8 Molecular Aspects of Microbial Growth
## Chapter 8 Molecular Aspects of Microbial Growth
**II Regulation of Development in Model *Bacteria***
Membrane Vesicles: Nano Vehicles Transporting Important Cargo
Membrane vesicles are nano-sized structures (10–400 nm) released from cells of all three domains of life during membrane budding. These vesicles can contain a variety of biomolecules such as nucleic acids, carbohydrates, proteins, enzymes, toxins, and even viruses. Because of the lipid bilayer protecting the cargo, membrane vesicles are excellent vehicles for transferring substances between cells.
Remarkably, recent studies show that vesicle size and content are not random but are controlled by cellular signals. Thus, microbes are able to employ these tiny vehicles for an assortment of activities such as DNA exchange between cells, toxin delivery into host cells, reducing competition through antimicrobial release, and biofilm formation. The study here deals with the biofilm way of life.
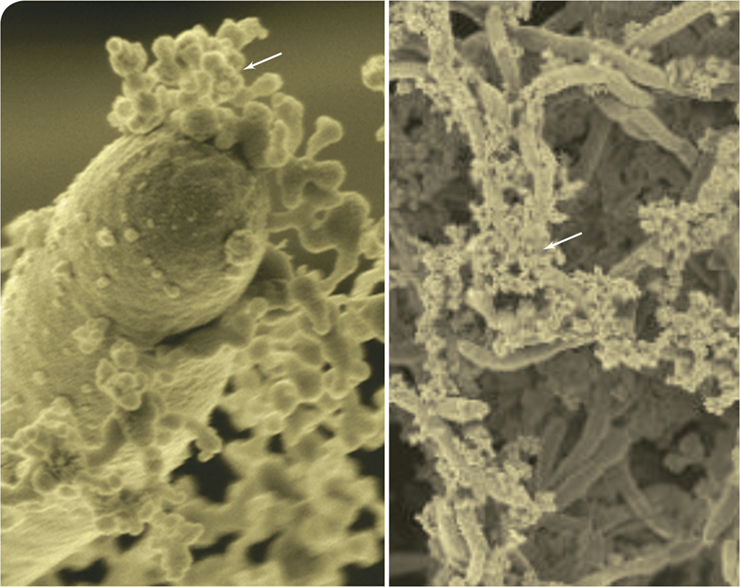
Biofilms are slimy, three-dimensional microbial communities formed to withstand environmental challenges. One of the major requirements for biofilm formation is the release of sticky extracellular polymeric substances (EPS) to help hold the biofilm matrix together. The image on the left shows a scanning electron micrograph of the eukaryotic microbe Candida albicans (elongated cell) releasing membrane vesicles (arrow) containing such polysaccharides for biofilm construction.
Pathogenic strains of C. albicans often form biofilms on indwelling medical devices, resulting in yeast infections that evade the immune system and are resistant to antifungal drugs. Microbiologists investigating methods to treat these impervious biofilms also discovered specific enzymes within the vesicles that modify the EPS in a manner that makes the biofilm resistant to antifungal agents (image to the right, arrow denotes EPS). By contrast, vesicles from free-living cells contain completely different cargo. Strains of C. albicans genetically modified to limit their vesicle production produced yeast biofilms sensitive to antifungal drugs.
Research on membrane vesicles highlights the importance of ongoing studies of microbial growth and how new targets for attack by therapeutic agents can be exposed by basic research to understand the molecular aspects of microbial growth.
Source: Zarnowski, R., et al. 2018. Candida albicans biofilm-induced vesicles confer drug resistance through matrix biogenesis. PLoS Biol. 16(10): e2006872.
In previous chapters we discussed the basic biology of microbes (Chapters 1, 2 and 3 and Chapter 5) and how prokaryotic cells grow and divide (Chapter 4), replicate their chromosome(s) (Chapter 6), and regulate gene expression (Chapter 7). Here we follow up on the major concepts that unfolded in Chapter 4 with a focus on key molecular processes that occur in the microbial growth cycle. We also consider cellular differentiation in some model bacteria, revisit the process of biofilm growth, and close by exploring how some common antibiotics affect microbial growth and the weapons bacteria deploy to counter such attacks.
I Bacterial Cell Division
Prokaryotic cell division is preceded by chromosome replication and the synthesis of new cell wall material in a way that defines cell shape. Cell division is orchestrated in a carefully controlled fashion by protein complexes whose activities can be visualized by powerful light microscopic techniques.
Most cells divide by binary fission (Section 4.6 and Figure 4.8), and this process occurs in a defined series of steps such that each daughter cell obtains a copy of the genome. During the division cycle, the cell must also produce new peptidoglycan and cytoskeleton elements to prevent bursting from osmotic forces. This cytoskeleton gives the cell its distinct morphology (Figure 1.8). To successfully orchestrate all of these events, various regulatory cascades are put into play. In this first part of the chapter we focus on the molecular mechanisms employed by two well-studied gram-negative bacteria, Escherichia coli and Caulobacter crescentus, and introduce advanced microscopic techniques that have revealed the major molecular events that underlie cell division and cell morphology.
8.1 Visualizing Molecular Growth
In Chapter 1 we discussed transmission electron microscopy and electron cryotomography as powerful techniques to visualize macromolecules such as chromosomes, proteins, and membranes. In addition to these, super-resolution microscopy is a powerful form of light microscopy that employs a suite of fluorescent molecules to reveal even the tiniest details within a cell. Super-resolution techniques can resolve structures as small as 5–50 nm in living cells, allowing for dynamic cellular behaviors to be observed in real time.
Fluorescent Tagging
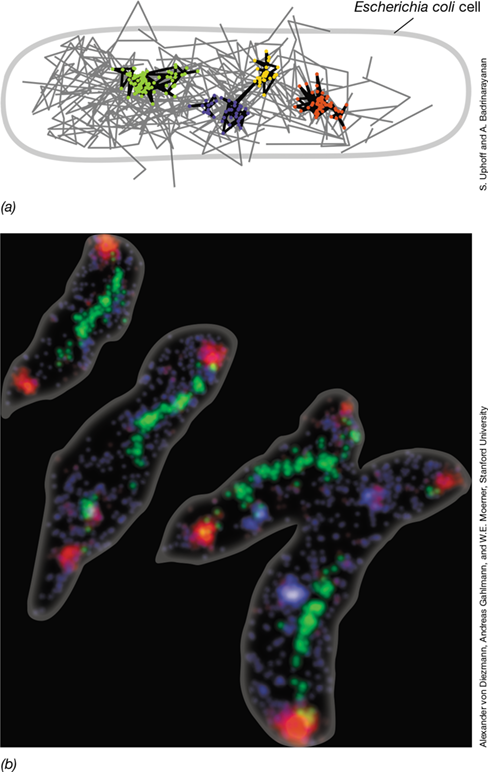
To visualize the localization of specific proteins and to monitor gene expression in cells, reporter genes are valuable tools. Reporter genes encode proteins that are easy to detect or assay and are used by fusing them to genes of interest in such a way that both the reporter gene and the gene of interest are coexpressed. For visualizing molecular events, reporter genes that encode fluorescent products such as the green fluorescent protein (GFP) are routinely used (Figure 8.1). Through the use of fluorescence microscopy (Section 1.8) and multiple fluorescent reporter proteins, the activity and localization of different macromolecules can be determined simultaneously. Figure 8.1a demonstrates use of GFP and another fluorescent protein to determine the cellular locations of σF and σE, two spore-specific sigma factors (Section 6.5) that play key roles in endospore development in Bacillus (Section 2.8 and Section 8.6).
Figure 8.1 Fluorescence micrographs illustrating molecular growth characteristics.

(a) During endospore formation in Bacillus, alternative sigma factors are localized to specific regions of the cell (Section 8.6). σF linked to GFP indicates activity of the σF protein in the developing endospore (at one end of each cell). σE linked to a reporter protein that fluoresces red indicates expression and activity of the σE protein throughout the mother cell prior to endospore formation. Regions correspond to the model depicted in Figure 8.18b. (b) Location and orientation of the Escherichia coli chromosome during cell cycle progression (Section 8.2). Regions are visible due to tagged DNA-binding proteins targeting the oriC (green), left arm (red), and right arm (cyan). Modified from Woldringh, C.L., Hansen, F.G., Vischer, N.O.E., and Atlung, T. 2015. Front. Microbiol. 6: 448. (c) Time-lapse series showing FtsZ (red) and MinD (green) in an elongated E. coli cell. Cell division was inhibited by antibiotic treatment and arrows indicate locations where unstable FtsZ proteins polymerize and depolymerize. Modified from Wu, F., Van Rijn, E., Van Schie, B.G.C., Keymer, J.E. and Dekker, C. 2015. Front. Microbiol. 6: 607. (d) Peptidoglycan synthesis in Staphylococcus aureus. The incorporation of amino acids is visible because a different fluorophore is linked to each D-amino acid present in peptidoglycan. The colors show that peptidoglycan synthesis begins at the septum region and continues in a concentric fashion around the growing cell. Modified from Pereira, A.R., et al., 2016. MBio 7(5): doi:10.1128/mBio.00908-16.
Fluorescent tagging can also be used to resolve different genetic elements in a cell, such as a chromosome and a plasmid (see Figure 8.7). In fact, even different loci within an individual nucleic acid molecule can be observed in a living cell. While DNA or RNA loci are not linked directly to a reporter gene like GFP, genes encoding nucleic acid–binding proteins specific for target loci can be fused to a reporter gene. If necessary, the corresponding DNA or RNA binding sites can be introduced into the nucleic acid region of interest using recombinant techniques (Chapter 12). Figure 8.1b illustrates how separate “arms” of the Escherichia coli chromosome can be visualized in a living cell by engineering a strain to express different fluorescently tagged proteins that bind to different regions of the DNA.
Advances in protein labeling and microscopy techniques have yielded new fluorescent variant proteins that fold quickly and allow for visualization of both spatial and temporal protein dynamics in live cells. Figure 8.1c illustrates the spatiotemporal dynamics of MinD and FtsZ (proteins of the divisome, see Section 8.3) in a growing cell over a time-lapse series. Other cellular aspects can be visualized by linking individual fluorophores (small fluorescent chemical compounds) to specific molecules such as amino acids. For example, Figure 8.1d illustrates the dynamic synthesis of peptidoglycan in growing Staphylococcus aureus cells through the incorporation of three separately fluorophore-labeled amino acids present in the cell wall.
Super-Resolution Techniques
Although fluorescence microscopy enables observation of tagged internal cellular structures, resolution is insufficient to observe the molecular events occurring during the cell cycle. By contrast, super-resolution techniques can reveal and quantify elements as small as single molecules in living cells. Super-resolution techniques employ photoactivated probes that switch from bright to dark emission states depending on the wavelength of light that strikes them; this allows for only one fluorescent molecule (or target) to be excited at a time. By recording the position of individual molecules over time, a super-resolution image is generated (**Figure 8.2*a***).
Figure 8.2 Super-resolution imaging of molecular growth characteristics.

(a) Photoactivated localization micrograph (PALM) mapping the movement of the nucleoid-binding protein MukB in an individual Escherichia coli cell. The four different colors represent clusters of nucleoid-bound MukB molecules, while tracks corresponding to their movement are represented by black lines. Gray lines represent tracks of free MukB (Section 8.2). (b) Three-dimensional quantitative multicolor imaging of Caulobacter cellular structures. PopZ is tagged with a red fluorescent reporter, while crescentin (Section 8.4) is tagged with a green fluorescent reporter.
Super-resolution techniques can reveal the details of cellular superstructures, for example by precisely mapping the diffusion and movement of individual proteins. Figure 8.2a illustrates the use of photoactivated localization microscopy, a form of super-resolution microscopy, to map the movement of individual MukB molecules in a living E. coli cell. MukBEF is a protein complex that binds to the nucleoid and localizes to distinct regions in the cell to assist with unknotting daughter chromosomes after replication (Section 8.2). Not only can super-resolution microscopy be used to observe structural changes to cellular complexes over time, the micrographs can also be modified to construct three-dimensional (3D) images. Figure 8.2b illustrates the use of a super-resolution method called 3D subdiffraction imaging to observe the arrangement of different protein complexes in the budding bacterium Caulobacter crescentus.
By providing spatiotemporal information about individual proteins, super-resolution techniques have been instrumental in proving that like their eukaryotic counterparts, prokaryotic cells contain a highly dynamic subcellular structural organization whose activities during the cellular growth process can be tracked at the molecular level. We now focus on some of these key molecular events as we revisit the growth cycle of prokaryotic cells in detail.
Check Your Understanding
What are the advantages of using super-resolution microscopy versus standard fluorescence microscopy?
8.2 Chromosome Replication and Segregation
A prerequisite for bacterial growth is replication of the genome. Chromosome replication must be tightly regulated to coincide with cell division. After genome replication and prior to cell division, molecular mechanisms must also ensure that the daughter chromosomes are segregated (Figure 8.3). Because the process of cell division in bacterial and archaeal cells requires both temporal and spatial control elements, the process is controlled by a cell cycle (Figure 8.3). Temporally, two copies of the cell’s genome must exist prior to cell division. Spatially, the two copies must be equally segregated and the septum must form at the correct location in the cell for a successful cell cycle to occur.
Figure 8.3 Overview of the bacterial cell cycle.
Phases analogous to the growth 1 (G1), synthesis (S), and growth 2 (G2) phases of the eukaryotic cell cycle are represented.
Regulation of Chromosome Replication Initiation
How does the cell ensure that the genome is completely replicated before cell division while also preventing multiple rounds of replication? Several different proteins play a role in initiating and inhibiting chromosome replication in Escherichia coli. Here we focus on a key protein in this regard, DnaA, along with a handful of accessory proteins necessary to complete replication initiation.
As we discussed in Section 6.3, binding of DnaA to specific DNA sequences within the oriC region of the chromosome leads to unwinding of the DNA and loading of the replisome for chromosome replication in Escherichia coli (Figure 8.3; to review DNA replication, see Figure 6.16). DnaA is most active when it is linked to a molecule of ATP, forming DnaA–ATP. To tightly control chromosome replication, multiple regulatory mechanisms come into play to inactivate DnaA–ATP once replication has initiated. These mechanisms include competition for oriC binding, repression of dnaA expression, titration of DnaA–ATP away from oriC, and inactivation of DnaA–ATP.
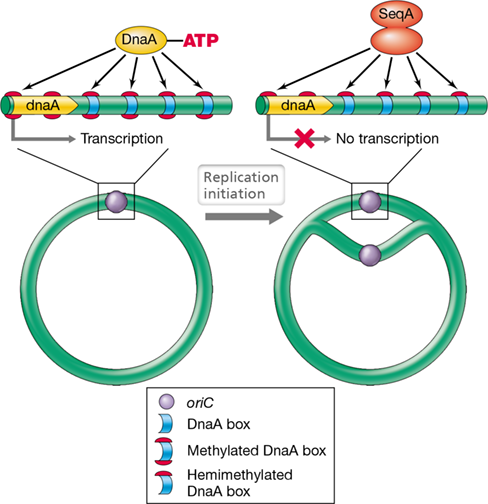
Before DNA replication initiates, both strands of the chromosome contain methyl groups on the adenine residue of –GATC– sequences within the chromosome. However, immediately after replication has initiated, only the parental strand remains methylated. This results in hemimethylated DNA and facilitates a competition for origin binding between DnaA–ATP and a DNA-binding protein called SeqA (Figure 8.4). Because hemimethylated oriC sequences are strongly bound by SeqA, DnaA–ATP is blocked from binding and reinitiating chromosome replication (Figures 8.3 and 8.4). Approximately 10 min after replication has initiated, GATC sequences within the newly synthesized daughter strand are methylated by a DNA adenine methylase.
Figure 8.4 Binding of the ***Escherichia coli oriC*** by DnaA and SeqA proteins.

DnaA–ATP only binds to fully methylated DnaA boxes, while SeqA binds to hemimethylated DNA. The binding of SeqA upstream of the dnaA gene leads to repression of transcription.
As a result of the chromosomal proximity of the dnaA gene to oriC and of the promoter region (Section 6.5) of dnaA possessing a GATC sequence, binding of SeqA also plays a role in repressing the expression of dnaA. Once replication has initiated, the promoter region for dnaA is quickly hemimethylated, and the binding of SeqA prevents RNA polymerase from binding and transcribing dnaA (Figure 8.4). Subsequently, expression of dnaA is also autoregulated by its corresponding protein binding to DnaA boxes within its promoter region.
The final mechanism of preventing DnaA–ATP from binding oriC is to decrease the ratio of DnaA–ATP to DnaA–ADP. How does the cell increase the level of DnaA–ADP when ATP dominates over ADP in a growing cell? This is controlled by the ATPase HdaA, which associates with DNA in the proximity of the replisome and specifically targets DnaA–ATP. This enzyme promotes the hydrolysis of ATP associated with DnaA in a replication-dependent manner and thus functions as a final method of regulating the initiation of chromosome replication. As a result of these combined mechanisms, the cellular level of DnaA–ATP oscillates during the cell cycle, reaching the maximum active amount when initiation of chromosome replication is needed and waning thereafter.
Genome Replication in Fast-Growing Cells
As we learned in Chapter 6, the circular nature of the chromosome of E. coli (and that of most other Bacteria and Archaea) creates an opportunity for speeding up DNA replication. This is because replication of circular genomes is bidirectional from the origin of replication. During bidirectional replication, synthesis occurs in both leading and lagging directions on each template strand, and this allows DNA to replicate as rapidly as possible (Figure 6.15). Studies of chromosome replication in E. coli have shown that about 40 min is required for genome replication and that this value is independent of the generation time, which in E. coli can be as little as 20 min. If an E. coli cell is growing at twice the rate that its chromosome is replicating, how does the cell resolve this conundrum?
In cells growing at doubling times shorter than 40 min, multiple DNA replication forks are present in each cell. That is, a new round of DNA replication begins before the last round has been completed (Figure 8.5); as a result, some genes are present in more than one copy. This can occur after the DNA in the oriC region of the newly synthesized DNA has been methylated, which releases SeqA from the DNA and allows DnaA–ATP to be recruited to reinitiate another round of replication (Figure 8.4). This ensures that at generation times shorter than the time required for replication (a process that occurs at a constant and maximal speed), each daughter cell receives a complete genome at the time of septum formation.
Figure 8.5 Genome replication in cells of ***Escherichia coli*** growing at 60-min or 20-min generation times.

In cells doubling every 20 min, multiple replication forks ensure that each daughter cell gets a complete copy of the genome, which takes 40 min to replicate. In cells doubling every 60 min, multiple replication forks are unnecessary.
Chromosome Segregation
During cell division, segregation of the chromosomes to the cell poles is needed not only to ensure that each daughter cell gets a copy of the genome, but also to allow for septum formation (Figure 8.3). If the two copies of the nucleoid remained in the center of the cell, nucleoid occlusion (a process that prevents the cell from dividing across the nucleoids) would impede proper cell division. In eukaryotes, mitotic spindles separate replicated chromosomes in mitosis (Section 2.13). In many bacteria including the budding bacterium Caulobacter, a similar mechanism called the Par (partitioning) system is used to distribute chromosomes and plasmids equally to progeny cells during growth (Figure 8.6). This system is composed of the ParA ATPase, the ParB chromosome-binding protein, and the PopZ complex, as well as a centromere-like parS sequence located near oriC.
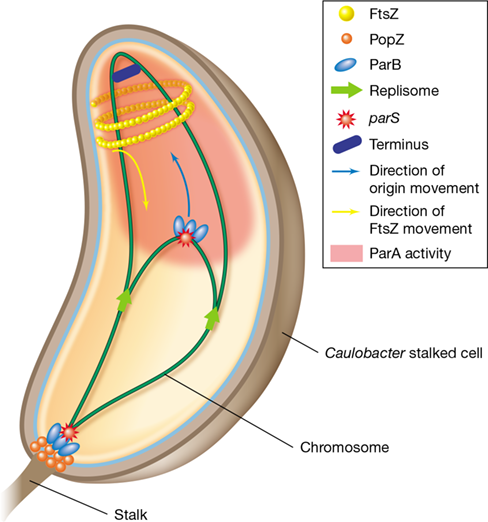
Figure 8.6 Chromosome partitioning in ***Caulobacter***.

After replication initiates, PopZ localizes to the stalked pole of the cell. Next, ParB binds to the parS sequence on the parental chromosome and the complex associates with PopZ at the pole. As replication continues, the parS sequence on the daughter chromosome is bound by ParB and moved to the nonstalked cell pole by the activity of diffuse ParA molecules. The location of FtsZ during partitioning is indicated.
Unlike eukaryotic mitotic spindles, the Par system does not segregate fully replicated chromosomes. Instead, PopZ proteins localize to the stalked pole of the cell and facilitate anchoring of the chromosome to this location by interacting with ParB bound to the parS sequence (Figure 8.6). Once chromosome replication initiates, the newly synthesized parS sequence binds to another molecule of ParB, which is then pulled to the stalked pole by the ATPase activity of ParA. Not only does PopZ help anchor parS of the parent chromosome to the stalked pole of the cell, it also helps recruit ParA to transfer the newly synthesized parS sequence bound to ParB to the nonstalked cell pole (Figure 8.6).
While E. coli lacks the Par system, the daughter chromosomes must still be segregated prior to cell division. After replication the resulting circular chromosomes remain interlinked or tangled, much like the links in a chain. This linkage is broken by the structural maintenance of chromosome complex, which is composed of a topoisomerase (IV) (Section 6.1) and MukBEF proteins. Super-resolution images show that the MukBEF proteins move to discrete locations within the nucleoid (Figure 8.2a) and recruit a topoisomerase to separate replicated sister chromosomes (a process called decatenation) prior to segregation. While the actual segregation process is still not completely understood, the chromosome “arms” appear to remain distinctly separated from one another during replication and the chromosomal loci are pushed to the cell poles in their order of replication (Figure 8.1b). This separation of daughter chromosomes in E. coli thus appears to be independent of specific proteins and proceeds instead by the physical action of replication and DNA accumulation.
How are extrachromosomal genetic elements segregated between daughter cells? Although plasmids are not considered essential for cell survival under all conditions, they are replicated using the same cellular machinery as the chromosome (Figure 8.7). Various control mechanisms exist to ensure that a relatively constant copy number of a given plasmid is disseminated to progeny cells. Replication of large ColE1-type plasmids occurs at the poles instead of in the center of the cell where the nucleoid exists (Figure 8.7). This location helps ensure that efficient transfer to daughter cells occurs during cell division and that stable inheritance is achieved over generations. Other mechanisms for segregating plasmids include partitioning systems similar to the Par system in Caulobacter (Figure 8.6).
Figure 8.7 Cellular location of ColE1 plasmid in ***Escherichia coli*** during replication.

The plasmid (yellow) localizes to the cell poles, while the nucleoid remains in the center of the cell during DNA replication. The separate DNA molecules are visible due to fluorescently labeled DNA-binding proteins.
Check Your Understanding
How does SeqA prevent DnaA–ATP from binding to the oriC regions immediately after chromosome replication initiates?
Explain how the minimum generation time for the bacterium Escherichia coli can be less than the time needed to replicate its chromosome.
8.3 Cell Division and Fts Proteins
Cell division is both spatially and temporally controlled to ensure that each daughter cell has a copy of the genome before the septum seals off the two cells (Figure 8.3). Here we consider regulation of septum formation and a series of key proteins that identify the site of cell division and control the overall process.
The Divisome
Several essential proteins play roles in cell division in Bacteria. Collectively, these proteins are called Fts proteins and a key one, FtsZ, plays a crucial role in binary fission. FtsZ is related to tubulin, the important cell-division protein in eukaryotes (Section 2.15) and is also found in virtually all Archaea. Other Fts proteins are found only in Bacteria and not in Archaea, so our discussion here will be restricted to the Bacteria. The gram-negative Escherichia coli and the gram-positive Bacillus subtilis have been the model bacterial species for the study of cell-division events.
Mastering Microbiology
Art Activity: Figure 8.8a The FtsZ ring and cell division
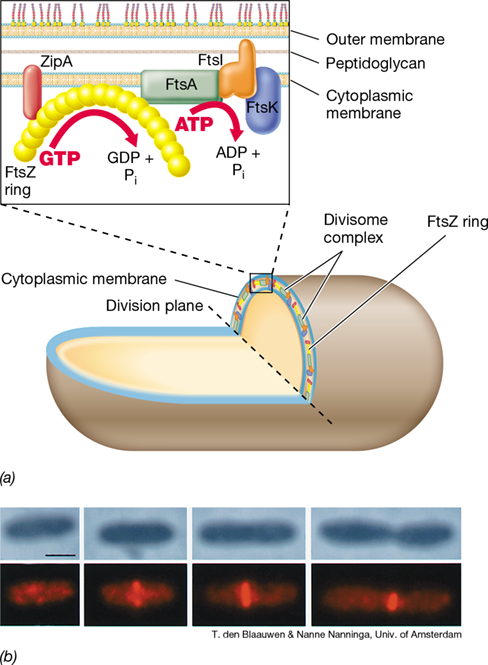
Fts proteins interact in the cell to form a division apparatus called the divisome. In rod-shaped cells, formation of the divisome begins with the attachment of molecules of FtsZ in a ring precisely around the center of the cell; this ring becomes the cell-division plane. In a cell of E. coli, about 10,000 FtsZ molecules polymerize to form the ring; once formed, the FtsZ ring attracts other divisome proteins, including FtsA and ZipA (Figure 8.8). ZipA is an anchor that connects the FtsZ ring to the cytoplasmic membrane and stabilizes it. FtsA, a protein related to actin, an important cytoskeletal protein in eukaryotes (Section 2.15), both recruits FtsZ and other divisome proteins and helps connect the ring to the cytoplasmic membrane. The divisome forms about three-quarters of the way into the cell-division cycle. However, long before the divisome forms, the cell has already begun to elongate and DNA replication has begun (see Figure 8.9).
Figure 8.8 The FtsZ ring and cell division.

(a) Cutaway view of a rod-shaped cell showing the ring of FtsZ molecules around the division plane. Blowup shows the arrangement of individual divisome proteins. ZipA is an FtsZ anchor, FtsI is a peptidoglycan biosynthesis protein, FtsK assists in chromosome separation, and FtsA is an ATPase. (b) Appearance and breakdown of the FtsZ ring during the cell cycle of Escherichia coli. Microscopy: upper row, phase-contrast; bottom row, cells stained with a specific reagent that binds to FtsZ. Cell division events: first column, FtsZ ring not yet formed; second column, FtsZ ring appears as nucleoids start to segregate; third column, full FtsZ ring forms as cell elongates; fourth column, breakdown of the FtsZ ring and cell division. Marker bar in upper left photo, 1 μm
The divisome also contains Fts proteins needed for peptidoglycan synthesis, such as FtsI (Figure 8.8). FtsI is one of several penicillin-binding proteins present in the cell. Penicillin-binding proteins are so named because their activities are inhibited by the antibiotic penicillin (Section 8.5). The divisome orchestrates synthesis of new cytoplasmic membrane and cell wall material, called the division septum, at the center of a rod-shaped cell until the cell reaches twice its original length. Following elongation, the septum forms and the cell divides, yielding two daughter cells (Figure 8.3).
Min Proteins and Cell Division
DNA replicates before the FtsZ ring forms (Figure 8.9) because the ring forms in the space between the duplicated nucleoids; before the nucleoids segregate, they effectively block formation of the FtsZ ring in a process known as nucleoid occlusion (Section 8.2). The proteins MinC, MinD, and MinE interact to help guide FtsZ to the cell midpoint. MinD forms a spiral structure on the inner surface of the cytoplasmic membrane and helps to localize MinC to the cytoplasmic membrane. The MinD spiral oscillates back and forth along the long axis of the growing cell and functions to inhibit cell division by preventing the FtsZ ring from forming (Figure 8.9). Simultaneously, however, MinE also oscillates from pole to pole, and as it does, it functions to sweep MinC and MinD aside. Hence, because MinC and MinD dwell longer at the poles than anywhere else during their oscillation cycle, the center of the cell will have, on average, the lowest concentration of these proteins. As a result, the cell center becomes the most permissive site for FtsZ binding and so the FtsZ ring forms there. In this unusual series of events, the Min proteins ensure that the divisome forms only at the cell center and not at the cell poles (Figure 8.9).
Figure 8.9 DNA replication and cell-division events.

The protein MinE directs formation of the FtsZ ring and divisome complex at the cell-division plane. Shown is a schematic for cells of Escherichia coli growing with a doubling time of 80 min. MinC and MinD are most abundant at the cell poles during FtsZ ring formation.
As cell elongation continues and septum formation begins, the two copies of the chromosome are pulled apart, each to its own daughter cell (Figure 8.9). The Fts protein FtsK and several other proteins assist in this process. As the cell constricts, the FtsZ ring begins to depolymerize, triggering the inward growth of wall materials to form the septum and seal off one daughter cell from the other. The enzymatic activity of FtsZ also hydrolyzes guanosine triphosphate (GTP, an energy-rich compound) to yield the energy necessary to fuel the polymerization and depolymerization of the FtsZ ring (Figures 8.8 and 8.9).
In the budding bacterium Caulobacter, MinC and MinD are absent. Instead, a protein called MipZ (midcell positioning of FtsZ) controls the assembly site of FtsZ. During chromosome segregation, FtsZ localizes to the nonstalked pole of the cell with the chromosome terminus (Figure 8.6). MipZ forms a gradient throughout the cell by directly binding to the ParB–parS complex that facilitates chromosome segregation to the nonstalked cell pole as indicated in Figure 8.6. Once MipZ reaches the nonstalked pole, it displaces polar-localized FtsZ to the center of the cell for septum formation. The timeline for this process in Caulobacter is the same as for the Min system of E. coli (Figure 8.9).
Why do we need to know such intricate details of the cell division process? Besides understanding the molecular biology of microbial growth for purely scientific reasons, there are significant practical reasons for understanding the details of bacterial cell division. Such knowledge could lead to the development of new drugs that target specific steps in the growth cycle of pathogenic bacteria. As with penicillin (a drug that targets bacterial cell wall synthesis), the discovery of drugs that interfere with the function of specific Fts or other cell-division proteins could have broad applications in clinical medicine. In a world where the efficacy of many widely used antibiotics is rapidly fading because of exploding pathogen resistance, the need for new antibacterial antibiotics becomes ever more critical.
Check Your Understanding
How does FtsZ find the cell midpoint of a rod-shaped cell?
8.4 Determinants of Cell Morphology
Just as specific proteins direct cell division in bacteria, other specific proteins form the cell cytoskeleton. The cytoskeleton is the scaffolding that directs cell shape, yielding the spheres, spirals, crescents, and other morphologies typical of bacterial cells (Figure 1.8). Interestingly, these shape-determining proteins, known as SEDS (shape, elongation, division, and sporulation) proteins, show significant homology to key cytoskeletal proteins in eukaryotic cells (Section 2.15). Thus, like eukaryotes, bacteria possess a dynamic and multifaceted cell cytoskeleton.
Cell Shape and MreB
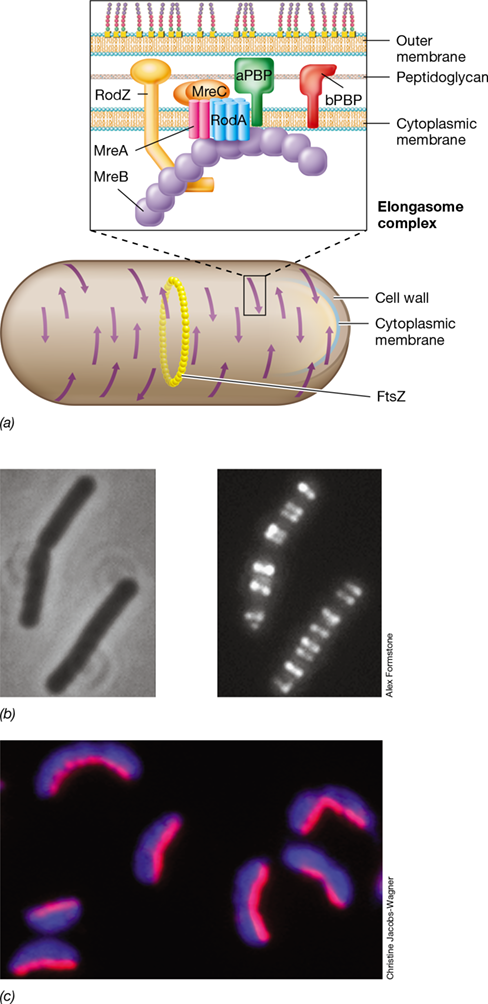
The major shape-determining factor in Bacteria is the protein MreB that forms a simple cytoskeleton in Bacteria and in a few species of Archaea. MreB forms patchlike filaments just below the cytoplasmic membrane that follow the natural curves of the cell (Figure 8.10). The MreB cytoskeleton defines cell shape by recruiting other proteins that function in cell wall growth, such as RodA (peptidoglycan synthesis; Figure 8.10a and Section 8.5), to form the elongasome. Inactivation of the gene encoding MreB or other proteins in the elongasome causes rod-shaped bacteria to become coccus-shaped. Moreover, most naturally occurring coccoid bacteria lack the mreB and rodZ genes and thus do not make MreB or RodZ. This implies that the “default” morphology for a bacterium is a sphere. Variations in the arrangement of MreB filaments in cells of nonspherical bacteria are likely responsible for the different common morphologies of prokaryotic cells.
Figure 8.10 MreB and crescentin as determinants of cell morphology.

(a) The cytoskeletal protein MreB is an actin analog that moves in tracks perpendicular to the long axis of a rod-shaped cell, making contact with the cytoplasmic membrane in several locations (indicated by arrows). The blowup shows the arrangement of individual elongasome proteins with MreB. RodZ anchors the complex, while MreA and MreC are structural proteins. RodA and class A and B penicillin-binding proteins (aPBP and bPBP) coordinate to synthesize new peptidoglycan. (b) Photomicrographs of the same cells of Bacillus subtilis. Left, phase-contrast; right, fluorescence. The cells contain a substance that makes the MreB protein fluoresce, shown here as bright white. (c) Cells of Caulobacter crescentus, a naturally curved (vibrio-shaped) cell. Cells are stained to show the shape-determining protein crescentin (red), which lies along the concave surface of the cell, and with DAPI, which stains DNA and thus the entire cell (blue).
How does MreB define a cell’s shape? The filament structures formed by MreB (Figure 8.10a) are not static, but instead passively move from one side to the other within the cytoplasm of a growing cell. MreB filaments and other associated proteins localize the synthesis of peptidoglycan (Section 8.5) at points where the rod-shaped filaments contact the cytoplasmic membrane (Figure 8.10a). This allows new cell wall to form at several points along the cell rather than from a single location at the FtsZ site outward, as in spherical bacteria (see Figure 8.13). By moving progressively in tracks perpendicular to the cell cylinder and initiating cell wall synthesis at multiple sites along the cell wall, MreB helps direct new wall synthesis in such a way that a rod-shaped cell elongates only along its long axis with cell wall synthesis dispersed at intervals (Figure 8.10b).
Crescentin
In the vibrio-shaped bacterium Caulobacter, a shape-determining protein called crescentin is present in addition to MreB. Copies of crescentin organize into filaments about 10 nm wide that localize onto the concave face of the curved cell (Figure 8.2b). The arrangement and localization of crescentin filaments impart the characteristic curved morphology to the Caulobacter cell (Figure 8.10c). Caulobacter is an aquatic bacterium that undergoes a life cycle in which swimming cells, called swarmers, eventually form a stalk and attach to surfaces. Attached cells then undergo cell division to form new swarmer cells that are released to colonize new habitats (see Section 8.8 and Figure 8.20). The steps in this life cycle are highly orchestrated at the genetic level, and because of this, Caulobacter has been used as a model system for the study of gene expression in cellular differentiation. Although crescentin seems to be unique to Caulobacter, proteins similar to crescentin have been found in other curved cells, such as the bacterium that causes peptic ulcers, Helicobacter pylori. This suggests that these proteins may be necessary for the formation of curved cells.
Evolution of Cell Division and Cell Shape
How do the determinants of cell shape and cell division in Bacteria compare with those in eukaryotes? While MreB functions similarly to the eukaryotic protein actin, FtsZ is both structurally and functionally related to the eukaryotic protein tubulin. Actin forms structures called microfilaments that function as scaffolding in the eukaryotic cell cytoskeleton and in cell division, whereas tubulin forms microtubules that are important in mitosis and other processes. In addition, the shape-determining protein crescentin in Caulobacter is related to the keratin proteins that make up intermediate filaments in eukaryotic cells. Intermediate filaments form part of the eukaryotic cytoskeleton, and genes encoding similar proteins have been found in some other Bacteria. It thus appears that several proteins that control cell division and the cell cytoskeleton in eukaryotic cells (Section 2.15) have evolutionary roots in the Bacteria. However, with the exception of FtsZ, genes encoding homologs of these proteins appear to be absent from most Archaea.
While our focus has been on the filament proteins MreB and crescentin in bacteria that grow by synthesizing new peptidoglycan at the center of the cell (or dispersively), a diversity of morphologies exists in the bacterial world. This is especially evident in species of Alphaproteobacteria (Figure 8.11), gram-negative bacteria that show not only significant morphological diversity but also significant metabolic diversity (Chapters 14 and 16). Hence, Alphaproteobacteria fill a host of diverse ecological niches in nature, from soil and water to symbiotic plant and animal associations.
Figure 8.11 Phylogeny and morphology of select diverse ***Alphaproteobacteria***.

The tree was constructed from comparative sequences of rpoC, a gene encoding one of the subunits of RNA polymerase. Escherichia coli, a member of the Gammaproteobacteria, is included for comparison. Colored shading represents areas of new peptidoglycan synthesis as indicated. Note the extensive morphological diversity within the Alphaproteobacteria. Modified from Randich, A.M., and Brun, Y.V. 2015. Front. Microbiol. 6: 580.
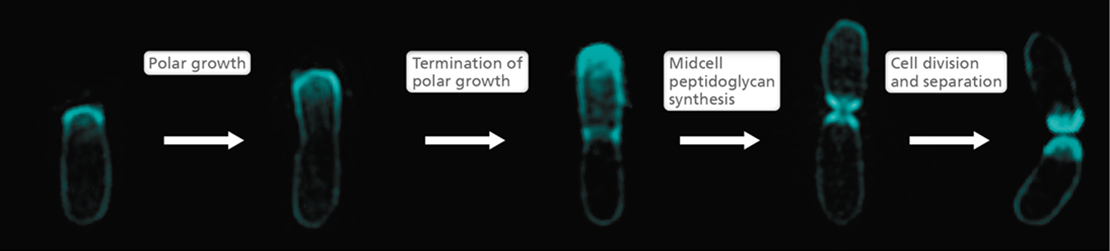
Besides growing as rod-shaped cells and producing new cell material as we have seen in Figures 8.8–8.10, the Alphaproteobacteria contain species that can grow by synthesizing peptidoglycan at the poles (polar elongation). Agrobacterium tumefaciens, a plant pathogen, elongates at one pole in a process called unipolar elongation (Figure 8.12). Although rod-shaped, Agrobacterium lacks MreB. Instead peptidoglycan synthesis occurs at one pole before initiating at the midcell for cell division. For cell division to occur at the midcell, Agrobacterium does possess FtsZ. However, FtsZ resides in the growing pole during unipolar growth and then moves to midcell to recruit the peptidoglycan synthesis machinery and form the ring where the septum forms. It is thought that proteins within the divisome (Section 8.3) help sequester FtsZ in the growing pole during unipolar growth.
Figure 8.12 Polar growth of ***Agrobacterium tumefaciens***.

A time series of fluorescent micrographs showing an A. tumefaciens cell following addition of a fluorescently labeled d-amino acid. The cell cycle progression shows addition of new peptidoglycan to one cell pole prior to septum formation and cell division.
Other genera of Alphaproteobacteria grow in a process known as budding. Budding results in diverse cell morphologies (Figure 8.11; Section 4.10) and is a consequence of synthesizing peptidoglycan at various regions within the cell. However, the precise positioning of the peptidoglycan-synthesizing machinery in budding cells has yet to be discovered. Figure 8.11 also illustrates a principle we will see repeated many times in this book: The phylogenetic position of a bacterium cannot be predicted from its morphology and vice versa. With only rare exception (for example, the spirochetes, Section 15.17), morphology and phylogeny are unrelated characteristics.
Check Your Understanding
How does MreB control the shape of a rod-shaped bacterium?
What protein is thought to control the shape of cells of Caulobacter?
What relationships exist between cytoskeletal proteins in Bacteria and those in eukaryotes?
8.5 Peptidoglycan Biosynthesis
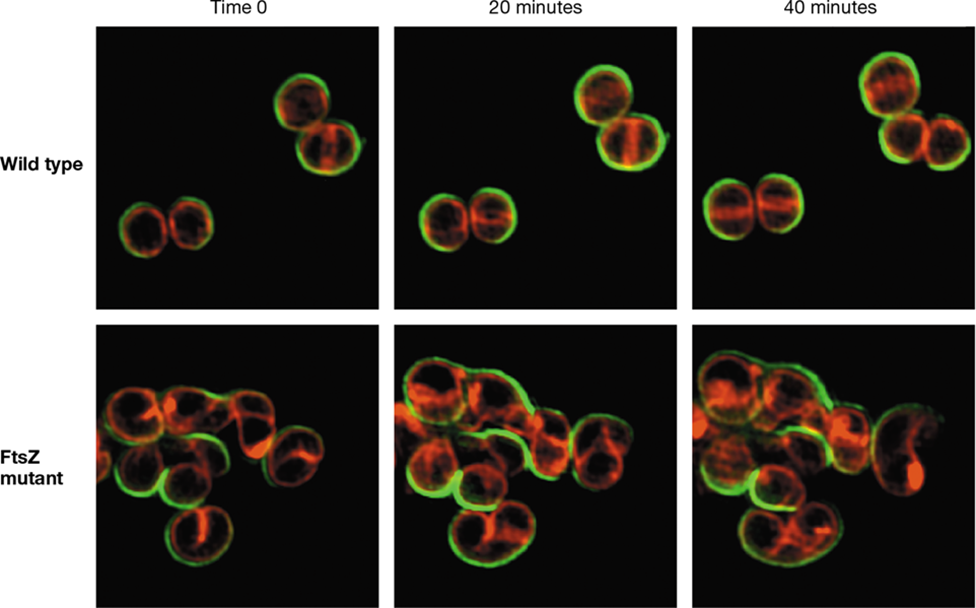
In cells of Bacteria that contain peptidoglycan—and virtually all species do—preexisting peptidoglycan has to be temporarily severed to allow newly synthesized peptidoglycan to be inserted during the growth process. As we have just seen in rod-shaped cells, new cell wall grows at several locations along the length of the cell (Figure 8.10a, b), and it is localized in those cells that divide by polar elongation or budding. By contrast, new cell wall material in cocci grows out in opposite directions from the FtsZ ring (Figure 8.13). Because cocci do not possess the SEDS proteins MreB and RodZ (Section 8.4), FtsZ plays a critical role in maintaining cell shape in cocci. Figure 8.14 illustrates cell division and peptidoglycan synthesis in the gram-positive coccus Staphylococcus aureus, as well as the irregular elongation that occurs in an S. aureus FtsZ mutant. Without a fully functional FtsZ, S. aureus cells begin to elongate and form curved cytoskeletons that illustrate the importance of SEDS proteins in maintaining cellular morphology.
Figure 8.13 Cell wall synthesis in gram-positive ***Bacteria***.
(a) Localization of cell wall synthesis during cell division. In cocci, cell wall synthesis (shown in green) is localized at only one point (compare with Figure 8.10a). (b) Scanning electron micrograph of cells of Streptococcus pyogenes showing wall bands (arrows). A single cell is about 1 μm in diameter.
Figure 8.14 ***Staphylococcus aureus*** cell division and FtsZ.

Time-lapse series of fluorescence micrographs illustrating cell division in the wild type and an FtsZ mutant strain. Cell wall material is stained green, while membrane material is stained red. Wild-type cells form a septum for cell division at midcell and retain a coccus shape, while the mutant cells form an irregular elongated morphology. Modified from Pereira, A.R., et al. 2016. MBio 7(5): doi:10.1128/mBio. 00908-16.
Regardless of the mechanism a bacterium uses to maintain its cytoskeleton, a growing bacterial cell must both synthesize new peptidoglycan and export it outside the cytoplasmic membrane to link up with existing cell wall material, and we consider this problem here.
Insertion of New Peptidoglycan
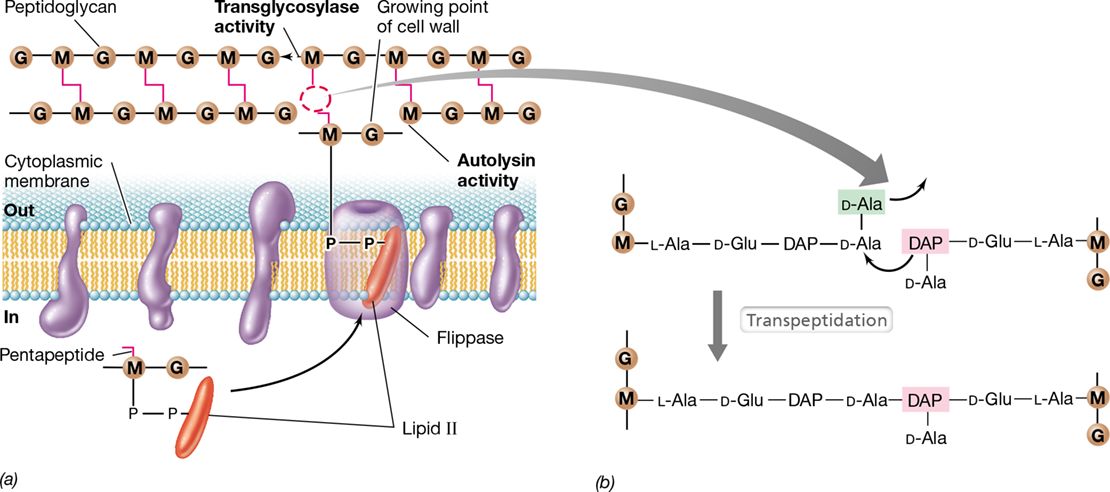
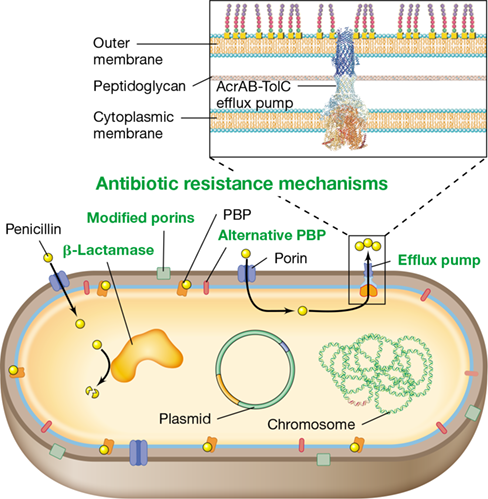
Peptidoglycan can be thought of as a stress-bearing fabric, much like a thin sheet of rubber. Synthesis of new peptidoglycan during growth requires the controlled cutting of preexisting peptidoglycan along with the simultaneous insertion of peptidoglycan precursors. A lipid carrier molecule called bactoprenol plays a major role in the latter process. Bactoprenol is a hydrophobic C55 alcohol that is bonded to an N-acetylglucosamine/N-acetylmuramic acid/pentapeptide peptidoglycan precursor to form lipid II (Figure 8.15). Bactoprenol transports peptidoglycan precursors across the cytoplasmic membrane by rendering lipid II sufficiently hydrophobic to pass through a transmembrane ABC transporter (Figure 2.6) known as flippase (**Figure 8.16*a***).
Figure 8.15 Bactoprenol (undecaprenol diphosphate).

This highly hydrophobic molecule carries cell wall peptidoglycan precursors through the cytoplasmic membrane. The entire molecule is called lipid II.
Figure 8.16 Peptidoglycan synthesis.

(a) Flippase transports lipid II across the cytoplasmic membrane to the growing point of the cell wall. Autolysin breaks glycolytic bonds in preexisting peptidoglycan, while transglycosylase synthesizes them, linking old peptidoglycan with new. (b) The transpeptidation reaction that leads to the final cross-linking of two peptidoglycan chains. Penicillin inhibits this reaction. G, N-acetylglucosamine; M, N-acetylmuramic acid.
Mastering Microbiology
Once outside the cell, lipid II interacts with peptidoglycan polymerases called transglycosylases that insert peptidoglycan precursors into a growing point in the cell wall and catalyze glycosidic bond formation (the remaining bactoprenol is then recycled to the cytoplasm to begin a new round of precursor transport). However, prior to insertion of the peptidoglycan precursor, small gaps in the existing peptidoglycan are made by enzymes called autolysins, enzymes that function to hydrolyze the bond that connects N-acetylglucosamine with N-acetylmuramic acid in the peptidoglycan backbone. The peptidoglycan precursor is then added across the gaps (Figure 8.16a). The junction between new and old peptidoglycan forms a ridge on the cell surface of gram-positive bacteria that can be observed by electron microscopy as a wall band (Figure 8.13b).
Peptidoglycan synthesis must be a highly coordinated process, and peptidoglycan precursors must be readily available during autolysin activity. This is because tetrapeptide units must be spliced into existing peptidoglycan immediately after autolysin activity in order to prevent a breach in the peptidoglycan fabric at the splice point; a breach could cause osmotic issues for the cell and lead to spontaneous cell lysis, called autolysis.
Transpeptidation
The final step in peptidoglycan synthesis is transpeptidation. Transpeptidation forms the peptide cross-links between muramic acid residues in adjacent glycan chains (Section 2.3 and Figures 2.8 and 2.9). In gram-negative bacteria such as Escherichia coli, cross-links form between diaminopimelic acid (DAP) on one peptide and d-alanine on the adjacent peptide. Although there are two d-alanine residues at the end of the peptidoglycan precursor, only one remains in the final molecule, as the other is removed during transpeptidation (Figure 8.16b). This reaction is exergonic (energy-releasing, Section 3.1) and supplies the energy necessary to drive transpeptidation forward. In E. coli, the protein FtsI (Figure 8.8a) functions as the transpeptidase.
Transpeptidation is medically noteworthy because it is the reaction inhibited by the antibiotic penicillin. Several penicillin-binding proteins have been identified in bacteria, including FtsI (Figure 8.8a). When penicillin is bound to penicillin-binding proteins, the proteins are inactivated. If transpeptidation is blocked in an otherwise growing cell, the continued activity of autolysins (Figure 8.16a) so weakens the peptidoglycan that the cell eventually bursts. The absence of peptidoglycan in eukaryotes (such as humans) is the basis of the clinical efficacy of penicillin—the antibiotic destroys only growing bacteria and thus targets pathogenic bacteria that often grow rapidly in an active infection.
Check Your Understanding
What is transpeptidation, and why is it important both to the cell and to clinical medicine?
II Regulation of Development in Model *Bacteria*
**The formation of endospores in Bacillus and cell differentiation in Caulobacter are excellent models of developmental events in individual cells. The transitions between swimming and biofilms in Pseudomonas and Vibrio are valuable models of developmental events in cell populations.**
Now that we have an understanding of the basic molecular processes that occur during bacterial growth, we move on to explore how certain bacteria differentiate to form specialized cells and how bacteria can transition from growing in suspensions (planktonic growth) to form multicellular biofilms.
Development and differentiation are largely characteristics one associates with multicellular organisms. Because most Bacteria and Archaea grow as single cells, few species show differentiation. But a few important examples are known and are classical cases of differential gene expression yielding two genetically identical descendants whose functions differ. Here we discuss three well-studied examples of development and differentiation: the formation of endospores in the gram-positive soil bacterium Bacillus; the formation of two cell types—motile and stationary—in the gram-negative aquatic bacterium Caulobacter; and the formation of heterocysts in the nitrogen-fixing cyanobacterium Anabaena. We conclude by considering the formation of biofilms in the gram-negative and pathogenic bacteria Pseudomonas aeruginosa and Vibrio cholerae.
8.6 Regulation of Endospore Formation
Many microorganisms respond to adverse conditions by converting growing (vegetative) cells into rigid and resistant spores (Section 2.8 and Figure 2.29). Once favorable conditions return, the spore germinates, and the organism returns to the vegetative state (see Figure 8.19 and Section 8.7). Among Bacteria, the genus Bacillus is well known for the formation of endospores, that is, spores formed inside a mother cell (Figure 8.17). Prior to endospore formation, the cell divides asymmetrically. The smaller cell develops into the endospore, which is surrounded by the larger mother cell (Figure 8.17). Once development is complete, the mother cell bursts, releasing the endospore.
Figure 8.17 ***Bacillus*** endospore elasticity map.

Atomic force microscopy combined with a nanochemical surface map illustrating rigidity and release of endospore from mother cell. Flat white regions are indicative of rigidity, while dark regions illustrate elasticity and near bursting of the mother cell. Inset: The heat map bar is a measure of elasticity with lower numbers (black) representing the highest level of elasticity.
Wang, C., Stanciu, C., Ehrhardt, C.J., Yadavalli, V.K. 2015. Journal of Microscopy 258: 49−58
Endospore Formation: Sporulation Factors
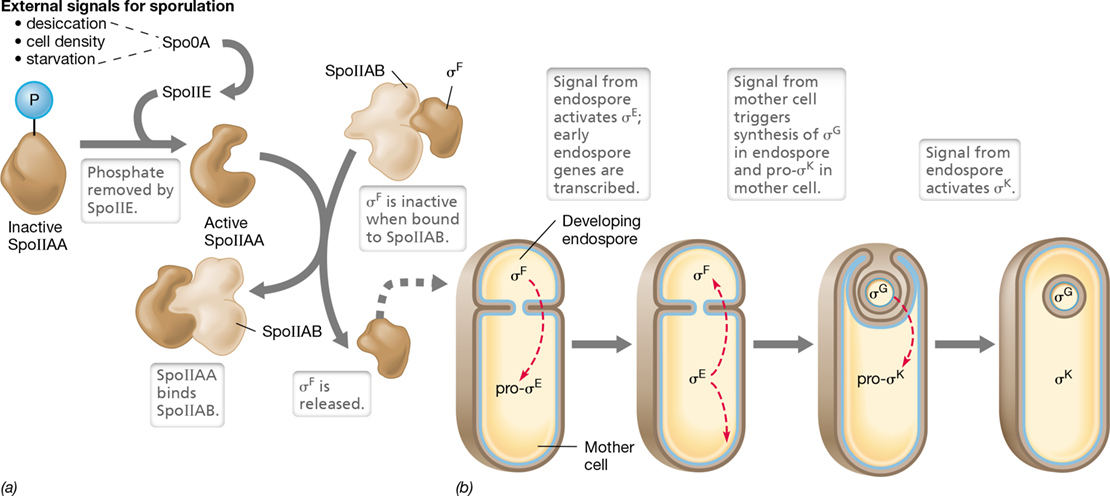
Endospore formation in Bacillus subtilis, a model species for endospore studies, is triggered by unfavorable conditions, such as starvation, desiccation, or growth-inhibitory temperatures. A cell of B. subtilis monitors its environment through a group of five sensor kinases. These enzymes function via a phosphotransfer relay system whose mechanism resembles that of a two-component regulatory system (Section 7.5) but is considerably more complex (Figure 8.18). The net result of multiple adverse conditions is the successive phosphorylation of several proteins called sporulation factors, culminating with sporulation factor Spo0A. When Spo0A is highly phosphorylated, sporulation proceeds. Spo0A controls the expression of several sporulation-specific genes. The product of one of these, SpoIIE, removes the phosphate from SpoIIAA, and this triggers the latter to remove the anti-sigma factor SpoIIAB; this liberates the RNA polymerase sigma factor (Section 6.5) σF, a key step in the sporulation process.
Figure 8.18 Control of endospore formation in ***Bacillus***.

After external signals for sporulation are received, a cascade of sigma (σ) factors controls differentiation. (a) Active SpoIIAA binds the anti-σ factor SpoIIAB, thus liberating the first σ factor, σF. (b) σF initiates a cascade of sigma factors, some of which already exist and need to be activated, others of which are not yet present and whose genes must be expressed. These σ factors then promote transcription of genes needed for endospore development.
Endospore Formation: Alternative Sigma Factors
Once a cell of B. subtilis commits to sporulation, endospore development is controlled by four different σ factors, two of which, σF and σG, activate genes needed inside the developing endospore (called the forespore) and two of which, σE and σK, activate genes needed in the mother cell surrounding the forespore (Figure 8.18b). The sporulation signal, transmitted via Spo0A, activates σF in the forespore (σF is already present in the forespore but is inactive because it is bound by an anti-sigma factor, Figure 8.18a). Once free, σF is active and can bind to RNA polymerase and promote transcription of genes whose products are needed for the next stage of sporulation (Figures 8.1a and 8.18b). These include the gene encoding the sigma factor σG and the genes for proteins that cross into the mother cell and activate σE.
Active σE is required for transcription of yet more genes inside the mother cell, including the gene for σK. The sigma factors σG (in the forespore) and σK (in the mother cell) are required for transcription of genes needed even later in the sporulation process (Figure 8.18b). Eventually, the many spore coats and other unique structures and conditions typical of the endospore (Section 2.8 and Table 2.1) are formed, and the mature endospore is released.
Nutrients for Endospore Formation
Nutrient limitation is the major trigger of sporulation in Bacillus (Section 2.8). But if this is the case, how do cells obtain sufficient nutrients to complete the formation of endospores? One fascinating aspect of the regulation of endospore formation is another regulatory event in which sporulating cells cannibalize cells of their own species. Those Bacillus cells in which Spo0A has already become activated secrete a toxic protein that lyses nearby Bacillus cells whose Spo0A protein has not yet become activated. This lytic protein is produced along with a second protein that functions to delay sporulation of neighboring cells. Cells committed to sporulation also make an antitoxin protein to protect themselves against the effects of their own toxic protein. When lysed, their sacrificed sister cells are used as a source of nutrients for developing endospores. Shortages of certain key nutrients, in particular phosphate, increase transcription of the gene that encodes the toxic protein.
In sporulation we thus see not only a strategy by which cellular differentiation allows the species to form cells that can withstand adverse conditions, but a strategy in which survival of a few (as opposed to all) cells of the species in a population is a priority and is facilitated by the sacrifice of other cells of the same species (see Figure 8.30 to visualize this strategy).
Check Your Understanding
How are different sets of genes expressed in the developing endospore and the mother cell?
What is an anti-sigma factor and how can its effect be overcome?
8.7 Regulation of Endospore Germination
Once cells have proceeded from cell to endospore, they are not committed to dormancy indefinitely. While endospores are not metabolically active and can remain dormant for years, they are still able to sense and respond to nutrients and favorable growth conditions. Such circumstances allow endospores to convert back to vegetative cells by triggering a highly ordered developmental process called germination. This process essentially reverses the steps in sporulation, and in Bacillus, it is orchestrated by over 600 proteins and occurs in three consecutive stages: activation, germination, and outgrowth (Figure 8.19).
Figure 8.19 Germination of ***Bacillus*** endospores.

The binding of nutrients to germinant receptors triggers three stages that result in conversion of an endospore to an actively dividing vegetative cell. Photos show time-lapse images of expression of a red fluorescent protein as endospores (green) become metabolically active (red). DPA, dipicolinic acid. Images adapted from Mutlu, A., et al. 2018. Nat. Commun. 9: 69.
Triggers for Activating Endospore Germination
Although an endospore is considered to be dormant, there are receptors called germinant receptors (GR) within the inner membrane surrounding the core of the endospore (Figure 8.19). GRs exist in clusters called the “germinosome,” which functions to sense and bind nutrients such as amino acids, sugars, and cell wall peptides from vegetative cells. Germinants trigger stage I of endospore germination—activation. While it is unknown how specific GR binding can be, the protein GerA is known to bind the amino acid alanine, and GerB and GerK bind a mixture of asparagine, glucose, fructose, and potassium ions as germinants. Temperature increases also help trigger germination by altering endospore inner membrane properties and GR conformation, processes that lead to more efficient germinant binding.
How does binding of a germinant by a GR trigger activation of an endospore? This is not completely understood; however, it is known that a cascade of events occurs following germinant binding that results in the release of monovalent cations and calcium–dipicolinic acid (DPA, Section 2.8) from the spore core. This release is initiated by reversing sporulation factor activity (Figure 8.19), a process that allows for partial rehydration of the endospore core (the endospore core contains a small fraction of the water content of a vegetative cell, Table 2.1). Besides these physiological events, expression of genes encoding the cellular machinery for major molecular processes such as transcription and translation are increased along with those encoding chaperones for protein folding (Section 6.11) necessary for the cell to enter the germination and outgrowth stages, to be considered now.
Germination and Outgrowth Stages
Once stage I is completed, the next step to spore germination is the triggering of cortex lytic enzymes (CLE) to degrade peptidoglycan in the endospore cortex (Figure 8.19). Cortex peptidoglycan is modified from normal peptidoglycan such that CLE can recognize it and not degrade cell wall peptidoglycan produced during outgrowth of vegetative cells. This removal of the endospore cortex is the major event of stage II—germination. Once the endospore cortex has been degraded, complete hydration is achieved and this sets the stage for more active metabolism.
During stage III of endospore germination—outgrowth—germinated cells begin near-normal rates of metabolism and commence events necessary to initiate the cell cycle and binary fission (Figure 8.3). Cell elongation during outgrowth results in escape of the newly emerging vegetative cell from spore coats (Figure 8.19). Outgrowth requires the increased expression of genes encoding energy-generating processes, such as the citric acid cycle and respiratory enzymes (Chapter 3). Outgrowth is also triggered by the increased expression of genes encoding enzymes that biosynthesize peptidoglycan, teichoic and lipoteichoic acids (components of the gram-positive cell wall, Section 2.3 and Figure 2.10) and the cell division protein FtsZ (Section 8.3).
Check Your Understanding
Where in the endospore are germinant receptors found, and what types of molecules do they detect?
How does reversing the activity of sporulation factors lead to hydration of the endospore?
What type of enzyme is necessary to remove the endospore cortex?
8.8 *Caulobacter* Differentiation
In addition to endospore formation in Bacillus, the gram-negative aquatic bacterium Caulobacter provides a second example of a cell that divides into two genetically identical daughter cells that are both structurally and functionally distinct and express different sets of genes.
Caulobacter is a genus of Alphaproteobacteria (Figure 8.11) that undergoes a simple life cycle and is a common bacterium in oligotrophic (nutrient-poor) aquatic environments (Section 15.18). In the Caulobacter life cycle, free-swimming (swarmer) cells alternate with cells that lack flagella and instead are attached to surfaces by a stalk with a holdfast at its end. The role of swarmer cells is strictly dispersal because swarmers cannot divide to form new swarmer cells nor replicate their DNA. Conversely, the role of the stalked cell is strictly reproduction. In order to divide, swarmer cells must first differentiate into stalked cells, and to swim, stalked cells must first produce swarmers; this is the nature of the Caulobacter life cycle (Figure 8.20; Section 7.9 and Figure 7.25b).
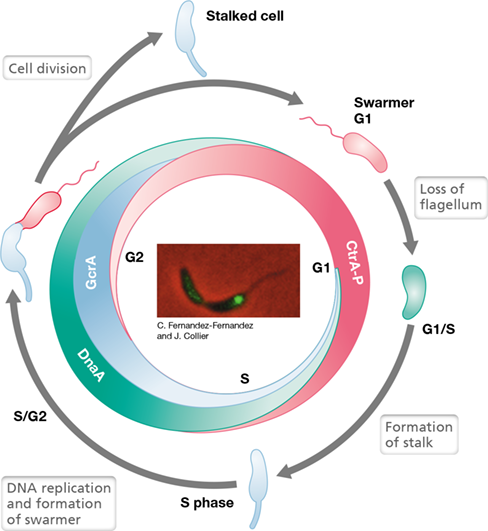
Figure 8.20 Cell cycle regulation in ***Caulobacter***.

Levels of three global regulators, CtrA, DnaA, and GcrA, oscillate through the cycle as shown. G1 and G2 are the two growth phases and S is the DNA synthesis phase. In G1 swarmer cells, CtrA represses initiation of DNA replication and expression of GcrA. At the G1/S transition, CtrA is degraded and DnaA levels rise. DnaA binds to the origin of replication and initiates replication (see inset photo). GcrA also rises and activates genes for cell division and DNA synthesis. At the S/G2 transition, CtrA levels begin to rise again and shut down GcrA expression. GcrA levels slowly decline in the stalked cell but are rapidly degraded in the swarmer (the daughter cell). CtrA is degraded in the stalked cell. Inset: Fused to the green fluorescent protein as a reporter, a subunit of DNA polymerase is localized in the end of the stalked Caulobacter cell where DNA replication occurs. Each cell of the dividing Caulobacter pair is about 2 μm long.
Regulatory Features
The Caulobacter cell cycle is controlled by three major regulatory proteins whose concentrations oscillate in succession. Two of these are the transcriptional regulators GcrA and CtrA. The third is DnaA, a protein that functions both in its normal role in initiating DNA replication (Section 8.2) and also as a transcriptional regulator. Each of these regulators is active at a specific stage in the cell cycle, and each controls many other genes needed at that particular stage in the cycle.
CtrA is activated by phosphorylation by a sensor kinase (Section 7.5) in response to cell cycle signals. Environmental starvation signals such as carbon and ammonia limitation also trigger the stringent response and ppGpp production (Section 7.9), which prevent degradation of CtrA-P in emerging swarmer cells. Once phosphorylated, CtrA-P activates genes that encode synthesis of the flagellum and other functions specific to swarmer cells. Conversely, CtrA-P represses the synthesis of GcrA and also inhibits the initiation of DNA replication in swarmer cells by binding to and blocking the origin of replication (Figure 8.20).
As the cell cycle proceeds, CtrA is degraded by a specific protease, and as a consequence, levels of DnaA begin to rise. The absence of CtrA-P allows access to the chromosomal origin of replication, and, as in all Bacteria, DnaA binds to the origin and triggers the initiation of DNA replication (Section 8.2). In addition, Caulobacter DnaA activates several other genes needed for chromosomal replication. The level of DnaA then falls due to protease degradation, and the level of GcrA rises. The GcrA regulator promotes the elongation phase of chromosome replication, cell division events, and the growth of the stalk on the immobile daughter cell. Eventually, GcrA levels fall and CtrA reappears (in the daughter cell destined to swim away) (Figure 8.20), and the cell cycle is repeated.
Mastering Microbiology
Art Activity: Figure 8.20 Cell cycle regulation in Caulobacter
*Caulobacter* and the Eukaryotic Cell Cycle
Both external stimuli and internal factors such as nutrient and metabolite levels coordinate the events of the Caulobacter cell cycle (Section 7.9). Since its genome has been sequenced and good genetic transfer systems are available, the Caulobacter cell cycle has been used as a model for studying cell developmental processes in other organisms as well. This focus is primarily due to the strict cell cycle followed by Caulobacter, which resembles the cell cycle of eukaryotic cells in many respects. In fact, the resemblance is so striking that terminology used to describe the eukaryotic cell cycle has been adapted to the Caulobacter system (Figure 8.20).
In eukaryotic cells, phase G1 of cell division is where growth and normal metabolic events occur, and in phase G2 the cell prepares for subsequent mitotic events, which occur in the M phase. Between G1 and G2 is the S phase, where DNA replication occurs. In the Caulobacter life cycle there is no mitosis, of course, but analogs of the G1, G2, and S phases are apparent (Figure 8.20), making this bacterium an excellent model for studying cell-division events in higher organisms.
We now consider one other well-studied cell differentiation process, this time in a metabolically robust oxygenic phototrophic bacterium in which photosynthetic O2 production has driven the evolution of a specialized structure for nitrogen metabolism.
Check Your Understanding
Why are the levels of DnaA protein controlled during the Caulobacter cell cycle?
When do the regulators CtrA and GcrA carry out their main roles during the Caulobacter life cycle?
8.9 Heterocyst Formation in *Anabaena*
Cyanobacteria are oxygenic phototrophs; that is, their photosynthetic metabolism yields oxygen (Sections 14.6 and 15.3). They are also able to perform nitrogen fixation—the reduction of N2 to NH3 as a nitrogen source (Section 3.12). Nitrogen fixation is a highly energy-demanding process catalyzed by nitrogenase, an extremely oxygen-sensitive enzyme. How then is it possible for both nitrogen fixation and oxygenic photosynthesis to occur simultaneously in the same bacterium? To solve this problem, some filamentous cyanobacteria such as Anabaena and Nostoc undergo a developmental process to form specialized cells called heterocysts that are dedicated to nitrogen fixation.
Heterocyst Formation
Because heterocysts lack photosystem II—the pigment–protein complex that produces O2 during oxygenic photosynthesis (Section 3.11 and Section 14.6)—they are anoxic cells. This anaerobic lifestyle provides a hospitable environment for nitrogenase and thus nitrogen fixation. Heterocysts arise from the differentiation of vegetative cells that contain photosystem II and thus produce O2, and typically develop in a regular pattern along a filament (**Figure 8.21*a***). As we will see, this patterning separates two incompatible metabolic processes while still allowing for necessary nutrient exchanges and growth.
Figure 8.21 Regulation of heterocyst formation.

(a) Fluorescence microscopy showing Anabaena filaments expressing the green fluorescent protein linked to heterocyst-specific genes; vegetative cells are red from chlorophyll a fluorescence. (b) Molecule dispersion in heterocysts. Fixed carbon from photosynthesis in the vegetative cells is transferred to the heterocyst, while fixed nitrogen produced in the heterocyst is shared with the vegetative cells. The protein PatS, which is synthesized by heterocysts, is also dispersed to neighboring vegetative cells where it inhibits expression of genes necessary for heterocyst formation. (c) Cascade of events in the activation of genes necessary for heterocyst formation. The cascade is initiated by an increase in levels of α-ketoglutarate, the carbon skeleton of the amino acids glutamate and glutamine.
Heterocyst formation requires several morphological and metabolic changes that are regulated by a network of systems that sense both external conditions and intracellular signaling molecules. These changes include the formation of a thickened cell wall to prevent O2 diffusion into the cell, inactivation of photosystem II, expression of nitrogenase, and the patterning of heterocyst differentiation along the filament (Figure 8.21a). Because nutrients can be exchanged between heterocysts and adjacent vegetative cells (Figure 8.21b), other regulatory steps are initiated to prevent nearby vegetative cells from undergoing the developmental conversion to heterocysts.
Regulation of Heterocyst Formation
The cascade of events leading to heterocyst formation is triggered by a limitation of “fixed” nitrogen (nitrate, ammonia, etc.); the limitation is sensed in the vegetative cell as an elevation in levels of α-ketoglutarate, the acceptor molecule for formation of the amino acid glutamate (Section 3.14). When the cell is starved for fixed nitrogen, α-ketoglutarate accumulates and activates the transcriptional global regulator NtcA. NtcA then activates transcription of the gene hetR, which encodes HetR, the major transcriptional regulator controlling heterocyst formation. HetR activates a cascade of genes necessary for differentiation of the heterocyst, expression of cytochrome c oxidases to remove traces of O2 that may still be present, and expression of the nif operon for the synthesis and regulation of nitrogenase (Figure 8.21c).
Only specific cells along the filament form heterocysts, and the consistent pattern observed (Figure 8.21a) is under strict control. Intercellular connections between cells in an Anabaena filament allow vegetative cells to provide fixed carbon to the heterocyst as an electron donor (for N2 reduction by nitrogenase) in exchange for some of the NH3 produced. However, the cell connections also allow for intercellular communication by regulatory molecules. In this regard, differentiating cells produce a small peptide called PatS that diffuses away from the developing heterocyst to form a gradient along the vegetative cells in the filament (Figure 8.21b). PatS inhibits differentiation in vegetative cells by preventing HetR from activating genes necessary for heterocyst formation. A second regulator called PatA, a response regulator analogous to the chemotaxis response regulator CheY (Sections 7.5 and 7.6 and Figure 7.17), also participates in heterocyst pattern development. PatA promotes the activity of HetR, decreases the activity of PatS, and may also participate in cell division.
While other regulatory links in heterocyst formation remain active areas of study, the differentiation of vegetative cells to heterocysts in heterocystous cyanobacteria is a unique example of multicellular patterning and intercellular communication in prokaryotic cells. We now consider a final developmental model system, one in which cell populations—in contrast to individual cells—cooperate to dramatically change their growth characteristics.
Check Your Understanding
Vegetative cells in Anabaena produce oxygen, while its heterocysts do not. Why?
What is the major transcriptional regulator that controls heterocyst formation?
8.10 Biofilm Formation
In Section 4.9 we briefly discussed the basic characteristic of bacterial biofilms—attached polysaccharide matrices containing embedded bacterial cells—along with their environmental and medical significance. We revisit the concept of biofilms here with a focus on the molecular mechanisms that control biofilm development.
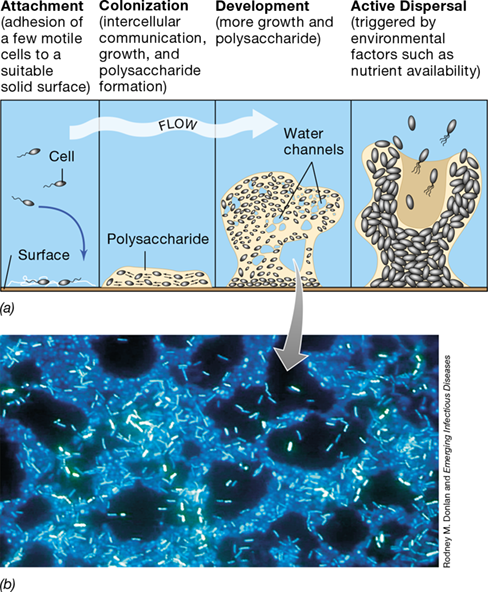
Biofilm formation can be controlled in different ways, and the process has been particularly well studied in two gram-negative Bacteria, Pseudomonas aeruginosa and Vibrio cholerae. However, the process is not limited to Bacteria, as biofilms have also been observed in some Archaea and yeasts. Biofilm formation can be considered a type of developmental cycle that has four basic stages: (1) attachment, (2) colonization, (3) development, and (4) dispersal. Random collision of cells with a surface accounts for the initial cell attachment, which is facilitated by cell surface structures such as flagella and pili or surface proteins. Attachment of a cell to a surface (Figure 8.22) is a signal for the expression of biofilm-specific genes. The latter include genes encoding proteins that produce intercellular signaling molecules and extracellular polymeric substances (abbreviated as EPS), which includes polysaccharide, DNA, and protein, that initiate matrix formation. Once committed to biofilm formation, a previously suspended (planktonic) cell typically loses its flagella and becomes nonmotile. However, biofilms are not static entities and cells can be released from the biofilm through an active process of dispersal.
Figure 8.22 Biofilm formation.
(a) Biofilms begin with the attachment of a few cells that then grow and communicate with other cells. The matrix is formed and becomes more extensive as the biofilm grows, eventually releasing cells. (b) Photomicrograph of a DAPI-stained biofilm that developed on a stainless steel pipe. Note the water channels.
Cyclic Di-GMP and Biofilm Formation
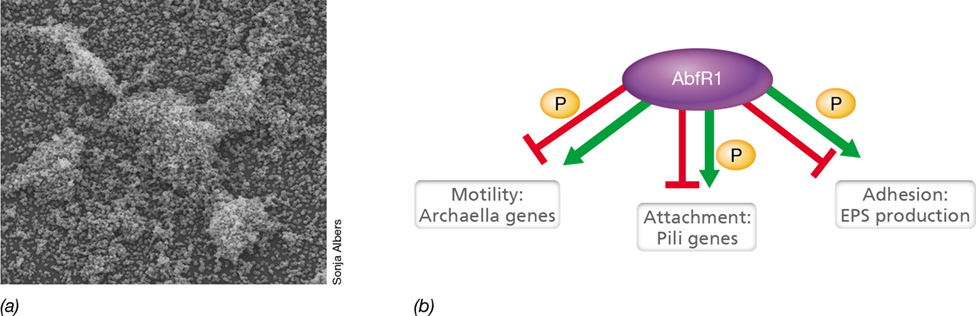
Several signals guide bacteria in transitioning from planktonic growth to life in a semisolid matrix. The actual switch from planktonic to biofilm growth in many bacteria is triggered by the cellular accumulation of the regulatory nucleotide cyclic di-guanosine monophosphate (c-di-GMP) (Figure 8.23). Although various nucleotides play important regulatory roles in all domains of life (Section 7.8), c-di-GMP is widely distributed only in Bacteria. The synthesis or degradation of c-di-GMP depends on both environmental and cellular cues, and its synthesis either promotes or inhibits a variety of physiological events including cell differentiation, bacterial development, and biofilm-relevant functions (Figure 8.23).
Figure 8.23 The signaling molecule cyclic di-guanosine monophosphate.

This molecule is used as an intracellular signaling molecule by many bacteria to control specific physiological processes by binding to various effectors. Two molecules of GTP are converted to c-di-GMP by the activity of proteins with GGDEF (diguanylate cyclase) domains, while the activity of proteins with phosphodiesterase domains converts c-di-GMP to pGpG and GMP.
Enzymes that synthesize c-di-GMP from GTP are called diguanylate cyclases, and these enzymes often possess an allosteric binding site for c-di-GMP. Thus, these enzymes are sensitive to feedback inhibition (Section 7.15) and c-di-GMP production is reduced once elevated levels exist in the cell. Conversely, proteins with phosphodiesterase domains degrade the signaling nucleotide to the guanine derivatives pGpG and GMP (Figure 8.23). These two types of proteins coordinate antagonizing activities to yield cellular levels of c-di-GMP necessary to influence cellular activities. This regulatory molecule directly affects a host of cellular activities by binding to structural proteins and enzymes as well as to transcription factors and riboswitches (Sections 7.2 and 7.13, respectively). In the specific context of biofilms, c-di-GMP binds to proteins that reduce the activity of the flagellar motor, regulates cell surface proteins required for attachment, and mediates the biosynthesis of extracellular matrix polysaccharides of the biofilm.
*Pseudomonas aeruginosa* and Biofilms
Pseudomonas aeruginosa can form a tenacious biofilm (Section 4.9) containing specific polysaccharides that subsequently increase its pathogenicity and prevent the penetration of antibiotics. P. aeruginosa is a classic opportunistic pathogen and from its primary reservoir in soil can infect the blood, lungs, urinary tract, ears, skin, and other tissues of humans. The major symptoms of the human genetic disease cystic fibrosis are caused by thick biofilms of P. aeruginosa that develop in the lungs, and the bacterium is a significant nosocomial (hospital-acquired) pathogen.
Besides the intracellular activities triggered by c-di-GMP, intercellular communication by quorum sensing (Section 7.7) is critical for the development and maintenance of P. aeruginosa biofilms. As the regulatory molecules called acyl homoserine lactones (AHLs, Figure 7.18b) accumulate, they signal P. aeruginosa cells that the population of this species is enlarging and thus the opportunity for biofilm formation exists. The production of AHLs also triggers expression of a subset of the genes necessary for biofilm formation including those that increase extracellular polysaccharide and c-di-GMP synthesis.
Elevated c-di-GMP levels initiate the production of EPS, including a polysaccharide called Pel, which functions as both a primary scaffold for the microbial community and a mechanism for resisting the penetration of antibiotics; c-di-GMP also leads to decreased flagellar function. Over time in nutrient-rich conditions, P. aeruginosa cells can develop mushroom-shaped microcolonies that can be more than 0.1 mm high and contain millions of cells (Figure 8.22; Figure 4.16). The final architecture of a biofilm is determined by several factors in addition to signaling molecules, and these include both nutritional factors and the flow rate of the biofilm environment.
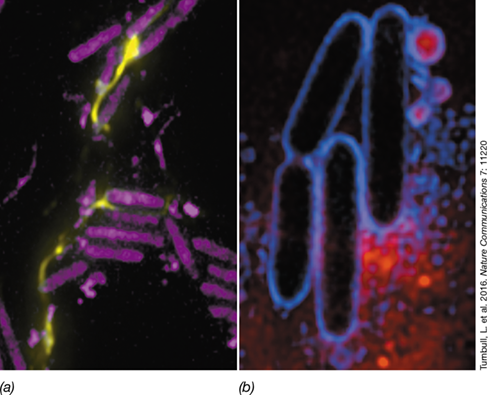
Besides the contribution of decreased flagellar movement and Pel production, DNA release by lysed cells can also promote biofilm formation in P. aeruginosa (**Figure 8.24*a***). This event—termed “explosive death”—occurs in a subpopulation of P. aeruginosa cells. Explosive death is caused by the expression of a lysis protein encoded in the P. aeruginosa genome by an inactive prophage; the protein is produced in response to stress conditions such as DNA-damaging reagents and antibiotics and is occasionally a random event under normal conditions. When this lysis enzyme is expressed, the bacterial cells break open and release their DNA (Figure 8.24a). This highly viscous DNA, now extracellular, is integrated into the EPS that forms the biofilm scaffold. Besides the release of DNA, explosive death also leads to shattered cell membrane fragments that curl together to form vesicles surrounding cellular proteins and DNA. These vesicles also contribute to EPS formation and biofilm development (Figure 8.24b). Explosive death likely explains why antibiotic treatment of P. aeruginosa infections often enhances biofilm formation; this protective mechanism in bacterial cells thwarts antibiotic penetration.
Figure 8.24 Explosive death and biofilm formation in ***Pseudomonas aeruginosa***.

This gram-negative gammaproteobacterium (Section 16.4) exists in soil as a harmless saprophyte, but can also be a vicious opportunistic human pathogen that can cause infections of blood, burns, and related wounds, and ears, lungs, and eyes. P. aeruginosa is of particular concern in individuals suffering from the genetic disease cystic fibrosis because of the organism's ability to form biofilms that affect lung capacity. (a) Cell lysis and DNA release. Production of the lysis enzyme causes the bacterial cells (purple) to break open, releasing their DNA (yellow). This DNA is critical to forming the EPS that holds the biofilm together. (b) Cell bursting and biofilm formation. Cell lysis leads not only to the release of DNA, but also to shattered cell membrane fragments that curl together to form vesicles surrounding proteins and DNA that contribute to EPS formation (small blue circles with red centers). Images adapted from Turnbull, L., et al. 2016. Nat. Commun. 7: 1120.
Biofilm formation strategies can differ among species of the same genus. In Pseudomonas fluorescens, increases in c-di-GMP also promote biofilm formation. However, the biofilm machinery regulated by c-di-GMP in P. fluorescens differs from that of P. aeruginosa. In P. fluorescens, changes in c-di-GMP levels affect secretion and cell surface localization of a large adhesion protein called LapA that helps attach cells to the surface. For example, when environmental levels of a key nutrient such as phosphate are high, c-di-GMP levels are high, LapA localizes to the cell membrane, and biofilm synthesis is promoted. However, in response to low levels of phosphate, P. fluorescens cells maintain a low c-di-GMP level that prevents localization of LapA to the outer membrane, thereby disabling the attachment mechanism required to initiate biofilm formation. If phosphate levels continue to fall within a P. fluorescens biofilm, the associated reduction in c-di-GMP levels results in the release of a specific protease normally sequestered by a c-di-GMP-binding protein. This protease cleaves LapA, resulting in the release of attached cells and promoting their dispersal to explore for available nutrients in nearby habitats.
*Vibrio cholerae* and Biofilms
Vibrio cholerae, the causative agent of cholera (Section 33.3), forms biofilms containing polysaccharide and the carbohydrate-binding matrix proteins RbmA, RbmC, and Bap1. Figure 8.25 illustrates the impressive production of these matrix proteins during biofilm growth. Synthesis of these biofilm proteins is controlled by both intercellular and intracellular signaling. Two transcriptional activators, VpsR and VpsT, are required to activate expression of the matrix protein genes, while a repressor protein, HapR, negatively regulates biofilm formation by decreasing the activity of the two activator proteins VpsT and VpsR. The activity of these key transcription factors is controlled by a host of other factors including two-component regulatory systems, alternative sigma factors, catabolite repression, quorum sensing, and regulatory RNAs (Chapter 7).
Figure 8.25 ***Vibrio cholerae*** biofilm formation.
(a) Time-course multiresolution fluorescence micrographs illustrating the production and release of the carbohydrate-binding matrix proteins RbmA, RbmC, and Bap1 during biofilm formation. Each protein is detected by the binding of a specific fluorescently labeled antibody. (b) Low-cell-density-dependent activation of genes encoding carbohydrate-binding matrix proteins for biofilm formation. A low concentration of quorum-sensing molecules promotes transcription of a small regulatory RNA that binds to the mRNA encoding the HapR repressor. This results in lower levels of HapR protein and thus synthesis of the transcriptional activators VpsR and VpsT; these activate transcription of the genes rbmA, rbmC, and bap1. Note that VpsT is active when bound to c-di-GMP. (c) High-cell-density-dependent inhibition of biofilm formation. A high concentration of quorum-sensing molecules results in decreased expression of the sRNA that binds to the mRNA encoding HapR. Thus, high levels of HapR protein exist in the cell, inhibiting the activity of VpsR and VpsT. This signal cascade shuts off transcription of rbmA, rbmC, and bap1.
While c-di-GMP is the inducer molecule that activates VpsT and thus expression of genes for biofilm formation (Figure 8.23), quorum sensing in V. cholerae acts in an opposite manner from that in P. aeruginosa. At low cell densities, small regulatory RNA molecules bind to the mRNA encoding HapR, thus preventing its translation. As V. cholerae cell density increases, the level of quorum signaling molecules also increases and this results in decreasing levels of the regulatory RNAs that target HapR mRNA. The latter results in an increased concentration of the HapR repressor, decreased expression of the activator proteins VpsT and VpsR, and the dispersal of attached cells. Thus, unlike in P. aeruginosa, in V. cholerae, biofilm formation is triggered by low cell densities and repressed by high cell densities. Or, said another way, in V. cholerae, the accumulation of quorum sensing molecules triggers a regulatory cascade that ultimately results in the repression of biofilm formation genes and the activation of flagellar genes, just the opposite of what occurs in P. aeruginosa.
The ecological significance of this phenomenon is that biofilm formation is more likely to occur when V. cholerae is found in its natural marine environment where nutrients are typically scarce, thus leading to smaller populations than those found inside the close quarters of an intestinal cell where nutrients are more plentiful. Biofilm formation allows the V. cholerae cell to attach to marine surfaces such as plankton (Section 33.3), crustaceans, and sediments for greater access to nutrients and protection from perturbations. The final step in biofilm formation, dispersal, ultimately aids in the transmission of V. cholerae to new host cells.
Because the biofilm lifestyle of many pathogenic bacteria often leads to either persister cells (Section 8.12) or antibiotic-resistant cells (or both), and because biofilms themselves function as barriers to the penetration of antimicrobial drugs, there is great medical interest in understanding the molecular biology of biofilm development and pursuing new classes of drugs that might interfere with biofilm formation.
Biofilms and *Archaea*
While our understanding of biofilms in Archaea is just beginning, at least some of the events in archaeal biofilm formation appear to share features with those of bacterial biofilms. Most of our knowledge comes from studying biofilm formation in the motile hyperthermophilic sulfur chemolithotroph Sulfolobus acidocaldarius (**Figure 8.26*a***).
Figure 8.26 Biofilm formation in the crenarchaeon ***Sulfolobus acidocaldarius***.

Sulfolobus is an acidophilic and hyperthermophilic sulfur-oxidizing archaean (Section 17.10). (a) Scanning electron micrograph of a 6-day-old biofilm. (b) Regulation of biofilm formation by the transcription factor AbfR1. Activation and repression are dependent on the phosphorylation state of AbfR1 with green arrows indicating activation and red lines indicating repression. As can be seen, when AbfR1 is phosphorylated, motility is repressed and attachment activated. Image adapted from Koerdt, A., et al. 2010. PLoS ONE 5(11): e14104.
A transcription factor called AbfR1plays a major role in the Sulfolobus biofilm. In the phosphorylated state, AbfR1 activates biofilm formation through the increased expression of genes for EPS and pili synthesis and decreased expression of genes encoding archaella (Section 2.9) synthesis (Figure 8.26b). This ultimately leads to increased cell attachment and decreased motility. The opposite occurs when AbfR1 is in the unphosphorylated state—archaella production is activated and pili and EPS synthesis repressed. While the signal and sensor kinase (Section 7.5) responsible for phosphorylating AbfR1 remain unknown, biofilm formation in Sulfolobus appears to be a response to significant changes in pH or temperature.
Although the role that quorum sensing (Section 7.7) may play in Sulfolobus biofilm formation is also unclear, Archaea have been shown to produce quorum-sensing-like molecules. Archaea do not produce c-di-GMP—a major regulator in Bacteria (Figure 8.23)—but genomic analysis suggests that many species can produce c-di-AMP. This regulatory molecule, which is also found in Bacteria, may therefore play a role in archaeal biofilm formation (Figure 8.26) and other events in Archaea where quorum-sensing molecules may participate.
We will continue our discussion of biofilms and strategies to control their formation when we consider the ecology of biofilms in Chapter 20. But for now, we move on to Part III where we consider antibiotics and bacterial growth and focus on the alarming practical problem of antibiotic resistance.
Check Your Understanding
What are the four basic stages of biofilm formation?
Besides autoinducer synthesis, what intracellular molecule promotes biofilm formation in many Bacteria?
III Antibiotics and Microbial Growth
Antibiotics target specific bacterial growth processes and have been a boon to clinical medicine. However, antibiotic efficacy has been compromised by bacterial resistance mechanisms that include mutations that alter the targets, drug destruction or efflux, and entering a transient dormant state.
Throughout this chapter we have discussed how cells coordinate their molecular biology to optimize steps in microbial growth. But what about mechanisms that short-circuit these synchronized processes? Here we focus on how antibiotics target growth processes and the bacterial responses that can lead to resistance and persistence. In Chapter 28 we will focus on the clinical significance of antibiotics and consider their spectrum of activity and spread of resistance.
8.11 Antibiotic Targets and Antibiotic Resistance
Antibiotics are antimicrobial agents produced by microorganisms, primarily certain bacteria and fungi. These agents are characterized by their ability to either kill or inhibit the growth of bacteria, and all target molecular processes in the cell that are essential to growth and survival. In this section we consider how antibiotics work and some key resistance mechanisms that bacteria have evolved to counter their effects.
Antibiotics That Target Major Molecular Processes
Because all steps within the central dogma (the flow of genetic information; Chapter 6) are essential for growth, many antibiotics specifically target enzymes that catalyze DNA replication, RNA synthesis, and protein synthesis (Figure 8.27). For example, quinolones such as ciprofloxacin target DNA gyrase in gram-negative bacteria and topoisomerase IV in gram-positive bacteria. Thus quinolones lead to cell death by interfering with DNA unwinding and replication (Section 6.1). Likewise, when transcription is inhibited in a growing cell, mRNA cannot be made and new protein synthesis is interrupted. The antibiotics rifampin and actinomycin prevent RNA synthesis by either blocking the RNA polymerase active site (rifampin) or blocking RNA elongation by binding to the major groove in DNA (Section 6.5).
Figure 8.27 Growth targets of select antibiotics.
Many antibiotics inhibit bacterial growth by targeting some aspect of protein synthesis (Figure 8.27). Recall that ribosomes in Bacteria are 70S structures while eukaryotic ribosomes are 80S (Section 6.10). The antibiotic puromycin contains a region that mimics the 3′ end of a tRNA, and this structural mimicry results in specific binding of the antibiotic to the A site in the 70S ribosome; this induces chain termination and effectively shuts down protein synthesis. Aminoglycoside antibiotics such as streptomycin specifically target the 16S rRNA of the 30S ribosome and result in the ribosome misreading mRNAs, thus leading to error-filled proteins that accumulate in the cell and ultimately inhibit growth.
Antibiotics That Target the Cell Membrane and Wall
Other common antibiotic targets include the cell membrane, the cell wall, and specific metabolic processes (Figure 8.27). Daptomycin is a lipopeptide produced by certain Streptomyces species that specifically binds to phosphatidylglycerol residues of the bacterial cytoplasmic membrane (Section 2.1); this leads to pore formation and depolarization of the membrane, ultimately resulting in cell death. Some antibiotics target the gram-negative cell outer membrane (Section 2.4). For example, polymyxins are cyclic peptides whose long hydrophobic tails specifically target the LPS layer and ultimately disrupt membrane structure, causing leakage and cell death.
Several antibiotics target the synthesis of peptidoglycan in bacteria, including the β-lactams penicillin, cephalosporin, and their derivatives. These antibiotics inhibit growth by interfering with proteins that catalyze transpeptidation (penicillin-binding proteins, Section 8.5), the process by which cross-links are made between muramic acid residues that contribute to the structural strength of peptidoglycan (Section 2.3). Similarly, the antibiotic vancomycin inhibits peptidoglycan synthesis in gram-positive bacteria by binding to the pentapeptide of peptidoglycan precursors and preventing the formation of peptide interbridges by transpeptidases. The topical antibiotic bacitracin prevents peptidoglycan synthesis by binding to the peptidoglycan precursor transport system (bactoprenol, Section 8.5) and preventing new peptidoglycan precursors from reaching the site of peptidoglycan synthesis. As autolysins continue to introduce small gaps in the existing peptidoglycan (Figure 8.16a), a shortage of precursors to patch the gaps weakens the cell wall and leads to cell lysis.
Many other antibiotics and related antimicrobials are known, some of which target other aspects of a bacterium’s biology, such as particular metabolic reactions. But the bulk of clinically useful antibiotics strike one of the targets shown in Figure 8.27. We finish this section with a look at how bacteria have countered the activity of antibiotics by evolving major mechanisms of antibiotic resistance.
Antibiotic Resistance: Spontaneous Mutations and Antibiotic Modification
If bacteria are to survive the onslaught of antibiotics produced by other microbes (or their own antibiotics if they are an antibiotic-producing organism), they require resistance mechanisms of one sort or another. Resistance mechanisms are genetically encoded and fall into four classes: (1) modification of the drug target, (2) enzymatic inactivation, (3) removal from the cell via efflux pumps, and (4) metabolic bypasses (Figure 8.28).
Figure 8.28 Mechanisms of antibiotic resistance.

Penicillin (a β-lactam antibiotic) is used to illustrate the mechanisms of modified target (modified porin), drug inactivation (β-lactamase), efflux pump, and metabolic bypass (alternative penicillin-binding protein). Genes for efflux pumps and β-lactamase enzymes can be plasmid or chromosomally encoded, while the alternative penicillin-binding protein is encoded only on the chromosome. PBP: Penicillin-binding protein. Mechanisms of antibiotic resistance are in green bold. Insert: Electron micrograph of the ArcAB-TolC efflux system from Escherichia coli positioned in an illustration of a gram-negative cell envelope.
Random chromosomal mutations can lead to antibiotic resistance. For example, spontaneous mutants of Escherichia coli or other bacteria resistant to the antibiotic rifampin—which inhibits the activity of RNA polymerase—can be obtained by simply exposing a large cell population to the antibiotic. Under such conditions, spontaneous mutants that produce an altered RNA polymerase unaffected by rifampin are strongly selected for.
Resistance genes can also exist on a variety of mobile genetic elements (Chapter 6), and such genes can be readily transmitted between bacteria of the same or different species by horizontal gene flow (Chapter 9). Unlike a spontaneous mutation that affects the target of the antibiotic, many of these mobile resistance genes, especially ones transmitted by R plasmids, encode enzymes that inactivate the antibiotic by altering its structure, either by chemical modification or cleavage. As examples, β-lactamase binds to β-lactam antibiotics and cleaves a key ring structure in the molecule, and an acetylating enzyme adds acetyl groups to free hydroxy groups of chloramphenicol; once cleaved or chemically modified, the drugs can no longer bind to their target and microbial growth can continue.
Antibiotic Resistance: Efflux Pumps and Metabolic Bypasses
Another effective resistance strategy is to pump out antibiotics that have entered the cell. Efflux pumps are ubiquitous in gram-negative bacteria and function by transporting various molecules, including antibiotics, out of the cell (Figure 8.28). Efflux lowers the intracellular concentration of an antibiotic and thus allows the cell to survive at higher external concentrations. Many efflux pumps act promiscuously by transporting different classes of antibiotics outside of the cell, which contributes to the problem of multidrug resistance. The AcrAB-TolC efflux system of E. coli is one of the best-characterized efflux pumps and can pump out several antibiotics including rifampin, chloramphenicol, and fluoroquinolones (Figure 8.28, inset).
Growth as a biofilm (Section 8.10) leads to increased antibiotic resistance, a characteristic that makes infections caused by biofilm-forming bacteria difficult to treat. Although the exopolysaccharide matrix of a biofilm decreases the permeability of antibiotics, efflux pumps also play a major role. For example, genes encoding the AcrAB-TolC efflux pump in E. coli are upregulated when cells enter a biofilm growth mode. Pseudomonas aeruginosa, a classic biofilm former, encodes several multidrug efflux pumps that are also more active when cells grow in an attached state.
A final form of antibiotic resistance occurs when the target of the antibiotic is no longer essential to the cell’s metabolism or survival. A classic example of this is methicillin-resistant Staphylococcus aureus (MRSA), the causative agent of a variety of serious, even life-threatening, infections (Section 31.9). Methicillin is a β-lactam antibiotic that, like other penicillins, targets the activity of penicillin-binding proteins. However, unlike many penicillin derivatives, methicillin is resistant to cleavage by β-lactamase. Although many strains of S. aureus are killed by methicillin, MRSA strains contain a 20- to 60-kilobase DNA chromosomal (or genomic) island (Section 13.10) called the Staphylococcus chromosomal cassette for methicillin resistance (SCCmec); these genes encode an alternative penicillin-binding protein called MecA that is not recognized by methicillin or other β-lactams (Figure 8.28).
Interestingly, MRSA strains synthesize MecA only in the presence of methicillin or other β-lactams. This regulation is due to the presence of the repressor protein MecI and the β-lactam sensor MecR1. In the absence of a β-lactam antibiotic, MecI binds to the operator site (Section 7.2) of mecA and represses transcription. If the cell is treated with methicillin or other β-lactams, it triggers the MecR1 protease to degrade the MecI repressor. This induces transcription of mecA (and thus the production of MecA), which allows MRSA strains to synthesize peptidoglycan in the presence of methicillin. The chromosomal island of MRSA strains also possesses a transposable element (Section 6.2) that encodes resistance to the antibiotics erythromycin and spectinomycin. The island is thus likely subject to horizontal transfer to confer multidrug resistance. This reality makes MRSA a serious problem for clinical medicine.
Check Your Understanding
Describe two targets of antibiotics and discuss why the drugs are effective.
Some antibiotics target peptidoglycan synthesis. What is a molecular growth target of an antibiotic that inhibits peptidoglycan synthesis?
Why are efflux pumps capable of conferring multidrug resistance?
8.12 Persistence and Dormancy
Besides being sensitive or resistant to antibiotics, a third growth response is possible—persistence. Persistence occurs when a population of antibiotic-sensitive bacteria produces rare cells that transiently become tolerant to multiple antibiotics (**Figure 8.29*a***). These persisters, as they are called, are genetically identical to their antibiotic-susceptible siblings, and thus persistence is not inheritable or conferred by genetic transfer. Instead, persisters, which form at a rate of 10−6 to 10−4 in exponentially growing cultures, derive from a state of dormancy in which the cells are viable but do not grow.
Figure 8.29 Persistence and the HipAB toxin–antitoxin module.

(a) Antibiotic selection of persister cells and recovery from dormancy. (b) HipAB toxin–antitoxin module. During normal conditions, HipB sequesters the toxin HipA. The presence of Lon–PolyP leads to inactivation of the HipB antitoxin and free HipA toxin. HipA leads to slow growth and persistence and positively regulates the hipAB operon. (c) Mechanism of HipA toxin. HipA phosphorylates GltX, preventing the tRNA synthetase from charging amino acids. This leads to an uncharged tRNA entering the A site of the ribosome and the stimulation of RelA to produce (p)ppGpp.
Because antibiotics target active processes, the dormant state prevents the antibiotic from killing the cell. When antibiotic treatment is stopped, the dormant cells emerge from dormancy and grow. Persistence is believed to be the cause of recurring infections of Mycobacterium tuberculosis in those with chronic tuberculosis (Section 31.4) and of Pseudomonas aeruginosa in those suffering from the genetic disease cystic fibrosis.
How does a subpopulation of cells go dormant while genetically identical daughter cells do not? For a cell to become a persister and be able to later reverse the process, coordinated molecular events that affect cell growth must take place. The keys to this unusual phenomenon are chromosomally encoded toxin–antitoxin modules, the stringent response (Section 7.9), and a phenomenon called phenotypic heterogeneity. Sporulation is a classic example of cells in a population displaying different phenotypes (Section 8.6). This phenotypic heterogeneity occurs because gene expression is not equal in all cells of a population. As Figure 8.30 illustrates, a subpopulation of vegetative cells remains metabolically active after other cells have committed to forming endospores.
Figure 8.30 Phenotypic heterogeneity during sporulation.

Fluorescence micrograph illustrating a population of Bacillus subtilis cells with some members committing to developing spores, while other cells remain vegetative and metabolically active. Cells yellow or green in color are sporulating cells expressing GFP linked to a promoter for the sporulation factor Spo0A (Figure 8.18) (developing endospores are green). Cells red in color are vegetative cells expressing a red fluorescent protein linked to tryptophan synthesis genes.
Mutlu, A., Trauth, S., Ziesack, M., Nagler, K., Bergeest, J.P., Rohr, K., Becker, N., Höfer, T., and Bischofs, I.B. 2018. Nat. Commun. 9: 69
Toxin–Antitoxin Modules
Toxin–antitoxin (TA) modules are genes that encode two components: a toxin whose production inhibits cell growth and an antitoxin that counteracts the activity of the toxin. TA modules are found in almost all Bacteria and in many Archaea. The identification of over 30 TA systems in Escherichia coli and 60 TA systems in M. tuberculosis suggests that TA modules play a role not only in normal physiology but also in pathogenicity. It is thought that toxin activity promotes cellular adaptation to ever-changing environments by slowing down cell growth to help ensure survival during stressful conditions.
The hipAB genes encode a TA module that has been shown to trigger persistence in E. coli (Figure 8.29b). In this module, HipA is a toxin that inhibits translation and HipB is an antitoxin that is susceptible to a protease called Lon. Under normal cellular conditions, the HipA toxin and HipB antitoxin form a stable complex that prevents HipA from exerting its toxicity. However, if Lon is activated by its signal molecule polyphosphate (PolyP), the HipB antitoxin is degraded. Without the HipB antitoxin present to neutralize the HipA toxin, translation is inhibited and cell growth is arrested (Figure 8.29b).
How does the HipA toxin inhibit translation? HipA acts as a kinase (phosphorylating protein) targeting glutamyl-tRNA synthetase (GltX). If HipA is free to phosphorylate GltX, GltX can no longer charge its respective tRNAs with glutamine. This leads to uncharged tRNAs entering the A site of the ribosome (Section 6.10) and ultimately to stalling of the ribosome and activation of RelA (Figure 8.29c). Stalling, in turn, inhibits translation and thus protein synthesis. Free HipA also binds upstream of the hipAB operon, further activating transcription and thus increasing the concentration of cellular HipA (Figure 8.29b).
Steps to Dormancy
As we have seen, free HipA induces ribosome stalling. This stalling not only inhibits translation but also leads to the most important criteria for the development of persistence—production of the alarmone (p)ppGpp (guanosine tetraphosphate and guanosine pentaphosphate) by RelA and induction of the stringent response pathway (Figure 8.29c).
Recall that the stringent response leads to decreased rRNA and tRNA synthesis, and thus protein synthesis is inhibited (Section 7.9). The pathway also leads to decreased DNA replication and cell division. Thus, those cells in a population that have triggered the stringent response no longer actively grow; they enter a state of dormancy. Although this cascade of events only occurs in a small subpopulation of cells, antibiotic treatment strongly selects and enriches for multidrug-tolerant persister cells. Once the antibiotic exposure has ended, the persisters can exit the stringent response pathway and resume producing antitoxin to neutralize the effect of the toxin. When this happens, protein synthesis returns to normal levels, allowing cells to grow.
Why does this cascade of events only occur in a subpopulation of cells without any sort of genetic direction? HipA-induced persistence occurs in cells that randomly produce higher amounts of the signal molecule PolyP. While it is often assumed that cells within a clonal population are homogeneous, cell-to-cell differences in gene expression and in metabolite and protein content do occur, resulting in phenotypic heterogeneity (Figure 8.30). These differences can be the result of RNA polymerase producing a subset of mRNAs through random interactions with DNA or of unequal distribution of macromolecules between daughter cells during cell division. These cell-to-cell variations can have many outcomes, but a major one for clinical medicine is the persistence of difficult-to-eradicate pathogenic bacteria whose dormant state can falsely signal a cure, only to be followed by reinfection by the same pathogen at a later date.
We move on now to a new chapter, one whose focus is on genetics and the mechanisms for genetic exchange that Bacteria and Archaea have exploited to rapidly obtain new metabolic and other properties that boost their chances of survival and reproduction.
Check Your Understanding
What prevents the toxin component of TA modules from killing the cell under normal conditions?
What occurs in the cell that results in freed HipA toxin?
Chapter Review
Go to Mastering Microbiology for videos, animations, practice tests, and more.
I Bacterial Cell Division
8.1 Macromolecules such as proteins and nucleic acids can be visualized in living cells using fluorescent tagging and microscopy techniques. Super-resolution microscopy can even resolve two individual molecules, generate 3D images, and track the movement of molecules throughout the cell.
Q What type of probes must be used for super-resolution microscopy and why?
8.2 Before cell division occurs in prokaryotic cells, two complete copies of the genome must be present. While multiple replication forks can exist to decrease generation times, the process must be regulated. The nucleoid must also segregate to the poles of the cell to allow the septum to form.
**Q What is the role of PopZ in segregating the Caulobacter daughter chromosomes?**
8.3 Cell division and chromosome replication are coordinately regulated, and the Fts proteins are keys to these processes. With the help of MinE, FtsZ defines the cell-division plane and helps assemble the divisome, the protein complex that orchestrates cell division.
Q What is the role of the penicillin-binding protein FtsI in cell division?
8.4 MreB helps define cell shape, and in rod-shaped cells, MreB forms patchlike filaments that direct cell wall synthesis along the long axis of the cell. The protein crescentin plays an analogous role in Caulobacter, leading to formation of a curved cell. The eukaryotic cell proteins actin and tubulin that affect shape and cell division have prokaryotic cell counterparts.
Q What morphology do prokaryotic cells have that lack MreB or crescentin?
8.5 During bacterial growth, new cell peptidoglycan is synthesized by the insertion of peptidoglycan precursors into the preexisting peptidoglycan fabric. Bactoprenol facilitates transport of these units through the cytoplasmic membrane. Transpeptidation completes the process of cell wall synthesis by cross-linking adjacent ribbons of peptidoglycan at muramic acid residues.
Q What are autolysins and why are they necessary?
II Regulation of Development in Model *Bacteria*
8.6 Sporulation in Bacillus during adverse conditions is triggered by a complex phosphotransfer relay system that monitors multiple aspects of the environment. The sporulation factor Spo0A then sets in motion a cascade of regulatory responses under the control of several alternative sigma factors.
**Q What is a common trigger for sporulation in Bacillus?**
8.7 Endospore germination occurs when nutrients become available. This developmental process occurs in three stages: activation, germination, and outgrowth. Activation is triggered by nutrients binding to germinant receptors within the endospore. This binding triggers a cascade of events that ultimately leads to rehydration of the endospore, normal metabolism, growth, and cell division.
Q What type of genes are upregulated during the different stages of endospore germination?
8.8 Differentiation in Caulobacter consists of the alternation between motile cells and those that are attached to surfaces. Three major regulatory proteins—CtrA, GcrA, and DnaA—act in succession to control the three phases of the cell cycle. Each in turn controls many other genes needed at specific times in the cell cycle.
**Q How is the Caulobacter life cycle similar to the eukaryotic cell cycle?**
8.9 Heterocyst formation requires expression of the major regulatory protein HetR in the protoheterocysts. However, the protein must be inactivated in vegetative cells by diffusion of the PatS peptide along the filament.
Q What is meant by “patterning” during heterocyst formation?
8.10 During biofilm formation, cells make contact with a surface using cell structures such as flagella and pili. Once cells are attached to a surface, both intra- and intercellular signaling molecules lead to the production of an extracellular matrix and colonization. Biofilms are not static entities, and the final step in their development is dispersal and release of cells.
Q What type of genes does c-di-GMP activate during biofilm formation?
III Antibiotics and Microbial Growth
8.11 Antibiotics are antimicrobial agents produced by microorganisms that target processes critical for growth such as DNA replication, transcription, protein synthesis, and cell wall synthesis. Specific mechanisms of antibiotic resistance include modification of the drug target, enzymatic inactivation, removal via efflux pumps, and metabolic bypass. These mechanisms are either chromosomally or plasmid encoded.
Q Why do antibiotics that target bacterial ribosomes not make humans sick?
8.12 Persistence is a dormant growth state in which cells are not susceptible to antibiotics. This lack of sensitivity is transient and thus not considered antibiotic resistance. Persistence is the result of toxin–antitoxin modules and the induction of the stringent response. Cells in the persistent state can resume growth once antibiotic exposure ceases.
Q What are the steps to inducing the stringent response during the development of persistence?
Application Questions
If DnaA was not regulated in Escherichia coli and multiple rounds of replication were completed before cell division, what would be the consequence to the daughter cell and why? Would the resulting cell still be considered haploid?
Explain how cells exhibiting different phenotypes under the same conditions can possess the same genotype. Give examples in your explanation.
Describe how you would genetically design a “superbug” resistant to β-lactams, methicillin, streptomycin, daptomycin, and trimethoprim.
Discuss why so many different sigma factors can be produced by bacteria such as Bacillus.
Chapter Glossary
a chemical substance produced by a microorganism that kills or inhibits the growth of another microorganism FtsZ
a protein that forms a ring at the future site of septum formation in a prokaryotic cell Green fluorescent protein (GFP)
a protein that fluoresces green and is widely used in genetic analysis Heterocysts
specialized cells dedicated to nitrogen fixation Persistence
a dormant growth state in which a few members of a genetically identical population are transiently tolerant to multiple antibiotics Reporter gene
a gene used in genetic analysis because the product it encodes is easy to detect Super-resolution microscopy
a form of light microscopy that employs special fluorescent molecules to resolve structures as small as 5–50 nm Toxin–antitoxin (TA) module
a genetic locus encoding a toxin that inhibits growth and an antitoxin that neutralizes the activity of the toxin