12 Biotechnology and Synthetic Biology
II Making Products from Genetically Engineered Microbes: Biotechnology
An Ingestible Biosensor: Using Bacteria to Monitor Gastrointestinal Health
By constantly monitoring and responding to molecules in their environment, microbes possess a remarkable ability to sense and adapt to a range of conditions. Synthetic biologists have exploited this resiliency, along with genetic tractability, to develop microbially driven biosensors for detecting specific stimuli. Now the technology behind these biosensors has been modified to detect markers of human disease.

At the top of the image shown here is a tiny ingestible biosensor that is able to monitor gastrointestinal bleeding (the coin below indicates the scale). The device detects the presence of blood through the response of Escherichia coli cells that have been genetically modified to express luminescence genes when exposed to heme, a component released from lysed red blood cells. The square image in the bottom right of the photo shows four quadrants of the device’s semipermeable membrane that confines millions of the engineered bacteria. Once loaded with bacteria, the membrane is attached to the capsule such that the bacteria are both protected and exposed to surrounding molecules as the device travels through the digestive tract.
As the engineered bacteria encounter heme, they modify their gene expression to emit light. Photo transmitters located under the membrane then communicate this response to a microprocessor that ultimately transmits the signal wirelessly to a cell phone. While this biosensor has been successfully used to detect the presence of blood in the stomachs of swine, engineers are working to further miniaturize the electronics. Small as it is, the biosensor’s current size (∼3.8 cm long) is difficult for humans to swallow.
With current medical procedures for analyzing upper gastrointestinal health requiring expensive and unpleasant procedures, such as endoscopy, the exciting fields of biotechnology and synthetic biology promise future noninvasive and real-time monitoring with as little as a biosensor capsule and a cell phone!
Source: Mimee, M., et al. 2018. An ingestible bacterial-electronic system to monitor gastrointestinal health. Science 360: 915.
Industrial microbiology uses microbes on a large scale to produce products such as enzymes, foods, and beverages. These microbes are typically not genetically modified. Instead, naturally overproducing strains are selected from wild-type strains and used for industrial purposes. In contrast, biotechnology uses genetically modified microorganisms to produce high-value products that the organisms do not naturally produce. In this chapter we discuss the basic techniques of genetic engineering that underlie biotechnology, in particular those used to clone, alter, and express genes efficiently in host organisms. We also explore how genetic engineering and biotechnology can be used for industrial, medical, and agricultural applications (Figure 12.1) and introduce the exciting new field of synthetic biology. The latter is an emerging science that assembles bits and pieces of DNA—aptly called biobricks—into entirely new chromosomes or other genetic elements that can modify microbes or even macroorganisms in ways that benefit humans (Figure 12.1).
Figure 12.1 Genetic engineering and biobrick assembly.

Molecular cloning techniques are used to modify organisms for the production of high-value products. RBS, ribosome-binding site; sRNAs, small RNAs; ORFs, open reading frames.
I Tools of the Genetic Engineer
DNA from any source can be manipulated in the laboratory in unprecedented ways using the powerful tools of PCR, restriction enzymes, molecular cloning and recombineering, nucleic acid hybridization, and a host of gene expression systems.
Performing genetics in vivo (in living organisms) has many limitations that can be overcome by manipulating DNA in vitro (in a test tube). Genetic engineering refers to the use of in vitro techniques to alter genes in the laboratory. Such altered genes may be reinserted into the original source organism or into some other host organism. Expression of a gene from one organism in a different host organism that does not normally possess the gene is called heterologous expression.
Genetic engineering requires that specific DNA be isolated, purified, and further manipulated. We begin by considering some of the basic tools of the genetic engineer, including amplification of DNA, the separation of nucleic acids by electrophoresis, nucleic acid hybridization, and molecular cloning. We also describe methods for expressing foreign genes in bacteria and targeted mutagenesis.
12.1 Manipulating DNA: PCR and Nucleic Acid Hybridization
12.1 Manipulating DNA: PCR and Nucleic Acid Hybridization
12.1 Manipulating DNA: PCR and Nucleic Acid Hybridization
The first objective of genetic engineering is to isolate copies of specific genes in pure form, and the key method for doing so is the polymerase chain reaction (PCR) (Figure 12.2). Simply put, the polymerase chain reaction is DNA replication in vitro, as segments of target DNA are multiplied by up to a billionfold in the process of amplification. During each round of amplification, the amount of DNA doubles, leading to an exponential increase in the target DNA. Using an automated PCR machine called a thermocycler, a large amount of amplified DNA can be produced from only a few molecules of target DNA. In some cases it is desirable to quantify the initial amount of target DNA, and a variation on PCR called quantitative PCR (qPCR) is used for this purpose (Section 29.8). A second variation on the original PCR technique allows for amplification of RNA (following its conversion to DNA, as discussed later in this section).
Figure 12.2 The polymerase chain reaction (PCR).
The PCR amplifies specific DNA sequences. (a) Target DNA is heated to separate the strands, and a large excess of two oligonucleotide primers, one complementary to each strand, is added along with DNA polymerase. (b) Following primer annealing, primer extension yields a copy of the original double-stranded DNA. (c) Two additional PCR cycles yield four and eight copies, respectively, of the original DNA sequence. (d) Effect of running 20 PCR cycles on a DNA preparation originally containing 1 copy of a target gene. Note that the plot is semilogarithmic.
Mastering Microbiology
Art Activity: Figure 12.1 The polymerase chain reaction (PCR)
PCR and Polymerases
To synthesize DNA, PCR requires DNA polymerase, the enzyme that naturally copies DNA molecules (Section 6.3), and artificially synthesized oligonucleotide primers (Section 12.4) made of DNA (rather than the RNA primers used by cells to replicate DNA). PCR does not actually copy whole DNA molecules but amplifies stretches of up to a few thousand base pairs (the target) from within a larger DNA molecule (the template) during the following steps (Figure 12.2):
Template DNA is heated to denature it (that is, to separate the two strands), and then two DNA oligonucleotide primers complementary to sequences flanking the target DNA on each strand are added in excess. This ensures that most template strands anneal to a primer, and not to each other, as the mixture cools (Figure 12.2a).
DNA polymerase then extends the primers using the original DNA as the template (Figure 12.2b).
After an appropriate incubation period, the mixture is heated again to separate the strands, but now the target gene is present in twice the original amount. The mixture is then cooled to allow the primers to hybridize with complementary regions of newly synthesized and original DNA, and the process is repeated (Figure 12.2c). In practice, 20–30 cycles are typically run, yielding a 106-fold to 109-fold increase in the target sequence (Figure 12.2d).
Mastering Microbiology
Art Activity: Figure 12.2 Reverse transcription PCR
Because high temperatures are used to denature the double-stranded copies of DNA in vitro, use of a thermostable DNA polymerase is critical. Taq polymerase, a DNA polymerase isolated from the thermophilic hot spring bacterium Thermus aquaticus (Section 16.20), is stable to 95 °C and thus is unaffected by the denaturation step employed in the PCR (Figure 12.2). A DNA polymerase from Pyrococcus furiosus, a hyperthermophilic species of Archaea with a growth temperature optimum of 100 °C (Section 17.4), is called Pfu polymerase and is even more thermostable than Taq polymerase. Moreover, unlike Taq polymerase, Pfu polymerase has proofreading activity (Section 6.4), making it especially useful when high accuracy is crucial. To supply the demand for thermostable DNA polymerases in biotechnology, the genes encoding these enzymes have been cloned into Escherichia coli, allowing the enzymes to be produced commercially in large quantities.
PCR Applications and RT-PCR
PCR is extremely valuable for obtaining DNA for gene cloning or for sequencing purposes because the gene or genes of interest can easily be amplified if flanking sequences are known. PCR is also used routinely in comparative or phylogenetic studies to amplify genes from various sources. In these cases the primers are made commercially to base-pair with regions of the gene that are conserved in sequence across a wide variety of organisms. Because small ribosomal subunit (SSU) rRNA—a molecule used for phylogenetic analyses—has both highly conserved and highly variable regions (Section 13.11 and Figure 13.24), primers specific for the SSU rRNA gene from different taxonomic groups can be used to survey habitats for their microbial communities (Section 19.6). Also, because it is so sensitive, PCR can be used to amplify even very small quantities of DNA. For example, PCR has been used to amplify DNA from sources as varied as mummified human remains, fossilized plants and animals, and even single microbial cells (Section 10.11). PCR is also a common tool of medical diagnostics in clinical microbiology laboratories (Section 29.8) and is widely used in forensic science to link crime scene evidence such as blood, semen, or tissue samples to a specific suspect. In clinical laboratories, especially, time is of the essence, and the faster an infectious disease is diagnosed, the sooner an effective treatment can be chosen and administered.
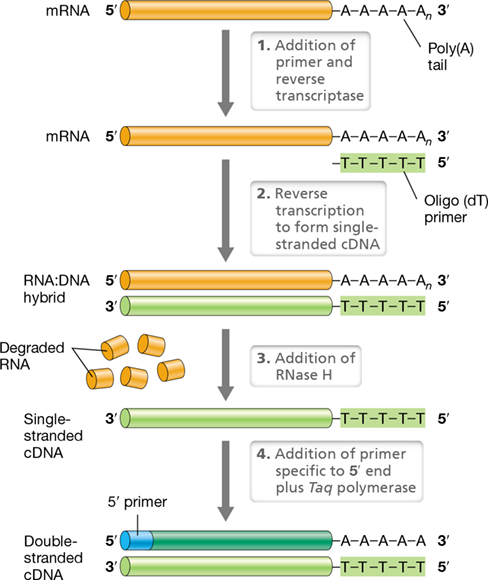
An important extension of the standard PCR procedure is reverse transcription PCR (RT-PCR), used to make DNA from an mRNA template (Figure 12.3). This procedure can be used to detect if a gene is expressed or to produce an intron-free eukaryotic gene for expression in bacteria (Section 12.3). RT-PCR uses the retroviral enzyme reverse transcriptase to convert RNA into complementary DNA (cDNA) (Sections 11.1 and 11.11). Figure 12.3 illustrates how reverse transcriptase makes a single strand of cDNA using RNA as a template. To make cDNA, a primer complementary to the 3′ end of the target RNA is used by the enzyme reverse transcriptase to initiate DNA synthesis. If the template is eukaryotic mRNA, a primer complementary to the poly(A) tail (Section 6.6) of the mRNA can be used. The activity of reverse transcriptase results in a hybrid nucleic acid molecule containing both DNA and RNA. RNase H, a ribonuclease specific for the hybrid molecule, hydrolyzes the RNA, leaving the single-stranded cDNA as template for standard PCR using an additional 5′ primer complementary to the 3′ end of the cDNA. Note that some commercial reverse transcriptase enzymes used for RT-PCR also possess an RNase H domain.
Figure 12.3 Reverse transcription PCR.


Steps in the synthesis of complementary DNA (cDNA) from a eukaryotic mRNA. Reverse transcriptase synthesizes a hybrid molecule containing both RNA and DNA using the mRNA as a template and oligo(dT) primer as a substrate. Next, the enzyme RNase H hydrolyzes the RNA portion of the hybrid molecule, yielding a single-stranded molecule of complementary DNA (cDNA). Following the addition of a 5′ primer complementary to the 3′ end of the cDNA, Taq polymerase produces a double-stranded cDNA.
Gel Electrophoresis and Nucleic Acid Hybridization
To verify that amplification of a nucleic acid was successful and for other nucleic acid manipulation steps, DNA or RNA fragments can be separated from each other by gel electrophoresis, a technique that employs an agarose gel to separate nucleic acid fragments based on differences in their size and charge (**Figure 12.4*a***). When an electrical current is applied, nucleic acids move through the gel toward the positive electrode because of their negatively charged phosphate groups, and small molecules migrate more rapidly than large molecules. After the gel has been run for a time sufficient to separate the molecules, the gel is stained with a dye that binds to nucleic acids and makes them fluoresce, such as ethidium bromide (Figure 12.4b). To determine the size of the DNA or RNA of interest, the migration is compared to a standard sample consisting of nucleic acid fragments of known sizes, called a ladder. DNA fragments can then be purified from gels and used for a variety of purposes, such as cloning or hybridization.
Figure 12.4 Agarose gel electrophoresis of DNA.

(a) DNA samples are loaded into wells in a submerged agarose gel. (b) A photograph of a stained agarose gel. The DNA was loaded into wells toward the top of the gel (negative pole) as shown, and the positive electrode is at the bottom. The standard sample in lane A (DNA ladder) has fragments of known size that may be used to determine the sizes of the fragments in the other lanes. Bands stain less intensely at the bottom of the gel because the fragments are smaller, and thus there is less DNA to stain.
When DNA is denatured, the single strands can be used to form hybrid double-stranded molecules with other single-stranded DNA (or RNA) molecules by complementary base pairing (Section 6.1) in a process called nucleic acid hybridization, or hybridization for short. Hybridization is widely used in detecting, characterizing, and identifying segments of DNA and RNA. Single-stranded nucleic acids whose identity is already known and that are used in hybridization are called nucleic acid probes, or simply probes. To allow detection, probes are made radioactive or are labeled with chemicals that are colored or yield fluorescent products (Section 19.5), and by varying the hybridization conditions, the “stringency” of the hybridization can be adjusted such that complementary base pairing is somewhat flexible or, alternatively, must be nearly exact.
Hybridization is useful for finding related sequences in different genomes or other genetic elements and to determine if a gene is expressed into an RNA transcript. In Southern blotting, probes of known sequence are hybridized to target DNA fragments that have been separated by gel electrophoresis. The hybridization procedure in which DNA is the target sequence in the gel, and RNA or DNA is the probe, is called a Southern blot. By contrast, a Northern blot uses RNA as the target sequence and DNA or RNA as the probe to detect gene expression. In both techniques, the nucleic acid fragments must be in a single-stranded form and are transferred to a synthetic membrane. The membrane is then exposed to the labeled probe. If the probe is complementary to any of the fragments, hybrids form, and the probe attaches to the membrane at the locations of the complementary fragments. Figure 12.5 shows how a Southern blot can be used to identify fragments of DNA containing sequences that hybridize to the probe and how the intensity of a signal on a Northern blot gives a rough estimate of mRNA abundance from the target gene.
Figure 12.5 Nucleic acid hybridization.

(a) Southern blotting. (Left panel) Purified molecules of DNA from several different plasmids were treated with restriction enzymes and then subjected to agarose gel electrophoresis. (Right panel) Blot of the DNA gel shown to the left. After blotting, DNA in the gel was hybridized to a radioactive probe. The positions of the bands were visualized by X-ray autoradiography. Note that only some of the DNA fragments (circled in yellow in the left panel) have sequences complementary to the labeled probe. Lane 6 contained DNA used as a size marker, and none of its bands hybridized to the probe. (b) Northern blotting. (Top panel) Hybridization and detection of a radioactive gene-specific probe to a blot of total RNA. The probe only bound to RNA from biofilm-grown cells, indicating that the target gene is not expressed during planktonic (suspended) growth. (Bottom panel) Hybridization and detection of a radioactive probe corresponding to the 5S rRNA to the same blot. The signal intensity indicates that equal amounts of RNA from each sample were loaded into the gel.
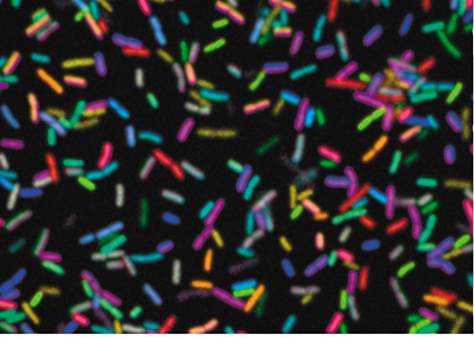
Nucleic acid hybridization has many other uses. Hybridization is the basis of the fluorescence in situ hybridization (FISH) technique (Section 19.5) (Figure 12.6), in which fluorescent probes are used to target specific DNA (or RNA) sequences in cells. This approach allows the identification of pathogens in clinical samples or bacteria of interest in environmental samples. For example, Figure 12.6 demonstrates the simultaneous use of eight different oligonucleotide probes in combinations to distinguish between 28 different strains of E. coli whose SSU (small subunit) rRNA sequences varied only slightly from strain to strain. The variations in color give a visual indication of the specificity and power of nucleic acid probes. Hybridization is also important in various “omics,” in particular transcriptomics and metatranscriptomics, where genome-wide gene expression can be monitored in pure cultures and natural populations, respectively, using microarray technology (Section 10.8).
Figure 12.6 Fluorescence spectral image of 28 differently labeled strains of *Escherichia coli.*
Cells were labeled with combinations of fluorophore-conjugated oligonucleotides that are complementary to E. coli 16S rRNA. (Figure 13.24).

Alex Valm and Gary Bonsy, Marine Biological Laboratory, Woods Hole, MA
In the next two sections we consider the important processes of gene cloning and the expression of cloned genes, respectively. If either of these events fail, the desired outcome of the genetic engineering will be in doubt.
Check Your Understanding
Why is a primer needed at each end of the DNA segment being amplified by PCR?
What are some applications of nucleic acid hybridization in molecular biology?
12.2 Molecular Cloning
The movement of desired genes from their original source to a small and manipulable genetic element (the vector) is called molecular cloning. Molecular cloning results in recombinant DNA, a molecule containing DNA from different sources. Once cloned, the gene(s) of interest can be manipulated, and when the recombinant vector is placed in an appropriate host, the cloned DNA is replicated, providing the foundation for much of genetic engineering.
An Overview of Gene Cloning and Restriction Enzymes
Following isolation of the source DNA, the major steps in gene cloning are (1) inserting the DNA into a cloning vector (Figure 12.7), and (2) inserting the vector into a host. The source DNA can be a gene or genes amplified by the polymerase chain reaction (Section 12.1), DNA synthesized from an RNA template by reverse transcriptase (Section 12.1), or even completely synthetic DNA made in vitro (Section 12.4). Cloning vectors are small, independently replicating genetic elements that can both carry and replicate cloned DNA segments (see Figure 12.10). Cloning vectors are typically designed to allow for easy insertion of foreign DNA. One way to insert foreign DNA into a vector is by using a restriction site (Figures 12.7 and 12.8). Restriction endonucleases, or restriction enzymes for short, recognize specific base sequences (restriction sites) within DNA and cut the phosphodiester backbone, resulting in double-stranded breaks (Figure 12.8). The recognition sequences—which are unique to each restriction enzyme—are typically inverted repeats and are called palindromes.
Figure 12.7 Major steps in gene cloning using restriction enzymes.

By cutting the foreign DNA and the vector DNA with the same restriction enzyme, complementary sticky ends are generated that allow foreign DNA to be inserted into the vector.
Figure 12.8 Restriction and modification of DNA.

Sequences of DNA recognized by the restriction endonucleases EcoRI and EcoRV. The red arrows indicate the bonds cleaved by the enzyme, and the dashed line indicates the axis of symmetry of the sequence. After cutting DNA with these restriction enzymes, note the single-stranded “sticky” ends generated by EcoRI versus the “blunt” ends generated by EcoRV.
Restriction enzymes with different sequence specificities are widespread among Bacteria, where they help protect cells from attack by viral DNA (Section 9.12). The cell is protected from its own restriction enzyme(s) by chemical modification (typically by methylation) of one of the bases in any potential restriction sites that exist in its genome. The restriction enzyme EcoRI makes staggered cuts, leaving short, single-stranded overhangs called “sticky” ends at the termini of the two fragments. Other restriction enzymes such as EcoRV cut both strands of the DNA directly opposite each other, resulting in blunt ends (Figure 12.8). If the source DNA and the vector are both cut with the same restriction enzyme that yields complementary sticky ends, the two molecules can be joined (annealed) using DNA ligase, an enzyme that covalently links the strands of the vector and the source DNA. If the source DNA is PCR generated, DNA ligase is used to join the amplified DNA to specialized vectors (see Figure 12.11a, b).
In the final step of gene cloning, recombinant DNA molecules are introduced into suitable host organisms where they can replicate. But in practice, this often yields a mixture of recombinant constructs, where only some of the cells contain the desired cloned gene. To identify a host colony containing the correct recombinant DNA, one can select host cells expressing a vector-encoded marker such as antibiotic resistance. Colonies can then be screened for recombinant vectors by looking for the inactivation of a vector gene due to insertion of foreign DNA (see Figures 12.10 and 12.11a, b).
Recombineering
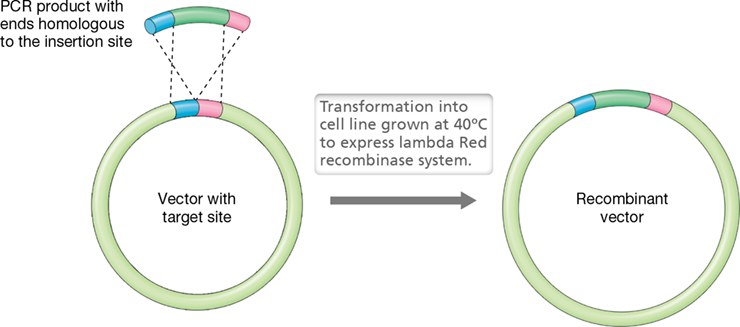
While cloning with restriction enzymes has been the standard of molecular biology (Figure 12.7), its biggest limitation is that modifications can only be made at restriction enzyme sites. To avoid this requirement, specialized strains of Escherichia coli have been designed to allow for gene cloning using homologous recombination (Section 9.5). Through the use of recombineering (recombination-mediated genetic engineering), foreign DNA can be inserted into vectors (or the chromosome) by designing the insert DNA to possess short regions (30–50 bases) of sequence homology to the target DNA molecule and activating recombinase enzymes. These recombinase enzymes cleave, exchange, and rejoin strands of DNA between areas of homology. Thus, if the ends of a DNA insert are constructed through PCR to share homology with a vector at the target insertion position, recombinase enzymes will “flip” the DNA insert into the vector (Figure 12.9).
Figure 12.9 Recombineering.
Enzymes from the lambda Red recombinase system coupled with sequence homology can be used to insert foreign DNA into target sites of vectors and chromosomes. The sequence homology can be generated by PCR and primer design.
Recombineering was originally developed in the yeast Saccharomyces cerevisiae; this microbe, widely used in genetic engineering (see Figure 12.12), possesses native recombinase enzymes that can recombine DNA regions of sequence homology as short as 20 bases (see Figure 12.40). While homologous recombination does occur naturally in E. coli, the efficiency is very low with short regions of homology. Thus, the engineered strains for cloning express the bacteriophage lambda Red recombinase system. The lambda Red (identified from recombination defective phage mutants) system is a mutant form of the system used by the bacteriophage to insert its genome into the E. coli chromosome during lysogeny (Section 11.4). The genes encoding this modified recombinase system are located on either a plasmid or the chromosome of the engineered strain, and their transcription is controlled by a repressor protein (Chapter 7) that is sensitive to temperature (Figure 12.9). Thus, the recombinase enzymes are only expressed and active when the temperature is shifted to 40ºC. This allows a molecular biologist to control the time point when the recombinases are produced to recombine the foreign DNA in the vector (or chromosome) and helps avoid unwanted recombination events.
Recombineering has revolutionized cloning in E. coli because compatible restriction enzyme sites in the vector and the DNA to be cloned are not needed. PCR products or synthesized DNA fragments (Section 12.3) can be recombined into a target site as long as they possess at least 30 base pairs of homology to the desired insertion site. Because the Red recombinase system can also handle single-stranded DNA molecules, oligonucleotide primers can also be synthesized with homology to the target site. Thus, recombineering can be used not only to clone foreign genes into E. coli, but also to generate point mutations and gene fusions (see Figure 12.18). After recombineering, recombinant DNA molecules are selected in the same way as gene cloning with restriction enzymes (see Figures 12.10 and 12.11).
Cloning Vectors
Several types of cloning vectors exist, including viruses, cosmids (plasmids with a lambda phage cos site, Figure 11.11), and artificial chromosomes, and their use is dependent on the size of the DNA fragment to be cloned and the host in which the vector will be inserted. Plasmids are widely used cloning vectors that often exist as multiple copies in bacterial cells. The plasmid pUC19 (Figure 12.10) is a standard cloning plasmid that contains an ampicillin resistance gene for selection and a blue–white color-screening system to select for recombinants. It also contains a short segment of artificial DNA containing cut sites for many different restriction enzymes. This segment, called a multiple cloning site (MCS), is inserted into the lacZ gene encoding the lactose-degrading enzyme β-galactosidase (Section 7.8 and Figure 7.23). The presence of the short MCS does not inactivate lacZ, and cut sites for restriction enzymes present in the MCS are absent from the rest of the vector.
Figure 12.10 Cloning into the plasmid vector pUC19.

Essential features include an ampicillin resistance marker and the multiple cloning site (MCS) with multiple restriction enzyme cut sites. The cloning vector and foreign DNA are cut with compatible restriction enzymes at positions indicated by the arrows. Insertion of DNA within the MCS inactivates β-galactosidase, allowing blue–white screening for the presence of the insert. The photo on the bottom shows colonies of Escherichia coli on an X-gal plate. The enzyme β-galactosidase can cleave the normally colorless X-gal to form a blue product.
The use of pUC19 in gene cloning is shown in Figure 12.10. A suitable restriction enzyme with a cut site within the MCS is chosen, and both the vector and the foreign DNA to be cloned are cut with this enzyme. The vector is linearized, and segments of the foreign DNA are inserted into the open cut site and ligated into position with the enzyme DNA ligase. This insertion disrupts the lacZ gene—a phenomenon called insertional inactivation—and is used to detect the presence of foreign DNA within the vector or recombinant vector. After DNA ligation, the resulting plasmids are transformed (Section 9.6) into cells of Escherichia coli and the cells plated on media containing both ampicillin (to select for cells containing the plasmid) and a lactose analog called X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside), to detect β-galactosidase activity. X-gal, which is colorless, can be cleaved by β-galactosidase to generate a blue product. Thus, cells containing the vector without cloned DNA form blue colonies (that is, β-galactosidase is active), whereas cells containing the vector with an insert of cloned DNA do not form β-galactosidase and are therefore white and are the focus of further analyses.
Plasmids developed specifically for cloning DNA products synthesized by the polymerase chain reaction (PCR; Section 12.1) have also been designed (**Figure 12.11a,*b***). The enzymatic activity of Taq polymerase adds a template-independent adenosine residue to the 3′ ends of its products. Linearized vectors are commercially available that contain overhanging thymidine residues that allow for base pairing with the Taq PCR product and subsequent ligation using DNA ligase. Ligation of the PCR product results in a circular vector with an interrupted lacZ gene for selection of a transformant (Section 9.6) with a recombinant vector (Figure 12.11a).
Figure 12.11 Specialized vectors.

(a) PCR vector with overhangs and lacZ. The linearized cloning vector contains overhanging thymidine residues that base-pair with the adenosine residues present on the 3′ ends of Taq-polymerase-generated PCR. Ligation of the two pieces of DNA yields a circular plasmid containing an interrupted lacZ and thus white colonies on plates containing X-gal (see Figure 12.10). (b) PCR vector with blunt ends and ccdB. Ligation of a blunt PCR product generated by Pfu polymerase yields a circular plasmid containing an interrupted ccdB, and thus all transformants that survive contain a recombinant plasmid. (c) Genetic map of a shuttle vector used in yeast. The vector contains components that allow it to shuttle between Escherichia coli and yeast and be selected in each organism: oriC, origin of replication in E. coli ; oriY, origin of replication in yeast; MCS, multiple cloning site; ESM, eukaryotic selectable marker; CEN, yeast centromere sequence; promoter; t/pa, transcription termination/polyadenylation signals. Ampicillin resistance allows for selection of the vector in E. coli. Arrows indicate the direction of transcription. (d) A yeast artificial chromosome (YAC) containing inserted DNA. The foreign DNA was cloned into the vector at a NotI restriction site. The telomeres are labeled TEL and the centromere CEN. The origin of replication is labeled ARS (for autonomous replication sequence). The URA3 gene is used for selection. The host into which the clone is transformed has a mutation in URA3 and requires uracil for growth (Ura−). Host cells containing this YAC become Ura+. The diagram is not to scale; vector DNA is only 10 kbp whereas cloned DNA can be up to 800 kbp.
Because Pfu DNA polymerase does not add template-independent adenosine residues, separate linear vectors with blunt ends are available for cloning. These vectors typically rely on insertional inactivation of the ccdB gene to select against vectors that rejoin without an insert (Figure 12.11b). CcdB is the toxin component of a toxin–antitoxin module (Section 8.12) that inhibits the enzyme DNA gyrase, and when CcdB is expressed, it prevents DNA replication. Thus, if the vector is not recombinant, the resulting transformed E. coli cells will die from expressing CcdB. This is a strong selection tool because all the resulting colonies from the transformation should contain recombinant vectors (Figure 12.11b).
For manipulating genes in E. coli and subsequent cloning into other organisms, shuttle vectors are used. These vectors can replicate and be stably maintained in two distinct organisms, such as E. coli and yeast or E. coli and mammalian cells. The importance of a shuttle vector is that DNA cloned in one organism can be replicated in a second host without having to modify the vector. Thus, the vectors must contain replication, transcription, and selection features for each host, and this is shown for a shuttle vector used in yeast in Figure 12.11c. However, genes can also be cloned directly into the yeast Saccharomyces cerevisiae using yeast artificial chromosomes (YACs) (Figure 12.11d). YACs are linear vectors that replicate in yeast like normal chromosomes but have sites where very large fragments of DNA can be inserted. To function like normal eukaryotic chromosomes, YACs have an origin of DNA replication, telomeres for replicating DNA at the ends of the chromosome, and a centromere for segregation during mitosis. YACs also contain a cloning site and a gene for selection following transformation into the host (Figure 12.11d).
Hosts for Cloning Vectors
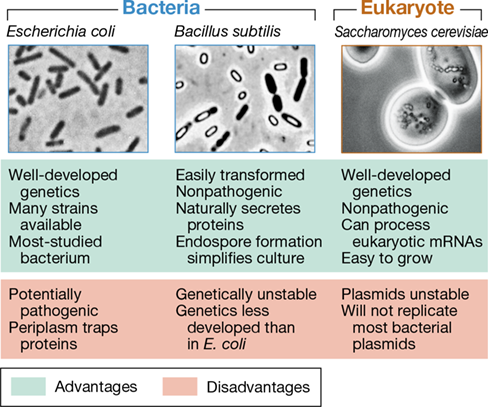
The most useful hosts for cloning are nonpathogenic microorganisms that are easy to grow and transform with engineered DNA. They must also be genetically stable in culture and have the appropriate enzymes to allow replication of the vector. It is also helpful if considerable background information on the host and a wealth of tools for its genetic manipulation exist. Hosts that meet these conditions include the bacteria E. coli and Bacillus subtilis, and the yeast S. cerevisiae (Figure 12.12). Complete genome sequences are available for all of these organisms, and they are widely used as cloning hosts.
Figure 12.12 Hosts for molecular cloning.

A summary of the advantages and disadvantages of some common cloning hosts.
Although E. coli is found in the human intestine and some wild-type strains are potentially harmful (Section 33.11), several modified E. coli strains have been developed specifically for cloning purposes. However, if cloned gene expression is desired, the outer membrane of this gram-negative bacterium (Section 2.4) can hinder protein secretion. This problem can be overcome using the gram-positive bacterium B. subtilis as a cloning host (Figure 12.12). Cloning of DNA from eukaryotic sources into eukaryotic rather than prokaryotic cells is often done since eukaryotic hosts already possess the complex RNA and post-translational processing systems required for the production of eukaryotic proteins (Section 6.6). Because it is easy to grow and manipulate, the workhorse for cloning in eukaryotic cells is the yeast S. cerevisiae. However, some cloning applications require the use of plant tissues, insect cell lines, or cultured mammalian cells. Regardless of host type, it is necessary to get cloned DNA into the host. While chemical transformation and electroporation (Section 9.6) are often used to introduce DNA into cells of Bacteria and Archaea, methods such as transfection (see Figure 12.21), microinjection, and electroporation can be used for eukaryotic cells.
Check Your Understanding
What do the terms multiple cloning site, insertional inactivation, and recombineering mean?
When would it be beneficial to use a eukaryotic host for molecular cloning?
12.3 Expressing Foreign Genes in *Bacteria*
Once genes are cloned, they can be transcribed and translated (expressed) to produce their encoded proteins. Obstacles to the expression of genes from mammalian or other eukaryotic sources in bacteria include the following: (1) The genes must be placed under control of a bacterial promoter; (2) any introns (Section 6.6) must be removed; (3) codon usage (codon bias, Section 6.9) may require edits to gene sequences; and (4) many eukaryotic proteins require host modification after translation to yield the active form and bacteria cannot perform most such modifications. Here we consider solutions to these challenges.
Transcription and Translation of Cloned Genes Using Expression Vectors
Expression vectors are designed to allow the experimenter to control the expression of cloned genes. However, the native promoter of a cloned gene may work poorly in a new host, and the overproduction of foreign proteins may also damage the host cell. Therefore, it is important to regulate the expression of cloned genes. Typically, the regulation is at the transcriptional level, and in practice, high levels of transcription require strong promoters that bind RNA polymerase efficiently (Section 6.5). An example of this is the use of the bacteriophage T7 promoter and T7 RNA polymerase to regulate gene expression. When T7 infects Escherichia coli, it encodes its own RNA polymerase that recognizes only T7 promoters (Section 11.4). In T7 expression vectors, cloned genes are placed under control of the T7 promoter. To achieve this, the gene for T7 RNA polymerase must also be present in the cell under the control of an easily regulated system, such as lac (Section 7.3; Figure 12.13). This is usually done by integrating the gene for T7 RNA polymerase with a lac promoter into the chromosome of a specialized host strain.
Figure 12.13 The T7 expression system.

The gene for T7 RNA polymerase is in a gene fusion under control of the lac promoter and is inserted into the chromosome of a special host strain of Escherichia coli (BL21). Addition of IPTG induces the lac promoter, causing expression of T7 RNA polymerase. This transcribes the cloned gene, which is under control of the T7 promoter and is carried by the pET plasmid. RBS, ribosome-binding site.
The BL21 series of E. coli host strains are especially designed to work with the pET series of T7 expression vectors (Figure 12.13). The cloned genes are expressed shortly after T7 RNA polymerase transcription has been switched on by a lac inducer, such as the chemical IPTG (Section 7.3). Because it recognizes only T7 promoters, the T7 RNA polymerase transcribes only the cloned genes. The T7 RNA polymerase is so highly active that it uses most of the RNA precursors, thereby limiting transcription to the cloned genes. Consequently, host genes that require host RNA polymerase are for the most part not transcribed and thus the cells stop growing; translation in such cells then yields primarily the protein of interest. The T7 control system is thus very effective for generating large amounts of a specific protein.
Expression vectors must also be designed to ensure that the mRNA produced is efficiently translated. To synthesize protein from an mRNA molecule, it is essential for the ribosomes to bind at the correct site and begin reading in the correct frame. In bacteria this is accomplished by having a ribosome-binding site (RBS, Section 6.9) and a nearby start codon on the mRNA. Bacterial RBSs are not found in eukaryotic genes and must be engineered into the vector if high levels of expression of the eukaryotic gene are to be obtained.
Other adjustments to a cloned gene may be necessary to ensure high-efficiency translation. For example, codon usage can be an obstacle. Codon usage is related to the concentration of the appropriate tRNA in the cell (Section 10.2 and Table 10.3). Because of the redundancy of the genetic code, more than one tRNA exists for most amino acids (Section 6.9). Therefore, if a cloned gene has a codon usage pattern distinct from that of its expression host, it will probably be translated inefficiently in that host. Site-directed mutagenesis (Section 12.4) can be used to change selected codons in the gene, making it more amenable to the codon usage pattern of the host.
Cloning the Gene via mRNA or Artificial Synthesis
If a cloned gene contains introns, as eukaryotic genes typically do (Section 6.6), the correct protein product will not be made in a bacterial host unless modifications are made. This can be done via mRNA. In a typical mammalian cell, less than 5% of the total RNA is mRNA. However, eukaryotic mRNA is unique because of the poly(A) tails found at the 3′ end (Section 6.6), and this makes it easy to isolate, even though it is of low abundance. If a cell extract is passed over a chromatographic column containing strands of poly(T) linked to a cellulose support, most of the mRNA separates from other RNAs by sticking to the support by specific pairing of As and Ts. The RNA is then released from the column by a low-salt buffer, which gives a preparation greatly enriched in mRNA.
Once mRNA has been isolated, the genetic information is converted into complementary DNA (cDNA) by RT-PCR as was illustrated in Figure 12.3. This double-stranded cDNA contains the coding sequence but lacks introns (Figure 12.14), and thus it can be inserted into a plasmid or other vector for cloning in bacteria. However, because the cDNA contains only coding sequences, it lacks a promoter and other upstream regulatory sequences necessary for expression. Thus expression vectors containing bacterial promoters and ribosome-binding sites (RBS) are used to obtain high-level expression of genes cloned in this way (see Figure 12.15).
Figure 12.14 Complementary DNA (cDNA).

Steps illustrating the synthesis of an intron-lacking cDNA corresponding to a eukaryotic gene generated by reverse transcription PCR (RT-PCR; see Figure 12.3).
For small proteins it is possible to artificially synthesize the entire gene (Section 12.4). Many mammalian proteins such as high-value peptide hormones are made by protease cleavage of large precursor molecules. Thus, in order to produce a short peptide such as insulin in its active form, construction and cloning of an artificial gene that encodes just the final hormone rather than the larger precursor protein from which it was derived may have several advantages. Constructed genes are naturally free of introns and thus the mRNA does not need processing. Also, promoters and other regulatory sequences can be inserted into the gene upstream of the coding sequences, and codon bias (Sections 6.9 and 10.2) can be adjusted to best suit the expression host.
Protein Stability and Purification
The synthesis of a protein in a new host may spawn additional problems. For example, some proteins are susceptible to degradation by protease enzymes and others may be toxic to their host. Also, when proteins are massively overproduced, they sometimes aggregate into insoluble inclusions. Although inclusions are relatively easy to purify, the protein they contain is often difficult to solubilize and may be partially denatured. Protein purification can be simplified if the target protein is made as a fusion protein along with a carrier protein encoded by the vector. To do this, the two genes are fused to yield a single coding sequence. A short segment that is recognized and cleaved by a commercially available protease is included between them. After transcription and translation, a single protein is made that is purified by methods designed for the carrier protein. The fusion protein is then cleaved by the protease to release the target protein from the carrier protein. Fusion proteins simplify purification of the target protein because a carrier protein is chosen that will not form inclusions and is easy to purify.
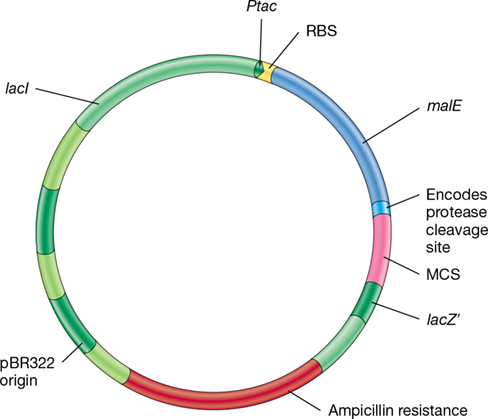
Several vectors are available to generate fusion proteins, and Figure 12.15 shows an example of a fusion vector that is also an expression vector. In this example, the carrier protein is the E. coli maltose-binding protein (encoded by malE, Figure 12.15), a protein that is easily purified by methods based on its high affinity for maltose. Once purified, the two portions of the fusion protein are separated by protease or chemical treatment. One other advantage of making a fusion protein is that the carrier protein can be chosen to contain the bacterial signal sequence, a peptide rich in hydrophobic amino acids that enables transport of the protein across the cytoplasmic membrane (Section 6.12). This makes possible a bacterial expression system that not only makes and secretes mammalian proteins, but also allows for the heterologously expressed protein to be separated from all of the other proteins secreted by the cell using binding resins specific for the maltose-binding protein. Thus, carrier proteins can be used to save time, money, and effort in obtaining a desired product.
Figure 12.15 An expression vector for gene fusions.

The gene to be cloned is inserted into the multiple cloning site (MCS) to be in frame with the malE gene, which encodes maltose-binding protein. The insertion inactivates the gene for the alpha fragment of lacZ, which encodes β-galactosidase. The fused gene is under control of the hybrid tac promoter (Ptac) and an Escherichia coli ribosome-binding site (RBS). The plasmid also contains the lacI gene, which encodes the lac repressor. Therefore, an inducer must be added to turn on the tac promoter. The plasmid contains a gene conferring ampicillin resistance on its host.
In the next two sections we will see how some of the DNA manipulations we have already considered can be used to generate mutants of interest and how some genes can be exploited as signals of gene expression.
Check Your Understanding
How can the bacteriophage T7 promoter be used to control expression of a eukaryotic gene in Escherichia coli?
What major advantage does cloning mammalian genes from mRNA or using synthetic genes have over PCR amplification and cloning of the native gene?
12.4 Molecular Methods for Mutagenesis
Conventional mutagens introduce mutations at random in the DNA of the intact organism (Section 9.4). In contrast, site-directed mutagenesis uses synthetic DNA plus DNA cloning techniques to introduce mutations into genes at precisely determined sites. In addition to changing one or just a few bases, mutations may also be engineered by inserting large segments of DNA at precisely determined locations.
Site-Directed Mutagenesis
Site-directed mutagenesis is now a fairly straightforward process due to the ease in obtaining 12- to 40-base DNA oligonucleotides of precise sequence through chemical synthesis; primers or probes for use in the polymerase chain reaction (PCR) and hybridization are obtained in the same manner (Section 12.1). In fact, DNA oligonucleotides over 100 bases long can be obtained. Thus, PCR can be used to obtain a gene with a specific mutation depending on the position of the desired change. If the target gene is part of the Escherichia coli chromosome or has already been cloned into a vector, a PCR product (or even an oligonucleotide) containing the desired mutation can be exchanged through recombineering using an appropriate strain of E. coli (Figure 12.9 and **Figure 12.16*a***). This scheme allows any base pair in a specific gene to be changed. Progeny bacteria are then screened through PCR and subsequent Sanger sequencing of the product to detect those carrying the mutation (Figure 10.4). When the mutated gene is expressed, a protein with an altered amino acid sequence will be produced. Site-directed mutagenesis can thus be used to manipulate proteins to test the functional importance of specific amino acids at specific sites in the protein.
Site-directed mutagenesis has many applications. The technique has been widely used by enzymologists to change a specific amino acid in the active site of an enzyme to see how the modified enzyme compares with the wild-type enzyme. In such experiments, the vector encoding the mutant enzyme is inserted into a mutant host strain unable to make the original enzyme. Consequently, the activity measured is due to the mutant version of the enzyme alone. Using in vitro mutagenesis, enzymologists can link virtually any aspect of an enzyme’s activity—catalysis, resistance, susceptibility to chemical or physical agents, interactions with other proteins—to specific amino acids in the enzyme. In genetic engineering, site-directed mutagenesis has been used to improve the properties of specific proteins, and we discuss some examples in Section 12.6.
Cassette Mutagenesis and Gene Disruption
To make more than a few base-pair changes or replace entire sections of a gene of interest, synthetic fragments called DNA cassettes (or cartridges) can be used to mutate DNA in a process known as cassette mutagenesis. These cassettes can be synthesized using the polymerase chain reaction (PCR) or by direct DNA synthesis, the cost of which is now a minor laboratory expense. Once the appropriate cassette has been obtained, it can then replace sections of the DNA of interest using restriction sites or recombineering (Section 12.2) to facilitate the manipulations. Cassettes used to replace sections of genes are typically the same size as the wild-type DNA fragments they replace.
Another type of cassette mutagenesis is called gene disruption. In this technique, cassettes are inserted into a gene, thus disrupting the coding sequence (Figure 12.16b). Cassettes used for making insertion mutations can be almost any size and can even carry an entire gene. To facilitate selection, cassettes that encode antibiotic resistance are commonly used. For example, a DNA cassette containing a gene conferring kanamycin resistance is inserted at a restriction site in a cloned gene. The vector carrying the disrupted gene is then converted from a circular to a linear form by cutting it with a different restriction enzyme. Finally, the linear DNA is transformed into the host and kanamycin resistance is selected. The linear plasmid cannot replicate, and so resistant cells arise mostly by homologous recombination (Section 9.5) between the mutated gene on the plasmid and the wild-type gene on the chromosome (Figure 12.16b). Because the gene disruption cassette is designed to contain regions of homology with the target gene that are longer than 50 base pairs, special recombinase systems used for recombineering are not needed (Section 12.2).
Figure 12.16 Site-directed mutagenesis and gene disruption by cassette mutagenesis.

(a) Site-directed mutagenesis. A short stretch of DNA containing a single base change is inserted into a target gene such that the target gene product is altered in a known way at a known position in the polypeptide. (b) Gene disruption by a cassette insertion. (1) A cloned wild-type copy of gene X, carried on a plasmid, and a kanamycin cassette are cut with EcoRI, mixed, and ligated. (2) The resulting plasmid contains the kanamycin cassette as an insertion mutation within gene X. This new plasmid is cut with BamHI and transformed into a cell. (3) The transformed cell contains the linearized plasmid with a disrupted gene X and its own chromosome with a wild-type copy of the gene. In some cells, homologous recombination occurs between the wild-type and mutant forms of gene X. Cells that can grow in the presence of kanamycin have only a single, disrupted copy of gene X. Site-directed mutagenesis is widely used to create "knockout" mutants to identify essential genes (Section 10.4).
When a cassette is inserted, the cells not only gain antibiotic resistance but also lose the function of the gene into which the cassette is inserted. Such mutations are called knockout mutations and are widely used in biology. Knockouts are similar to insertion mutations made by transposons (Section 9.11), but here the experimenter chooses which gene will be mutated. Knockout mutations in haploid organisms yield viable cells only if the disrupted gene is nonessential. Thus, gene knockouts are commonly used for determining whether a gene of interest is essential (Section 10.4).
Check Your Understanding
How can site-directed mutagenesis be useful to enzymologists?
What is used to alter more than a few base pairs in a gene of interest?
12.5 Reporter Genes and Gene Fusions
DNA manipulation has revolutionized the study of gene regulation, and gene fusions have been a major tool for studying regulatory events. We briefly mentioned gene fusions in our discussion of the utility of fusion proteins for stabilizing and purifying cloned gene products in Section 12.3. In a reporter gene fusion, a coding sequence from one source (the reporter) is fused to a regulatory region from another source to form a hybrid gene. Regulation of gene expression is then studied by assaying for the product of the reporter as a function of different conditions sensed by the regulator.
Reporter Genes
The key property of a reporter gene is that it encodes a protein that is easy to detect and assay. Reporter genes are used for a variety of purposes. They may be used to report the presence or absence of a particular genetic element (such as a plasmid) or DNA inserted within a vector. They can also be fused to other genes or to the promoter of other genes so that gene expression can be studied (Figure 8.1).
The first widely used reporter gene was lacZ from Escherichia coli, a gene that encodes the enzyme β-galactosidase, required for lactose catabolism (Section 7.3). Cells expressing β-galactosidase can be detected easily by the color of their colonies on indicator plates that contain the artificial substrate X-gal (Section 12.2); X-gal is cleaved by β-galactosidase to yield a blue color (see Figure 12.10).
The green fluorescent protein (GFP) is widely used as a reporter (Figure 12.17). Although the gene encoding GFP was originally cloned from the jellyfish Aequorea victoria, GFP can be expressed in both prokaryotic and eukaryotic cells (Figure 12.17) because it is stable and causes little or no disruption of host cell metabolism. If expression of a cloned gene is linked to the expression of GFP, the latter signals (reports) that the cloned gene has also been expressed (Figure 12.17). Since the discovery of the GFP, many similar but differently colored fluorescent proteins have been developed as reporters (Section 8.1).
Figure 12.17 Green fluorescent protein (GFP).

GFP can be used as a general tag or as a specific tag for protein localization. (a) Using prokaryotic cells, a plate shows uniformly green bacterial colonies because several genes were fused with the gene encoding GFP. (b) Using a eukaryotic example, the gene encoding the DNA-binding protein Pho2 from the yeast Saccharomyces cerevisiae was fused to the gene encoding GFP. The recombinant gene was transformed into budding yeast cells that expressed the fluorescent fusion protein only in the nucleus. Budding cell division (Section 18.10 and Figures 18.27 and 18.28) is in progress in the two cells on the lower right.
Gene Fusions
Gene fusions are genetic constructs that consist of segments from two different genes. If the promoter that controls a coding sequence is removed, the coding sequence can be fused to a different regulator to place the gene under the control of a different promoter. Alternatively, the promoter region can be fused to a gene whose product is easy to assay. There are two different types of gene fusions. In operon fusions, a coding sequence that retains its own translational start site and signals is fused to the transcriptional signals of another gene. In protein fusions, genes that encode two different proteins are fused together so that they share the same transcriptional and translational start and stop signals. Following translation, protein fusions yield a single hybrid polypeptide (Section 12.3).
Gene fusions are often used in studying gene regulation, especially if measuring the levels of the natural gene product is difficult, expensive, or time consuming. The regulatory region of the gene of interest is fused to the coding sequence for a reporter gene, such as that for β-galactosidase or GFP. The reporter is then made under the conditions that would trigger expression of the target gene (Figure 12.18). The expression of the reporter is assayed under a variety of conditions to determine how the gene of interest is regulated (Chapter 7). Transcriptional control is assayed by fusing the transcriptional start signals of the gene of interest to a reporter gene, whereas translational control is assayed by fusing translational start signals of a gene of interest to a reporter gene under the control of a known promoter.
Figure 12.18 Construction and use of gene fusions.

The promoter of the target gene is fused to the reporter coding sequence. Consequently, the reporter gene is expressed under those conditions where the target gene would normally be expressed. The reporter shown here is an enzyme (such as β-galactosidase) that converts a substrate to a colored product that is easy to detect. This approach greatly facilitates the investigation of regulatory mechanisms.
Gene fusions may also be used to test for the effects of regulatory genes. Mutations that affect regulatory genes are introduced into cells carrying gene fusions, and expression is measured and compared to cells lacking the regulatory mutations. This allows the rapid screening of multiple regulatory genes that are suspected of controlling the target gene. Besides the use of fusions to monitor for the presence or expression of a gene, fusions can also be used to join proteins that are easily purified to proteins of interest to aid in purification of the latter (Section 12.3).
We now move on from considering how genes can be manipulated to the application of gene technologies for the synthesis of valuable commercial products by genetically engineered organisms.
Check Your Understanding
What is a reporter gene? The product of which reporter gene yields a green color?
Why are gene fusions useful in studying gene regulation?
II: Making Products from Genetically Engineered Microbes: Biotechnology
II: Making Products from Genetically Engineered Microbes: Biotechnology
II Making Products from Genetically Engineered Microbes: Biotechnology
Microbes can be altered by the genetic engineer to manufacture products such as therapeutic human proteins, vaccines, and biofuels, and genetic tools obtained from microbes can be used to create genetically novel transgenic plants and animals.
Genetic engineering can transform microorganisms into tiny factories for the production of valuable products including fuels, chemicals, drugs, and human hormones, such as insulin. This is the science of biotechnology. Up to this point we have only considered the techniques used for manipulating, cloning, and expressing DNA. We now consider how these techniques are applied in biotechnology to produce valuable proteins and genetically modified plants, animals, vaccines, and metabolic pathways.
12.6 Somatotropin and Other Mammalian Proteins
One of the most economically profitable areas of biotechnology has been the production of human proteins. Many mammalian proteins have high pharmaceutical value but are typically present in very low amounts in normal tissue, and it is therefore extremely costly to purify them. Even if the protein can be produced in cell culture, this is much more expensive and difficult than growing microbial cultures that produce the protein in high yield. Therefore, the biotechnology industry has developed genetically engineered microorganisms to produce many different mammalian proteins.
Somatotropin
Although insulin was the first human protein to be produced by bacteria, the genetic engineering required was complicated because insulin consists of two short polypeptides held together by disulfide bonds. A more straightforward example is human somatotropin (growth hormone), which consists of a single polypeptide encoded by a single gene; a deficiency of somatotropin in the body results in hereditary dwarfism. Because the human somatotropin gene has been successfully cloned and expressed in bacteria, children showing stunted growth can be treated with recombinant human somatotropin to correct this. However, some forms of dwarfism are caused by a lack of the somatotropin receptor, and in such cases, administration of somatotropin has no effect.
The human somatotropin gene was cloned as complementary DNA (cDNA) from mRNA as described in Section 12.3 (see Figure 12.19). The cDNA was then expressed in a bacterial expression vector. The main problem with producing relatively short polypeptide hormones such as somatotropin is their susceptibility to protease digestion, but this problem was overcome by using bacterial host strains lacking key protease enzymes. Today recombinant human growth hormone taken by injection is marketed under several brand names in the United States and has successfully treated thousands of children afflicted with any of several different syndromes that result in short stature. Recombinant somatotropin has also been used to treat some cases of tissue atrophy in adults. However, use in adults is not a common practice, and growth hormone is banned by the International Olympic Committee and by some professional sports leagues for its alleged performance-enhancing capabilities.
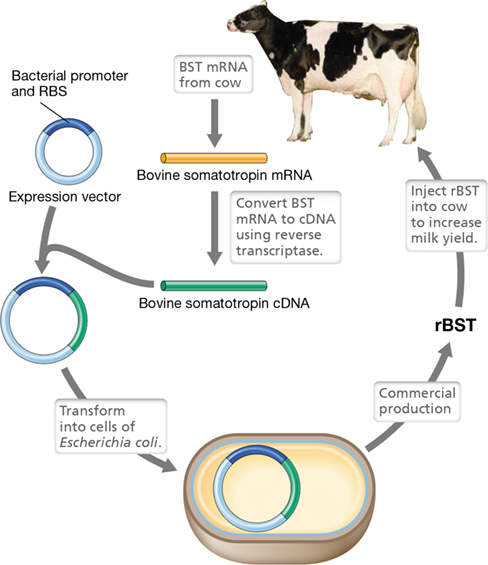
Recombinant bovine somatotropin (rBST) is used in the dairy industry (Figure 12.19). Injection of rBST into cows does not make them grow larger but instead stimulates milk production. This is because somatotropin has two binding sites; one is the somatotropin receptor that stimulates growth while the other is the prolactin receptor that promotes milk production. Thus, cows treated with rBST produce more milk. However, when human somatotropin is used to treat short stature conditions, it is desirable to avoid any side effects from the hormone’s prolactin activity. To alleviate this problem, site-directed mutagenesis (Section 12.4) of the human somatotropin gene was used to alter those amino acids of somatotropin that bind to the prolactin receptor, thus ensuring that the hormone would only target growth. As this example shows, it is possible not only to make genuine human hormones but also to alter their specificity and activity to make them better pharmaceuticals.
Figure 12.19 Cloning and expression of bovine somatotropin.

The mRNA for bovine somatotropin (BST) is obtained from a cow, and the mRNA is converted to cDNA by reverse transcriptase. The cDNA version of the somatotropin gene is then cloned into a bacterial expression vector that has a bacterial promoter and ribosome-binding site (RBS). The construct is transformed into cells of Escherichia coli, and recombinant bovine somatotropin (rBST) is produced. Milk production increases in cows treated with rBST.
Mastering Microbiology
Art Activity: Figure 12.18 Cloning and expression of bovine somatotropin
Other Mammalian Proteins
Many other mammalian proteins are produced today by genetic engineering (Table 12.1). These include, in particular, an assortment of hormones and proteins for blood clotting and other blood processes. For example, tissue plasminogen activator (TPA) is a protein that dissolves blood clots in the bloodstream that may form in the final stages of the healing process. TPA is primarily used in heart patients or others suffering from poor circulation to prevent the development of clots that can be life-threatening. Heart disease is a leading cause of death in many developed countries, especially in the United States, so microbially produced TPA is in high demand.
Table 12.1 A few human medical products made by genetic engineering

In contrast to TPA, the blood clotting factors VII, VIII, and IX are critically important for the formation of blood clots. Hemophiliacs suffer from a deficiency of one or more clotting factors and can therefore be treated with microbially produced clotting factors. In the past hemophiliacs have been treated with clotting factor extracts from pooled human blood, some of which was contaminated with viruses such as HIV and hepatitis C, putting hemophiliacs at high risk for contracting AIDS, hepatitis, or liver cancer. Recombinant clotting factors have eliminated this problem.
Some mammalian proteins made by genetic engineering are enzymes rather than hormones (Table 12.1). For instance, human DNase I is used to treat the buildup of DNA-containing mucus in the lungs of patients with cystic fibrosis. The mucus forms because cystic fibrosis is often accompanied by life-threatening lung infections by the bacterium Pseudomonas aeruginosa. The bacterial cells form biofilms (Section 8.10 and Section 20.4) within the lungs that make drug treatment difficult. DNA is released when the bacteria lyse, and this fuels mucus formation, making it difficult to breathe. DNase digests the DNA and greatly decreases the viscosity of the mucus.
Check Your Understanding
What is the advantage of using genetic engineering to make insulin?
What are the major problems when manufacturing proteins in bacteria?
Explain how an enzyme can be useful in treating a bacterial infection, such as that which occurs with cystic fibrosis.
12.7 Transgenic Organisms in Agriculture and Aquaculture
12.7 Transgenic Organisms in Agriculture and Aquaculture
12.7 Transgenic Organisms in Agriculture and Aquaculture
Genetic improvement of plants and animals by traditional selection and breeding has a long history, but recombinant DNA technology has led to revolutionary changes. Although the genetic engineering of higher organisms is not truly microbiology, much of the DNA manipulation leading up to the genetically engineered plant or animal is carried out using bacteria and the techniques we have learned thus far. Hence, we consider the genetic manipulation of plants and animals here with a focus on the microbiology that supported it.
Because genetically engineered plants or animals contain a gene from another organism—called a transgene—they are transgenic organisms. The public knows these as genetically modified organisms (GMOs). Strictly speaking, the term genetically modified refers to genetically engineered organisms whether or not they contain foreign DNA. In this section we discuss how foreign genes are inserted into plant and fish genomes and how transgenic organisms may be used.
The Ti Plasmid and Transgenic Plants
While recombinant DNA can be transformed into plant cells by electroporation or transfection, the Ti plasmid from the gram-negative bacterium Agrobacterium tumefaciens, a plant pathogen, can be used to transfer DNA directly into the cells of certain plants. This plasmid is responsible for A. tumefaciens virulence and encodes genes that mobilize DNA for transfer to the plant, which as a result contracts crown gall disease (Section 23.6). The segment of the Ti plasmid DNA that is actually transferred to the plant is called T-DNA. The sequences at the ends of the T-DNA are essential for transfer, and the foreign DNA to be transferred must reside between these ends.
One common Ti-vector system that has been used for the transfer of genes to plants is a two-plasmid system called a binary vector, which consists of a cloning vector plus a helper plasmid. The cloning vector is a shuttle vector (Section 12.2) that contains the two ends of the T-DNA flanking a multiple cloning site, two origins of replication so that it can replicate in both Escherichia coli (the host for cloning) and A. tumefaciens, and two antibiotic resistance markers, one for selection in plants and the other for selection in bacteria. The foreign DNA is inserted into the vector, which is transformed into E. coli and then moved to A. tumefaciens by conjugation (Figure 12.20).
Figure 12.20 Production of transgenic plants using a binary vector system in *Agrobacterium tumefaciens*.

(a) Plant cloning vector containing ends of T-DNA (red), foreign DNA, origins of replication, and resistance markers. (b) The vector is transformed into cells of Escherichia coli for cloning and then (c) transferred to A. tumefaciens by conjugation. The resident Ti plasmid (D-Ti) has been genetically engineered to remove key pathogenesis genes. (d) D-Ti can still mobilize the T-DNA region of the vector for transfer to plant cells grown in tissue culture. (e) From the recombinant plant cell, a whole plant can be grown. (Details of Ti plasmid transfer from bacterium to plant are shown in Figure 23.26.)
This cloning vector lacks the genes needed to transfer T-DNA to a plant. However, when placed in an A. tumefaciens cell that contains a suitable helper plasmid, the T-DNA can be transferred to a plant. The “disarmed” helper plasmid, called D-Ti, contains the virulence (vir) region of the Ti plasmid but lacks the T-DNA. It also lacks the genes that initiate disease but supplies all the functions needed to transfer the T-DNA from the cloning vector. The cloned DNA and the kanamycin resistance marker of the vector are mobilized by D-Ti and transferred into a plant cell where they enter the nucleus (Figure 12.20d). Following integration into a plant chromosome, the foreign DNA can be expressed and confer new properties on the plant.
A number of transgenic plants have been produced using the Ti plasmid of A. tumefaciens. The Ti system works well with broadleaf plants (dicots), including crops such as tomato, potato, tobacco, soybean, alfalfa, and cotton. It has also been used to produce transgenic trees, such as walnut and apple. The Ti system does not work with plants from the grass family (monocots, including the important crop plant corn), but other methods of introducing DNA, such as transfection by microprojectile bombardment with a particle gun (Figure 12.21), have been used successfully for them.
Figure 12.21 DNA gun for transfection of eukaryotic cells.

The inner workings of the gun show how metal pellets coated with nucleic acids (microprojectiles) are propelled at target cells. (a) Before firing and (b) after firing. A shock wave due to gas release propels the disk carrying the microprojectiles against the fine screen. The microprojectiles continue on into the target tissue.
Herbicide- and Insect-Resistant Plants
Major areas targeted for genetic improvement in plants include herbicide, insect, and microbial disease resistance, as well as improved product quality. The main genetically modified (GM) crops today are soybeans, corn, cotton, and canola. Almost all the GM soybeans and canola planted in the United States are herbicide resistant, whereas the corn and cotton are herbicide resistant or insect resistant, or both.
Herbicide resistance is genetically engineered into a crop plant to protect it from herbicides applied to kill weeds. Many herbicides inhibit a key plant enzyme or protein necessary for growth. For example, the herbicide glyphosate (RoundupTM, made by Monsanto) kills plants by inhibiting an enzyme necessary for making aromatic amino acids. Some bacteria contain an equivalent enzyme and are also killed by glyphosate. However, mutant bacteria were selected that were resistant to glyphosate and contained a resistant form of the enzyme. The gene encoding this resistant enzyme from A. tumefaciens was cloned, modified for expression in plants, and transferred (Figure 12.20) into important crop plants, such as soybeans. When sprayed with glyphosate, plants containing the bacterial gene are not killed (Figure 12.22). Thus glyphosate can be used to kill weeds that compete for water and nutrients with the growing crop plants. Herbicide-resistant soybeans are now widely planted in the United States, including plants resistant to both glyphosate and a second widely used herbicide, dicamba.
Figure 12.22 Transgenic plants: herbicide resistance.

The photograph shows a portion of a field of soybeans that has been treated with RoundupTM, a glyphosate-based herbicide manufactured by Monsanto Company (St. Louis, Missouri, USA). The remnants of plants on the right are normal soybeans; the plants on the left have been genetically engineered to be glyphosate resistant.
Transgenic plants resistant to damage by certain insects have also been produced by genetic engineering (Figure 12.23). One widely used approach is based on introducing genes encoding the toxic proteins of the gram-positive, endospore-forming bacterium Bacillus thuringiensis into plants. As it sporulates, B. thuringiensis produces a crystalline protein called Bt toxin (Section 16.8) that is toxic to moth and butterfly larvae. Many variants of Bt toxin exist that are specific for different insects. Certain strains of B. thuringiensis produce additional proteins toxic to beetle and fly larvae and mosquitoes.
Figure 12.23 Transgenic plants: insect resistance.

The results of an assay to determine the effect of beet armyworm larvae on tobacco leaves. (a) Leaf from a wild-type plant. (b) Leaf from a transgenic plant expresses Bt toxin in its chloroplasts and the toxin kills the larvae.
The Bt transgene is normally inserted directly into the plant genome. For example, a natural Bt toxin gene was cloned into a plasmid vector under control of a chloroplast ribosomal RNA promoter and then transfected into tobacco plant chloroplasts by microprojectile bombardment (Figure 12.21). This yielded transgenic plants that expressed Bt toxin at levels that were extremely toxic to larvae of several insect species (Figure 12.23). Binding Bt triggers a change in the toxin’s conformation that disrupts the insect digestive system, causing death. Bt toxin is harmless to mammals (including humans) because any toxin ingested is destroyed in the stomach and the specific Bt receptors in the insect intestine are absent from the intestines of other groups of organisms.
Despite the toxicity and availability of numerous Bt variants, insect resistance to the toxin is emerging. This is especially problematic with corn rootworms. To combat resistance issues, plant engineers are looking at other soil microbes for the production of different types of insecticidal proteins. This has resulted in the discovery of a small insecticidal peptide from the gram-negative soil-dwelling Pseudomonas chlororaphis. Transfer of the gene encoding the peptide into corn using an Agrobacterium-mediated system (Figure 12.20) resulted in protection from the rootworm. While the mechanism by which the P. chlororaphis peptide kills rootworms is not completely understood, the peptide kills Bt-resistant rootworms but does not affect other common insects such as bees and other pollinators. Thus, a new weapon is now available to specifically combat corn rootworm and may help to retard the development of resistance in this potentially devastating crop pest.
Transgenic Fish
Many foreign genes have been incorporated and expressed in laboratory research animals and in commercially important animals. The genetic engineering uses microinjection to deliver cloned genes to fertilized eggs; genetic recombination then incorporates the foreign DNA into the genomes of the eggs. More recently, farm animals and fish have been genetically modified to improve yields.
An interesting practical example of a transgenic animal is the AquAdvantage salmon developed by AquaBounty Technologies (Figure 12.24). These transgenic salmon do not grow to be larger than normal salmon but simply reach market size much faster—18 months versus 3 years. The gene for growth hormone in native salmon is activated by light. Consequently, salmon grow rapidly only during the summer months. In the genetically engineered salmon, the promoter for the growth hormone gene was replaced with the promoter from another fish that grows at a more or less constant rate all year round. The result was salmon that make growth hormone continuously and thus grow faster. Such transgenic salmon can be grown commercially in aquaculture operations and harvested more quickly than with non-GMO farm-raised salmon.
Figure 12.24 Fast-growing transgenic salmon.

The AquAdvantageTM salmon (top) was engineered by AquaBounty Technologies (Maynard, Massachusetts, USA). The transgenic and control fish are both 18 months old but weigh 4.5 kg and 1.2 kg, respectively.
In 1995 AquaBounty applied to the U.S. Food and Drug Administration (FDA) for approval to distribute the fast-growing salmon. After two decades of debate regarding the potential risks of consuming genetically modified fish, approval for its distribution occurred in 2015. However, in response to remaining concerns regarding the sale of the modified salmon, the United States enacted a law at the end of 2015 that prohibited its sale until labeling could be agreed upon. However, in early 2019, the FDA removed the final hurdle to the sale of GMO salmon in the United States, and it can now be sold (marketed as AquaAdvantage® salmon and containing a label indicating that it is a bioenginered product).
Check Your Understanding
Give an example of a genetically modified plant and describe how its modification benefits agriculture.
How have transgenic salmon been engineered to reach market size faster?
12.8 Engineered Vaccines and Therapeutic Agents
Genetic engineering is used to manufacture certain vaccines and medical therapeutic agents. Of interest to human health and biotechnology are the two types of relationships microbes form within the human body. While some microbes form beneficial relationships within humans by aiding digestion, producing nutrients, and “educating” the immune system (Section 24.2), other microbes cause disease through their ability to infiltrate cells and release virulence factors such as toxins and destructive enzymes. With this in mind, scientists have explored the use of both types of microbial communities for making novel vaccines and for delivering drugs or toxic agents to specific cell types. We consider both of these potential medical miracles here.
Recombinant Vaccines, Vaccinia Virus, and Subunit Vaccines
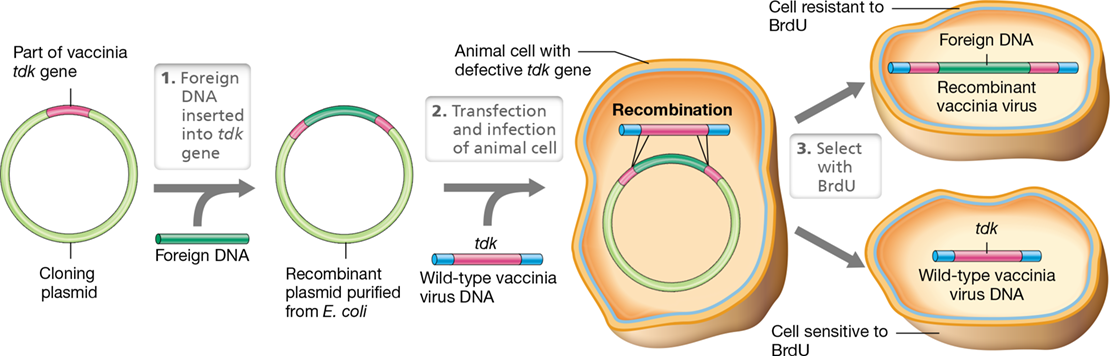
Vaccines are substances that elicit immunity to a disease when injected into an animal (Section 28.3). Genetic engineering can modify a pathogen by deleting genes from its genome that encode virulence factors (Section 25.5) while retaining those whose products elicit an immune response. This yields a recombinant and infective vaccine that is considered attenuated because it is less virulent than the original strain (Section 25.3 and Figure 25.10). Conversely, one can add genes from a pathogenic virus to the genome of a relatively harmless virus, called a carrier virus. Such vaccines are called vector vaccines and induce immunity to the pathogenic virus. Indeed, one can even combine the two approaches by disarming one pathogen and adding back to it immunity-inducing genes from a second pathogen. This yields a polyvalent vaccine, a vaccine that immunizes against two different diseases at the same time.
Vaccinia virus (Section 11.6) is widely used to prepare recombinant vaccines for human use; however, cloning into vaccinia requires a selective marker, which is provided by the gene encoding the enzyme thymidine kinase. Vaccinia virus contains a gene encoding thymidine kinase, an enzyme that converts thymidine into thymidine triphosphate. However, this enzyme also converts the base analog 5-bromodeoxyuridine (BrdU) into a nucleotide that is incorporated into DNA, causing a lethal reaction. Thus, cells that express either host- or virus-encoded thymidine kinase are killed when treated with BrdU.
Genes to be put into the vaccinia virus genome are first inserted into an Escherichia coli plasmid that contains part of the vaccinia thymidine kinase (tdk) gene (Figure 12.25). The foreign DNA is inserted into the tdk gene, which is therefore disrupted. This recombinant plasmid is then transformed into animal cells whose own tdk genes have been inactivated. These cells are also infected with wild-type vaccinia virus. The two versions of the tdk gene—one on the plasmid and the other on the virus—then recombine. Some viruses gain a disrupted tdk gene plus its foreign insert (Figure 12.25). Cells infected by wild-type vaccinia virus (with an active thymidine kinase) are killed by BrdU. By contrast, cells infected by recombinant vaccinia virus (with a disrupted tdk gene) grow long enough to yield a new generation of virus particles (Figure 12.25). This protocol thus selects for viruses whose tdk gene contains a cloned insert of foreign DNA. Vaccinia viruses can also be engineered to carry genes from multiple viruses, forming polyvalent vaccines. Currently, several vaccinia vector vaccines have been developed and licensed for veterinary use, including one for rabies, while many other vaccinia vaccines are at the clinical trial stage.
Figure 12.25 Production of recombinant vaccinia virus.

Foreign DNA is inserted into a short segment of the thymidine kinase gene (tdk) from vaccinia virus carried on a plasmid. Following replication of this plasmid in Escherichia coli, both the recombinant plasmid and wild-type vaccinia virus are put into the same animal host cell to promote recombination. The animal cells are treated with 5-bromodeoxyuridine (BrdU), which kills only cells with an active thymidine kinase. Only recombinant vaccinia viruses whose tdk gene is inactivated by insertion of foreign DNA survive.
Subunit vaccines, vaccines that contain only a specific protein or two from a pathogen, are also produced by recombinant means. For a pathogenic virus, the gene encoding its coat or capsid protein is often the best vaccine candidate because capsid proteins are typically highly immunogenic (Section 5.1 and Section 28.3). Subunit vaccines are popular because large amounts of immunogenic proteins are produced that can be administered at high dosage without the risk that exists with attenuated or killed-cell vaccines that may inadvertently contain viable pathogen cells or viruses. However, some subunit vaccines, such as that prepared against a surface protein of human hepatitis B (Section 31.11), require that the immunogenic proteins be glycosylated by the host before they are immunologically active. To solve this problem, the subunit hepatitis B vaccine was produced in a eukaryotic host (yeast), which generated the glycosylated and immunologically active form of the vaccine.
Commensal *Bacteria* and Therapeutic Delivery
While new therapeutic agents to treat different diseases are constantly being developed, delivery to the target tissues or cells can be problematic. This is especially true for drugs that are rapidly degraded in the bloodstream or by the acidity of the stomach. To alleviate these problems, members of the microbiome that form positive relationships in the body—such as the gut normal flora—have been exploited to produce and release drugs while inside a host (Figure 12.26). Because of their commensal lifestyle, these microbes do not illicit an immune response and are tolerated well by the body. For therapeutic delivery of anticancer drugs, members of the microbiome that flourish in anaerobic niches such as the intestinal tract and in tumors are of particular interest. Tumors become anaerobic because of rapid cell proliferation, which results in the tumor rapidly outgrowing its blood supply. Dying tumor cells also release nutrients that attract members of the microbiome. Through genetic engineering, some of these bacteria that are naturally tolerated by the body and colonize tumors have been harnessed to not only deliver therapeutic substances specifically to diseased or cancerous cells (Figure 12.26a), but also to release tumor-specific antigens within circulating immune cells to facilitate the immune system’s targeting of tumors (see below and Figure 12.26b).
Figure 12.26 Strategies for bacterial delivery of therapeutic agents.

(a) Pathogens that grow in the low-oxygen environment of a tumor can be attenuated and genetically modified to release drugs directly inside tumors. (b) Pathogens that elicit a strong immune response can be attenuated and engineered to release antigens normally found on tumor cells. This antigen release stimulates antibody production and immune system destruction of tumors. (c) Probiotic strains normally found in the healthy gut can be engineered to release therapeutic molecules to treat various diseases as well as help train the immune system to tolerate beneficial microbes.
An exciting example of the targeted delivery of a beneficial drug is the use of engineered commensal bacteria to convert intestinal cells into glucose-responsive insulin-secreting cells. In type 1 (juvenile) diabetes mellitus, the immune system destroys insulin-producing beta cells in the pancreas (Section 28.1). Studies on how to generate new beta cells are under way; meanwhile, different studies have focused on ways to convert other cell types into insulin producers. From these studies, a therapeutic molecule known as the full-length glucagon-like peptide 1 (GLP1) has been shown in a rat diabetes model to be able to reprogram intestinal epithelial cells to respond to glucose and produce insulin. However, GLP1 has a short half-life in the bloodstream and the target epithelial cells are naturally shed from the intestine over the course of days. Thus, to deliver GLP1 to intestinal epithelial cells on a continuous basis, genetic engineers modified a strain of Lactobacillus, a gram-positive bacterium that can propagate in the intestine, to constitutively secrete GLP1 in the gut (Figure 12.26c). While this work has only been done in an experimental animal, it offers promise for the future of treating diabetes in humans.
Probiotics are live commensal microorganisms administered to a human to benefit health (Section 24.12), and engineered probiotics have been used to sense and kill biofilm-forming strains of Pseudomonas aeruginosa. The ability to sense P. aeruginosa is based on its production of the quorum-sensing inducer molecule N-acyl homoserine lactone (AHL; Section 7.7). A probiotic strain of Escherichia coli has been modified not only to sense P. aeruginosa, but also to release a cocktail of therapeutic substances in response to AHL levels. These include a dispersin molecule to destroy P. aeruginosa biofilms, as well as bacteriocin (Section 6.2) and lysis proteins to specifically kill P. aeruginosa cells. Figure 12.27 illustrates the success of this engineered probiotic in a worm experimental model infected with P. aeruginosa producing green fluorescent protein (GFP, Section 12.5 and Figure 12.17). While P. aeruginosa can successfully colonize the host worm in the presence of a wild-type strain of E. coli (Figure 12.27a), the pathogen cannot colonize the host worm in the presence of an engineered strain of E. coli releasing therapeutic molecules in response to P. aeruginosa AHL levels (Figure 12.27b). A major goal of such research is to develop new tools to fight the human genetic disease cystic fibrosis, a disease in which P. aeruginosa biofilms in the lungs are a life-threatening complication.
Figure 12.27 Probiotic *Escherichia coli* engineered to kill *Pseudomonas*.

Infection of the worm Caenorhabditis elegans with biofilm-forming P. aeruginosa expressing the green fluorescent protein (GFP) and treatment with an engineered strain of E. coli. (a) Active P. aeruginosa infection (visualized by GFP production) in the presence of a probiotic (attenuated) strain of E. coli (top, fluorescent micrograph; bottom, phase-contrast micrograph). (b) Limited P. aeruginosa survival occurs in response to the addition of the engineered version of the E. coli probiotic strain. The engineered strain was genetically modified to release therapeutic molecules that disrupt biofilms and kill P. aeruginosa cells when a quorum-sensing molecule produced by Pseudomonas is detected. Image adapted from Hwang, I.Y., Koh, E., Wong, A., March, J.C., Bentley, W.E., Lee, Y.S., and Chang, M.W. 2017. Nat. Commun. 8: 15028.
Pathogens as Engineered Anticancer Therapies
While many cancers are treatable with radiation and chemotherapy, how to specifically target the immune system, drugs, or radiation to tumor cells has been a long-standing problem, and biotechnology may have a solution. Listeria monocytogenes is a pathogenic bacterium that causes listeriosis, a serious foodborne illness (Section 33.13 and Figures 33.14 and 33.15). L. monocytogenes grows within human cells, which allows wild-type strains to evade the immune system. By contrast, attenuated strains of L. monocytogenes can be attacked by the immune system. These traits have been harnessed to engineer recombinant L. monocytogenes strains that express antigens found on tumor cells. The presence of these tumor-associated antigens triggers the immune system to produce tumor-specific antibodies (Figure 12.26b), and these ultimately lead to destruction of not only the bacterium but also the tumor. An example of such a tumor-targeting bacterial vaccine is an engineered strain of L. monocytogenes that expresses antigens specific to tumors caused by the human papillomavirus (HPV, Section 31.14).
Mastering Microbiology
Art Activity: Figure 12.26 Engineered anthrax toxin
While attenuated L. monocytogenes strains in healthy cells are cleared by the immune system, clearance does not occur in tumor cells. This observation hinted that weakened strains of L. monocytogenes might also be turned into anticancer vehicles to deliver toxic drugs or radioisotopes specifically to tumor cells. This was accomplished by coupling the radionuclide 188rhenium to a recombinant strain of L. monocytogenes. In experiments with mice, this therapeutic strain infected and multiplied in pancreatic tumor cells (Figure 12.28) without harming normal pancreatic cells. In Section 12.11 we will pick up on this theme again and describe how synthetic biology has been used not only to attenuate a pathogen to persist in tumor cells, but also to contain a synthetic biological circuit for the cyclical delivery of an antitumor drug to minimize drug toxicity to the rest of the body (see Figure 12.39).
Figure 12.28 Therapeutic *Listeria*.
L. monocytogenes cells (pink) linked to the radionuclide 188rhenium enter and multiply inside mouse pancreatic tumor cells (blue, cells; green, extracellular matrix) that have spread from the primary tumor. Radiation from the 188rhenium slowly kills the tumor cells.
Specific Delivery of Antibodies as an Anticancer Therapy
Another mechanism for treating cancer is by using antibodies, proteins produced by the immune system to attack foreign substances (Section 27.3). It has been found that the binding of antibodies to specific targets inside cancer cells can trigger the host’s immune system to kill the cancer cell. However, antibodies do not freely enter cells and thus a transport mechanism has been genetically engineered using a toxin produced by Bacillus anthracis, the bacterium that causes anthrax (Sections 30.9 and 32.8) (Figure 12.29). Anthrax toxin contains three components: edema factor, lethal factor, and protective antigen; the latter is essential for carrying the toxic edema and lethal factors into the cell.
Figure 12.29 Engineered anthrax toxin.

The protective antigen component of the anthrax toxin is engineered to carry a synthetic antibody. This engineered protective antigen specifically binds to a cell receptor on target cancer cells. After binding to the receptor, the engineered complex is taken up into the cell through an endosome. Following release into the cytoplasm, the synthetic antibody binds to an essential cellular protein, triggering the cancer cell’s death through the host’s immune response. (Anthrax toxin is discussed in more detail in Sections 30.9 and 32.8.)
Using genetic engineering, scientists have modified the B. anthracis protective antigen to carry a synthetic anticancer antibody instead of the toxic edema and lethal factors. Injection of the modified and now harmless toxin results in the protective antigen recognizing and binding to a receptor on the outside of the cancer cell (Figure 12.29). The protective antigen:antibody complex is then taken up into the cancer cell through the formation of an endosome. Once the antibody is released into the cytoplasm, it specifically binds a protein essential to tumor viability. This binding then triggers the cell’s immune system, which recognizes the antibody:cellular protein complex as foreign and kills the cell (Figure 12.29). Any antigen:antibody complex incorporated by normal cells is harmless because both the toxic portions of the toxin and the specific cancer proteins targeted by the antibody are absent from normal cells.
Stimulating the immune system to fight cancer may well turn out to be a mechanism in a whole new line of anticancer therapies. The novel system devised here—combining antitumor antibodies with a delivery system crafted from a highly toxic bacterium—shows both the power and the promise of genetic engineering to accelerate the war on cancer. The immunological principles behind this and other important new anticancer therapies will be developed in Chapter 28.
We now transition from medical applications of genetic engineering to consider commercial applications in the final two sections of Part II of the chapter.
Check Your Understanding
What are the important differences among a recombinant live attenuated vaccine, a vector vaccine, and a subunit vaccine?
How can beneficial members of the microbiome be used as therapeutic agents?
What feature of some pathogenic bacteria makes them attractive for use as engineered cancer treatments?
12.9 Mining Genomes and Engineering Pathways
Complex environments, such as fertile soil, contain vast numbers of uncultured microbes and their genes that are ripe for harvesting by genetic engineering. In addition, microbial metabolic pathways can be altered, embellished, or otherwise modified to change their characteristics and improve their efficiency. Here we explore how genetic engineering can both mine environmental genomes and alter metabolic pathways.
Environmental Gene Mining
Just as the total gene content of an organism is its genome, the collective genomes of an environment are its metagenome (Section 10.7 and Section 19.8). Gene mining is the process of identifying and isolating potentially useful genes from the environment without the need to culture the organisms that contained them. In gene mining, DNA (or RNA) isolated directly from environmental samples is cloned into suitable vectors to construct a metagenomic library (Figure 12.30). If RNA is isolated, it must first be converted to cDNA by reverse transcriptase (Figure 12.3).
Figure 12.30 Metagenomic search for useful genes in the environment.

DNA samples are obtained from different environments, such as seawater, forest soil, and agricultural soil. A metagenomic library is constructed using bacterial artificial chromosomes (BACs) and screened for genes of interest. Possibly useful clones are analyzed further.
Screening of environmental metagenomic libraries has identified novel genes encoding enzymes that can degrade various pollutants and enzymes that make novel antibiotics. Retrieval of gene clusters encoding entire metabolic pathways—such as for antibiotic synthesis—requires vectors such as bacterial artificial chromosomes (BACs) (we discussed the general principles of artificial chromosomes in Section 12.2). BACs are similar to plasmids except that they can carry large inserts of DNA. BACs are especially useful for screening samples from rich environments, such as soil, where vast numbers of unknown genomes are present and correspondingly large numbers of genes are available to screen (Figure 12.30).
Several lipases, chitinases, esterases, and other degradative enzymes with novel substrate ranges and other properties have been isolated by this approach, and such enzymes have many industrial applications. Enzymes with improved resistance to industrial production conditions, such as high temperature, high or low pH, and oxidizing conditions, are especially valuable and desirable. Metagenomics can also target products with a particular combination of properties, such as a heat-stable lipase. Lipases hydrolyze fats, but their industrial production and use often require that they remain active at high temperatures. To isolate a thermostable lipase, a metagenomic library was prepared from a hot spring sample and the DNA was transformed into cells of Escherichia coli. Recombinant colonies expressing lipase activity were then selected and analyses indicated that certain of them remained active at 90 °C. The gene encoding the heat-stable lipase was then introduced into an expression vector for commercial production of the enzyme.
By metagenomic mining of extreme environments, several useful heat- and acid-stable enzymes have been isolated for cleaning food-processing equipment in the food industry (Figure 12.31). To prevent foodborne infections, food-processing equipment must be rigorously cleaned, and cleaning protocols typically employ rigorous acid and base treatments, detergents, and sanitizers, all of which consume large amounts of chemicals and generate large volumes of wastewater that must be treated. By contrast, cleaning equipment with enzymes that function optimally near the boiling point of water in dilute acids (Figure 12.31) requires fewer chemicals and less water and more effectively removes microbial biofilms than standard cleaning practices.
Figure 12.31 Application of CinderBio hyperstable enzymes for the cleaning of creamery equipment.
These heat-stable enzymes clean industrial food-processing equipment as well as or better than traditional cleaning methods and do not generate large amounts of toxic wastewater.
Pathway Engineering: The Indigo Synthesis Example
Pathway engineering is the process of assembling a new or improved biochemical pathway using genes from one or more organisms. Engineered microbes are used to make alcohols, solvents, food additives, dyes, antibiotics, and many other products. They may also be used to degrade agricultural waste, pollutants, herbicides, and other toxic or undesirable materials. Here we discuss improving or modifying existing pathways, and later we explore the use of synthetic biology to create entirely new pathways (Section 12.11).
An interesting example of pathway engineering is the production of indigo by E. coli (Figure 12.32). Indigo is an important dye used for treating wool and cotton; blue jeans, for example, are made of cotton dyed with indigo. Although indigo can be synthesized chemically, the heavy demand for indigo by the textile industry has spawned new approaches for its synthesis, including a biotechnological approach using pathway engineering.
Figure 12.32 Engineered pathway for production of the dye indigo.

Escherichia coli naturally expresses tryptophanase, which converts tryptophan into indole. Naphthalene oxygenase (originally from Pseudomonas) converts indole to dihydroxy-indole, which spontaneously dehydrates to indoxyl. Upon exposure to air, indoxyl dimerizes spontaneously to form indigo.
Because the structure of indigo is very similar to that of the aromatic hydrocarbon naphthalene, enzymes that oxygenate naphthalene also oxidize indole to its dihydroxy derivative; the latter oxidizes spontaneously in air to yield indigo, a bright blue pigment. Enzymes for oxygenating naphthalene are encoded by several plasmids found in Pseudomonas and other soil bacteria. When genes from such plasmids were cloned into E. coli, the cells turned blue because they had incorporated the genes encoding the enzyme naphthalene oxygenase.
The indigo pathway consists of four steps, two enzymatic and two spontaneous (Figure 12.32). E. coli naturally synthesizes the enzyme tryptophanase that carries out the first of these steps, the conversion of tryptophan to indole. In the engineered E. coli, a second step converts indole to the product that converts to indigo spontaneously (Figure 12.32). For indigo production, tryptophan must be supplied to the recombinant E. coli cells, and for commercial application, this was accomplished by affixing cells to a solid support in a bioreactor and then continuously trickling a tryptophan solution obtained from waste protein sources over the cells. If the tryptophan solution is recirculated over the cells several times, indigo levels steadily increase until the dye can be harvested.
Although bioproduction of indigo is clearly possible, at present it is difficult to compete with the chemical production of more than 20 kilotons of indigo per year. Indeed, the two greatest challenges of pathway engineering are controlling the pathway and producing the desired compound at the yields necessary to be cost effective.
Check Your Understanding
Explain why metagenomic cloning gives large numbers of novel genes.
What types of environments are often sampled to prospect for industrial enzymes and why?
How was Escherichia coli modified to produce indigo?
12.10 Engineering Biofuels
With the global supply of fossil fuels limited and environmental concerns growing about how climate change will affect our planet, renewable energy sources such as biologically produced fuels—biofuels—are in demand. The major biofuels in use today are ethanol, biodiesel, hydrogen, and methane. Select microorganisms can produce biofuels; however, the yield from wild-type organisms is often hindered by side reactions, toxic by-products, and missing enzymes for critical steps. For example, hydrogen (H2) is produced by nitrogen-fixing bacteria through the activity of a nitrogenase enzyme complex (Figure 3.28). However, most of this hydrogen is subsequently consumed by hydrogenase enzymes in wild-type nitrogen fixers. Thus, to enhance the production of biofuels, microorganisms have been genetically modified to optimize production. Here we discuss how genetic engineering has allowed for the use of alternative biofuel feedstocks, how enzyme replacement can yield new biofuels, and how phototrophic microorganisms can be harnessed as biofuel factories.
Bacterial Conversion of Switchgrass to Ethanol
Over 16 billion gallons of ethanol are produced per year in the United States from the fermentation of corn sugar by yeast (**Figure 12.33*a***). However, because corn requires considerable cost and energy inputs to grow and harvest and is a major food source for both humans and domesticated animals, alternative nonedible and low-resource-input plant materials are more desirable biofuel feedstocks. Much attention has been focused on fast-growing grasses such as switchgrass (Panicum virgatum) (Figure 12.33b) as a source of cellulose for the production of ethanol. However, switchgrass cellulose is integrated with other plant polymers such as hemicellulose and lignin and requires high-temperature, chemical, and enzymatic pretreatment to break the polymers down to fermentable sugars.
Figure 12.33 Biofuels.

(a) A bioethanol plant in Nebraska (USA). In the plant, glucose from cornstarch is fermented by yeast to ethanol plus CO2. The large tank in the foreground is the ethanol storage tank, and the pipes in the background are for distilling the alcohol from the fermentation broth. (b) Switchgrass, a source of cellulose as a feedstock for ethanol production. (c) Fluorescence photomicrograph of acridine orange–stained cells of the thermophilic and cellulolytic bacterium Caldicellulosiruptor kronotskyensis growing on switchgrass (a single cell measures about 0.6 μm×3 μm). The green color is from the switchgrass plants.
By tapping into both microbial diversity and genetic engineering, a bacterium has been discovered and genetically modified not only to break down switchgrass cellulose to fermentable sugars but also to ferment this sugar to ethanol. Caldicellulosiruptor, a gram-positive anaerobic and thermophilic bacterium, naturally produces cellulase and hemicellulase enzymes that can convert cellulose and hemicellulose to glucose. Unique proteins called taˉpirins that extrude from the outer layer of the bacterium’s peptidoglycan allow the cell to directly bind to raw switchgrass during the conversion process (Figure 12.33c). While Caldicellulosiruptor bescii grows optimally at 80 °C and can ferment sugars, it naturally yields mostly acetate, lactate, and hydrogen as fermentation products. To directly convert switchgrass to ethanol, genetic engineers altered the terminal steps of the C. bescii glycolytic pathway (Section 3.6 and Figure 3.11) by replacing genes encoding lactate dehydrogenase and other acidic fermentation products with a bifunctional acetaldehyde/alcohol dehydrogenase from another thermophile, Clostridium thermocellum. This shifted 70% of the C. bescii fermentation products to ethanol.
Use of thermophilic microorganisms for biofuel production has several advantages such as reduced risk of contamination with mesophiles and improved substrate solubility. It also makes the collection of any volatile products easier. For example, separating small amounts of desired products from large amounts of growth media (such as collecting ethanol during yeast fermentation of corn sugar) requires significant energy inputs to cool the bioreactors for growth of the organism and then later to heat and distill off the ethanol (Figure 12.33a). By contrast, since C. bescii grows optimally at 80 °C—just above the boiling point of ethanol—commercial production of alcohol from cellulose by this genetically engineered bacterium requires little or no cooling and saves energy by the continuous emission of the desired product, ethanol.
Engineered Alkenes and Alkanes
Petroleum contains a mixture of hydrocarbons of varying chain length. Propane (C3H8), produced from natural gas processing and petroleum refining, is a widely used heating and cooking fuel and a key fuel for agricultural applications. Using genetic engineering, scientists have modified strains of Escherichia coli to convert glucose into propane and some other petroleum hydrocarbons.
Hydrocarbon production in E. coli begins with the synthesis of a fatty aldehyde. This was done by heterologously expressing in E. coli the Photorhabdus luminescens luxCED genes, which encode enzymes that reduce fatty acids to their corresponding aldehydes (Figure 12.34). The activity of these fatty acid reductase, synthetase, and transferase enzymes yields a fatty aldehyde that can be converted to hydrocarbons by the enzyme aldehyde decarbonylase. However, E. coli lacks this enzyme as well. To overcome this limitation, genetic engineers cloned the aldehyde decarbonylase gene from the cyanobacterium Nostoc punctiforme into E. coli, allowing the engineered E. coli to convert linear fatty acids added to its growth medium into linear hydrocarbons (Figure 12.34).
Figure 12.34 Hydrocarbon-producing *Escherichia coli*.

E. coli naturally produces fatty acids from acetyl-CoA. By engineering a strain to express the LuxC (fatty acid reductase), LuxE (fatty acid synthetase), and LuxD (fatty acid transferase) proteins from the fluorescent bacterium Photorhabdus luminescens (inset: fluorescent colony), a fatty aldehyde intermediate is produced. This fatty aldehyde can then be converted to a hydrocarbon if the same strain has also been engineered to express the aldehyde decarbonylase enzyme from the filamentous cyanobacterium Nostoc punctiforme (inset photo).
Because branched-chain hydrocarbons yield higher octane numbers (which is good for gasoline engine performance), scientists expanded this engineered pathway by heterologously expressing enzymes that participate in the first step of the fatty acid elongation cycle (Figure 3.35) from Bacillus subtilis. This allowed the engineered E. coli to use more branched-chain fatty acids as starting substrates and by doing so generate higher-octane fuels.
Microalgae and Biodiesel
Microalgae are unicellular phototrophic eukaryotes (Section 18.16) that produce an abundance of bioactive compounds including lipids, fatty acids, and carotenoids. These products are made using only sunlight, CO2, a few minerals, and water. Microalgae of interest to biotechnology include the green algal genera Chlorella and Chlamydomonas. These organisms produce significant amounts of storage lipids known as triacylglycerides (TAG), substances that can be chemically or enzymatically treated to yield biodiesel, a fuel for use in the diesel engines present in many trucks and heavy transport vehicles.
Improving TAG synthesis in the microalgal biosynthetic pathway required some genetic engineering breakthroughs, and **Figure 12.35*a*** illustrates the successful design of vectors that allow proteins to be targeted to the nucleus or chloroplast of cells of Chlamydomonas; other vectors have been developed that allow the mitochondrion or the endoplasmic reticulum to be targeted. Organelle targeting is facilitated through the fusion of signal sequences (Section 6.12) to the protein of interest. A vector for the simultaneous expression of two foreign genes in separate cellular locations has also been generated (Figure 12.35b). These targeting vectors are critical for manipulating compartmentalized TAG biosynthetic activities such as control of gene expression by transcription factors (encoded by the nuclear genome), ATP-producing enzymes (encoded by the mitochondrial genome), and enzymes for initial fatty acid synthesis reactions (encoded by the chloroplast genome). Translation of proteins by ribosomes on the endoplasmic reticulum is also important for protein secretion.
Figure 12.35 Genetic tools for engineering microalgae.

(a) Fluorescent micrographs showing the expression of reporter genes in targeted regions of cells of Chlamydomonas. Top panel: A reporter gene encoding a red fluorescent protein that targets the nucleus (left) and chloroplasts (right). Bottom panel: both images overlaid with a green fluorescent stain for the chloroplast. (b) Dual expression of two reporter genes to different cellular locations using a single vector. A gene encoding a blue fluorescent protein is localized to the nucleus, while a gene encoding a red fluorescent protein is targeted to the endoplasmic reticulum in cells of Chlamydomonas. Adapted from Rasala, B.A., S-S. Chao, M. Pier, D.J. Barrera, and S.P. Mayfield. 2014 PLoS ONE 9 (4): e94028.
Using microalgal triacylglycerides as a feedstock for biodiesel increases the possibilities for biofuel production, and the fact that the process is driven by the energy of sunlight makes it an environmentally attractive process. However, the major obstacle facing all biofuel production schemes—especially those that require sunlight—is the expense of the equipment and engineering necessary to scale production up to levels necessary to compete with the petroleum industry. Currently, about 90 million barrels of oil a day arrive on the highly volatile world energy market, and until oil supplies diminish to the point where a significant price hike is encountered, any biofuel will have a difficult time competing.
The last part of this chapter considers the “final frontier” for genetic engineering: the ability to synthesize cell parts or even entire cells from scratch in the revolutionary new field of synthetic biology.
Check Your Understanding
How has Caldicellulosiruptor been modified to produce ethanol directly from switchgrass?
What are the advantages of using thermophiles to produce biofuels?
What has been the limiting factor in engineering microalgae to produce greater amounts of lipids?
III Synthetic Biology and Genome Editing
Scientists can stitch together bits and pieces of DNA from various sources to create artificial genes, operons, or entire genomes and can edit these new genetic constructs at will to change the properties of existing organisms or even to fabricate an entirely synthetic organism.
The term synthetic biology refers to the use of genetic engineering to create novel biological systems out of available biological parts, often taken from several different organisms. These biological parts (promoters, enhancers, operators, riboswitches, regulatory proteins, enzyme domains, signal receivers, etc.) have been termed biobricks. Synthetic biology links biobricks together in various combinations to form modules capable of generating complex behaviors (Figure 12.1). While we will discuss some amazing examples of synthetic biology (including the formation of synthetic cells) in Sections 12.11 and 12.12, college students are also trying their hand at synthetic biology. An undergraduate competition called the International Genetically Engineered Machine (iGEM; https://igem.org/) occurs annually worldwide. Teams in this competition have used synthetic biology to engineer products ranging from probiotic bacteria that secrete the anti-inflammatory cytokine interleukin-10 to help treat irritable bowel disease (cytokines and interleukins are discussed in Chapters 26 and 27), to microfluidic systems that “print” (assemble) genetic circuits and transform them into cells of Escherichia coli.
Another powerful new and rapidly developing technology allows the precise “editing” of genomes in living cells, a technique that has revolutionized biotechnology. In Section 12.13 we explore how the microbial immune system has been leveraged to edit any genome and the remarkable applications of this genome editing technology.
12.11 Synthetic Metabolic Pathways, Biosensors, and Genetic Circuits
12.11 Synthetic Metabolic Pathways, Biosensors, and Genetic Circuits
12.11 Synthetic Metabolic Pathways, Biosensors, and Genetic Circuits
A major focus of synthetic biology thus far has been the construction or modification of metabolic pathways. High-value products are often expensive because purifying them from their original source, typically plants, is costly. The pathways responsible for these products include proteins and RNAs that regulate the synthesis, export, and protection from harmful effects of the product. Thus the DNA encoding pathways for high-value products can range from 1000 to 100,000 base pairs of DNA, which makes it impossible to clone the necessary genes into a model system.
However, by using various enzyme and regulatory biobricks, synthetic biologists can reconstruct or refactor natural pathways into artificial ones that allow for easy expression in model systems. This refactoring ultimately leads to the efficient conversion of cheap and abundant substrates into high-value products. Figure 12.36 illustrates refactoring of the 20-gene pathway encoding nitrogen fixation (Section 3.12) in Klebsiella oxytoca; the result is a streamlined and optimized five-gene pathway. While genetically modified organisms (GMOs) are used for refactoring and the production of high-value products, no foreign DNA is present in the products.
Figure 12.36 Synthetic biology and pathway refactoring.

(a) Chromosomal operons encoding the nitrogen fixation pathway in Klebsiella oxytoca. Direction of arrows indicates direction of transcription and color-coding is as indicated. H, nifH; D, nifD; K, nifK; Y, nifY; E, nifE. (b) Refactored “minimalist” nitrogen fixation pathway. RBS, ribosome-binding site. Data modified from Smanksi, M.J., Zhou, H., Claesen, J., Shen, B., Fischbach, M.A., and Voigt, C.A. 2016. Nat. Rev. Microbiol. 14, 135.
Engineering a Major Food Product
Vanillin, one of the most popular flavoring agents in the world, is a secondary metabolite extracted from the seedpods (vanilla beans) of orchids of the genus Vanilla. Natural vanillin is expensive because of the slow growth of orchids, their relatively low output, and the high production costs of cultivating and harvesting the beans. By analyzing the natural metabolic pathway for the production of vanillin, genetic engineers have synthesized strains of Escherichia coli and yeast that can synthesize the flavoring agent from glucose. Vanillin synthesis in E. coli requires five heterologously expressed enzymes, and once the necessary biobricks were incorporated into the bacterium, it produced vanillin identical in structure and taste to naturally produced vanillin. (However, some argue that naturally produced vanilla contains additional compounds extracted from the vanilla beans that contribute to its unique taste.)
Mastering Microbiology
Art Activity: Figure 12.33 Artemisinin synthesis through synthetic biology
“Synbio vanillin,” as the E. coli product has been called, is commercially available as an inexpensive source of vanillin and has been used primarily for flavoring ice cream and baked goods. As the second most expensive spice in the world (behind saffron), natural vanilla is an especially costly ingredient for bulk use such as in ice cream. Synbio vanillin is a good example of how synthetic biology can be used to convert inexpensive feedstocks (corn sugar) into high-value products.
Synthetic Pharmaceuticals: Artemisinin and Malaria
Pharmaceuticals are often derived from natural products; aspirin, for example, was originally obtained from willow bark. While aspirin is now chemically synthesized, not all pharmaceuticals can be synthesized economically. One example is the antimalarial drug artemisinin. Malaria, which is caused by protozoans of the genus Plasmodium, is transmitted by mosquitoes and infects nearly 500 million people each year, primarily in tropical and subtropical countries (Section 34.5). While various traditional antimalarials are in use, the parasite has evolved resistance to many of these and thus new antimalarial drugs are constantly needed. Artemisinin is an alternative antimalarial that is produced in limited amounts by the cultivated sweet wormwood plant Artemisia annua. To ensure availability of the drug, the Semi-Synthetic Artemisinin Project was initiated with the goal of engineering a microorganism for the synthesis of artemisinic acid, used for production of artemisinin (Figure 12.37).
Figure 12.37 Artemisinin synthesis through synthetic biology.

A summary of the engineering steps used to modify a Saccharomyces cerevisiae yeast strain to produce artemisinic acid. Colored arrows represent expression of genes from the plant Artemisia annua and other genetic modifications.
A. annua naturally produces artemisinic acid by converting acetyl-CoA to farnesyl diphosphate (FPP; a 15-carbon intermediate) using the mevalonate biosynthetic pathway. FPP is then oxidized to artemisinic acid and dihydroartemisinic acid through an intermediate called amorphadiene. Initially, the plan was to have E. coli produce artemisinic acid through fermentation. Synthetic biologists made numerous attempts to engineer E. coli with the correct biobricks to produce artemisinic acid through permutations of the natural pathway, inhibiting natural competing enzymes, mutating genes for codon optimization (Section 12.3), and modifying fermentation conditions, but no strategy emerged that could yield the amorphadiene intermediate at sufficient levels. However, by transferring the necessary metabolic pathway biobricks to a modified strain of the baker’s yeast Saccharomyces cerevisiae along with metabolic adjustments to divert carbon flux toward the final product, synthetic biologists designed a yeast strain that produces large amounts of artemisinic acid that can then be chemically converted to artemisinin (Figure 12.37).
Synthetic biology has also been successful at synthesizing powerful painkilling drugs. For example, genetic engineers modified yeast to produce the chemical thebaine, a precursor to morphine and hydrocodone, from glucose. This was accomplished by the heterologous expression in yeast of 21 different genes that originated from sources as diverse as plants, a rat, and the gram-negative bacterium Pseudomonas. The synthesized thebaine can then be chemically converted to a suite of pain-relieving drugs and marketed by pharmaceutical companies.
Biosensors and Genetic Circuits: Photographic *Escherichia coli*
An early excursion into the world of synthetic biology was the use of genetically modified E. coli to produce photographs using light sensing and a genetic circuit, another name for a gene regulatory network. The engineered bacteria are grown as a lawn on agar plates, and when an image is projected onto the lawn, unilluminated bacteria make a dark pigment while illuminated bacteria do not. The result is a primitive photograph of the projected image (Figure 12.38).
Figure 12.38 Bacterial photography.

(a) Light-detecting Escherichia coli cells were genetically engineered using components from cyanobacteria and E. coli itself. Red light inhibits phosphate (P) transfer to the DNA-binding protein OmpR; phosphorylated OmpR is required to activate lacZ transcription (lacZ encodes β-galactosidase). (b) Procedure for making a bacterial photograph. The opaque portions of the mask correspond to zones where β-galactosidase is active and thus to the dark regions of the final image. (c) A bacterial photograph of a portrait of the eminent biologist Charles Darwin.
Construction of the photographic E. coli required synthetic biologists to create three key biobricks to form a sensor linked to a genetic circuit: (1) a light sensor or detector and signaling module; (2) a pathway to convert heme (already present in E. coli) into the photoreceptor pigment phycocyanobilin (an accessory light-harvesting pigment of cyanobacteria, Section 14.4); and (3) an enzyme encoded by a gene whose transcription can be switched on and off to make the dark pigment (Figure 12.38a). The photoreceptor is a fusion protein (Section 12.5) in which the sensing half is the light-detecting part of the phytochrome protein from the cyanobacterium Synechocystis. This required phycocyanobilin is not naturally made by E. coli; hence the need to install the biobrick that contained the pathway to make phycocyanobilin.
The other half of the fusion protein is the signal transmission domain of the EnvZ sensor protein from E. coli. EnvZ is part of a two-component regulatory system, its partner being OmpR (Section 7.5). Normally, EnvZ activates the DNA-binding protein OmpR, and the latter in turn activates target genes by binding to the promoter. The hybrid protein was designed to activate OmpR in the dark but not in the light. This is because phosphorylation of OmpR is required for activation, and red light converts the sensor to a state in which phosphorylation is inhibited. Consequently, the target gene is off in the light and on in the dark. When a mask is placed over the Petri plate containing a lawn of the engineered E. coli cells (Figure 12.38b), cells in the dark make a pigment that cells in the light do not, and in this way a “photograph” of the masked image develops (Figure 12.38c).
The pigment made by the E. coli cells results from the activity of the lactose-degrading enzyme β-galactosidase, naturally present in E. coli. The target gene, lacZ, encodes this enzyme. In the dark, lacZ is expressed and β-galactosidase is made. The enzyme cleaves the lactose analog S-gal (3,4-cyclohexenoesculetin-β-d-galactopyranoside; similar to X-gal in Section 12.2) present in the growth medium to release galactose and a black dye. In the light, the lacZ gene is not expressed, no β-galactosidase is made, and so no dye is released. Contrast in the photograph is controlled by how much light cells see, which is governed by the nature of the mask that is used (Figure 12.38c).
Although bacterial photographs can hardly compare with digital photographs, the knowledge gained from assembling the biobricks required for bacterial photographs—work that is now many years old—helped form a foundation for the deployment of synthetic approaches in more complex biological systems, to which we turn now.
Biosensors and Genetic Circuits: Synchronized and Cyclical Drug Delivery
In Section 12.8 we discussed how features of the human microbiome (Chapter 24) can be used to deliver therapeutic agents to treat diseases including cancer. Synthetic biology is revolutionizing these treatment approaches through the design of synchronized genetic circuits that not only control the level of therapeutic delivery, but also the population level of the engineered microbe. We demonstrate this using the human pathogen Salmonella enterica (typhimurium), a bacterium that targets the gastrointestinal tract (Section 33.10).
We have already seen how certain genetically modified gastrointestinal bacteria can grow and release anticancer drugs in the hypoxic environment of tumor cells (Section 12.8 and Figure 12.26a), and a similar anticancer strategy using S. enterica (typhimurium) has been developed. Advanced genetics can also be performed on this bacterium, allowing for the strain to be manipulated not only to deliver drugs to tumors but to do so using an “internal clock.” To design a S. enterica (typhimurium) strain that releases an anticancer drug in a controlled and cyclical manner, genetic engineers took advantage of the natural ability of some microbes to communicate with others of their own kind through quorum sensing (Section 7.7). Because a transcriptional activator (LuxR) is able to use the quorum-sensing molecule N-acyl homoserine lactone (AHL; Figure 7.18) as its inducer to activate transcription (Section 7.3), a genetic circuit could be developed to link promoter regions of DNA that bind LuxR to genes that encode not only the production of AHL, but also the production of an anticancer drug and a cell lysis enzyme (**Figure 12.39*a***). Because AHL can freely diffuse into and out of cells, this genetic circuit results in AHL levels accumulating as the cell density increases. As AHL levels increase, the inducer molecule binds to LuxR and this activator–inducer complex binds to the promoters of genes encoding four components: LuxI (AHL synthesis), the anticancer drug, a fluorescent protein (GFP) to monitor the dynamics of the circuit, and the lysis protein E from bacteriophage ϕX174 (Figure 11.6).
Figure 12.39 Genetic circuits and synchronized therapeutic agents.

(a) A tumor-targeting bacterial strain of Salmonella was modified to produce and sense the quorum-sensing molecule N-acyl homoserine lactone (AHL) in response to population increase. Because AHL diffuses freely in and out of cells (red arrows), the cellular concentration increases as the surrounding cell density increases. Increased AHL levels lead to production of a lysis protein (E), an anticancer drug, and more AHL due to their corresponding genes being linked to a promoter that binds to the activator LuxR-AHL complex. This genetic circuit results in a positive feedback loop leading to cell lysis and drug release in the subpopulation of cells that encounter a specific threshold concentration of AHL. This cell lysis occurs in synchrony. Cells that did not encounter the required level of AHL survive to repopulate and initiate another cycle. (b) Time series of a coculture of Salmonella enterica (typhimurium) containing the part a genetic circuit, which is also linked to GFP for visualization (bottom chamber of panel), and HeLa cancer cells (top chamber of panel). The panels show an increase in bacterial cell concentration (42 minutes) and lysis and cell death (180 minutes). Bacterial cell lysis and drug release coincide with HeLa cell death (white cells) at 180 minutes. Intact bacterial cells at 180 minutes are able to grow and reinitiate the time course cycle. HeLa cells are an immortal cell line used in medical research.
As the population of the engineered strain reaches a critical threshold, almost all cells in the population produce the components of the genetic circuit (Figure 12.39a). This leads to not only production of the anticancer drug, but also its release due to the production of the lysis E protein. However, a few outliers will exist in the population because of phenotypic heterogeneity (Section 8.12), and these survivors will repopulate the tumor region (Figure 12.39a). The end result of this elegant genetic circuit is periodic bacterial lysis and drug delivery in response to a quorum-sensing clock.
Figure 12.39b illustrates the synchronized drug delivery circuit in action using a laboratory coculture system that allows cervical cancer HeLa cells to grow in a chamber above the engineered S. enterica (typhimurium) strain. The two cell types are separated and not allowed to mix, but molecules and drugs are able to move freely between the two growth chambers. As the engineered S. enterica (typhimurium) cell density increases, so does the level of GFP used to monitor the cells. This level of GFP decreases as the engineered microbes lyse from activation of the genetic circuit. Cell lysis leads to release of the anticancer drug and ultimately to HeLa cell death (Figure 12.39b).
By using biobricks encoding products and regulatory regions from various microorganisms, synthetic biology has produced a synchronized and cyclical therapeutic agent. However, while modified, this engineered strain of S. enterica (typhimurium) still possesses most of its wild-type genome. We turn now to approaches to engineering cells with synthetic genomes.
Check Your Understanding
What organism has been genetically modified to produce precursors to the drugs artemisinin and morphine?
How was Escherichia coli modified to produce a photograph?
12.12 Synthetic Cells
The synthesis of an entire cell from scratch can be considered the pinnacle of synthetic biology. This has not happened yet, but a related feat was announced in 2010: A group of scientists from the J. Craig Venter Institute (JCVI, California, USA) had produced a “synthetic” bacterium. However, the organism produced was not the result of assembling various biobricks to form a living organism—true de novo cell synthesis—but instead was the product of the artificial construction of a small bacterial genome from a known genome sequence and the insertion of this synthetic genome into a different bacterial species to yield viable cells (Figure 12.40).
Figure 12.40 Formation of a synthetic *Mycoplasma* cell.

DNA fragments corresponding to the desired Mycoplasma mycoides chromosome are synthesized with overlapping ends (represented by the same colors). These DNA fragments are linked together using the homologous recombination machinery of Saccharomyces cerevisiae, with sites of homologous recombination indicated by lines. Once assembled, the synthetic chromosome is transformed into a cell of Mycoplasma capricolum. This results in a cell with two separate chromosomes. After cell division, one cell possesses the synthetic chromosome (considered the synthetic M. mycoides) and the other remains a wild-type M. capricolum. The cell containing the synthetic chromosome was identified because it contained the reporter gene lacZ and thus produced blue colonies (see Figure 12.10).
Construction of a synthetic cell was accomplished by synthesizing a 1.08-million-base-pair (Mbp) genome based on the known genome sequence of the bacterium Mycoplasma mycoides. This small circular chromosome was pieced together from linear fragments of DNA containing homologous ends and the homologous recombination complex of yeast cells. The fully assembled 901-gene synthetic chromosome was then purified and transformed into a cell of Mycoplasma capricolum (Figure 12.40). The synthetic chromosome contained a reporter gene, lacZ, absent from the M. capricolum genome, which caused colonies containing the synthetic chromosome to turn blue (see Figure 12.10); this was needed to distinguish M. capricolum cells containing the M. mycoides genome from those containing the M. capricolum genome after cell division (Figure 12.40). When cells in the blue colonies were examined, they showed all of the properties of the original M. mycoides cell. This new cell was named JCVI-syn1.0. Although JCVI-syn1.0 was not constructed solely from biobricks (the M. capricolum host cell contributed ribosomes, various enzymes, and other important cytoplasmic components), the experiment did prove that an entire genome could be transplanted from one species to another.
Since the original creation of JCVI-syn1.0, synthetic biologists have expanded their work to determining the minimal genome necessary for a free-living cell. Using comparative genomics and prior knowledge about specific gene sequences, microbiologists at JCVI designed and synthesized several minimal genomes that they hypothesized would sustain life. To their dismay, none of these resulted in a viable cell. So instead, they generated modules of DNA corresponding to a Mycoplasma genome and sewed different combinations together to form synthetic genomes. Once viable cells were obtained from transplanting these genomes as described above, nonessential genes from the smallest genome were identified by transposon mutagenesis. After removing these unnecessary genes, a synthetic minimal cell coined JCVI-syn3.0 was created (Figure 12.41). This autonomous life form possesses a 531-kilobase genome encoding just 473 genes; JCVI-syn3.0 thus contains a genome smaller than that of any other free-living cell.
Figure 12.41 Synthetic biology and the design of a minimal cell.

Scanning electron micrograph of cells of the autonomous strain JCVI-syn3.0 possessing a synthetic genome containing 473 genes.
While this work showcases the amazing advancements in synthetic biology and the potential for creating designer cells with novel functions, a surprising mystery surrounds this minimal cell: the roles for almost a third of JCVI-syn3.0’s genes remain unknown, highlighting how much we still need to learn about the genetic foundation of a living cell.
Check Your Understanding
How were linear synthetic fragments of DNA assembled to form a Mycoplasma genome?
How were Mycoplasma transformants with the synthetic chromosome selected for?
12.13 Genome Editing and CRISPRs
In Chapter 9 we discussed clustered regularly interspaced short palindromic repeat (CRISPR) systems and their role in protecting Bacteria and Archaea from foreign DNA and maintaining genome integrity. Microbiologists studying a CRISPR system called CRISPR/Cas9 (Cas9 refers to CRISPR associated protein 9) in the bacterium Streptococcus pyogenes discovered that the system could also recognize and cleave a specific DNA sequence within other cells and that foreign DNA could be inserted into the cut site. This discovery has revolutionized biology.
The CRISPR/Cas9 system provides the most powerful and precise tool yet for altering eukaryotic genomes in living cells. Indeed, genome editing, as it has come to be known, has been used successfully to edit the genomes of plants, animal embryos, and human cell lines. Here we explore how this system works, its benefits to synthetic biology, and how it can be used as a gene drive to propagate specific genes throughout a population of organisms.
Sequence Targeting by the Cas9 Protein
As was illustrated in Figure 9.37, CRISPR systems possess Cas proteins that function as endonucleases when guided to a piece of nucleic acid by the complementary binding of CRISPR RNAs (crRNAs). By designing a synthetic RNA molecule that both recruits the Streptococcus Cas9 protein and binds to the desired target DNA sequence, genetic engineers have harnessed the power of the CRISPR/Cas system to cut specific DNA sequences in the genome of virtually any cell. At the cut site, the DNA can either be ligated (yielding a gene deletion) or used for inserting new DNA (**Figure 12.42a,*b***).
Figure 12.42 CRISPR/Cas9 genome editing.

sgRNA represents the synthetic guide RNA, while PAM represents a protospacer adjacent motif. Note that each genome target site must possess a PAM sequence for DNA cleavage to occur. (a) Insertion of foreign DNA into a targeted site of the genome. An sgRNA is synthesized to bind to a single target site on the genome through complementarity. This binding of the sgRNA to the DNA stimulates the Cas9 protein to cleave the genome at the target site. Foreign DNA with ends homologous to the cleavage site can be incorporated into the cut site through homologous recombination. This results in a genomic insertion. (b) Deletion of a genomic region. Two separate target sites flanking the DNA to be deleted are selected. After the design, addition, and binding of sgRNAs corresponding to these regions, Cas9 protein–dependent DNA cleavage occurs. This results in a double-stranded break in the target chromosome and a free piece of DNA. The double-strand break is then ligated by the cell’s DNA double-strand break repair pathway, while the free piece of genomic DNA is degraded. This results in a genomic deletion. (c) Crystal structure of the Streptococcus pyogenes Cas9 protein. The target DNA is shown in green and the sgRNA in orange.
The synthetic RNA molecule used for gene editing is called a synthetic guide RNA (sgRNA), and Figure 12.42c shows a model of the Cas9 protein from S. pyogenes binding both the sgRNA and target DNA. For complete Cas9 endonuclease activity, a short protospacer adjacent motif (PAM; Figure 9.37) must also occur on the target DNA. Without this PAM sequence, the Cas9 protein will bind to the region where the sgRNA binds but will not cut. When the Cas9 protein does cut DNA, its two endonuclease domains (shown in purple in Figure 12.42c) cooperate to cut both DNA strands, and this generates double-stranded breaks (for simplicity, Figure 12.42a and b have single strands—shown as tubes—representing double strands). Depending on the target cell, various methods of delivering the CRISPR system can be used. Genes corresponding to the designed sgRNA and Cas9 protein are often cloned into a plasmid under regulatory control of a strong promoter. Alternatively, the sgRNA and mRNA corresponding to the Cas9 protein can be generated in vitro. In either case, the materials are injected directly into target cells to trigger the gene editing process.
The presence of a PAM sequence close to the desired cleavage site is often the only limitation to cutting a specific DNA sequence. However, the PAM sequence is just three nucleotides in length and thus occurs with frequency in most genomes. CRISPR systems from other bacteria also recognize different PAMs, so alternative Cas proteins may be employed if needed. To delete a region of DNA, two target cleavage sites flanking the DNA sequence to be deleted must be identified and corresponding sgRNAs designed (Figure 12.42b). By contrast, only one cleavage site and corresponding sgRNA are needed to insert DNA (Figure 12.42a). To edit a region of a chromosome, sgRNAs are designed to bind to target DNA. This binding stimulates the Cas9 protein to cleave the target site if a PAM sequence is nearby (Figure 12.42a, b).
While a Cas9 protein and sgRNA can be used to cut DNA at specific sites (Figure 12.42), how is new DNA inserted at the cleavage site and how does the DNA get ligated back together? These tasks are accomplished by harnessing the cell’s own DNA repair machinery. If a piece of DNA containing sequences with homology to the cut site is added to the system, homologous recombination will be used to incorporate the DNA (Section 9.5), yielding a genomic insertion (Figure 12.42a). If the goal is only to delete the chromosomal region between two cut sites, the nonhomologous double-strand DNA break repair pathway will be employed to ligate the DNA following the deletion event (Figure 12.42b). The mechanism used by this DNA repair pathway makes it possible for genetic engineers to create an insertion or a deletion of as few as one nucleotide in a gene sequence through the cleavage of a single target site. No additional piece of DNA is needed because one nucleotide is either added or removed during the re-ligation of the two strands of DNA.
CRISPR Editing in Practice
Applications of CRISPR genome editing have seen a meteoric rise since its discovery in 2013. By designing a Cas9-encoding gene to possess codons optimized for the organism of interest (Section 12.3) and engineering sgRNA to target the gene of interest, almost any organism’s DNA can be edited. In fact, CRISPR gene editing has exploded in crop plants including rice, sorghum, wheat, corn, and soybeans. The CRISPR/Cas9 system has also been employed in tomato plants by targeting a gene region in which a mutation leads to leaves that are needle-like or wiry (Figure 12.43). In this study, DNA encoding the Cas9 protein and sgRNA was introduced into the plant cells using Agrobacterium and the Ti plasmid system (Figure 12.20). Because the resulting mutations are stable and heritable, CRISPR genome editing has also been used to engineer numerous beneficial vegetable and fruit modifications including drought-resistant lettuce, grapes with increased levels of vitamin C, and disease-resistant oranges.
Figure 12.43 CRISPR editing of tomato genome.

Interruption of the tomato SlAGO7 gene encoding an argonaute homolog results in plants that have wiry or spindly leaves (left) compared to a normal tomato plant (right).
While a Cas9 system has been used to excise the genome of the retrovirus HIV (the causative agent of AIDS, Section 11.11) from the genome of infected human cells in vitro, Cas proteins from other bacteria can be used to target HIV or other specific viral RNAs. These RNA-targeting Cas proteins have been harnessed to not only create highly sensitive and portable diagnostic tools for detecting viruses such as Zika (see Chapter 10 Explore the Microbial World, “DNA Sequencing in the Palm of Your Hand”), but also to target RNA viruses inside of cells. For example, the Csy4 CRISPR-associated protein from the bacterium Pseudomonas is an endoribonuclease that processes the long CRISPR transcript (Figure 9.37). The Csy4 protein has been modified to recognize and destroy free HIV RNA in infected cells (Figure 12.44). Similarly, the CRISPR/Cas9 system from the bacterium Francisella novicida has been reprogrammed to target the single-stranded RNA genome of hepatitis C virus (a liver pathogen and a cause of liver cancer) in a human cell line.
Figure 12.44 CRISPR-mediated inhibition of HIV infection.

The results of using the Pseudomonas Csy4 endoribonuclease CRISPR protein to target RNA-based HIV in infected human embryonic kidney cells. Red indicates HIV provirus integration into infected cells, while free HIV virions are indicated in green. Top panel: expression of an HIV–Csy4 targeting vector. Bottom panel: expression of a mutated vector containing a nonfunctional Csy4 protein. Adapted from Guo, R., H. Wang, J. Cui, G. Wang, W. Li, and J-F Hu. 2015. PLoS ONE 10 (10): e0141335.
Not only has CRISPR genome editing been used to delete, interrupt, and insert DNA sequences into a single location, it can also be used to target multiple genetic loci. An impressive example of this is the removal of 62 copies of the porcine endogenous retrovirus from swine cells. This retrovirus generates an immune response in humans and is one of the factors preventing the use of swine organs for transplants in humans (swine are anatomically very similar to humans). Although other swine proteins also induce an immune response in humans, genetic engineers predict that in the foreseeable future they will be able to edit away all of these factors to produce immune-friendly pig embryos for human organ production.
Currently, CRISPR genome editing appears to have very few limitations. In fact, fertility clinic human embryos that were nonviable and could not result in a live birth have even been modified. This landmark accomplishment and reports that scientists in China may have successfully created CRISPR-modified babies resistant to HIV, smallpox, and cholera have raised serious ethical questions regarding the use of CRISPR editing in humans. But the technique may be the key to eradicating a host of devastating genetic diseases before a baby carrying the genes for one or more of these diseases is born. However, more studies are needed to verify gene target specificity and the response of the human immune system to Cas proteins.
Gene Drives and Engineered Mosquitoes
Not only can Cas proteins be used to edit almost any gene, the system can also be used as a gene drive to spread specific mutations throughout entire populations over generations. Gene drives, which only occur in sexually reproducing organisms, result in an allele (different forms of a gene) being inherited more than the normal frequency of 50%. Gene drives do occur in nature, but they are often facilitated by selfish DNA such as transposons (Section 9.11). Thus, the spread and chromosomal location of the mutation is difficult to control. By contrast, CRISPR and Cas9 gene editing allows for the inheritance of the allele to be specifically controlled. The spread of a specific allele is based on diploid organisms possessing two copies of most genes, the ability of the Cas9 protein to be targeted to a specific site by a synthetic guide RNA (sgRNA), the endonuclease activity of Cas9 resulting in double-stranded DNA breaks, and the homology-directed DNA repair pathway.
**Figure 12.45*b*** illustrates the spread of an allele through a mosquito population by normal Mendelian inheritance. By contrast, Figure 12.45c shows how linking genes for Cas9 and the sgRNA to a specific allele results in movement of the allele from one chromosomal copy to the other. Once the cell expresses the Cas9 and sgRNA genes, the gene drive results in a double-stranded break in the wild-type chromosomal copy. This break in the DNA is lethal unless repaired by the homology-directed repair system, which works by copying DNA from the intact chromosome into the broken region. Thus, the Cas9-based gene drive ultimately results in the spread of the allele throughout the entire population as heterozygotes become homozygous for both the mutant allele and the Cas9 gene drive (Figure 12.45d).
Figure 12.45 Spread of gene drives by Cas9 technology.

(a) The mosquito Anopheles stephensi containing a green fluorescent protein (GFP) allele linked to a Cas9 gene drive (left) compared to wild type (right). (b) Normal inheritance of an altered allele (in this example, the gene encoding GFP) in mosquitoes through Mendelian genetics. Mating of a modified insect with a wild-type insect results in a heterozygote and only a 50% chance of the allele being transferred from parent to offspring. (c) By linking the allele to genes for Cas9 and synthetic guide RNA (sgRNA), the genetic modification in one copy of the chromosome spreads to a wild-type chromosome through the activity of Cas9 gene editing. The sgRNA guides the Cas9 endonuclease (solid red arrow) to cut the wild-type chromosome at a specific location, and the doublestrand break (dashed red arrow) results in DNA repair of the cut chromosome using the altered allele as a template for DNA replication. (d) The altered allele linked to the Cas9 system becomes a gene drive as its inheritance occurs over 50% of the time, which eventually leads to the genetic modification occurring in the entire population.
The applications for gene drives are countless, but one that is showing promise for public health is the modification of mosquitoes. While Figure 12.45a shows a mosquito carrying a Cas9 gene drive linked to an allele for the green fluorescent protein (GFP) for easy visualization, gene drives are being developed to prevent mosquitoes from carrying diseases such as malaria. The system can also be used to prevent the reproduction of disease-carrying mosquitoes by targeting an allele that results in female development. By linking this allele to a Cas9 gene drive, scientists have produced a population of mosquitoes unable to lay eggs after eight generations. While more scientific work is needed to determine the safety of releasing gene drives into nature, synthetic biologists now have the potential to eliminate malaria as a human disease and save millions of lives.
However, because the applications of gene drives are not just limited to pests like mosquitoes and have the potential to affect whole species, scientists have turned their attention to exploring ways to control the movement and reverse the activity of gene drives. Questions such as “Should scientists be altering genomes in ways that could have negative ecological consequences?” and “Can gene drives in one species be transmitted to other species?” are serious ones that need to be addressed. One way of protecting against these possibilities—effective biocontainment—is considered next to conclude this chapter.
Check Your Understanding
What characteristics of the Cas9 protein make it an efficient DNA editing tool?
What is the role of the sgRNA in genome editing? How is recombinant DNA inserted into a genome using CRISPR editing?
Give an example of how a gene drive can slow or halt transmission of an infectious disease.
12.14 Biocontainment of Genetically Modified Organisms
12.14 Biocontainment of Genetically Modified Organisms
12.14 Biocontainment of Genetically Modified Organisms
Throughout this chapter we have focused on genetically engineering microbes as factories for the synthesis of high-value products, including specific enzyme systems for gene editing. While these applications of genetic engineering are clearly beneficial, environmental concerns remain that genetically modified organisms (GMOs) may spread their modified genes to wild-type populations with possible adverse consequences. Such concerns have slowed the acceptance of GMOs, and have been used to ban the sale of GMOs in certain countries. How can synthetic biology help solve these problems? Containment is one obvious answer and we consider this now.
Early Containment Schemes
Through the years, various schemes have been proposed for containing GMOs. For example, both the use of auxotrophic strains and the induction of genes encoding self-toxins have been attempted with GMOs, but for both strategies, successful and reliable implementation has been a problem. Recall that an auxotroph is a mutant derivative of a microbial species that has a nutritional requirement; without the nutrient, the auxotroph cannot grow (Section 9.1). This dependency is the theory behind the proposal to use auxotrophs to contain GMOs. However, in nature, auxotrophic strains can often survive by cross-feeding off the metabolites of other organisms; in addition, the possibility always remains that an auxotrophic GMO could revert to the wild type by back mutation and lose its nutritional dependencies.
In Section 8.12 we saw how certain bacteria produce a growth-inhibiting toxin that slows cell growth to help ensure survival of the population under stressful conditions. This has also been explored as a mechanism for biocontainment. That is, if a GMO were to escape confinement within a bioreactor (where all growth conditions are ideal for production of the desired product), the toxin would kick in and trigger the dormant state, from which the escaped GMO could not recover. However, in nature, where a cell has to compete not only with cells of its own kind but also with other microbial species, survival strongly selects for toxin gene mutations; if such a mutation occurred quickly, the toxin system could be disarmed and the GMO might survive. Similarly to modifying bacteria for toxin production, a modified bacterial strain can be rewired to produce a lysis protein to control population levels, as we discussed in Section 12.11. However, a subset of cells will always be out of sync during expression of the lysis protein and persist to repopulate (Figure 12.39a).
Neither the auxotroph nor the toxin and lysis approaches adequately solve the problem of GMO containment. Moreover, none of these mechanisms address the possibility of engineered DNA being released through industrial waste streams and finding its way into other microorganisms by horizontal gene transfer (Chapter 9). Thus, to more thoroughly and safely address the containment problem, genetic engineers have tapped into synthetic biology itself to devise novel methods of controlling GMOs in the environment, and we consider one now.
Rewiring the Genetic Code
A novel approach to prevent genetically modified bacteria from surviving outside of their bioreactor or other containment is to recode the genome of the GMO so the bacterium can only grow if supplied with a synthetic amino acid (Figure 12.46). This recoding results not only in biocontainment, but also in the ability to make novel proteins not found anywhere in nature. This requires rewiring the organism’s genetic code and translational machinery (Chapter 6) to synthesize proteins containing the synthetic amino acid; a significant feat, but synthetic biology has already accomplished it.
Figure 12.46 Recoding and control of genetically modified *Escherichia coli*.

(a) The chromosome of an E. coli cell is genetically modified (recoded) to replace all TAG stop codons with TAA stop codons. The recoded E. coli stably maintains a vector expressing a tRNA with an AUC anticodon and an aminoacyl-tRNA synthetase (Figure 6.32) that charges the tRNA with a synthetic amino acid (sAA). (b) Control of cell growth by inserting a UAG codon into mRNAs of essential proteins. If the synthetic amino acid is added to the growth medium, the recoded E. coli will translate functional essential proteins. If the synthetic amino acid is not present, an uncharged tRNA will bind to UAG codons engineered into essential proteins. This results in truncated essential proteins and ultimately cell death.
In the first step of recoding a cell, scientists replaced all the TAG (UAG on the RNA) stop codons associated with open reading frames on the Escherichia coli chromosome with the TAA (UAA on the RNA) stop codon. They also deleted the gene for release factor 1 that terminates translation when the ribosome encounters a UAG on the mRNA. Because this manipulation placed an alternative stop codon at the end of genes that had TAG codons, proteins of the correct length are still produced during translation and the recoded cell grows normally. But this genetic manipulation also freed up the UAG stop codon to be reassigned another translational function.
To engineer dependence on a synthetic amino acid, genes for an aminoacyl-tRNA synthetase (Section 6.8) that recognizes the synthetic amino acid (sAA) and a corresponding tRNA with an AUC anticodon were expressed from a vector (Figure 12.46a). This resulted in tRNAs with the AUC anticodon carrying the sAA. A set of essential genes was then modified to contain the TAG codon in positions where incorporation of the sAA would not affect protein activity. Thus for the recoded bacterium to translate mRNAs possessing the UAG codon, the cell must be fed the artificial amino acid (Figure 12.46a). If the growth environment does not have the sAA (as would be the case if the GMO escaped to nature), uncharged tRNAs will enter into the ribosome when a UAG codon is encountered; if this occurs during the translation of an essential protein, translation will stall and a truncated (and nonfunctional) protein will result (Figure 12.46b). This will lead to death of the cell and puts ultimate control of the recoded GMO into human hands. Because the TAG codon was placed in three essential genes, it is extremely unlikely that sufficient mutations could arise to remove the GMO’s dependence on the presence of the sAA.
While this strategy for biocontainment is dependent on the incorporation of the synthetic amino acid in regions of the protein that do not affect activity, synthetic biologists are using the same strategy to make proteins with novel activities and characteristics. By using amino acids with unique binding properties, there is no limit to the type of “alien” proteins that can be produced by synthetic biology. Some practical examples include modified drugs that are assimilated more easily or are less toxic, and improvements in the catalysis or specificity of enzymes for use in biotechnology. In addition to rewriting the genetic code, synthetic biology can also harness the CRISPR genome editing system to modify organisms to prevent their unwanted spread.
Continued advances in synthetic biology and the control of GMOs will not only allow for more widespread use of these organisms for synthesizing desired products and therapeutic agents in carefully controlled production settings but may also trigger the more extensive use of GMOs in general to solve urgent problems that remain in medicine, agriculture, and the environment.
Check Your Understanding
Why is the use of auxotrophy not a good method for controlling the growth of a genetically modified organism?
How can a tRNA be engineered to encode for a synthetic amino acid?
Why is it unlikely that GMOs recoded to depend on a synthetic amino acid will mutate to no longer depend on the exogenously supplied synthetic amino acid?
Chapter Review
Go to Mastering Microbiology for videos, animations, practice tests, and more.
I Tools of the Genetic Engineer
12.1 The polymerase chain reaction is a procedure for amplifying DNA in vitro and employs heat-stable DNA polymerases. This amplified DNA is often used for cloning purposes and can be visualized by gel electrophoresis. Complementary nucleic acid sequences may be detected by hybridization.
Q Describe the basic principles of gene amplification using the polymerase chain reaction (PCR). How have thermophilic and hyperthermophilic microbes simplified the use of PCR?
12.2 The isolation of a specific gene or region of a chromosome by molecular cloning is done using a cloning vector. Plasmids are useful cloning vectors because they are easy to isolate and purify and are often able to multiply to high copy numbers in bacterial cells. The choice of a cloning host depends on the final application. In many cases the host can be a bacterium, but in others, it is essential that the host be a eukaryote.
Q How does the insertional inactivation of β-galactosidase (LacZ) allow the presence of foreign DNA in a plasmid vector such as pUC19 to be detected?
12.3 Many cloned genes are not expressed efficiently in a foreign host. Expression vectors have been developed that both increase transcription of the cloned gene and control the level of transcription. To achieve very high levels of expression of eukaryotic genes in bacteria, the expressed gene must be free of introns. This can be accomplished by synthesizing cDNA from the mature mRNA encoding the protein of interest or by making an entirely synthetic gene. Protein fusions are often used to stabilize or solubilize the cloned protein.
Q What is the significance of reverse transcriptase in the cloning of animal genes for expression in bacteria?
12.4 Synthetic DNA molecules of desired sequence can be made in vitro and used to construct a mutated gene directly or to change specific base pairs within a gene by site-directed mutagenesis. Also, genes can be disrupted by inserting DNA fragments, called cassettes, into them, generating knockout mutants.
Q What does site-directed mutagenesis allow you to do that normal mutagenesis does not?
12.5 Reporter genes are genes whose products are easy to assay or detect. They are used to simplify and increase the speed of genetic analysis. In gene fusions, segments from two different genes, one of which is usually a reporter gene, are spliced together.
II Making Products from Genetically Engineered Microbes: Biotechnology
12.6 The first human protein made commercially using engineered bacteria was human insulin. Recombinant bovine somatotropin is sometimes used in the United States to increase milk yield in dairy cows.
Q What classes of mammalian proteins are produced by biotechnology? How are the genes for such proteins obtained?
12.7 Genetic engineering can make plants resistant to disease and improve product quality. The Ti plasmid of the bacterium Agrobacterium tumefaciens can transfer DNA into plant cells. Genetically engineered commercial plants are called genetically modified organisms (GMOs).
Q What is the Ti plasmid and how has it been of use in genetic engineering?
12.8 Many recombinant vaccines have been produced or are under development. These include live recombinant, vector, and subunit vaccines. Properties associated with pathogens can be used to develop cancer-treating therapeutic agents.
Q What is a subunit vaccine and why are subunit vaccines considered a safer way of conferring immunity to viral pathogens than attenuated virus vaccines?
12.9 Genes for useful products may be cloned directly from DNA or RNA in environmental samples without first isolating the organisms that carry them. In pathway engineering, genes that encode the enzymes for a metabolic pathway are assembled. These genes may come from one or more organisms, but the engineering must achieve regulation of the coordinated sequence of expression required in the pathway.
Q How has metagenomics been used to find novel useful products?
12.10 While select microorganisms can produce biofuels in small amounts, others can be modified to produce various biofuels through pathway engineering. This modification often requires genes from multiple microorganisms. New tools for genetically manipulating microalgae have also been developed to facilitate biofuel production.
Q What do microalgae produce that can be chemically or enzymatically converted to biodiesel?
III Synthetic Biology and Genome Editing
12.11 Instead of modifying or improving a single existing pathway, synthetic biology focuses on engineering novel biological systems by linking known biological components together in various combinations. These modifications can result in the production of high-value products and synchronized biofactories.
**Q How does synthetic biology differ from engineering of Escherichia coli to produce indigo?**
12.12 Through the use of recombineering techniques, linear fragments of DNA can be synthesized and assembled into a genome. These synthesized genomes can then be transplanted into bacterial cells, displacing the parent genome and thus creating a synthetic cell.
Q How has the creation of a synthetic cell allowed microbiologists to probe the minimal genome required for autonomous life?
12.13 Not only can CRISPR systems be used as a prokaryotic immune system, they can also be modified to edit the genomes of eukaryotes. CRISPR requires a guide RNA to locate the genes to be cut and Cas proteins to cut the DNA. The resulting double-stranded breaks are suitable for the insertion of foreign DNA.
Q How has CRISPR editing technology been applied to targeting virus-infected eukaryotic cells?
12.14 With the advancements in genetic engineering, methods for controlling genetically modified organisms are imperative. One promising method is the use of synthetic biology to recode organisms for dependence on the presence of synthetic amino acids.
Q What are some mechanisms for controlling a genetically modified organism other than making it dependent on synthetic amino acids?
Application Questions
Suppose you have just determined the DNA base sequence for an especially strong promoter in Escherichia coli and you are interested in incorporating this sequence into an expression vector. What steps would you take and which tools would you use to be sure that this promoter actually worked as expected in its new location?
You have just discovered a protein in mice that may be an effective cure for cancer, but it is present only in tiny amounts. Describe the steps you would use to produce this protein in therapeutic amounts. Which host would you want to clone the gene into and why? Which host would you use to express the protein in and why?
Describe how you could use a soil sample and metagenomics to isolate a new antifungal protein that protects wheat from fungal infection. What steps could you take in the laboratory to modify this antifungal protein to be more effective against a broad range of fungal plant pathogens? Suppose you want to create genetically modified wheat that expresses your improved antifungal protein; what general steps would you take to introduce the foreign gene using CRISPR technology?
Describe how you could recode Escherichia coli to produce novel proteins containing more than the standard 22 amino acids.
Chapter Glossary
to decrease or eliminate the virulence of a pathogen or virus Bacterial artificial chromosome (BAC)
a circular artificial chromosome with a bacterial origin of replication Biotechnology
the use of organisms, typically genetically altered, in industrial, medical, or agricultural applications Cassette mutagenesis
creating mutations by the insertion of a DNA cassette Complementary DNA (cDNA)
DNA made from an RNA template during the reverse transcription PCR (RT-PCR) procedure DNA cassette
an artificially designed segment of DNA that usually carries a gene for resistance to an antibiotic or some other convenient marker and is flanked by convenient restriction sites Expression vector
a cloning vector that contains the necessary regulatory sequences to allow transcription and translation of cloned genes Gel electrophoresis
a technique for separation of nucleic acid molecules by passing an electric current through a gel made of agarose or polyacrylamide Gene disruption
(also called gene knockout) the inactivation of a gene by insertion of a DNA fragment that interrupts the coding sequence Gene fusion
a structure created by joining together segments of two separate genes, in particular when the regulatory region of one gene is joined to the coding region of a reporter gene Genetically modified organism (GMO)
an organism whose genome has been altered using genetic engineering; the abbreviation GM is also used in terms such as GM crops and GM foods Genetic engineering
the use of in vitro techniques in the isolation, alteration, and expression of DNA or RNA and in the development of genetically modified organisms Green fluorescent protein (GFP)
a protein that fluoresces green and is widely used in genetic analysis Heterologous expression
the transcription and translation of a gene or genes from one organism in a different organism Hybridization
the joining of two single-stranded nucleic acid molecules by complementary base pairing to form a double-stranded hybrid DNA or DNA–RNA molecule Molecular cloning
the isolation and incorporation of a fragment of DNA into a vector where it can be replicated Northern blot
a hybridization procedure where RNA is the target and DNA or RNA is the probe Nucleic acid probe
a strand of nucleic acid that can be labeled and used to hybridize to a complementary molecule from a mixture of other nucleic acids Operon fusion
a gene fusion in which a coding sequence that retains its own translational signals is fused to the transcriptional signals of another gene Pathway engineering
the assembly of a new or improved biochemical pathway using genes from one or more organisms Polymerase chain reaction (PCR)
the artificial amplification of a DNA sequence by repeated cycles of strand separation and replication Polyvalent vaccine
a vaccine that immunizes against more than one disease Protein fusion
a gene fusion in which two coding sequences are fused so that they share the same transcriptional and translational start sites Recombinant DNA
a DNA molecule containing DNA originating from two or more sources Reporter gene
a gene used in genetic analysis because the product it encodes is easy to detect Restriction enzyme
an enzyme that recognizes a specific DNA sequence and then cuts the DNA; also known as a restriction endonuclease Shuttle vector
a cloning vector that can replicate in two different organisms; used for moving DNA between unrelated organisms Site-directed mutagenesis
the construction in vitro of a gene with a specific mutation Southern blot
a hybridization procedure where DNA is the target and RNA or DNA is the probe Subunit vaccine
a vaccine that contains only a specific protein or two from a pathogen Synthetic biology
arranging biobricks to form new genetic elements for insertion into microbes or higher organisms T-DNA
the segment of the Agrobacterium tumefaciens Ti plasmid that is transferred into plant cells Ti plasmid
a conjugative plasmid in the bacterium Agrobacterium tumefaciens that can transfer genes into plants Transgenic organism
a plant or an animal with foreign DNA inserted into its genome Vector
(as in cloning vector) a DNA molecule that is replicated by a host cell and used to carry cloned genes or other DNA segments for genetic engineering Vector vaccine
a vaccine made by inserting genes from a pathogenic virus into a relatively harmless carrier virus Yeast artificial chromosome (YAC)
an artificial chromosome with a yeast origin of replication and a centromere sequence