Chapter 7: Thermochemistry

Chapter 7: Thermochemistry
SCIENCE MASTERY ASSESSMENT
Every pre-med knows this feeling: there is so much content I have to know for the MCAT! How do I know what to do first or what's important?
While the high-yield badges throughout this book will help you identify the most important topics, this Science Mastery Assessment is another tool in your MCAT prep arsenal. This quiz (which can also be taken in your online resources) and the guidance below will help ensure that you are spending the appropriate amount of time on this chapter based on your personal strengths and weaknesses. Don't worry though— skipping something now does not mean you'll never study it. Later on in your prep, as you complete full-length tests, you'll uncover specific pieces of content that you need to review and can come back to these chapters as appropriate.
How to Use This Assessment
If you answer 0–7 questions correctly:
Spend about 1 hour to read this chapter in full and take limited notes throughout. Follow up by reviewing all quiz questions to ensure that you now understand how to solve each one.
If you answer 8–11 questions correctly:
Spend 20–40 minutes reviewing the quiz questions. Beginning with the questions you missed, read and take notes on the corresponding subchapters. For questions you answered correctly, ensure your thinking matches that of the explanation and you understand why each choice was correct or incorrect.
If you answer 12–15 questions correctly:
Spend less than 20 minutes reviewing all questions from the quiz. If you missed any, then include a quick read-through of the corresponding subchapters, or even just the relevant content within a subchapter, as part of your question review. For questions you answered correctly, ensure your thinking matches that of the explanation and review the Concept Summary at the end of the chapter.
- Consider the cooling of an ideal gas in a closed system. This process is illustrated in the pressure–volume graph shown in the following figure.
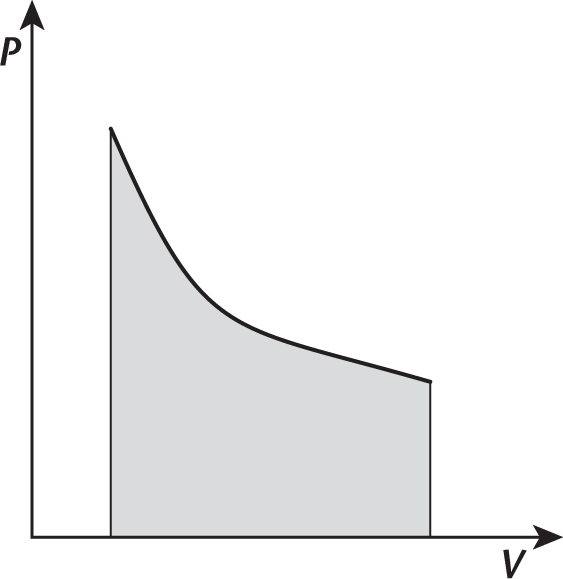
Based on this information, the process may be:
- adiabatic.
- isobaric.
- isothermal.
- isochoric.
-
A pot of water at 100 °C sits on a heating element and boils. Which of the following best characterizes this phase change process?
- Isothermal expansion
- Adiabatic expansion
- Isovolumetric heating
- Adiabatic compression
-
Pure sodium metal spontaneously combusts upon contact with room temperature water. What is true about the equilibrium constant of this combustion reaction at 25 °C?
- Keq < 0
- 0 < Keq < 1
- Keq = 1
- Keq > 1
-
Which of the following processes has the most exothermic standard heat of combustion?
- Combustion of ethane
- Combustion of propane
- Combustion of n-butane
- Combustion of n-pentane
-
Methanol reacts with acetic acid to form methyl acetate and water.
Type of Bond Bond Dissociation Energy ( kJ mol )
C – C 348
C – H 415
C = O 805
O – H 463
C – O 360
Based on the values in the table above, what is the heat of reaction in kJ mol ?
- 0
- 464
- 824
- 1288
-
Which of the following refers to the temperature and pressure at which all three phases exist in equilibrium?
- Critical point
- Triple point
- Deposition
- State function
-
A 10 g sample of water is brought from 40oC to boiling and is completely boiled to the gas phase. Which of the following expressions represents the total amount of energy required if the final temperature of the vapor is 100oC? (The heat of vaporization of water is 2,260 J/g.)
- (10 g)(40oC)(4.2 J/goC) + (2,260 J/g)(10 g)
- (10 g)(40oC)(4.2 J/goC) + (2,260 J/oC)(60oC)
- (10 g)(60oC)(4.2 J/goC) + (2,260 J/g)(10 g)
- (10 g)(60oC)(4.2 J/goC) + (2,260 J/oC)(60oC)
-
Consider the chemical reaction in the vessel depicted in the following diagram.
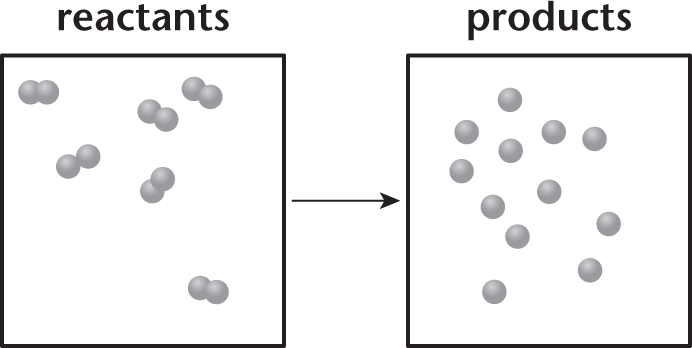
- The reaction is spontaneous.
- The reaction is nonspontaneous.
- The reaction is at equilibrium.
- There is not enough information to determine if the reaction is spontaneous.
-
Suppose Δ G rxn ∘ = − 2000 J mol for a chemical reaction. At 300 K, what is the change in Gibbs free energy in J mol ?
- ΔG = –2000 + (300 K)(8.314)(ln Q)
- ΔG = –2000 – (300 K)(8.314)(ln Q)
- ΔG = –2000 + (300 K)(8.314)(log Q)
- ΔG = –2000 – (300 K)(8.314)(log Q)
-
A chemical reaction has a negative enthalpy and a negative entropy. Which of the following terms necessarily describes this reaction?
- Exothermic
- Endothermic
- Exergonic
- Endergonic
-
Which of the following statements is true of a process that is spontaneous in the forward direction?
- ΔG > 0 and Keq > Q
- ΔG > 0 and Keq < Q
- ΔG < 0 and Keq > Q
- ΔG < 0 and Keq < Q
-
Which of the following reactions has the greatest decrease in entropy?
- 2 NH3 (g) → 3 H2 (g) + N2 (g)
- 2 Na (s) + Cl2 (g) → 2 NaCl (s)
- 2 H2O2 (l) → 2 H2O (l) + O2 (g)
- Zn (s) + CuSO4 (aq) → ZnSO4 (aq) + Cu (s)
-
A reaction coordinate for a chemical reaction is displayed in the graph below.
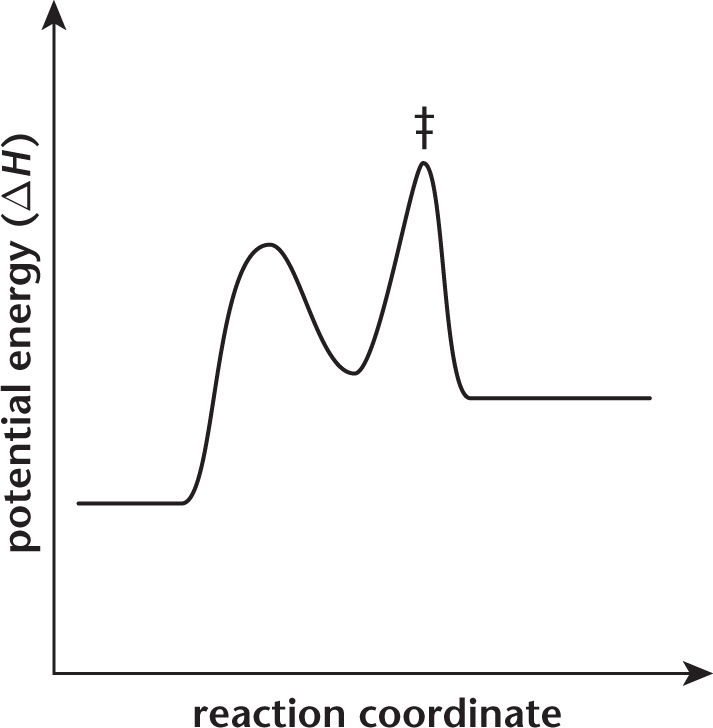
Which of the following terms describes the energy of this reaction?
- Endothermic
- Exothermic
- Endergonic
- Exergonic
-
The process of melting NaCl requires a large amount of energy due to the strong attraction between ions. A researcher finds that 58 g of salt melts at 1060 K and requires a heat input of 30,000 J. What is the change in entropy?
- 28 J/K
- 1,580 J/K
- 61,000 J/K
- Not enough information is provided
-
Explosions are necessarily characterized by:
- ΔG < 0.
- ΔH > 0.
- ΔS < 0.
- T < 0.
Answer Key
- A
- A
- D
- D
- A
- B
- C
- D
- A
- A
- C
- B
- A
- A
- A
Chapter 7: Thermochemistry
CHAPTER 7
THERMOCHEMISTRY
In This Chapter
7.1 Systems and Processes
Thermodynamic Terminology
7.2 States and State Functions
Overview
Phase Changes
Phase Diagrams
7.3 Heat
Overview
Constant-Pressure and Constant-Volume Calorimetry
Heating Curves
7.4 Enthalpy
Standard Heat of Formation
Standard Heat of Reaction
Hess’s Law
Bond Dissociation Energy
Standard Heat of Combustion
7.5 Entropy 7.6 Gibbs Free Energy
Overview
Standard Gibbs Free Energy
Free Energy, Keq, and Q
Concept Summary
CHAPTER PROFILE
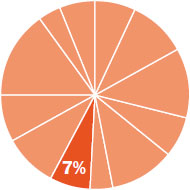
The content in this chapter should be relevant to about 7% of all questions about general chemistry on the MCAT.
This chapter covers material from the following AAMC content categories:
1D: Principles of bioenergetics and fuel molecule metabolism
5E: Principles of chemical thermodynamics and kinetics
Introduction
Styrofoam cups are such good insulators that they can be used as holding containers for certain calorimetry experiments. Coffee-cup calorimetry, which uses Styrofoam cups to measure heats of solution and specific heats of metals and other materials, is low-tech, yet it can produce remarkably accurate results as long as care has been taken to calibrate the calorimeter and to minimize heat loss through the top of the container. The next time you are at your favorite coffee chain, think about what occurs when cold cream is added to hot coffee. If we took the time to measure the masses and temperatures of the hot coffee and the cold cream before mixing them, measured the drink’s temperature after it had been stirred, and looked up the specific heats of water and cream, we would have enough information to calculate the amount of heat exchanged between the hot coffee and the cold cream.
This chapter will review the basic principles of thermochemistry, which is the study of the energy changes that accompany chemical and physical processes. Starting with the first law of thermodynamics, which states that energy is never created nor destroyed but—at most—simply changed from one form to another, we will quantify the various exchanges in energy as a system moves from some initial state to a final state. As we go along, we will define what is meant by system and surroundings, state functions, heat, enthalpy, entropy, and Gibbs free energy.
7.1 Systems and Processes
LEARNING OBJECTIVES
After Chapter 7.1, you will be able to:
- Identify the system and its surroundings given a situation involving transfer of heat
- Recall the features of isothermal, adiabatic, isobaric, and isovolumetric processes
Students often have some anxiety over what constitutes a system and what—by exclusion from the system—constitutes the surroundings or environment. Perhaps the problem isn’t so much the definitions themselves but the way in which the boundary between the two can be shifted to suit the needs of the experimenter or observer. Simply put, the system is the matter that is being observed—the total amount of reactants and products in a chemical reaction. It could be the amount of solute and solvent used to create a solution. It could be the gas inside a balloon. Then, the surroundings, or environment, are everything outside of that system. However, the boundary between system and surroundings is not permanently fixed and can be moved. For example, one might consider the mass of coffee in a coffee cup to be the system and the cup containing it to be part of the environment. This setup would likely be used if someone was interested in determining the amount of heat transferred from the hot coffee to the cooler coffee cup. Alternatively, one might define the system as the hot coffee and the cup together, and the environment as the air surrounding the coffee cup. This setup would likely be used if someone was interested in calculating the heat exchange between the hot coffee and cup system and the cooler surrounding air. The boundary can be extended out farther and farther, until the entire mass of the universe is ultimately included in the system. At this point, there are no surroundings. Again, where the boundary is placed is a decision based on what phenomenon one is interested in studying.
Thermodynamic Terminology
Systems can be characterized by whether or not they can exchange heat or matter with the surroundings. A system may be characterized as follows:
- Isolated: The system cannot exchange energy (heat and work) or matter with the surroundings; for example, an insulated bomb calorimeter.
- Closed: The system can exchange energy (heat and work) but not matter with the surroundings; for example, a steam radiator.
- Open: The system can exchange both energy (heat and work) and matter with the surroundings; for example, a pot of boiling water.
When a system experiences a change in one or more of its properties (such as concentrations of reactants or products, temperature, or pressure), it undergoes a process.
While processes, by definition, are associated with a change of the state of a system, some processes are uniquely identified by some property that is constant throughout the process. Many of these processes create special conditions because they allow us to simplify the first law of thermodynamics:
ΔU = Q – W
Equation 7.1
where ΔU is the change in internal energy of the system, Q is the heat added to the system, and W is the work done by the system.
For example, isothermal processes occur when the system’s temperature is constant. Constant temperature implies that the total internal energy of the system (U) is constant throughout the process. This is because temperature and internal energy are directly proportional. When U is constant, ΔU = 0 and the first law simplifies to Q = W (the heat added to the system equals the work done by the system). An isothermal process appears as a hyperbolic curve on a pressure–volume graph (P–V graph). Work is represented by the area under such a curve, as shown in Figure 7.1.
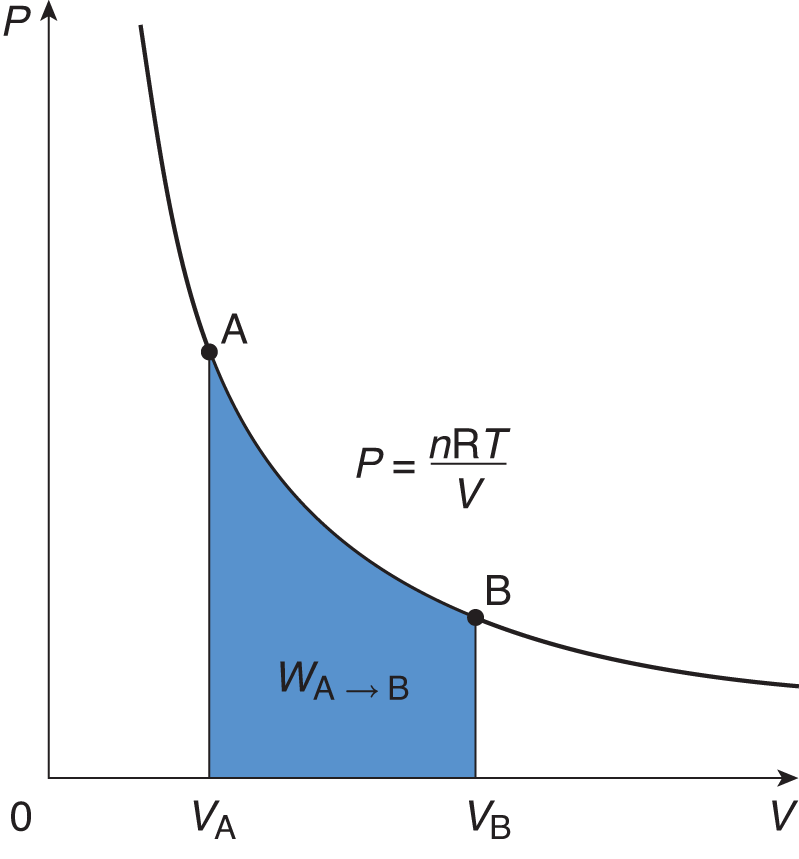
Figure 7.1. Graph of an Isothermal Expansion Temperature is constant in an isothermal process; thus, the area under the curve represents not only the work performed by the gas, but also the heat that entered the system.
Adiabatic processes occur when no heat is exchanged between the system and the environment; thus, the thermal energy of the system is constant throughout the process. When Q = 0, the first law simplifies to ΔU = –W (the change in internal energy of the system is equal to work done on the system [the opposite of work done by the system]). An adiabatic process also appears hyperbolic on a P–V graph, as shown in Figure 7.2.
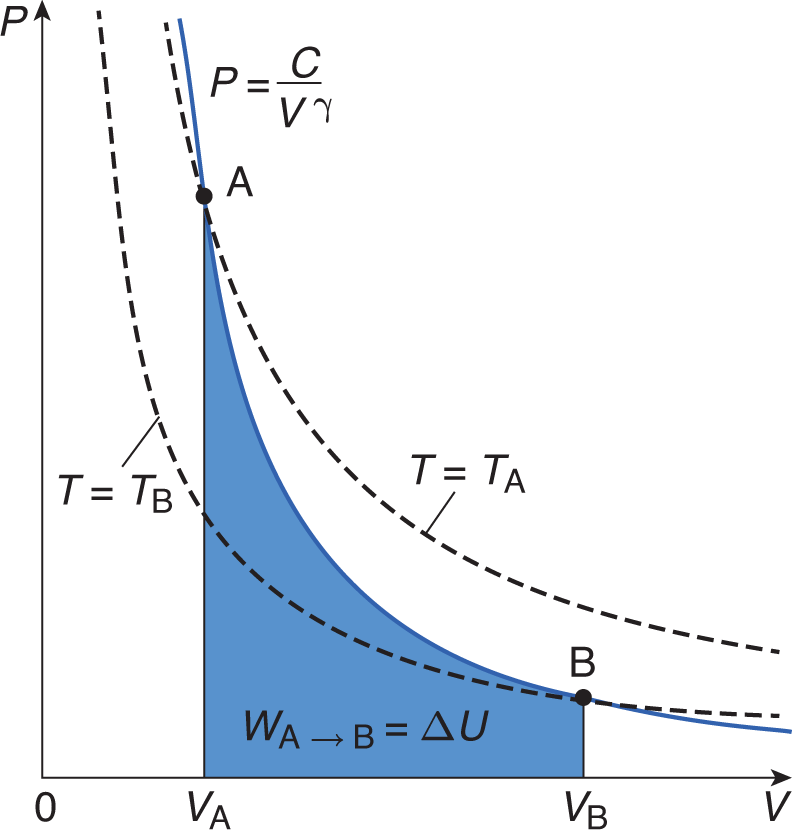
Figure 7.2. Graph of an Adiabatic Expansion Heat exchange is zero in an adiabatic process; temperature is not constant (as shown by the dotted lines).
Isobaric processes occur when the pressure of the system is constant. Isothermal and isobaric processes are common because it is usually easy to control temperature and pressure. Isobaric processes do not alter the first law, but note that an isobaric process appears as a flat line on a P–V graph, as shown in Figure 7.3.
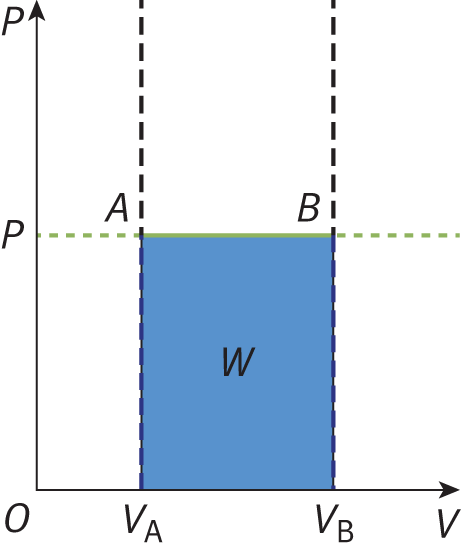
Figure 7.3. Graph of an Isobaric Expansion Pressure is constant in an isobaric process; the slope of the line is therefore zero.
Finally, isovolumetric (isochoric) processes experience no change in volume. Because the gas neither expands nor compresses, no work is performed in such a process. Thus, the first law simplifies to ΔU = Q (the change in internal energy is equal to the heat added to the system). An isovolumetric process is a vertical line on a P–V graph; the area under the curve, which represents the work done by the gas, is zero.
BRIDGE
The terms isothermal, adiabatic, isobaric, and isovolumetric (isochoric) may seem familiar because they are also discussed in Chapter 3 of MCAT Physics and Math Review.
Processes themselves can also be classified as spontaneous or nonspontaneous. A spontaneous process is one that can occur by itself without having to be driven by energy from an outside source. Calculating the change in the Gibbs free energy (ΔG) for a process, such as a chemical reaction, allows us to predict whether the process will be spontaneous or nonspontaneous. As discussed later in the chapter, the same quantities that are used to calculate the change in the Gibbs free energy, ΔH and ΔS, can also tell us whether the process will be temperature dependent; that is, spontaneous at some temperatures and nonspontaneous at others.
Spontaneous reactions, as mentioned in Chapters 5 and 6 of MCAT General Chemistry Review, will not necessarily happen quickly and may not go to completion. Many spontaneous reactions have very high activation energies and, therefore, rarely take place. For example, when was the last time you saw a match ignite itself? However, providing a quantity of thermal energy (generated by the friction associated with striking the match) that equals or exceeds the activation energy will allow the match to light and burn spontaneously. At this point, the combustion of the chemical components of the match using molecular oxygen in the air will not need any additional external energy once the activation energy has been supplied.
Some spontaneous reactions proceed very slowly. The role of enzymes—biological catalysts—is to selectively enhance the rate of certain spontaneous (but slow) chemical reactions so that the biologically necessary products can be formed at a rate sufficient for sustaining life. As we discussed in Chapter 6 of MCAT General Chemistry Review, some reactions do not go to completion but settle into a low-energy state called equilibrium. Spontaneous reactions may go to completion, but many simply reach equilibrium with dynamically stable concentrations of reactants and products. A common method for supplying energy for nonspontaneous reactions is by coupling nonspontaneous reactions to spontaneous ones, as shown in Figure 7.4.
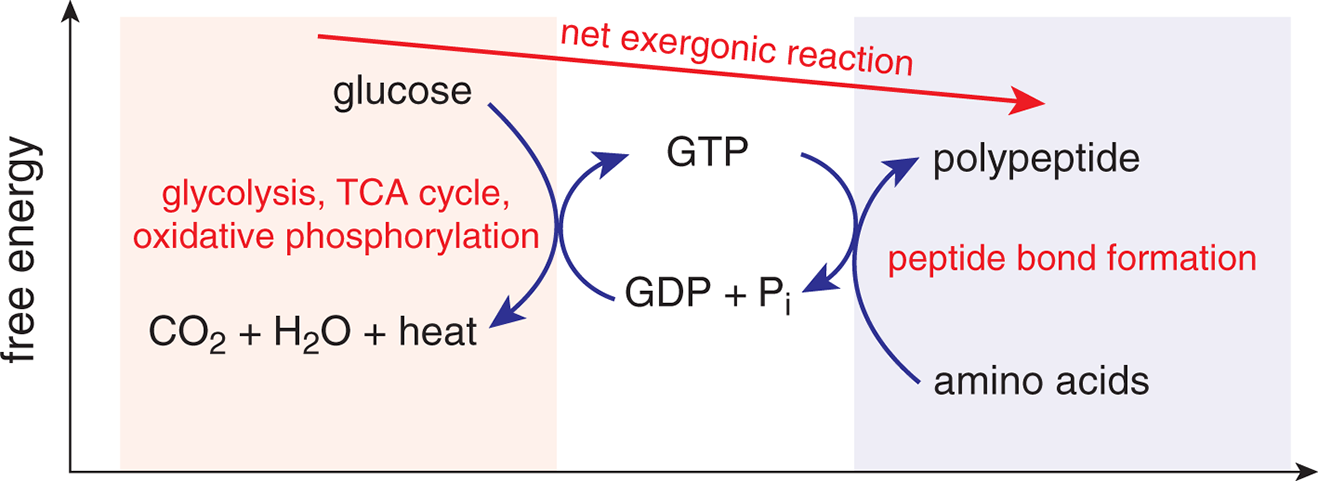
Figure 7.4. Coupling of Reactions The combustion of glucose is exergonic; the formation of peptide bonds is endergonic. Energy from the combustion of glucose can be stored in the bonds in GTP, which are then lysed to provide the energy for forming peptide bonds.
MCAT CONCEPT CHECK 7.1:
Before you move on, assess your understanding of the material with these questions.
-
A person snaps an ice pack and places it on one leg. In terms of energy transfer, what would be considered the system and what would be the surroundings in this scenario?
- System: ________________________________
- Surroundings: __________________________
-
What is unique about each of the following types of processes?
- Isothermal: ________________________________
- Adiabatic: ________________________________
- Isobaric: ________________________________
- Isovolumetric (isochoric): ________________________________
7.2 States and State Functions
LEARNING OBJECTIVES
After Chapter 7.2, you will be able to:
- Recall standard conditions and the calculations they are used for
- Distinguish between a state function and a process function
- List the common state functions
- Identify the triple point and critical point on a phase diagram:
The state of a system can be described by certain macroscopic properties. These properties, or state functions, describe the system in an equilibrium state. They cannot describe the process of the system; that is, how the system got to its current equilibrium. They are useful only for comparing one equilibrium state to another. The pathway taken from one equilibrium state to another is described quantitatively by the process functions, the most important of which are work (W) and heat (Q).
Overview
The state functions include pressure (P), density (ρ), temperature (T), volume (V), enthalpy (H), internal energy (U), Gibbs free energy (G), and entropy (S). When the state of a system changes from one equilibrium to another, one or more of these state functions will change. In addition, while state functions are independent of the path (process) taken, they are not necessarily independent of one another. For example, Gibbs free energy is related to enthalpy, temperature, and entropy.
MNEMONIC
State functions: When I’m under pressure and feeling dense, all I want to do is watch TV and get HUGS.
Pressure (P), density (ρ), temperature (T), volume (V), enthalpy (H), internal energy (U), Gibbs free energy (G), and entropy (S).
Because systems can be in different equilibrium states at different temperatures and pressures, a set of standard conditions has been defined for measuring the enthalpy, entropy, and Gibbs free energy changes of a reaction. The standard conditions are defined as 25 °C (298 K), 1 atm pressure, and 1 M concentrations. Don’t confuse standard conditions with standard temperature and pressure (STP), for which the temperature is 0 °C (273 K) and pressure is 1 atm. Standard conditions are used for kinetics, equilibrium, and thermodynamics problems; STP is used for ideal gas calculations.
MCAT EXPERTISE
On the MCAT, be sure that you do not confuse standard conditions in thermodynamics with standard temperature and pressure (STP), which is used in gas law calculations:
- Standard conditions: 25 °C (298 K), 1 atm pressure, 1 M concentrations
- STP: 0 °C (273 K), 1 atm pressure
Under standard conditions, the most stable form of a substance is called the standard state of that substance. You should recognize the standard states for some elements and compounds commonly encountered on the MCAT. For example, H2 (g), H2O (l), NaCl (s), O2 (g), and C (s,graphite) are the most stable forms of these substances under standard conditions. Recognizing whether or not a substance is in its standard state is important for thermochemical calculations, such as heats of reactions and—in particular—heats of formation. The changes in enthalpy, entropy, and free energy that occur when a reaction takes place under standard conditions are called the standard enthalpy, standard entropy, and standard free energychanges, respectively, and are symbolized by ΔH°, ΔS°, and ΔG°. The degree sign in these variables represents zero, as the standard state is used as the “zero point” for all thermodynamic calculations.
Phase Changes
Phase diagrams are graphs that show the standard and nonstandard states of matter for a given substance in an isolated system, as determined by temperatures and pressures. Phase changes (solid ⇌ liquid ⇌ gas) are reversible, and an equilibrium of phases will eventually be reached at any given combination of temperature and pressure. For example, at 0 °C and 1 atm in an isolated system, ice and water exist in an equilibrium. In other words, some of the ice may absorb heat (from the liquid water) and melt, but because that heat is being removed from the liquid water, an equal amount of the liquid water will freeze and form ice. Thus, the relative amounts of ice and water remain constant. Equilibrium between the liquid and gas states of water will be established in a closed container at room temperature and atmospheric pressure, such as a plastic water bottle with the cap screwed on tightly. Most of the water in the bottle will be in the liquid phase, but a small number of molecules at the surface will gain enough kinetic energy to escape into the gas phase; likewise, a small number of gas molecules will lose sufficient kinetic energy to reenter the liquid phase. After a while, equilibrium is established, and the relative amounts of water in the liquid and gas phases become constant—at standard conditions, equilibrium occurs when the air above the water has about 3 percent water vapor by mass. Phase equilibria are analogous to the dynamic equilibria of reversible chemical reactions: the concentrations of reactants and products are constant because the rates of the forward and reverse reactions are equal.
KEY CONCEPT
As with all equilibria, the rates of the forward and reverse processes will be the same when considering phase changes.
Gas–Liquid Equilibrium
The temperature of any substance in any phase is related to the average kinetic energy of the molecules that make up the substance. However, not all of the molecules have exactly the same instantaneous speeds. Therefore, the molecules possess a range of instantaneous kinetic energy values. In the liquid phase, the molecules are relatively free to move around one another. Some of the molecules near the surface of the liquid may have enough kinetic energy to leave the liquid phase and escape into the gaseous phase. This process is known as evaporation or vaporization. Each time the liquid loses a high-energy particle, the temperature of the remaining liquid decreases. Evaporation is an endothermic process for which the heat source is the liquid water. Of course, the liquid water itself may be receiving thermal energy from some other source, as in the case of a puddle of water drying up under the hot summer sun or a pot of water on the stovetop. Given enough energy, the liquid will completely evaporate.
Boiling is a specific type of vaporization that occurs only under certain conditions. Any liquid will lose some particles to the vapor phase over time; however, boiling is the rapid bubbling of the entire liquid with rapid release of the liquid as gas particles. While evaporation happens in all liquids at all temperatures, boiling can only occur above the boiling point of a liquid and involves vaporization through the entire volume of the liquid.
In a covered or closed container, the escaping molecules are trapped above the solution. These molecules exert a countering pressure, which forces some of the gas back into the liquid phase; this process is called condensation. Condensation is facilitated by lower temperature or higher pressure. Atmospheric pressure acts on a liquid in a manner similar to that of an actual physical lid. As evaporation and condensation proceed, the respective rates of the two processes become equal, and equilibrium is reached. The pressure that the gas exerts over the liquid at equilibrium is the vapor pressure of the liquid. Vapor pressure increases as temperature increases because more molecules have sufficient kinetic energy to escape into the gas phase. The temperature at which the vapor pressure of the liquid equals the ambient (also known as external, applied, or incident) pressure is called the boiling point.
Liquid–Solid Equilibrium
We’ve already illustrated the equilibrium that can exist between the liquid and the solid phases of water at 0 °C. Even though the atoms or molecules of a solid are confined to specific locations, each atom or molecule can undergo motions about some equilibrium position. These vibrational motions increase when heat is applied. From our understanding of entropy, we can say that the availability of energy microstates increases as the temperature of the solid increases. In basic terms, this means that the molecules have greater freedom of movement, and energy disperses. If atoms or molecules in the solid phase absorb enough energy, the three-dimensional structure of the solid will break down, and the atoms or molecules will escape into the liquid phase. The transition from solid to liquid is called fusion or melting. The reverse process, from liquid to solid, is called solidification, crystallization, or freezing. The temperature at which these processes occur is called the melting point or freezing point, depending on the direction of the transition. Whereas pure crystalline solids have distinct, very precise melting points, amorphous solids, such as glass, plastic, chocolate, and candle wax, tend to melt (or solidify) over a larger range of temperatures due to their less-ordered molecular structure.
Gas–Solid Equilibrium
The final phase equilibrium is that which exists between the gaseous and solid phases. When a solid goes directly into the gas phase, the process is called sublimation. Dry ice (solid CO2) sublimes at room temperature and atmospheric pressure; the absence of the liquid phase makes it a convenient dry refrigerant. The reverse transition, from the gaseous to the solid phase, is called deposition. In organic chemistry laboratories, a device known as a cold finger may be used to purify a product that is heated under reduced pressure, causing it to sublime. The desired product is usually more volatile than the impurities, so the gas is purer than the original product and the impurities are left in the solid state. The gas then deposits onto the cold finger, which has cold water flowing through it, yielding a purified solid product that can be collected.
Phase Diagrams
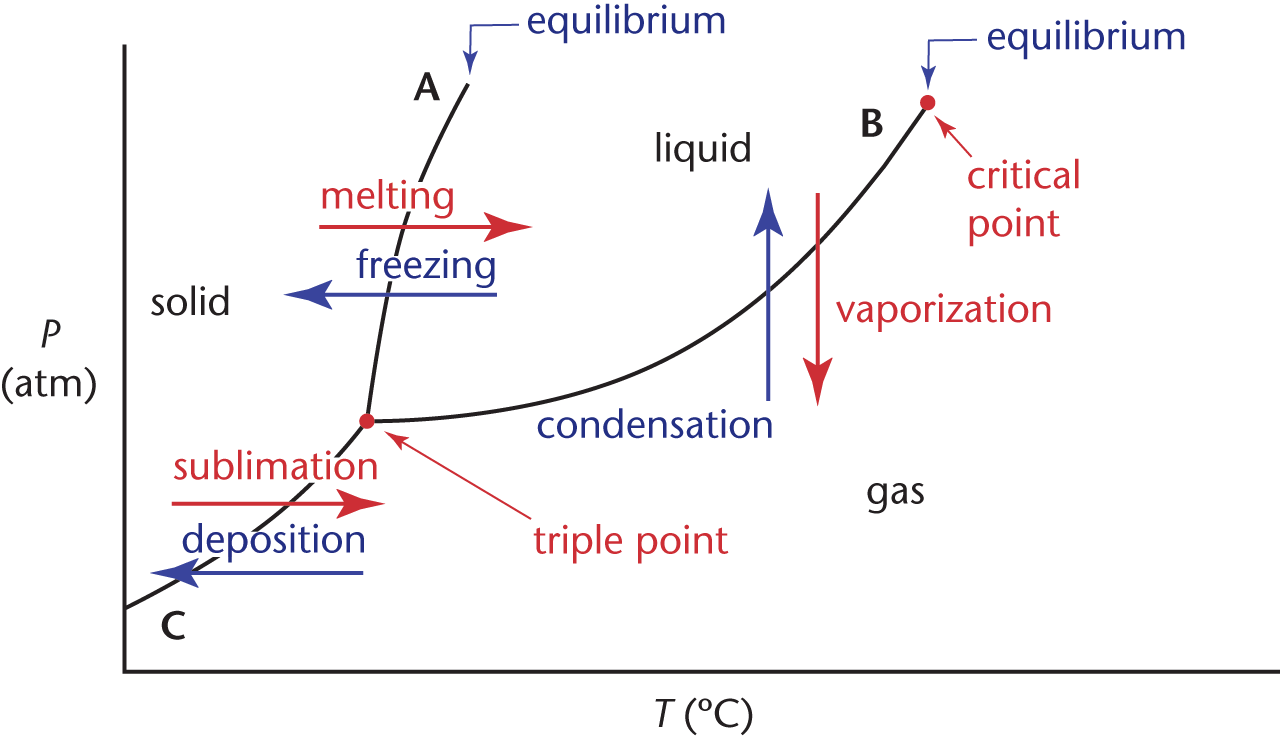
Phase diagrams are graphs that show the temperatures and pressures at which a substance will be thermodynamically stable in a particular phase. They also show the temperatures and pressures at which phases will be in equilibrium.
The lines on a phase diagram are called the lines of equilibrium or the phase boundaries and indicate the temperature and pressure values for the equilibria between phases. The lines of equilibrium divide the diagram into three regions corresponding to the three phases—solid, liquid, and gas—and they themselves represent the phase transformations. The phase diagram for a single compound is shown in Figure 7.5.
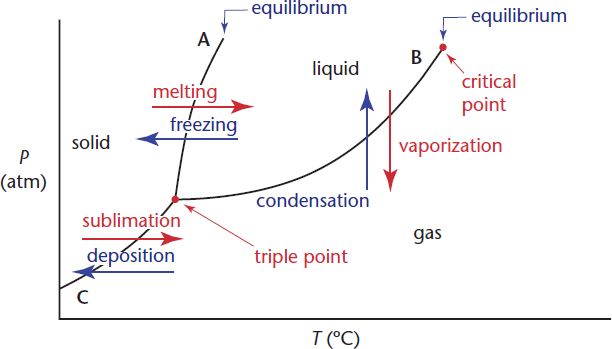
Figure 7.5. Phase Diagram for a Single Compound
MCAT EXPERTISE
On the MCAT, you should be able to identify and understand each area and every line of a phase diagram.
Line A represents the solid–liquid interface, line B the liquid–gas interface, and line C the solid–gas interface. In general, the gas phase is found at high temperatures and low pressures, the solid phase is found at low temperatures and high pressures, and the liquid phase is found at moderate temperatures and moderate pressures. The point at which the three phase boundaries meet is called the triple point. This is the temperature and pressure at which the three phases exist in equilibrium. The phase boundary that separates the solid and the liquid phases extends indefinitely from the triple point. The phase boundary between the liquid and gas phases, however, terminates at a point called the critical point. This is the temperature and pressure above which there is no distinction between the phases. Although this may seem to be an impossibility—after all, it’s always possible to distinguish between the liquid and the solid phase—such supercritical fluids are perfectly logical. As a liquid is heated in a closed system its density decreases and the density of the vapor sitting above it increases. The critical point is the temperature and pressure at which the two densities become equal and there is no distinction between the two phases. The heat of vaporization at this point and for all temperatures and pressures above the critical point values is zero.
REAL WORLD
Because of water’s unique properties, ice floats and skates flow smoothly over ice rinks. This all “boils” down to the negative slope of the solid–liquid equilibrium line in its phase diagram. Because the density of ice is less than that of liquid water, an increase in pressure (at a constant temperature) will actually melt ice (the opposite of what is seen for the substance in Figure 7.5).
MCAT CONCEPT CHECK 7.2:
Before you move on, assess your understanding of the material with these questions.
-
What are standard conditions? When are standard conditions used for calculations?
_____________________________
-
What is the definition of a state function? A process function?
- State function: ________________________________
- Process function: _______________________________
-
List at least five common state functions:
- ________________________________
- ________________________________
- ________________________________
- ________________________________
- ________________________________
-
Identify the triple point and critical point on the diagram below. What is the definition of the triple point? The critical point?
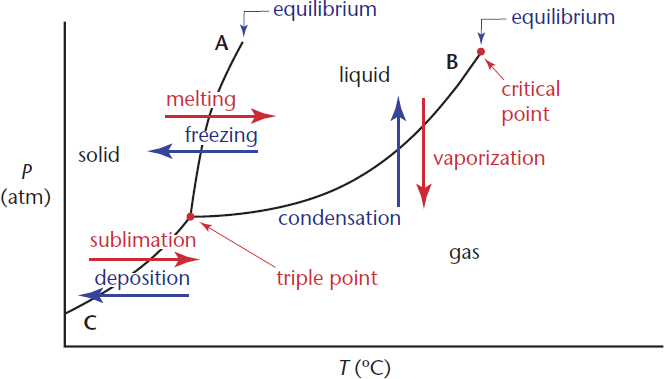
- Triple point: ________________________________
- Critical point: ______________________________
7.3 Heat
LEARNING OBJECTIVES
After Chapter 7.3, you will be able to:
- Differentiate between temperature and heat
- Compare specific heat and heat capacity
- Recall the specific heat of water
- Describe the processes for constant-volume and constant-pressure calorimetry:
Before we can examine the first of the four state functions that are the focus of this chapter, we must address the topic of heat, which is a source of confusion for many students. Perhaps the greatest barrier to a proper understanding of heat is the semantic conflation of the terms heat and temperature. Many people use these terms interchangeably in everyday conversation, but this obscures the lexicon of thermodynamics. Temperature (T) is related to the average kinetic energy of the particles of a substance. Temperature is the way that we scale how hot or cold something is. We are familiar with a few temperature scales: Fahrenheit, Celsius, and Kelvin. The average kinetic energy of the particles in a substance is related to the thermal energy (enthalpy) of the substance, but because we must also include consideration of how much substance is present to calculate total thermal energy content, the most we can say about temperature is that when a substance’s thermal energy increases, its temperature also increases. Nevertheless, we cannot say that something that is hot necessarily has greater thermal energy (in absolute terms) than a substance that is cold. For example, we might determine that a large amount of lukewarm water has a greater total heat content than a very small amount of hot water.
KEY CONCEPT
Remember that heat and temperature are different. Heat is a specific form of energy that can enter or leave a system, while temperature is a measure of the average kinetic energy of the particles in a system.
The absolute temperature scale, Kelvin, was determined via the third law of thermodynamics, which elucidated that there is a finite limit to temperature below which nothing can exist. There can be no temperature below 0 K because, by definition, the system is said to be unable to lose any more heat energy. Quantum mechanics describes a state of molecular motions possible below absolute zero, but this is beyond the scope of the MCAT.
Overview
Heat (Q) is the transfer of energy from one substance to another as a result of their differences in temperature. In fact, the zeroth law of thermodynamics implies that objects are in thermal equilibrium only when their temperatures are equal. Heat is therefore a process function, not a state function: we can quantify how much thermal energy is transferred between two or more objects as a result of their difference in temperatures by measuring the heat transferred.
Remember that the first law of thermodynamics states that the change in the total internal energy (ΔU) of a system is equal to the amount of heat (Q) transferred to the system minus the amount of work (W) done by the system: ΔU = Q − W.
Because heat and work are measured independently, we can assess the transfer of energy in the form of heat through any process regardless of the amount of work done. Processes in which the system absorbs heat are called endothermic (ΔQ > 0), while those processes in which the system releases heat are called exothermic (ΔQ < 0). The unit of heat is the unit of energy: joule (J) or calorie (cal), for which 1 cal = 4.184 J. Enthalpy (ΔH) is equivalent to heat (Q) under constant pressure, which is an assumption the MCAT usually makes for thermodynamics problems.
REAL WORLD
One of the most important ways that the body works to prevent overheating is through the production of sweat—an exocrine secretion of water, electrolytes, and urea. However, it is not the production of sweat that is the cooling mechanism. It’s the evaporation of the sweat that helps cool the body. Evaporation (vaporization) from the liquid to gas phase is an endothermic process: energy must be absorbed from the body for the particles of the liquid to gain enough kinetic energy to escape into the gas phase. Hot, arid desert air has a lower partial pressure of water vapor than humid, tropical air, so sweat vaporizes more readily in the dry air than it does in the humid air. Accordingly, most people will feel more comfortable in dry heat than in humid heat.
When substances of different temperatures are brought into thermal contact with each other—that is, some physical arrangement that allows heat transfer—energy will move from the warmer substance to the cooler substance. When a substance undergoes an endothermic or exothermic reaction, heat energy will be exchanged between the system and the environment.
The process of measuring transferred heat is called calorimetry. Two basic types of calorimetry include constant-pressure calorimetry and constant-volume calorimetry. The coffee-cup calorimeter, introduced at the beginning of this chapter, is a low-tech example of a constant-pressure calorimeter, while a bomb calorimeter is an example of a constant-volume calorimeter.
MNEMONIC
The equation for heat transfer, given a specific heat, is the same as the test you’re studying for! q = mcΔT looks a lot like “q equals MCAT.”
The heat (q) absorbed or released in a given process is calculated via the equation:
q = mcΔT
Equation 7.2
where m is the mass, c is the specific heat of the substance, and ΔT is the change in temperature (in kelvin or degrees Celsius). Specific heat is defined as the amount of energy required to raise the temperature of one gram of a substance by one degree Celsius (or one kelvin). Specific heat values will generally be provided on Test Day, but one constant to remember is the specific heat of H 2 O ( l ) : c H 2 O = 1 cal g ⋅ K .
REAL WORLD
When walking barefoot, a blacktop feels much hotter than a wooden walkway even when they are the same temperature. This is because they have different specific heats.
It requires less heat to raise the temperature of a glass of water the same amount as a swimming pool. While these two items have the same specific heat, c, they have different heat capacities—the product mc (mass times specific heat).
Constant-Pressure and Constant-Volume Calorimetry
To picture the setup of a constant-pressure calorimeter, just think of the coffee-cup calorimeter: an insulated container covered with a lid and filled with a solution in which a reaction or some physical process, such as dissolution, is occurring. The incident pressure, which is atmospheric pressure, remains constant throughout the process, and the temperature can be measured as the reaction progresses. There should be sufficient thermal insulation (such as Styrofoam) to ensure that the heat being measured is an accurate representation of the reaction, without gain or loss of heat to the environment. Other commercial applications of these same principles include home insulation, padded clothing, and certain food containers such as thermoses.
REAL WORLD
Tests looking at plasma proteins or cancer diagnostics in medicine have utilized differential scanning calorimetry (DSC), which is a constant-pressure device, to identify blood components. Such results have shown that thermal properties of major plasma proteins are altered from early- to late-stage tumors.
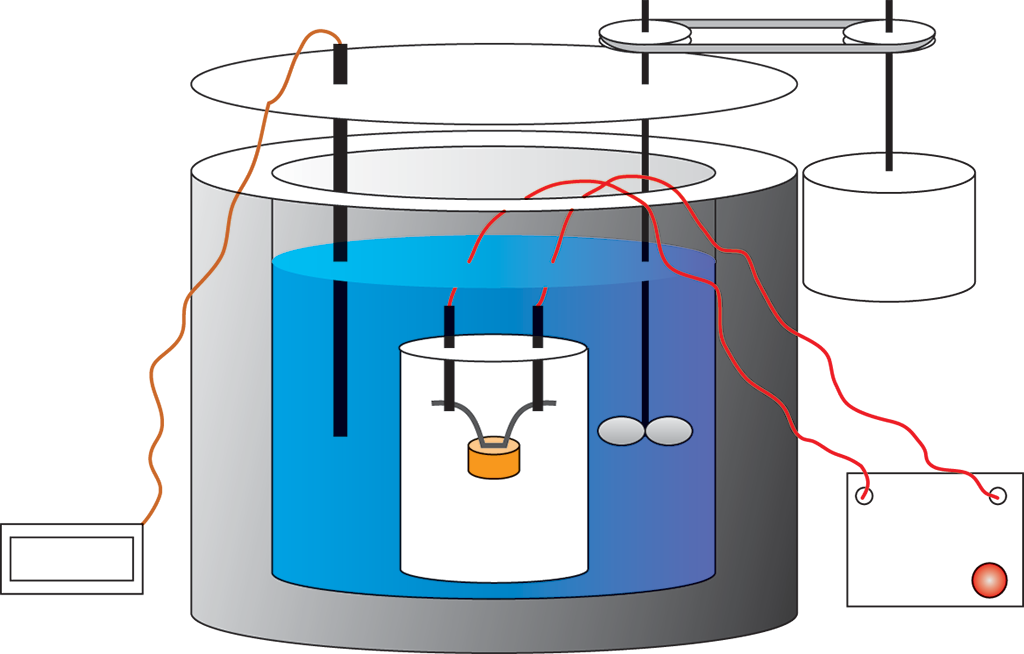
The term bomb calorimeter may sound rather ominous, but a more accurate descriptive term is decomposition vessel. This better reflects what is actually taking place in constant-volume calorimetry. As shown in Figure 7.6, a sample of matter, typically a hydrocarbon, is placed in the steel decomposition vessel, which is then filled with almost pure oxygen gas. The decomposition vessel is then placed in an insulated container holding a known mass of water. The contents of the decomposition vessel are ignited by an electric ignition mechanism. The material combusts (burns) in the presence of the oxygen, and the heat that evolves is the heat of the combustion reaction. Because W = PΔV, no work is done in an isovolumetric process (ΔV = 0), so Wcalorimeter = 0. Furthermore, because of the insulation, the whole calorimeter can be considered isolated from the rest of the universe, so we can identify the system as the sample plus the oxygen and steel vessel, and the surroundings as the water.
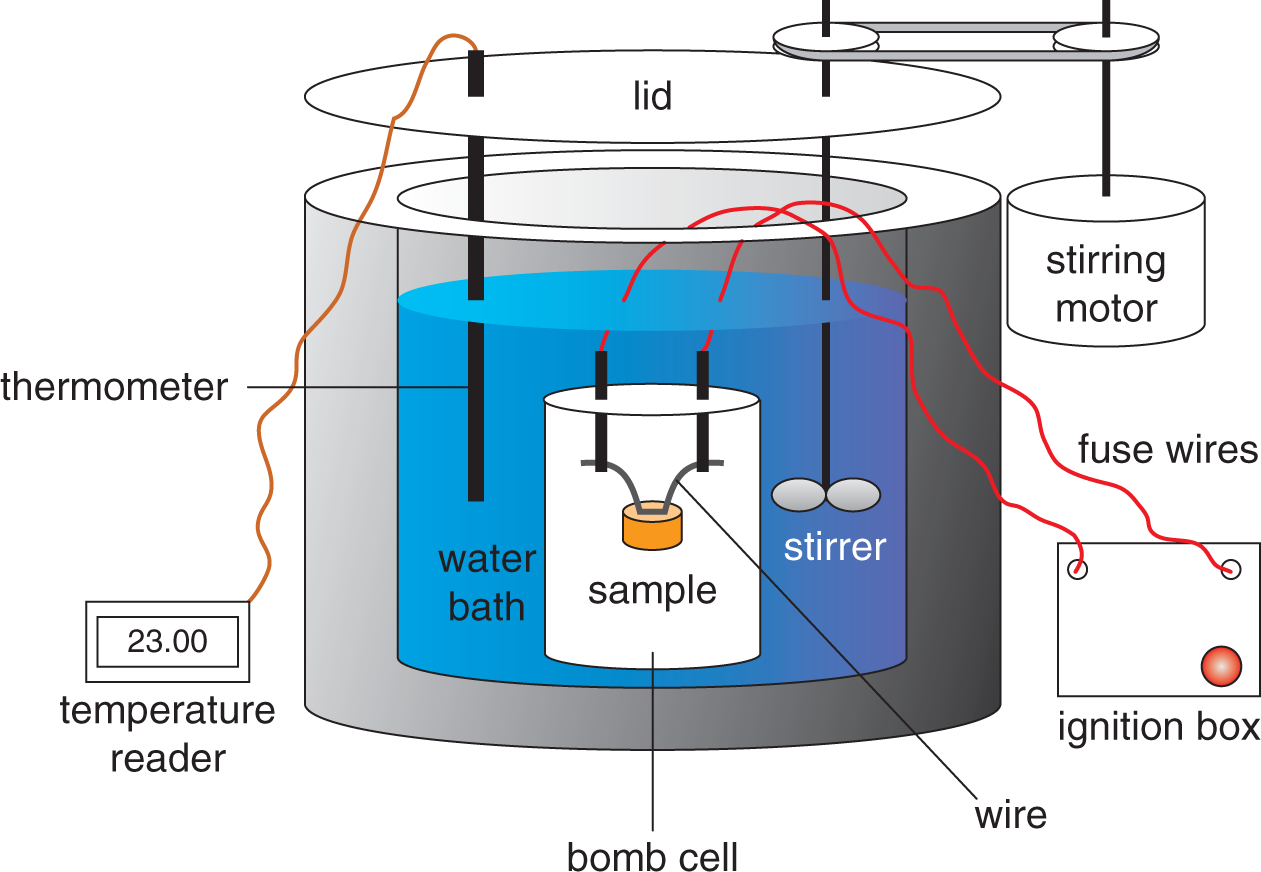
Figure 7.6. Diagram of a Bomb Calorimeter
REAL WORLD
Bomb calorimeters have helped elucidate the thermodynamic properties of various chemical compounds, including food additives, to determine nutritional value (the caloric content of the additive).
Because no heat is exchanged between the calorimeter and the rest of the universe, Qcalorimeter is 0. So,
ΔUsystem + ΔUsurroundings = ΔUcalorimeter = Qcalorimeter – Wcalorimeter = 0
Therefore,
ΔUsystem = –ΔUsurroundings
and because no work is done, q system = – q surroundings m steel c steel Δ T + m oxygen c oxygen Δ T = – m water c water Δ T
Note that by using the layer of insulation to isolate the entire calorimeter from the rest of the universe, we’ve created an adiabatic process. This means that no heat is exchanged between the calorimeter and the rest of the universe, but it is exchanged between the steel decomposition vessel and the surrounding water. As the previous derivation shows, heat exchange between the system and its surroundings makes it possible for us to calculate the heat of combustion.
MCAT EXPERTISE
Knowing that heat can transfer energy from a system to the surroundings is a key concept tested on calorimetry questions. Whenever you are asked about equilibrium questions regarding the final temperature of a two-liquid (or liquid–solid) system, remember that the colder object gains thermal energy and the hotter object loses it. You should instinctively realize that a metal bar at 1000 K is hotter than a bath of water at 298 K even though water has a high specific heat. Thus, set up the equation as qcold = –qhot. This form of the equation avoids the pesky sign notation issues in the ΔT equation encountered in most general chemistry texts.
Example: One cup containing 100 grams of water at 300 K is mixed into another cup containing 200 g of water at 450 K. What is the equilibrium temperature of the system? (Note: Assume that the pressure is sufficiently high to avoid boiling.)
Solution: The two liquids undergo thermal exchange; thus, the heat given off by one liquid will be equal to the heat absorbed by the other. q cold = − q hot m cold c H 2 O Δ T cold = m hot c H 2 O ( − Δ T hot )
Now plug in the values from the question. Because we are solving for final (equilibrium) temperature of a mixture, we can use any value of c so long as we are consistent for both liquids (in this case we have two quantities of water and will use c H 2 O = 1 cal g · K ). m cold c H 2 O Δ T cold = m hot c H 2 O ( − Δ T hot ) 100 g ( 1 cal g · K ) T f − 300 K = ( 200 g ) ( 1 cal g · K ) ( 450 K − T f ) 100 T f − 30 ,000 cal = 90,000 cal – 200 T f 300 T f = 120 ,000 T f = 120 ,000 300 = 400 K
Heating Curves
When a compound is heated, the temperature rises until the melting or boiling point is reached. Then, the temperature remains constant as the compound is converted to the next phase (liquid or gas, respectively). Once the entire sample is converted, then the temperature begins to rise again. This is depicted in the heating curves in Figure 7.7.
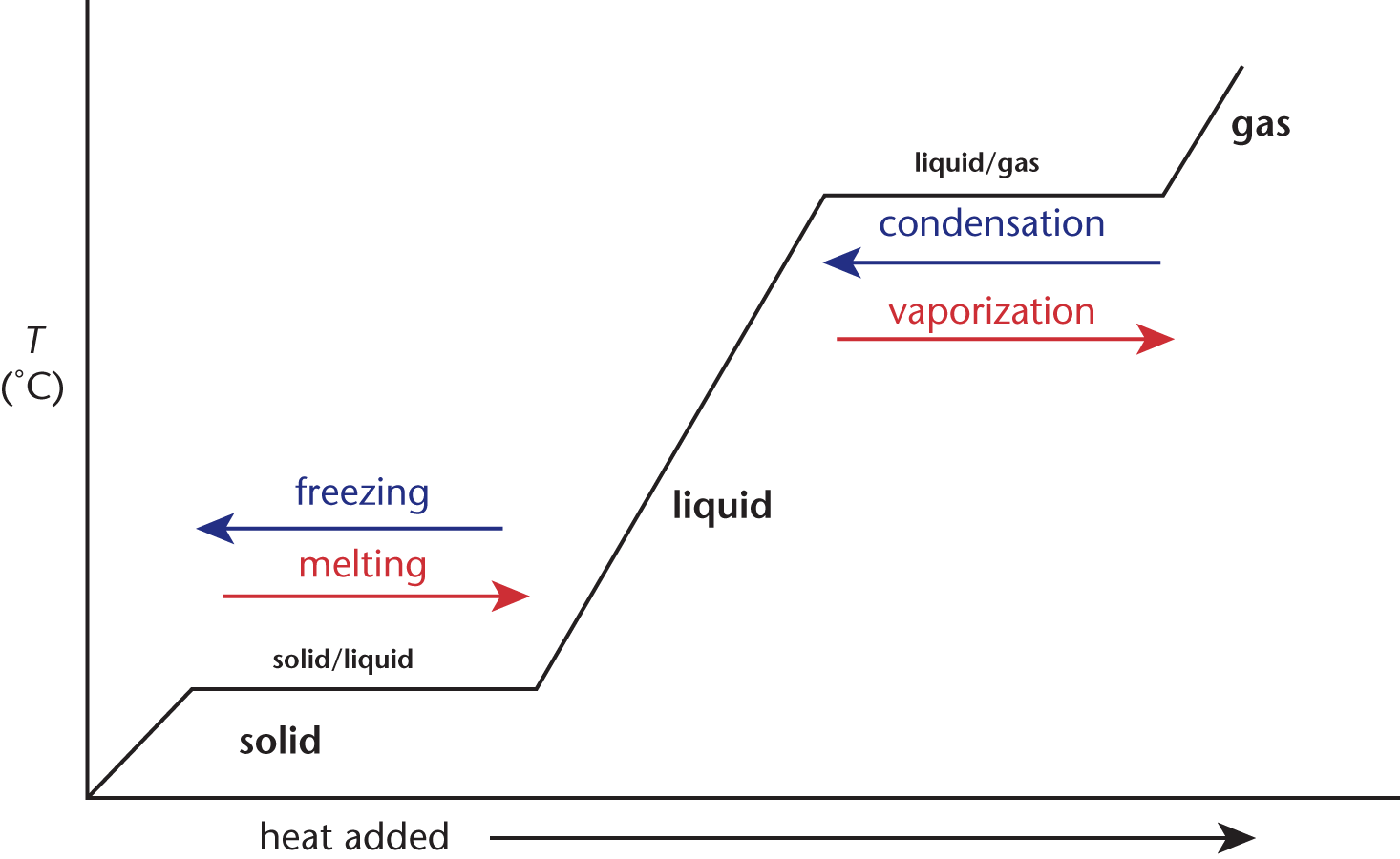
Figure 7.7. Heating Curve for a Single Compound
Heating curves show that phase change reactions do not undergo changes in temperature. For this reason, we cannot use q = mcΔT during this interval because ΔT = 0. We know intuitively that heat must continue to be added in order for the whole solid to melt, so where does this heat go? The solid absorbs energy, which allows particles to overcome the attractive forces that hold them in a rigid, three-dimensional arrangement. When melting an ice cube, all of the heat added during the process is used to overcome the intermolecular forces between water molecules in ice, forming liquid water. Once all of the ice has been turned into liquid water, the temperature of the liquid water can then increase again. The converse is also true: removing heat from a liquid at the solid–liquid phase transition temperature will cause the formation of a rigid lattice of water molecules.
During phase changes, we must use values based on enthalpy. When transitioning at the solid–liquid boundary, the enthalpy (or heat)of fusion (ΔHfus) must be used to determine the heat transferred during the phase change. When transitioning from solid to liquid, the change in enthalpy will be positive because heat must be added; when transitioning from a liquid to a solid, the change in enthalpy will be negative because heat must be removed. At the liquid–gas boundary, the enthalpy (or heat)of vaporization (ΔHvap) must be used, and its sign convention also follows a similar pattern. These are utilized in the equation
q = mL
Equation 7.3
where m is the mass and L is the latent heat, a general term for the enthalpy of an isothermal process, given in the units cal g .
KEY CONCEPT
We need a different formula to calculate q during phase changes when ΔT = 0. If we used q = mcΔT, we’d erroneously think q = 0.
The total amount of heat needed to cross multiple phase boundaries is simply a summation of the heats for changing the temperature of each of the respective phases and the heats associated with phase changes.
Example: What amount of energy is required to change a 90 gram ice cube at –10 °C to vapor at 110 °C? (Note: c H 2 O ( l ) = 4.18 J g · K , c H 2 O ( s ) = 2.18 J g · K , c H 2 O ( g ) = 2.00 J g · K , Δ H fus = 6.02 kJ mol , Δ H vap = 40.67 kJ mol )
Solution: Some of the constants given are in terms of mass (g), and some are in terms of moles, so we should convert the mass (90 g) to moles:
n = 90 g H 2 O 18 g mol = 10 2 = 5 moles H 2 O
Because we are beginning in the ice phase, we must heat the ice cube to the solid–liquid phase transition, which occurs at 0 °C. This first step involves a change in temperature, so we must use the heat formula that contains ΔT and all the pertinent variables for ice (solid water). Also, it is important to match all results in terms of J and kJ for the different steps of the calculation.
q 1 = m ice c ice Δ T 1 q 1 = 90 g ( 2.18 J g · K ) 10 K ≈ 90 × 2 × 10 = 1800 J = 1 . 8 kJ
In step 2, we must convert the ice into liquid form. During this phase change, there will be no temperature change.
q 2 = m L = n H 2 O Δ H fus q 2 = 5 mol ( 6.02 kJ mol ) ≈ 5 × 6 = 30 kJ
In step 3, we heat the water to its liquid–gas phase transition temperature at 100 °C.
q 3 = m water c water Δ T 3 q 3 = 90 g ( 4.18 J g · K ) 100 K ≈ 90 × 4 × 100 = 36 , 000 J = 36 kJ
In step 4, we vaporize the water. Again, no temperature change will occur during this phase change.
q 4 = m L = n H 2 O Δ H vap q 4 = 5 mol ( 40.67 kJ mol ) ≈ 5 × 40 = 200 kJ
In step 5, we must finally heat the water to the target temperature of 110 °C.
q 5 = m steam c steam Δ T 5 q 5 = [ 90 g ] ( 2.00 J g · K ) [ 10 K ] = 90 × 2 × 10 = 1800 J = 1 . 8 kJ
The total heat required for this whole phase change from beginning to end is:
q tot = q 1 + q 2 + q 3 + q 4 + q 5 q tot = 1 . 8 + 30 + 36 + 200 + 1 . 8 q tot ≈ 2 + 30 + 36 + 200 + 2 = 270 kJ (actual value =274.8 kJ)
A question thisinvolved is unlikely to be seen on the MCAT because so many steps must be calculated. However, understanding the significance and rationale of this calculation is definitely within the scope of the test.
MCAT EXPERTISE
It is not in your best interest to memorize all the possible values for c and ΔH for Test Day. The MCAT will provide constants as needed, especially if the system is not water. That being said, practicing with heat calculations for water solutions and gaining familiarity with the heat capacities of water will help on Test Day.
MCAT CONCEPT CHECK 7.3:
Before you move on, assess your understanding of the material with these questions.
-
Contrast temperature and heat.
- Temperature:__________________________
________________________________
- Heat:
-
Contrast specific heat and heat capacity.
________________________________
- Specific heat:
____________________________
- Heat capacity:
-
Contrast constant-volume and constant-pressure calorimetry.
________________________________
- Constant-volume:
____________________________
- Constant-pressure:
-
What is the specific heat of liquid water (in calories)?
7.4 Enthalpy
LEARNING OBJECTIVES
After Chapter 7.4, you will be able to:
- Distinguish between endothermic and exothermic reactions
- Determine the enthalpy of a molecule or atom given reaction data:
C ( s , graphite ) + 2 H 2 ( g ) → CH 4 ( g )
CH 4 ( g ) + 2 O 2 ( g ) → CO 2 ( g ) + 2 H 2 O ( l ) Δ H a = − 890.4 kJ mol C ( s , graphite ) + O 2 ( g ) → CO 2 ( g ) Δ H b = − 393.5 kJ mol 2 × [ H 2 ( g ) + 1 2 O 2 ( g ) → H 2 O ( l ) ] Δ H g = 2 × − 285.8 kJ mol
Most reactions in the laboratory occur under constant pressure (at 1 atm) in closed thermodynamic systems. To express heat changes at constant pressure, chemists use the term enthalpy (H). Enthalpy is a state function, so we can calculate the change in enthalpy (ΔH) for a system that has undergone a process—for example, a chemical reaction—by comparing the enthalpy of the final state to the enthalpy of the initial state, irrespective of the path taken. The change in enthalpy is equal to the heat transferred into or out of the system at constant pressure. To find the enthalpy change of a reaction, ΔHrxn, one must subtract the enthalpy of the reactants from the enthalpy of the products:
ΔHrxn = Hproducts – Hreactants
Equation 7.4
A positive ΔHrxn corresponds to an endothermic process, and a negative ΔHrxn corresponds to an exothermic process. It is not possible to measure enthalpy directly; only ΔH can be measured, and only for certain fast and spontaneous processes. Thus, several methods have been developed to calculate ΔH for any process.
Standard Heat of Formation
The standard enthalpy of formation of a compound, ΔH°f, is the enthalpy required to produce one mole of a compound from its elements in their standard states. Remember that standard state refers to the most stable physical state of an element or compound at 298 K and 1 atm. Note that ΔH°f of an element in its standard state, by definition, is zero. The ΔH°f values of most known substances are tabulated. You do not need to memorize these values because they will be provided for you.
Standard Heat of Reaction
The standard enthalpy of a reaction, ΔH°rxn, is the enthalpy change accompanying a reaction being carried out under standard conditions. This can be calculated by taking the difference between the sum of the standard heats of formation for the products and the sum of the standard heats of formation of the reactants:
ΔH°rxn = Σ ΔH°f,products − Σ ΔH°f,reactants
Equation 7.5
Hess’s Law
Enthalpy is a state function and is a property of the equilibrium state, so the pathway taken for a process is irrelevant to the change in enthalpy from one equilibrium state to another. As a consequence of this, Hess’s law states that enthalpy changes of reactions are additive. When thermochemical equations (chemical equations for which energy changes are known) are added to give the net equation for a reaction, the corresponding heats of reaction are also added to give the net heat of reaction, as shown in Figure 7.8.
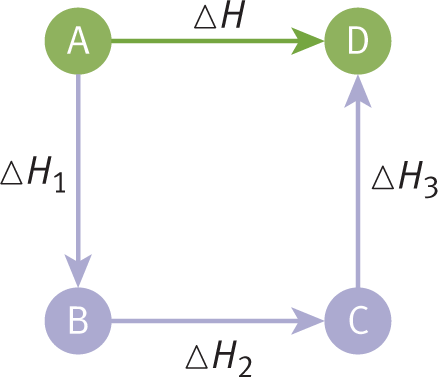
Figure 7.8. Illustration of Hess’s Law: Forming Product (D) from Reactant (A) Because enthalpy is a state function, ΔH= ΔH1 + ΔH2 + ΔH3
KEY CONCEPT
State functions are always path independent.
Hess’s law is embodied in the enthalpy equations we’ve already introduced. For example, we can describe any reaction as the result of breaking down the reactants into their component elements, then forming the products from these elements. The enthalpy change for the reverse of any reaction has the same magnitude, but the opposite sign, as the enthalpy change for the forward reaction. Therefore,
ΔHreactants → elements = –ΔHelements → reactants
The ΔHrxn can be written as:
ΔHrxn = ΔHreactants → elements + ΔHelements → products
which is another way of writing
ΔH°rxn = Σ ΔH°f,products − Σ ΔH°f,reactants
Consider the following phase change:
Br 2 ( l ) → Br 2 ( g ) Δ H rxn ° = 31 kJ mol
The enthalpy change for the phase change is called the heat of vaporization (ΔH°vap). As long as the initial and final states exist at standard conditions, the ΔH°rxn will always equal the ΔH°vap, irrespective of the particular pathway that the process takes. For example, it’s possible that Br2 (l) could first decompose to Br atoms, which then recombine to form Br2 (g), rather than simply boiling from the liquid to gaseous state. However, because the net reaction is the same, the change in enthalpy will be the same.
Example: Given the following thermochemical equations:
(a) CH 4 ( g ) + 2 O 2 ( g ) → CO 2 ( g ) + 2 H 2 O ( l ) Δ H a = − 890.4 kJ mol
(b) C ( s , graphite ) + O 2 ( g ) → CO 2 ( g ) Δ H b = − 393.5 kJ mol
(c) H 2 ( g ) + 1 2 O 2 ( g ) → H 2 O ( l ) Δ H c = − 285.8 kJ mol
Calculate ΔH for this reaction:
(d) C (s,graphite) + 2 H2 (g) → CH4 (g)
Solution: Equations (a), (b), and (c) must be combined to obtain equation (d). Because equation (d) contains only C, H2, and CH4, we must eliminate O2, CO2, and H2O from the first three equations. Equation (a) is reversed to move CH4 to the product side (equation (e) below). Next, equation (b) is left as is (we will call this equation (f) for consistency below) and (c) is multiplied by 2 (equation (g) below). Then, (d) can be calculated from (e) + (f) + (g):
(e) CO 2 ( g ) + 2 H 2 O ( l ) → CH 4 ( g ) + 2 O 2 ( g ) Δ H e = 890.4 kJ mol
(f) C ( s , graphite ) + O 2 ( g ) → CO 2 ( g ) Δ H f = − 393.5 kJ mol
(g) 2 × [ H 2 ( g ) + 1 2 O 2 ( g ) → H 2 O ( l ) ] Δ H g = 2 × − 285.8 kJ mol
C ( s , graphite ) + 2 H 2 ( g ) → CH 4 ( g ) Δ H d = Δ H e + Δ H f + Δ H g Δ H d = 890 . 4 + - 393 . 5 + 2 × - 285 . 8 ≈ 900 + - 400 - 2 × 285 Δ H d = 900 - 400 - 570 = - 70 kJ mol ( actual value = − 74.7 kJ mol )
It is important to realize that Hess’s law applies to any state function, including entropy and Gibbs free energy.
MCAT EXPERTISE
When doing a problem like this on the MCAT, make sure to switch signs when you reverse the equation. Also, make sure to multiply by the correct stoichiometric coefficients when performing your calculations.
Bond Dissociation Energy
Hess’s law can also be expressed in terms of bond enthalpies, also called bond dissociation energies. Bond dissociation energy is the average energy that is required to break a particular type of bond between atoms in the gas phase—remember, bond dissociation is an endothermic process. Bond dissociation energy is given in the units kJ mol of bonds broken and is often given in tables on the MCAT in a format similar to Table 7.1.
Table 7.1 Sample Bond Enthalpies
BOND ENTHALPY
(
ΔH,
kJ
mol
)
O=O 498
C–H 415
H–H 436
Bond enthalpies are the averages of the bond energies for the same bond in many different compounds. For example, the C–H bond enthalpy ( 415 kJ mol ) is averaged from measurements of the individual C–H bond enthalpies of thousands of different organic compounds. Note that bond formation, the opposite of bond breaking, has the same magnitude of energy but is negative rather than positive; that is, energy is released when bonds are formed. Remember that atoms generally form bonds to become more stable (often by completing an octet). Thus, it makes sense that bond formation is exothermic and bond dissociation is endothermic. The enthalpy change associated with a reaction is given by
ΔH°rxn = Σ ΔHbonds broken − Σ ΔHbonds formed = total energy absorbed − total energy released
Equation 7.6
KEY CONCEPT
Because it takes energy to pull two atoms apart, bond breakage is generally endothermic. The reverse process, bond formation, is generally exothermic.
Example: Calculate the enthalpy change for the following reaction:
C ( s ) + 2 H 2 ( g ) → CH 4 ( g ) Δ H = ?
Bond dissociation energies of H–H and C–H bonds are 436 kJ mol and 415 kJ mol , respectively. The ΔHf of C (g) is 715 kJ mol .
Solution: CH4 is formed from free elements in their standard states (C in solid state and H2 in gaseous state). Thus, here ΔHrxn = ΔHf. The reaction can be written in three steps:
- C ( s ) → C ( g ) Δ H 1
- 2 [ H 2 ( g ) → 2 H ( g ) ] 2 × Δ H 2
- C ( g ) + 4 H ( g ) → CH 4 ( g ) Δ H 3
with ΔHf = ΔH1 + (2 × ΔH2 )+ΔH3.
ΔH1 = ΔHf of C ( g ) = 715 kJ mol
ΔH2 is the energy required to break the H–H bond of one mole of H2, so ΔH2 = bond enthalpy of H 2 = 436 kJ mol . Note that reaction (b) is doubled in order to produce 4 atoms of H from two molecules of H2.
ΔH3 is the energy released when 4 C–H bonds are formed. Because energy is released when bonds are formed, ΔH3 is negative. Δ H 3 = − ( 4 × bond energy of C − H ) = − ( 4 × 415 kJ mol ) = - 1660 kJ mol
Therefore, for the entire reaction, Δ H rxn = Δ H f = 715 kJ mol + ( 2 × 436 kJ mol ) − 1660 kJ mol Δ H rxn ≈ 700 + 2 × 450 - 1660 = 700 + 900 - 1660 = - 60 kJ mol actual value = − 73 kJ mol
Standard Heat of Combustion
As the name implies, the standard heat of combustion, Δ***H*°comb**, is the enthalpy change associated with the combustion of a fuel. Because measurements of enthalpy change require a reaction to be spontaneous and fast, combustion reactions are the ideal processes for such measurements. Most combustion reactions presented on the MCAT occur in the presence of atmospheric oxygen, but keep in mind that there are other combustion reactions in which oxygen is not the oxidant. Diatomic fluorine, for example, can be used as an oxidant. In addition, hydrogen gas will combust with chlorine gas to form gaseous hydrochloric acid and, in the process, will evolve a large amount of heat and light as is characteristic of combustion reactions. The reactions listed in the CH4 (g) example shown earlier are combustion reactions with O2 (g) as the oxidant. Therefore, the enthalpy change listed for each of the three reactions is the ΔHcomb for each of the reactions.
The glycolytic pathway, described in Chapter 9 of MCAT Biochemistry Review, is also a combustion reaction that utilizes a fuel (glucose) mixed with an oxidant (oxygen) to produce carbon dioxide and water.
C6H12O6 + 6 O2 → 6 CO2 + 6 H2O
The heat of combustion for this reaction is found in a similar fashion to that of Hess’s Law. Given the numerous reactions and pathways involved, we can determine the overall enthalpy of the reaction, as shown in Figure 7.9.
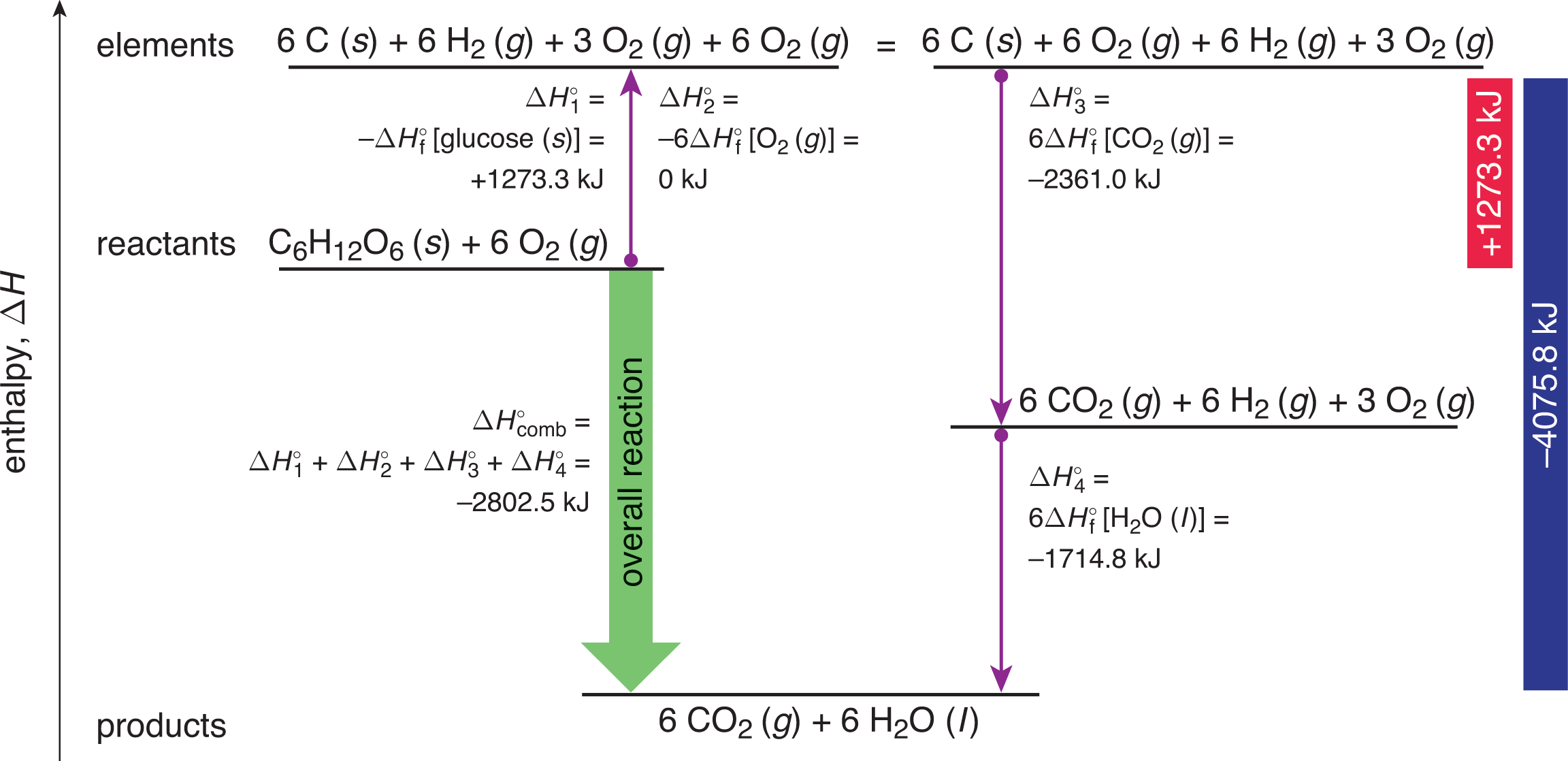
Figure 7.9. Determining the Enthalpy of Glycolysis
KEY CONCEPT
The larger the alkane reactant, the more numerous the combustion products.
MCAT CONCEPT CHECK 7.4:
Before you move on, assess your understanding of the material with these questions.
-
Define endothermic and exothermic processes.
________________________________
- Endothermic:
________________________________
- Exothermic:
-
Given the following reactions, determine the enthalpy of:
C (s,graphite) + 1 2 O 2 → CO . C ( s , graphite ) + O 2 → CO 2 Δ H = − 393.5 kJ mol CO + 1 2 O 2 → CO 2 Δ H = − 283 kJ mol
__________________________________
-
What is the enthalpy of reaction for the reaction: 2 H2O (g) → 2 H2 (g) + O2 (g), given the following bond enthalpies: H − H = 436 kJ mol , O = O = 498 kJ mol , O − O = 146 kJ mol , and O − H = 463 kJ mol
__________________________________
7.5 Entropy
LEARNING OBJECTIVES
After Chapter 7.5, you will be able to:
- Order the phases of matter from lowest to highest entropy
- Define entropy in terms of its relation to energy distribution and disorder
- Predict the direction of change in entropy within a given reaction
Many students are perplexed by the concept of entropy. Enthalpy makes intuitive sense, especially when the energy change from reactants to products is large, fast, and dramatic (as in combustion reactions involving explosions). Entropy seems to be less intuitive—except that it isn’t. Consider, for example, how “normal” each of the following seems: hot tea cools down, frozen drinks melt, iron rusts, buildings crumble, balloons deflate, living things die and decay, and so on.
These examples have a common denominator: in each of them, energy of some form is going from being localized or concentrated to being spread out or dispersed. The thermal energy in the hot tea is spreading out to the cooler air that surrounds it. The thermal energy in the warmer air is spreading out to the cooler frozen drink. The chemical energy in the bonds of elemental iron and oxygen is released and dispersed as a result of the formation of the more stable, lower-energy bonds of iron oxide (rust). The potential energy of the building is released and dispersed in the form of light, sound, and heat as the building crumbles and falls. The energy of the pressurized air is released to the surrounding atmosphere as the balloon deflates. The chemical energy of all the molecules and atoms in living flesh is released into the environment during the process of death and decay.
The second law of thermodynamics states that energy spontaneously disperses from being localized to becoming spread out if it is not hindered from doing so. Pay attention to this: the usual way of thinking about entropy as “disorder” must not be taken too literally, a trap that many students fall into. Be very careful in thinking about entropy as disorder. The old analogy between a messy (disordered) room and entropy is deficient and may not only hinder understanding but actually increase confusion.
Entropy is the measure of the spontaneous dispersal of energy at a specific temperature: how much energy is spread out, or how widely spread out energy becomes, in a process. The equation for calculating the change in entropy is: Δ S = Q rev T
Equation 7.7
where ΔS is the change in entropy, Qrev is the heat that is gained or lost in a reversible process, and T is the temperature in kelvin. The units of entropy are usually J mol · K . When energy is distributed into a system at a given temperature, its entropy increases. When energy is distributed out of a system at a given temperature, its entropy decreases.
KEY CONCEPT
Entropy changes that accompany phase changes can be easily estimated, at least qualitatively. For example, freezing is accompanied by a decrease in entropy, as the relatively disordered liquid becomes a well-ordered solid. Meanwhile, boiling is accompanied by a large increase in entropy, as the liquid becomes a much more disordered gas. For any substance, sublimation will be the phase transition with the greatest increase in entropy.
Notice that the second law states that energy will spontaneously disperse; it does not say that energy can never be localized or concentrated. However, the concentration of energy will rarely happen spontaneously in a closed system. Work usually must be done to concentrate energy. For example, refrigerators work against the direction of spontaneous heat flow (that is, they counteract the flow of heat from the “warm” exterior of the refrigerator to the “cool” interior), thereby “concentrating” energy outside of the system in the surroundings. As a result, refrigerators consume a lot of energy to accomplish this movement of energy against the temperature gradient.

Figure 7.10. Entropy in the kitchen
The second law has been described as time’s arrow because there is a unidirectional limitation on the movement of energy by which we recognize before and after or new and old, as shown in Figure 7.10. For example, you would instantly recognize whether a video recording of an explosion was running forward or backward. Another way of understanding this is to say that energy in a closed system will spontaneously spread out, and entropy will increase if it is not hindered from doing so. Remember that a system can be variably defined to include the entire universe; in fact, the second law ultimately claims that the entropy of the universe is increasing.
ΔSuniverse = ΔSsystem + ΔSsurroundings > 0
Equation 7.8
Entropy is a state function, so a change in entropy from one equilibrium state to another is pathway independent and only depends upon the difference in entropies of the final and initial states. Further, the standard entropy change for a reaction, ΔS°rxn, can be calculated using the standard entropies of the reactants and products—much like enthalpy:
ΔS°rxn = Σ ΔS°f,products − Σ ΔS°f,reactants
Equation 7.9
MCAT CONCEPT CHECK 7.5:
Before you move on, assess your understanding of the material with these questions.
-
Rank the phases of matter from lowest to highest entropy.
_______________________________________
-
Describe entropy in terms of energy dispersal and disorder.
_____________________________
-
Do the following situations result in an increase or decrease in entropy?
**Reaction ΔS H2O (l) → H2O (s) Dry ice sublimates into carbon dioxide NaCl (s) → NaCl (aq) N2 (g) + 3 H2 (g) → 2 NH3 (g) An ice pack is placed on a wound**
7.6 Gibbs Free Energy
LEARNING OBJECTIVES
After Chapter 7.6, you will be able to:
- Determine the Gibbs free energy for a reaction at varying temperatures
- Predict the temperature necessary for a temperature-dependent reaction to be at equilibrium
- Identify how changing concentrations of reactant or product will alter the progress of a reaction
The final state function that we will examine in this chapter is Gibbs free energy, G. This state function is a combination of the three that we’ve just examined: temperature, enthalpy, and entropy. The change in Gibbs free energy, ΔG, is a measure of the change in the enthalpy and the change in entropy as a system undergoes a process, and it indicates whether a reaction is spontaneous or nonspontaneous. The change in the free energy is the maximum amount of energy released by a process—occurring at constant temperature and pressure—that is available to perform useful work. The change in Gibbs free energy is defined as follows:
ΔG = ΔH – TΔS
Equation 7.10
where T is the temperature in kelvin and TΔS represents the total amount of energy that is absorbed by a system when its entropy increases reversibly.
MNEMONIC
Gibbs free energy: ΔG = ΔH – TΔS
Goldfish are (equals sign) Horrible without (minus sign) Tartar Sauce.
Overview
A helpful visual aid for conceptualizing Gibbs free energy is to think of it as a valley between two hills. Just as a ball would tend to roll down the hill into the valley and eventually come to rest at the lowest point in the valley, any system—including chemical reactions—will move in whichever direction results in a reduction of the free energy of the system. The bottom of the valley represents equilibrium, and the sides of the hill represent the various points in the pathway toward or away from equilibrium. This is shown graphically in Figure 7.11, which was also discussed in the previous chapter.
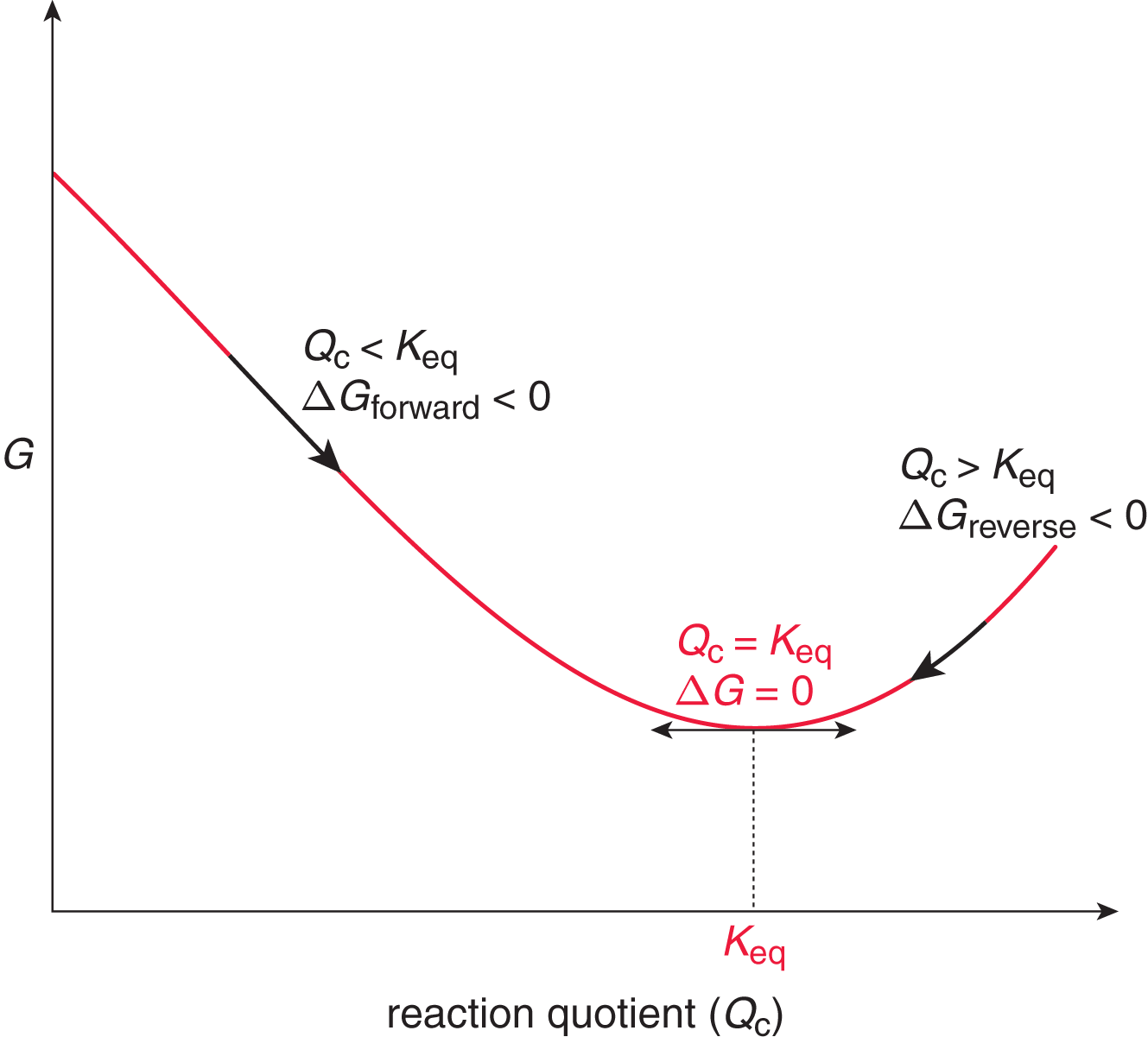
Figure 7.11. Gibbs Free Energy and Spontaneity A decrease in Gibbs free energy indicates that a reaction is spontaneous. When an equilibrated system is disturbed, it will spontaneously act to restore equilibrium.
Movement toward the equilibrium position is associated with a decrease in Gibbs free energy (ΔG < 0) and is spontaneous. When a system releases energy, it is said to be exergonic, as shown in Figure 7.12.
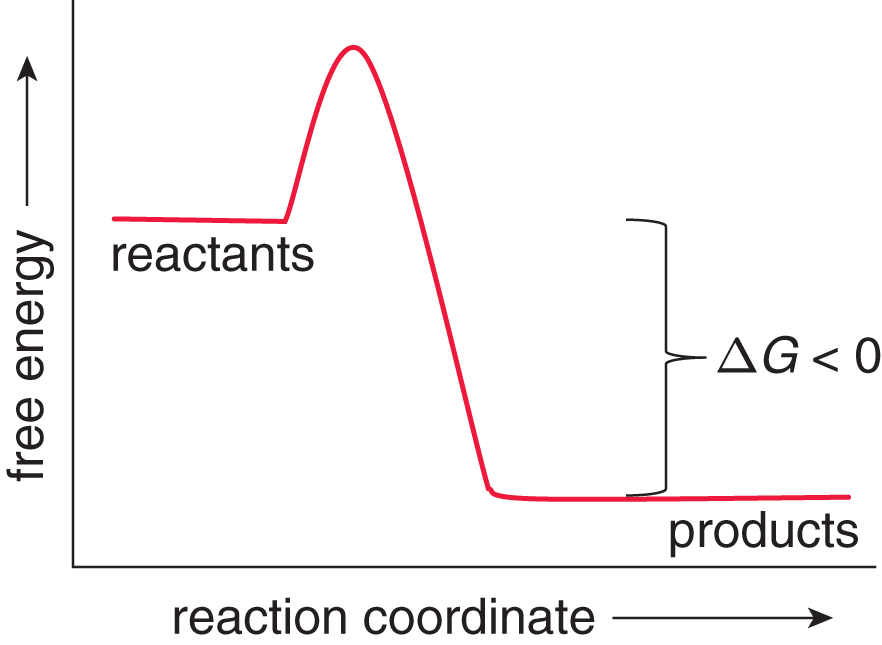
Figure 7.12. Exergonic Reaction Profile Exergonic reactions release energy and are spontaneous (ΔGrxn < 0).
MCAT EXPERTISE
Be careful not to confuse endergonic/exergonic (describing Gibbs free energy) with endothermic/exothermic (describing enthalpy).
On the other hand, movement away from the equilibrium position is associated with an increase in Gibbs free energy (ΔG > 0) and is nonspontaneous. Such a reaction is said to be endergonic, as shown in Figure 7.13.
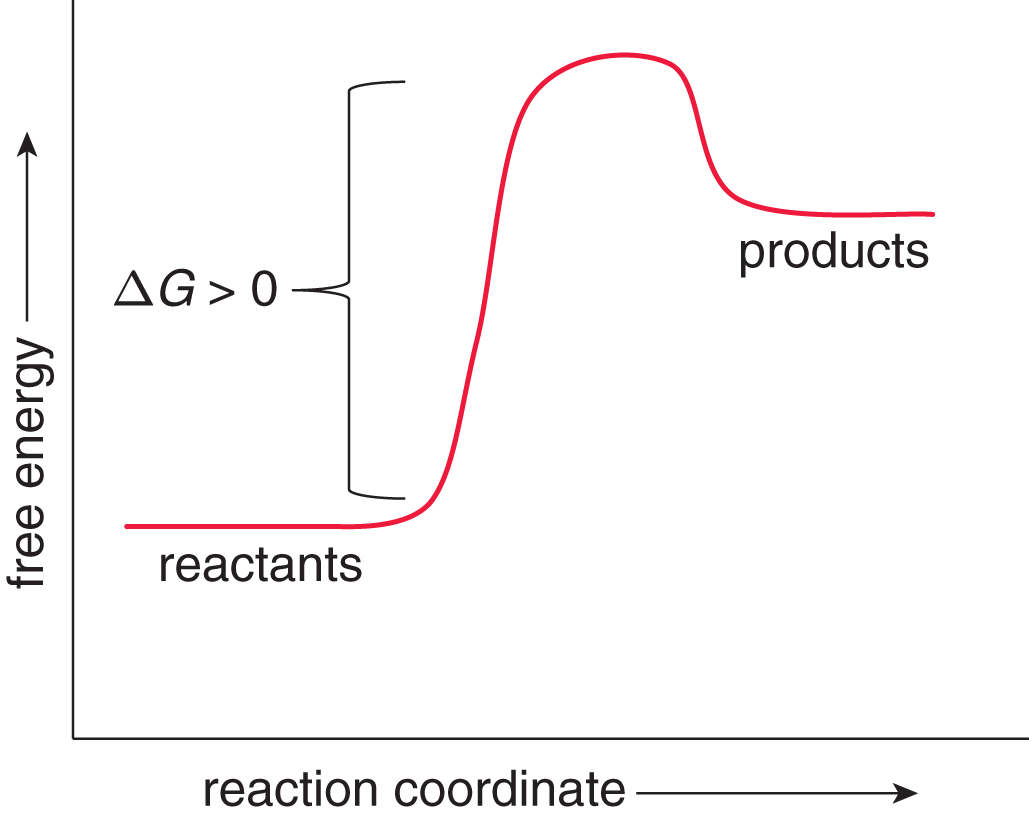
Figure 7.13. Endergonic Reaction Profile Endergonic reactions absorb energy and are nonspontaneous (ΔGrxn > 0).
Once at the energy minimum state—equilibrium—the system will resist any changes to its state, and the change in free energy is zero. To summarize:
- If ΔG is negative, the reaction is spontaneous.
- If ΔG is positive, the reaction is nonspontaneous.
- If ΔG is zero, the system is in a state of equilibrium; ΔH = TΔS.
You should recall that phase equilibria are states in which more than one phase exists. As with all equilibria, the change in Gibbs free energy must be equal to zero (ΔG = 0). For an equilibrium between a gas and a solid,
ΔG = G (g) – G (s) = 0
Therefore,
G (g) = G (s)
KEY CONCEPT
Recall that thermodynamics and kinetics are separate topics. When a reaction is thermodynamically spontaneous, it has no bearing on how fast it goes. It only means that it will proceed eventually without external energy input.
Because the temperature in Gibbs free energy is in kelvin, it is always positive. Therefore, the effects of the signs of ΔH and ΔS on the spontaneity of a process can be summarized as in Table 7.2.
Table 7.2 Effects of ΔH, ΔS, and T on Spontaneity
**ΔH ΔS OUTCOME**
+ + Spontaneous at high T
+ − Nonspontaneous at all T
− + Spontaneous at all T
− − Spontaneous at low T
KEY CONCEPT
ΔG is temperature dependent when ΔH and ΔS have the same sign.
Phase changes are examples of temperature-dependent processes. The phase changes of water should be familiar to you; have you ever wondered why water doesn’t boil at, say, 20°C instead of 100°C? When water boils, hydrogen bonds are broken, and the water molecules gain sufficient energy to escape into the gas phase. Thus, boiling is an endothermic process, and ΔH is positive. As thermal energy is transferred to the water molecules, energy is distributed through the molecules entering the gas phase. Thus, entropy is positive and the term TΔS is positive. If both ΔH and TΔS are positive, the reaction will only be spontaneous if TΔS is greater than ΔH, resulting in a negative ΔG. These conditions are met only when the temperature of the system is greater than 373 K (100 °C). Below 100 °C, the free energy change is positive, and boiling is nonspontaneous; the water remains a liquid. At 100 °C, ΔH – TΔS = 0, and an equilibrium is established between the liquid and gas phases in such a way that the water’s vapor pressure equals the ambient pressure. This is the definition of the boiling point: the temperature at which the vapor pressure equals the ambient pressure.
It is important to remember that the rate of a reaction depends on the activation energy Ea, not ΔG. Spontaneous reactions may be fast or slow. Sometimes a reversible reaction may produce two products that differ both in their stability (as measured by the change in the free energy associated with their production) and in their kinetics (as measured by their respective activation energies). Sometimes, the thermodynamically more stable product will have the slower kinetics due to higher activation energy. In this situation, we talk about kinetic vs. thermodynamic reaction control, which is discussed in Chapter 6 of MCAT General Chemistry Review. For a period of time after the reaction begins, the major product will be the one that is produced more quickly as a result of its lower activation energy. The reaction can be said to be under kinetic control at this time. Given enough time, however, and assuming a reversible reaction, the dominant product will be the thermodynamically more stable product as a result of its lower free energy value. The reaction can then be said to be under thermodynamic control. Eventually, the reaction will reach equilibrium, as defined by its Keq.
Standard Gibbs Free Energy
The free energy change of reactions can be measured under standard state conditions to yield the standard free energy, **ΔG°rxn**. For standard free energy determinations, the concentrations of any solutions in the reaction are 1 M. The standard free energy of formation of a compound, ΔG°f, is the free energy change that occurs when 1 mole of a compound in its standard state is produced from its respective elements in their standard states under standard state conditions. The standard free energy of formation for any element under standard state conditions is, by definition, zero. The standard free energy of a reaction, ΔG°rxn, is the free energy change that occurs when that reaction is carried out under standard state conditions; that is, when the reactants are converted to the products at standard conditions of temperature (298 K) and pressure (1 atm). Like enthalpy and entropy, the free energy of the reaction can be calculated from the free energies of formation of the reactants and products:
Δ G rxn ∘ = ∑ Δ G f,products ∘ − ∑ Δ G f,reactants ∘
Equation 7.11
Free Energy, *K*eq, and *Q*
We can derive the standard free energy change for a reaction from the equilibrium constant Keq for the reaction using the equation:
ΔG°rxn = –RT ln Keq
Equation 7.12
where R is the ideal gas constant, T is the temperature in kelvin, and Keq is the equilibrium constant. This equation allows us to make not only quantitative evaluations of the free energy change of a reaction, but also qualitative assessments of the spontaneity of the reaction. The greater the value of Keq, the more positive the value of its natural logarithm. The more positive the natural logarithm, the more negative the standard free energy change. The more negative the standard free energy change, the more spontaneous the reaction.
Once a reaction begins, however, the standard state conditions (specifically 1 M solutions) no longer apply. The value of the equilibrium constant must be replaced with another number that is reflective of where the reaction is in its path toward equilibrium. To determine the free energy change for a reaction that is in progress, we relate ΔGrxn (not ΔG°rxn) to the reaction quotient, Q:
Δ G rxn = Δ G rxn ∘ + R T ln Q = R T ln Q K eq
Equation 7.13
KEY CONCEPT
Note that the right side of this equation is similar to Equation 7.12. The use of the Q indicates that the system is not at equilibrium.
As described in Chapter 6 of MCAT General Chemistry Review, if the ratio of Q K eq is less than one (Q < Keq), then the natural logarithm will be negative, and the free energy change will be negative, so the reaction will spontaneously proceed forward until equilibrium is reached. If the ratio of Q K eq is greater than one (Q > Keq), then the natural logarithm will be positive, and the free energy change will be positive. In that case, the reaction will spontaneously move in the reverse direction until equilibrium is reached. Of course, if the ratio is equal to one, the reaction quotient is equal to the equilibrium constant; the reaction is at equilibrium, and the free energy change is zero (ln 1 = 0).
Reaction profiles of free energy can be altered by the presence of catalysts. While the overall free energy change of the reaction is not altered, the activation energy required to accomplish the reaction is reduced significantly in the presence of a catalyst, as shown in Figure 7.14.
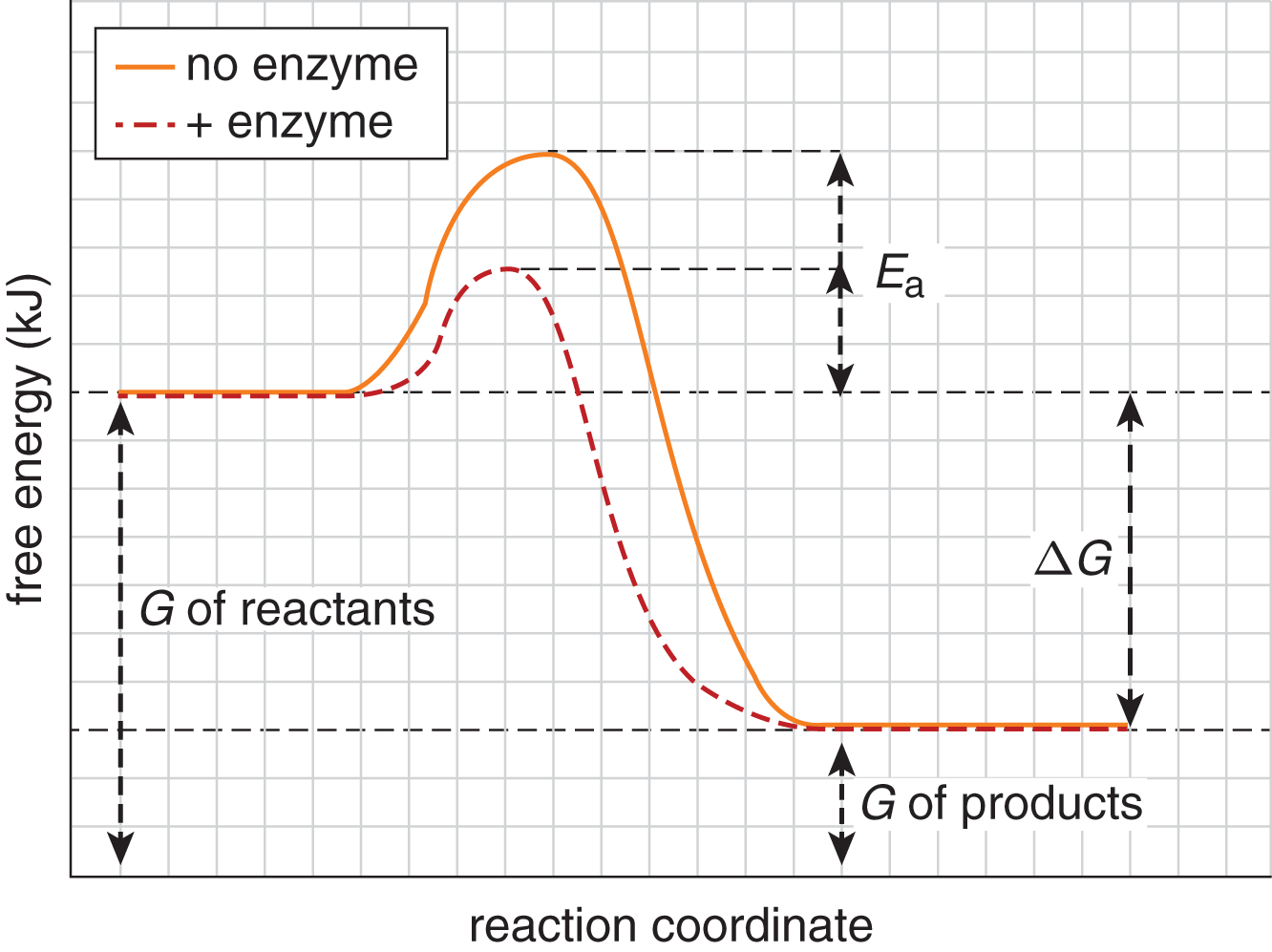
Figure 7.14. Catalysts Alter Kinetics but Not Equilibrium or Free Energy Change
MCAT CONCEPT CHECK 7.6:
Before you move on, assess your understanding of the material with these questions.
-
The Haber–Bosch process creates ammonia through several reactions, the final step of which is N 2 + 3 H 2 → 2 NH 3 ( Δ H rxn = − 93 kJ mol , Δ S rxn = − 198 J mol · K )
Determine what the Gibbs free energy of this reaction is at standard conditions and at 500 K:
________________________________
- At standard conditions:
____________________________
- At 500 K:
-
At what temperature would the reaction described above be at equilibrium?
_____________________________
-
If you were to suddenly flood the reaction vessel with significant amounts of ammonia, what would occur?
__________________________________
Conclusion
We began our discussion of thermochemistry with a review of different ways in which we characterize systems (open, closed, and isolated) and processes (isothermal, adiabatic, isobaric, and isovolumetric). We then further classified systems according to their state functions—system properties such as pressure, density, temperature, volume, enthalpy, internal energy, Gibbs free energy, and entropy that describe the equilibrium state. We examined the equilibria that exist between the different phases and noted that the change in Gibbs free energy for each phase change in equilibrium is zero, as is the case for all equilibria. We defined enthalpy as the heat content of the system and the change in enthalpy as the change in heat content of the system as it moves from one equilibrium state to another. Enthalpy is defined as the energy found in the intermolecular interactions and bonds of the compounds in the system. We explored the various ways Hess’s law can be applied to calculate the total enthalpy change for a series of reactions. Moving on to entropy, we described this property as a measure of the degree to which energy in a system becomes spread out through a process. There is danger in thinking too literally about entropy as “disorder” because a system’s entropy may be increasing even if there is no observable change in the system’s macroscopic disorder (such as ice warming from –10 °C to –5 °C). Gibbs free energy combines the effects of temperature, enthalpy, and entropy, and the change in Gibbs free energy determines whether a process will be spontaneous or nonspontaneous. When the change in Gibbs free energy is negative, the process is spontaneous, but when the change in Gibbs free energy is positive, the process is nonspontaneous.
Many reactions in the body must be spontaneous in order for cells to function. While there are some nonspontaneous reactions in our body, we are able to couple them to thermodynamically favorable (exergonic) reactions that allow the cell to perform even more complex functions.
GO ONLINE!
You've reviewed the content, now test your knowledge and critical thinking skills by completing a test-like passage set in your online resources!
CONCEPT SUMMARY
Systems and Processes
- Systems are classified based on what is or is not exchanged with the surroundings.
- Isolated systems exchange neither matter nor energy with the environment.
- Closed systems can exchange energy but not matter with the environment.
- Open systems can exchange both energy and matter with the environment.
- Processes can be characterized based on a single constant property.
- Isothermal processes occur at a constant temperature.
- Adiabatic processes exchange no heat with the environment.
- Isobaric processes occur at a constant pressure.
- Isovolumetric (isochoric) processes occur at a constant volume.
States and State Functions
- State functions describe the physical properties of an equilibrium state; they are pathway independent and include pressure, density, temperature, volume, enthalpy, internal energy, Gibbs free energy, and entropy.
- Standard conditions are defined as 298 K, 1 atm, and 1 M concentrations.
- The standard state of an element is its most prevalent form under standard conditions; standard enthalpy,standard entropy, and standard free energy are all calculated under standard conditions.
- Phase changes exist at characteristic temperatures and pressures.
- Fusion (melting) and freezing (crystallization or solidification) occur at the boundary between the solid and the liquid phases.
- Vaporization (evaporation or boiling) and condensation occur at the boundary between the liquid and the gas phases.
- Sublimation and deposition occur at the boundary between the solid and gas phases.
- At temperatures above the critical point, the liquid and gas phases are indistinguishable.
- At the triple point, all three phases of matter exist in equilibrium.
- The phase diagram for a system graphs the phases and phase equilibria as a function of temperature and pressure.
Heat
- Temperature and heat are not the same thing.
- Temperature is a scaled measure of the average kinetic energy of a substance.
- Heat is the transfer of energy that results from differences of temperature between two substances.
- The heat content of a system undergoing heating, cooling, or phase changes is the sum of all the respective energy changes.
Enthalpy
- Enthalpy is a measure of the potential energy of a system found in intermolecular attractions and chemical bonds.
- Hess’s law states that the total change in potential energy of a system is equal to the changes of potential energies of the individual steps of the process.
- Enthalpy can also be calculated using heats of formation, heats of combustion, or bond dissociation energies.
Entropy
- Entropy, while often thought of as disorder, is a measure of the degree to which energy has been spread throughout a system or between a system and its surroundings.
- Entropy is a ratio of heat transferred per mole per unit kelvin.
- Entropy is maximized at equilibrium.
Gibbs Free Energy
- Gibbs free energy is derived from both enthalpy and entropy values for a given system.
- The change in Gibbs free energy determines whether a process is spontaneous or nonspontaneous.
- ΔG < 0: reaction proceeds in forward direction (spontaneous)
- ΔG = 0: reaction is in dynamic equilibrium
- ΔG > 0: reaction proceeds in reverse direction (nonspontaneous)
- Gibbs free energy depends on temperature; temperature-dependent processes change between spontaneous and nonspontaneous, depending on the temperature.
ANSWERS TO CONCEPT CHECKS
**7.1**
- The boundary between system and surroundings could be placed anywhere. Most commonly, the ice pack would be considered the chemical system using up energy, and the person (and the remainder of the universe) constitutes the surroundings that are providing the heat for the ice pack to function.
-
- Isothermal: no change in temperature; ΔU = 0, Q = W
- Adiabatic: no heat exchange; Q = 0, ΔU = –W
- Isobaric: no change in pressure; line appears flat in a P–V graph
- Isovolumetric (isochoric): no change in volume; W = 0, ΔU = Q
**7.2**
- Kinetics, equilibrium, and thermodynamics calculations use standard conditions, which are 25 °C (298 K), 1 atm pressure, and 1 M concentrations.
- State functions are properties of a system at equilibrium and are independent of the path taken to achieve the equilibrium; they may be dependent on one another. Process functions define the path between equilibrium states and include Q (heat) and W (work).
- State functions include pressure (P), density (ρ), temperature (T), volume (V), enthalpy (H), internal energy (U), Gibbs free energy (G), and entropy (S).
- The triple point is the specific combination of temperature and pressure at which all three phases are in equilibrium. The critical point is the temperature and pressure above which the liquid and gas phases are indistinguishable and the heat of vaporization is zero.
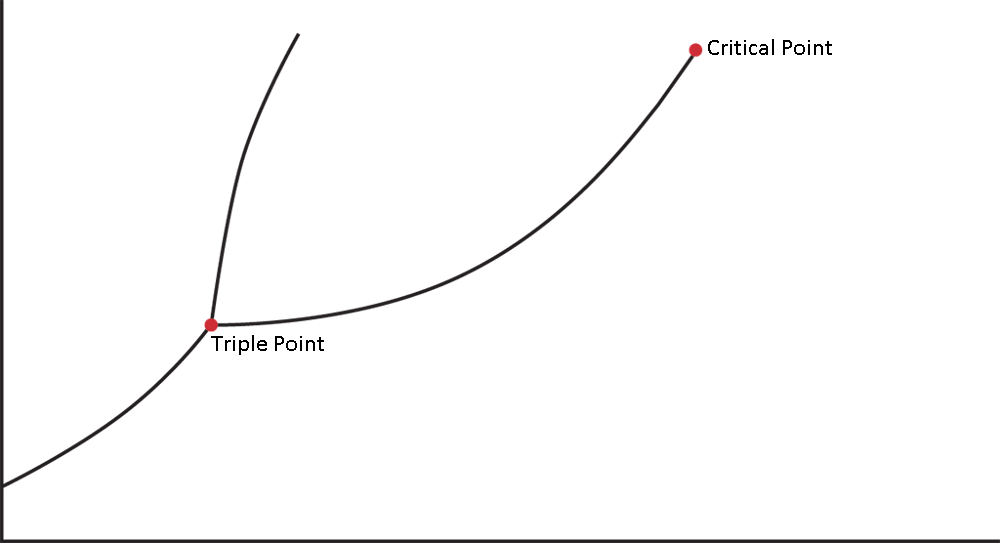
**7.3**
- Temperature is an indirect measure of the thermal content of a system that looks at average kinetic energy of particles in a sample. Heat is the thermal energy transferred between objects as a result of differences in their temperatures.
- Specific heat (c) is the energy required to raise the temperature of one gram of a substance by one degree Celsius. Heat capacity (mc) is the product of mass and specific heat and is the energy required to raise any given amount of a substance one degree Celsius.
- A constant-pressure calorimeter (coffee cup calorimeter) is exposed to constant (atmospheric) pressure. As the reaction proceeds, the temperature of the contents is measured to determine the heat of the reaction. A constant-volume calorimeter (bomb calorimeter) is one in which heats of certain reactions (like combustion) can be measured indirectly by assessing temperature change in a water bath around the reaction vessel.
- c H 2 O ( l ) = 1 cal g · K
**7.4**
- Endothermic reactions involve an increase in heat content of a system from the surroundings (ΔH > 0), while exothermic reactions involve a release of heat content from a system (ΔH < 0).
- To reach the net equation C ( s , graphite) + 1 2 O 2 → CO , the second reaction must be reversed along with the sign of its enthalpy of reaction.
C ( s , graphite ) + O 2 → CO 2 Δ H = − 393.5 kJ mol CO 2 → CO + 1 2 O 2 Δ H = + 283 kJ mol
Adding the enthalpies gives:
− 393 . 5 kJ mol + 283 kJ mol ≈ –400 +285 = - 115 kJ mol ( actual value = − 110.5 kJ mol )
- Enthalpy of reaction = bonds broken – bonds formed. There are four O–H bonds broken, two H–H bonds formed, and one O=O bond formed. Therefore,
Δ H rxn = bonds broken - bonds formed Δ H rxn = 4 × O─H - 2 × H─H + 1 × O=O Δ H rxn = 4 × 463 kJ mol − 2 × 436 kJ mol + 1 × 498 kJ mol Δ H rxn ≈ 4 × 450 - 2 × 435 + 500 = 1800 - 870 + 500 Δ H rxn = 1800 - 1370 = 430 kJ mol actual value = 482 kJ mol
**7.5**
- Solids have the lowest entropy, followed by liquids, with gases having the highest entropy.
- Entropy increases as a system has more disorder or freedom of movement, and energy is dispersed in a spontaneous system. Entropy of the universe can never be decreased spontaneously.
-
**Reaction ΔS H2O (l) → H2O (s)** Decrease (freezing)
Dry ice sublimates into carbon dioxide Increase (sublimation)
**NaCl (s) → NaCl (aq)** Increase (dissolution)
**N2 (g) + 3 H2 (g) → 2 NH3 (g)** Decrease (fewer moles of gas)
An ice pack is placed on a wound Increase (heat is transferred)
**7.6**
Δ G = Δ H − T Δ S = − 93 , 000 J mol − ( 298 K ) ( − 198 J mol · K ) ≈ − 93 , 000 - 300 × - 200 = − 93 , 000 + 60 , 000 = − 33 , 000 J mol = − 33 kJ mol ( actual = − 34.0 kJ mol )
- At standard conditions:
At 500 K: Δ G = Δ H − T Δ S = − 93 , 000 J mol − ( 500 K ) ( − 198 J mol · K ) ≈ − 93 , 000 - 500 × - 200 = − 93 , 000 + 100 , 000 = 7 , 000 J mol = 7 kJ mol ( actual = 6 kJ mol )
- The system is at equilibrium when Δ G = 0 :
Δ G = Δ H - T Δ S 0 = − 93 , 000 J mol − ( T ) ( − 198 J mol · K ) 93 , 000 ≈ 200 T T = 93 , 000 200 = 930 2 = 465 K ( actual = 470 K ) .
- The value of Q would increase significantly, causing the system to shift left, forming more reactants until the system again reached equilibrium.
SCIENCE MASTERY ASSESSMENT EXPLANATIONS
1. A
This process may be adiabatic. Given that the gas was cooled, it did not maintain constant temperature, eliminating (C). Isobaric and isovolumetric processes appear as horizontal and vertical lines in pressure–volume graphs, respectively, eliminating (B) and (D). Adiabatic processes appear hyperbolic on pressure–volume graphs, as illustrated here.
2. A
Since the temperature during a phase change is constant, all phase changes are isothermal processes. During boiling, heat is added to the system to break the intermolecular attractions between the water molecules. As the water molecules vaporize, they expand outward, occupying a larger volume. Thus, boiling is an isothermal expansion, consistent with (A).
3. D
Solve this question using the equation ΔG°rxn = −RT ln Keq. ΔG°rxn is negative (as it must be for a spontaneous reaction), and R and T are always positive. Therefore, lnKeq must also be positive for the sign convention to work out correctly. Since ln(1) = 0, the natural logarithm of any number greater than 1 will be positive, and the natural logarithm of any number less than 1 will be negative. In order for lnKeq to be a positive number, Keq must be greater than 1.
4. D
Combustion often involves the reaction of a hydrocarbon with oxygen to produce carbon dioxide and water. Longer hydrocarbon chains yield greater amounts of combustion products and release more heat in the process—that is, the reaction is more exothermic. Of the hydrocarbons listed here, n-pentane is the longest chain.
5. A
At first glance, this might seem like a math-heavy problem, but it really doesn’t require any calculations at all. We just have to keep track of which bonds are broken and which bonds are formed. Remember, breaking bonds requires energy, while forming bonds releases energy. Two bonds are broken: a C–O bond between the carbonyl carbon and oxygen of acetic acid
, and an O–H bond between the hydroxyl oxygen and hydrogen of methanol
. Two bonds are also formed: a C–O bond between the carbonyl carbon and the oxygen of methyl acetate
, and an O–H bond between a hydroxyl group and a hydrogen to form water
. Given that the same two bonds are broken and formed in this reaction, the energy change must be 0 kJ mol .
6. B
At the triple point (triple for three phases), the solid, liquid, and gas phases exist in equilibrium, consistent with (B). On the other hand, the critical point refers to the temperature and pressure above which gases and liquids are indistinguishable, so(A) can be eliminated.
7. C
There are two parts to this computation. First, the water must be heated from 40oC to 100oC, then it must be vaporized. To calculate the energy required to heat the water, use the formula q = mcΔT. Plugging in values yields (10 g)(60oC)(4.2 J/goC). Next, calculate the energy required to vaporize using the equation q = mL. Plugging in values yields (2,260 J/g)(10 g). Finally combine the two energies together: (10 g)(60oC)(4.2 J/goC) + (2,260 J/g)(10 g), matching (C).
8. D
There is not enough information available to determine the free energy of this reaction. While the entropy is clearly increasing (there are more particles in the system), it is unclear what the enthalpy change is. Because bonds are breaking, the reaction should be endothermic, meaning that both ΔS and ΔH are positive. In this case, it is a temperature-dependent process, and—without a temperature given—we cannot determine the sign on ΔG.
9. A
This problem asks for the free energy of a reaction at nonstandard conditions, which can be determined with the equation ΔG = ΔG° + RTln Q.
10. A
A reaction with a negative enthalpy is, by definition, exothermic. Because both enthalpy and entropy are negative, this is a temperature-dependent process, and the reaction will be both endergonic and exergonic—but only at particular temperatures, eliminating (C) and (D).
11. C
For a process to progress forward spontaneously, Q must be less than Keq and will therefore have a tendency to move in the direction toward equilibrium. A spontaneous reaction’s free energy is negative by convention.
12. B
This question is asking for a reaction with the greatest decrease in entropy, so the phases of the reactants and products must be evaluated. In general, the entropy of gases is the highest, while the entropy of solids is the lowest. Thus, a reaction in which gases react to produce solids will have a significant entropy decrease. In reaction (B), a gaseous reactant forms a solid product, so the entropy decreases, making (B) correct. By contrast, the other reactions have either a minimal change or increase in entropy. In reaction (A), two moles of gaseous reactants become four moles of gaseous product, so the entropy increases. In (C), a liquid reactant forms a gaseous product, so entropy likewise increases. In reaction (D), each side of the equation has a solid and an aqueous component, so there is approximately no change in entropy.
13. A
Eliminate (C) and (D), which describe the free energy of reaction and cannot be determined from this graph. While most reaction coordinate graphs we’ve explored in this book use free energy for the y-axis, this one uses potential energy (enthalpy). If the heat of formation of the products is greater than that of the reactants, the reaction is endothermic. We can determine this information from their relative positions on the graph: because the products are higher than the reactants, this is an endothermic reaction.
14. A
The entropy change can be calculated using the formula ΔS = q/T. Note that more information is given in the question than is necessary, so ignore the mass. Plugging in values yields: ΔS = 30,000 J / 1,060 K = 28 J/K. Thus, (A) is correct.
15. A
In an explosion, a significant amount of heat energy is released, meaning that the reaction is exothermic (ΔH < 0), eliminating (B). The entropy change associated with an explosion is positive because energy is dispersed over a much larger area, eliminating (C). If this is true, the expression ΔH – TΔS must be negative, indicating that this is an exergonic process (ΔG < 0). Absolute temperature can never be negative, eliminating (D).
GO ONLINE
Consult your online resources for additional practice.
EQUATIONS TO REMEMBER
(7.1) First law of thermodynamics: ΔU = Q – W
(7.2) Heat transfer (no phase change): q = mcΔT
(7.3) Heat transfer (during phase change): q = mL
(7.4) Generalized enthalpy of reaction: ΔHrxn = Hproducts – Hreactants
(7.5) Standard enthalpy of reaction:ΔH°rxn = Σ ΔH°f,products − Σ ΔH°f,reactants
(7.6) Bond enthalpy: ΔH°rxn = Σ ΔHbonds broken − Σ ΔHbonds formed = total energy absorbed − total energy released
(7.7) Entropy: Δ S = Q rev T
(7.8) Second law of thermodynamics:ΔSuniverse = ΔSsystem + ΔSsurroundings > 0
(7.9) Standard entropy of reaction:ΔS°rxn = Σ ΔS°f,products − Σ ΔS°f,reactants
(7.10) Gibbs free energy: ΔG = ΔH – TΔS
(7.11) Standard Gibbs free energy of reaction:ΔG°rxn = Σ ΔG°f,products − Σ ΔG°f,reactants
(7.12) Standard Gibbs free energy from equilibrium constant:ΔG°rxn = –RT ln Keq
(7.13) Gibbs free energy from reaction quotient: Δ G rxn = Δ G rxn ∘ + R T ln Q = R T ln Q K eq
SHARED CONCEPTS
General Chemistry Chapter 3
Bonding and Chemical Interactions
General Chemistry Chapter 4
Compounds and Stoichiometry
General Chemistry Chapter 5
Chemical Kinetics
General Chemistry Chapter 6
Equilibrium
Physics and Math Chapter 2
Work and Energy
Physics and Math Chapter 3
Thermodynamics