19 Taking the Measure of Microbial Systems
## Chapter 19 Taking the Measure of Microbial Systems
I Culture-Dependent Analyses of Microbial Communities
II Culture-Independent Microscopic Analyses of Microbial Communities
III Culture-Independent Molecular Analyses of Microbial Communities
Touring Microbial Biogeography Using Combinatorial Imaging
Virtually all microorganisms exist as parts of complex communities that interact through metabolic cooperation. Since complex metabolic interactions are not easily resolved, the microscope is an essential tool for first identifying possible cooperation based on colocalization of different species. However, even the simplest of microbial communities is composed of tens if not hundreds of different species. How it is possible to visualize the distribution of individual species in such a mixture?

As we will see in this chapter, microscopic identification of species commonly uses fluorescence in situ hybridization (FISH), a technique in which cells of individual species are identified by hybridization of fluorescent DNA probes to ribosomal RNA sequences unique to each species. However, the method can visualize only a few different species simultaneously, being limited by the number of dyes that fluoresce in different colors. A variation on standard FISH technology that circumvents this limitation is called CLASI-FISH (combinatorial labeling and spectral imaging–FISH), which can image more than 100 different species simultaneously. CLASI-FISH hybridizes each cell with a combination of probes specific for that species but labeled with different fluorescent dyes, giving each cell a unique fluorescent spectral signature. Since the resulting fluorescence at each wavelength is a linear combination of emissions from each fluorescent dye, statistical analysis can determine what combination of dyes produced the emission spectrum and therefore identify the contributing species.
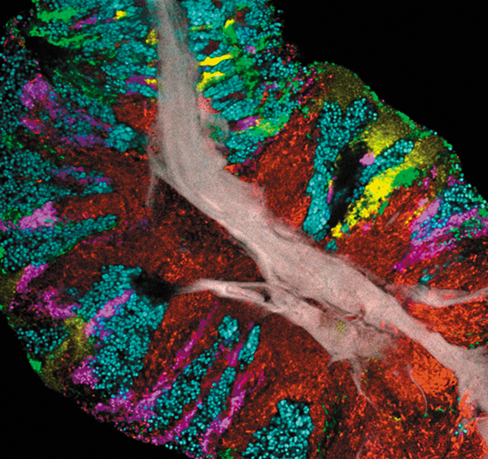
The photo shown here was taken from a scraping of the human tongue (the specimen is about 140 μm in diameter, left to right), showing a remarkable organization of microbial species that almost certainly reflects metabolic interactions that can now be further explored using the powerful tools we discuss in this chapter. Brown in the photo is human tissue; the bacteria are: red, Actinomyces spp.; green, Streptococcus spp.; blue, Rothia spp.; yellow, Neisseria spp.; and magenta, Veillonella spp.
Source: Welch, J.L.M., Dewhirst, F.E., and Borisy, G.G. 2019. Biogeography of the oral microbome: The site-specialist hypothesis. Annu. Rev. Microbiol. 73. doi: 10.1146/annurev-micro-090817-062503.
We now begin a new unit devoted to microorganisms in their natural habitats. We learned in Chapter 1 that microbial communities consist of cell populations living in association with other populations in nature. The science of microbial ecology is focused on how microbial populations assemble to form communities and how these communities interact with each other and their environments.
The major components of microbial ecology are biodiversity and microbial activity. To study biodiversity, microbial ecologists must identify and quantify microorganisms in their habitats. Knowing how to do this is often helpful for isolating organisms of interest as well, which is another goal of microbial ecology. To study microbial activity, microbial ecologists must measure the metabolic processes that microorganisms carry out in their habitats. In this chapter we consider modern methods for assessing microbial diversity and activity. Chapter 20 will outline the basic principles of microbial ecology and examine the types of environments that microorganisms inhabit. Chapters 21, 22, 23 and 24 will complete our coverage of microbial ecology by exploring nutrient cycles, applied microbiology, and the role microbes play in symbiotic associations with other life forms, including humans.
We begin with the microbial ecologist’s toolbox, which includes a collection of powerful tools for dissecting the structure and function of microbial communities in relation to their natural habitats.
I: Culture-Dependent Analyses of Microbial Communities
I: Culture-Dependent Analyses of Microbial Communities
I Culture-Dependent Analyses of Microbial Communities
Major advances in molecular, microscopic, and analytical methods have revealed important properties of microorganisms as they exist in nature. However, to fully characterize a microbe, there is no substitute for isolating it in pure culture. In addition to traditional isolation methods, single-cell and high-throughput cultivation methods have greatly advanced success in culturing the “uncultured majority” of microorganisms.
The vast majority of microorganisms, more than 99% of all species by most estimates, have never been grown in laboratory cultures. To date, about 17,000 species of Archaea and Bacteria have been formally described. By contrast, culture-independent molecular diversity surveys (Sections 19.4, 19.5, 19.6, 19.7 and 19.8) indicate that many millions—possibly even trillions—of species exist in nature and have yet to be cultured and formally described. This recognition has stimulated the development of new methods for isolating microbes from nature in order to establish pure cultures. Even though a host of sophisticated methods are available for studying microbes in their native environments, culturing a microorganism remains the only way to fully characterize its properties and predict its impact on its environment.
In the first part of this chapter we cover the enrichment approach, a time-honored and useful method for isolating microorganisms from nature, but a method with significant limitations. Enrichment is based on culturing in a selective growth medium, and thus the tools and methods used in this approach are considered culture-dependent analyses. As we will see, considerable progress has been made in culturing the more elusive microorganisms in natural populations by using robotics and associated microfabrication technology to establish large numbers of enrichment cultures that can be monitored simultaneously. In the second and third parts of this chapter we consider culture-independent analyses, techniques that can tell us much about the structure and function of microbial communities in the absence of actual laboratory cultures. In the final part of this chapter, we consider methods for measuring microbial activities in nature and linking them to specific organisms. Collectively, these methods allow the microbial ecologist to ask both “Who is there?” and “What are they doing?”
19.1 Enrichment Culture Microbiology
For an enrichment culture, a medium and a set of incubation conditions are established that are selective for the desired organism and counterselective for undesired organisms. Effective enrichment cultures duplicate as closely as possible the resources and conditions of a particular ecological niche. Hundreds of different enrichment strategies have been devised, and Tables 19.1 and 19.2 summarize some simple and direct ones.
Table 19.1 Some enrichment culture methods for phototrophic bacteria (main C source, CO2)

Table 19.2 Some enrichment culture methods for nonphototrophic bacteriaa


aAll media must contain an assortment of mineral salts including N, P, S, Mg2+, Mn2+, Fe2+, Ca2+, and other trace elements (Sections 4.1, 4.2). Certain organisms may have requirements for vitamins or other growth factors. This table is a general overview of enrichment methods focused on substrates and atmospheric conditions and has not considered the effects that temperature, pH and salinity can have on enrichment culture outcomes.
bComammox is the complete oxidation of ammonia all the way to nitrate (Section 20.3).
Inocula
Successful enrichment requires an appropriate inoculum containing the organism of interest. Thus, the making of an enrichment culture begins with collecting a sample from the appropriate habitat to serve as the inoculum (Tables 19.1 and 19.2). Enrichment cultures are established by placing the inoculum into selective media and incubating under specific conditions. In this way, many common microbes can be isolated. For example, the great Dutch microbiologist Martinus Beijerinck, who conceptualized the enrichment culture technique (Section 1.13), used enrichment cultures to isolate the nitrogen-fixing bacterium Azotobacter (Figure 19.1). Because Azotobacter is a rapidly growing bacterium capable of N2 fixation in air (Sections 3.12 and 15.9), enrichment using media devoid of fixed nitrogen (for example, ammonia or nitrate) and incubation in air selects strongly for this bacterium and its close relatives. Non-nitrogen-fixing bacteria and anaerobic nitrogen-fixing bacteria are counterselected in this technique.
Figure 19.1 The isolation of *Azotobacter*.

Selection for aerobic nitrogen-fixing bacteria usually results in the isolation of Azotobacter or its relatives. The selective basis of the enrichment is the absence of fixed nitrogen (NH4 + in this case) in the culture medium in the upper flask. Thus the medium selects from the microbial community those species that can fix N2 aerobically, of which Azotobacter is one of the most rapidly growing. See Section 1.13 and Figure 1.37 for more on the historical importance of Azotobacter.
Enrichment Culture Outcomes
For success with enrichment cultures, attention to both the culture medium and the incubation conditions is important. That is, the resources (nutrients) and conditions (temperature, pH, osmotic considerations, aerobic or anaerobic, and the like) must closely mimic those of the habitat to offer the best chance of obtaining the organism of interest (Table 20.1).
Some enrichment cultures yield nothing. This may be because an organism capable of growing under the enrichment conditions established is absent from the habitat. Alternatively, even though the organism of interest exists in the habitat sampled, the resources and conditions employed in the enrichment may simply be incompatible with its growth. Thus, enrichment cultures can yield a firm positive conclusion (that is, that an organism with certain capacities exists in a particular environment because it was enriched) but never a firm negative conclusion (that such an organism is not present because the enrichment failed). Moreover, the isolation of the desired organism from an enrichment culture says nothing about the abundance or ecological significance of the organism in its habitat. A positive enrichment proves only that the organism was present in the sample, and in practice, this can result from even a single viable cell.
Mastering Microbiology
Art Activity: Figure 19.1 The isolation of Azotobacter
The Winogradsky Column
The Winogradsky column is an artificial microbial ecosystem and a long-term source of various bacteria for enrichment cultures. Winogradsky columns have been used to isolate phototrophic purple and green bacteria, sulfate-reducing bacteria, and many other anaerobes. Named for the famous Russian microbiologist Sergei Winogradsky (Section 1.13), the column was first used by Winogradsky in the late nineteenth century in his classic studies of soil microorganisms.
Mastering Microbiology
Art Activity: Figure 19.2a The Winogradsky column

A Winogradsky column is prepared by filling a glass cylinder about half full with organic-rich, preferably sulfidic mud into which carbon substrates have been mixed. The substrates determine which organisms are enriched. Fermentative substrates, such as glucose, that can lead to acidic conditions and excessive gas formation (which can create gas pockets that disrupt the enrichment culture and let in air) are avoided. The mud is supplemented with small amounts of calcium carbonate (CaCO3) as a buffer and gypsum (CaSO4) as a source of sulfate. The mud is packed tightly in the cylinder, taking care to avoid trapping air, and then covered with lake, pond, or ditch water (or seawater if it is a marine column). The top of the cylinder is covered to prevent evaporation, and the container is placed near a window that receives diffuse sunlight for a period of months.
In a typical Winogradsky column, a diverse community of microbes develops (**Figure 19.2*a***). Algae and cyanobacteria develop quickly in the upper portions of the water column; by producing O2 these organisms help to keep this zone of the column oxic much as they do in the upper zones of a lake. Fermentative processes in the mud lead to the production of organic acids, alcohols, and H2, suitable substrates for sulfate-reducing bacteria (Section 15.11). Hydrogen sulfide (H2S) from the sulfate reducers triggers the development of purple and green sulfur bacteria (anoxygenic phototrophs, Sections 14.3 and 15.4, 15.5, 15.6, 15.7 and 15.8) that use sulfide as a photosynthetic electron donor. These organisms typically grow in patches in the mud on the sides of the column but may bloom in the water itself if oxygenic phototrophs are scarce (Figure 19.2b). The pigmented cells of the anoxygenic phototrophs can be sampled with a pipette for microscopy, isolation, and characterization (Table 19.1).
Figure 19.2 The Winogradsky column.

(a) Schematic view of a typical column used to enrich phototrophic bacteria. The column is incubated in a location that receives subdued sunlight. Anoxic decomposition leading to SO4 2− reduction creates the gradient of H2S. (b) Photo of Winogradsky columns that have remained anoxic up to the top; each column had a bloom of a different phototrophic bacterium. Left to right: Thiospirillum jenense, Chromatium okenii, both of which are purple sulfur bacteria, and Chlorobium limicola (green sulfur bacterium).
Winogradsky columns have been used to enrich both aerobic and anaerobic Bacteria and Archaea. Besides supplying a ready source of inocula for enrichment cultures, columns can also be supplemented with a specific compound to enrich an organism in the inoculum that can degrade it. Once a crude enrichment has been established in the column, culture media can be inoculated for the isolation of pure cultures, as discussed in Section 19.2.
Enrichment Bias
Although the enrichment culture technique is quite useful and still widely practiced, there exists a bias, and sometimes a very severe bias, in the outcome of enrichments. This bias is typically most profound in liquid enrichment cultures where the most rapidly growing organism(s) for the chosen set of conditions dominate. However, using molecular techniques to be described later, we now know that the most rapidly growing organisms in laboratory cultures are often only minor components of the microbial community rather than the most abundant and ecologically relevant organisms carrying out the process of interest. This could be for several reasons including the fact that the levels of resources available in laboratory cultures are typically much higher than those in nature, and the conditions in the natural habitat, including both the types and proportions of different organisms present as well as the physical and chemical conditions, are nearly impossible to reproduce and maintain for long periods in laboratory cultures.
This problem of enrichment bias can be demonstrated by comparing the results obtained in dilution cultures (Section 19.2) with classical liquid enrichment. Dilution of an inoculum followed by liquid enrichment or plating often yields different organisms than liquid enrichments established with the same but undiluted inocula. It is thought that dilution of the inoculum eliminates quantitatively insignificant but rapidly growing “weed” species, allowing development of organisms that are more abundant in the community but slower growing. Dilution of the inoculum is thus a common practice in enrichment culture microbiology today. As discussed below, the problem of overgrowth by “weed” species can also be circumvented by physical isolation of the desired organism before introducing it into a growth medium. This can be accomplished by dilution and a variety of classical isolation procedures that we turn to in the next section. However, more recently, sophisticated methods have been developed to physically isolate single cells of interest (or a single type of cells) and place them in a growth medium that is free of undesired cells. We consider these techniques in Section 19.3. Finally, as we will discuss later in this chapter, culture-independent molecular methods have most clearly revealed the limitations of cultivation in capturing the full microbial diversity of most environments.
Check Your Understanding
Describe the enrichment strategy behind Beijerinck’s isolation of Azotobacter.
Why is sulfate (SO4 2−) added to a Winogradsky column?
What is enrichment bias? How does dilution reduce enrichment bias?
19.2 Classical Procedures for Isolating Microbes
Once a positive enrichment culture has been obtained, the next step is typically to attempt to get the enriched organism in pure culture—one containing a single kind of microorganism. Pure cultures are valuable because genomes can be quickly isolated and analyzed and experiments can be done under controlled laboratory conditions to clearly define the physiology of the isolate. Pure cultures have been studied since the days of Robert Koch (Section 1.12), and we considered some of these methods earlier (Section 4.2).
Agar Dilution Tubes and the Most-Probable-Number Technique
Common isolation procedures include the streak plate, agar dilution, and liquid dilution. For organisms that form colonies on agar plates, the streak plate is quick, easy, and the method of choice (**Figure 19.3*a***); if a well-isolated colony is selected and restreaked several successive times, a pure culture is usually obtained. With proper incubation facilities (for example, anoxic jars or anoxic chambers for anaerobes, Section 4.16), it is possible to purify both aerobes and anaerobes on agar plates by the streak plate method.
Figure 19.3 Pure culture methods.

(a) Organisms that form distinct colonies on plates are usually easy to purify. (b) Colonies of phototrophic purple bacteria in agar dilution tubes; the molten agar was cooled to approximately 45 °C before inoculation. A dilution series was established from left to right, eventually yielding well-isolated colonies. The tubes were sealed with a 1:1 mixture of sterile paraffin and mineral oil to maintain anaerobiosis.
In the agar dilution tube method, a mixed culture is diluted in tubes of molten agar medium, resulting in colonies embedded in the agar. This method is useful for purifying anaerobic organisms such as phototrophic sulfur bacteria and sulfate-reducing bacteria from samples taken from Winogradsky columns or other sources. A culture is purified by successive dilutions of cell suspensions in tubes of molten agar medium (Figure 19.3b, Figure 15.28g). Repeating this procedure using a colony from the highest-dilution tube as inoculum for a new set of dilutions usually yields pure cultures. A related procedure called the roll tube method uses tubes containing a thin layer of agar on their inner surface. The agar can then be streaked for isolated colonies. Because the tubes can be flushed with an oxygen-free gas during streaking, the roll tube method is primarily used for the isolation of anaerobic microbes.
Another purification procedure is the serial dilution of an inoculum in a liquid medium until the final tube in the series shows no growth. When a 10-fold serial dilution is used, for example, the last tube showing growth should have originated from ten or fewer cells. Besides being a method for obtaining pure cultures, serial dilution techniques are widely used to estimate viable cell numbers in the most-probable-number (MPN) technique (Figure 19.4). MPN methods have been used for estimating the numbers of microorganisms in foods, wastewater, and other samples in which cell numbers need to be assessed routinely. An MPN count of a natural sample can be done using highly selective media and incubation conditions to target one or a small group of organisms or a particular pathogen. Alternatively, a count can be done using complex media to get a general estimate of viable cell numbers (but see Section 4.4 for a caveat that applies to such estimates). Use of several replicate tubes at each dilution improves accuracy of the final MPN obtained.
Figure 19.4 Procedure for a most-probable-number (MPN) analysis.

Growth in the 10−4 but not the 10−5 dilution means that cell numbers were at least 104 cells/ml in the sample used for inoculation. Because particle-attached microorganisms can skew numbers significantly, gentle methods to disassociate microorganisms from particles are often used prior to dilution. In addition, each dilution tube is mixed thoroughly before removing a sample for the next dilution.
Criteria for Culture Purity
Regardless of the methods used to purify a culture, once a putative pure culture has been obtained, it is essential to verify its purity. This is typically done through a combination of (1) microscopy, (2) observation of colony characteristics on plates or in dilution tubes, and (3) tests of the culture for growth in other media. In the latter, it is important to test the culture for growth in media and under growth conditions in which the desired organism is predicted to grow poorly or not at all but in which contaminants will grow vigorously. In the final analysis, the microscopic observation of a single morphological type of cell that displays uniform staining characteristics (for example, in a Gram stain) coupled with uniform colony characteristics and the absence of contamination in growth tests with various culture media is strong evidence that a culture is a pure (axenic) culture.
Certain molecular methods described in this chapter for characterizing natural microbial communities can also be applied to the verification of culture purity. However, these techniques are complementary and do not substitute for the more fundamental observations of culture characteristics and cellular morphology.
Check Your Understanding
What is a pure culture and why is obtaining one useful in microbial ecology?

How does the agar dilution method differ from streaking to obtain isolated colonies?
Why would microscopic examination alone not be sufficient to establish that a culture is pure?
19.3 Selective Single-Cell Isolation: Laser Tweezers, Flow Cytometry, Microfluidics, and High Throughput Methods
19.3 Selective Single-Cell Isolation: Laser Tweezers, Flow Cytometry, Microfluidics, and High Throughput Methods
19.3 Selective Single-Cell Isolation: Laser Tweezers, Flow Cytometry, Microfluidics, and High-Throughput Methods
The problem of enrichment bias has fueled the development of new methods for culturing microbes from nature. These advancements have emerged from the understanding that every microbe has a fundamental niche and a realized niche. The fundamental niche refers to the range of environments in which a species will be sustained when it is not resource-limited, such as may result from competition with other species. By contrast, the realized niche refers to the range of natural environments supporting a species when it is confronted with factors such as resource limitation, predation, and competition from other species.
Establishing laboratory conditions that fall within the fundamental niche may be sufficient to support an organism once it is in pure culture but may fail to selectively enrich the same organism from a natural sample. Because the realized niche of most microorganisms is unknown, there has been an increasing emphasis on developing methods that physically isolate single cells into separate compartments free from competition with other microbes. These include both manual and robotic methods that function to sort individual cells from an environmental sample, and we consider these methods now.
Laser Tweezers and Flow Cytometry
Laser tweezers consist of an inverted light microscope equipped with a strongly focused infrared laser and a micromanipulation device. Trapping a single cell is possible because the laser beam creates a force that pushes down on a microbial cell (or other small object) and holds it in place (**Figure 19.5*a***). Then when the laser beam is moved, the trapped cell moves along with it. If a mixed sample is in a capillary tube, a single cell can be optically trapped and moved away from contaminating organisms (Figure 19.5b). The cell can then be isolated by breaking the tube at a point between the cell and the contaminants and flushing the cell into a small tube of sterile medium. Laser tweezers, when coupled with staining techniques that identify particular organisms (Sections 19.4 and 19.5), can be used to select organisms of interest from a mixture for purification and further laboratory study.
Figure 19.5 The laser tweezers for the isolation of single cells.

(a) Mechanism by which individual cells can be isolated. (b) Once a cell has been isolated in a capillary tube, it can be tested for subsequent growth in pure culture.
Flow cytometry is a technique for counting and examining a mixture of cells by suspending them in a stream of fluid and passing them through an electronic detector that sorts them according to defined criteria; for example, by cell size, shape, or fluorescent properties. This ability makes cell sorting useful not only for isolating single cells but also for enriching a particular cell type from a mixture. Cell sorters can deposit individual cells into wells of a microtiter plate where each well contains the same growth medium or a slightly different growth medium. Because the growth requirements of some organisms include organic compounds and metabolites produced by other organisms that share their environment, addition of filter-sterilized source water (for aquatic organisms) or soil water extract (for soil organisms) can be used to supplement the media tested. Each well in the microtiter plate can then be monitored for growth or some other property either manually or using robotic methods (high-throughput culture, see next subsection). We explore the mechanism and uses of flow cytometry in more detail in Section 19.12 (see Figure 19.42).
High-Throughput Culture and Microfluidic Devices
Continuing innovations in single-cell isolation methodology have spawned high-throughput culturing methods and related methods for use on an even smaller scale. High-throughput methods require dilution (or cell sorting) of a sample to yield a single cell in each well of a microtiter plate (Figure 19.6). From there, each well is robotically monitored over time for cell growth or a specific target gene. High-throughput methods allow the experimenter to test many alternative sets of resources and growth conditions systematically in an attempt to replicate the realized niche or, alternatively, to allow the organism to occupy its fundamental niche by relieving it from competition. Microtiter wells that are positive for cell growth or a target gene of interest identify the acceptable resources and conditions for growth of a particular microbe and supply valuable clues for the design of laboratory culture media to obtain its growth in pure culture.
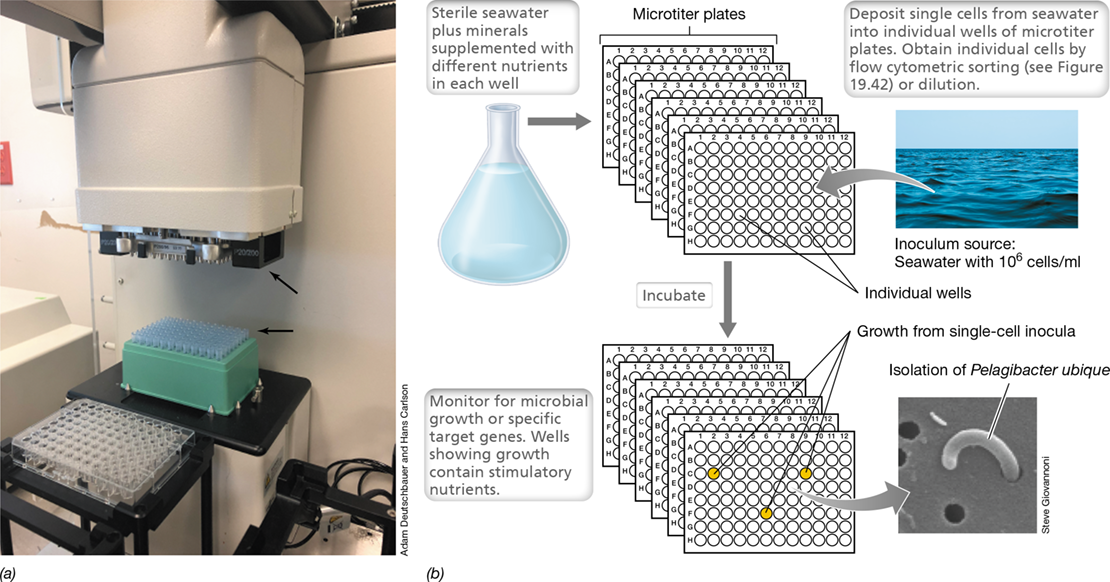
Figure 19.6 Methodological pipeline for high-throughput cultivation of previously uncultured microorganisms.

The method shown here was used to isolate the marine bacterium Pelagibacter ubique. (a) Robotic system for high-throughput multiplexed pipetting of growth medium into microtiter plates (arrows point to pipette tips and robotic pipette holder). (b) Following the addition of filter-sterilized seawater and low nutrient concentrations to the individual wells, and deposition of single cells into individual wells of the microtiter plate, pure cultures of Pelagibacter and other novel marine Bacteria were obtained. Pelagibacter is the most abundant bacterium in the open oceans (Section 20.12).
High-throughput cultivation has shown increasing success in isolating unique bacteria. For example, high-throughput methods were used for the isolation of one of the most abundant bacteria on Earth, the small marine planktonic bacterium Pelagibacter ubique (Figure 19.6). This bacterium thrives on the very dilute pool of dissolved organic matter present in the open oceans and eluded classical enrichment methods for years. But with high-throughput technology, this ecologically important bacterium was brought into laboratory culture where its biology could be studied in more detail.
Microfluidic devices carry the high-throughput concept even further by using microfabrication technology to combine channels and wells for fluid transfer and collection on a miniaturized platform. One such device is less than 10 centimeters long yet holds 3200 nanoliter-sized wells, with each well serving as a small culture vessel (Figure 19.7). An environmental sample is introduced into the microfluidic device such that each well receives a single cell. Different medium formulations can be tested, and the media supplemented with a small amount of filter-sterilized water or soil extract collected from the sampled environment (these additions may stimulate growth by providing trace nutrients missing in the culture medium).
Figure 19.7 Microfluidic platform for cultivation.

An environmental inoculum is suspended in a cultivation medium and loaded onto this microfluidic device, enabling confinement of as many as 3200 single cells in nanoliter wells to promote the growth of microcolonies. Following different periods of incubation, cultured populations are collected at the outlet and further grown under conditions demonstrated to support growth on the microfluidic device. The device is about 7 cm wide.
Mastering Microbiology
Art Activity: Figure 19.6 Methodological pipeline for high-throughput cultivation of previously uncultured microorganisms
Both cell growth and target genes can be assessed in each well of the microfluidic device; growth is assessed by direct microscopic examination of cell numbers in a well under the microscope. If insufficient cellular biomass is available for molecular characterization of specific target genes by methods such as PCR (see Section 19.6), genome amplification as described for single-cell genomics (Section 10.11) can be used prior to further analysis.
A variation on the microfluidics technique employs a microchamber device modified such that each of the tiny chambers is separated from the external environment by a membrane that traps the microbes but allows soluble nutrients to diffuse in and out. Following the introduction of a single cell into each chamber, the device is placed back into the environment from which the inoculum was obtained. Then, after incubation for a month or more, microbes that initiate growth only when incubated under the conditions and resources present in their habitats can often be isolated and subsequently propagated in the laboratory.
Although technology is rapidly advancing the art of isolating new microbes, patience is still needed in any cultivation effort, as the discovery of slow-growing or dormant organisms may require months of incubation. Also, many microbes in nature are likely adapted to extremely low nutrient concentrations and may be inhibited by levels of nutrients used to grow organisms commonly studied in the laboratory. Both high-throughput and microfluidic methods overcome these problems by their ability to separate individual cells from other cells that may release inhibitory materials and by surveying a nearly limitless variety of nutrient conditions. Currently, these methods offer the best opportunity for culturing the most interesting (and likely ecologically relevant) microorganisms from nature.
Check Your Understanding
How might you isolate a morphologically unique bacterium present in an enrichment culture in relatively low numbers?
What is meant by “high-throughput” in culturing microorganisms? How has it benefited microbiology?
II: Culture-Independent Microscopic Analyses of Microbial Communities
II: Culture-Independent Microscopic Analyses of Microbial Communities
II Culture-Independent Microscopic Analyses of Microbial Communities
The microscope has been the microbiologist’s foremost tool for studying microbial structure. Today the microscope can also assist in probing microbial diversity and activity, thanks to a suite of fluorescent techniques. These advances have greatly improved our understanding of microbial community structure and provided unprecedented insight into microbial symbiotic relationships with plants, animals, and other microbes.
Microbial ecologists quantify cells in a microbial habitat to estimate abundance of the entire community or, more specifically, relative abundances of the different species in the community. Cell stains are necessary to obtain these types of data, and we detail these methods here. Organisms in natural environments can also be detected by assaying their genes. Genes encoding either ribosomal RNA (rRNA, Section 13.11) or enzymes that support a specific physiology are the usual targets in these studies. A rapidly developing approach to the study of microbial ecology, called multi-omics, combines multiple molecular, analytical, and omics methods (Chapter 10), and is introduced in Section 19.8.
19.4 General Staining Methods
Several general staining methods are suitable for quantifying microorganisms in natural samples. Although these methods do not reveal the physiology or phylogeny of the cells, they are nonetheless reliable and widely used by microbial ecologists for measuring total cell numbers. One method also allows cell viability to be assessed.
Fluorescent Staining with Dyes That Bind Nucleic Acids or Reveal Viability
Fluorescent dyes can be used to stain microorganisms from virtually any microbial habitat. DAPI (4′, 6-diamidino-2-phenylindole) is a popular stain for this purpose, as is the dye acridine orange. There is also increasing use of SYBR Green I, a dye that confers very bright fluorescence to all microorganisms, including viruses. These stains bind to DNA and are strongly fluorescent when exposed to ultraviolet (UV) radiation (DAPI absorption maximum, 400 nm; acridine orange absorption maximum, 500 nm; SYBR Green I absorption maximum, 497 nm), making the microbial cells in the sample readily visible and easy to enumerate. Cells stained with DAPI fluoresce blue, cells stained with acridine orange fluoresce orange or greenish-orange, and cells stained with SYBR Green I fluoresce green (Figure 19.8).
Figure 19.8 Nonspecific fluorescent stains.

(a) DAPI and (b) acridine orange staining showing microbial communities inhabiting activated sludge in a municipal wastewater treatment plant. With acridine orange, cells containing low RNA levels stain green. (c) SYBR Green–stained sample of Puget Sound (Washington, USA) surface water showing green-fluorescing bacterial cells. The large cells near the center of the field are 0.8–1.0 μm in diameter.
Dyes that stain DNA are widely used for the enumeration of microorganisms in environmental, food, and clinical samples. Depending on the sample, background staining is occasionally a problem with fluorescent stains, but because these dyes specifically stain nucleic acids, they are for the most part nonreactive with inert matter. Thus, for many samples, from soil as well as aquatic sources, they can give a reasonable estimate of the cell numbers present. Staining with the brightly fluorescent SYBR Green I also provides excellent enumeration of aquatic virus populations (Section 20.13). For dilute aquatic samples, cells can be stained following collection on a membrane surface by filtration.
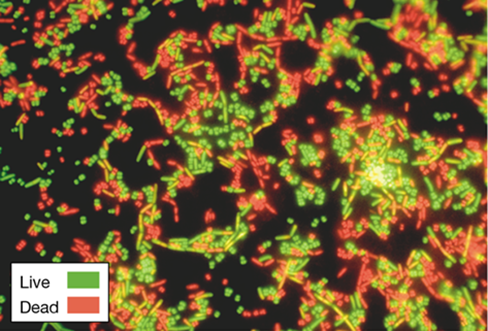
DNA staining is a nonspecific process; all microorganisms in a sample are stained. Although this may at first seem desirable, it is not necessarily so. For example, DAPI and acridine orange fail to differentiate between living and dead cells or between different species of microorganisms, so they cannot be used to assess cell viability or to track specific microorganisms in an environment. Viability staining solves one of these problems because it differentiates live cells from dead ones. Hence, viability stains yield both abundance and viability data at the same time. The basis of differentiating between live and dead cells lies not with a cell’s DNA but whether its cytoplasmic membrane is intact or not. Two dyes that fluoresce green and red are added to a sample; the green-fluorescing dye penetrates all cells, viable or not, whereas the red dye, which contains the chemical propidium iodide, penetrates only those cells whose cytoplasmic membrane is no longer intact and that are therefore dead. Thus, when viewed microscopically, green cells are scored as alive and red cells as dead, yielding an instant assessment of both abundance and viability (Figure 19.9).
Figure 19.9 Viability staining.

Live (green) and dead (red) cells of Micrococcus luteus (cocci) and Bacillus cereus (rods) stained by the LIVE/DEAD BacLight Bacterial Viability Stain.
Although useful for research that uses laboratory cultures, the live/dead staining method is not suitable for use in the direct microscopic examination of samples from many natural habitats because of problems with nonspecific staining of background materials. However, procedures have been developed to overcome this problem in analyses of aquatic environments; the water sample is filtered, and the filters are stained with the live/dead stain and examined microscopically. Thus in aquatic microbiology, live/dead staining is often used to measure the viability of cell populations in the water column of lakes or oceans, or in the flowing waters of streams, rivers, and other aquatic environments.
Fluorescent Proteins as Cell Tags and Reporter Genes
Bacterial cells can be altered by genetic engineering to make them autofluorescent. As discussed earlier, a gene encoding the green fluorescent protein (GFP) can be inserted into the genome of virtually any cultured bacterium (Sections 8.1 and 12.5). When the gene encoding the GFP (gfp) is expressed, cells fluoresce green when observed with ultraviolet microscopy (Figure 19.10; Figure 12.17). Although GFP is not useful for the study of natural populations of microorganisms (because these cells lack the GFP gene), GFP-tagged cells can be introduced into an environment, such as plant roots, and then tracked over time by microscopy. Using this method, microbial ecologists can study competition between the native microbiota and a GFP-tagged introduced strain and can assess the effect of perturbations of an environment on the survivability of the introduced strain.
Figure 19.10 Fluorescent protein reporters.

(a) Twelve different fluorescent proteins (FP1–FP12) are known that have distinct excitation (Excite) and emission (Emit) properties. (b) Cells of Sinorhizobium meliloti (arrows) carrying a plasmid with an α-galactoside-inducible promoter fused to the GFP (FP5); the cells are on clover seedling roots. Green fluorescence indicates that α-galactosides are released and available to support the growth of this bacterium. (c) S. meliloti cells (arrows) carrying a plasmid with a succinate-inducible promoter fused to GFP; green fluorescence indicates that succinate or other C4 dicarboxylic acids have been secreted by the plant root hairs.
The gene gfp and those encoding other fluorescent proteins have also been used extensively in laboratory cultures of various bacteria and in controlled environments as reporter genes. When the gene is fused with an operon under the control of a specific regulatory protein, transcription can be studied by using fluorescence as the indicator (a “reporter”) of activity. That is, when genes containing the fused fluorescent protein gene are transcribed and translated, both the protein of interest and the fluorescent protein are made, and cells fluoresce the characteristic color. For example, expression of gfp was used to demonstrate that colonization of alfalfa roots by the nitrogen-fixing symbiotic bacterium Sinorhizobium meliloti (legume–root nodule symbiosis, Section 23.4) is promoted by sugars and dicarboxylic acids released by the plant (Figure 19.10b, c). The emission properties of GFP and other fluorescent proteins isolated from different marine invertebrates (jellyfish, corals, anemones) have since been altered through mutation to yield a broad palette of fluorescent proteins of varying spectral properties (Figure 19.10a), offering the experimenter the capability to monitor several species simultaneously.
One drawback to the use of GFP is that to become fluorescent it requires O2, and thus it is not suitable for tracking cells introduced into strictly anoxic habitats. However, flavin-based fluorescent proteins that do not require O2 are available to overcome this limitation. These proteins are derived from bacterial and plant photosensory flavoproteins and are more thermally stable than the GFP, making them useful for tracking mildly thermophilic species. In addition to these fluorescent tags for tracking microbes, phylogenetic stains (Section 19.5) are widely used for identifying microbes, and a wide variety of new fluorescent “super-resolution” microscopy techniques are available for tracking individual molecules within a microbial cell (Section 8.1). Thus, fluorescence technology has come a long way since the days when only DAPI and acridine orange were available for visualizing microbial cells in nature.
Check Your Understanding
How does viability staining differ from stains like DAPI?
What types of environments limit the application of GFP?
Why is it incorrect to say that the GFP is a “staining” method?
19.5 Microscopic Specificity: Fluorescence In Situ Hybridization (FISH)
19.5 Microscopic Specificity: Fluorescence In Situ Hybridization (FISH)
19.5 Microscopic Specificity: Fluorescence In Situ Hybridization (FISH)
As we have just seen, the microscope is an essential tool for enumerating and assaying microorganisms in a general way—their morphology, their Gram reaction, their viability, and the like. However, the biggest limitation with the methods we have discussed thus far is that none of them reveal the phylogeny of the microorganisms observed under the microscope. That is, how many species, or phyla, or even domains, are present in the sample? More specific fluorescent staining methods are required to answer these questions.
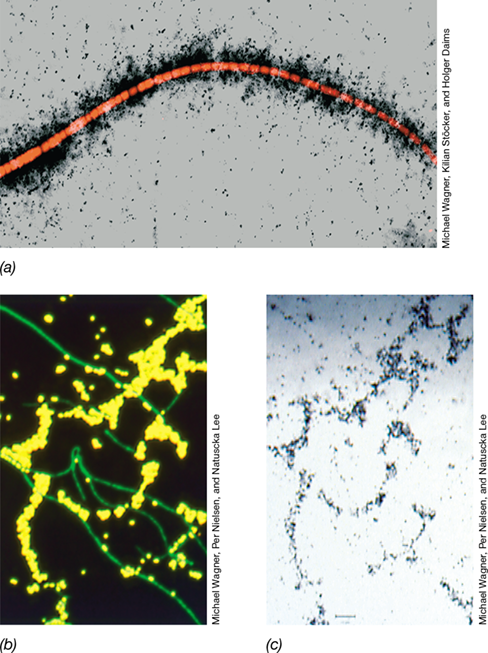
When observing unstained or nonspecifically stained natural populations of microorganisms under the microscope (Section 19.4), one should remember that the sample likely contains a phylogenetically diverse community, even if many cells “look” the same. This is because the simple shapes of bacteria can conceal their remarkable diversity, and Figure 19.11 shows an example of this. The two photos are of the same microscope field, one taken in black and white of cells in a microbial community and the other taken in color of the same cells stained in a way that reveals their phylogeny. If only unstained cells (Figure 19.11a) were viewed, it would be easy to conclude that the large ovoid-shaped cells are all of a single species. But staining to reveal phylogeny (Figure 19.11b) shows that they are not; the blue and yellow fluorescent stains target different species of large ovoid cells while the green stain reveals yet a third species in the community. Microscopic assessments of phylogenetic diversity such as this can only be made using phylogenetic stains, and we consider these here along with some related staining techniques.
Figure 19.11 Morphology and genetic diversity.

The photomicrographs shown here, produced by (a) phase contrast and (b) a technique called phylogenetic FISH (Section 19.5), are of the same field of cells. Although the large oval cells are of a rather unusual size for prokaryotic cells and all look similar by phase-contrast microscopy, the phylogenetic stains reveal that there are two genetically distinct types (one stains yellow and one stains blue). Both cell types are about 2.3 μm in diameter. The smaller, green cells in pairs or clusters are about 1 μm in diameter.
Phylogenetic Identification Using FISH
Because of their great specificity, nucleic acid probes can be harnessed as powerful tools for identifying and quantifying microorganisms. Recall that a nucleic acid probe is a DNA or RNA oligonucleotide complementary to a sequence in a target gene or RNA; when the probe and the target come together, they hybridize (Section 12.1). Nucleic acid probes can be made fluorescent by attaching fluorescent dyes to them. The fluorescent probes can then be used to identify organisms that contain a nucleic acid sequence complementary to the probe. This technique is called fluorescence in situ hybridization (FISH), and different applications are described here, including methods that target phylogeny (Figure 19.12 and see Figure 19.13) or gene expression (see Figure 19.14).
Figure 19.12 Fluorescently labeled rRNA probes: Phylogenetic stains.

(a) Phase-contrast photomicrograph of cells of Bacillus megaterium (rod, Bacteria) and the yeast Saccharomyces cerevisiae (oval cells, Eukarya). (b) Same field; cells stained with a yellow-green universal rRNA probe (this probe hybridizes with rRNA from organisms of any phylogenetic domain). (c) Same field; cells stained with a eukaryal probe (only cells of S. cerevisiae react). Cells of B. megaterium are about 1.5 μm in diameter and cells of S. cerevisiae are about 6 μm in diameter.
Phylogenetic FISH stains are fluorescing oligonucleotides complementary in base sequence to sequences in ribosomal RNA (16S or 23S rRNA in Bacteria and Archaea or 18S or 28S rRNA in eukaryotes, Chapter 13). Phylogenetic stains penetrate cells without lysing them and hybridize with rRNA directly in the ribosomes. The number of fluorescent probes bound to a cell reflects the number of its ribosomes. As single microbial cells can contain tens of thousands of ribosomes, strong signals can be achieved. And, because ribosomes are scattered throughout the cell in prokaryotic cells, the entire cell becomes fluorescent (Figures 19.11b and 19.12). One limitation of the FISH technique is that slow-growing organisms often have very low numbers of ribosomes and mRNAs, and therefore fluoresce very poorly. However, new staining methods have been developed that measure protein synthesis directly, and provide essential information about the viability and activity of cells in their natural environments, even when growing slowly (see Figure 19.15).
By targeting sites in the rRNA sequence that are variable between different organisms, phylogenetic stains can be designed to be very specific and react with only one species or a handful of related microbial species. Alternatively, by targeting conserved sequences in the rRNA they can be made more general and react with, for example, all cells of a given phylogenetic domain (Bacteria, Archaea, Eukarya) or a given phylum within a domain. Using FISH, an investigator can identify or track a specific organism of interest or an entire domain of interest in a natural sample. For example, if one wished to determine the percentage of a given microbial population that are Archaea, an archaeal-specific phylogenetic stain could be used in combination with DAPI (Section 19.4) to assess Archaea and total numbers, respectively, and a percentage derived by calculation.
FISH technology can also employ multiple phylogenetic probes at once. With a suite of probes, each designed to react with a particular organism or group and each containing its own fluorescent dye, FISH can image multiple taxa in a habitat in a single experiment (Figure 19.13 and see MicrobiologyNow). Similarly, the method easily distinguishes between genetically distinct cells of similar morphology (Figure 19.11b). If FISH is combined with confocal microscopy (Section 1.9), it is possible to explore microbial populations with depth, as, for example, in a biofilm (Section 20.4). In addition to microbial ecology, FISH is also an important tool in the food industry and in clinical diagnostics for the microscopic detection of specific pathogens in food products or clinical specimens.
Figure 19.13 FISH analysis of activated sludge from a wastewater treatment plant.

(a) Nitrifying bacteria. Red, ammonia-oxidizing bacteria; green, nitrite-oxidizing bacteria. (b) Confocal laser scanning micrograph of a sewage sludge sample treated with three phylogenetic FISH probes, each containing a fluorescent dye (green, red, or blue) that identifies a particular group of Proteobacteria (Chapter 16). Green-, red-, or blue-stained cells reacted with only a single probe; other cells reacted with two (turquoise, yellow, purple) or three (white) probes.
Single-Cell Metabolic Activity Staining Using CARD-FISH and BONCAT-FISH
FISH methods have been developed for measuring the metabolic activity of single cells by using probes for specific messenger RNAs (transcriptional activity) or for protein synthesis (translational activity). Besides characterizing different taxa in a habitat as phylogenetic stains do, these forms of FISH measure gene expression. These microscopic methods thus complement phylogenetic staining—techniques that answer the microbial ecologist’s important question of “Who is there?”—by answering the other important question in microbial ecology: “What are they doing?”
Measuring transcription requires targeting messenger RNA (mRNA), which is much less abundant in cells than rRNA; because of this, standard FISH techniques cannot be applied. Instead, the signal (fluorescence) must be amplified. A FISH method that enhances the signal is called catalyzed reporter deposition FISH (CARD-FISH). In CARD-FISH the specific nucleic acid probe contains a molecule of the enzyme peroxidase conjugated to it instead of a fluorescent dye. After there has been time for hybridization between the probe and a specific mRNA, the preparation is treated with a fluorescently labeled compound called tyramide, which is a substrate for peroxidase. Within cells containing the nucleic acid probe, the tyramide is converted by the activity of peroxidase into a very reactive intermediate that covalently binds to adjacent proteins; this amplifies the signal sufficiently to be detected by fluorescence microscopy (Figure 19.14). Each molecule of peroxidase activates many molecules of tyramide so that even mRNAs present at very low abundance can be visualized. Besides detecting mRNA, CARD-FISH is also useful in phylogenetic studies of microbes that may be growing very slowly, such as organisms inhabiting the open oceans where cold temperatures and low nutrient concentrations limit growth rates (Figure 19.14). Because such cells have few ribosomes compared with more actively growing cells, standard FISH methodology often yields only a weak signal.
Figure 19.14 Catalyzed reporter deposition FISH (CARD-FISH) labeling of *Archaea*.

Archaeal cells in this preparation fluoresce intensely (green) relative to DAPI-stained cells (blue).
Although cells that fluoresce brightly by FISH are likely actively synthesizing proteins (as indicated by their high number of ribosomes), a direct measure of translational activity can be made using bioorthogonal noncanonical amino acid tagging (BONCAT) combined with FISH (BONCAT-FISH). Bioorthogonal compounds are synthetic molecules that mimic natural metabolites and when assimilated and metabolized by a living cell do not interfere with normal physiological processes. Similarly, bioorthogonal analysis refers to a chemical reaction that can occur inside a living cell without interfering with normal function.
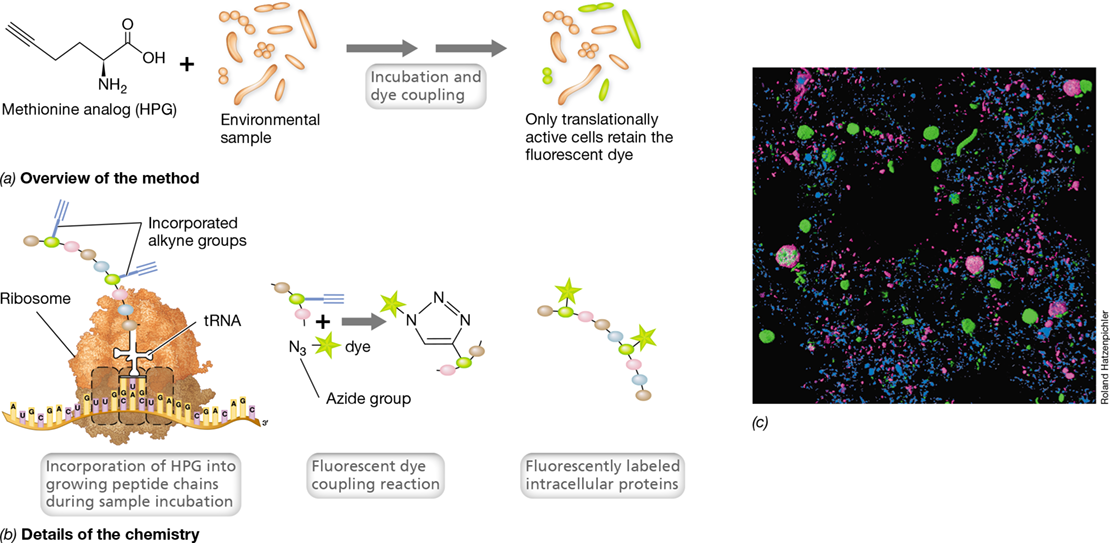
In BONCAT (Figure 19.15), cells are first incubated with a modified version of a normal cellular metabolite, such as the amino acid methionine containing a reactive alkyne group (Figure 19.15a). This methionine analog is called a noncanonical (meaning nonstandard) amino acid. Following incorporation of this analog into proteins of actively growing cells, the cells are treated with a fluorescent reporter molecule containing a chemically reactive azide group that bonds specifically to the methionine analog by way of its reactive alkyne group (Figure 19.15b). The phylogenetic identity of translationally active cells is then determined using standard FISH technology in which the rRNA probe is labeled with a different fluorescent dye from that attached to the azide group. An example of BONCAT-FISH to identify translationally active cells in sediments of a marsh estuary is shown in Figure 19.15c. Note that only some of the cell clusters stained green by a bacteria-specific FISH probe are also translationally active, as indicated by BONCAT magenta staining. Thus, both a phylogenetic snapshot and an activity snapshot are obtained in a single experiment.
Figure 19.15 BONCAT labeling of translationally active microbial cells.

(a) Overview. Cells in the environmental sample take up the methionine analog l-homopropargylglycine (HPG). Following incubation to allow active cells to take up the HPG and incorporate it into growing peptide chains, cells are treated with a dye that makes translationally active cells fluoresce. (b) Chemistry of the process. HPG is incorporated by the cell as methionine in growing polypeptides. When treated with the fluorescent dye-labeled azide, the azide group on the dye binds to the alkyne group on HPG to yield a fluorescent protein. (c) BONCAT-FISH analysis of activity and diversity of microbial cells in Little Sippewissett Salt Marsh (Massachusetts, USA). Blue, cells stained with the nonspecific nucleic acid dye DAPI (Section 19.4). Green, cells stained by FISH for the phylogenetic domain Bacteria. Magenta, translationally active cells stained using BONCAT. The large green ovoid cells are about 4μm in diameter, the large magenta cells are about 6μm in diameter, and the small magenta rod-shaped cells are about 1.5μm long.
After BONCAT-FISH, further genetic analysis is possible by using cell sorting to recover labeled cells for genomic analysis of sorted populations or of single cells (see Section 19.12). Once cells have been concentrated by cell sorting techniques, sequencing can be done on single genes, specific gene families, or entire genomes to yield an increasingly more detailed picture of the genetic makeup of the targeted cells.
Although the techniques we have just discussed do not require laboratory culture, in Part III we consider what microbial ecologists typically mean when they say their microbial community analyses are “culture-independent.”
Check Your Understanding
What structure in the cell is the target for fluorescent probes in phylogenetic FISH?
FISH and CARD-FISH can be used to reveal different things about cells in nature. Explain.
Compare the utility of CARD-FISH versus BONCAT-FISH for evaluating cellular activity.
III: Culture-Independent Molecular Analyses of Microbial Communities
III: Culture-Independent Molecular Analyses of Microbial Communities
III Culture-Independent Molecular Analyses of Microbial Communities
Revolutionary changes in DNA sequencing technology have made it possible to rapidly recover vast amounts of gene and genome sequence information from the environment. However, fundamental ecological understanding is possible only when that sequence information is paired with appropriate measures of cellular and community activities.
Microbial biodiversity studies often forgo isolating organisms or even quantifying or identifying them microscopically. Instead, complementary molecular methods are used to measure the biodiversity, potential activities, and realized activities of complex communities. As we now review in this section, gene and genome sequencing serve as an important measure of diversity and potential activity, with the caveat that annotation of all novel gene functions remains a daunting challenge (Section 10.2). However, detection of a gene is not the same as gene expression. As we will discuss in the following two sections, metabolic activity can be measured at the level of both the community and single cells by combining analyses of gene expression (transcription and translation) with measurements of active processes using the tools of metabolomics (Section 10.10) and isotopic tracers to unravel the food webs sustaining complex microbial communities.
Many surveys of microbial diversity and potential activity are performed at the gene level using the polymerase chain reaction (PCR), DNA fragment analysis by gel electrophoresis (DGGE, T-RFLP, ARISA, each discussed below) or molecular cloning, and DNA sequencing and analysis (Chapters 10 and 12). Increasingly, as a result of changing technologies in DNA sequencing, entire genomes extracted from cells present in an environmental sample are analyzed as a more comprehensive measure of the biodiversity of microbial communities. We begin with an overview of single gene analysis and then introduce multi-omics, the integration of data from multiple omics platforms to more fully characterize microbial community structure and function (see Figure 19.21). The term omics (Chapter 10) is increasingly used to describe individual or various combinations of genomics, transcriptomics, proteomics, and metabolomics for analysis of organisms and communities of organisms. When these methods are used for the study of complex microbial communities, the terminology is often modified with the prefix “meta” (e.g., metagenomics, metatranscriptomics, metaproteomics) as discussed in Section 19.8.
19.6 PCR Methods of Microbial Community Analysis
We discussed the principle of the polymerase chain reaction (PCR) in Section 12.1. Recall the major steps involved: (1) Two nucleic acid primers are hybridized to a complementary sequence in a target gene; (2) DNA polymerase copies the target gene; and (3) multiple copies of the target gene are made by repeated melting of complementary strands, hybridization of primers, and new synthesis (Figure 12.2). From a single copy of a gene, several million copies can be made for subsequent analyses. PCR finds wide applications in microbial ecology.
Mastering Microbiology
Art Activity: Figure 19.15 Steps in single-gene biodiversity analysis of a microbial community
PCR and Microbial Community Analysis
Which genes are best suited to be target genes for microbial community analyses? Because genes encoding the small subunit ribosomal (SSU) rRNAs are phylogenetically informative and techniques for their analysis are well developed (Section 13.11), they are widely used in community analyses. Moreover, because rRNA genes are universal and contain several regions of high sequence conservation, it is possible to amplify them from all organisms using only a few different PCR primers, even though the organisms may be phylogenetically distantly related. In addition to rRNA genes, genes that encode enzymes for metabolic functions unique to a specific organism or group of related organisms can be the target genes (Table 19.3). Many of the organisms that harbor these genes are described in Chapter 15.
Table 19.3 Genes commonly used for evaluating specific microbial processes in the environment using PCR

aAll of these metabolic processes are discussed in Chapter 14.
Genes such as those encoding rRNAs that have retained ancestral function while changing in sequence over time as species have diverged are called orthologs (Sections 13.8 and 13.11 and Figure 13.16). Assuming absence of horizontal gene transfer (Section 13.9), organisms that share the same or very closely related orthologous genes are called a phylotype. In microbial ecology, the phylotype concept is primarily used to provide a natural (phylogenetic) framework for describing the microbial diversity of a given habitat, regardless of whether the identified phylotypes are cultured organisms or not. Thus, the word phylotype is widely used to describe the microbial diversity of a habitat based solely on nucleic acid sequences. It is only when additional physiological and genetic information becomes available, typically after the organism is brought into laboratory culture (Sections 19.2 and 19.3), that proposing a genus and species name for a phylotype becomes possible.
In a typical molecular community analysis experiment, total DNA is isolated from a microbial habitat (Figure 19.16). Commercially available kits that yield high-purity DNA from soil or other complex habitats are available for this purpose. The DNA obtained is a mixture of genomic DNA from all of the microorganisms that were in the sample from the habitat (see Figure 19.17). From this mixture, PCR is used to amplify the target gene and make multiple copies of each variant (phylotype) of the target gene. If RNA is isolated instead of DNA (to detect those genes being transcribed), the RNA can be converted into complementary DNA (cDNA) by the enzyme reverse transcriptase (Section 11.11) and the cDNA subjected to PCR as for isolated DNA. However, regardless of whether DNA or RNA is originally isolated, the different phylotypes need to be sorted out following the PCR step before they can be sequenced. Sorting can be accomplished using one of three different methods: (1) physical separation by gel electrophoresis (Section 12.2), (2) clone library construction (Sections 12.2 and 12.9), and (3) next-generation sequencing technology (Section 10.2). We consider these methods now.
Figure 19.16 Steps in single-gene biodiversity analysis of a microbial community.

From total community DNA, 16S rRNA genes are amplified using primers that target only Firmicutes, a group of gram-positive Bacteria that includes the endospore-forming genera Bacillus and Clostridium. The 16S rRNA gene products obtained from PCR are then either separated by DGGE or sequenced directly by next-generation sequencing; from the sequence data, a phylogenetic tree is generated. “Env” indicates an environmental sequence (phylotype). Alternatively, in T-RFLP analyses, the PCR-amplified products are labeled with a fluorescent dye and fragmented by cutting with a restriction enzyme before electrophoresis. The number of peaks identified by fluorescence corresponds to the number of phylotypes.
Denaturing Gradient Gel Electrophoresis: Separating Very Similar Genes
One method to resolve phylotypes is denaturing gradient gel electrophoresis (DGGE), which separates genes of the same size that differ in their melting (denaturing) profile because of differences in their base sequence (**Figure 19.17a,*b***). DGGE employs a gradient of a DNA denaturant, typically a mixture of urea and formamide. When a double-stranded DNA fragment moving through the gel reaches a region containing sufficient denaturant, the strands begin to “melt”; at this point, their migration stops (Figure 19.17b, c). Differences in base sequence cause differences in the melting properties of DNA. Thus, the different bands observed in a DGGE gel are phylotypes that can differ in base sequence significantly or by as little as a single base change.
Figure 19.17 PCR and DGGE gels.

Bulk DNA was isolated from a microbial community and amplified by PCR using primers for 16S rRNA genes of Bacteria (a, lanes 1 and 8). Six bands later resolved by DGGE (b, lanes 2–7) were excised and reamplified and each gave a single band at the same location on the PCR gel (a, lanes 2–7). However, by DGGE analysis, each band migrated to a different location on the gel (b, lanes 2–7). Note that all bands migrate to the same location in the nondenaturing PCR gel because they are all of the same size, but they migrate to different locations on the DGGE gel because they have different sequences. (c) DGGE profiles of microbial communities from different wastewater treatment facilities amplified using primers for the 16S rRNA genes of Bacteria.
Once DGGE has been performed, the individual bands are excised and sequenced (Figure 19.17). With 16S rRNA as the target gene, for example, the DGGE pattern immediately reveals the number of phylotypes (distinct 16S rRNA genes) present in a habitat (Figure 19.17c). Although not suited for the analysis of highly complex communities, since individual bands may not be fully resolved, the method provides an excellent mechanism to quickly evaluate temporal and spatial shifts in microbial community structure (Figure 19.17c). If PCR primers specific for genes other than 16S rRNA are used, such as a metabolic gene (Table 19.3), the variants of this specific gene that exist in the sample can also be assessed. Thus, although the number of bands on a DGGE gel is an overview of the biodiversity in a habitat (Figure 19.17c), sequence analysis is still required for identification and to infer phylogenetic relationship.
T-RFLP and ARISA
A rapid method of microbial community analysis is terminal restriction fragment length polymorphism (T-RFLP). In this method a target gene (usually an rRNA gene) is amplified by PCR from community DNA using a primer set in which one of the primers is end-labeled with a fluorescent dye. The PCR products are then treated with a restriction enzyme (Section 12.2) that cuts the DNA at specific sequences. This generates a series of DNA fragments of varying length, the number of which depends on how many restriction cut sites exist in the DNA. The fluorescently labeled terminal fragments are then separated by size on an automated DNA sequencing instrument that detects fluorescent fragments (thus, only the terminal dye-labeled fragments are detected). The pattern obtained shows the rRNA sequence variation and general abundance of different sequence types (fragment fluorescence intensity) in the microbial community sampled (Figure 19.16).
DGGE and T-RFLP both measure single-gene diversity, but in different ways. The pattern of bands on a DGGE gel reflects the number of same-length sequence variants of a single gene (Figure 19.17), whereas the pattern of bands on a T-RFLP gel reflects variants differing in DNA sequence of a single gene as measured by differences in restriction enzyme cut sites. The information obtained from a T-RFLP analysis, in addition to providing insight into the diversity and population abundances of a microbial community, can also be used to infer phylogeny. Diagnostic information for each fragment includes knowledge of sequences near both ends (primer sequence and restriction enzyme cut site), knowledge that a second restriction site does not exist within the fragment, and fragment length. Using specialized software, this information can be used to search for matching 16S rRNA sequences in public databases. Although this is of some predictive value, closely related sequences are often not differentiated by these criteria. Thus, T-RFLP generally underestimates the diversity within a microbial community.
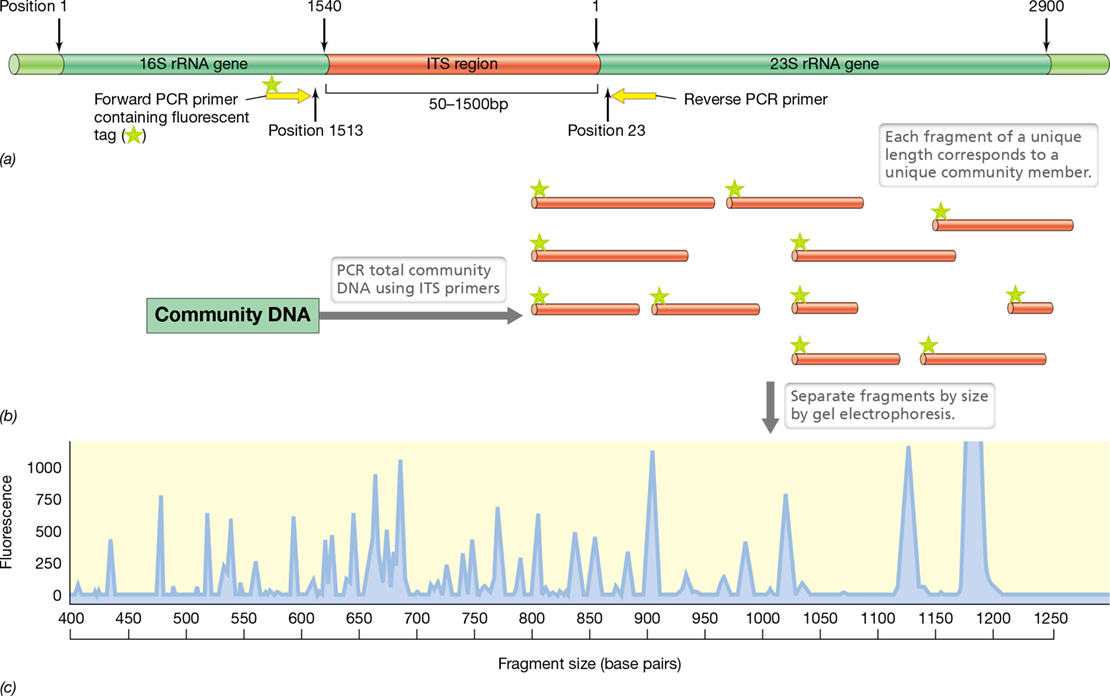
A technique related to T-RFLP that provides more detailed analysis of microbial communities is automated ribosomal intergenic spacer analysis (ARISA), which exploits the typical proximity of 16S and 23S rRNA genes in the genomes of Bacteria and Archaea. The DNA separating these two genes, called the internal transcribed spacer (ITS) region, differs in length among species and often also differs in length among the multiple rRNA operons of a single species (**Figure 19.18*a***). The PCR primers for ARISA are complementary to conserved sequences in the 16S and 23S rRNA genes that flank the spacer region. Amplification (Figure 19.18b) and analysis (Figure 19.18c) are conducted as described for T-RFLP, resulting in a complex pattern of bands that can be used for community analysis. However, ARISA differs from T-RFLP in that ARISA does not require a restriction enzyme digestion following PCR amplification. The word “automated” in the ARISA acronym refers to the use of a DNA sequencing instrument that automatically identifies and assigns sizes to each dye-labeled fragment (Figure 19.18c), as can also be done in T-RFLP analyses. ARISA has received greatest application in the study of microbial community dynamics by monitoring, for example, changes in the presence and relative abundance of a specific community member through time and space.
Figure 19.18 Automated ribosomal intergenic spacer analysis (ARISA).

(a) Structure of rRNA operon spanning the 16S rRNA gene (positions 1–1540), an internal transcribed spacer (ITS) region of variable length, and the 23S rRNA gene (positions 1–2900). The PCR primers, one labeled with a fluorescent dye, are complementary to conserved sequences near the ITS region. (b) Amplified DNA fragments of different lengths, each corresponding to a community member. (c) Fragment analysis determined by an automated DNA sequencer. The peaks, which correspond to different ITS regions, can be identified by cloning and sequencing the amplified products.
Diversity Studies Using Clone Libraries or Next-Generation Sequencing
Early molecular microbial diversity research relied on the construction of clone libraries to separate individual amplified DNA molecules (amplicons); each clone in the library contained a unique sequence that was then used as a template for sequence determination (Sections 10.2 and 12.2). Figure 19.17a shows that a 16S rRNA gene amplicon mixture appears as a single band when run on a nondenaturing gel. However, because the amplified target gene came from a mixture of different cells, the different phylotypes in the single band have different sequences and need to be sorted out before they are sequenced. Today this is almost exclusively done by next-generation sequencing systems (Section 10.2) rather than by DGGE or by cloning. There are many such sequencing platforms available (Figure 19.19), and each has advantages and disadvantages. Long sequence reads are particularly useful for environmental metagenomic studies where the goal is to reconstruct genomes from members of the microbial community (Section 19.8). The MinION (nanopore technology, Explore the Microbial World, “DNA Sequencing in the Palm of Your Hand,” Chapter 10) sequencing system is widely useful for this purpose because it can generate the long sequence reads necessary for genome assembly purposes. Once a genome is assembled, sequence accuracy can be improved using other platforms better capable of sequencing short DNA fragments at high fidelity (Figure 19.19a).
Figure 19.19 Community diversity analyses using next-generation sequencing technology.

(a) Current sequencing platforms (Section 10.2) have the capacity to generate 1012 nucleotides (nt) of sequence in a single sequencing run (requiring a week or less), with individual read lengths varying from 100 to 800 nucleotides. The two segments in the rightmost bar show that technologies generating longer reads have lower throughput per sequencing run. (b) This enormous sequencing capacity revealed many unique phylotypes that were not detected using DGGE or clone library sequencing. Fewer than 100 unique phylotypes would be detected by Sanger (first-generation) sequencing of 1000 clones in a library of 16S rRNA gene PCR amplicons. Jed Fuhrman is acknowledged for input to part b.
MinION and these other next-generation sequencing systems do not require a cloning step, as individual DNA fragments are separated and amplified on the sequencing device itself; thus, PCR products can be used directly for sequencing. Since millions of amplification reactions are then conducted simultaneously, the total number of sequencing reads vastly exceeds what is possible by sequencing individual clones obtained in a clone library one at a time (Figure 19.19). This tremendous volume of sequence data allows for what has been called deep sequence analyses, meaning that minor phylotypes that were possibly missed by the more limited and expensive clone library method are now revealed (Figure 19.19b). For example, if a particular phylotype were present at only 0.01% in a library of cloned sequences, sequencing of a thousand clones or more would be needed to ever detect it. By contrast, next-generation sequencing would detect this low-abundance phylotype along with its more abundant neighbors. This collection of minor phylotypes, which represent a substantial fraction of total diversity but only a minor component of total organism abundance in most environments, has been called the rare biosphere (Figure 19.19). Once next-generation sequencing reveals these rare but potentially very important components of a microbial community, various tools in the microbial ecologist’s toolbox can be deployed to learn more about their activities and overall role in their ecosystem.
Results of PCR Phylogenetic Analyses
Phylogenetic analyses of microbial communities have yielded surprising results. For example, using the gene encoding 16S rRNA as the target, analyses of natural microbial communities typically show that many phylogenetically distinct Bacteria and Archaea (phylotypes) are present whose rRNA gene sequences differ from those of all known laboratory cultures. Moreover, using quantitative PCR (qPCR, a variant of PCR that allows each phylotype to be quantified as well as amplified, Section 12.1), it has often been observed that the most abundant phylotypes in a natural microbial community are ones that have thus far defied laboratory culture. These sobering results make it clear that our knowledge of microbial diversity from enrichment cultures is far from complete and that enrichment bias (Section 19.1) is a serious problem in culture-dependent biodiversity studies. Obviously, much work remains to put our abilities to culture microbes on a par with our existing abilities to detect and identify them in nature.
Check Your Understanding
What could you conclude from PCR/DGGE analysis of a sample that yielded one size band by PCR and one band by DGGE? One size band by PCR and four bands by DGGE?
What surprising finding has come out of many molecular studies of natural habitats using 16S rRNA as the target gene?
How has next-generation sequencing technology altered our understanding of microbial community diversity?
19.7 Microarrays for Analysis of Microbial Phylogenetic and Functional Diversity
19.7 Microarrays for Analysis of Microbial Phylogenetic and Functional Diversity
19.7 Microarrays for Analysis of Microbial Phylogenetic and Functional Diversity
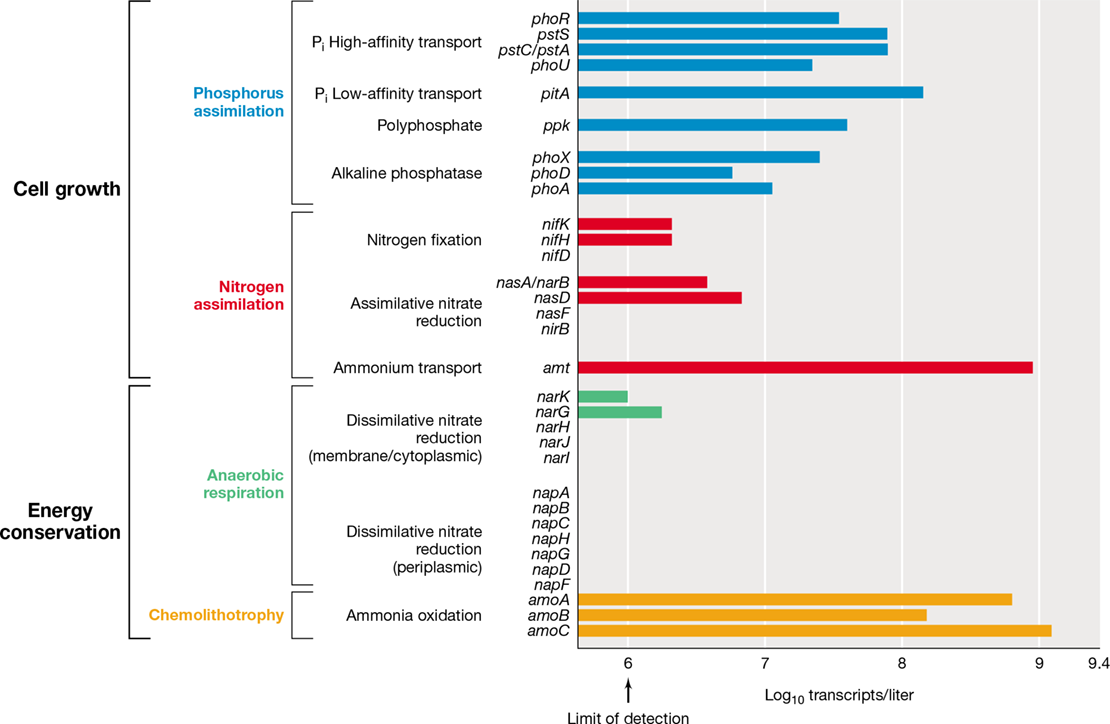
We previously considered the use of DNA chips—a type of microarray—for assessing overall gene expression in a microbial pure culture (Section 10.8). Specific microarrays can also be constructed for rapid gene-based analyses of biodiversity and the functional potential of natural microbial communities. Microarrays designed to exclusively target different 16S rRNA sequence types (phylotypes), called PhyloChips, have been used in the past, but microarrays employed for environmental studies now focus on metabolic genes of biogeochemical significance. This type of microarray, the GeoChip, detects genes required for selected metabolic processes, including sulfate reduction, ammonia oxidation, denitrification, and nitrogen fixation (Figure 19.20 and Table 19.3).
Figure 19.20 GeoChip analysis of functional gene diversity.

The GeoChip contains over 161,000 probes covering more than 365,000 gene sequences in public databases, encompassing most major biogeochemical processes. The image shows green fluorescence of varying intensity (approximating gene abundance) following hybridization of fluorescent dye–labeled environmental DNA to the individual probes in one region of the high-density array of probes. The red spots correspond to repeated applications of a known amount of a reference DNA standard. A red-dye-labeled probe complementary to the reference standard was added to the environmental DNA prior to hybridization. Red fluorescence of equal intensity among the reference standard spots confirms that hybridization was uniform throughout the array.
The GeoChip—also called a functional gene microarray—targets genes that function in a measurable environmental process. However, because genes encoding enzymes of similar function can vary significantly in sequence, the arrays must contain many thousands of probes in order to achieve reasonable coverage of natural diversity. Even then, such arrays may only sample a fraction of the actual functional diversity in a habitat. Successful use of a GeoChip requires several steps: (1) isolation of total community DNA from the sample, (2) random sequence amplification (as is also used for amplification of DNA from a single cell, Section 10.11), (3) fluorescence labeling of the environmental DNA, and (4) hybridizing the environmental DNA to probes on the GeoChip. The GeoChip functional gene microarray contains over 160,000 probes covering more than 1400 gene families encoding proteins that participate in carbon, nitrogen, and sulfur cycling processes. By measuring the hybridization signal conferred by binding of environmentally derived DNA to each probe on the microarray (Figure 19.20), a rapid appraisal of the potential metabolisms operating in a particular habitat can be obtained.
GeoChips circumvent many of the time-consuming steps of molecular microbial ecology—PCR, DGGE, cloning, and sequencing (Figure 19.16). In addition, the GeoChip is useful for detecting low-abundance genes that would be difficult to identify by sequencing and also offers some information on actual gene abundance. However, an important caveat to the use of microarrays is the possibility of nonspecific hybridization. That is, gene variants that are closely related in sequence may not be resolved in a microarray because of overlapping hybridization patterns. Moreover, totally unrelated genes may yield false positive results if they are sufficiently complementary in base sequence to hybridize to the probe. And finally, unlike nucleic acid sequencing, whose costs keep plummeting yearly, designing and manufacturing gene chips are not yet inexpensive endeavors. Combined with culture-independent phylogenetic diversity studies (Section 19.6), functional gene arrays offer the microbial ecologist a detailed picture of both the diversity of organisms and the diversity of metabolisms in any given environment. However, what the GeoChip and related functional gene arrays do not reveal is actual metabolic activity; they show only the genetic potential for metabolic activity. We got a taste of how metabolic activity can be measured using BONCAT, a microscopic method of detecting metabolic activity (Figure 19.15). In the next section, we explore other methods of assessing microbial activity that use omics, the most powerful tools in the microbial ecologist’s toolbox.
Check Your Understanding
What are some caveats of interpreting a hybridization signal on the GeoChip?
What are the advantages and disadvantages of microarray technology compared to sequencing PCR products?
19.8 Environmental Multi-omics: Integration of Genomics, Transcriptomics, Proteomics, and Metabolomics
19.8 Environmental Multi-omics: Integration of Genomics, Transcriptomics, Proteomics, and Metabolomics
19.8 Environmental Multi-omics: Integration of Genomics, Transcriptomics, Proteomics, and Metabolomics
As was presented in Chapter 10, a more complete understanding of how a microorganism functions requires an integrated accounting of all central cellular processes. These include gene expression, functional knowledge of all gene products and product activities, and all metabolites produced during growth. The analytical tools used to characterize those processes in pure cultures (Sections 10.11, 10.12 and 10.13) are now being applied to the study of complex microbial communities to yield an unprecedented view of microbial ecology.
Characterization of a natural system is a much more complex problem than the analysis of a single organism. Not only must processes of each organism be measured, but the many changing biotic and abiotic interactions that modify those processes must also be resolved. Here we consider the methods of genomic, transcriptomic, proteomic, and metabolomic analysis required to unveil patterns of microbial diversity that will ultimately lead to a more predictive understanding of microbial community function and response to environmental change. Collectively called omics or, when used in combination, multi-omics, these rapidly evolving methods are having a dramatic impact on our understanding of the structure and function of natural microbial communities. We begin with an overview of individual omics (Figure 19.21), and then present exemplar case studies of their application.
Figure 19.21 Multi-omics methods overview.

The combined use of advanced genetic, molecular, microscopic, and isotopic methods is revolutionizing the study of natural microbial communities. Assignment of protein and transcript sequences to specific community members is accomplished by mapping their sequences on unique genomes assembled from metagenomic sequences. Abbreviations: FISH, fluorescence in situ hybridization (Figures 19.11–19.14); BONCAT, bioorthogonal noncanonical amino acid tagging (Figure 19.15); FACS, fluorescence-activated cell sorting (see Figure 19.42); MAR, microautoradiography (see Figure 19.41); SIMS, secondary ion mass spectrometry (see Figure 19.39).
Metagenomics and Reconstructing Environmental Genomes
An encompassing approach to the genetic characterization of microbial communities is environmental genomics, also called metagenomics. Before the metagenomics era, microbial community analyses typically focused on the diversity of a single gene in an environmental sample. By contrast, in environmental genomics, all genes in a given microbial community can be sampled and, if the experiment is properly designed, the information obtained can support a much deeper understanding of the structure and function of the community than can single-gene analyses.
The goal of a metagenomics study today is to use next-generation DNA sequencing (Section 10.2) to identify as many genes as possible from an environmental DNA sample and determine the phylogeny of the organism(s) to which the genes belong. Although complete and finished genomes are often not the goal of metagenomics, there is increasing interest in assembling individual genomes from large metagenomic datasets to at least the draft stage. Rather than simply generating a list of all genes present in an environment, nearly complete genomes can better connect functional and phylogenetic aspects of a microbial habitat. An example of this is shown in the “connection graph” of Figure 19.22 that depicts an assembly of genomes from a coastal marine water sample. A total of 58.5 billion nucleotides in the metagenome were used to stitch together these complete and near-complete genomes. Such massive metagenomic undertakings can be highly complex enterprises but often reveal links between physiologies and phylogenies not obtainable in the absence of reconstructed genomes.
Figure 19.22 Genome assembly from a coastal marine metagenome consisting of 58.5 billion nucleotides of sequence.

This “connection graph” is intended as a visual representation of the complexity and abundance of partial and complete genomes assembled from the water sample. The long strands, colored by differences in the percentage of guanine plus cytosine content, correspond to prokaryotic genomes and the small circular strands are most likely from viruses or plasmids.
A problem with genome assembly from a mixture of environmental DNA sequence reads, however, is that the genomes obtained are unlikely to be either complete or clonal, instead being composed of fragments of DNA from closely related strains of a species (Figure 19.23). This has proven to be a major problem in the assembly of soil microbial genomes, for example, from large pools of metagenomic data. A single gram of fertile soil contains about 1012 bacterial and archaeal genes and 109 genomes; complete coverage of these genes is not yet feasible with available technology, even with the sequencing of 300 billion nucleotides in one soil study! Single-cell genomics (Section 10.11 and Figure 10.30) may ultimately overcome this problem (see also Section 19.12), but by definition, this method only provides information from a single microbial cell.
Figure 19.23 Single-gene versus environmental genomic approaches to microbial community analysis.

In the environmental genomic approach, all community DNA is sequenced, but the assembled genomes may not all be complete. Total gene recovery is variable and depends on several factors including the complexity of the habitat and the amount of sequence determined. Recovery is typically better when diversity is low and sequence redundancy is high.
Of critical importance in evaluating a genome reconstructed from metagenomic DNA is to assess whether it contains all of the genes required by a cell (for example, all necessary tRNA and rRNA genes and genes encoding essential proteins such as DNA and RNA polymerases) and is therefore a legitimate genome candidate. In addition, an assessment of the relative abundance of genes encoding specific functions is equally valuable, since abundance changes suggest interactions among species or a common response to a particular environmental variable. For example, if a high number of genes were recovered in the pathway for nitrogen fixation, this would suggest that the environment sampled was limited in NH4 +, NO3 −, and other forms of fixed nitrogen, thus selecting for nitrogen-fixing bacteria. Figure 19.23 contrasts the environmental genomic approach with single-gene analysis of microbial communities.
Although metagenomics can reveal much about a microbial habitat, there are many things metagenomics cannot tell us about environmental microbial communities. Currently no methods are available for translating metagenomic sequence data into fundamental physiological information about the microbial community, such as the maximum specific growth rate of different species, their affinities for nutrients, their optimum, minimum, and maximum pH or temperature for growth, or their speed of recovery from starvation. Moreover, any metabolisms never before encountered are unlikely to be deduced from nucleotide sequence data alone. These realities once again underscore why it is important to culture new microbes from nature; at present, there is simply no substitute for culture-based characterization to define many critical aspects of a microbe’s functional biology.
Some Examples of Environmental Genomics
Environmental genomics can detect both new genes in known organisms and known genes in new organisms. A large number of interesting microbial communities have been probed using early metagenomic tools. In an early study of bacterial and archaeal diversity in the Sargasso Sea (a low-nutrient region of the Atlantic Ocean near Bermuda), over one billion nucleotides were sequenced and from this over 1800 bacterial and archaeal species were detected, including 148 previously unknown phylotypes and many novel genes. Many of these species had previously been missed using rRNA community analyses that employed PCR and cloning or PCR and DGGE (Section 19.6). Genes that fail to amplify, of course, remain undetected in community analyses, and cloning efficiency is far from 100%. Metagenomics sidesteps these problems by sequencing DNA directly without the need to amplify it or resolve different phylotypes before sequencing.
The Sargasso Sea metagenome study revealed several novel findings such as the presence of candidate ammonia-oxidizing genes in archaeal genomes, a result that was later confirmed with the isolation and description of a new group of Archaea, the Thaumarchaeota (Section 17.5). Moreover, genes encoding proteorhodopsin, a light-sensitive proton pump present in certain Proteobacteria and related to bacteriorhodopsin of extreme halophiles (Section 17.1), were found in the genomes of several new phylogenetic lineages of Bacteria. However, despite this major sequencing undertaking, much was missed. This is because 1 milliliter of seawater contains approximately 5 trillion base pairs (bp) of bacterial genomic DNA and would therefore require 5000 times the sequencing effort just to cover each base pair once on average! Hence, even with current technology (see Table 10.2)—which can generate over a trillion bp of sequence in a few days (Figure 19.19)—no one natural environment has yet been sequenced completely.
Genomic/metagenomic approaches have also revealed variations in genes associated with a single phylotype; that is, in strains that contain identical, or nearly identical, rRNA genes. For example, in studies of Prochlorococcus, the most abundant cyanobacterium (oxygenic phototroph) in the ocean (Section 20.11), comparison of the genome sequences of cultured strains with Prochlorococcus genes obtained from metagenomic analyses of ocean water identified extensive regions shared between the cultured and environmental populations (Figure 19.24). This high level of gene conservation confirms that the organisms in culture are typical of environmental populations. In addition, however, these analyses also identified several highly variable regions in which the genomes of cultured strains differed significantly from those of environmental populations. These variable regions were clustered in the genome as genomic islands (chromosomal islands, Section 13.10), and likely encode functions that control the growth response of particular Prochlorococcus populations to environmental variables unique to their habitat, such as temperature or light quality and intensity (Section 20.11 and Figure 20.26).
Figure 19.24 Metagenomic analysis.

Sequences (represented as green dashes) from the Sargasso Sea metagenome that align to the genome sequence of a cultured Prochlorococcus, showing regions where the cultured strain has genes of high similarity (high percent identity) with sequences in the metagenome, and other regions (shaded) where it lacks genes in common (genomic islands, ISL1–ISL5). Since the DNA sequence contained within the genomic islands is thought to encode niche-specific functions, the cultured strain would likely not exhibit the same environmental distribution as strains containing all the island genes. Fold coverage is a measure of how completely the various regions in the Prochlorococcus genome are accounted for by similar sequences in the metagenome.
Metatranscriptomics, Metaproteomics, and Metabolomics
As we discussed in Chapter 10, the genomics era has spawned several additional “omics,” in particular, metatranscriptomics, metaproteomics, and metabolomics. Metatranscriptomics is analogous to metagenomics but analyzes the sequences of community RNA rather than community DNA. The isolated RNA is converted into cDNA by reverse transcription (Section 11.11) before sequencing and analysis as for DNA. Whereas metagenomics describes the functional capacities of the community (for example, the relative abundance of specific genes), metatranscriptomics reveals which genes in the community are actually being expressed and the relative level of that expression, at a specific time and place (Figure 19.21). Because the expression of most genes in Bacteria and Archaea is controlled at the level of transcription (Chapter 7), mRNA abundance can be taken as a census of individual gene expression levels. Thus, gene transcript abundance determined for an entire community can be used to infer the operation of major metabolic processes catalyzed by that community at the time of sampling (Figure 19.25).
Figure 19.25 Metatranscriptomic analysis of coastal marine surface waters.

Expression of genes for key steps in the N and P cycle in a seawater sample determined by sequencing environmental mRNA. These data showed that the microbial community was using both inorganic (high expression of P transporters) and organic (alkaline phosphatase) forms of phosphate (PO4 3−). Low levels of transcripts for genes required for nitrate (NO3 −) assimilation contrasted with the high expression of genes for ammonia (NH3) transport and chemolithotrophic NH3 oxidation. Also, as expected for oxic marine surface waters, there was little expression of genes for NO3 − respiration. Data courtesy of Mary Ann Moran, University of Georgia Marine Sciences.
Metaproteomics, the measure of the diversity and abundance of different proteins in a community, is an even more direct measure of cell function than is metatranscriptomics. This is because different mRNAs have different half-lives and efficiencies of translation, and thus will not all yield the same number of protein copies. However, metaproteomics is a greater technical challenge than either metagenomics or metatranscriptomics. Protein identification, usually by mass spectrometric characterization of peptides released from enzymatic digestion of the total protein pool by specific proteases (Section 10.9), relies on naturally available material since it is not possible to amplify protein sequences as one does using PCR to amplify nucleic acids for sequencing. Protein identification also requires at least partial separation of the individual peptides in order to reduce complexity of the sample and a reference genome or metagenome to identify potential coding sequences (see Figure 19.21). Metabolomics is the comprehensive analysis of cellular and extracellular metabolites (also called exometabolites) of a microbial community and is the most direct measure of the food web structure of a community. As discussed in Chapter 10 (Section 10.10), advanced methods in mass spectrometry are now providing the analytical power required to resolve and quantify the many hundreds of metabolites that form the food webs of natural microbial communities.
The utility and power of integrating omics methods for the study of natural microbial communities has been on display in many ecological studies published in the scientific literature in recent years and was evident in the metatranscriptomic study shown in Figure 19.25. We continue this theme here with two case studies, one employing metaproteomics and the other metabolomics.
Utility of Metaproteomics: An Environmental Case Study of Permafrost
Permafrost covers an estimated 20% of the land on Earth and sequesters large amounts of organic carbon. Because climate change will likely lead to the release of two major “greenhouse gases”—CO2 and CH4—from permafrost, identifying the microbes inhabiting actively melting permafrost along with their metabolic potential is essential for climate predictions. Using a combination of metagenomics (Section 10.7) and metaproteomics (Section 10.9), a revealing snapshot of the genes and proteins of the microbial community present in a permafrost meltwater bog has been obtained (Figure 19.26).
Figure 19.26 Meta-omics of permafrost.

(a) Metagenomic and metaproteomic analysis of an environmental sample. Total DNA and protein are extracted from a sample and analyzed to identify microorganisms and their associated proteins. Following sequencing, the DNA is assembled into partial and complete individual genomes. The 16S rRNA gene can also be profiled to determine the phylogenetic affiliation of Bacteria and Archaea present in the sample. After identifying the digested proteins by mass spectrometry, their microbial source can be determined by searching for corresponding DNA sequences in the metagenomic data. (b) Visualization of a subset of the proteins identified from metagenomic and metaproteomic analysis of a bog sample. Ccp: cytochrome c peroxidase, DsrA: sulfite reductase subunit A, GlnA/GlnB: nitrogen storage, Hup: heterosulfide reductase, Lem: peptidoglycan-associated lipoprotein, Mtd/MtrA/MtrB/MtrH/McrA/McrB/McrG/Mer/Mo: methane metabolism, NapC: NapC/NirT cytochrome c, Opa: opacity protein, PSP: phosphate-selective porin, Ptol: periplasmic component of the Tol biopolymer transport system, Thr: thermosome, SusD: sulfate ABC transport system ATP-binding protein, TonB: periplasmic protein. Data adapted from Hultman, J., et al. 2015. Nature 521: 208.
During the metagenomic analysis of the bog, DNA sequences were assembled and annotated into complete and partial genomes (Figure 19.26a). This analysis indicated that Bacteria of the phyla Proteobacteria, Actinobacteria, and Chloroflexi predominated (Chapters 15 and 16), while methanogens (Euryarchaeota) were the dominant Archaea (Chapter 17). From the large amount of sequence data obtained, the genomes of three novel (and as yet uncultured) methanogens could be assembled without any cultures of the organisms having been obtained. This indicated that unique methanogens resided in the permafrost—most likely species that function well in the cold (psychrophiles)—and therefore that the potential for increasing rates of methanogenesis as the permafrost melted was high. Using the annotated DNA sequences and the output from the mass spectrometry detection of peptides, the identity of proteins extracted from the bog sample was determined along with their microbial sources (Figure 19.26b). The proteomics detected an abundance of functionally diverse proteins including those that participate in cellular housekeeping, transport, and organic carbon respiration. But importantly, and in agreement with the metagenomics data, several proteins associated with C1 metabolism, including those necessary for both making methane (methanogenesis, Section 14.15) and oxidizing methane (methanotrophy, Section 14.16) as well as oxidizing and reducing carbon monoxide (CO dehydrogenases), were also detected in the bog microbial community.
This combined metagenomic and metaproteomic snapshot of a major microbial community highlights the power of omics for resolving the “who” and “how” of complex microbial communities and for using the results to predict the response(s) of these communities to environmental changes. In the case of permafrost melting, the potential for the release of large amounts of CO2 (due to increased respiration of organic carbon) and CH4 (due to increased activities of methanogens) from permafrost as climate change progresses (see Section 21.9) was clearly apparent (Figure 19.26).
Utility of Metabolomics: An Environmental Case Study of Biocrusts
A study of the microbial ecology of biological soil crusts (biocrusts) offers an example of how metabolomics can provide new understanding of food webs that sustain natural microbial communities. Biocrusts are microbe-dominated communities that develop in arid environments of limited vegetation that in part function to reduce soil erosion (Figure 19.27; Section 20.6). They are relatively simple communities in which a dominant filamentous cyanobacterium, in this case Microcoleus vaginatus (a member of the Oscillatoriales, Section 15.3), is the primary producer and the ultimate source of dissolved carbon used by associated heterotrophic microbes. These communities persist in a desiccated, dormant state for extended periods but rapidly become active after a rainfall. Analysis of the biocrust metabolome using advanced methods of mass spectrometry identified and quantified hundreds of metabolites (e.g., adenine, aspartate, uracil, methylguanine, 2-isopropylmalate, methionine, guanidinobutanoate, glycine betaine, malate, succinate). Many of these metabolites are produced by Microcoleus and released to the environment for consumption by other members of the soil crust community. Since this cyanobacterium is unable to fix nitrogen, its nitrogen requirement is largely supplied by nitrogen-containing metabolites supplied by heterotrophic members of the community.
Figure 19.27 Desert biocrust of the Colorado Plateau.

(a) The thin desert biocrust is stabilized by the filamentous cyanobacterium Microcoleus, which binds together soil particles and reduces erosion. (b, c) Visible Microcoleus filaments (arrows) show the fine architecture of the crust. Microcoleus nourishes a rich community of primarily heterotrophic bacteria, releasing metabolites consumed by community members, which in turn produce metabolites consumed by the cyanobacterium. Environmental metabolomics (see Figure 19.28) has the power to unravel the complex food web that sustains these critical microbial communities in arid lands.
Characterization of the intracellular metabolites and exometabolites of a pure culture of Microcoleus showed that a significant fraction of its intracellular metabolites were released during growth and thus made available to heterotrophic community members as exometabolites (**Figure 19.28*a***). In turn, the heterotrophic community produced metabolites used selectively by the Microcoleus or by other heterotrophs (Figure 19.28b). Bacterial cultures isolated from the crust were found to have strong substrate preferences, each depleting a certain fraction of the metabolites while ignoring others. There was also little overlap in utilization; only two metabolites, glutamate and a second, unidentified compound, were used by all members of a group of bacterial isolates. Thus, each isolate from the biocrust likely occupies a specialized nutritional niche, using only a subset of the total pool of metabolites as nutrients. Analysis of the patterns of metabolite uptake and release suggested 80 potential cross-feeding patterns. In addition to providing an example of the power of metabolomics for untangling complex food webs, the use of this exometabolomic approach for identifying metabolites used by uncultured community members yields a second scientific benefit: It can guide researchers in formulating selective culture media for enrichment of heterotrophic bacteria that eluded initial cultivation efforts.
Figure 19.28 Metabolic network supporting the desert biocrust microbial community.

(a) Abundance of each metabolite arranged in decreasing order (upper panel) The primary producer, the cyanobacterium Microcoleus, releases a large fraction of its intracellular metabolites (green) into the environment (blue). (b). These metabolites nourish a complex heterotrophic community that in turn produces metabolites nourishing Microcoleus. In addition, hundreds of additional metabolites consumed by the crust community were identified indicating additional metabolic interactions and cross-feeding among heterotrophs (arrows between bacterial cells). Data adapted from Baran, R., et al. 2015. Nat. Commun. 6: 8289, doi: 10.1038/ncomms9289.
In the final part of this chapter, up next, we look at some other tools in the microbial ecologist’s toolbox that can measure total microbial community activities.
Check Your Understanding
What is a metaproteome, and how does it differ from a metagenome and from a metatranscriptome?
How do environmental multi-omics approaches differ from environmental genomics or single-gene analyses, such as that based on 16S rRNA gene analysis for microbial community characterization?
How can the most metabolically active cell populations in a community be identified using environmental omics methods?
IV Measuring Microbial Activities in Nature
Microbial communities manifest their presence through the concerted activities of individual microorganisms that together drive major biogeochemical processes. A powerful suite of isotopic and genetic methods combined with high-resolution microscopy and sensor technology have made it possible for the microbial ecologist to map these processes from the level of individual cells to entire environmental systems.
We wrap up this chapter by considering methods for measuring microbial activities in natural samples beyond those already discussed in Parts II and III. The techniques we consider here include the use of radioisotopes, microsensors, nanosensors, stable isotopes, and genomic methods. Activity measurements in a natural sample are collective estimates of the physiological reactions occurring in the entire microbial community, although several techniques to be discussed later (Sections 19.10, 19.11 and 19.12) allow for a more targeted assessment of physiological activity. Activity measurements reveal both the types and rates of the major metabolic reactions occurring in a habitat, and the various techniques can be used alone or in combination in microbial community analyses. In conjunction with multi-omics analyses, these methods help define the structure and function of microbial ecosystems—the ultimate goal of microbial ecology—and often inform the design of enrichment cultures to bring microbial community members into the laboratory.
19.9 Chemical Assays, Radioisotopic Methods, Microsensors, and Nanosensors
19.9 Chemical Assays, Radioisotopic Methods, Microsensors, and Nanosensors
19.9 Chemical Assays, Radioisotopic Methods, Microsensors, and Nanosensors
In many studies, direct chemical measurements of microbial reactions are sufficient for assessing microbial activity in an environment. For example, the fate of lactate oxidation by sulfate-reducing bacteria in a sediment sample can be tracked easily. If sulfate-reducing bacteria are present and active in a sediment sample, then lactate added to the sediment will be consumed and SO4 2− will be reduced to hydrogen sulfide (H2S). Since lactate, SO4 2−, and H2S can all be measured specifically and with high sensitivity by chemical assay, the transformations of these substances relative to one another in a sample can easily be followed (**Figure 19.29*a***). However, when very high sensitivity is required, or when turnover rates need to be determined, radioisotopes are more useful than strictly chemical assays.
Figure 19.29 Microbial activity measurements.

(a) Chemical measurements of lactate and H2S transformations during SO4 2− reduction. Radioisotopic measurements: (b) photosynthesis measured with 14CO2; (c) SO4 2− reduction measured with 35SO4 2− conversion to H2 35S. (d) production of 14CO2 from 14C-glucose.
Another approach to the sensitive measurement of certain microbial process rates takes advantage of selective chemical inhibition of an enzyme specific to the process. For example, a widely employed chemical inhibitor uses acetylene to inhibit some processes, such as nitrification, or as an alternative substrate in another, such as nitrogen fixation. The final set of tools we will describe in this section are different types of microsensors and nanosensors used to measure an active microbial process by quantifying changes in the concentration of specific chemicals in time and space.
Radioisotopes
When very high sensitivity is required, or turnover rates need to be determined, or the fate of portions of a molecule needs to be followed, radioisotopes are more useful than strictly chemical assays. For instance, if measuring photoautotrophy is the goal, the light-dependent uptake of radioactive carbon dioxide (14CO2) into microbial cells can be measured (Figure 19.29b). If sulfate reduction is of interest, the rate of conversion of 35SO4 2− to H235S can be assessed (Figure 19.29c). Heterotrophic activities can be measured by tracking the release of 14CO2 from 14C-labeled organic compounds (Figure 19.29d), and so on.
Both isotopic and chemical methods are widely used in microbial ecology. To be valid, however, these must employ proper controls because some isotopic transformations might be due to abiotic processes. The killed cell control is an essential control in such experiments. That is, it is essential to show that the transformation being measured stops when chemical agents or heat treatments that kill microorganisms are applied to the sample. Formalin at a final concentration of 4% is commonly used as a chemical sterilant in microbial ecology studies. This kills all cells, and transformations that are insensitive to the presence of 4% formalin can be ascribed to abiotic processes (Figure 19.29a). Additional controls are also necessary. For example, if H2-driven sulfate reduction is to be measured in a bottle, an essential control is to have a separate bottle identical in every way except that it lacks H2 (Figure 19.29c). If photosynthesis is to be measured, a dark control is essential (Figure 19.29b), and so on. The art of doing good experimental science is in designing experiments with the correct controls, and this is especially true of bulk assays of microbial activity where particular subsets of the entire microbial community are targeted.
Acetylene as an Inhibitor and an Alternative Substrate
In some activity measurements, it is useful to inhibit the activities of certain organisms in order to focus on the activities of others. For example, the short-chain hydrocarbon acetylene (HC≡CH) is an inhibitor of several metabolisms, including nitrification (Section 14.9), denitrification (Section 14.11), and methanogenesis (Section 14.15). In nitrification, acetylene inhibits the first step, ammonia oxidation to nitrite, while in denitrification, acetylene inhibits the terminal step, the reduction of nitrous oxide (N2O) to N2. Thus, acetylene can be used to focus on particular steps in these processes with the knowledge that the inhibited steps are not occurring. For example, the use of acetylene to block reduction of N2O is a common assay for denitrification, using the production rate of N2O (an easily assayed gas) in the presence of acetylene as a measure of the denitrification rate in an environmental sample. Acetylene and other metabolic inhibitors are useful in microbial ecology but must be used with the caveat that the inhibitor might also affect some unrecognized metabolic process that contributes to the activity being measured.
Acetylene is also commonly used for another purpose. The capacity for nitrogen fixation is widely distributed among Bacteria and Archaea (Table 19.4; Section 3.12). Nitrogenases are not specific for N2 and also reduce other triply bonded compounds, such as acetylene, as alternative substrates. Unlike the reduction of N2 to 2 NH3—a six-electron process—the reduction of acetylene by nitrogenase is only a two-electron process, ethylene (H2C═CH2) being the final product. Hence, the reduction of acetylene to ethylene provides a straightforward method for measuring nitrogenase activity (Figure 19.30). This technique, known as the acetylene reduction assay, is widely used in microbiology to detect and quantify nitrogen fixation. Nitrogen fixation can also be assayed directly using the stable (nonradioactive) isotope 15N2 and measuring its reduction to 15NH3; however, acetylene reduction is a more rapid and sensitive method for measuring N2 fixation and can easily be used in laboratory studies of pure cultures or ecological studies of nitrogen-fixing bacteria directly in their habitat. To do this, a sample, which may be soil, water, or a culture, is incubated in a vessel with HC ≡ CH and the gas phase is later analyzed by gas chromatography for the production of H2C═CH2 (Figure 19.30). The unique enzymatic activity of nitrogenase—reduction of a triple bond—provides high confidence that acetylene reduction to ethylene measured in a natural sample is due to the activity of nitrogen-fixing bacteria.
Table 19.4 Some nitrogen-fixing organismsa

aOnly some common genera are listed in each category; many other genera are known. Except where indicated, all organisms are Bacteria.
bNitrogen fixation occurs only under anoxic conditions.
Figure 19.30 The acetylene reduction assay of nitrogenase activity in nitrogen-fixing bacteria.

The results show no ethylene (C2H4) at time 0 but increasing production of C2H4 as the assay proceeds. As C2H4 is produced, a corresponding amount of C2H2 is consumed. Because the reduction of N2 to 2 NH3 requires 6 e− and the reduction of C2H2 to C2H4 only 2 e−, each three C2H4 produced is equivalent to the reduction of one N2.
Microsensors
Microsensors in the form of glass needles containing a sensing mechanism at the tip have been used to study the activity of microorganisms in nature. Microsensors have been constructed that measure many chemical species including pH, O2, NO2 −, NO3 −, nitrous oxide (N2O), CO2, H2, and H2S. As the name microsensor implies, these devices are very small, their tips ranging in diameter from 2 to 100 μm (Figure 19.31). The sensors are carefully inserted into the habitat in small increments to follow microbial activities over very short distances.
Figure 19.31 Microsensors.

(a) Schematic drawing of an oxygen (O2) microsensor. Oxygen diffuses through the silicone membrane in the microsensor tip and reacts with electrons on the gold surface of the cathode, forming hydroxide ions (OH−); the latter generates a current proportional to the O2 concentration in the sample. Note the scale of the electrode. (b) Biological microsensor for the detection of nitrate (NO3 −). Bacteria immobilized at the sensor tip denitrify NO3 − or NO2 − to N2O, which is detected by electrochemical reduction to N2 at the cathode. Based on drawings by Niels Peter Revsbech.
Microsensors have many applications. For example, O2 concentrations in microbial mats (Figure 20.8c), aquatic sediments, or soil particles (Figure 20.3) can be accurately measured over extremely fine intervals using microsensors. A micromanipulator is used to insert the sensors gradually through the sample such that measurements can be taken every 50–100 μm (Figure 19.32). Using a bank of microsensors, each sensitive to a different chemical, simultaneous measurements of several transformations in a habitat can be made.
Figure 19.32 Depth profiles of O2 and NO3 −.

Data obtained using the lander shown in Figure 19.33 equipped with microelectrode sensors for remote chemical characterization of deep-sea sediments. Note the zones of nitrification and denitrification and the fine scale resolution. DNRA, dissimilative nitrate (NO3 −) reduction to ammonia (NH4 +). Based on data and drawings by Niels Peter Revsbech.
Microbial processes in the sea are extensively studied because they have a profound impact on nutrient cycles and the overall health of the planet. As it is difficult to reproduce in the laboratory the conditions found at great depths, it is useful to use microsensors on robotic devices to analyze microbial activities on the seafloor. Figure 19.33 shows deployment of an instrument “lander” equipped with a suite of microsensors to detect various chemicals in the sediment. These data can then be analyzed and compared with that in overlying ocean water to explore the exchange of nutrients and other chemicals between water and sediment.
Figure 19.33 Deployment of a deep-sea lander.

The lander is equipped with a bank of microsensors (arrow) to measure distribution of chemicals in marine sediments.
One of the biologically most important chemical species in the oceans is nitrate (NO3 −), but electrochemical sensors cannot measure NO3 − in seawater, as the high concentrations of salts interfere. To circumvent this problem, a “living” microsensor was designed that contains bacteria within its tip that reduce NO3 − (or NO2 −) to N2O. The N2O produced by the bacteria is then detected following its abiotic reduction to N2 at the cathode of the microsensor (Figure 19.31b); this provides an electrical impulse signaling the presence of NO3 −. In the oxic layer of marine sediments, NO3 − is produced from the oxidation of NH4 + (nitrification, Section 14.9), so there is often a peak of NO3 − in the sediment surface layer (Figure 19.32). In the deeper, anoxic layers of the sediment, NO3 − is consumed by denitrification and dissimilative nitrate reduction to ammonia (DNRA) (Section 14.11), and NO3 − therefore disappears a few millimeters below the oxic–anoxic interface (Figure 19.32).
Sensor Nanoparticles for Spatially Resolved Chemical Imaging
An entirely different sensor approach to measuring biologically significant processes in the environment uses nanotechnology. Traditional microsensors only generate a single point measurement, and because of this, multiple measurements must be made, for example, when measuring depth-related changes in O2 concentration in a marine sediment (Figure 19.32). Hence, this approach is essentially unusable for examining chemical gradients in more than one dimension, such as on an uneven biological surface. To explore such microbial habitats, sensor nanoparticles have been developed that can be sprayed in a thin layer over the uneven surface. These tiny particles (from a few to several hundred nanometers in diameter) made from silica or synthetic polymers incorporate fluorescent or phosphorescent compounds that have a concentration-dependent luminescent response to specific chemicals. Biological nanosensors have been developed for assaying temperature, pH, O2, and a few ions of biological importance, such as Ca2+ and K+.
A study to measure changing O2 distribution over a coral surface demonstrates the power of sensor nanotechnology (Figure 19.34). As we will learn in Chapter 23, stony corals are marine animals whose cells contain the symbiotic dinoflagellate Symbiodinium, an alga that nourishes the coral cells by carrying out photosynthesis. However, Symbiodinium is extremely sensitive to changes in temperature and irradiation associated with climate change. When growth conditions become unfavorable, the symbiont can be expelled and the coral bleached, and this has resulted in the extensive loss of living coral reefs worldwide in recent years (Figure 23.41). To document these changes experimentally, ecological studies of oxygen-producing coral tissues were undertaken to measure the response of the symbiotic alga to changes in light intensity. To do this, a coral surface was covered with a thin layer of nanoparticles containing a fluorescent indicator for oxygen. Following calibration using an inert fluorescent reference dye also incorporated in the particles, the spatial distribution of O2 concentration was determined by taking fluorescent images of the coral under different levels of illumination, where oxygen levels can be seen to change quickly (Figure 19.34). When combined with nanosensors that respond to temperature differences, similar experiments should also be possible to measure coral responses to increasing ocean temperatures, an environmental variable thought to be a major trigger of the massive coral die-offs seen in recent years. By establishing links between coral structural organization, light, temperature, and chemical microenvironments, these analyses should yield important insights into factors that control the coral symbiosis and allow for more accurate predictions of how this important marine symbiosis will respond to climate change.
Figure 19.34 Oxygen nanosensor analysis of coral photosynthetic activity.

(a) Calibration of nanosensor response to different dissolved oxygen concentrations using a fragment of a coral skeleton painted with the nanosensor octaethylporphine ketone platinum (II). (b) Living coral response to transition from darkness (top panel) to light (bottom panel). Note how the oxygen-depleted region due to coral respiration in the dark panel (arrow) quickly becomes oxygenated in the light. (c) Upper panel, photomicrograph of a coral polyp showing the polyp mouth (arrow). The red color is not from nanoparticles but is due to autofluorescence of chlorophyll in Symbiodinium cells present in the polyp and surrounding coral tissue. Lower panel, high-resolution profile from nanosensor data of an oxygen gradient across a polyp similar to that shown in the upper panel following transition to darkness. The scale on the abscissa indicates the small size of a polyp. The figure data and components in parts a and b were generated by Dr. Klaus Koren (Department of Bioscience, University of Aarhus) and Dr. Michael Kühl (Department of Biology, University of Copenhagen) with technical assistance from Sofie Lindegaard Jakobsen (Department of Biology, University of Copenhagen).
Other applications of nanosensor technology have been deployed or are on the horizon. For example, nanoparticles sensitive to both O2 and pH have been used to examine the delivery of oxygen to the root systems of the seagrass Zostera marina. The observation of reduced pH and elevated oxygen close to the seagrass roots was attributed to oxygen delivered by the plant to support the growth of sulfur-oxidizing chemolithotrophs (Sections 14.7 and 15.12), bacteria that oxidize the toxic hydrogen sulfide (H2S); the resulting H2SO4 acidifies the roots, lowering the pH.
Although ecological applications of nanotechnology are still in their infancy, it is likely that in coming years more chemically diverse sensor particles will be developed and their use will become more widespread because of their unique ability to track gradients of microbial activity along uneven surfaces.
Check Your Understanding
How can a microbial ecologist be sure that transformation of a radioisotope is actually caused by microbes?
If a large pulse of organic matter entered the sediment, how would that change the profiles of NO3 − and O2 shown in Figure 19.32?
Suggest additional applications of sensor nanoparticles in environmental microbiology.
19.10 Stable Isotopes and Stable Isotope Probing
Many of the chemical elements have more than one isotope, which differ in their number of neutrons. Certain isotopes are unstable and break down as a result of radioactive decay. Others, called stable isotopes, are not radioactive, but are metabolized differently by microorganisms and can be used to study microbial transformations in nature. There are two methods in which stable isotopes can yield information on microbial activities, isotopic fractionation and stable isotope probing.
Isotopic Fractionation
The two elements most useful for stable isotope studies in microbial ecology are carbon (C) and sulfur (S), although the heavy isotope of nitrogen, 15N, is also widely used. Carbon (C) exists in nature primarily as 12C, but about 5% exists as 13C. Likewise, S with its four stable isotopes exists primarily as 32S. Some S is found as 34S, and very small amounts as 33S and 36S. The relative abundance of these isotopes changes when certain C or S compounds are metabolized by microorganisms because the enzymes that act on these compounds typically favor the lighter isotope of C or S. That is, relative to the lighter isotope, the heavier isotope is discriminated against when both are metabolized by an enzyme (Figure 19.35).
Figure 19.35 Mechanism of isotopic fractionation with C as an example.

Enzymes that fix CO2 preferentially fix the lighter isotope (12C). This results in fixed carbon being enriched in 12C and depleted in 13C relative to substrate CO2. The size of the arrows indicates the relative abundance of each isotope of carbon.
For example, when CO2 is fixed into cell material by an autotrophic organism, the cellular C becomes enriched in 12C and depleted in 13C, relative to an inorganic carbon standard of known isotopic composition. Likewise, the S atom in H2S produced from the bacterial reduction of SO4 2− is isotopically lighter than H2S that has formed geochemically. These discriminations are called isotopic fractionations (Figure 19.35) and are typically the result of biological activities. Thus this technique can be used as a measure of whether a particular transformation has been catalyzed by microorganisms.
The isotopic fractionation of C in a sample is calculated as the extent of 13C depletion relative to a standard having an isotopic composition of geological origin. The standard for C isotope analysis is rocks from a Cretaceous (65- to 150-million-year-old) limestone formation (the Pee Dee belemnite). Because the magnitude of fractionation is usually very small, depletion is calculated as “per mil” (‰, or parts per thousand) and reported as the δ13C (pronounced “delta C 13”) of a sample using the following formula: δ13C=(13C/12C sample)−(13C/12C standard)(13C/12C standard)×1000‰
The same formula is used to calculate the fractionation of S isotopes, in this case using iron sulfide (FeS) mineral from the Canyon Diablo meteorite as the standard: δ34S=(34S/32S sample)−(34S/32S standard)(34S/32S standard)×1000‰
Use of Isotopic Fractionation in Microbial Ecology
The isotopic composition of a material can reveal its biological or geological past. For example, plant material and petroleum (which is derived from plant material) have similar isotopic compositions (Figure 19.36). Carbon from both plants and petroleum is isotopically lighter than the CO2 from which it was formed because the biochemical pathway used to fix CO2 discriminated against 13CO2 (Figures 19.35 and 19.36). Moreover, methane (CH4) produced by methanogenic Archaea (Section 17.2) is isotopically extremely light, indicating that methanogens discriminate strongly against 13CO2 when they reduce CO2 to CH4 (Section 14.15). By contrast, carbon in isotopically heavier marine carbonates is clearly of geological origin (Figure 19.36).
Figure 19.36 Isotopic geochemistry of 13C and 12C.

Note that C fixed by autotrophic organisms is enriched in 12C and depleted in 13C. Methane formed from the reduction of CO2 with H2 by methanogenic Archaea shows extreme isotopic fractionation.
Because of the differences in the proportion of 12C and 13C in carbon of biological versus geological origin, the 13C/12C ratio of rocks of different ages has been used as evidence for or against past biological activity in Earth’s ancient environments. Organic C in rocks as old as 3.5 billion years shows evidence of isotopic fractionation (Figure 19.36), supporting the idea that autotrophic life existed at this time. Indeed, we now believe that the first life on Earth appeared somewhat before this, about 3.8–3.9 billion years ago (Sections 1.5 and 13.1).
The activity of sulfate-reducing bacteria is easy to recognize from their fractionation of stable S isotopes in sulfides (Figure 19.37). As compared with an H2S standard, sedimentary H2S is highly enriched in 32S (depleted in 34S, Figure 19.37). Fractionation during sulfate reduction allows one to identify biologically produced S and has been widely used to trace the activities of sulfur-cycling Bacteria and Archaea through geological time. Sulfur isotopic analyses have also been used as evidence for the lack of life on the Moon. For example, the data in Figure 19.37 show that the isotopic composition of sulfides in lunar rocks closely approximates that of the H2S standard, which represents primordial Earth, and differs from that of microbially produced H2S.
Figure 19.37 Isotopic geochemistry of 34S and 32S.

Note that H2S and S0 of biogenic origin are enriched in 32S and depleted in 34S.
Stable Isotope Probing
Beyond stable isotope fractionation, an alternative stable isotope method called stable isotope probing (SIP) can be used to identify an organism or organisms carrying out the transformation of a nutrient labeled with a specific stable heavy isotope, such as 13C or 15N or even 18O (the lighter, more common isotopes of these elements are 12C, 14N, and 16O, respectively). The idea behind SIP is that the label will be selectively incorporated only into the cellular material of organisms actively metabolizing the nutrient. Then, following the isolation and sequencing of isotopically labeled DNA, the organisms carrying out the transformation can be identified.
Stable isotope probing can be used to ask general to more specific questions. For example, if 13C-labeled benzoate were added to a sediment sample and the sample incubated for an appropriate period, the 13C label would end up in the DNA of the organism (or organisms) that metabolized the benzoate (Figure 19.38). Thus, although all DNA from the sample would be isolated, the 13C-DNA is heavier, albeit only slightly heavier, than 12C-DNA, and this difference is sufficient to separate the DNAs by a special type of centrifugation technique (Figure 19.38). Once the 13C-DNA is isolated, it can be analyzed for phylogenetic genes or metabolic genes to yield genomic information about the benzoate degrader(s) in the sample. If instead of an organic compound, the experimental study focused on nitrogen fixation (conversion of N2 to cell nitrogen, Section 3.12), 15N2 could be supplied to a sample. When nitrogen fixers in the sample incorporate this, they will produce slightly heavier DNA than organisms that cannot fix nitrogen, and the heavier DNA can be isolated and analyzed for genes of interest.
Figure 19.38 Stable isotope probing.

The microbial community in an environmental sample is fed a 13C substrate. Organisms that can metabolize the substrate produce 13C-DNA as they grow and divide; 13C-DNA can be separated from lighter 12C-DNA by density gradient centrifugation (photo). The isolated DNA is then subjected to specific gene analysis or entire genomic analysis.
SIP can also be used in combination with metagenomics to pinpoint organisms carrying out a specific metabolism (from SIP results) in the context of all the other species and metabolisms present in the sample (as revealed by metagenomic results). Besides a phylogenetic “hit” from the SIP results, additional genomic analyses of the labeled DNA could reveal functional genes required for the specific metabolism. Moreover, the phylogenetic and functional results from the SIP experiment could be further confirmed from the metagenomic profile. Clearly many opportunities exist for blending SIP and metagenomics to study important ecological problems.
Check Your Understanding
What is the simplest explanation for why lunar sulfides are isotopically similar to those of the primordial Earth?
What is the expected isotopic composition of carbon in methanotrophs (bacteria that consume CH4)?
How might exchange of metabolites among members of a microbial community complicate interpretation of a stable isotope probe experiment?
19.11 Linking Functions to Specific Organisms
The isotopic methods described thus far used samples containing large numbers of cells to infer that specific metabolisms were occurring within a community or in particular species within the community. These methods give an overview of community activities but do not reveal the contribution of individual cells. To do this, new methods have been developed that can measure the activity and the elemental and isotopic composition of single cells. These are powerful methods for connecting cells of a specific microbial population with a specific activity or ecological niche, but in most cases, the phylogeny of the organisms of interest must be known in order to develop the necessary FISH probes (Section 19.5). Single-cell methods have been of particular importance to the study of metabolite exchange among microorganisms, such as exchanges involved in syntrophic relationships (Section 14.22), where close physical association is essential for efficient exchange.
Single-Cell Metabolisms Imaged by Secondary Ion Mass Spectrometry (SIMS)
Secondary ion mass spectrometry (SIMS) is based on the detection of ions released from a sample placed under a focused high-energy primary ion beam, for example, of cesium (Cs+); from the data generated, the elemental and isotopic composition of released materials can be obtained. When the primary ion beam impacts the sample, most chemical bonds are broken and atoms or polyatomic fragments are ejected from a very thin layer (1–2 nm) of the surface as either neutral or charged particles (secondary ions), a process called sputtering. These secondary ions are directed to a mass spectrometer, an instrument that can determine their mass-to-charge ratio.
NanoSIMS instruments are SIMS devices designed to yield information on single cells. The instrument is equipped with Cs+ and O2 primary beam sources with a resolution of 50 nm for the Cs+ ion beam and 200 nm for the O2 beam. The O2 beam generates positive secondary ions and is used to analyze metals (e.g., Fe, Na, Mg) while the Cs+ beam generates negative secondary ions for the analysis of major cellular elements (C, N, P, S, O, H) and halogens. The NanoSIMS instrument also records where on the specimen the ion beam is directed such that a two-dimensional image of the distribution of specific ions on the sample surface is obtained. In addition, by focusing the ion beam on the same spot during repeated cycles of sputtering, material can be slowly burned away to expose deeper regions of the sample. This high-resolution SIMS analysis is where the term NanoSIMS got its name. NanoSIMS instruments have multiple detectors that provide for the simultaneous analysis of ions of different mass-to-charge ratios originating from the same sample location (**Figure 19.39*a***).
Figure 19.39 NanoSIMS technology.

(a) Schematic of NanoSIMS operation showing the beams of primary (red) and secondary (blue) ions and five different detectors, each of which identifies ions of a different mass-to-charge ratio. (b–d) Demonstration of interspecies nutrient transfer from a filamentous cyanobacterium (Anabaena) to a Rhizobium species attached to the cyanobacterial heterocyst. The coculture was incubated with 15N2, and the transfer of 15N-labeled compounds from Anabaena to Rhizobium was imaged using a combination of enhanced elemental labeling–FISH (EL-FISH, a technique that allows for simultaneous fluorescence and halogen labeling) and NanoSIMS. (b) Total 12C abundance shown by gray tones. (c) 15N enrichment. (d) 19F abundance conferred by a probe that hybridizes only to the attached rhizobial cells (EL-FISH).
When NanoSIMS is combined with FISH (Section 19.5) in a technology called FISH-SIMS, the incorporation of different elements, natural isotopes, or isotope-labeled substrates can be tracked into individual cells of specific cell populations. A variation on the FISH-SIMS method that simplifies the identification of cells scanned by NanoSIMS uses probe-conferred deposition of a halide (Br, F, I), either through direct incorporation of the halide into an oligonucleotide probe or by using a halide-containing tyramide substrate (see CARD-FISH, Section 19.5). Halogens possess a high ionization yield compared with other elements and are thus easy to detect and are typically of low natural abundance. Using this technology, one of the NanoSIMS detectors is dedicated to identifying cells to which the probe has hybridized (Figure 19.39d) by halogen ionization while the remaining detectors are used for assessing elemental composition (Figure 19.39c).
Because of its excellent spatial resolution, NanoSIMS technology is being increasingly used to examine metabolite transfer among single cells of interacting microorganisms. For example, labeling with 15N2 followed by NanoSIMS was used to demonstrate the transfer of N2 fixed by filamentous cyanobacteria to attached heterotrophic bacteria (Figure 19.39c). Labeling with 15NH4 and 13C-labeled CO2 or organic substrates is also being used to explore the assimilation of key nutrients and the transfer of metabolites among microbial species in both aquatic and soil microbial communities.
Raman Microspectroscopy
Raman microspectroscopy can be used to characterize the molecular and isotopic composition of single cells by nondestructive illumination with monochromatic light generated by a laser. Raman is a form of spectroscopy that measures light scattering and can yield both qualitative and quantitative results. Compositional analysis is based on photon scattering after interaction with different cellular components. Although most of the scattered photons have the same energy as the incident photons, a small fraction of scattered photons are shifted in wavelength (relative to the incident wavelength) to either a longer wavelength (a phenomenon known as Stokes Raman scattering) or shorter wavelength (anti-Stokes scattering).
Raman spectrometers separate the more abundant Stokes scattered photons for analysis and, when combined with confocal microscopy (Section 1.9), can generate a compositional spectrum of a single microbial cell (Figure 19.40). Although the spectrum is complex, several compounds and molecules have characteristic peaks that can identify specific cell types, physiological states, or metabolic activities following incorporation of compounds labeled with stable isotopes.
Figure 19.40 Raman microspectroscopic analysis of single cells.

A cell of Acanthamoeba containing FISH-stained chlamydial symbionts (inset photo, arrows point to blue chlamydial cells) and Raman spectroscopy of isolated chlamydial cells. After incubation for 72 and 120 h in medium containing 13C phenylalanine, symbionts were released by lysis of the amoeba and their Raman spectra recorded. Peaks diagnostic for labeled phenylalanine are shown in red. Since the Raman wave number of 13C phenylalanine at 967 cm−1 is well resolved from other spectral features, the ratio of 1003 cm−1 (unlabeled phenylalanine, at *) to 967 cm−1 (labeled phenylalanine) peak areas corresponds to the relative amount of labeled phenylalanine incorporated by single cells (note how this increases with time). Other red peaks correspond to spectral peaks specific for different chemical bonds in 13C phenylalanine. Also shown are selected peaks and their wave numbers for other cellular components of the symbiont.
Major advantages of Raman microspectroscopy include the following: It is nondestructive and can be used on living cells; water does not cause interference; it can be combined with FISH; and cells of interest can be further manipulated, for example, through capture by laser tweezers (Section 19.3). In the example of Raman shown in Figure 19.40, the incorporation of phenylalanine (an amino acid) by an obligate chlamydial symbiont of an amoeba has been tracked following addition of phenylalanine labeled with 15N and 13C to the amoeba growth medium. A less specific but still useful application of Raman is based on the incorporation of heavy protons from deuterated water (2H2O). Since water is a reactant in many biochemical reactions, metabolically active microorganisms that incorporate the heavy hydrogen atom can be identified by Raman microspectroscopy for further processing, including characterization by single-cell genomics (Section 10.11).
Radioisotopes in Combination with FISH: Microautoradiography-FISH
Radioisotopes are used as measures of microbial activity in a microscopic technique called microautoradiography (MAR). In this method, cells from a microbial community are exposed to a substrate containing a radioisotope, such as an organic compound or CO2. Heterotrophs take up the radioactive organic compounds and autotrophs take up the radioactive CO2. Following incubation in the substrate, cells are affixed to a slide and the slide is dipped in photographic emulsion. While the slide is left in darkness for a period, radioactive decay from the incorporated substrate induces formation of silver grains in the emulsion; these appear as black dots above and around the cells. **Figure 19.41*a*** shows a MAR experiment in which an autotrophic cell has taken up 14CO2.
Figure 19.41 MAR-FISH.

Fluorescence in situ hybridization (FISH) combined with microautoradiography (MAR). (a) An uncultured filamentous cell belonging to the Gammaproteobacteria (as revealed by FISH) is shown to be an autotroph (as revealed by MAR-measured uptake of 14CO2). (b) Uptake of 14C-glucose by a mixed culture of Escherichia coli (yellow cells) and Herpetosiphon aurantiacus (filamentous green cells). (c) MAR of the same field of cells shown in part b. The radioactivity of incorporated glucose exposes the film and shows that glucose was assimilated mainly by cells of E. coli.
Microautoradiography can be done simultaneously with FISH (Section 19.5) in MAR-FISH, a powerful technique that combines identification with activity measurements. MAR-FISH allows a microbial ecologist to determine (by MAR) which organisms in a natural sample are metabolizing a particular radiolabeled substance while at the same time identifying these organisms (by FISH) (Figure 19.41b, c). MAR-FISH thus goes a step beyond phylogenetic identification by revealing physiological information about the organisms, as is also true of NanoSIMS. Such data are useful not only for understanding the activity of the microbial ecosystem but also for guiding enrichment cultures. For example, knowledge of the phylogeny and morphology of an organism metabolizing a particular substrate in a natural sample can be used to design an enrichment protocol to isolate the organism. In addition, MAR-FISH results can be quantified by counting the silver grains as a measure of the amount of substrate consumed by single cells, allowing the activity distribution in a community to be described. The technique is limited only by the availability of suitable radioactive isotopes. For example, although C-labeled substrates work well, it is not feasible to track N incorporation using MAR-FISH because the radioactive isotope 13N has a very short half-life. However, it is feasible to track N incorporation using the nonradioactive stable isotope 15N with NanoSIMS, as we saw earlier (Figure 19.39).
Check Your Understanding
How could NanoSIMS be used to identify a nitrogen-fixing bacterium?
Why is Raman microspectroscopy suited for the selective isolation of microorganisms and NanoSIMS is not?
How does MAR-FISH link microbial diversity and activity?
19.12 Linking Genes and Cellular Properties to Individual Cells
19.12 Linking Genes and Cellular Properties to Individual Cells
19.12 Linking Genes and Cellular Properties to Individual Cells
We have seen in the previous section how the combination of FISH with MAR or FISH with NanoSIMS allows for analyses of both microbial diversity and activity. Coupled with advanced DNA sequencing methods that can determine a genome sequence from the DNA contained in a single cell (Section 10.11), these techniques are at the cutting edge of microbial ecology today. Improvements in single-cell DNA sequencing technology, combined with high-throughput analysis and isolation of single cells by flow cytometry, now allow for gene identification and selected physiological analyses (e.g., size and intrinsic fluorescence) to be performed on selected populations and single cells in the environment.
Flow Cytometry and Multiparametric Analyses
Because of the large population sizes of natural microbial communities, methods that rely on microscopy can examine only a very small part of a whole community. It is difficult to assess cell numbers by counting cells microscopically, and this problem is compounded if populations are present in low numbers. However, flow cytometry (Section 19.3) offers an alternative to more labor-intensive microscopic methods.
Mastering Microbiology
Art Activity: Figure 19.36 Flow cytometric cell sorting
Flow cytometers can examine specific cell parameters such as size, shape, or fluorescent properties as the cells pass through a detector at rates of many thousands of cells per second (Figure 19.42). Fluorescence may be intrinsic (for example, chlorophyll fluorescence of phototrophic microorganisms); or it may be conferred by DNA staining, by differential staining of live versus dead cells (vital stains), or by fluorescent DNA probes (FISH or BONCAT-FISH), all methods discussed earlier in this chapter.
Figure 19.42 Flow cytometric cell sorting.

As the fluid stream exits the nozzle, it is broken into droplets containing no more than a single cell. Droplets containing desired cell types (detected by fluorescence or light scatter) are charged and collected by redirection into collection tubes or microtiter plates by positively or negatively charged deflection plates. DNA recovered from a specific population of cells or amplified from a single cell (Figure 10.30) is characterized by PCR amplification and sequencing of specific genes or by full genome sequence analysis.
A major advantage of flow cytometry is the ability to carry out multiparametric analyses, that is, the capacity to combine multiple parameters in the analysis of a microbiological sample or to sort cells in order to find a specific population. A good example of this was the discovery in the late 1980s of a novel and abundant community of marine cyanobacteria, all species of the genus Prochlorococcus. Prochlorococcus cells are smaller and have different fluorescent properties than another common marine cyanobacterium, Synechococcus. Based on differences in size and fluorescence, flow cytometry resolved these two populations and Prochlorococcus was subsequently shown to be the predominant oxygenic phototroph in ocean waters between 40°S and 40°N latitude, reaching concentrations greater than 105 cells/ml. Metagenomics has also been used to identify the unique genomic features of different natural populations of Prochlorococcus (Figure 19.24). These findings have led to the conclusion that Prochlorococcus is the most abundant phototrophic organism on Earth. We discuss the biology of Prochlorococcus in more detail in Section 20.11.
Single-Cell Genomics
A major stumbling block in a PCR-based gene recovery method is the requirement that a specific gene that will react with the primers used in the amplification be identified prior to analysis. Newer methods of DNA amplification now provide an alternative method for associating specific genes with a specific organism without the problems and biases associated with PCR. These methods employ single-cell genomics (Section 10.11), one of the more recent tools to enter the microbial ecologist’s toolbox. As discussed in Chapter 10, when combined with methods for recovery of single cells and high-throughput DNA sequencing methods, single-cell genome sequencing provides a powerful tool for linking specific metabolic functions to individual cells that have never been grown in laboratory culture.
Multiple displacement amplification (MDA) (Figure 19.43) is key to single-cell genomics because it can amplify chromosomal DNA from a single cell isolated from a natural environment using a cell sorting technique, such as flow cytometry (Figure 19.42). MDA uses a specific bacteriophage DNA polymerase to initiate replication of cell DNA at random points in the chromosome, displacing the complementary strand as each polymerase molecule synthesizes new DNA. The phage polymerase has strong strand displacement activity, resulting in the synthesis of numerous high-molecular-weight DNA products at very high fidelity. The number of genome copies produced by MDA is usually sufficient to assemble the complete, or nearly complete, genome of the cell that yielded the DNA using next-generation sequencing platforms and powerful sequence analysis software. In this way, both phylogenetic and metabolic functions can be inferred from the genome sequence and PCR is not required. PCR can be a problem if there exists a bias in the genome against the primers used. MDA avoids this by annealing random rather than specific primers to the genomic DNA such that amplification of the entire genome is highly likely.
Figure 19.43 Genetic analyses of sorted cells.

DNA is recovered from a specific population of cells following FISH labeling and flow cytometric sorting (Figure 19.42). DNA is characterized by PCR amplification and sequencing of specific genes, or by amplification of the entire genome by multiple displacement amplification (MDA) followed by sequencing. For MDA, an amount of DNA sufficient for full genome sequence determination is produced using short DNAs of random sequence as primers (A) to initiate genome replication by a bacteriophage DNA polymerase. The bacteriophage polymerase copies DNA from multiple points in the genome and also displaces newly synthesized DNA (B, C), thereby freeing additional DNA for primer annealing and (D) initiation of polymerization.
Not surprisingly, MDA requires stringent control over purity to eliminate contaminating DNA, but when combined with high-throughput DNA sequencing methods, MDA provides a powerful tool for linking specific metabolic functions to individual cells that have thus far eluded laboratory culture. Information about the metabolic capacities of these uncultured organisms can then be used to develop strategies to recover them by either classical enrichment and isolation methods (Sections 19.1 and 19.2) or by any of the several single-cell isolation culturing techniques now available to tease out individual cells and get them growing in the laboratory (Section 19.3 and Figure 19.42).
A Preview of What’s to Come
Now that we have some background on how we can track and identify microbes in nature and assess their activities, we will use these tools as we move on to the next chapter to consider where microbes actually live in nature. In Chapter 20 we will link microbial diversity with major habitats to answer the questions “Who is where?” and “Why are they there?” In Chapter 21 we will connect diversity with metabolism to explore the major nutrient cycles in nature that are driven by microbial activities. In Chapter 22 we will consider microbes and their habitats once again but in the context of microbes that reside in our buildings, subways, and other major structures constructed by humans. Chapters 23 and 24 also link microbes to habitats but in the context of the many symbiotic associations that microbes have developed with plants and animals, including ourselves. Background on where microbes live in nature and why is the natural introduction to the final two units in this text where we focus on microbe–human relationships, both the highly beneficial and the life-threatening.
Mastering Microbiology
Art Activity: Figure 19.37 Genetic analysis of sorted cells
Check Your Understanding
Compared with microscopy, what are the advantages and disadvantages of flow cytometry for characterizing a microbial community?
What key method is required to do genomics on a single cell?
Chapter Review
Go to Mastering Microbiology for videos, animations, practice tests, and more.
I Culture-Dependent Analyses of Microbial Communities
19.1 The enrichment culture technique is a means of obtaining microorganisms from natural samples. Successful enrichment and isolation prove that an organism of a specific metabolic type was present in the sample, but do not indicate its ecological importance or abundance. Enrichments following dilution of the sample often yield different organisms than enrichments with undiluted samples.
Q Why do the results of a direct enrichment of an environmental sample and enrichment following dilution of the sample often differ with respect to the types of populations recovered?
19.2 Once a successful enrichment culture has been established, pure cultures can often be obtained by conventional microbiological procedures, including streak plates, agar dilution, and liquid dilution methods.
Q What criteria serve to demonstrate that a culture of a previously undescribed microorganism is pure?
19.3 Several methods are available to isolate and culture single cells. Laser tweezers allow one to isolate a cell from a microscope field and move it away from contaminants. Flow cytometric sorting combined with high-throughput culturing technology allow for isolated cells to be cultured in a large variety of culture media simultaneously to identify the resources and conditions best suited to the growth of the isolated cell.
Q What feature of high-throughput culturing relieves the human demands that would otherwise be required to set up and monitor huge numbers of individual cultures?
II Culture-Independent Microscopic Analyses of Microbial Communities
19.4 DAPI, acridine orange, and SYBR Green are general stains for quantifying microorganisms in natural samples. Some stains can differentiate live versus dead cells. The GFP makes cells autofluorescent and is a means for tracking cells introduced into the environment and reporting gene expression. In natural samples, morphologically identical cells may actually be genetically distinct.
Q What limits the application of GFP for general studies of microbial activity and distribution in the environment?
19.5 FISH methods have combined the power of nucleic acid probes with fluorescent dyes and are thus highly specific in their staining properties. FISH methods include phylogenetic stains, CARD-FISH, and BONCAT-FISH. Using fluorescence microscopy, CARD-FISH can identify transcriptionally active cells while BONCAT-FISH can identify translationally active cells.
Q Compare and contrast CARD-FISH and BONCAT-FISH. Why are these methods more suitable than FISH for characterizing very slowly growing microorganisms in the environment?
III Culture-Independent Molecular Analyses of Microbial Communities
19.6 PCR can be used to amplify specific target genes such as rRNA genes or key metabolic genes for subsequent analysis of community structure and potential functions. DGGE and T-RFLP can identify the different variants of these genes among the species in a community. Application of ARISA is limited to amplification of the internal transcribed spacer region separating the 16S and 23S rRNA genes.
Q Which method, ARISA or T-RFLP, would provide more detail about microbial community complexity? Why?
19.7 In current application, microarrays used for environmental studies primarily consist of thousands of DNA probes that hybridize to specific genes encoding key biochemical processes.
Q Why might a microarray be superior to using high-throughput sequencing to quantify both abundant and rare gene sequences in a complex microbial community? What are the limitations of functional microarrays, such as the GeoChip, for analysis of microbial communities?
19.8 Environmental multi-omics uses different combinations of metagenomics, metatranscriptomics, metaproteomics, and metabolomics to derive deeper understanding of how microbial diversity relates to the complex metabolic food webs sustaining natural microbial communities and associated biogeochemical processes.
Q Give an example of how multi-omics has contributed to a better understanding of microbial food web structure. Why is the use of environmental genomic information alone often insufficient to discover new biochemical properties?
IV Measuring Microbial Activities in Nature
19.9 The activity of microorganisms in natural samples can be assessed very sensitively using radioisotopes or microsensors, or both. The measurements obtained give the net activity of the microbial community.
Q What are the major advantages of radioisotopic methods in the study of microbial ecology? What type of controls (discuss at least two) would you include in a radioisotopic experiment to show 14CO2 incorporation by cyanobacteria or to show 14C-glucose incorporation by anaerobic bacteria?
19.10 Natural isotopic composition, the result of isotopic fractionation by enzymes that discriminate against the heavier form of an element, can reveal the biological origin and/or biochemical mechanisms involved in the formation of various substances. Stable isotope probing (SIP) uses compounds labeled with isotopes not naturally abundant to identify microorganisms metabolizing and assimilating the compound added to a community.
Q Will autotrophic organisms contain more or less 12C in their organic compounds than was present in the CO2 that fed them? Why would SIP using 15NO3 − not be useful for identifying bacteria carrying out nitrate respiration?
19.11 A variety of advanced technologies such as NanoSIMS, MAR-FISH, BONCAT-FISH, and Raman microspectroscopy make it possible to examine metabolic activity, gene content, and gene expression of single cells in natural microbial communities. NanoSIMS employs secondary ion mass spectrometry technology. MAR-FISH combines the uptake of radiolabeled substrates (MAR) along with phylogenetic identification (FISH). Raman microspectroscopy is a nondestructive method (retaining cell viability).
Q What can MAR-FISH tell you that FISH alone cannot? How might you combine SIP and NanoSIMS to identify novel methane-consuming cells in a natural community?
19.12 Flow cytometry combined with cell sorting can rapidly evaluate many thousands of single cells in a natural environment for basic cellular properties (size, shape) or gene content (using specific fluorescent probes). Single-cell genomics incorporates methods for use on individual cells from a natural microbial community, for example, by flow cytometric isolation of cells and multiple displacement amplification to obtain genome sequences of single cells.
Q How would you use cytometric cell sorting to evaluate genome sequence variation among a population of marine bacteria present in low abundance?
Application Questions
Design an experiment for measuring the activity of sulfur-oxidizing bacteria in soil. If only certain species of the sulfur oxidizers present were metabolically active, how could you tell this? How would you prove that your activity measurement was due to biological activity?
You wish to know whether Archaea exist in a lake water sample but are unsuccessful in culturing any. Using techniques described in this chapter, how could you determine whether Archaea exist in the sample, and if they do, what proportion of the cells in the lake sample are Archaea?
Design an experiment to solve the following problem: Determine the rate of methanogenesis (CO2+4 H2→CH4+2 H2O) in anoxic lake sediments and whether or not it is H2-limited. Also, determine the morphology of the dominant methanogen (recall that these are Archaea, Section 17.2). Finally, calculate what percentage the dominant methanogen is of the total archaeal and total prokaryotic populations in the sediments. Remember to specify necessary controls.
Design a SIP experiment that would allow you to determine which organisms in a lake water sample were capable of oxidizing the hydrocarbon hexane (C6H14). Assume that four different species could do this. How would you combine SIP with other molecular analyses to identify these four species?
Chapter Glossary
a nonspecific fluorescent dye used to stain DNA in microbial cells in a natural sample BONCAT-FISH
a method to identify translationally active cells labeled by FISH by incorporating a noncanonical amino acid into their proteins followed by coupling it to a fluorescent reporter molecule (BONCAT stands for bioorthogonal noncanonical amino acid tagging) DAPI
a nonspecific fluorescent dye that stains DNA in microbial cells; used to obtain total cell numbers in natural samples Denaturing gradient gel electrophoresis (DGGE)
an electrophoretic technique capable of separating nucleic acid fragments of the same size that differ in base sequence Enrichment bias
a problem with enrichment cultures in which “weed” species tend to dominate in the enrichment, often to the exclusion of the most abundant or ecologically significant organisms in the inoculum Enrichment culture
a culture that employs highly selective laboratory methods for obtaining microorganisms from natural samples Environmental genomics (metagenomics)
the use of genomic methods (sequencing and analyzing genomes) to characterize natural microbial communities Exometabolite
a product of metabolism released from a cell that may be metabolized by other organisms Flow cytometry
a technique for counting and examining microscopic particles by suspending them in a stream of fluid and passing them by an electronic detection device Fluorescence in situ hybridization (FISH)
a method employing a fluorescent dye covalently bonded to a specific nucleic acid probe for identifying or tracking organisms in the environment Fluorescent protein
any of a large group of proteins that fluoresce different colors, including the green fluorescent protein, for tracking genetically modified organisms and determining conditions that induce the expression of specific genes Fundamental niche
the range of environments in which a species will be sustained when it is not resource-limited, such as may result from competition with other species Green fluorescent protein (GFP)
a protein that fluoresces green and is widely used in genetic analysis High-throughput culturing methods
the use of microtiter plates whose wells contain various culture media that can be inoculated with single cells whose growth or target gene content is measured robotically Isotopic fractionation
the discrimination by enzymes against the heavier isotope of the various isotopes of C or S, leading to enrichment of the lighter isotopes Laser tweezers
a device for obtaining pure cultures by optically trapping a single cell with a laser beam and moving it away from surrounding cells into sterile growth medium MAR-FISH
a technique that combines identification of microorganisms by FISH with measurement of metabolic activities by microautoradiography (MAR) Metabolomics
the comprehensive analysis of cellular and extracellular metabolites of a cell, organism, or microbial community Metaproteomics
the measurement of whole-community protein expression using mass spectrometry to assign peptides to the amino acid sequences encoded by unique genes Metatranscriptomics
the measurement of whole-community gene expression using RNA sequencing Microautoradiography (MAR)
the measurement of the uptake of radioactive substrates by visually observing the cells in an exposed photographic emulsion Microbial ecology
the study of the interaction of microorganisms with each other and their environment Microfluidic devices
miniaturized systems for fluid handling that are increasingly used for high-throughput culturing of microorganisms Microsensor
a small glass sensor or electrode for measuring pH or specific compounds such as O2, H2S, or NO3 − that can be immersed into a microbial habitat at microscale intervals Most-probable-number (MPN) technique
the serial dilution of a natural sample to determine the highest dilution yielding growth Multi-omics
the integration of data from multiple omics platforms to more fully characterize microbial community structure and function Multiple displacement amplification (MDA)
a method to generate multiple copies of chromosomal DNA from a single organism Nucleic acid probe
a strand of nucleic acid that can be labeled and used to hybridize to a complementary molecule from a mixture of other nucleic acids Phylotype
one or more organisms with the same or related sequences of a phylogenetic marker gene Realized niche
the range of natural environments supporting a species when that organism is confronted with factors such as resource limitation, predation, and competition from other species Stable isotope probing (SIP)
a method for characterizing an organism that incorporates a particular substrate by supplying the substrate in 13C or 15N form and then isolating heavy isotope–enriched DNA and analyzing the genes Winogradsky column
a glass column packed with mud and overlaid with water to mimic an aquatic environment, in which various bacteria develop over a period of months