Omics Tools Unravel Mysteries of “Fettuccine” Rocks
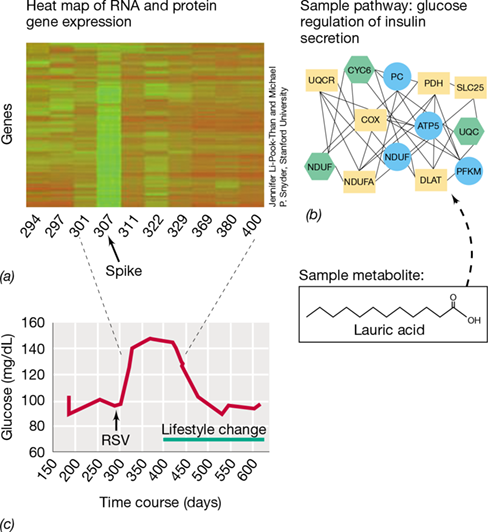
Mammoth Hot Springs (MHS) in Yellowstone National Park (USA) displays many geochemical traits similar to those on Mars. Because of this, geomicrobiologists study these hot, oxygen-limited and sulfur-rich springs for microbial fossils whose fingerprints of life (biomarkers) could be used to detect life on other planets. The photo on the left shows filamentous microbial mats from MHS that resemble fettuccine pasta as a result of mineral encrustation. Tens of centimeters long, these streamers contain extremophilic microbes that entomb themselves in 5 millimeters of travertine (calcium carbonate) per day, a striking biomarker that can be easily observed. How are these pasta-looking rocks formed?
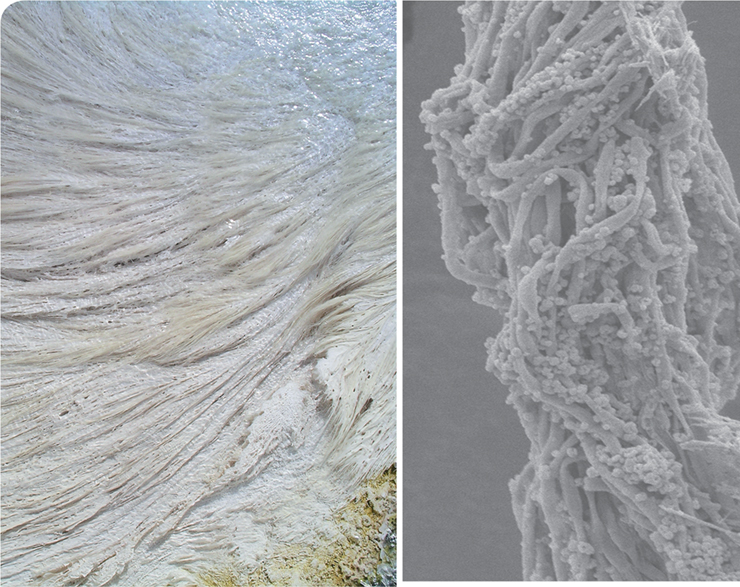
Using a combination of geochemical measurements, microscopy, and metagenomic, metatranscriptomic, and metaproteomic approaches, scientists have unraveled many of the mysteries surrounding the MHS streamers. These mats are dominated by the thermophilic, chemolithotrophic bacterium Sulfurihydrogenibium yellowstonense, which uses CO2 and reduced sulfur as carbon and energy sources, respectively. Multi-omic data sets indicate that these remarkable cells produce pili as well as extracellular polymeric substances to latch onto one another and form streamers in the fast-moving spring water. Once the cells have formed microbial filaments (see scanning electron micrograph on the right), proteins expressed on the surface of S. yellowstonense catalyze the formation of travertine over a billion times faster than it would occur abiotically.
What is the advantage of forming these mineral-encrusted filamentous streamers? By undulating within travertine ridges, S. yellowstonense is able to maximize its access to the minimal oxygen and sulfide—key substrates for its energy metabolism—available within MHS waters. Thus, this organism has evolved an ingenious way to optimize chemolithotrophic growth.
Besides revealing fascinating microbiology, this study illustrates the power of omics tools to characterize microbes and their physiology in natural environments. It also highlights the utility of combined omics approaches for detecting biomarkers useful for searching for life beyond Earth.
Source: Dong, Y., et al. 2019. Physiology, metabolism, and fossilization of hot-springs filamentous microbial mats. Astrobiology doi:10.1089 /ast.2018.1965.
Traditional approaches to studying microbial physiology and biochemistry have focused on the analysis of individual biochemical pathways or molecular responses under specific conditions. While informative, this reductionist approach can only target a specific gene or subset of genes or gene products—RNAs and proteins—and fails to address the dynamic nature of microorganisms and how a network of biological molecules controls their behavior in a coordinated fashion. By contrast, “omics” is a broad discipline that integrates different methodologies to characterize and quantify large pools of biological molecules. Through the power of combining various omics, a detailed picture of an organism’s response to its environment can be generated. Because the ability to store and analyze massive amounts of biological information by computer is essential to omics, the understanding of entire biological systems is evolving in parallel with computing power and storage and retrieval capabilities.
I Genomics
Buried within a genome sequence lies the heart of a cell’s biology, and with the sequencing technology available today, microbiology—indeed all of biology—is moving ahead faster than ever as genomes unveil their secrets at a breathtaking pace.
The foundation of omics biology lies in nucleic acid and protein sequences, characteristics ultimately controlled by the cell’s genome. The genome is an organism’s entire complement of genetic information, including genes that encode proteins, RNAs, and regulatory sequences, as well as any noncoding DNA that may be present. The genome sequence of an organism not only reveals its genes, but also yields important clues to how the organism functions. While new omics are coined with regularity, this chapter focuses on the major omic themes of biological molecules—genomics, transcriptomics, proteomics, and metabolomics—and describes how these various pieces of the puzzle are integrated to yield important information about the biology of a single organism, or even an entire microbial community in the case of metagenomics, the comprehensive analysis of specific genes or genomes in an environment (Figure 10.1).
Figure 10.1 Utility of microbial genome sequences.

A genome sequence allows for the development of omics approaches and tools for understanding, investigating, and monitoring microorganisms, both in culture and in nature. It can also provide targets for drug and vaccine design.
The word genomics refers to the discipline of mapping, sequencing, analyzing, and comparing genomes. Here we review how genomes are sequenced and some techniques used to analyze these genomes and their gene content.
10.1 Introduction to Genomics
Advances in genomics rely heavily on improvements in molecular technologies and computing power. The automation of DNA sequencing and the development of powerful computational tools for DNA and protein sequence analysis have reduced the cost and increased the speed at which genomes are analyzed. Thus the number of sequenced genomes has grown rapidly, with the major genomics bottleneck being the digestion of vast amounts of nucleic acid sequence data.
Genomics: Then and Now
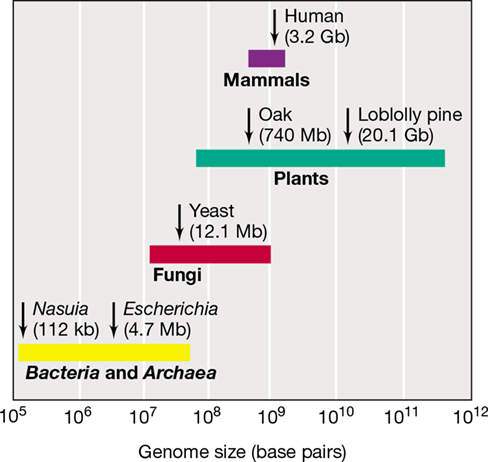
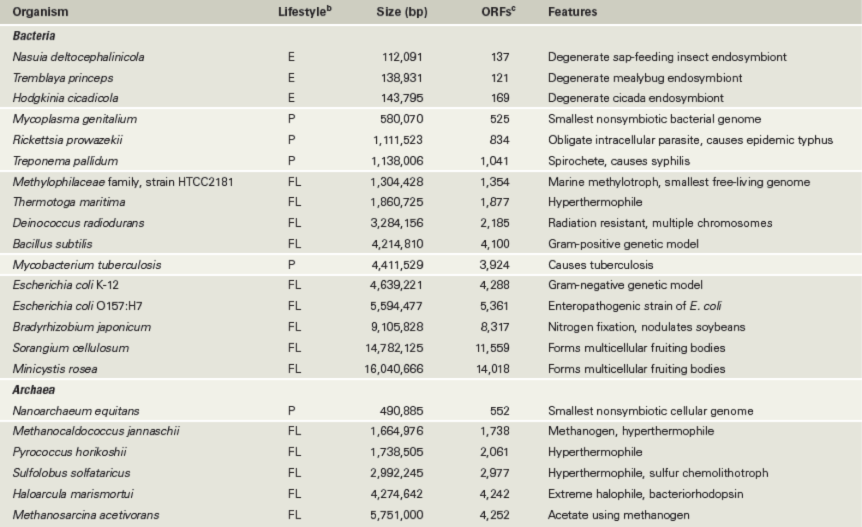
The first genomes sequenced were those of small viruses over 40 years ago, and the first bacterial genome sequence was published in 1995. Today, DNA sequences from over 125,000 Bacteria, Archaea, and viruses, as well as datasets from metagenomic projects (Section 10.7), are available in public databases such as the Genomes Online Database (GOLD; see https://gold.jgi.doe.gov for an up-to-date list). With the goal of using genome sequences to advance systems and ecosystems biology, the United States Department of Energy’s Joint Genome Institute (JGI) sponsors GOLD. The JGI has also joined the Genomic Encyclopedia of Bacteria and Archaea (GEBA) in an effort to expand the coverage of genome sequences across the phylogenetic tree of life (Figure 1.41b). At the time of this writing, GEBA had added over 1000 new genome sequences to the databases. These genome sequences represent a phylogenetically diverse range of Bacteria and Archaea. Despite these additions, there is plenty of “space” within the genomic tree of life (Figure 1.41) for new microbial genome sequencing projects. Table 10.1 lists some representative genomes from Bacteria and Archaea. The genomes of many eukaryotic organisms have also been sequenced, including the haploid human genome, which contains about 3.2 billion bp (∼21,000 protein-encoding genes, Figure 10.2). As astonishing as this large number might seem, the human genome is far from the largest genome known. Of all life forms on Earth, certain plants are known to contain the largest genomes (Figure 10.2).
Table 10.1 Genomes of select species of *Bacteria* and *Archaea*a

aInformation on prokaryotic genomes can be found at https://gold.jgi.doe.gov.
cOpen reading frames. Genes encoding known proteins are included, as well as ORFs that could encode a protein greater than 100 amino acid residues. Smaller ORFs are not included unless they show similarity to a gene from another organism or unless the codon bias is typical of the organism being studied.
Figure 10.2 Genome sizes of microbial cells and higher organisms.

Compare with viral genome sizes in Chapter 11 (Figure 11.1). The flowering plant Paris japonica has the largest known genome, some 50 times that of humans.
What Can Genomes Tell Us?
As we will discuss throughout this chapter, modern microbiology thrives on genome sequences; indeed, little in microbiology has been left untouched by genomic sequences. Microbial genome sequencing has discovered everything from genes encoding heat-stable enzymes in microbes that thrive in boiling water (Figure 4.25b) to genes that encode virulence factors in the most vicious pathogens. Genome sequencing has also been instrumental in developing microarrays for studying gene expression (Section 10.8), detecting horizontal transfer events (between microbes of different species, genera, and even phyla; Chapters 9 and 13), monitoring and diagnosing disease outbreaks (based on the presence of “signature genes” of different pathogens), discovering CRISPRs (Section 9.12) and other anti-bacteriophage systems (Section 10.5), understanding metabolic pathways, and discerning the growth requirements of microbes that have thus far defied laboratory culture.
The ability to sequence genomes has also been used to solve obscure medical mysteries. An excellent example is the genomics that revealed the causative agent of the “Black Death,” which swept through Europe in the middle of the fourteenth century (**Figure 10.3*a***). While it was believed that the Black Death was caused by a massive outbreak of bubonic plague, a typically fatal disease caused by the bacterium Yersinia pestis (Section 32.7), scientists could not be positive until they recovered and sequenced DNA samples from the teeth and bones of corpses of people known to have died from the Black Death. By comparisons of this ancient DNA with the genome of Y. pestis, the mystery behind this devastating medieval disease was unraveled: The Black Death was indeed bubonic plague.
Figure 10.3 Diverse examples of what genomes can tell us.

(a) Genomics helped solve an ancient medical mystery surrounding plague. The blackened skin on the toes of this modern plague victim originates from hemorrhaging due to systemic infection with the pathogenic bacterium Yersinia pestis, shown in inset. (b) Genome sequencing was used to assign the marine ammonia-oxidizing archaeon Nitrosopumilus to a new phylum of Archaea, the Thaumarchaeota.
Microbial genomics has also been used to identify new microbial phyla. For example, until recently, only three phyla of Archaea were known—Euryarchaeota, Crenarchaeota, and Nanoarchaeota (Chapter 17). Because every cultured species was isolated from an extreme environment, many microbiologists concluded that Archaea were mainly extremophiles and that they did not inhabit oceans, lakes, and soil in significant numbers. However, based on environmental 16S rRNA gene sequencing, Archaea only marginally affiliated with Crenarchaeota were detected in marine and freshwater samples. Who were these organisms, and how were they making a living? Subsequently, Nitrosopumilus, the first ammonia-oxidizing (nitrifying) archaeon known, was isolated (Figure 10.3b; Section 14.9). Using the powerful analytical tools of genomics, the genomes of two distinct ammonia-oxidizing Archaea were compared with those of all other Archaea. This genomic analysis clearly showed that these ammonia-oxidizing Archaea belonged in a new phylum, now called the Thaumarchaeota (Section 17.5).
The above is just a taste of how genomics has impacted microbiology. Other relevant examples will appear regularly as you make your way through this text. The major message here is twofold: (1) We are clearly living in the era of microbial genomics, and (2) the genomics revolution has spawned a wealth of powerful tools to attack old problems in new ways. Indeed, in the past 40 years or so, microbiology as a science has leapt forward farther and faster than at any time in its history.
Check Your Understanding
How many protein-encoding genes are in the human genome?
List three examples of how genomics has led to major new discoveries in microbiology.
10.2 Sequencing and Annotating Genomes
In biology, the term sequencing refers to determining the precise order of subunits in a macromolecule. In the case of DNA (or RNA), the sequence is the order in which the nucleotides are aligned. DNA sequencing today forms the heart of the omics revolution and its technology is advancing so quickly that new methods appear every year. Yet despite the technological breakthroughs that have catapulted us into the omics age, some of the earliest sequencing methodologies—born of simple yet brilliant basic science—form the foundation of the latest methods today (see next subsection).
After sequencing and assembly of the gene fragments, the next step is genome annotation, the conversion of raw sequence data into a list of the genes and other functional sequences present in the genome. The term bioinformatics refers to the use of computers to store and analyze the sequences and structures of nucleic acids and proteins. Improved sequencing methods are now generating data almost faster than it can be properly analyzed. Thus, although automated annotation software exists and powerful new versions are being developed at a rapid pace, annotation remains the major “bottleneck” in genomics. Here we focus on the process of genome sequencing, assembly, and annotation.
DNA Sequencing
The first widely used method for sequencing DNA was the dideoxy method developed by the British scientist Fred Sanger, who won a Nobel Prize (his second) for this accomplishment. In the Sanger procedure the sequence is determined by making a copy of the original single-stranded DNA in a process similar to the polymerase chain reaction (PCR, Section 12.1). The secret behind the Sanger method was the addition of a mixture of normal deoxyribonucleotides (dNTPs) and small amounts of the corresponding dideoxyribonucleotides (ddNTPs), one for each of the four bases—adenine, guanine, cytosine, and thymine—to the mixture used to make the DNA copy (**Figure 10.4*a***). The dideoxy analog is a specific chain-terminator; because it lacks a 3′-hydroxyl, the analog prevents further elongation of the chain after its insertion. Because ddNTPs insert randomly, DNA chains of varying length are produced in the synthesis reaction (Figure 10.4a). Sanger sequencing originally used radioactive labels, but automated systems were quickly developed that used a separate fluorescent label for each different ddNTP and detected the DNA products (separated by passing through a sizing column) with a laser (Figure 10.4a).
Figure 10.4 DNA sequencing.
(a) Sanger sequencing. When a polymerase incorporates a ddNTP during synthesis, the chain of DNA is terminated. The identity of the terminal ddNTP can be determined by capillary electrophoresis and fluorescence detection. (b) Pyrosequencing. Whenever a new dNTP is inserted into the growing strand of DNA (red arrows), pyrophosphate (PPi) is released and is used to make ATP from AMP by the enzyme sulfurylase. The ATP is consumed by the enzyme luciferase, which releases light. Unused dNTPs are degraded by the enzyme apyrase (gray arrow).
Mastering Microbiology
Because the original Sanger method was dependent on primers binding to a known sequence and was limited to around 800 nucleotides per reaction, entire chromosomes or large DNA molecules could not be sequenced in a single reaction. Instead large DNA molecules had to be cut into smaller fragments and cloned into vectors for sequencing. This led to the development of new sequencing technologies, which appear now with such regularity that the term “next-generation sequencing” is commonly used to describe the latest and greatest in nucleic acid sequencing. For example, pyrosequencing, a second-generation sequencing method still widely used today, is based on the Sanger method and employs the light-emitting enzyme luciferase to detect incorporation of dNTPs by emitting a pulse of light (Figure 10.4b). It should be noted that despite the advent of new sequencing technologies, the Sanger method is still routinely used to sequence plasmid constructs and PCR products. Table 10.2 summarizes modern sequencing methods and illustrates how the cost of sequencing 1 megabase (Mbp, million base pairs) of DNA dropped over 100,000-fold in 15 years.
Genome Assembly and Annotation
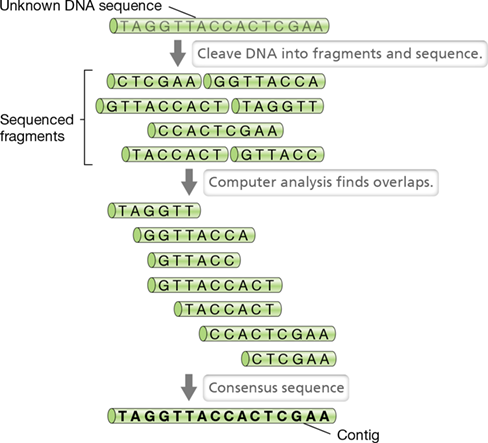
Regardless of which sequencing system is used—even hand-held systems are available today (see Explore the Microbial World, DNA Sequencing in the Palm of Your Hand, on page 300)—the sequences obtained must be assembled before they can be analyzed. Genome assembly consists of putting the fragments in the correct order to reconstruct the chromosome and eliminating any overlaps that may appear. Then, for assembled genomic sequences to be useful, they must be annotated in order to identify genes and other functional regions. Many of the tasks surrounding genome assembly and annotation are highly computational. For genome assembly, a computer examines many short DNA fragments that have been sequenced and deduces their order by detecting all of the instances where two fragments of DNA possess overlapping sequence (Figure 10.5). These overlaps are used to merge sequencing reads into contigs, or contiguous consensus sequences. Individual contigs with overlapping ends are then aligned to form scaffolds (contigs as well as gaps) that are ultimately used to generate a map representing the complete genome.
Figure 10.5 Computer assembly of DNA sequence.

Most DNA sequencing methods generate vast numbers of short sequences (30 to several hundred bases) that must be assembled. The computer searches for overlaps in the short sequences and then arranges them to form contigs, or a consensus sequence.
From the genome map, the annotation process can begin. Because the genomes of Bacteria and Archaea possess very few intervening sequences (introns, Section 6.6), their genomes essentially consist of a series of open reading frames (ORFs) separated by short regulatory regions and transcriptional terminators. A functional ORF is one that actually encodes a protein (Section 6.9) and can be identified from a computer search of the sequence (Figure 10.6). Although any given cellular gene is always transcribed from one DNA strand, a gene can actually be located on either strand and thus computer inspection of both strands of DNA is required.
Figure 10.6 Computer identification of possible ORFs.

The computer scans the DNA sequence looking first for start and stop codons. It then counts the number of codons in each uninterrupted reading frame and rejects those that are too short. The probability of a genuine ORF is made stronger if a likely ribosomal binding site (RBS) is found the correct distance in front of the reading frame. Codon bias calculations are used to test whether an ORF complies with the codon usage of the organism being examined.
Finding and Identifying ORFs
The first step in finding an ORF is to locate start and stop codons in the sequence (Section 6.9 and Table 6.4). However, in-frame start and stop codons appear randomly with reasonable frequency; thus, further clues are needed. In Bacteria, translation begins at start codons located immediately downstream of a ribosome-binding sequence (RBS or Shine–Dalgarno site) on the mRNA (Section 6.9). Thus, locating potential ribosome-binding sequences in addition to start and stop codons helps decide both whether an ORF is functional and which start codon is actually used. In addition, an ORF is more likely to be functional if its sequence is similar to those of ORFs in the genomes of other organisms (regardless of whether they encode known proteins) or if the ORF includes a sequence known to encode a protein functional domain. This is because proteins with similar functions in different cells tend to share a common evolutionary origin and thus share sequence and structural features (Section 13.8). A computer can search for sequence similarities in major databases such as GenBank (http://www.ncbi.nlm.nih.gov/Genbank/) using BLAST (Basic Local Alignment Search Tool), an algorithm that can compare a nucleic acid or protein sequence with all other such sequences in the database.
Other issues must be considered in a genome annotation as well. For example, more than one codon exists for many of the 20 common amino acids (Table 6.4), and some codons are used more frequently than others. The latter is known as codon bias (codon usage) and differs greatly between organisms. For example, Table 10.3 shows the different usage of the six arginine codons in Escherichia coli compared to their usage in humans and fruit flies. If the codon bias in a given ORF differs greatly from the consensus for the organism containing it, that ORF may be nonfunctional or may be functional but obtained by horizontal gene transfer (Section 13.9).
Genomic Analyses: The Final Tally
No genome sequence project ends with 100% of the genome identified. In fact, this is one of the most exciting and challenging findings of genomic analyses: Many genes in microbes almost certainly encode proteins whose function(s) remain unknown. Although there are differences among organisms, in most genomes the percentage of genes whose role can be clearly identified is approximately 70% of the total number of ORFs detected. Uncharacterized (or unknown) ORFs are said to encode hypothetical proteins, proteins that probably exist although their function is unknown. These ORFs have uninterrupted reading frames of reasonable length and the necessary start and stop codons and ribosome-binding site (Figure 10.6); however, the proteins they encode lack sufficient amino acid sequence homology with any known protein to be unambiguously identified. Some gene annotations can only assign a gene to a protein family or to a general function (such as “transport protein”) without being more specific. Many of the unidentified genes in E. coli are thought to encode proteins that play a role in some unidentified regulatory process or are proteins required only for special nutritional or environmental conditions. A few may also function as “backups” of key enzymes. Later in this chapter we will discuss methods, which are only possible because of the omics revolution, to help identify the function of these hypotheticals (Sections 10.5 and 10.7).
In addition to protein-encoding genes, some genes encode RNA molecules that are not translated. Such genes therefore lack start codons and may well have multiple stop codons within the gene. Some noncoding RNAs, such as tRNAs and rRNAs, are easy to detect because they are well characterized and are highly conserved. However, many noncoding regulatory RNA molecules (Section 7.12) are conserved only in their three-dimensional structure, with little sequence homology. Thus transcriptomics, specifically RNA-Seq (Section 10.8), has become instrumental in identifying these noncoding genes.
With this general background in nucleic acid sequencing and the coding features of genomes, we move on to compare the nature of genomes in various microbial groups. We begin with the Bacteria and Archaea where thousands of genome sequences are available for comparative analyses.
Check Your Understanding
What key molecules are essential for Sanger sequencing?
What is an open reading frame (ORF)? What is a hypothetical protein?
What is the major problem in identifying genes encoding nontranslated RNA?
10.3 Genome Size and Gene Content in Bacteria and Archaea
10.3 Genome Size and Gene Content in Bacteria and Archaea
10.3 Genome Size and Gene Content in *Bacteria* and *Archaea*
Once a genome has been assembled, comparative genomics using databases such as MicrobesOnline (http://www.microbesonline.org)—which contains nearly 4000 microbial genome sequences—can be used to probe its biological secrets. By using comparative genomics, it has been determined that genomes of Bacteria and Archaea show a strong correlation between genome size and open reading frame (ORF) content (**Figure 10.7*a***). Regardless of the organism, each megabase pair of DNA in a prokaryotic cell encodes about 1000 ORFs, and as the size of these genomes increases, the gene number also increases proportionally. This contrasts markedly with the genomes of eukaryotes, in which noncoding DNA (introns, Section 6.6) may constitute a large fraction of the genome, especially in organisms with large genomes (Figure 10.2).
Figure 10.7 Bacterial and archaeal genome size and content.

(a) Correlation between genome size and ORF content in prokaryotic cells. Analyses of 115 completed genomes from species of both Bacteria and Archaea. Data from Konstantinidis, K.T., and Tiedje, J.M. 2004. Proc. Natl. Acad. Sci. (USA) 101: 3160. (b) Distribution of genome sizes among a representative sample of Archaea (blue) and Bacteria (green). Genomes were chosen based on even distribution across the two phylogenetic domains. Shared frequency of occurrence data points are represented in red. Data adapted from Kellner, S., Spang, A., Offre, P., Szöllosi, G.J., Petijean, C., and Williams, T.A. 2018. Emerging Top. Life Sci. doi: 10.1042/ETLS20180021.
While the sizes of bacterial genomes vary 100-fold, from the insect symbiont Tremblaya princeps with 121 protein-encoding genes to the soil-dwelling Minicystis rosea with over 14,000 genes, archaeal genomes only vary in size 10-fold (Figure 10.7b). This archaeal range encompasses the ectosymbiont Nanoarchaeum equitans with 552 protein-encoding genes to the methanogen Methanosarcina acetivorans with 4252 genes (Figure 10.2 and Table 10.1). Whereas about 1300 genes have been the predicted minimum for the number necessary for a cell to have a free-living existence, recent environmental metagenomic (Figure 1.1 and Section 10.7) datasets have called this earlier estimate into question; individual genomes have been assembled from various environments that contain only 600 to 800 genes (Chapter 1 Explore the Microbial World, “Tiny Cells”). Representative organisms for these genomes are not yet in culture to determine if they are free-living bacteria, but if they are, the genetic requirements for a free-living cell are far lower than originally suspected.
Small Genomes
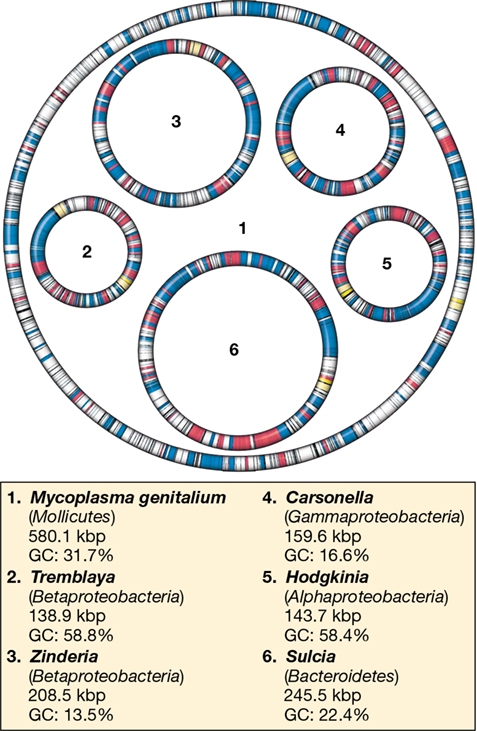
The smallest cellular genomes belong to bacteria that are parasitic or endosymbiotic (cells that live inside other cells), with the insect symbionts Tremblaya (in mealybugs) and Hodgkinia (in cicadas) possessing some of the smallest genomes (around 140 kbp, Table 10.1 and Figure 10.8). The absolute smallest genome discovered thus far is that of Nasuia deltocephalicola, a sap-feeding insect symbiont whose genome is only 112 kbp. Because of their reduced genome size, such symbionts are totally dependent on their insect host cells for survival and nutrients. In turn, the symbionts provide the insect with essential amino acids and other nutrients that the insect cannot synthesize.
Figure 10.8 Symbiont genomes.

Five insect symbiont genomes are shown drawn to scale inside the circle representing the genome of a Mycoplasma. Blue: genes encoding genetic information processing; red: genes encoding amino acid and vitamin biosyntheses; yellow: rRNA genes; white: other genes; gaps indicate noncoding DNA. Kbp, kilobase pairs. GC indicates percentage of nucleotides that are guanine or cytosine.
With genomes of around 500 kbp, Mycoplasma (Bacteria) and Nanoarchaeum equitans (Archaea) have the smallest genomes among parasitic prokaryotic cells (Table 10.1). N. equitans is a hyperthermophile and a parasite of another hyperthermophile, the archaeon Ignicoccus (Section 17.11). N. equitans lacks virtually all genes that encode metabolic proteins and presumably depends on its host for most catabolic as well as anabolic functions. While some pathogens such as Mycobacterium tuberculosis have quite large genomes (4.4 Mbp), the genomes of most human pathogens, such as Mycoplasma, Chlamydia, and Rickettsia, are smaller than the largest known viral genome, that of Pandoravirus (2.5 Mbp, Section 11.1).
Using Mycoplasma, which has around 525 genes, as a starting point, it has been estimated that around 250–300 genes are the minimum number possible for a viable cell. These estimates rely partly on comparisons with other small genomes. In addition, systematic mutagenesis has been performed to identify essential genes. For example, experiments with Escherichia coli and Bacillus subtilis, both of which have about 4000 genes, indicate that approximately 300–400 genes are essential depending on the growth conditions. In fact, synthetic biologists have been able to create a minimal Mycoplasma bacterium with 473 genes (Section 12.12). While this is only 52 genes fewer than that of the Mycoplasma genitalium genome (Table 10.1), the designer microbe has a doubling time of 3 hours versus the weeks of the original M. genitalium. Thus the trimmed-down genome benefits the synthetic bacterium in the laboratory.
Large Genomes
Some Bacteria have genomes that are as large as those of some eukaryotic microbes. In fact, because eukaryotes tend to have significant amounts of noncoding DNA and bacteria do not (Section 10.4), some bacterial genomes actually have more genes than microbial eukaryotes, despite having less DNA. For example, the genome of Bradyrhizobium japonicum, a bacterium that forms nitrogen-fixing root nodules on leguminous plants such as soybeans (Section 23.4), has 9.1 Mbp of DNA and 8300 ORFs, whereas the genome of the baker’s yeast Saccharomyces cerevisiae, a eukaryote, has 12.1 Mbp of DNA and only 5400 ORFs (see Tables 10.1 and 10.5).
The second largest bacterial genome known is that of Sorangium cellulosum, a species of the gliding myxobacteria (Section 15.16; little has been reported on the larger M. rosea genome, Table 10.1). With just under 14.8 Mbp on a single circular chromosome, S. cellulosum has more DNA than several eukaryotes including yeast and the pathogenic protozoans Cryptosporidium and Giardia (see Table 10.5). The S. cellulosum genome is composed of roughly 10.5% noncoding DNA and 11,559 protein-encoding genes, making it over three times larger than the genome of E. coli. Interestingly, the S. cellulosum genome encodes 508 kinases (enzymes that phosphorylate other proteins to regulate their activity), which is over three times that of any other genome including those of eukaryotes. This suggests that the lifestyle of S. cellulosum is highly diverse and that its ecological success requires extensive regulation. In contrast to Bacteria, the largest genomes found in species of Archaea thus far are only about 5 Mbp (Table 10.1).
Gene Content of Bacterial Genomes
The complement of genes in a particular organism reveals its capabilities. Conversely, genomes are molded by adaptation to particular lifestyles. Comparative analyses are useful when searching for genes that encode enzymes that probably exist because of the lifestyle of an organism, and in some cases these searches yield big surprises. For example, Vampirovibrio chlorellavorus is a predatory bacterium that attacks its host, the green alga Chlorella, by surface attachment and ultimate ingestion of its cellular contents (thus the terms “vámpír” from Hungarian, meaning “blood sucker,” and “vorus” from Latin, meaning “to devour,” in the organism’s name). Isolates of V. chlorellavorus existed only as 36-year-old freeze-dried samples that had not been successfully revived. However, by using advanced sequencing techniques, the genome of V. chlorellavorus was recovered, and surprisingly, genomic analyses indicated that V. chlorellavorus falls within the phylum Cyanobacteria (Section 15.3) even though it lacks genes for photosynthesis.
Figure 10.9, reprinted from a scientific journal, is included here to give you an idea of why a microbe’s genome should be sequenced and the amazing amount of information that can be gleaned from annotation, though the details are beyond the scope of this chapter. The figure summarizes some of the metabolic pathways and transport systems of V. chlorellavorus deduced from analysis of its genome. These include an electron transport chain for microaerobic growth (Section 4.16), the ability to ferment (Sections 3.6 and 3.7), chemotaxis abilities (Section 2.11), and the synthesis of 15 of the 20 essential amino acids (Figure 6.27). Comparative genomics also indicated that V. chlorellavorus used a conjugative type IV secretion system (Section 6.13) to attack its prey, the first discovery of this strategy in a predatory bacterium.
Figure 10.9 Functional and metabolic predictions for *Vampirovibrio chlorellavorus* based on genomic annotation.

Although the details are beyond our discussion, the figure illustrates the power of genomic sequencing and annotation on piecing together the physiology of an organism. Within the cytoplasmic membrane, the following systems are highlighted: secretion (green), chemotaxis and movement (blue), electron transport (red), ATP-binding cassette transporters (yellow), and permeases/pumps/transporters (orange). Black ovals indicate substrates that enter the glycolysis pathway, while fermentation end products are indicated as black rectangles. Colors of internal compounds correspond to the following: green (amino acids), red (cofactors and vitamins), purple (nucleotides), and orange (non-mevalonate pathway products). Note that genes for synthesis of serine (highlighted in blue) are not present, so presumably it is transported into the cell. Adapted from Soo, R.M., et al. 2015. Peer J. 3: e968.
Soo, R.M., Woodcroft, B.J., Parks, D.H., Tyson, G.W., and P. Hugenholtz. 2015, PeerJ 3: e968
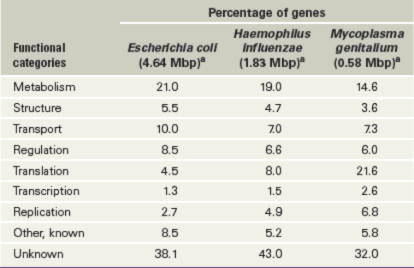
A functional analysis of genes and their activities in several bacteria is given in Table 10.4. Thus far, a distinct pattern of gene distribution in Bacteria has emerged. Metabolic genes are typically the most abundant class in bacterial genomes, although genes for protein synthesis overtake metabolic genes on a percentage basis as genome size decreases (Table 10.4 and Figure 10.10). Although many genes can be dispensed with, genes that encode the protein-synthesizing apparatus cannot. Thus, the smaller the genome, the greater the percentage of genes that encode translational processes. Conversely, the larger the genome, the more genes there are for transcriptional regulation and signal transduction (Chapter 7).
Table 10.4 Gene function in some genomes of *Bacteria*

aChromosome size, in megabase pairs. Each organism listed contains only a single circular chromosome.
Figure 10.10 Functional category of genes as a percentage of the genome.

The percentage of genes encoding products for translation or DNA replication is greater in organisms with small genomes, whereas the percentage of transcriptional regulatory genes is greater in organisms with large genomes.
Analyses of gene categories have also been done for several Archaea. On average, Archaea devote a higher percentage of their genomes to energy and coenzyme production than do Bacteria (this result is undoubtedly skewed a bit due to the large number of novel coenzymes produced by methanogenic Archaea, Section 14.15 and Figure 14.36). On the other hand, Archaea appear to contain fewer genes for carbohydrate metabolism and membrane functions (such as transport and membrane biosynthesis) than do Bacteria. However, this conclusion may also be skewed a bit because the corresponding pathways have been less studied in Archaea than in Bacteria and many of the relevant archaeal genes remain unidentified.
We now transition to look at the genomes of eukaryotes and their major organelles, structures whose evolutionary roots lie in the Bacteria.
Check Your Understanding
What lifestyle is typical of Bacteria and Archaea that contain fewer than 500 protein-encoding genes?
Which is likely to have more genes, a species of Bacteria with 8 Mbp of DNA or a eukaryote with 10 Mbp? Explain.
In prokaryotic cells with the largest genomes, which gene category contains the largest percentage of genes?
10.4 Organelle and Eukaryotic Microbial Genomes
Mitochondria and chloroplasts are eukaryotic cell organelles derived from endosymbiotic bacteria (Section 2.14 and Section 18.1) and thus share many fundamental traits with Bacteria to which they are phylogenetically related. The genomes of both organelles encode the machinery necessary for protein synthesis including ribosomes, transfer RNAs, and the other components necessary to drive translation. The genomes of several microbial eukaryotes have also been sequenced (Table 10.5), and their size varies widely (Figure 10.2). Certain single-celled protozoans, including the free-living ciliate Paramecium (40,000 genes) and the pathogen Trichomonas (60,000 genes), have significantly more genes than do humans (Table 10.5). In this section we focus on organellar genomes and the genomes of a few select microbial eukaryotes.
Table 10.5 Some eukaryotic nuclear genomesa
aAll data are for the haploid nuclear genomes of these organisms in megabase pairs. For most large genomes, both size and ORFs listed are best estimates due to large numbers of repetitive sequences and/or introns in the genomes.
The Chloroplast Genome
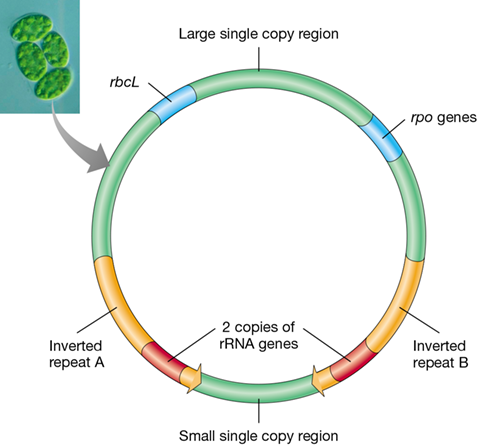
Green plant and algae cells contain chloroplasts, the organelles that perform photosynthesis (Section 14.3 and Figure 14.9). Each chloroplast contains several identical copies of the genome. Until recently, it was accepted that all chloroplast genomes were circular DNA molecules. However, with the power of next-generation sequencing, linear and single-stranded plasmid-like chloroplast genomes have also been detected. In fact, most of the chloroplast genomes in corn (maize) are linear in structure. Based on the over 800 chloroplast genomes in the databases, the typical chloroplast genome is about 100–200 kbp and contains two inverted repeats of 6–76 kbp that each encode copies of the three rRNA genes (Figure 10.11). As might be expected, many chloroplast genes encode proteins for photosynthetic reactions and autotrophy. For example, the enzyme RuBisCO, which is composed of a small and large subunit, catalyzes the first step in CO2 fixation in the Calvin cycle (Section 3.12 and Section 14.2). The rbcL gene encoding the large subunit of RuBisCO is present on the chloroplast genome (Figure 10.11), whereas the gene for the small subunit, rbcS, resides in the plant cell nucleus and its protein product must be imported from the cytoplasm into the chloroplast after synthesis.
Figure 10.11 Map of a typical chloroplast genome.

The inverted repeats each contain a copy of the three genes for rRNA (5S, 16S, and 23S). The large subunit of RuBisCO is encoded by rbcL and the chloroplast RNA polymerase by rpo genes. Inset: Photo of four cells of the green alga Makinoella with chloroplasts clearly visible.
The chloroplast genome also encodes tRNAs used in translation, several proteins used in transcription and translation, and some other proteins. Not all chloroplast proteins are encoded by the chloroplast genome; some are nuclear encoded. These are likely genes that migrated to the nucleus as the chloroplast evolved from an endosymbiotic cell into a photosynthetic organelle. Introns, the hallmark of genes in eukaryotes, are common in chloroplast genes and are primarily of the self-splicing type (Section 6.6).
Mitochondrial Genomes and Proteomes
Mitochondria are the eukaryotic cell’s respiratory organelles and are present in all but a few eukaryotes (Section 2.14 and Section 18.1). Mitochondrial genomes primarily encode proteins for oxidative phosphorylation and, like chloroplast genomes, also encode proteins, rRNAs, and tRNAs for protein synthesis. However, most mitochondrial genomes encode far fewer proteins than those of chloroplasts. The largest mitochondrial genome known has only 62 protein-encoding genes, but others contain as few as three. The mitochondria of almost all mammals, including humans, encode only 13 proteins in addition to 22 tRNAs and 2 rRNAs. **Figure 10.12*a*** shows a map of the 16,569-bp human mitochondrial genome. While human mitochondrial genomes are circular, diverse arrangements exist in other organisms. For example, some mitochondrial genomes are linear, including those of certain algae, protozoans, and fungi. Finally, the mitochondria of many fungi and flowering plants contain, in addition to the mitochondrial genome, small circular or linear plasmids (Section 6.2).
Figure 10.12 Map of the human mitochondrial genome and the mitochondrial proteome.

(a) The genome encodes rRNAs, 22 tRNAs, and several proteins. Arrows show direction of transcription for genes of a given color, and the three-letter amino acid designations for tRNA genes are also shown. The 13 protein-encoding genes are in green. Cytb, cytochrome b; ND1–6, components of the NADH dehydrogenase complex; COI–III, subunits of the cytochrome oxidase complex; ATPase 6 and 8, polypeptides of the mitochondrial ATPase complex. The two promoters are in the region called the D loop, which is also involved in DNA replication. Inset: Transmission electron micrograph of a mitochondrion (credit, D.W. Fawcett). (b) Mitochondrial proteomes. The numbers in each colored bar are the number of proteins encoded on the mitochondria of some model eukaryotes.
Mitochondria require many more proteins than their genome encodes (in particular, proteins needed for translation), and thus many mitochondrial proteins are encoded by genes in the nucleus. The yeast mitochondrion contains as many as 800 different proteins in its proteome (all the proteins encoded by a genome; Section 10.9). However, only eight (∼1%) of them are encoded by the yeast mitochondrial genome, the remaining proteins being encoded by nuclear genes (Figure 10.12b). The nuclear-encoded proteins required for translation and energy generation in mitochondria are more closely related to their counterparts in Bacteria than to those in the eukaryotic cytoplasm, consistent with both the evolutionary history of the mitochondrion (Section 13.4) and with a scenario—like that seen in the chloroplast—of genes having migrated from the original endosymbiont to the host cell nucleus.
Genomes and Introns in Some Microbial Eukaryotes
Apart from the human pathogenic protozoan Trichomonas, which contains almost three times more genes than human cells, parasitic eukaryotic microorganisms typically have relatively small genomes of 10–40 Mbp containing between 4000 and 11,000 genes. For example, Trypanosoma brucei, the agent of African sleeping sickness (Section 34.6), has 11 chromosomes, 35 Mbp of DNA, and almost 11,000 genes. The four species of Plasmodium that infect humans (causing malaria, Section 34.5) have genomes ranging from 23 to 27 Mbp arranged in 14 chromosomes containing a total of about 5500 genes.
As in Bacteria, the smallest eukaryotic genome belongs to an endosymbiont. Known as a nucleomorph, it is the degenerate remains of a eukaryotic endosymbiont of a certain green alga that has acquired the ability to photosynthesize by secondary endosymbiosis (Section 18.1). Nucleomorph genomes range from about 0.37 to 0.85 Mbp. The smallest genome in a parasitic eukaryote belongs to Encephalitozoon intestinalis, an intracellular pathogen of humans and other animals. E. intestinalis even lacks mitochondria, and although its haploid genome contains 11 chromosomes, the genome size is only 2.3 Mbp with approximately 1800 genes (Table 10.5); this is smaller than many bacterial genomes (Table 10.1).
The baker’s yeast Saccharomyces cerevisiae is widely used as a model eukaryote and its genome contains 16 chromosomes (13.4 Mbp of DNA). Yeast has approximately 6000 ORFs, which is fewer than that of some genomes of Bacteria (Tables 10.1 and 10.5). How many of these yeast genes are actually essential? This question has been addressed by systematically inactivating each gene in turn with knockout mutations (mutations that completely inactivate genes, Section 12.4). Knockout mutations cannot normally be obtained in essential genes in a haploid organism. However, yeast can be grown in both diploid and haploid states (Section 18.10). By generating knockout mutations in diploid cells and then investigating whether they can also exist in haploid cells, it is possible to determine whether a particular gene is essential for cell viability. Using knockout mutations, it has been shown that around 900 yeast ORFs (17% of its genome) are absolutely essential. Note that this number of essential genes is much greater than the approximately 300 genes estimated to be the minimal number required in a bacterial cell (Section 10.3).
Being a eukaryote, the yeast genome contains introns (Section 6.6). However, the total number of introns in the protein-encoding genes of yeast is a mere 225. Most yeast genes that contain introns have only a single small intron near the 5′ end of the gene. This situation differs greatly from that seen in more complex eukaryotes (Figure 10.13). For example, in the worm Caenorhabditis elegans, the average gene has five introns, and in the fruit fly Drosophila, the average gene has four. Introns are also common in the genes of plants, averaging around four per gene. The model flowering plant Arabidopsis averages five introns per gene, and over 75% of Arabidopsis genes have introns. In humans almost all protein-encoding genes have introns, and it is common for a single gene to have 10 or more. Moreover, introns in human genes are typically much longer than exons, the DNA that actually encodes proteins. Indeed, exons make up only about 1% of the human genome, whereas introns account for 24%. The remaining DNA is made up of repetitive sequences, noncoding RNA, and regulatory regions.
Figure 10.13 Intron frequency in the genes of different eukaryotes.

The average number of introns per gene is shown for a range of eukaryotic organisms; microbial species tend to have fewer introns per gene, whereas plants and animals have the most.
We will discuss how comparative genomics can be used to determine evolutionary relationships and how genomes evolve in Chapter 13. For now, we turn our focus to how various omic approaches can be used to determine the function of each gene product. The dynamic nature of microbes and how they interact with their environment can be characterized through the use of functional omics.
Check Your Understanding
What is unusual about the genes that encode mitochondrial proteins?
What is unusual about the genome of the eukaryote Encephalitozoon?
II Functional Omics
Knowing an organism’s genome sequence may not reveal what all the genes encode and when and why they are transcribed and translated. These topics require functional analyses of molecular events downstream of the genome itself.
Despite the major effort required to generate an annotated genome sequence, the net result is simply a “list of parts.” To understand how a cell functions, we need to know more than which genes are present. We must also understand (1) gene expression, (2) the function of gene products, (3) the activity of the proteins made, and (4) the metabolites produced during growth.
In analogy to the term “genome,” the entire complement of RNA, proteins, or metabolites produced under a given set of conditions is called the transcriptome, proteome, and metabolome, respectively. The suffix “omic” denotes their corresponding areas of study. Table 10.6 summarizes some of the “omics” terminology used in microbiology today.
10.5 Functional Genomics
As previously discussed (Section 10.2), genome sequencing, assembly, and annotation yields an abundance of information. However, the roles of many open reading frames (ORFs) remain unknown after annotation and are thus classified as encoding “hypothetical proteins.” The percentage of hypotheticals in a given microbial genome averages 30% of the total annotated ORFs. This value even holds true for the minimal Mycoplasma genome created by synthetic biologists, which possesses a trim 473 genes (Section 12.12). In fact, the function of less than 1% of the approximately 120 million protein sequences that exist in public databases is known! Thus, obtaining a genome sequence is only the beginning of teasing apart how a microbe functions and survives in its environment.
In this unit of the chapter we discuss how to gain insight into gene function through the analysis of RNA, protein, and metabolites, and we begin with how comparative genomics, genetic tools, and next-generation sequencing can be used to determine gene function.
Functional Genomics and Heterologous Expression
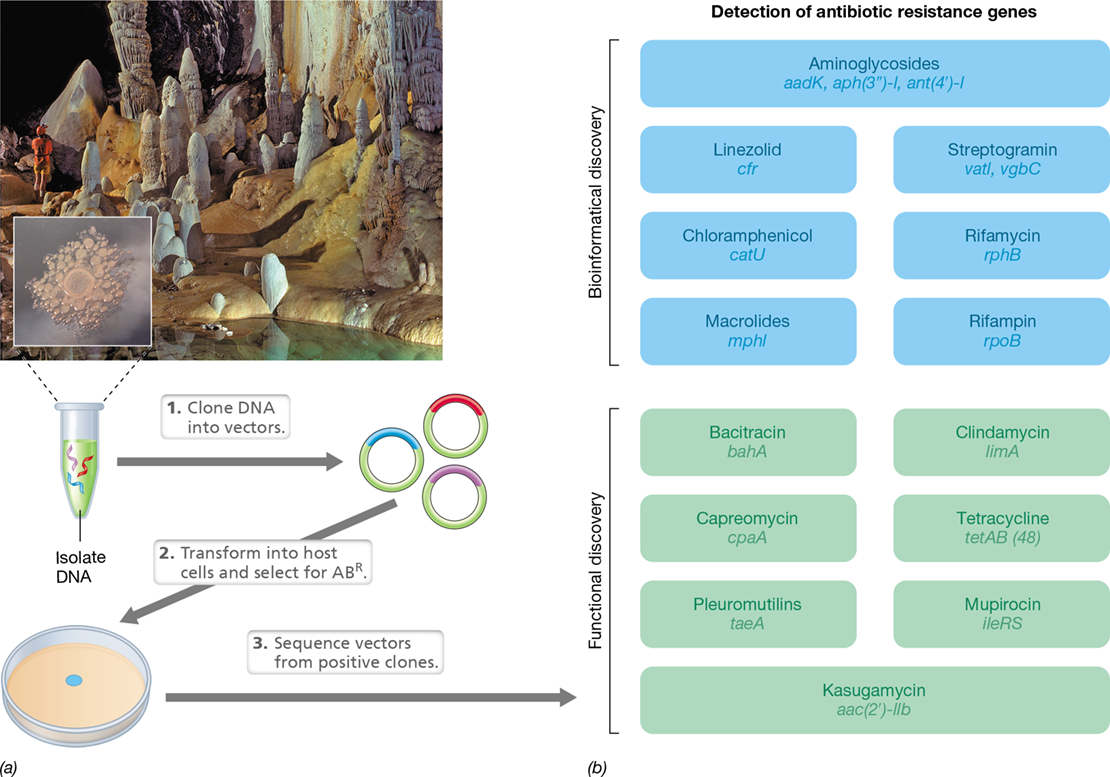
How do microbes get selected for genome sequencing? This selection is usually a result of an interesting phenotypic trait displayed by the microbe. Such was the case with the multi-antibiotic-resistant, gram-positive bacterium Paenibacillus species strain LC231 (Figure 10.14). While Bacteria displaying resistance to multiple drugs is not unique (Section 28.7), strain LC231 was cultured from an underground cave ecosystem that has been isolated from the surface for over 4 million years (Figure 10.14a). With no exposure to current pathogens (where it could have picked up genes by horizontal transfer, Chapter 9) or to antibiotics used in clinical or veterinary medicine, this bacterium displayed resistance—surprisingly—to at least 14 different classes of antibiotics! How did such resistance come about?
Figure 10.14 Functional genomics and discovery of new antibiotic resistance genes.

(a) Heterologous expression of Paenibacillius strain LC231 DNA and selection of antibiotic-resistant transformants for sequence analysis. DNA from Paenibacillus strain LC231, isolated from Lechuguilla Cave, Carlsbad Caverns, New Mexico, USA (inset photo of Paenibacillus colonies courtesy of L. Ejim, C. Groves, and G. Wright), is extracted and inserted into plasmids for expression in Escherichia coli. Plasmid DNA from antibiotic-resistant E. coli colonies is sequenced and analyzed for the presence of new genes conferring antibiotic resistance (ABR). (b) Discovery of genes conferring resistance to 14 different types of antibiotics in Paenibacillus strain LC231. Gene names are listed under the type of antibiotic they confer resistance to. Genes discovered by genome database searches are in blue, while genes identified by functional genomics and heterologous expression in E. coli from part a are in green. Data adapted from Pawlowski, A.C., Wenliang, W., Koteva, K., Barton, H.A., McArthur, A.G., and Wright, G.D. 2016. Nat. Commun. 7: 13803.
In an effort to understand the gene products or resistome (Table 10.6) responsible for the multidrug resistance of strain LC231, bioinformatics revealed ten ORFs known to encode resistance to seven different types of antibiotics (Figure 10.14b). This comparative genome analysis was facilitated by an online program called Resistance Gene Identifier, which allows for genome sequences to be searched against a database of known antibiotic resistance genes from other bacteria. However, the mechanism by which LC231 was resistant to seven other antibiotic types was not evident from comparative genomics. To attack this question, microbiologists used a common functional genomics approach that employs the model bacterium Escherichia coli. This approach is based on heterologous expression, which is the process of expressing a gene from one organism in a different, host organism (Chapter 12). To heterologously express LC231 genes in E. coli, the LC231 genome was fragmented and inserted into plasmids. The resulting plasmids were transformed into E. coli to create a clone library of transformants, with each transformant containing a plasmid with a different piece of the LC231 genome (Figure 10.14a and Chapter 12). E. coli colonies that resulted from this clone library were then screened and selected for resistance to the antibiotics of interest. To determine the identity of the LC231 genome fragment responsible for conferring antibiotic resistance to the transformant, the corresponding plasmid DNA was isolated and sequenced (Figure 10.14a).
The functional genomic screen in E. coli resulted in “mining” five new antibiotic resistance genes that had not been previously discovered (Figure 10.14b). Two of these genes encode novel enzymes that modify the antibiotics bacitracin and capreomycin by an amidohydrolase (an enzyme that removes an amine group) and acetyltransferase (an enzyme that adds an acetyl group), respectively; such modifications inactivate the antibiotics. While its environmental isolation limits strain LC231 from spreading its resistome to pathogens—assuming that deep subsurface materials are not exposed to Earth’s surface from natural catastrophes or human activities—comparative genomics has shown that the genomes of at least some subsurface-dwelling bacteria contain novel antibiotic resistance determinants that represent an untouched pool of antibiotic resistance. This discovery highlights the importance not only of monitoring antibiotic use in clinical and veterinary microbiology, but also of developing mechanisms to inhibit newly discovered antibiotic-modifying enzymes. With the abundance of mobile genetic elements, or mobilome components (Table 10.6 and Section 13.9), that exist in microbial genomes in nature, given enough time, these new antibiotic resistance genes will undoubtedly be transferred to other species and could someday emerge in human pathogens.
The Utility of Comparative Genomics: Discovery of New Antiphage Systems
In Chapter 9 we discussed some barriers to horizontal gene transfer that are employed by Bacteria and Archaea. While the role of many of these microbial “immune systems” was discovered quite by accident, the omics era has allowed for similar defense systems to be identified in thousands of genomes by searching for similar gene sequences in the databases. In the process of annotating these restriction modification, phage exclusion, and CRISPR systems (Section 9.12), computational biologists have noticed that many of the genes encoding these systems are clustered next to each other within the genome on what are called chromosomal islands (Section 13.10). Besides containing the systems mentioned above, these chromosomal islands also contain numerous open reading frames (ORFs) of unknown function (Section 10.2).
Using comparative genomics, molecular biologists have aligned the sequences in these chromosomal islands using a process similar to that described for genome assembly and annotation in Section 10.2. This alignment resulted in the discovery of homologs (ORFs possessing similar sequence and evolutionary origin; Section 13.8) to be enriched next to genes encoding known defense systems in the genomes of many species (**Figure 10.15*a***). Besides the similar gene sequence and length, these unknown homologs are often found surrounded by other ORFs oriented in the same direction in many genomes. Both proximity and shared direction of transcription are strong predictors of an operon and suggest that the gene products are dependent on one another. To test if these conserved ORFs did encode antiphage systems, scientists expressed the genes in model bacteria and tested the new host’s resistance to bacteriophage infection by using a range of viruses. This study resulted in the discovery of over ten new antiphage systems and highlights both the constant arms race between microbes and their viruses and the previously hidden antiviral capacities present in bacterial genomes.
Figure 10.15 Comparative genomics and discovery of the DISARM antiphage system.

(a) Alignment of a chromosomal island region from four separate genomes containing genes encoding known antiphage systems (yellow). A region containing unannotated homologs sharing strong sequence similarity (denoted by red dashed box) with adjacent ORFs in the same orientation is selected for functional analysis in a model bacterium. (b) Expression of the DISARM (defense island system associated with restriction–modification) antiphage system in Bacillus subtilis. Cells expressing the DISARM system are labeled blue, while those without DNA encoding the resistance system are labeled red. The genome of the bacteriophage added to the cells contains a DNA binding site for a fluorescently tagged protein present in both cell types. As the bacteriophage genome is replicated, more fluorescent protein binds and the blue-white foci enlarge (white arrows; 60- and 80-minute time points). Over time, bacteriophage genomes are prevented from replicating in cells expressing the DISARM system (blue cells) as indicated by the relative absence of blue-white foci.
Figure 10.15b illustrates the antiphage activity of the DISARM (defense island system associated with restriction–modification) system that was isolated from Bacillus paralicheniformis. Heterologous expression of this system in Bacillus subtilis renders the modified bacterium resistant to eight distinct tailed bacteriophages (Section 5.2) because their corresponding viral DNA does not get replicated in cells expressing the DISARM system. Conversely, cells not expressing DISARM are still replicating bacteriophage DNA 80 minutes postinfection (Figure 10.15b). While the exact mechanism behind the DISARM system is still being elucidated, it does encode a methylase—an enzyme that modifies the host’s DNA—and hence likely functions in a way similar to other restriction modification systems by degrading the unmethylated bacteriophage DNA (Section 9.12).
Check Your Understanding
How can genes encoding antibiotic resistance be identified using heterologous expression systems?
10.6 High-Throughput Functional Gene Analysis: Tn-Seq
10.6 High-Throughput Functional Gene Analysis: Tn-Seq
10.6 High-Throughput Functional Gene Analysis: Tn-Seq
Besides the heterologous expression and related systems just discussed, if genetic studies can be performed in a microbe whose genome has been sequenced, the functional role of its genes can be characterized by constructing a library of gene mutants that allow for gene analysis on an enormous scale. The most common and random way to generate these mutants is through transposon (Tn) mutagenesis (Section 9.11).
Because transposons encode antibiotic resistance genes along with their insertion sequences, Tn mutants with gene interruptions can be selected using an antibiotic that allows growth of only cells containing the transposon (such cells are resistant to the antibiotic). The application of this technique was illustrated in Figure 9.35 where a gene product that plays a role in biofilm formation was identified in Pseudomonas aeruginosa. However, this application required the screening of individual transposon mutants. By contrast, the development of next-generation sequencing technology has allowed for high-throughput screening of mutants. To be considered high-throughput screening, automated devices are used to gather sequence data on a large set of strains or mutants in parallel. This advancement in screening capabilities combined with transposon mutagenesis has been instrumental in the development of a powerful functional genomics tool called transposon insertion site sequencing (Tn-Seq). The utility of Tn-Seq is based on three premises: (1) a transposable element exists that is flanked by a specific nucleotide sequence called a restriction site (Section 12.2) that guides a nuclease enzyme to cut at a specific chromosomal sequence outside of the transposon insertion locus; (2) distinct Tn mutants can be generated that display different fitness profiles in a pooled culture depending on the genomic insertion, location, and growth condition screened; and (3) it is possible to determine the chromosomal location of each transposon insertion and quantify the number of each Tn mutant in a mixed pool (Figure 10.16).
Figure 10.16 Tn-Seq and fitness analysis of *Caulobacter crescentus* mutants during bacteriophage infection.

(a) Electron micrograph of bacteriophage ϕCbK binding to the flagellated cell pole of a cell of C. crescentus. (b) Replication cycle of ϕCbK. ϕCbK adsorbs to the flagellum and pili of C. crescentus and then injects its genome through the bacteriophage-specific receptor. The host cell then replicates the ϕCbK genome and new viral particles are made prior to cell lysis. (c) A library of transposon (Tn) mutants (Section 9.11) is constructed and exposed to increasing numbers of ϕCbK particles. These mutants contain a Tn that possesses terminal DNA sequences that are recognized by a special restriction enzyme (RE). This RE cuts the chromosomal DNA at sites outside of the Tn insertion. As the pool of Tn mutants is exposed to increasing concentrations of ϕCbK, mutants that are resistant to the bacteriophage survive at a greater rate and thus display increased fitness. Cell colors denote mutants in gene products necessary for the corresponding ϕCbK replication cycle in part b. (d) Tn-Seq analysis of Tn mutant pools following infection with ϕCbK. Total DNA from the Tn mutant pools is extracted, cut with the RE mentioned in part c, and sequenced by next-generation methods. The chromosomal location of the Tn insertion site determined from sequencing is plotted versus the concentration of ϕCbK. The data show that phage receptor mutants are most resistant. Line colors correspond to Tn mutants in the ϕCbK replication cycle as depicted in part b. Data adapted from Christen, M., et al. 2016. J. Mol. Biol. 428: 419.
If a gene product of unknown function is beneficial or essential for the survival of a cell under a specific growth condition, its corresponding Tn mutant will display decreased fitness and be present at low frequency in a mixed culture of competitive transposon mutants. Conversely, if elimination of a gene product is beneficial under certain growth conditions, the corresponding Tn mutant will display increased fitness and be present at a high ratio compared to less competitive Tn mutants within the pool. Mutant fitness or abundance is measured by extracting genomic DNA from the pooled culture after exposure to a certain growth condition and digestion with a restriction enzyme (Figure 10.16c; Section 12.2) that recognizes a nucleotide sequence 20 base pairs outside of the transposon. Next-generation sequencing methods are then used to add adapters, amplify, and sequence the short region of chromosomal DNA next to the Tn insertion site (Figure 10.16c), which can then be mapped back to the genome.
Tn-Seq has been used to characterize novel genes encoding competence (Section 9.6) in Streptococcus mutans, persistence (Section 8.12) in pathogenic Escherichia coli, antibiotic resistance in Enterococcus, and amino acid biosynthesis in the anaerobic sulfate-reducing bacterium Desulfovibrio. Figure 10.16 illustrates a Tn-Seq scheme used to characterize cellular components in Caulobacter crescentus, a model bacterium for studying the cell cycle (Section 8.8), that lead to susceptibility to bacteriophage ϕCbK infection. Tn-Seq studies of a pool of Tn mutants subjected to increasing concentrations of ϕCbK resulted in the recovery of Tn mutants with insertions in genes that encode the phage receptor protein with the highest frequency (Figure 10.16c, d). This phage receptor is essential for the transport of the phage DNA into the host and ultimate infection (Figure 10.16b and Section 5.4). However, mutants with interruptions in genes that encode proteins that help phage to initially “find” and adsorb to the bacterium, such as those encoding the flagellum and pili, decreased in abundance as more phage were added to the culture (Figure 10.16c, d). Tn mutants recovered at an intermediate level in the pool possessed insertions in genes needed to replicate the ϕCbK genome (Figure 10.16 c, d). These mutants have the same adsorption and infection rates as the wild type but are less efficient at producing new ϕCbK.
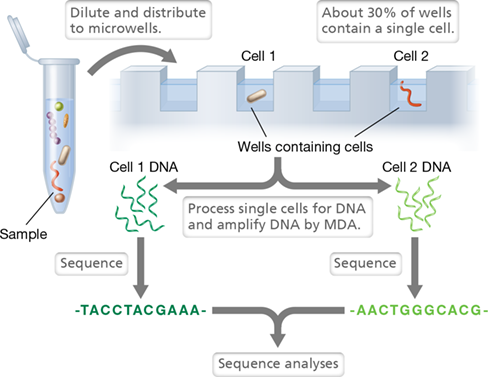
Because of the power of Tn-Seq, scientists have modified the technique for new discoveries. Current fitness studies can miss single-cell phenotypes as some of these Tn mutants can obtain resources from other mutants in the pool. To prevent mutants from sharing cellular metabolites, a technique called microfluidics is used to encapsulate individual cells in a manner similar to that discussed for single-cell genomics in Section 10.11. In this way, cross-feeding is prevented, and previously unrecognized phenotypes can be detected.
With this background in functional genomics, we now switch gears to study how multiple—as compared to single—genomes can be studied simultaneously using a powerful form of omics that can reveal the diversity of single target genes or the entire genomic complement of virtually any environmental sample.
Check Your Understanding
What are the three premises that Tn-Seq is based on?
How can a gene essential to a bacterium’s survival be identified using Tn-Seq?
10.7 Metagenomics
Microbial communities contain many microbial species, many of which have never been cultured or formally identified. Metagenomics is the science that analyzes pooled DNA or RNA from an environmental sample containing organisms that have not been isolated and identified (Figure 10.17). Just as the total gene content of an organism is its genome, so the total gene content of a microbial community is its metagenome (Table 10.6). In addition to metagenomic analyses based on DNA sequencing, analyses based on RNA or proteins—metatranscriptomics (Section 10.8) or metaproteomics (Section 10.9), respectively—may be used to explore the patterns of gene expression in natural microbial communities. With today’s molecular technology, these studies can even be done on individual cells (Section 10.11).
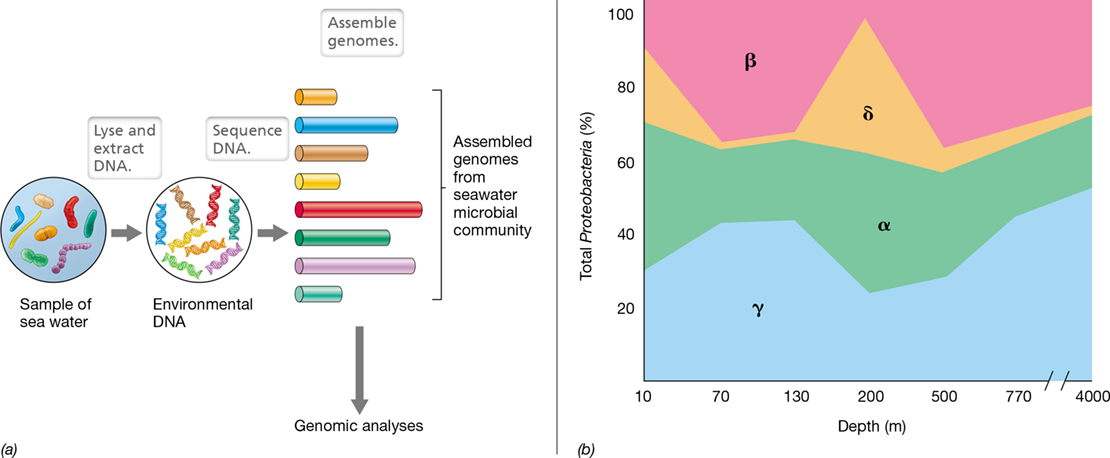
Figure 10.17 Metagenomics and the microbiome.

(a) Isolation, sequencing, and identification of DNA from a sample of seawater. (b) Proteobacteria in the ocean. The distribution with depth of the major subgroups (alpha α, beta β, gamma γ, and delta δ) of Proteobacteria in the Pacific Ocean is shown. Many other types of bacteria are also present (not shown). Data adapted from Kembel, S.W., Eisen, J.A., Pollard, K.S., and Green, J.L. 2011. PLoS One 6: e23214.
Examples of Metagenomic Studies
While several examples of metagenomic studies are presented in this text, two simple examples are highlighted here. Several environments have been surveyed by large-scale metagenome sequencing projects. Extreme environments, such as highly acidic runoff waters from mining operations (Section 22.2), tend to have low microbial species diversity. Consequently it has been possible to isolate community DNA (and metabolites, Section 10.10) and assemble much of it into nearly complete individual genomes. Conversely, complex environments such as fertile soils or aquatic environments are much more challenging, and complete genome assemblies here are more difficult, yet possible (Section 19.8). A surprising finding that has emerged from metagenomic studies is that the majority of genes recovered from natural habitats do not originate from cells but from viruses.This is discussed further in Chapter 11 where we consider the genomics and phylogeny of viruses.
Even if complete genomes cannot be assembled from environmental DNA, much useful information can be derived from metagenomic surveys (Section 19.8). For example, environments can be analyzed for the presence and distribution of specific microbial groups. These vary greatly in relative abundance in different environments, and Figure 10.17b illustrates this for subgroups of Proteobacteria (a major phylum of gram-negative Bacteria, Chapter 16) at a sampling site in the Pacific Ocean near the Hawaiian Islands. Light, oxygen, nutrients, and temperature all change with depth in a water column, and these factors can be correlated with proteobacterial subgroups to show which are most competitive at each depth (Figure 10.17b). One curious observation that has emerged from such metagenomic studies is that much DNA in natural habitats does not reside in living cells. Around 50–60% of the DNA in the oceans is extracellular DNA present in deep-sea sediments. Presumably this was DNA deposited when dead organisms from the upper layers of the ocean sank to the bottom and lysed. Because nucleic acids are major reservoirs of phosphate, marine sediment DNA is thought to be a major component of the global phosphorus cycle.
Metagenomics and “Biome” Studies
The human body is estimated to contain about 10 trillion (1013) cells, but each of us also carries around ten times more prokaryotic cells than human ones. This collection of prokaryotic cells is called the human microbiome (Chapter 24). Most of these organisms inhabit the large intestine, with the majority belonging to one of two phylogenetic groups of Bacteria, the Bacteroidetes and the Firmicutes (Chapter 16). A fascinating finding is that the composition of the gut microbiome correlates with obesity in both humans and experimental mouse models. The data show that the higher the proportion of Firmicutes (mostly species of Clostridium and relatives) in the gut, the more obese is the human or mouse. A suggested mechanism that explains this finding is that fermentative species of Firmicutes convert more dietary fiber into fatty acids that can be absorbed by the host (Section 24.9). In this way, the obese host gets more usable organic carbon than the thinner host from the same amount of food.
Recent surveys of the human and mouse gut microbiome have also revealed the rather surprising finding that over 60 species of fungi (eukaryotic microbes, Chapter 18) are present (Figure 10.18). These constitute the gut mycobiome (the prefix “myco” means fungal). Many fungi, typically nonpathogenic yeasts, inhabit the skin, the oral cavity, and virtually all moist surfaces on the human body. Many of these are common and generally harmless yeasts, such as Saccharomyces, Cladosporium, and most species of Candida. Most of these also are found in the gut, although some gut fungi—such as Aspergillus and Trichosporon—are potential serious pathogens (Chapter 34). Moreover, although gut fungi constitute less than 1% of the total human microbiome, it is known that certain conditions such as inflammatory bowel disease and some cases of obesity correlate strongly with specific fungal populations. Hence, metagenomics holds great promise for exploring possible connections between specific microbial populations and specific diseases in humans and other animals. Moreover, in cases where a clear cause-and-effect relationship is strongly suspected, metagenomics also holds great promise as a clinical tool for making medical diagnoses.
Figure 10.18 The mouse mycobiome.
The data shown represent the relative amount of different fungal genera of the mouse intestine. The pie chart shows the most common fungi present are yeasts. Data adapted from Iliev, I.D., et al. 2012. Science 336: 1314.
In the next two sections we travel beyond the genome to explore the technology for examining gene expression: omic methods that can reveal the RNAs and proteins encoded by the genome.
10.8 Gene Chips and Transcriptomics
Once a genome sequence is available, the sequence can be used to synthesize miniature devices that can be used to detect genes from specific microbes, determine genome differences between closely related strains of the same species (for example, the presence of chromosomal islands), identify genome sequences that are bound by specific DNA-binding proteins, and measure gene expression. Transcriptomics refers to the study of a cell’s global transcription and is done by monitoring the transcriptome, the total RNA generated under a chosen growth condition. Besides the aforementioned reasons for doing transcriptomics, the technique can also put a functional label on genes that have been annotated as simply encoding “hypothetical proteins.” In these cases, discovering the conditions under which these genes are transcribed often yields clues to their function. Two main approaches are used in transcriptomics: microarrays and RNA-Seq.
Microarrays and the DNA Gene Chip
Microarrays are small, solid supports to which genes or, more often, oligonucleotides corresponding to segments of genes are fixed and arrayed spatially in a known pattern; they are often called gene chips (**Figure 10.19*a***). Microarrays measure the DNA or RNA that hybridizes to the DNA sequences on the chip. When DNA is denatured (that is, the two strands are separated), the single strands can form hybrid double-stranded molecules with other nucleic acid molecules by complementary or almost complementary base pairing (Figure 10.19b; Section 12.1). This process is called nucleic acid hybridization, or hybridization for short, and is widely used in detecting, characterizing, and identifying segments of DNA or RNA. The single-stranded segments of nucleic acid, whose identity is already known, are called nucleic acid probes or, simply, probes. To detect hybridization to the probes, the nucleic acid added to the chip must be labeled with a fluorescent dye and then the hybridized chip is scanned with a laser fluorescence detector that measures which of the probes contain hybridized DNA (Figure 10.19b, c).
Figure 10.19 DNA chip design and application.

(a) DNA chip design. Short single-stranded oligonucleotides (probes) corresponding to each gene in an organism or to diagnostic sequences corresponding to numerous organisms are synthesized and affixed at known locations to make a microarray. (b) Microarray hybridization. The presence of specific DNA or RNA (in the form of cDNA) is assayed by hybridizing fluorescently labeled samples (DNA or cDNA) to the DNA probes on the chip. Labeled DNA or cDNA will bind to the probes on the chip if they possess sequence complementarity. (c) Analysis of microarray hybridization. A scanning laser is used to identify regions of the chip where labeled nucleic acid has bound to the probes.
Mastering Microbiology
Art Activity: Figure 10.19 DNA chip design and application
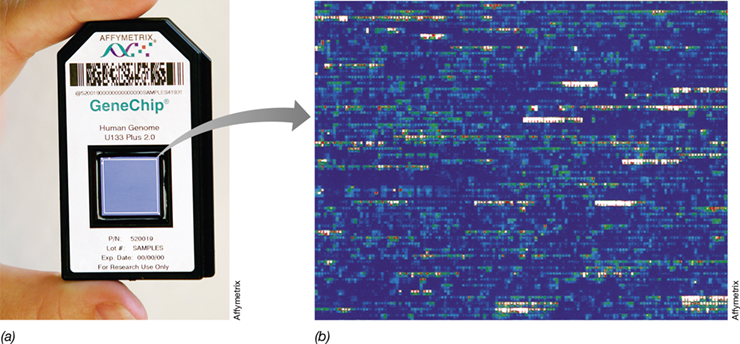
Gene chips are typically about 1 to 2 cm and are inserted into a plastic holder that can easily be manipulated (**Figure 10.20*a***); each chip holds thousands of different DNA fragments in a known order. In practice, each gene is usually represented more than once in the array to increase reliability. Whole genome arrays contain DNA segments that cover the entire genome of an organism. For example, a chip that covers the entire human genome (Figure 10.20a) can analyze over 47,000 human transcripts and has room for 6500 additional oligonucleotides for use in clinical diagnostics.
Figure 10.20 Using gene chips to assay gene expression.

(a) The human genome chip contains over 47,000 gene fragments. Blowup from part a to part b indicates location of actual microarray. (b) A hybridized yeast chip shows fragments from a quarter of the genome of baker’s yeast, Saccharomyces cerevisiae. Each gene is present in several copies and has been probed with fluorescently labeled cDNA (derived from mRNA) from yeast cells grown under a specific condition. The background of the chip is blue. Locations where the cDNA has hybridized are indicated by a gradation of colors up to a maximum number of hybridizations, which shows as white. Because the location of each gene on the chip is known, when the chip is scanned, it reveals which genes were expressed.
Measuring Gene Expression and Other Uses of Gene Chips
In a gene expression microarray, the probes are designed and synthesized for each gene based on the genomic sequence. Once attached to the solid support, the DNA segments can be hybridized with labeled RNA from cells grown under specific conditions, and the microarray is then scanned and analyzed by computer. Because mRNA levels are typically too low for use directly, the mRNA sequences are first amplified and converted into DNA using a modified version of the polymerase chain reaction (PCR) that converts RNA to complementary DNA (cDNA, Section 12.1).
To monitor global gene expression, total RNA (or cDNA) from a test sample is hybridized to an array of oligonucleotides corresponding to the entire genome. Figure 10.20b shows part of a chip containing probes for over 6000 protein-encoding genes of the yeast Saccharomyces cerevisiae. After hybridizing yeast cDNA to the chip, a distinct hybridization pattern is observed, and the fluorescence and its intensity reveal both which genes were expressed and at what level (Figure 10.20b); these data yield the transcriptome of the yeast culture grown under specified conditions (Table 10.6). Using such analyses, gene expression under different growth conditions can be measured. For example, in yeast—which can grow by either fermentation or respiration (Chapter 3)—transcriptome analyses have shown that genes that control production of ethanol (a key yeast fermentation product) are strongly repressed while genes encoding citric acid cycle functions (needed for aerobic growth) are strongly activated when the organism is shifted from fermentative to respiratory conditions. Overall, over 700 genes are turned on and more than 1000 turned off during this metabolic transition. In “shift” experiments of this type, the expression pattern of genes of unknown function is also revealed, and analysis of these expression patterns sometimes yields valuable clues to the cellular function of these unknown proteins.
Microarrays can also be used to identify specific microbes. For example, identification (ID) chips have been used in the food industry to detect DNA sequences unique to specific pathogens, such as the gastrointestinal pathogen Escherichia coli O157:H7 (Table 10.1), an occasional foodborne pathogen (Section 33.11). In environmental work, microarrays called PhyloChips have been used to assess microbial diversity. These contain oligonucleotides complementary to the 16S rRNA of different bacterial species, a molecule widely used in microbial systematics (Sections 13.11 and 13.12). After extraction of bulk DNA or RNA from an environment, the presence or absence of a given species can be assessed by the hybridization response on the chip (Section 19.7). Although ID chips and PhyloChips can be made highly specific, the inexpensive nature of DNA isolation, sequencing, and analysis have made metagenomic approaches (Section 10.7) to the identification of specific pathogens or phylogenetic groups in natural samples the preferred method of assessment.
RNA-Seq Analysis
RNA-Seq analysis is a transcriptomic method in which all the RNA molecules from a cell are converted to DNA (cDNA, Section 12.1) and then sequenced. Provided that the genome sequence is available for comparison, RNA-Seq reveals both which genes were transcribed and how many RNA copies of each gene were made. Because RNA-Seq targets all transcripts, it is ideal for measuring the expression of mRNA, to identify long untranslated regions, and to discover noncoding RNAs. RNA-Seq requires high-throughput sequencing (second-generation sequencing, Section 10.2) and is complicated a bit by the fact that the most abundant RNA in a cell is ribosomal RNA (rRNA). Nevertheless, methods are available to remove rRNA or enrich mRNA and primary transcripts from a total RNA pool. In addition, recent advancements in sequencing technology may allow sequencing without removing rRNA.
RNA-Seq has overtaken microarray analysis as the method of choice for global studies of gene expression. The data from transcriptomic experiments can be presented in the form of a heat map, which uses different colors to show the level of gene expression. For example, **Figure 10.21*a*** identifies gene clusters that are upregulated (more intensely transcribed) during nitrogen deprivation in the heterocyst-forming cyanobacterium Anabaena, a phototroph that can use N2 as its nitrogen source (nitrogen fixation, Section 3.12). Gene clusters 1–4 represent increased expression of nitrogen fixation and heterocyst formation genes (heterocysts are the site of N2 fixation) as the time of nitrogen deprivation increases (Figure 10.21a). RNA-Seq data can also be used for transcript mapping by plotting the sequences read against the genome annotation. Figure 10.21b illustrates the sequencing coverage at each base along the open reading frame regions for psbB, petF, and nrrA, genes that encode two key proteins needed for photosynthesis and a regulator protein for heterocyst formation in Anabaena, respectively (Section 8.9). These plots demonstrated long 5′ untranslated regions present in these transcripts, which may be sites where specific DNA-binding proteins attach in regulatory events (Chapter 7).
Figure 10.21 RNA-Seq analysis of the heterocyst-forming cyanobacterium *Anabaena* during nitrogen starvation.

Cyanobacteria are oxygenic phototrophs and only some species, such as those that form heterocysts, can fix nitrogen under fully oxic conditions. (a) Heat map of gene expression 6, 12, and 21 h after cells were starved for fixed nitrogen. Genes that display increased expression are in red, whereas those that display decreased expression are in blue. Gene clusters 1–4 all encode proteins linked to nitrogen fixation. (b) Mapping of RNA-Seq reads. The arrows indicate the annotated open reading frames, and the plots underneath correspond to the number of sequencing reads detected for each chromosomal nucleotide position. Note that the negative numbers represent chromosomal positions upstream of the predicted start codon for the genes. The genes psbB and petF encode key proteins of photosynthesis, and nrrA encodes a protein that regulates heterocyst formation (the heterocyst is the site of N2 fixation, Section 3.12). The pigments, diversity, and general biology of cyanobacteria are discussed in Sections 14.3, 14.4, 14.6 and 15.3. Parts a and b modified from Flaherty, B.L., F. van Nieuwerburgh, S.R. Head, and J.W. Golden. 2011. BMC Genomics 12: 332.
Transcript abundance under different culture conditions can also be analyzed using RNA-Seq data, as indicated by a comparison of cultures of a Clostridium species in exponential and stationary phase (Figure 10.22). Clostridia are gram-positive bacteria that produce endospores, the highly resistant and dormant stage of the cell’s life cycle (Section 2.8); the general properties of sporulating clostridia are discussed in Section 16.8. As one might predict, transcription of genes of the glycolytic pathway (the major means by which the organism makes ATP) is elevated during exponential growth, whereas expression of sporulation genes increases in stationary phase, when nutrients become limiting (Section 8.6). RNA-Seq is also being used for microbial community analysis and can provide information on relative transcription levels when a genome sequence is not available for comparison. In this case the sequences detected must be identified by homology with sequences present in databanks.
Figure 10.22 Transcriptomic analysis of sporulation genes in *Clostridium*.

RNA-Seq analysis of cultures of a Clostridium species grown for 4.5 h (cells in exponential phase) or 14 h (cells in stationary phase). Two genomic regions are shown: (left) ∼5.4-kb segment surrounding the gap-pgk-tpi glycolytic operon, and (right) ∼1.2-kb segment surrounding the cotJC-cotJB sporulation operon. Production of endospores is triggered by nutrient starvation. Data from Wang, Y., X. Li, Y. Mao, and H.P. Blaschek. 2011. BMC Genomics 12: 479.
As discussed in Section 10.7, metagenomics is the genomic analysis of pooled DNA or RNA obtained from organisms in an environment. Metagenomic analysis using RNA-Seq can be exploited for culturing microbes from nature that had previously resisted laboratory culture. This can be accomplished by using RNA-Seq to identify highly transcribed genes in the microbial community that contain the microbe of interest, followed by sequence analyses to identify the proteins encoded by the highly transcribed genes. These data can be used by microbiologists to deduce which nutrients the microbe is most likely using given the predicted enzymatic activities of these proteins. Culture media can then be devised using this information as a guide to successfully culture the previously uncultured.
We transition now from nucleic acid analyses to protein analyses to reveal the importance of other omics to modern-day microbiology and biology in general.
Check Your Understanding
Why is it useful to survey expression of the entire genome under particular conditions?
What do microarrays tell you that studying gene expression by assaying individual enzymes cannot?
What technological advances does RNA-Seq depend on?
10.9 Proteomics and the Interactome
The genome-wide study of the structure, function, and activity of an organism’s proteins is called proteomics. The number and types of proteins in a cell change in response to the cell’s environment or other factors, such as developmental cycles. As a result, the term proteome has unfortunately become ambiguous. In its wider sense, a proteome refers to all the proteins encoded by an organism’s genome. In its narrower sense, however, it refers to those proteins present in a cell at any given time. The term translatome has also been used to describe the latter; that is, it refers to every protein made by a cell under specific conditions. Recall from Chapter 7 that the extensive regulatory systems of Bacteria and Archaea tightly control which proteins are made under any given set of nutrients and environmental conditions. An understanding of which proteins are synthesized under a given set of conditions—the goal of proteomics—complements transcriptomics data and is the “gold standard” for confirming that a transcript detected by transcriptomics was actually translated.
Methods in Proteomics
To identify individual proteins within a mixed pool of proteins, proteomics relies on some form of mass spectrometry, a method that can also be used to identify metabolites (Section 10.10). The mass of 12C is defined as exactly 12 molecular mass units (daltons). However, the masses of other atoms, such as 14N or 16O, are not exact integers. Mass spectrometry using extremely high mass resolution techniques, which can distinguish between slightly different mass-to-charge ratios, allows the unambiguous determination of the molecular formula of any small molecule. Thus mass spectrometry can be used to identify the amino acid sequence of several peptides in a sample.
Figure 10.23 illustrates a standard proteomic analysis, which begins with extraction of total protein from a sample. To increase sensitivity, liquid chromatography is typically used to separate protein mixtures. In high-pressure liquid chromatography (HPLC), a protein sample is dissolved in a suitable liquid and forced under pressure through a special column that separates proteins by differences in their chemical properties, such as size, charge, or hydrophobicity. Fractions are collected at the end of the column, the proteins in each fraction are digested by protease enzymes, and the resulting peptides are identified by mass spectrometry. The amino acid sequence of these peptides can then be searched against the translation of a genome to identify the presence of specific proteins (Figure 10.23).
Figure 10.23 Proteomics.

Total protein is isolated from a culture or environmental sample. This protein pool is converted to fractions of small peptides using various denaturation, separation, and digestion methods. Following ionization of the peptide fractions, the ions are characterized using mass spectrometry (M.S). Peaks from the M.S. are compared to standards, and the primary amino acid sequence of the resulting peptides is identified and quantified. The M.S. peaks can also be used to determine if the peptides possess post-translational modifications (Section 7.16 and see Figure 10.25).
MALDI (matrix-assisted laser desorption ionization) is an advanced version of mass spectrometry that does not require the protein separation and digestion step (Figure 10.23). Instead the sample is affixed to a matrix and then ionized and vaporized by a laser (Figure 10.24). The ions generated are accelerated along the column toward the detector by an electric field. The time of flight (TOF) for each ion depends on its mass/charge ratio—the smaller this ratio, the faster the ion moves. The detector measures the TOF for each ion, and the computer calculates the mass and hence the molecular formula. The combination of these two techniques is known as MALDI-TOF.
Figure 10.24 MALDI-TOF mass spectrometry.

In matrix-assisted laser desorption ionization (MALDI) spectroscopy, the sample is ionized by a laser and the ions travel down the tube to the detector. The time of flight (TOF) depends on the mass/charge (m/z) ratio of the ion. The computer identifies the ions based on their time of flight, that is, the time it takes to reach the detector.
Utility of Proteomics: Profiling Protein Modifications
Not only can proteomics identify proteins that are abundant under certain growth conditions, but it can also be modified to detect post-translational modifications (Section 7.16). For example, because mass spectrometry spectra are known for all proteins of the Mycobacterium tuberculosis (the causative agent of tuberculosis, Section 31.4) proteome, additional chemical groups attached to peptides can also be identified. These chemical modifications include phosphorylation, glycosylation, methylation, pupylation (addition of a ubiquitin-like molecule), lipidation, and acetylation.
Figure 10.25 illustrates the modified proteome of M. tuberculosis and the role of the modified proteins in the phenotype of the pathogen. This analysis shows that some modifications can be enriched in functional categories, such as lipidation of proteins that participate in cell wall biosynthesis (the cell wall of M. tuberculosis contains an abundance of lipids that assist in its virulence and play a major role in its acid-fast staining properties, Sections 16.11 and 31.4). Pupylation, first discovered to occur in Bacteria by studying M. tuberculosis, is believed to direct proteins to a cellular degradation pathway and is enriched with proteins associated with virulence and metabolism. These protein modifications may play a role in the pathogenesis of M. tuberculosis as well as the host’s immune response during infection by engaging eukaryotic cell receptors or antigen processing pathways. By studying the modified proteome, immunologists may also be able to identify novel epitopes (portions of an antigen where antibodies attach) as vaccine candidates (Section 28.3).
Figure 10.25 Modified proteome of *Mycobacterium tuberculosis*.

M. tuberculosis is the causative agent of the human disease tuberculosis, and its biology is discussed in Sections 16.11 and 31.4. Post-translational modifications (Section 7.16) found on different categories of M. tuberculosis proteins. The “M. tuberculosis specific” category of proteins were first detected in M. tuberculosis and possess conserved Pro-Glu/Pro-Pro-Glu amino acid sequence motifs (Section 10.12). Many of these M. tuberculosis–specific proteins are believed to be secreted into host cells because they contain an N-terminal signal sequence (Sections 6.12 and 6.13). Data adapted from Banaei-Esfahani, A., Nicod, C., Aebersold, R., and Collins, B.C. 2017. Curr. Opin. Microbiol. 39: 64.
While proteomic analyses can be performed on pure cultures of specific microbes, metaproteomics of environmental samples are particularly insightful for revealing the collective metabolic potential of a microbial community. As will be illustrated in Chapter 19, metaproteomics combined with metagenomic and metatranscriptomic approaches can provide a snapshot of ongoing physiological processes in a microbial community and highlights the power of omics for answering the key questions about the community of “who’s there” and “what are they doing” (Figure 19.8).
The Interactome
By analogy with the terms genome and proteome, the interactome is the complete set of interactions among the macromolecules within a cell (Figure 10.26). Originally, the word interactome was applied only to interactions between proteins, many of which assemble into complexes. However, it is also possible to consider interactions between different classes of macromolecules, such as between protein and RNA (see the protein–RNA interactome in Figure 10.34b).
Figure 10.26 Motility protein interactome for *Campylobacter jejuni*.

This network illustrates the way in which interactome data are depicted. (a) A subsection of the network highlighting the well-known proteins of the chemotaxis signal transduction pathway (CheW, CheA, and CheY) and their partners. MCP, methyl-accepting chemotaxis proteins (Section 7.6). (b) High-confidence interactions between all proteins known to have roles in motility. Note the six small networks that fall outside the single large network.
Interactome data are typically expressed in the form of network diagrams, with each node representing a protein and the connecting lines representing the interactions. Diagrams of whole interactomes can be extremely complex (see Figure 10.33), and thus more focused interactomes, such as the motility protein network from the bacterium Campylobacter jejuni (Figure 10.26), are more instructive. This figure shows the core interactions between well-known components of the chemotaxis system (Sections 2.11 and 7.6), including all other proteins that are known to interact with these.
There is an omics that deals specifically with metabolism, and we tackle that next.
Check Your Understanding
Why is the term “proteome” ambiguous, whereas the term “genome” is not?
What are the most common experimental methods used to survey the proteome?
10.10 Metabolomics
The metabolome is the complete set of metabolic intermediates and small molecules produced by an organism. In other words, the metabolome reflects the enzymatic pathways encoded by the genome. While transcriptomics and proteomics point to the activity of specific pathways, metabolomic data confirm that these potential reactions and pathways are actually occurring in the cell at any given time or under any given set of conditions.
Advances in Metabolomic Techniques: NIMS
Metabolomics has lagged behind other omics due largely to the immense chemical diversity of small metabolites present in cells (Chapters 3 and 14). This makes systematic metabolomic screening technically challenging. Early attempts used nuclear magnetic resonance (NMR) analysis of extracts from cells labeled with 13C-glucose (13C is a heavy isotope of carbon, most of which is 12C). However, this method is limited in sensitivity, and the number of metabolites that can be identified in a mixture simultaneously is too low for resolution of complete cell extracts.
While MALDI-TOF mass spectrometry (Section 10.9) can be used to detect and identify metabolites, nanostructure-initiator mass spectrometry (NIMS) is a more useful technique that can directly analyze biological samples without the need for special analytical preparation (Figure 10.27). Thus biofluids, tissues, cultures, or even individual cells can be analyzed. In NIMS, a laser is used to ionize the sample—just as in MALDI-TOF—but the silicon-coated surface used in NIMS does not generate the background interference seen during ionization of a matrix by MALDI-TOF. This allows for the accurate identification of small metabolites present at low concentrations and increased spatial resolution during tissue imaging. Modifications to the silicon surface can also be made to detect notoriously difficult molecules, such as structurally similar carbohydrates or steroids. These traits also make NIMS more sensitive than MALDI, which is illustrated by the ability of NIMS to detect a specific drug in a single human cell in the yoctomole (10−24 M) range (Figure 10.27).
Figure 10.27 NIMS identification of metabolites.

In nanostructure-initiator mass spectrometry (NIMS), a cell is placed on the silicon initiator surface and vaporized using a laser. The resulting ionized metabolites within a single cell are then detected using mass spectrometry as indicated by the mass-to-charge plots. Peak 342 represents the metabolite propafenone (1 yoctomole=1×10−24 mole). Because NIMS lacks a matrix, it has extremely high sensitivity and resolution. Ionized metabolites are represented rising from the surface.
Utility of Metabolomics
Metabolome analysis has been particularly useful for the study of plant biochemistry, since plants produce several thousand different metabolites—more than most other types of organisms. These compounds include many so-called secondary metabolites, chemicals such as scents, flavors, alkaloids, and pigments, many of which are commercially important. Metabolomic investigations have monitored the levels of several hundred metabolites in the model plant Arabidopsis, and significant changes were observed in the levels of many of these metabolites in response to changes in temperature, an alarming hint that global climate change will likely alter plant metabolism in major ways. Metabolomics can also be done on microbial cultures as well as natural microbial communities such as biofilms (Sections 4.9 and 8.10; Section 20.4). For example, microbiologists have detected over 3500 different metabolites in a relatively simple microbial biofilm growing in extremely acidic (pH ∼0.9) and heavy-metal-rich water draining from an abandoned iron mine site in northern California (USA). Many of these metabolites were suspected of being osmolytes and other protective molecules used by the microbial community inhabiting the runoff for combating the osmotic and other life stressors in this extreme environment.
Metabolomics has also been deployed to help characterize the human microbiome (Chapter 24). For instance, our skin epidermis is composed not only of human cells but also of microbes that contribute to epithelial health. Thus metabolites formed by skin cells, associated microorganisms, and personal hygiene products are present. Figure 10.28 shows results of an actual study of the pattern of metabolites and microbial diversity on the skin of a human male and a female plotted simultaneously on a three-dimensional topographical heat map of the human body. One of the goals of this type of research is to determine if the presence of particular metabolites can be linked to the presence of particular microbial species. Such a picture of the human skin could offer microbiologists as well as medical clinicians a better understanding of the diversity and ecology of the skin microbiome and pave the way for the future use of studies of this type in the diagnosis of skin health or disease.
Figure 10.28 Metabolomic and metagenomic mapping of skin.

Three-dimensional heat maps represent the diversity of metabolites and microorganisms detected on various areas of the skin on a human male and a female. Red indicates the highest level of diversity and purple indicates the lowest level of diversity.
As expected, the omics study of the skin (Figure 10.28) revealed many microbes known from previous studies of the skin microbiome (Section 24.5). But the study also discovered a diverse mixture of metabolites. While common chemicals produced by human skin cells such as triacylglycerides and diacylglycerides were readily detected, various metabolites resulting from microbial processing of these compounds were also detected. Overall, the level of metabolite diversity detected on specific areas of the body did not correlate well with microbial diversity. Instead, the complexity of the human skin metabolome significantly exceeded the diversity of the microbial profile, and this indicates that each species is likely producing several distinct metabolites. Interestingly, and somewhat surprisingly, the results also showed that personal hygiene products are a major source of metabolites on human skin.
Throughout this chapter we have discussed various omics and their applications as more or less individual entities. We now shift our focus to integrating multiple omics to better understand the entire organism—the major goal of systems biology.
Check Your Understanding
What techniques are used to monitor the metabolome?
III Systems Biology
The many omics born from the genomics revolution have been used in concert to attack complex problems in both single cells and entire microbial communities. The omics complement each other in ways that allow scientists to develop models that can be tested by laboratory experimentation.
The basic strategy of systems biology is to generate comprehensive models for predicting the behavior or properties of an organism that were not obvious from pre-omics era observations. These are referred to as the emergent properties of the organism. Understanding an organism’s emergent properties provides a deeper insight into its overall biology than can any single omics study by itself. Systems biology integrates the numerous omic datasets covered in Part II of this chapter to create insightful models of the biological workings of an individual organism or an entire microbial community (Figure 10.29). We begin by seeing how all of these can come together in ecological studies of individual cells in a microbial community.
Figure 10.29 The components of systems biology.

(a) The results of various “omics” analyses are combined and successively integrated into higher-level views of the entire biology of a pure culture, such as (b) that of the green sulfur bacterium Chlorobium ; or of a mixed microbial community, such as (c) that of phototrophic sulfur bacteria obtained from a lake; or of a single cell isolated from a microbial community (see Figure 10.30).
10.11 Single-Cell Genomics
Besides sequencing total environmental DNA as described for metagenomics (Section 10.7), the genomes of individual cells can also be sequenced—a technique called single-cell genomics (SCG) (Figure 10.30). This is possible because of the ability to amplify tiny amounts of DNA. Single-cell genomics is critical for studying the metabolic potential of microorganisms in natural microbial communities. While metagenomic analysis of a microbial community can detect the presence of genes specific to certain pathways, it is difficult to discern whether the pathways are contained within the same organism. Besides genome sequencing, transcriptome and proteome analyses can also be performed on individual cells, leading to a comprehensive omics study on one component of a microbial ecosystem.
Figure 10.30 Single-cell genomics.
A single cell isolated from an environmental sample can be the source of a diversified omics study. MDA, multiple displacement amplification (Section 19.12).
Cell Isolation and Sample Preparation
The ability to isolate cells is obviously essential for single-cell genomics, and various physical techniques including dilution in microwells (Section 19.3), encapsulation, and fluorescence-activated cell sorting (FACS) have been used to do so. For encapsulation, the sample is diluted and added to sterile oil to form microdroplets. Approximately 30% of the resulting droplets from this technique will contain only one cell (Figure 10.31). Combining droplet encapsulation with FACS, which is able to optically detect single cells, allows droplets that do not contain a cell to be rejected. This method of single-cell isolation has also been shown effective in isolating single virions for viral omics analyses.
Figure 10.31 Isolation and sequencing of single cells.

Droplets of a diluted sample are added to microfluidic wells at a concentration such that a single well will contain no more than a single cell. Wells containing cells are then subject to multiple displacement amplification (MDA) for DNA sequencing.
Sequencing DNA from single cells relies on a modified version of the polymerase chain reaction (PCR, Section 12.1) called multiple displacement amplification (MDA) (Figure 10.31; Section 19.12 and Figure 19.43). This PCR technique uses a highly sensitive viral DNA polymerase to amplify the femtogram (10−15 g) quantities of DNA present in a single bacterial cell into the micrograms (10−6 g) of DNA required for sequencing (a billionfold amplification). However, because of its very sensitivity, contamination is one of the biggest problems with MDA. Contaminating DNA can originate from the sample itself or from the laboratory equipment and reagents. If great care is not taken in isolating the cell and amplifying its genome, contaminating DNA can make up half or more of the reaction products and create major problems for genome assembly and further genomic analyses. In addition to analysis of a cell’s genome, its RNA can also be analyzed using RNA-Seq following amplification to form cDNA by a modified version of PCR (Section 10.8). Single-cell proteomic analyses are trickier than nucleic acid studies because amplification is not employed, but analyses using extremely sensitive fluorescence methods are available for this purpose (Figure 10.30).
Applications of Single-Cell Omics
Single-cell omics have the unique power to probe several facets of an organism’s biology in an individual cell rather than on a cell population basis. Using SCG, metabolic genes present in a microbial community (Figure 10.29c) not only can be identified but can be assigned to particular species; such information reveals which particular organisms are degrading which particular nutrients. For example, single-cell genomics has been used to analyze hydrocarbon degradation by bacteria in polluted environments, leading to a better understanding of which organisms are doing what in the overall process. Similarly, plasmids and viruses can be allocated to their correct host when the genome of a single cell is sequenced.
Single-cell genomics has also been used to explore the genomes of phyla of Bacteria and Archaea that are detected in environmental 16S rRNA gene surveys but for which no laboratory cultures are available (these are called candidate phyla, Section 13.12). Collectively, these yet-to-be-cultivated microbes have been called microbial dark matter. One dark matter study applied SCG to over 200 uncultivated archaeal and bacterial cells from nine different environments. The results revealed several surprising findings including the following: Some archaeal RNA polymerase sigma factors are similar to their bacterial counterparts (Sections 6.5 and 6.6); some mRNA stop codons (Section 6.9 and Table 6.4) have been reassigned to incorporate actual tRNAs with their attached amino acids; the genomes of some bacterial cells encode an oxidoreductase enzyme previously seen only in eukaryotes; some Archaea contain genes encoding a bacterial-like stringent response (Section 7.9); and some Archaea produce an enzyme that functions in peptidoglycan synthesis (recall that peptidoglycan is a “signature molecule” of Bacteria and is not known from any Archaea, Section 2.3). Many other examples have emerged as well.
Single-cell genomics is an excellent example of serendipity, the “pleasant surprise” of finding one thing while working on another. In this case, the serendipity occurred when omic methods designed with one goal in mind—analysis of the biology of cell populations—were refined to probe the biology of a single cell in ways never before thought possible. Single-cell genomics is poised to complement the Earth Microbiome Project, a sequencing endeavor to archive the genome sequence of all cultured bacterial and archaeal type strains (http://www.earthmicrobiome.org/). For those species of Bacteria and Archaea that reside in the microbial dark matter, SCG offers a way to include these species in the archive while simultaneously revealing valuable clues that will one day bring these organisms into laboratory culture.
We close out this chapter with two final sections, each focused on combining previously discussed omics methods to solve a specific problem. The first section explores the system biology of a notorious bacterial pathogen, whereas the second section is devoted to the entire human body.
Check Your Understanding
How are single cells isolated from a mixed population?
What must be done before minute amounts of DNA can be sequenced?
10.12 Integrating Mycobacterium tuberculosis Omics
10.12 Integrating Mycobacterium tuberculosis Omics
10.12 Integrating *Mycobacterium tuberculosis* Omics
Mycobacterium tuberculosis is a pathogen that infects one-third of the world’s population and kills approximately 2 million people every year (Section 31.4). Multidrug resistance and the ability to temporarily enter dormancy in response to stress (Section 8.12) are two characteristics that contribute to the persistence of M. tuberculosis. Thus, the identification of new treatment methods is critical for combating M. tuberculosis. This challenge is being tackled using a systems biology approach to understand how M. tuberculosis—an intracellular pathogen—adapts to the oxygen deficiency (hypoxia, a condition that develops in tuberculosis) in host cells and to identify potential drug targets for therapeutic design.
Tuberculosis Gene Expression and Regulatory Networks
As we discussed in Section 10.8, RNA-Seq can be used to characterize an organism’s complete expression profile. A modification of this technique termed dual RNA-Seq can be used to simultaneously profile the transcriptomes of a pathogen and its host cell. This approach allows for the responses of both the pathogen and host to be captured during the infection process and has been especially useful for understanding how M. tuberculosis evades the host’s defense systems. Data from dual RNA-Seq and the integration of over 600 separate expression experiments have facilitated the construction of gene expression models. These models have identified the primary energy source of intracellular M. tuberculosis cells as host cholesterol, and they have shown that the amino acid aspartate produced by the host is used by M. tuberculosis not only as a nitrogen source but also as protection against reactive oxygen species (Section 4.16) produced by macrophages (a type of phagocytic cell, Section 26.1) trying to kill M. tuberculosis cells.
The M. tuberculosis research has integrated transcriptome and other omics datasets including ChIP-Seq, a method where an antibody that combines with a specific DNA-binding protein is used to trap the protein bound to its DNA, after which the DNA is removed and sequenced. From ChIP-Seq analyses, several transcription factors and regulatory networks critical to the pathogenesis of M. tuberculosis have been identified. These include regulation by the major bacterial cell regulator LexA (Section 9.4) of certain genes for DNA damage response, and control by members of the DevR regulon of the entry of M. tuberculosis into dormancy (Figure 10.32). Additional potential drug targets have also been identified through the screening of over 1000 different mutant strains of M. tuberculosis. This analysis mapped 18 previously uncharacterized genes to the persistence response of M. tuberculosis (Section 8.12), illustrating the power of integrating omics with mutant analyses to identify important pathogenicity genes in M. tuberculosis.
Figure 10.32 Genes controlled by the regulators LexA and DevR in *Mycobacterium tuberculosis*.

(a) A snapshot of the interactome of some of the gene clusters that participate in DNA repair as identified from their expression pattern. Members of the LexA regulon are in red, while green connectors indicate interactions of other DNA repair genes. (b) Colorized scanning electron micrograph of cells of M. tuberculosis. (c) Venn diagram of results of various studies of the DevR regulon. Three methods of assessing genes in the regulon were employed and the number of genes identified from each detection method is indicated. A total of 622 genes were identified by ChIP-Seq analysis; of these, only 37 were identified by every method. Data adapted from van Dam, J.C.J., et al. 2014. BMC Syst. Biol. 8: 111.
Tuberculosis Proteomics and Metabolomics
As was illustrated in Figure 10.25, proteomics has been used to identify M. tuberculosis protein modifications under certain conditions. The power of proteomics has been used to identify proteins essential to the M. tuberculosis hypoxia response. While expected proteins that combat reactive oxygen species such as superoxide dismutase (Section 4.16) were detected, so were toxin–antitoxin systems (Section 8.12) and proteins that participate in the biosynthesis of the unique lipoproteins of the M. tuberculosis cell envelope (Section 16.11). Production of nitrate and nitrite transporters also increased as M. tuberculosis switched to anaerobic respiration (Section 3.10) for energy metabolism. Moreover, at least 160 different uncharacterized proteins containing the N-terminal amino acid sequence “Pro-Glu/Pro-Pro-Glu” (amino acids and their abbreviations are listed in Figure 6.27) were detected in the pathogen. If drugs could be developed that specifically target this peptide, they might be effective treatments for tuberculosis.
Online fundraising and social media have even been combined for the common goal of identifying new drugs to treat tuberculosis. The Connect to Decode initiative (http://c2d.osdd.net) is composed of more than 800 researchers whose collective goal is to identify new tuberculosis drug targets by mapping the entire interactome of M. tuberculosis. By analyzing over 10,000 M. tuberculosis experimental datasets, the researchers have now annotated 87% of the proteins encoded by the genome. This is a vast improvement over the 52% of genes annotated in the original genome sequence. This collective effort has also resulted in a map of the complete M. tuberculosis interactome, with over 1400 proteins connected by more than 2500 functional relationships (Figure 10.33). Thus far, nearly 20 potential drug targets and their interactomes have been identified by this initiative. These targets lack homology to human proteins or to proteins of the human oral and gastrointestinal microbiome, thus increasing the probability of identifying drugs with the least side effects.
Figure 10.33 *Mycobacterium tuberculosis* protein interactome.

Node color indicates proteins of the same category, connecting lines indicate interactions, and node size indicates relative number of interactions.
While tuberculosis metabolomics is in its infancy, studies comparing virulent versus attenuated Mycobacterium strains have identified over 1000 different lipids—many of them unique lipoproteins—that are likely quite important to the biology of M. tuberculosis. Thus, the M. tuberculosis lipid metabolism proteome may reveal several potential therapeutic targets for tuberculosis. Finally, by profiling the secretome (an inventory of metabolites secreted) of M. tuberculosis, molecules unique to virulent strains (such as the nucleotide 1-tuberculosinyladenosine) have been discovered. This modified nucleotide is present in the urine of humans infected with M. tuberculosis, and its discovery illustrates the power of omics to link a particular pathogen to a specific molecule—be it a gene, protein, or metabolite—and yield new disease markers for use in clinical diagnostics. Moreover, if this modified nucleotide turns out to be essential to the survival of M. tuberculosis cells, drugs that interfere with its synthesis or activity are potential anti-tuberculosis drugs.
Check Your Understanding
How does dual RNA-Seq differ from traditional RNA-Seq?
How has systems biology provided new approaches to treating the disease tuberculosis?
10.13 Systems Biology and Human Health
In 2003 the sequence of the 3-billion-base-pair human genome was completed and released in an international effort that cost nearly $3 billion. With the advent of next-generation sequencing, the cost to sequence a human genome has fallen under $1000, triggering an onslaught of human genome sequence data. How can the human genome and systems biology be tailored to benefit human health? By comparing genome sequences in over 2500 people from different continents, scientists have already identified over 88 million sites in the human genome that are subject to variation. These genomic variants include single nucleotide differences, insertions and deletions, and rearrangements, each of which may turn out to be harmless, to be beneficial, to contribute to noninfectious “lifestyle” diseases such as obesity, diabetes, and heart disease, or to govern susceptibility to certain cancers. Understanding if and how genomic variants contribute to disease can be used to improve diagnostics, treatments, and prevention.
The omics revolution has opened the era of personalized medicine, where genomic, transcriptomic, proteomic, metabolomic, and pharmacogenomic (omics responses to drugs) data are exploited to generate a snapshot of normal and disease states along with immune processes that occur in between. The first step in personalized medicine is the generation of an integrative Personal Omics Profile (iPOP). Tracking changes in this profile during healthy and disease states can aid in assessing medical risks and diagnosing and treating patients. The utility of personalized medicine can be illustrated by an actual study where blood samples taken from a male subject 20 times over a period of two years were subjected to proteomic and metabolic profiling of about 5000 proteins and 4000 metabolites (Figure 10.34). The results suggested that the subject was at risk for coronary artery disease—which was not surprising based on his medical history and familial incidence—but also indicated that he was at high risk for type 2 diabetes. While this was a surprising finding considering his overall condition and familial health history, the prediction was based on profiling the subject’s immune response to a viral infection. During this infection, changes in both gene expression and protein profiles related to the insulin response were detected. Just as this subject’s personalized medicine profile predicted, he developed diabetes over the course of the two-year study (Figure 10.34). By tracking his iPOP, the onset of diabetes was revealed by the increasing production of RNAs, proteins, and metabolites, such as hemoglobin A1c and lauric acid, related to glucose metabolism (Figure 10.34).
Figure 10.34 An integrated personal omics profile (iPOP).

(a) Partial heat map of RNA and protein expression. Data plotted are from days 294–400 of the two-year study, with an increase in RNAs and proteins related to diabetes spiking on day 307. (b) Glucose regulation interactome. RNAs are indicated with blue circles, proteins with yellow squares, and RNA and its corresponding protein with green hexagons. A metabolite from the interactome is also shown. (c) Blood glucose levels during the time course of the study. The time of contracting a respiratory virus (RSV) is indicated as well as the time frame in which the patient made lifestyle and diet changes. Parts b and c adapted from Jennifer Li-Pook-Than and Michael P. Snyder, Stanford University.
This case study illustrates the potential of iPOPs in monitoring the immune response and disease states. In fact, the omics revolution now allows for genomics, transcriptomics, proteomics, and metabolomics to be performed on different types of immune system cells. The wealth of data from these analyses has led to the development of new immunotherapy options (Sections 12.8 and 28.4). However, problems such as error rates, the analysis and storage of such large datasets, assessment of complicating factors (such as additional diseases during the course of the study, Figure 10.34), and ethical issues need to be addressed before iPOPs become routine tools in clinical medicine.
Microbiology as well as biology in general has been forever changed by the genomics age. Our journey through this chapter has only scratched the surface in describing how omics can be used to address previously intractable scientific questions. Much more is almost certainly in store, as major leaps forward in the omics field so far have typically been only one technological advancement away. In Chapter 11 we continue our genomics theme but change our focus from cells to viruses—the most abundant and genetically diverse microbes on Earth.
Explore the Microbial World DNA Sequencing in the Palm of Your Hand
Explore the Microbial World DNA Sequencing in the Palm of Your Hand
DNA Sequencing in the Palm of Your Hand
DNA sequencing technologies are revolutionizing microbiology at a remarkable pace. Innovations in next-generation sequencing have even tackled the difficult issues of cost and portability. The world’s first mobile nucleic acid sequencer—the MinION—is a palm-sized device that possesses 2000 tiny pore-containing proteins called nanopores. As single strands of nucleic acid travel through the nanopores, individual nucleotides are identified in real time based on changes in electrical current (Figure 1). These current changes are relayed to a computer through a USB connection, which also powers the MinION. This miniature but mighty machine can display nucleic acid sequences from critical field samples in real time on a computer screen.
Figure 1 Nanopore-MinION sequencing.

A DNA double helix is converted to a single strand for passage through a membrane-bound pore. As the DNA transits the nanopore, it causes changes in electric current that are base-specific and thus reveal the DNA sequence.
Besides its portability, the methodology behind the nanopore technology yields extremely long sequencing reads compared to other next-generation sequencing methods (Table 10.2). This feature is being exploited to quickly sequence complete microbial genomes as well as to “close” those genomes that have been difficult because of their troublesome nucleotide repeats. The identification of nucleotides based on changes in electrical current as they travel through a nanopore also allows for modified nucleotides, such as those with additional methyl groups, to be detected. This feature is of interest to microbiologists who study gene regulation, as the methylation state of gene promoters can control gene expression; such is the case with genes that encode mobile gene elements such transposases (Section 9.11) that play critical roles in horizontal gene transfer and genome evolution (Chapter 13).
The utility of the MinION’s portability was clearly on display during the 2014–2015 Ebola virus hemorrhagic fever outbreak in West Africa. Scientists traveled to Guinea with three MinIONs in their luggage, a feat in itself as most DNA sequencers are too large and delicate to travel in baggage. Once in Guinea, scientists were able to survey the spread of different Ebola virus strains by analyzing unique nucleotide sequences present in each strain’s genome. In as little as 48 hours after sample collection, Ebola virus genomes from 14 patients were determined using MinION sequencing.1 Figure 2 shows a researcher loading a patient’s sample onto a MinION set up in a mobile field laboratory (inset).
Figure 2 Use of the MinION in the field during the 2015 Ebola outbreak in West Africa.
Because the Ebola genome mutates rapidly, the astonishing turnaround time provided by the MinION allowed epidemiologists to track geographical movements of different strains of the virus. This real-time analysis indicated that two major viral strains were the cause of Ebola persistence and that cross-border transmission between Sierra Leone and Guinea severely prolonged the outbreak.2 Traditional sequencing methods would not have supported such surveillance, as it requires weeks to obtain results after shipment of samples to remote laboratories.
While biologists have envisioned numerous uses for the MinION, it has already been tested by NASA on the International Space Station. During this test, the MinION’s ability to sequence the genomes of Escherichia coli, bacteriophage lambda, and mouse DNA was not hampered by the microgravity of outer space.3 In addition, the MinION has proven durable and dependable even in such remote and extreme locations as Antarctica,4 opening up the possibility that the large sequencing requirements of modern-day microbial ecology may be fulfilled, at least in part, right in the field at the time of sample collection. Moreover, developers are currently attempting to modify the portable sequencer to operate from a smartphone instead of a computer. And, because of its size, relatively low cost, and ease of use, the next frontier for the MinION will undoubtedly be the classroom.
1 Quick, J., et al. 2016. Real-time, portable genome sequencing for Ebola surveillance. Nature 530: 228.
3 Castoro-Wallace, S.L., et al. 2017. Nanopore DNA sequencing and genome assembly on the International Space Station. Sci. Rep. 7: 18022.
4 Johnson, S.S., et al. 2017. Real-time DNA sequencing in the Antarctic Dry Valleys using the Oxford nanopore sequencer. J. Biomol. Tech. 28(1): 2.
Chapter Review
Go to Mastering Microbiology for videos, animations, practice tests, and more.
I Genomics
10.1 Small viruses were the first organisms whose genomes were sequenced, but now many prokaryotic and eukaryotic cellular genomes have been sequenced.
Q What is one discovery resulting from the availability of a microbial genome?
10.2 DNA sequencing technology is advancing quickly. These advances have greatly increased the speed of DNA sequencing. Computer analysis of resulting sequencing data is also a vital part of genomics. Computational tools are used not only to annotate genomes but also to analyze sequences and the structures of biological macromolecules.
Q How can protein homology assist in genome annotation?
10.3 Sequenced genomes of Bacteria and Archaea range in size from 0.11 to 16 Mbp. The smallest are smaller than those of the largest viruses, whereas the largest have more genes than some eukaryotes. Gene content in prokaryotic cells is typically proportional to genome size. Many genes can be identified by their sequence similarity to genes found in other organisms. However, a significant percentage of sequenced genes are of unknown function.
Q What is the relationship between genome size and open reading frame content in genomes from prokaryotic cells?
10.4 Virtually all eukaryotic cells contain mitochondria, and in addition, plant cells contain chloroplasts. Although the genomes of organelles are independent of the nuclear genome, the organelles themselves are not. Many genes in the nucleus encode proteins required for organelle function. The complete genomic sequence of many microbial eukaryotes has also been determined, and the number of genes ranges from 1000 (less than many bacteria) to 60,000 (more than twice as many as humans).
Q Which genomes are larger, those of chloroplasts or those of mitochondria? How does your genome compare with that of yeast in overall size and gene number?
II Functional Omics
10.5 Once a microbial genome has been sequenced, assembled, and annotated, the role of many genes remains unknown. To characterize new gene products, several functional genomics tools can be applied. These include in particular comparative genome analyses and heterologous gene expression.
Q How can the abundance of available microbial genomes be used to identify new systems that prevent viral infection or horizontal transfer of DNA?
10.6 Transposon mutagenesis has been harnessed for identifying gene functions using a high-throughput sequencing methodology. The technique, Tn-Seq, has been used to pinpoint genes in many different bacteria that would be more difficult and laborious to discover using other functional genomic methods.
**Q If a gene in Escherichia coli was absolutely essential for growth under all conditions, how could you determine this using Tn-Seq?**
10.7 Most microorganisms in the environment have never been cultured. Nonetheless, analysis of DNA samples has revealed enormous sequence diversity in most habitats. The concept of the metagenome embraces the total genetic content of all the organisms in a particular habitat.
Q How do the human microbiome and mycobiome differ?
10.8 Microarrays consist of oligonucleotide probes corresponding to genes or gene fragments attached to a solid support in a known pattern; mRNA, cDNA, or DNA can then be labeled and hybridized to the gene chip to determine patterns of gene expression or the presence or absence of specific organisms. RNA-Seq combined with sequencing of cDNA can be used to profile the entire transcriptome of an organism.
Q Besides gene expression, what else can be assayed using gene chips?
10.9 Proteomics is the analysis of all the proteins present in an organism. The ultimate aim of proteomics is to understand the structure, function, and regulation of these proteins. The interactome is the total set of interactions between macromolecules inside the cell.
Q Besides determining the identity of proteins present under a specific condition, what else can proteomics determine about a microbial proteome?
10.10 Metabolomics profiles the complete set of metabolic intermediates produced by an organism. This analysis can determine active metabolic pathways and potential cross-feeding (one organism supplies a nutrient for another organism) in community samples.
Q Why is investigation of the metabolome lagging behind that of the proteome?
III Systems Biology
10.11 With advances in molecular techniques, the genomes of single cells can be sequenced. Expression and protein profiles of single cells can also be determined. These techniques are instrumental for studying as yet uncultured microbes.
Q How can single-cell genomics be used to address microbial dark matter?
10.12 By integrating multiple omics datasets in a systems biology approach, computer models predicting molecular activities and interactions in cells can be generated. For example, potential drug targets for the treatment of Mycobacterium tuberculosis have been identified using systems biology.
Q How can systems biology be used to discover new diagnostic markers for disease?
10.13 Systems biology can also be applied to personal medicine. Besides detecting genetic variants, disease risks can be predicted by profiling a person’s genome, transcriptome, proteome, and metabolome.
Q How can omics be used to design immunotherapy strategies?
Application Questions
Apart from genome size, what factors make complete assembly of a eukaryotic genome more difficult than assembly of a genome from a species of Bacteria or Archaea?
Describe how one might determine which proteins in Escherichia coli are repressed when a culture is shifted from a minimal medium (containing only a single carbon source) to a rich medium containing many amino acids, bases, and vitamins. How might one study which genes are expressed during each growth condition?
Describe how you could use systems biology to discover a new biologically produced antibiotic.
Chapter Glossary
the use of computational tools to acquire, analyze, store, and access DNA and protein sequences Codon bias
the nonrandom usage of multiple codons encoding the same amino acid; the relative proportions of different codons encoding the same amino acid vary in different organisms. Same as codon usage Gene chip
small, solid supports to which genes or portions of genes are affixed and arrayed spatially in a known pattern (also called microarrays) Genome
the total complement of genes contained in a cell or virus Genomics
the discipline that maps, sequences, analyzes, and compares genomes Hybridization
the joining of two single-stranded nucleic acid molecules by complementary base pairing to form a double-stranded hybrid DNA or DNA–RNA molecule Interactome
the total set of interactions between proteins (or other macromolecules) in an organism Metabolome
the total complement of small molecules and metabolic intermediates of a cell or organism Metagenome
the total genetic complement of all the cells present in a particular environment Metagenomics
the genomic analysis of pooled DNA or RNA from an environmental sample containing organisms that have not been isolated; same as environmental genomics Microarray
small, solid supports to which genes or portions of genes are affixed and arrayed spatially in a known pattern (also called gene chips) Mobilome
the mobile genetic elements in a genome Nucleic acid probe
a strand of nucleic acid that can be labeled and used to hybridize to a complementary molecule from a mixture of other nucleic acids Open reading frame (ORF)
a sequence of DNA or RNA that could be translated to give a polypeptide Proteome
(1) the total set of proteins encoded by a genome or (2) the total protein complement of an organism under a given set of conditions, also called the translatome Proteomics
the genome-wide study of the structure, function, and regulation of the proteins of an organism Sequencing
deducing the order of nucleotides in a DNA or RNA molecule by a series of chemical reactions Systems biology
the integration of data from genomics and other “omics” areas to build an overall picture of a biological system Transcriptome
the complement of all RNA produced in an organism under a specific set of conditions Translatome
the total set of proteins produced by an organism under a specific set of conditions