the cell senses soluble molecules and follows them, along a concentration gradient, to their source — a process known as chemotaxis. For example, leukocytes are guided toward an infection by a tripeptide that is released by many bacterial cells (Figure 17-45). In another example, during the development of skeletal muscle, a secreted protein signal called scatter factor guides the migration of myoblasts to the proper locations in limb buds. One of the best studied examples of chemotaxis is the migration of Dictyostelium amoebae during their starvation response. When these soil amoebae are stressed, they begin to secrete cAMP, which is an extracellular chemotactic agent in this organism. Other Dictyostelium cells move up the cAMP concentration gradient toward its source (see Figure 17-45). Thus the amoebae move toward one another, aggregate into a migratory slug, and then differentiate into a fruiting body in which starvation-resistant spores are formed.
EXPERIMENTAL FIGURE 17-45 Chemotactic molecules guide migrating cells by signaling the actin cytoskeleton. Dictyostelium cells migrate toward a pipette of cAMP (left), and human neutrophils (a type of leukocyte) migrate toward a pipette of fMLP (formylated Met-Leu-Phe), a chemotactic peptide produced by bacteria (right). In the lower two panels are individual chemotaxing Dictyostelium and neutrophil cells, which look remarkably similar, despite about 800 million years of evolution separating them. [Republished with permission from Elsevier, from C. A. Parent, 2004, “Making All The Right Moves: Chemotaxis in Neutrophils and Dictyostelium,” Curr. Opin. Cell Biol. 16(1):4–13; permission conveyed through Copyright Clearance Center, Inc.]
Despite the variety of different chemotactic molecules — sugars, peptides, cell metabolites, cell-wall or membrane lipids — they all work through a common and familiar mechanism: binding to cell-surface receptors, activation of intracellular signaling pathways, and remodeling of the cytoskeleton through the activation or inhibition of various actin-binding proteins. What is quite amazing is that just a 2 percent difference in the concentration of chemotactic molecules between the front and back of the cell is sufficient to induce directed cell migration (see Classic Experiment 17-2). Equally amazing is the finding that the internal signal transduction pathways used in chemotaxis have been conserved between Dictyostelium amoebae and human leukocytes despite almost a billion years of evolution (Figure 17-45). KEY CONCEPTS OF SECTION 17.7 Cell Migration: Mechanism, Signaling, and Chemotaxis Cell migration involves the extension of an actin-rich leading edge at the front of the cell, the formation of focal adhesions that move backward with respect to the cell, and their subsequent release, combined with rear contraction to push the cell forward (see
Figure 17-38). Cell migration also involves a directed endocytic cycle, taking membrane and adhesion molecules from the rear of the cell and inserting them at the front. The assembly and function of actin filaments is controlled by signaling pathways through small GTP-binding proteins of the Rho family. Cdc42 regulates overall polarity and the formation of filopodia, Rac regulates actin network formation through the complex, and Rho regulates actin filament formation by formins as well as contraction through regulation of myosin II (see Figure 17-42). Chemotaxis, the directed movement of cells toward extracellular chemical cues, involves signaling pathways that contribute to the regulation of the actin cytoskeleton and direction of cell migration.
Key Terms
End of Chapter Visit Achieve to access study tools and to learn more about the content in this chapter. Perspectives for the Future Classic Experiment 17-1: Looking at Muscle Contraction Classic Experiment 17-2: Sensing Chemotactic Gradients Analyze the Data Chapter References Additional study tools, including videos, animations, and quizzes Key Terms actin actin cross-linking proteins complex CapZ protein Cdc42 protein cell migration cell polarity chemotaxis cofilin contractile bundles critical concentration cytoskeleton
duty ratio filopodia formin intermediate filament lamellipodium leading edge microfilament microtubule microvilli motor protein myofibrils myosin myosin light-chain kinase nucleation promoting factor (NPF) power stroke profilin Rac protein Rho protein sarcomere sarcoplasmic reticulum (SR) step size stress fibers thick filaments thin filaments thymosintreadmilling tropomodulin tropomyosin
Review the Concepts
WASp Review the Concepts 1. Three systems of cytoskeletal filaments exist in most eukaryotic cells. Compare them in terms of composition, function, and structure. 2. Actin filaments have a defined polarity. What is filament polarity? How is it generated at the subunit level? How is filament polarity detectable? 3. In cells, actin filaments form bundles or networks. How do cells form such structures, and what specifically determines whether actin filaments will form a bundle or a network? 4. Much of our understanding of actin assembly in the cell is derived from experiments using purified actin in vitro. What techniques can be used to study actin assembly in vitro? Explain how each of these techniques works. Which of these techniques would tell you whether the mass of actin filaments is made up of many short actin filaments or fewer longer filaments? 5. The predominant forms of actin inside a cell are ATP–G-actin and ADP–F-actin. Explain how the interconversion of the nucleotide state is coupled to the assembly and disassembly of actin subunits. What would be the consequence for actin filament assembly/disassembly if a mutation prevented actin’s ability to bind ATP? What would be the consequence if a mutation prevented actin’s ability to hydrolyze ATP?
6. Actin filaments at the leading edge of a crawling cell are believed to undergo treadmilling. What is treadmilling, and what accounts for this assembly behavior? 7. Although purified actin can assemble reversibly in vitro, various actin-binding proteins regulate the assembly of actin filaments in the cell. Predict the effect on a cell’s actin cytoskeleton if function-blocking antibodies against each of the following were independently microinjected into cells: profilin, thymosin- , CapZ, and the complex. 8. Predict how actin would polymerize on a myosin-decorated short actin filament in the presence of CapZ, tropomodulin, or profilin-actin. 9. Compare and contrast the ways in which formin and WASp are activated and explain how each stimulates actin filament formation. 10. There are at least 20 different types of myosin. What properties do all types share, and what makes them different? Why is myosin II the only myosin capable of producing contractile force? 11. The ability of myosin to walk along an actin filament may be observed with the aid of an appropriately equipped microscope. Describe how such assays are typically performed. Why is ATP required in these assays? How can such assays be used to determine the direction of myosin movement or the force produced by myosin? 12. Contractile bundles occur in nonmuscle cells; these structures are less organized than the sarcomeres of muscle cells. What is
the purpose of nonmuscle contractile bundles? Which type of myosin is found in contractile bundles? 13. How does myosin convert the chemical energy released by ATP hydrolysis into mechanical work? 14. Myosin II has a duty ratio of 10 percent, and its step size is 8 nm. In contrast, myosin V has a much higher duty ratio (about 70 percent) and takes 36-nm steps as it walks down an actin filament. What differences between myosin II and myosin V account for their different properties? How do the different structures and properties of myosin II and myosin V reflect their different functions in cells? 15. Contraction of both skeletal and smooth muscle is triggered by an increase in cytosolic . Compare the mechanisms by which each type of muscle converts a rise in concentration into contraction. 16. Phosphorylation of myosin light-chain kinase (MLC kinase) by protein kinase A (PKA) inhibits MLC kinase activation by . Drugs such as albuterol bind to the β-adrenergic receptor, which causes a rise in cAMP in cells and activation of PKA. Explain why albuterol is useful for treating the severe contraction of the smooth muscle cells surrounding airway passages involved in an asthma attack. 17. Several types of cells use the actin cytoskeleton to power their locomotion across surfaces. How are different assemblies of actin filaments involved in locomotion? 18. To move in a specific direction, a migrating cell must use extracellular cues to establish which portion of the cell will act as the front and which will act as the back. Describe how small
GTPase proteins appear to be involved in the signaling pathways used by migrating cells to determine direction of movement. 19. Cell motility has been described as being like the motion of tank treads. At the leading edge, actin filaments form rapidly into bundles and networks that make protrusions and move the cell forward. At the rear, cell adhesions are broken and the tail end of the cell is brought forward. What provides the traction for moving cells? How does cell-body translocation happen? How are cell adhesions released as cells move forward?