An Uncertain Future for Coral Reef Ecosystems
Carbon dioxide (CO2) released from the burning of fossil fuels is contributing to global warming and ocean acidification. Although all ecosystems will ultimately be impacted by this, coral reef ecosystems are particularly sensitive. About a quarter of the oceans’ coral reefs have already been lost to thermal stress and pollution, and ongoing acidification is impairing their formation of calcium carbonate skeletons. Although occupying less than 1% of the ocean floor, coral reefs support about one-quarter of all marine species that provide food for millions of people. Thus it is essential that we quantify current rates of coral reef loss and predict potential future losses.

In an experimental study of adding CO2 to ocean waters bathing a natural coral reef ecosystem, it was clear that even a minor reduction in pH greatly reduces the rate of biogenic calcification. This study, performed at a section of the Great Barrier Reef (Australia), used tidal flow from an upper to a lower lagoon to adjust the pH of water bathing the lower section of reef. A 4000-gallon floating tank of seawater was first acidified by bubbling with CO2 and then pumped from the edge of the upper lagoon during tidal change, reducing the pH in the lower lagoon from ambient pH (8.13) to a treatment level (7.98). A nonreactive red dye was added before release to follow the dilution of the acidified plume as it mixed with seawater (photo).
In this experiment, calcification rates were quantified by measuring changes in alkalinity (due to carbonate production) over a one-hour period in the pH-adjusted water compared with seawater maintained at the original pH. Remarkably, net calcification of this natural reef community was reduced 34% by this relatively small change in pH, showing that these remarkable and very sensitive ecosystems are in jeopardy unless a rapid reduction in global CO2 emissions is initiated.
Source: Rebecca Albright, Yuichiro Takeshita, and David A. Koweek, et al. 2018. Carbon dioxide addition to coral reef waters suppresses net community calcification. Nature 555: 516.
I Carbon, Nitrogen, and Sulfur Cycles
The microbial cycling of carbon, nitrogen, and sulfur compounds provides essential nutrients for autotrophs. The organic matter these organisms produce is oxidized by heterotrophs and by human activities to CO2, an excess of which contributes to the ongoing problem of global warming.
The key nutrients for life are cycled by both microorganisms and macroorganisms, but for each nutrient it is microbial activities that dominate. Understanding how microbial nutrient cycles work is important because the cycles and their many feedback loops are essential for plant agriculture and the overall health of a sustainable planet.
We begin our coverage of nutrient cycles with the carbon cycle. Major areas of interest here are the magnitude of carbon reservoirs on Earth, the rates of carbon cycling within and between reservoirs, and the coupling of the carbon cycle to other nutrient cycles. We emphasize the gases carbon dioxide (CO2) and methane (CH4) because they are major components of the carbon cycle and have major impacts on the global ecosystem. We return to the carbon cycle in Part III of this chapter to consider how human activities are affecting this critical nutrient cycle.
21.1 The Carbon Cycle
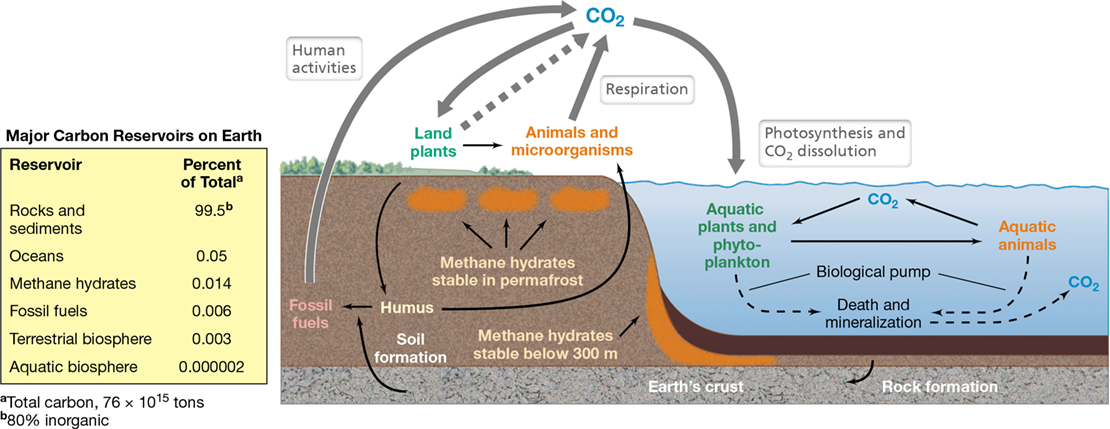
On a global basis, carbon (C) travels as CO2 through all of Earth’s major carbon reservoirs: the atmosphere, the land, the oceans, freshwaters, sediments and rocks, and biomass (Figure 21.1). As we have already seen for freshwater environments, the carbon and oxygen cycles are intimately linked (Section 20.9). All nutrient cycles link in some way to the carbon cycle, but the nitrogen (N) cycle links particularly strongly because, other than water (H2O), C and N make up the bulk of living organisms (Section 4.1 and see Figure 21.6).
Figure 21.1 The carbon cycle.


The carbon and oxygen cycles are closely connected, as oxygenic photosynthesis both removes CO2 and produces O2, and respiration both produces CO2 and removes O2. The accompanying table shows that the greatest reservoir of carbon on Earth is in rocks and sediments, and most of this is in inorganic form as carbonates.
Carbon Reservoirs
By far the largest C reservoir on Earth is the sediments and rocks of Earth’s crust (Figure 21.1), but the rate at which sediments and rocks decompose and carbon is removed as CO2 is so slow that flux out of this reservoir is insignificant on a human time scale. A large amount of C is found in land plants. This is the organic C of forests, grasslands, and agricultural crops—the major sites of phototrophic CO2 fixation. However, more C is present in dead organic material, called humus, than in living organisms. Humus is a complex mixture of organic materials that have resisted rapid decomposition and is derived primarily from dead plants and microorganisms. Some humic substances are quite recalcitrant, with a decomposition time of several decades, but certain other humic components decompose much more rapidly.

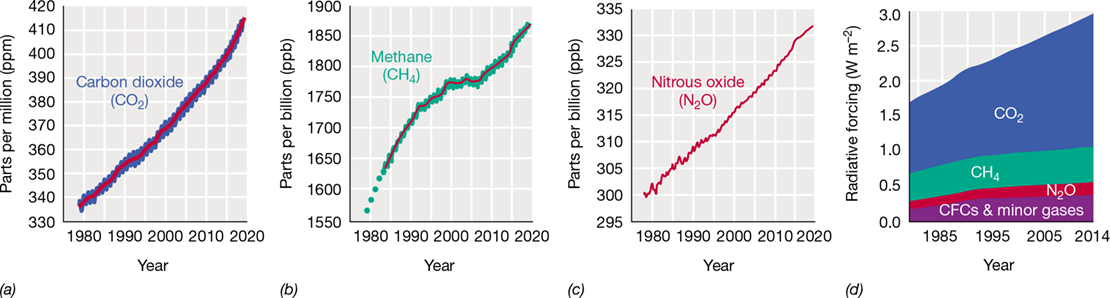
The most rapid means of transfer of C is via the atmosphere. Carbon dioxide is removed from the atmosphere primarily by photosynthesis of land plants and marine microorganisms and is returned to the atmosphere by respiration of animals and chemoorganotrophic microbes (Figure 21.1). The single most important contribution of CO2 to the atmosphere is by microbial decomposition of dead organic material. However, since the Industrial Revolution, human activities have increased atmospheric CO2 levels by nearly 40%, primarily from the combustion of fossil fuels. This rise in CO2, a major greenhouse gas, has triggered global changes in climate, the most notable feature of which is steadily increasing global temperatures, a condition called global warming (see Section 21.9 and Figure 21.24). That global warming will affect microbial nutrient cycling is a virtual certainty because everything we know about the physiology of microorganisms tells us that microbial activities change in response to temperature. Whether these changes will be favorable or unfavorable to higher organisms (including humans) is a major area of active research today (Section 21.9).
Photosynthesis and Decomposition
New organic compounds are biologically synthesized on Earth only by CO2 fixation by autotrophs—the phototrophs and chemolithotrophs. Most organic compounds originate in photosynthesis and thus phototrophic organisms are the foundation of the carbon cycle (Figure 21.1). However, phototrophic organisms are abundant in nature only in habitats where light is available. The deep sea, deep terrestrial subsurface, and other permanently dark habitats are devoid of indigenous phototrophs. There are two groups of oxygenic phototrophic organisms: plants and microorganisms. Plants are the dominant phototrophic organisms of terrestrial environments, whereas phototrophic microorganisms dominate in aquatic environments.
The redox cycle for C (Figure 21.2) begins with photosynthetic CO2 fixation, driven by the energy of light (Sections 3.11, 3.12, 14.5, and 14.6): CO2+H2O→(CH2O)+O2
Figure 21.2 Redox cycle for carbon.
The diagram contrasts autotrophic processes (CO2→organic compounds) and heterotrophic processes (organic compounds→CO2).
CH2O represents organic matter at the oxidation–reduction level of cell material. Phototrophic organisms also carry out respiration, both in the light and the dark. The overall equation for respiration is the reverse of oxygenic photosynthesis: (CH2O)+O2→CO2+H2O
For organic matter to accumulate, the rate of photosynthesis must exceed the rate of respiration. In this way, autotrophic organisms build biomass from CO2, and then this biomass in one way or another supplies the C heterotrophic organisms need. Anoxygenic phototrophs and chemolithotrophs also produce excess organic compounds, but in most environments the contributions of these organisms to the net accumulation of organic matter are minor compared to the inputs of oxygenic phototrophs. This is because the reductant used by oxygenic phototrophs, H2O (Section 14.3), is in virtually unlimited supply.
Organic compounds are degraded biologically to CH4 and CO2 (Figure 21.2). Carbon dioxide, most of which is of microbial origin, is produced by aerobic and anaerobic respirations (Sections 3.8 and 3.9). Methane is produced in anoxic environments by methanogens from the reduction of CO2 with hydrogen (H2) or from the splitting of acetate into CH4 and CO2 (Section 14.15). However, any naturally occurring organic compound can eventually be converted to CH4 from the cooperative activities of methanogens and various fermentative bacteria, as we will see in Section 21.2. Methane produced in anoxic habitats is insoluble and most often diffuses rapidly to oxic environments where it is either released to the atmosphere or oxidized to CO2 by methanotrophs (Sections 14.16 and 15.15; Figure 21.2). Hence, most of the C in organic compounds eventually returns to CO2, and the links in the carbon cycle are closed.
Methane Hydrates
Although present in the atmosphere at lower levels than CO2, CH4 is a potent greenhouse gas that is over 20 times more effective in trapping heat than is CO2. Some CH4 enters the atmosphere from methanogenic production, but not all biologically produced CH4 is immediately consumed or released to the atmosphere. Huge amounts of CH4 derived primarily from past microbial activities are trapped underground or under marine sediments as methane hydrates (Figure 21.1 and Figure 21.3), molecules of frozen CH4. Methane hydrates form when sufficient CH4 is present in environments of high pressure and low temperature, such as beneath the permafrost in the Arctic and in marine sediments (Figure 21.1). These deposits can be up to several hundred meters thick and are estimated to contain 700–10,000 petagrams (1 petagram×1015 g) of CH4. This exceeds other known CH4 reserves on Earth by several orders of magnitude.
Figure 21.3 Burning methane hydrate.
Frozen methane ice retrieved from marine sediments is ignited.
Mastering Microbiology
Methane hydrates are highly dynamic, absorbing and releasing CH4 in response to changes in pressure, temperature (Figure 21.4), and fluid movement. Methane hydrates also fuel deep-water ecosystems, called either cold seeps or methane seeps (Figure 20.24). Here, the slow release of CH4 from seafloor hydrates nourishes not only anaerobic methane-oxidizing Archaea (Section 14.16), but also animal communities that contain aerobic methane-oxidizing endosymbionts that oxidize CH4 and release organic matter to the animals (Section 23.11). Anaerobic oxidation of CH4 is coupled to the reduction of sulfate (SO4 2−), nitrate (NO3 −), and iron and manganese oxides, reducing the amount of free methane released. Hydrogen sulfide (H2S), produced by the oxidation of methane coupled to sulfate reduction (Section 14.16), can nourish dense microbial mats of aerobic filamentous sulfur-oxidizing bacteria (Section 14.7) on the seafloor overlaying methane hydrates (Figure 21.5; see also Section 20.5 and Figure 20.10).
Figure 21.4 Seasonal flares of methane bubbling from methane hydrates.

Methane hydrates in shallow coastal sediments are sensitive to seasonal changes in bottom water temperature. Flares of methane bubbles are observed when water temperatures warm by as little as 1–2 °C.
Figure 21.5 Sulfur-oxidizing microbial mats overlying a deep-sea methane seep.

This seep is located at 800 m depth in the Pacific Ocean, off Los Angeles, California. The oxidation of methane coupled to sulfate reduction in the subsurface produces abundant H2S, which feeds a sediment surface community of filamentous sulfide-oxidizing bacteria. The bacteria are filled with inclusions of elemental sulfur (S0), leading to the whitish color. The dark area near the bottom center is about 0.5 m long.
Although deep oceanic methane hydrates are stabilized by high pressure, hydrates in shallower coastal sediments are much more sensitive to small changes in temperature (Figure 21.4). Hydrates in shallow sediments are at the margin of what is called the gas hydrate stability zone (GHSZ). In these marginal regions, relatively small changes in temperature can destabilize the hydrates. For example, on-site monitoring of hydrate deposits in a marine coastal plain showed them to be sensitive to 1–2 °C seasonal changes in bottom-water temperature. During periods of seasonally elevated water temperature, methane bubbles freely (a phenomenon called methane flares) from the sediments (Figure 21.4). In addition to the release of methane from marine and terrestrial hydrates, as permafrost melts, its huge reserve of organic matter could be catabolized by microbes, leading eventually to the production of yet more methane (Section 21.9).
Carbon Balances and Coupled Cycles
Although it is convenient to consider carbon cycling as a series of reactions separate from those in other nutrient cycles, in reality, all nutrient cycles are coupled cycles; major changes in one cycle affect the functioning of others. But certain cycles, such as the carbon and nitrogen cycles (Figure 21.6), are extremely closely coupled because of the large amount of C and N in living organisms. The rate of primary productivity (CO2 fixation) is controlled by several factors, in particular by the magnitude of photosynthetic biomass and by available N, often a limiting nutrient. Thus, large-scale reductions in biomass, for instance by widespread deforestation, reduce rates of primary productivity and increase levels of CO2. High levels of organic C stimulate microbial nitrogen fixation (N2→NH3, Section 3.12), and this in turn adds more fixed N to the pool for primary producers; low levels of organic C have just the opposite effect (Figure 21.6). High levels of ammonia (NH3) stimulate primary production and nitrification (Section 14.9), but inhibit nitrogen fixation. High levels of nitrate (NO3 −), an excellent N source for plants and aquatic phototrophs, stimulate primary production but also increase the rate of denitrification (Section 14.11); the latter removes fixed forms of N from the environment and feeds back in a negative way on primary production (Figure 21.6).
Figure 21.6 Coupled cycles.

All nutrient cycles are interconnected, but the carbon and nitrogen cycles are intimately coupled. In the carbon cycle, CO2 supplies the C for carbon compounds. The N cycle, shown in more detail in Figure 21.9, supplies N for many biological compounds.
Mastering Microbiology
This simple example illustrates how nutrient cycles are anything but isolated entities. Instead, they are coupled systems that must maintain a delicate balance of inputs and outputs. Thus, one could expect the C and N cycles to respond to large inputs in specific components (for example, through inputs of CO2 or nitrogen fertilizers) in ways that are not always beneficial to the biosphere and that can have unintended consequences for all life, including humans (Section 21.9).
Check Your Understanding
In what ways are oxygenic photosynthesis and respiration related?
21.2 Syntrophy and Methanogenesis
Most organic compounds are oxidized in nature by aerobic microbial processes. However, because oxygen (O2) is a poorly soluble gas and is actively consumed when available, much organic carbon still ends up in anoxic environments. Methanogenesis, the biological production of CH4, is a major process in anoxic habitats and is catalyzed by a large group of Archaea, the methanogens, which are strict anaerobes. We discussed the biochemistry of methanogenesis in Section 14.15 and other aspects of methanogens in Section 17.2.
Most methanogens can use CO2 as a terminal electron acceptor in anaerobic respiration, reducing it to CH4 with H2 as electron donor. Only a very few other substrates, chiefly acetate, are directly converted to CH4 by methanogens (Section 14.15). To convert most organic compounds to CH4, methanogens must team up with partner organisms called syntrophs that function to supply them with precursors for methanogenesis.
Anoxic Decomposition and Syntrophy
In Section 14.22 we discussed the biochemistry of syntrophy, a process in which two or more organisms cooperate in the anaerobic degradation of organic compounds. Here we consider the interactions of syntrophic bacteria with their partner organisms and their significance for the carbon cycle. Our focus will be anoxic freshwater sediments and anoxic wastewater treatment, both of which are major sources of CH4.
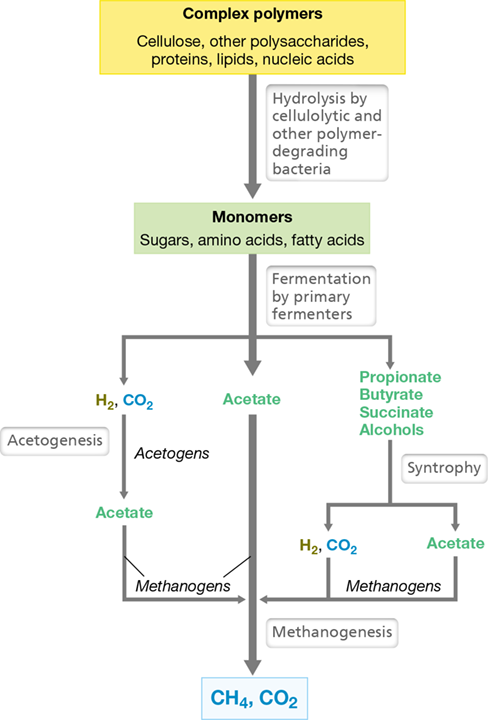
Polysaccharides, proteins, lipids, and nucleic acids from dead organisms find their way into anoxic habitats, where they are catabolized. The monomers released by hydrolysis of these polymers become major electron donors for energy metabolism. For the breakdown of a typical polysaccharide such as cellulose, the process begins with cellulolytic bacteria (Figure 21.7). These organisms hydrolyze cellulose into glucose, which is catabolized by fermentative organisms to short-chain fatty acids (acetate, propionate, and butyrate), alcohols such as ethanol and butanol, and the gases H2 and CO2. Hydrogen (H2) and acetate are consumed by methanogens directly, but the bulk of the carbon remains in the form of fatty acids and alcohols; these cannot be directly catabolized by methanogens and require the activities of syntrophic bacteria (Section 14.22; Figure 21.7).
Figure 21.7 Anoxic decomposition.

In anoxic decomposition, various groups of fermentative anaerobes cooperate in the conversion of complex organic materials to CH4 and CO2. This pattern holds for environments in which sulfate-reducing bacteria play only a minor role; for example, in freshwater lake sediments, sewage sludge, bioreactors, and the rumen.
Mastering Microbiology
Syntrophic bacteria are called secondary fermenters because they ferment the products of the primary fermenters, yielding H2, CO2, and acetate as products. For example, Syntrophomonas wolfei oxidizes C4 to C8 fatty acids, yielding acetate, CO2 (if the fatty acid was C5 or C7), and H2 (Table 21.1 and Figure 21.7). Other species of Syntrophomonas use fatty acids up to C18 in length, including some unsaturated fatty acids. Syntrophobacter wolinii specializes in propionate (C3) fermentation, generating acetate, CO2, and H2, and Syntrophus gentianae degrades aromatic compounds such as benzoate to acetate, H2, and CO2 (Table 21.1).
Table 21.1 Major reactions in the anoxic conversion of organic compounds to methanea

aData adapted from Zinder, S. 1984. Microbiology of anaerobic conversion of organic wastes to methane: recent developments. Am. Soc. Microbiol. News 50: 294.
bStandard conditions: solutes, 1 M; gases, 1 atm; 25 °C.
cConcentrations of reactants in typical anoxic freshwater ecosystems: fatty acids, 1 mM; HCO3 −, 20 mM; glucose, 10 μM; CH4, 0.6 atm; H2, 10−4 atm. Data from Gf 0 values in Table 3.3 and calculations described in Figure 3.7 and Section 3.3.
dCarbonic acid is formed by hydration of CO2 dissolved in water. Since the deprotonated form of this acid is the major species at near neutral pH, HCO3 − is commonly used in many microbial reactions involving CO2 as a reactant or product.
Despite rather extensive metabolic diversity, syntrophs are unable to carry out any of these reactions in pure culture. Instead, they depend on a H2-consuming partner organism because of the unusual bioenergetics linked to the syntrophic process. As described in Section 14.22, H2 consumption by a partner organism is absolutely essential for growth of syntrophic bacteria (in the absence of other electron acceptors), and the association of H2 producer and H2 consumer can be very intimate. In fact, H2 transfer in some syntrophic associations may not be directly via H2 transfer but through direct conduction, where electrons are transferred between species using electrically conductive wire-like structures (Section 21.5; Section 14.13 and Figure 14.31). But no matter the mechanisms, it is the transfer of H2 (or electrons) that makes the syntrophic association work. How is this so?
When the reactions listed in Table 21.1 for the fermentation of butyrate, propionate, ethanol, or benzoate are written with all reactants at standard conditions (solutes, 1 M; gases, 1 atm; 25 °C), the reactions yield free-energy changes (ΔG0′, Section 3.3) that are positive in arithmetic sign; that is, the reactions require rather than release energy. But the consumption of H2 dramatically affects the energetics, making the reaction favorable and allowing energy to be conserved. This can be seen in Table 21.1, where the ΔG values (free-energy change measured under actual conditions in the habitat) are negative in arithmetic sign if H2 concentrations are kept near zero by a H2-consuming partner organism. This allows the syntrophic bacterium to conserve a small amount of energy that is used to produce ATP.
The final products of the syntrophic partnership are CO2 and CH4 (Figure 21.7), and any naturally occurring organic compound that enters a methanogenic habitat will eventually be converted to these products. This includes even complex aromatic and aliphatic hydrocarbons. Additional organisms other than those shown in Figure 21.7 may participate in such degradations, but eventually fatty acids and alcohols will be generated, and they will be converted into methanogenic substrates by syntrophs. Acetate produced by syntrophs (as well as by the activities of acetogenic bacteria, Figure 21.7 and Section 14.14) is a direct methanogenic substrate and is converted to CO2 and CH4 by various methanogens.
Methanogenic Symbionts and Acetogens in Termites
A variety of anaerobic protists that thrive under strictly anoxic conditions, including ciliates and flagellates (Section 18.4), play a major role in the carbon cycle. Methanogenic Archaea live within some of these protist cells as H2-consuming endosymbionts. For example, methanogens are present within cells of trichomonal protists inhabiting the termite hindgut (Figure 21.8) where methanogenesis and acetogenesis are major metabolic processes. Methanogenic symbionts of protists are species of the genera Methanobacterium or Methanobrevibacter (Section 17.2 and Figure 17.5).
Figure 21.8 Termites and their carbon metabolism.

(a) A subterranean termite worker larva shown to the right of a hindgut dissected from another worker. The animal is about 0.5 cm long. (b) Partial structure of lignin, a component of wood. The methyoxy groups (highlighted in blue) are substrates for acetogenesis. Two views of the same microscopic field show termite hindgut protists photographed by (c) phase-contrast and (d) epifluorescence. Endosymbiotic methanogens in the protist cells fluoresce blue-green because of the methanogenic coenzyme F420 (compare with Figure 14.37). The average diameter of the protist cells is 15–20 μm.
In the termite hindgut, endosymbiotic methanogens along with acetogenic bacteria are thought to benefit their protist hosts by consuming H2 generated from glucose fermentation by cellulolytic protists. The acetogens are not endosymbionts but instead reside in the termite hindgut itself, consuming H2 from primary fermenters and reducing CO2 to make acetate. Unlike methanogens, acetogens can ferment glucose directly to acetate. Acetogens can also ferment methoxylated aromatic compounds to acetate. This is especially important in the termite hindgut because termites live on wood, which contains lignin, a complex polymer of methoxylated aromatic compounds (Figure 21.8b). The acetate produced by acetogens in the termite hindgut is consumed by the insect as its primary carbon and energy source.Microbial symbioses in the termite hindgut are discussed in more detail in Section 23.9.
Check Your Understanding
Why does Syntrophomonas need a partner organism in order to ferment fatty acids or alcohols?
What kinds of organisms are used in coculture with Syntrophomonas?
What substrates are used by acetogens to produce acetate?
21.3 The Nitrogen Cycle
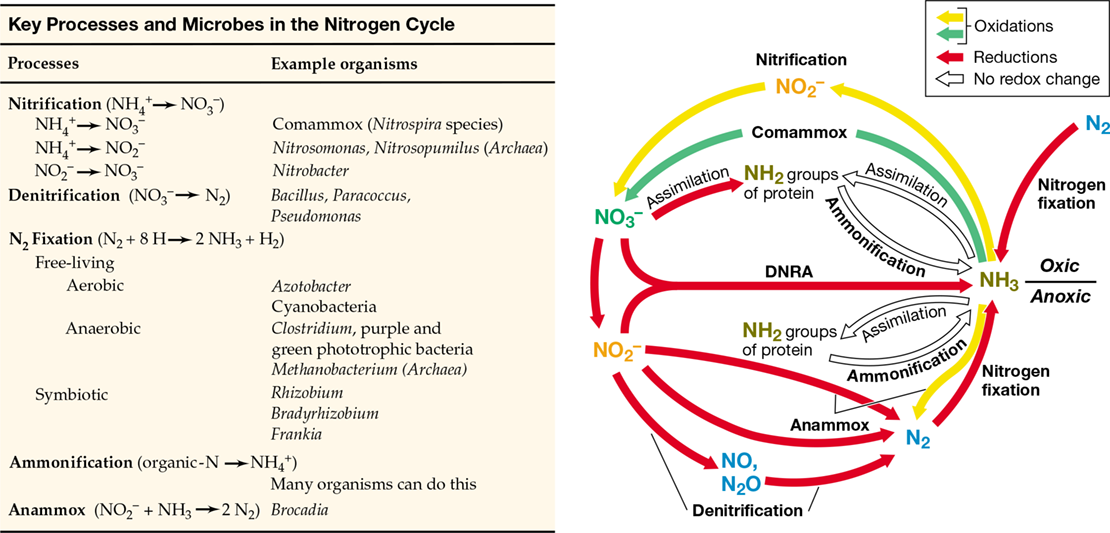
Nitrogen is an essential element for life (Section 3.1) and exists in a number of oxidation states that are cycled by specific redox reactions. We have discussed four major microbial N transformations thus far: nitrification, denitrification, anammox, and nitrogen fixation (Chapter 14). These and other key N transformations are summarized in the redox cycle shown in Figure 21.9.
Figure 21.9 Redox cycle for nitrogen.

The complete anammox reaction is NH4 ++NO2 −→N2+2 H2O (Figure 14.26). DNRA, dissimilative nitrate reduction to ammonia.
Nitrogen Fixation and Denitrification
Nitrogen gas (N2) is the most stable form of N and is a major reservoir for N on Earth. However, only a relatively small number of Bacteria and Archaea are able to use N2 as a cellular N source by the process of nitrogen fixation (N2+8 H→2 NH3+H2) (Section 3.12). The N recycled on Earth is mostly already “fixed N”; that is, N in combination with other elements, such as in ammonia (NH3) or nitrate (NO3 −). In many environments, however, the short supply of fixed N puts a premium on biological nitrogen fixation, and in these habitats, nitrogen-fixing bacteria flourish.
We discussed the role of NO3 − as an alternative electron acceptor in anaerobic respirationin Section 14.11. Under most conditions, the end product of NO3 − reduction is N2, nitric oxide (NO), or nitrous oxide (N2O). The reduction of NO3 − to these gaseous N compounds, called denitrification (Figure 21.9), is the main means by which N2 and N2O are formed biologically. Denitrification can be either beneficial or harmful. On the one hand, denitrification is a detrimental process. For example, if agricultural fields fertilized with NO3 − fertilizer become waterlogged following heavy rains, anoxic conditions can develop and denitrification can be extensive; this removes fixed N from the soil. On the other hand, denitrification can aid in wastewater treatment (Section 22.7). By converting NO3 − in wastewater to volatile forms of N, denitrification minimizes the load of fixed N in discharge waters that triggers algal growth and loss of water quality (Figure 20.19).
The production of N2O and NO by denitrification can have other environmental consequences. Nitrous oxide can be photochemically oxidized to NO in the atmosphere. Nitric oxide reacts with ozone (O3) in the upper atmosphere to form nitrogen dioxide (NO2), which is further oxidized to form nitric acid (HNO3) that returns to Earth as acid rain. In addition, N2O is a very potent greenhouse gas. Although N2O molecules persist on average only about 100 years because of their reactivity, on a per weight basis, the contribution of N2O to global warming is about 300 times that of CO2. Thus, denitrification contributes to global warming; to O3 destruction, which increases passage of ultraviolet radiation to the surface of Earth; and to acid rain, which increases acidity of soils. Increases in soil acidity change microbial community structure and function and, ultimately, soil fertility, impacting both plant diversity and agricultural yields of crop plants.
Ammonification and Ammonia Fluxes
Ammonia is released during the decomposition of organic N compounds such as amino acids and nucleotides, a process called ammonification (Figure 21.9). Another process contributing to the generation of NH3 is the respiratory reduction of NO3 − to NH3, called dissimilative nitrate reduction to ammonia (DNRA, Figure 21.9). DNRA dominates NO3 − and nitrite (NO2 −) reduction in electron donor–rich anoxic environments, such as highly organic marine sediments and the human gastrointestinal tract. Nitrate-reducing bacteria exploit this pathway primarily when NO3 − is limiting because DNRA consumes eight electrons per NO3 − reduced compared with the four and five electrons consumed when NO3 − is reduced to N2O or N2, respectively.
At neutral pH, NH3 exists as ammonium (NH4 +). Much of the NH4 + released by aerobic decomposition in soils is rapidly recycled and converted to amino acids in plants and microorganisms. However, because NH3 is volatile, some of it can be lost from alkaline soils by vaporization, and major losses of NH3 to the atmosphere occur in areas with dense animal populations (for example, cattle feedlots). On a global basis, however, NH3 constitutes only about 15% of the N released to the atmosphere, the rest being primarily N2 or N2O from denitrification.
Nitrification
Nitrification, the oxidation of NH3 to NO3 −, is a major process in well-drained oxic soils at neutral pH and is carried out by the nitrifying Bacteria and Archaea (Figure 21.9). Whereas denitrification consumes NO3 −, nitrification produces NO3 −. If materials high in NH3, such as manure or sewage, are added to soils, the rate of nitrification increases.
Nitrification was long considered an obligatory two-step process in which some species oxidize NH3 to NO2 − and then other species oxidize NO2 − to NO3 −. Many species of both Bacteria and Archaea can oxidize NH3 (Sections 14.9, 15.10, 17.5), whereas only species of Bacteria are known that oxidize NO2 −. However, some Bacteria, such as certain Nitrospira species, can oxidize ammonia completely to nitrate; these have been called comammox (for complete ammonia oxidizer) bacteria and seem to be especially important nitrifiers in freshwater lakes and rivers, drinking water systems, coastal sediments, and paddy soils.
Although NO3 − is readily assimilated by plants, it is soluble and therefore rapidly leached (or denitrified) from waterlogged soils; consequently, nitrification is not beneficial for plant agriculture. Ammonium, on the other hand, is positively charged and strongly adsorbed to negatively charged soils. Anhydrous NH3 is therefore used extensively as an agricultural fertilizer, but to prevent its conversion to NO3 − by nitrification, chemicals are added to the NH3 to inhibit the process. One common inhibitor is a pyridine compound called nitrapyrin (2-chloro-6-trichloromethylpyridine). Nitrapyrin specifically inhibits the first step in nitrification, the oxidation of NH3 to NO2 −. However, this effectively halts both steps in nitrification because the second step, NO2 −→NO3 −, depends on the first (Section 14.9). The addition of nitrapyrin to anhydrous NH3 has greatly increased the efficiency of crop fertilization and has helped prevent pollution of waterways by NO3 − leached from nitrified soils.
Anammox
In addition to oxygen-dependent nitrification, ammonia can be oxidized under anoxic conditions in a process called anammox (Section 14.10). Anammox bacteria affiliate with five genera (Brocadia, Kuenenia, Scalindua, Anammoxoglobus, and Jettenia) in a single phylogenetically cohesive family (Brocadiaceae) within the Planctomycetes (Section 16.16). Since anammox bacteria have yet to be isolated in pure culture, the genus and species names are prefaced by the term Candidatus to indicate their tentative taxonomic status (Section 13.12).
In the anammox reaction, NH3 is oxidized anaerobically with NO2 − as the electron acceptor, forming N2 as the final product (Figure 21.9; Figure 14.26c), which is released to the atmosphere. Although a major process in sewage and in anoxic marine basins and sediments, anammox is not a significant process in well-drained (oxic) soils. However, in wastewater processing plants (Section 22.8 and Figure 22.22), anammox is highly beneficial because like denitrification, it removes fixed nitrogen from sewage and reduces the ability of wastewater effluents to trigger algal and other microbial blooms (Figure 20.19a).
Check Your Understanding
What is nitrogen fixation and why is it important to the nitrogen cycle?
How do the processes of nitrification and denitrification differ? How do nitrification and anammox differ?
How does the compound nitrapyrin benefit both agriculture and the environment?
21.4 The Sulfur Cycle
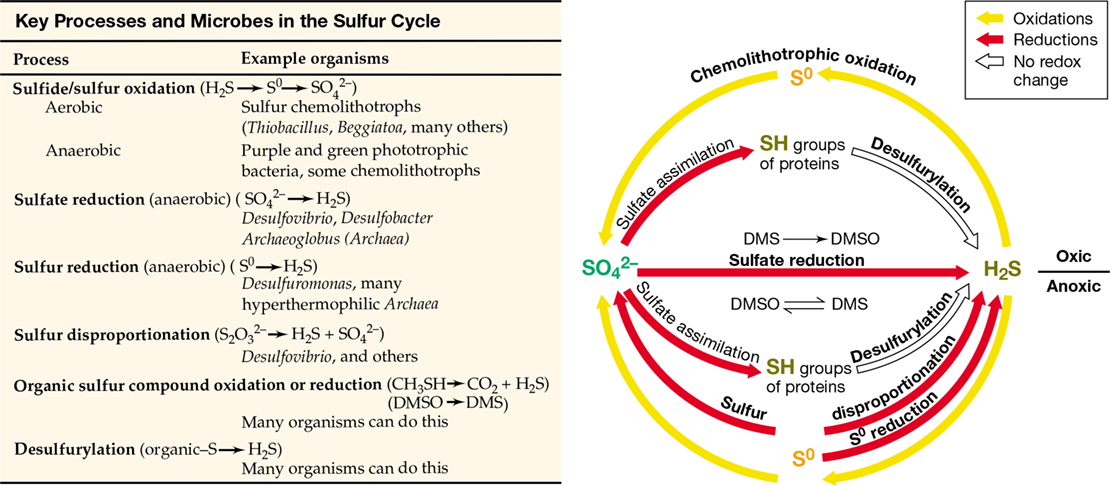
Microbial transformations of sulfur (S) are more complex than those of N because of the large number of oxidation states of S and the fact that several transformations of S also occur spontaneously (abiotically). The major reactions of the sulfur cycle—sulfide oxidation and sulfate (SO4 2−) reduction—were covered in Sections 14.7, 14.12, 15.11, and 15.12. The redox cycle for microbial sulfur transformations is shown in Figure 21.10.
Figure 21.10 Redox cycle for sulfur.

Although a number of oxidation states of S are possible, only three are significant in nature: −2 (sulfhydryl, R–SH; and sulfide, HS−), 0 (elemental sulfur, S0), and +6 (sulfate, SO4 2−). The bulk of S on Earth resides in sediments and rocks in the form of sulfate minerals, primarily gypsum (CaSO4), and sulfide minerals (pyrite, FeS2), but the oceans constitute the most significant reservoir of SO4 2− in the biosphere. A significant amount of S, in particular sulfur dioxide (SO2, a gas), enters the S cycle from human activities, primarily from the combustion of fossil fuels, especially coal.
Hydrogen Sulfide and Sulfate Reduction
Hydrogen sulfide (H2S) is a major volatile sulfur compound. Hydrogen sulfide is produced from bacterial sulfate reduction (SO4 2−+4 H2→H2S+2 H2O+2 OH−) (Figure 21.10) or emitted from sulfide springs and volcanoes. Although H2S is volatile, different forms exist depending on pH: H2S predominates below pH 7, and the nonvolatile HS− and S2− predominate above pH 7. Collectively, H2S, HS−, and S2− are referred to as “sulfide.”
Sulfate-reducing bacteria are a large and highly diverse group (Sections 14.12 and 15.11) and are widespread in nature. However, in anoxic habitats such as freshwater sediments and many soils, sulfate reduction is limited by SO4 2− availability. Moreover, because organic electron donors (or H2, which is a product of the fermentation of organic compounds) are needed to support sulfate reduction, it only occurs where significant amounts of organic material are present.
In marine sediments, the rate of sulfate reduction is typically carbon-limited and can be greatly increased by an influx of organic matter. This is important because the disposal of sewage or garbage in the oceans or coastal regions can trigger sulfate reduction. Hydrogen sulfide is toxic to many plants and animals and therefore its formation is potentially detrimental; sulfide is toxic because it combines with the iron of cytochromes and blocks respiration. Sulfide is commonly detoxified in nature by combination with iron, forming the insoluble minerals FeS (pyrrhotite) and FeS2 (pyrite). The black color of sulfidic sediments or sulfate-reducing bacterial cultures (Figure 15.28g) is due to these metal sulfide minerals.
Sulfide and Elemental Sulfur Oxidation–Reduction
Under oxic conditions, sulfide rapidly oxidizes spontaneously at neutral pH. Sulfur-oxidizing chemolithotrophic bacteria, most of which are aerobes (Sections 14.7 and 15.12), can catalyze the oxidation of sulfide. However, because of the rather rapid spontaneous reaction, microbial sulfide oxidation is significant only in areas where H2S emerging from anoxic environments meets the atmosphere. Where light is available, there can be anoxic oxidation of sulfide, catalyzed by the phototrophic purple and green sulfur bacteria (Sections 14.5, 15.4, and 15.6).
S0 is chemically stable but is readily oxidized by sulfur-oxidizing chemolithotrophic bacteria such as Thiobacillus and Acidithiobacillus. Because S0 is insoluble, the bacteria that oxidize it must attach to the S0 crystals to obtain their substrate (Figure 14.19). The oxidation of S0 forms sulfuric acid (H2SO4), and thus S0 oxidation characteristically lowers the pH in the environment, sometimes drastically. For this reason, small amounts of S0 can be added to alkaline agricultural soils as an inexpensive and natural way to lower the pH, relying on the ubiquitous sulfur chemolithotrophs to carry out the acidification process.
S0 can be reduced as well as oxidized. The reduction of S0 to sulfide (a form of anaerobic respiration) is a major ecological process of some Bacteria and hyperthermophilic Archaea (Chapter 17). Although sulfate-reducing bacteria can also reduce S0, most S0 reduction in nature occurs by the activities of the physiologically specialized sulfur reducers, organisms that are incapable of SO4 2− reduction (Section 15.11). However, the habitats of the sulfur reducers are generally those of the sulfate reducers, so from an ecological standpoint, the two groups form a metabolic guild unified by their formation of H2S.
Organic Sulfur Compounds
In addition to inorganic forms of S, multiple forms of organic S compounds are also cycled in nature. Many of these foul-smelling compounds are highly volatile and can thus enter the atmosphere. Hundreds of different dissolved organic sulfur compounds have been identified in surface waters of the oceans. Many of these are rapidly cycled and are increasingly thought to play important roles in ecosystem dynamics. However, only a handful have been fully chemically characterized.
The most abundant organic S compound in nature is dimethyl sulfide [DMS, (CH3)2S)]; it is produced primarily in marine environments as a degradation product of dimethylsulfoniopropionate [DMSP], a major osmoregulatory solute in marine algae (Section 4.15). As described previously (Section 20.12), DMSP is catabolized by marine bacteria yielding DMS; in the atmosphere, DMS becomes photooxidized to sulfate, and this promotes the formation of clouds that help cool our planet (Section 21.9).
By contrast, DMS produced in anoxic habitats can be microbially transformed in at least three ways: (1) by methanogenesis (yielding CH4 and H2S), (2) as an electron donor for CO2 fixation by certain phototrophic purple bacteria (yielding dimethyl sulfoxide, DMSO), and (3) as an electron donor in energy metabolism in certain chemoorganotrophs and chemolithotrophs (also yielding DMSO). DMSO can be an electron acceptor for anaerobic respiration (Section 14.13), producing DMS. Many other organic S compounds affect the global sulfur cycle, including methanethiol (CH3SH), dimethyl disulfide (CH3SSCH3), and carbon disulfide (CS2), but on a global basis, DMS is the most significant.
Check Your Understanding
Is H2S a substrate or a product of the sulfate-reducing bacteria? Of the chemolithotrophic sulfur bacteria?
Why does the bacterial oxidation of sulfur result in a pH drop?
What organic sulfur compound is most abundant in nature, and how does it help keep Earth cool?
II Other Nutrient Cycles
In addition to cycling major elements, microbes cycle many minor elements required for life—such as iron and phosphorus—as well as silicon and calcium required by certain marine phototrophs. Novel mechanisms of electron transfer are employed by microbes whose electron donors and acceptors are spatially distinct.
In Part II of this chapter we explore the interactions of microorganisms with metals—in particular the major processes of iron and manganese cycling—and with some nonmetals whose microbial transformations are also of major global significance.
21.5 The Iron and Manganese Cycles: Reductive Activities
21.5 The Iron and Manganese Cycles: Reductive Activities
21.5 The Iron and Manganese Cycles: Reductive Activities
Iron (Fe) is one of the most abundant elements in Earth’s crust. On the surface of Earth, Fe exists naturally in two oxidation states, ferrous [Fe2+, also Fe(II)] and ferric [Fe3+, also Fe(III)]. A third oxidation state, Fe0, is abundant in Earth’s core and is also a major product of human activities from the smelting of iron ores to form cast iron.
In nature, iron cycles primarily between the Fe2+ and Fe3+ forms, and these redox transitions are one-electron oxidations and reductions. Ferric iron is reduced both chemically and as a form of anaerobic respiration, and Fe2+ is oxidized both chemically and as a form of chemolithotrophic metabolism (Figure 21.11). Manganese (Mn), although present at 5- to 10-fold lesser abundance than Fe in the near-surface environment, is another redox-active metal of microbiological significance, existing primarily in two oxidation states (Mn2+ and Mn4+, see Figure 21.12).
Figure 21.11 Redox cycle for iron.

The major forms of iron in nature are Fe2+ and Fe3+. Fe0 is primarily a product of smelting of iron ores. Fe3+ forms various minerals such as ferric hydroxide, Fe(OH)3.
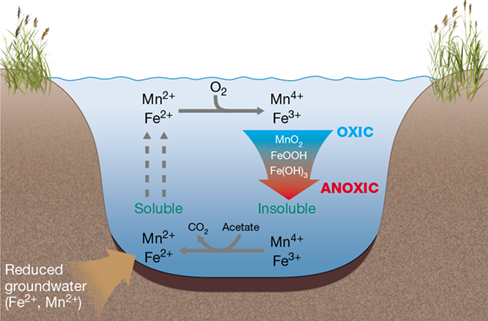
A key feature of the iron and manganese cycles is the different solubilities of these metals in their oxidized versus reduced forms. Reduced iron (Fe2+) and manganese (Mn2+) are soluble. In contrast, oxidized minerals of iron such as iron oxide-hydroxides [e.g., Fe(OH)3, FeOOH, and Fe2O3] and manganese oxide (MnO2) are insoluble and tend to settle out in aquatic environments. As a consequence, these strong oxidants can comprise several percent by weight of marine and freshwater sediments, making them among the most abundant of potential electron acceptors in many anoxic systems (see Figure 21.12).
Bacterial Reduction of Iron and Manganese Oxides
Some Bacteria and Archaea can use Fe3+ as an electron acceptor in anaerobic respiration (Section 15.13). These organisms also commonly have the capacity to use Mn4+ as an electron acceptor, and some have the capacity to reduce oxidized uranium (Section 22.3). Ferric iron and manganese oxide reduction is common in waterlogged soils, bogs, and anoxic lake sediments (Figure 21.12). When soluble reduced iron and manganese reach oxic regions, for example, through diffusion from anoxic regions of sediments, they are oxidized chemically [e.g., Fe2++14O2+212 H2O→Fe(OH)3+2 H+] or microbiologically (Figure 21.12). The chemical oxidation of Fe2+ is very rapid at near-neutral pH. Although the spontaneous oxidation of Mn2+ is very slow at neutral pH, the rate of oxidation can be increased up to five orders of magnitude by a variety of manganese-oxidizing bacteria and even fungi. The oxidized metal oxides and hydroxides then precipitate, returning the oxidized metals to the sediments where they can again serve as electron acceptors, completing the cycle.
Figure 21.12 Iron and manganese redox cycling in a typical freshwater system.

Iron and manganese oxides in sediments are used as electron acceptors by metal-reducing bacteria. The resulting reduced forms are soluble and diffuse into the oxic regions of the sediment or water column, where they are oxidized microbially or chemically. Precipitation of the insoluble oxidized metals then returns the metals to the sediments, completing the redox cycle.
Oxidized iron (Fe3+) and manganese (Mn4+) are chemically very reactive and can alter the availability of both nutrients and toxic metals. For example, phosphate is less available when it is trapped as insoluble ferric phosphate precipitates. Essential and toxic metals [e.g., copper (Cu), cadmium (Cd), cobalt (Co), lead (Pb), and arsenic (As)] also form insoluble complexes with iron and manganese oxides. When these oxides are subsequently microbiologically reduced, the bound phosphate is liberated along with the soluble forms of these metals. In addition to sequestering phosphate and metals, abiotic oxidation of refractory organic compounds by manganese oxide may yield more available sources of carbon for microbial growth.
Long-Range Electron Transfer to Insoluble Electron Acceptors
As we learned earlier (Chapters 3 and 14), in any form of respiration, electron disposal is necessary for energy conservation. When the electron acceptor is oxygen (O2), nitrate (NO3 −), or many other soluble substances used by bacteria as electron acceptors (Sections 14.11, 14.12, 14.13, 14.14 and 14.15), the final product diffuses away from the cell. Many bacteria reduce ferric iron (Fe3+) as an electron acceptor under anoxic conditions, including the obligate anaerobe Geobacter sulfurreducens and the facultative aerobe Shewanella oneidensis. However, in contrast to soluble electron acceptors, Fe3+ is typically present in nature as an insoluble mineral, such as an iron oxide, and thus the reduction of Fe3+ must occur outside the cell. Under such conditions, the ferric iron functions as an electrical anode, and the bacterial cell facilitates transfer of electrons from the electron donor to the anode.
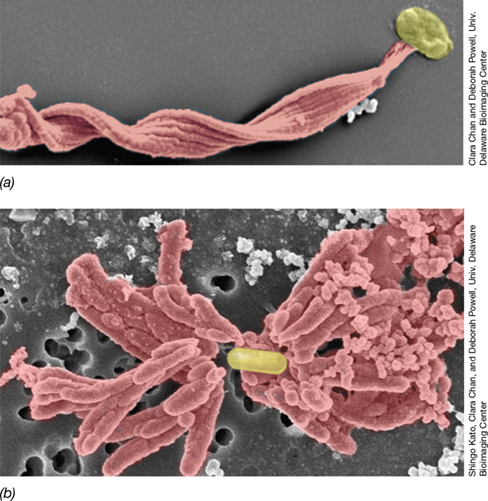
Regardless of the electron acceptor they use, when bacteria respire, they carry out oxidations and reductions that generate electricity. Thus, the surfaces and appendages of cells of bacteria that interact with iron and manganese oxides, such as Geobacter (**Figure 21.13*a***), are electrically conductive. Geobacter forms direct electrical connections with insoluble materials that can either accept or donate electrons. Electron transfer employs cytochromes localized along the length of pili that are generally 10–20 micrometers long (Figure 14.31). These electrically conductive structures function as electrical nanowires, much as copper wires do in a household electrical circuit. Being conductive structures, nanowires can form direct electrical connections with insoluble materials that either accept or donate electrons; alternatively, nanowires can form connections between cells. In this way, electrons obtained by Geobacter from the oxidation of organic electron donors or from H2 can be shuttled to a suitable electron acceptor.
Figure 21.13 Direct and long-range electron transfer.

(a) Cells of Geobacter attached to ferric iron precipitates (arrows) reduce Fe3+ to Fe2+. (b) Three-dimensional rendering of cabled filamentous sulfide-oxidizers, with inset transmission electron microscopic cross-section displaying presumptive electrically conductive filaments surrounding the cell perimeter. (c) Microsensor profiles of sediments colonized by cabled filamentous bacteria, showing wide separation of depths where sulfide oxidation and oxygen reduction occur. The profiles are explained as follows: Oxygen (O2) is quickly consumed by bacterial respiration in the upper regions of the sediment, whereas H2S, produced in anoxic regions, only accumulates deeper in the sediments. The pH is more acidic lower in the sediments because sulfide oxidation yields protons. As O2 is consumed near the sediment surface, protons are also consumed, and the pH rises.
Some electron transfers can occur over rather long distances, even longer than the cell itself. For example, hydrogen sulfide (H2S) oxidation in some anoxic marine sediments is facilitated by filamentous bacteria composed of many interconnected cells that transfer electrons from sulfide (the donor) in the sediments to O2 (the acceptor) in the overlying water, a distance of 2.5 cm (25000 μm, Figure 21.13b, c). The surface of the filamentous cells has ridges running along its entire length, and each microbial filament is surrounded by 15 to 17 such structures (Figure 21.13b). The structures function as electrical cables to transfer electrons from the sulfide oxidized at one end of the filament to the reduction of O2 at the other end of the filament.
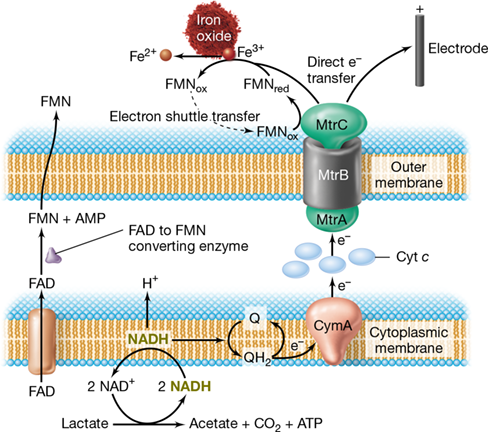
As for Geobacter, Shewanella is also capable of direct transfer of electrons to an insoluble exogenous electron acceptor. However, unlike Geobacter with its use of conductive pili, Shewanella uses cytochrome-rich extensions of its outer membrane to make physical contact with a mineral surface such as iron oxide. This system couples transfer of electrons, obtained from the oxidation of lactate or other organic compounds, to soluble cytochromes in the cell’s periplasm and then to the cell surface using the Mtr complex of membrane-spanning multiheme proteins (Figure 21.14). From the Mtr complex, electrons can be shuttled (see next subsection) to the iron oxide by way of FMN (an electron carrier, Section 3.8) or directly transferred from MtrC, the outermost part of the Mtr protein complex, to iron oxide. An electrode poised at an electropositive potential can also function as an electron acceptor for this organism (Figure 21.14).
Figure 21.14 *Shewanella* electron transfer to insoluble electron acceptors.

Electrons from lactate oxidation enter the quinone pool (Q, QH2) through a membrane-bound NADH dehydrogenase. Reduced quinone (QH2) is oxidized by a membrane-associated multiheme cytochrome (CymA), which transfers electrons to the outer membrane–spanning Mtr complex via periplasmic c-type cytochromes. The outer part of the Mtr complex (MtrC) then delivers electrons to an insoluble electron acceptor via direct interaction, such as to an iron oxide or an electrode, or via flavin (FMN) electron shuttles. FMNs are actively secreted by Shewanella. Although some proton efflux may be associated with electron transfer to CymA, ATP generation is primarily by substrate-level phosphorylation.
Extracellular Electron Shuttles
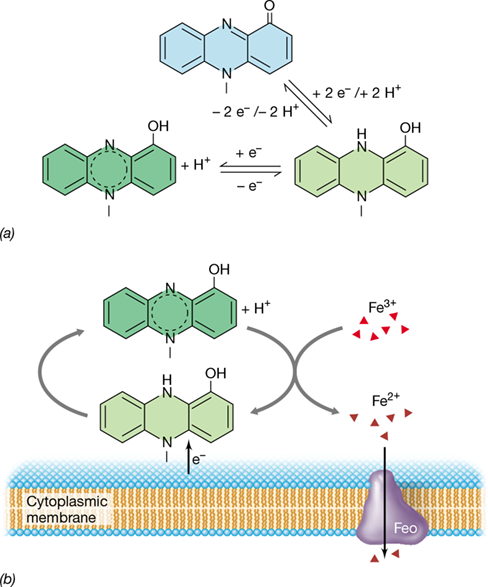
Another type of extracellular electron transfer is mediated by small, soluble, redox-active molecules called extracellular electron shuttles (EESs), typically aromatic organic compounds. The key functional attribute of an EES is the presence of conjugated double bonds that undergo redox transitions at a reduction potential (E0′) that facilitates transfer of electrons from the cell to an electron acceptor poised at a suitable potential difference. The acceptor can be an insoluble material such as oxidized metal or any other substance that can be reduced by the EES, including an actual electrode.
Naturally occurring humic substances (Section 21.1) often function as EESs. Since some constituents of humics can alternate between oxidized and reduced forms, they can function to shuttle electrons from the bacterium to reduce iron or manganese oxides (Figure 21.15). However, in addition to using shuttles that are available in the environment, a number of microorganisms secrete EESs to facilitate transfer of electrons to exogenous electron acceptors. These may be common cellular cofactors, such as flavin mononucleotide (FMN) secreted by Shewanella oneidensis (Figure 21.14), or more specialized shuttles such as the virulence factor pyocyanin (**Figure 21.16*a***), a derivative of phenazine, made by Pseudomonas aeruginosa. Pyocyanin has many effects, including enhancing substrate-level ATP synthesis by oxidizing excess intracellular redox cofactors such as NADH, promoting biofilm formation, generating peroxide by reduction of molecular oxygen, and a capacity to liberate ferrous iron from an infected host’s iron reserves. Pyocyanin is present in the lungs of patients suffering from the genetic disease cystic fibrosis, where vigorous biofilms of this bacterium develop (Section 8.10); maintenance of the bacterial infection is promoted in part by the organism’s ability to reduce ferric iron to the soluble ferrous form and sequester it through activity of the Feo iron transport membrane protein (Figure 21.16b).
Figure 21.15 Role of humic substances in humus as an electron shuttle in microbial metal reduction.

Quinone-like functional groups in humus are reduced by acetate-oxidizing bacteria. The reduced humus then donates electrons to metal oxides, releasing reduced iron (Fe2+) and oxidized humus. The cycle continues as oxidized humus is again reduced by the bacteria.
Figure 21.16 Pyocyanin electron shuttling.

(a) Pyocyanin undergoes single and double electron transfer reactions between cells and environmentally available electron acceptors. (b) In Pseudomonas species, the pyocyanin electron shuttle is a virulence factor, the reduced pyocyanin functioning to reduce ferric iron to acquire iron from the infected host and incorporate it through the Feo uptake system.
Both direct electron transfer (as seen in Section 23.3) and the use of EESs are also important for transfer of electrons between symbiotically associated microorganisms. In nature, electrical communication between bacterial cells may be a major way by which electrons generated from microbial metabolism in anoxic habitats are shuttled to oxic regions. Moreover, research on the microbiology of the process indicates that this microbial electricity could be harnessed in the form of microbial “fuel cells” that could oxidize toxic and waste carbon compounds in anoxic environments, with the resulting electrons coupled to power generation. In such a scheme, bacteria would be exploited to function as catalysts for diverting electrons from electron donors directly to artificial anodes (Figure 21.14), with the resulting electrical current being siphoned off to supply a portion of human power needs. Such a system would have to be developed on a very large scale, of course. However, large-scale systems for microbial growth are already well established in wastewater treatment facilities ( Sections 22.6, 22.7 and 22.8).
Check Your Understanding
In what oxidation states are Fe in Fe(OH)3 and Mn in MnO2?
What alternative mechanisms are used by iron-respiring microorganism to deliver electrons to insoluble electron acceptors?
21.6 The Iron and Manganese Cycles: Oxidative Activities
21.6 The Iron and Manganese Cycles: Oxidative Activities
21.6 The Iron and Manganese Cycles: Oxidative Activities
Besides functioning as electron acceptors in their oxidized forms, in their reduced forms, iron and manganese can also function as electron donors (Section 14.8). In this section, we explore this option as important links in the iron and manganese cycles.
Microbial Oxidation of Reduced Iron
At neutral pH, ferrous iron (Fe2+) is oxidized abiotically in oxic environments. By contrast, at acidic pH (pH <4), Fe2+ is not oxidized spontaneously. Thus, much of the research on microbial oxidation of iron has been focused on acidic, ferrous-iron-rich habitats, where acidophilic chemolithotrophs such as Acidithiobacillus ferrooxidans and Leptospirillum ferrooxidans oxidize Fe2+ to Fe3+ (Figure 21.17). As we will see in Chapter 22, this activity can be both beneficial and harmful. On the one hand, because the oxidation of ferrous iron generates acidity, this can be devastating if large amounts of iron and other metal sulfides are released into freshwaters or soils, such as from mining activities. The acidity produced from iron oxidation can make the environments unsuitable for plant and animal life (Figure 22.5). On the other hand, the generation of acidity and the ability of acidophilic chemolithotrophs to oxidize ferrous iron is exploited in the mining industry to extract metals such as copper from low-grade ores (Section 22.1 and Figure 22.2).
Figure 21.17 Oxidation of ferrous iron (Fe2+).

A microbial mat growing in the Rio Tinto, Spain. The mat consists of acidophilic green algae (eukaryotes) and various iron-oxidizing chemolithotrophic bacteria. The Rio Tinto has a pH of about 2 and contains high levels of dissolved metals, in particular Fe2+. The red-brown precipitates consist of Fe(OH)3 and other ferric minerals.
In addition to oxidation by acidophilic microorganisms, ferrous iron can be oxidized microbially at neutral pH. However, since the abiotic oxidation of reduced iron also occurs under these conditions, iron-oxidizing bacteria that inhabit nonacidic environments are restricted to a very narrow zone in which anoxic ferrous-iron-rich water mixes with oxygenated water. These microoxic habitats include freshwater and coastal sediments (Figure 21.12; Figure 20.22), seeps of iron-rich groundwater, slow-moving streams, ferrous-iron-rich waters from springs, and hydrothermal vents (Figure 21.18). When ferrous-iron-rich groundwaters are exposed to air, Fe2+ is oxidized at the interface of these two zones by iron-oxidizing bacteria such as Leptothrix and Gallionella (Figure 21.18b, c, d; Sections 14.8 and 15.14). Thus, their physiology dictates that they maintain a position within a narrow environment of low levels of O2 and high levels of reduced metals. Although O2 is the most environmentally significant electron acceptor, Fe2+ oxidation can also be coupled to NO3 − reduction by some anaerobic microbes (Sections 14.8 and 15.14), and Fe2+ functions as an electron donor in photosynthesis for some anoxygenic phototrophs (Sections 14.5, 15.2, and 15.5). Neutrophilic aerobic iron-oxidizing bacteria are members of two proteobacterial classes. The freshwater species are members of several closely related genera in the Betaproteobacteria whereas all known marine species are Zetaproteobacteria (Proteobacteria are discussed in Chapter 16). All characterized Zetaproteobacteria are autotrophic and aerobic iron-oxidizers, and all but the bacterium Ghiorsea bivora—which can oxidize either Fe2+ or H2 as electron donors—are obligate iron-oxidizers. Although primarily associated with marine environments, the Zetaproteobacteria have also been found in saline terrestrial aquifers and springs, and in biofilms associated with steel corrosion (Section 22.12).
Figure 21.18 Fe-oxidizing microbial mats.

(a) Freshwater microbial mat in a slow-moving stream where Fe2+-enriched groundwater is mixing with oxygenated surface water, triggering growth of Fe2+-oxidizing bacteria and precipitation of iron oxides. (b–d) Fe-oxidizing Bacteria. (b, c) Phase-contrast and epifluoresence photomicrographs of the sheath-forming Fe-oxidizer Leptothrix ochracea (the sheath is approximately 2 μm wide). (d) The stalk-forming Fe2+-oxidizer Gallionella ferruginea showing bean-shaped cells in the process of cell division at the end of the iron oxide–encrusted stalk (each bean-shaped cell is about 2 μm long). (e) An iron-oxidizing mat at a deep-sea hydrothermal vent (1000-meter depth) at Lō‘ihi Seamount. (f) TEM image of biogenic oxides produced at Lō‘ihi; note the variety of helical stalks and tubular sheathlike filaments (the filaments vary from 2 to 4 μm wide). (g) Phase-contrast photomicrograph of marine Fe2+-oxidizers growing at the ends of iron oxide filaments (cells denoted by arrows) from an experimental incubation at Lō‘ihi (filaments are approximately 2 μm wide).
As we saw in Section 21.5, organisms that reduce insoluble metal oxides can use extracellular conductors for electron transfer, such as electrically conductive pili or cell-surface-associated cytochromes. Cytochromes participate in both iron reduction and iron oxidation, and genomes of metal-oxidizing Gallionella and Sideroxydans species contain genes encoding periplasmic c-type cytochromes that resemble those encoding proteins known to reduce metal oxides in Shewanella (Figure 21.14), indicating that mechanistically related electron transfer pathways are used for both the reduction and oxidation of extracellular metals.
Although possibly sharing similar electron transfer mechanisms, metal-oxidizing bacteria are confronted with the problem that the precipitates they form can encase the cell in an iron oxide shell. To prevent this, metal oxidizers secrete organic substances that capture metal oxides and deposit them some distance away from the cell. For example, the iron oxidizer Gallionella produces extended organic stalks that become encrusted with metal oxides away from the cell (Figure 21.18d, f, and g; see also Figure 15.36). Similar structures are made by marine Zetaproteobacteria of the genus Mariprofundus; the species Mariprofundus ferrooxydans deposits iron oxides as a twisted stalk while Mariprofundus ferrinatatus makes shorter ropelike filaments of iron oxides (Figure 21.19). Laboratory experiments employing opposing gradients of Fe2+ and O2 showed that M. ferrooxydans uses its iron oxide stalk as both a holdfast and a dynamic structure to position cells at an optimum point within the gradient of oxygen and Fe2+. An alternative strategy is used by Leptothrix and Sphaerotilus species. These bacteria produce an organic sheath surrounding the cells that becomes encrusted with metal oxides (Figure 21.18b and Figure 15.55). Later, the cells move out of the sheath, leaving the metal oxide crust behind. Although not all metal oxidizers produce such morphologically conspicuous structures, it is thought that most, if not all, metal oxidizers are forced to produce some form of extracellular organic material in order to sequester the insoluble product of their energy metabolism.
Figure 21.19 Scanning electron micrograph of iron oxide minerals formed by species of marine *Zetaproteobacteria*.

(a) Twisted mineral stalk (colorized red) formed by a cell of Mariprofundus ferrooxydans (yellow). The cell is about 1.2 μm long. (b) Mariprofundus ferrinatatus forms short iron oxide structures (colorized red), which are shed from the single cell (outlined in yellow) that is covered by the mineral deposits. The cell is about 1 μm long. Images reproduced from McAllister, S.M., Moore, R.M., Gartman, A., Luther, G.W., III, Emerson, D., and C.S. Chan. 2019 FEMS Microbiol. Ecol. doi:10.1093/femsec/fiz015.
Microbial Oxidation of Reduced Manganese
Many bacteria catalyze Mn2+ oxidation, and the different species known are ubiquitous in nature, inhabiting fresh and marine waters, soils, and virtually anywhere significant levels of Mn2+ are present. Reduced manganese is present along with Fe2+ in anoxic waters that reach oxic zones, the Mn2+ resulting from the anaerobic respiration of Mn4+ (Figure 21.12; Figure 20.22); these anoxic/oxic interfaces establish a niche for the obligately aerobic Mn2+-oxidizing bacteria. Because the reduction potential of the Mn2+/Mn4+ couple is so high, very near to that of the O2/H2O couple (Figure 14.1), O2 is the only electron acceptor that can couple with Mn2+ oxidation; anaerobic respirations of Mn2+ are thus not possible.
Several genera of Bacteria can oxidize Mn2+ including Sphaerotilus and Leptothrix—both Betaproteobacteria—and Hyphomicrobium and Pedomicrobium—both Alphaproteobacteria (Figures 15.51, 15.55, and 15.56). Some Pseudomonas species can also oxidize Mn2+. Most of these organisms can be cultured and therefore Mn2+ oxidation easily measured. However, a major unanswered question surrounding this entire physiological group is whether they actually benefit from Mn2+ oxidation. Chemolithotrophic (and autotrophic) growth on Mn2+ has been achieved with one Pseudomonas species whose cells were shown to contain carboxysomes (Figure 15.30a). Carboxysomes are storage granules of the key enzymes of the Calvin cycle (Section 3.12), the main autotrophic pathway of aerobic chemolithotrophs. Convincing chemolithotrophic growth of other species of Mn2+ oxidizers has not been achieved. A possible alternative reason for Mn2+ oxidation is to convert soluble Mn2+—which can be a toxic metal when present in high enough concentrations—into nontoxic manganese oxides. Whatever the reason(s) for bacterial Mn2+ oxidation, the process can be a major one in nature and is typically linked to neutral pH habitats where extensive iron oxidation is occurring simultaneously.
Check Your Understanding
Both Acidithiobacillus and Mariprofundus are iron oxidizers, but their habitats differ dramatically. Explain.
Although some iron-oxidizing bacteria can grow on iron anaerobically, manganese-oxidizing bacteria cannot grow on manganese anaerobically. Explain.
21.7 The Phosphorus, Calcium, and Silicon Cycles
Many other chemical elements undergo microbial cycling and we focus on three key ones here: phosphorus (P), calcium (Ca), and silicon (Si). The cycling of these elements is important in aquatic environments, particularly in the oceans, which are major reservoirs of Ca and Si and where large amounts of Ca and Si are incorporated into the exoskeletons of certain microbes. However, unlike the C, N, and S cycles, in the Ca and Si cycles there are no redox changes or gaseous forms that can escape and alter Earth’s atmospheric chemistry. However, different redox states of P are biogeochemically significant. From a global perspective, keeping these cycles in balance—especially that of Ca—is important for maintaining sustainable life on Earth.
Mastering Microbiology
Art Activity: Figure 21.15 The marine calcium cycle
Phosphorus and Phosphonates
Phosphorus exists in nature primarily as organic and inorganic phosphates. Phosphorus reservoirs include phosphate-containing minerals in rocks, dissolved phosphates in freshwaters and marine waters, and the nucleic acids and phospholipids of living organisms. Although P has multiple oxidation states, most environmental phosphates are at the +5 oxidation state (for example, hydrogen phosphate, HPO4 2−). In nature, P cycles through living organisms (as cellular P), waters and soils (as inorganic and organic P), and Earth’s crust (as inorganic P). P is typically the limiting nutrient for photosynthesis in freshwaters, which receive it from the weathering of rocks.
In the oceans, a fraction of dissolved P is organic, in the form of phosphate esters and phosphonates. Phosphonates, also referred to as phosphonic acids when in the protonated forms, are organophosphate compounds that contain a direct bond between the P and C atoms. In this form the P atom is more reduced (+3 oxidation state) than in phosphate (+5 oxidation state). Phosphonates are produced by certain microorganisms and comprise about a quarter of the organic P pool in nature. For many organisms, phosphonates are a less available source of P than is HPO4 2− because the organisms lack the enzymes required to degrade phosphonates. Such organisms can be phosphorus-limited even when sufficient P is present as phosphonates.
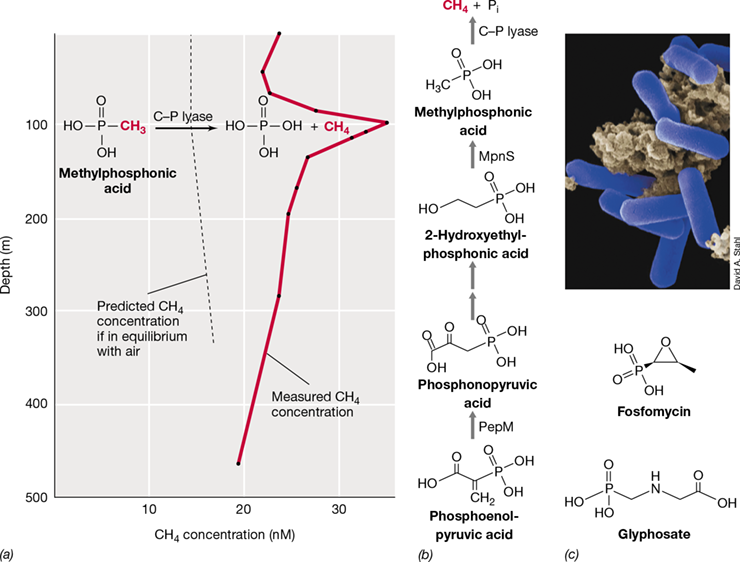
Phosphonates and reduced inorganic forms of P—phosphite (H2PO3 −, +3 oxidation state) and hypophosphite (H2PO2 −,+1 oxidation state)—are rapidly cycled in the marine environment by producers and consumers. This cycling of P is important for organisms that live in P-depleted environments and have the capacity to make or consume these alternative forms of phosphorus. Why marine organisms produce so much of these reduced phosphorus species is unknown. However, the fact that some marine microorganisms produce methylphosphonic acid may have solved another marine metabolic conundrum. The degradation of methylphosphonic acid (CH5O3P) by some marine microorganisms—a process that liberates methane (CH4)—likely explains the previously puzzling observation that relatively high levels of CH4 are present in the oxygenated surface waters of the ocean (see Explore the Microbial World, “Solving the Marine Methane Paradox”).
Calcium
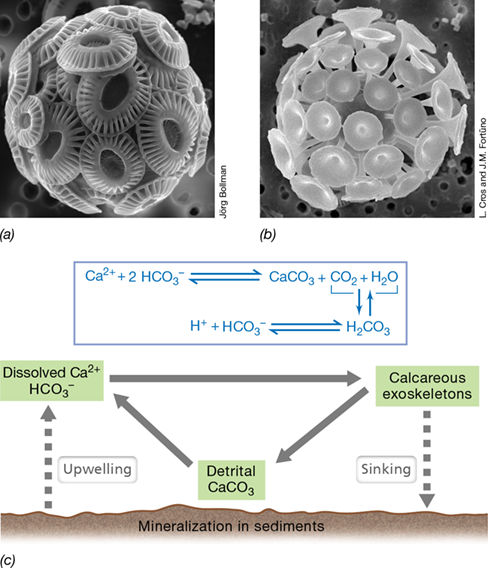
The major global reservoirs of calcium (Ca) are calcareous rocks and the oceans. In the oceans, where dissolved Ca exists as Ca2+, calcium cycling is a highly dynamic process although the concentration of Ca2+ in seawater remains constant at about 10 mM. Several marine eukaryotic phototrophic microorganisms take up Ca2+ to form their calcareous exoskeletons; these include in particular species of haptophytes called coccolithophores (Section 18.7 and Figure 18.15) and Rhizaria called foraminifera (Section 18.6) (Figure 21.20). The calcium-cycling activities of these planktonic phototrophs are also tightly coupled with inorganic components of the carbon cycle.
Figure 21.20 The marine calcium cycle.

Scanning electron micrographs of cells of the calcareous phytoplankton (a) Emiliania huxleyi and (b) Discosphaera tubifera. The exoskeletons of these coccolithophores are made of calcium carbonate (CaCO3). A cell of E. huxleyi is about 8 μm wide and a cell of D. tubifera is about 12 μm wide. (c) The marine calcium cycle; dynamic pools of Ca are shaded in green. Detrital CaCO3 is that in fecal pellets and other organic matter from dead organisms. Note how H2CO3 formation lowers ocean pH when it dissociates to form H+ and HCO3 −.
The precipitation of calcium carbonate (CaCO3) to form the shells of calcareous phytoplankton controls both CO2 flux into ocean surface water and inorganic C transport into deep ocean waters and the sediments. Moreover, the formation of CaCO3 both depletes surface dissolved bicarbonate (HCO3 −) and increases the level of dissolved CO2 (Figure 21.20c). The latter reduces the influx of atmospheric CO2 into surface ocean waters, and this helps maintain the slightly alkaline pH of the oceans. When these calcareous organisms die and sink toward the sediments, Ca2+ and inorganic and organic C are transported to the deep ocean from which they are only slowly released over long periods (see also page 585).
The formation of CaCO3 exoskeletons brings into play a delicate balance between Ca2+ and C and is a process sensitive to changes in atmospheric CO2 levels. This is because increased levels of atmospheric CO2 increase the formation of carbonic acid (H2CO3), and as this dissociates to form HCO3 − and H+, CaCO3 dissolves and seawater pH decreases (Figure 21.20c). Greater ocean acidity resulting from rising atmospheric CO2 is predicted to reduce the rate of formation of calcareous shells, which will likely have effects on other microbial nutrient cycles and plant and animal communities (Section 21.9).
Silicon
The marine Si cycle is controlled primarily by unicellular eukaryotes (diatoms and other Stramenopiles referred to as silicoflagellates, and radiolarians) that build ornate external cell skeletons called frustules (diatoms, **Figure 21.21*a***; Section 18.5 and Figure 18.12) or tests (radiolarians, Section 18.6). These structures are not constructed of CaCO3 as in the coccolithophores, but of silica (SiO2, also called opal), whose formation begins with the uptake by the cell of dissolved silicic acid (Figure 21.21b).
Figure 21.21 The marine silicon cycle.

(a) Dark-field photomicrograph of a collection of diatom shells (frustules). The frustules are made of silica (SiO2). (b) The marine silicon cycle; dynamic pools of Si are shaded in green.
Mastering Microbiology
Art Activity: Figure 21.16 The marine silica cycle
Diatoms are rapidly growing phototrophic eukaryotes and often dominate blooms of phytoplankton in coastal and open ocean waters. However, unlike other major phytoplankton groups, diatoms require Si and can become silicon-limited when blooms develop. Also, because of their large size, diatom cells tend to sink faster than other organic particles, and in this way, they contribute significantly to the return of Si and C to deeper ocean waters. The transport of organic material produced through primary production in near-surface waters to deeper ocean waters, primarily by sinking particles, is called the biological pump and is an important aspect of the carbon cycle in terms of carbon burial and mineralization in marine environments (Figure 21.1).
In addition to the major nutrient requirements of any phototrophic organism (CO2, N, P, Fe), diatoms require sufficient dissolved Si, and in nature this originates primarily from Si released from the skeletons of dead diatoms (Figure 21.21b). Although Si is released fairly rapidly following cell death, during periods of high diatom production in relatively shallow waters, a significant fraction of dissolved Si can become buried in sediments and remain there for millions of years. This has consequences for continued diatom growth and their phototrophic consumption of dissolved CO2 from ocean waters. The flux of CO2 into and out of ocean water affects its pH (Figure 21.20c), and through this link, the Si and C cycles are coupled in a manner similar to what we have seen with the Ca and C cycles (Figure 21.20).
Although the nutrient cycles we have seen thus far are mainly driven by microbial activities (plus plants in the case of the C cycle), humans can have major impacts on nutrient cycling through major inputs of C, N and toxic elements, and we consider these next.
Check Your Understanding
How does the formation of CaCO3 skeletons by calcareous phytoplankton retard movement of CO2 from the atmosphere to oceans and help maintain ocean water pH?
How might Si depletion in the photic zone influence the biological pump?
III Humans and Nutrient Cycling
In addition to steadily increasing levels of CO2, intensive agriculture and fossil fuel burning have increased levels of nitrous oxide and methane and have accelerated the mobilization of mercury. Human inputs into major nutrient cycles will likely affect all life forms, be they microbes, flora, or fauna.
Humans have a profound impact on microbial nutrient cycles by adding and removing specific components of the cycles in large amounts. Here we consider human inputs of three major chemical groups: mercury (Hg), CO2 and other atmospheric gases, and various fixed N compounds. These compounds originate primarily from the burning of fossil fuels or from agricultural activities, and either cause toxicity problems (Hg) or affect Earth in globally significant ways (gases and N compounds). We begin with the very toxic metal Hg, which is transformed by bacteria in many different ways.
21.8 Mercury Transformations
Mercury is not a microbial nutrient (Figure 4.1), but microbial transformations of various mercuric compounds are natural processes and help to detoxify some of the most toxic forms. Mercury is a widely used industrial product, especially in the electronics industry. Mercury is also an active ingredient in many pesticides, a pollutant from the chemical and mining industries and from the combustion of coal and municipal wastes, and a common contaminant of aquatic ecosystems and wetlands. Because of its propensity to concentrate in living tissues, Hg is of considerable environmental importance. The major form of Hg in the atmosphere is elemental mercury (Hg0), which is volatile and is oxidized to mercuric ion (Hg2+) photochemically. Most Hg thus enters aquatic environments as Hg2+ (Figure 21.22).
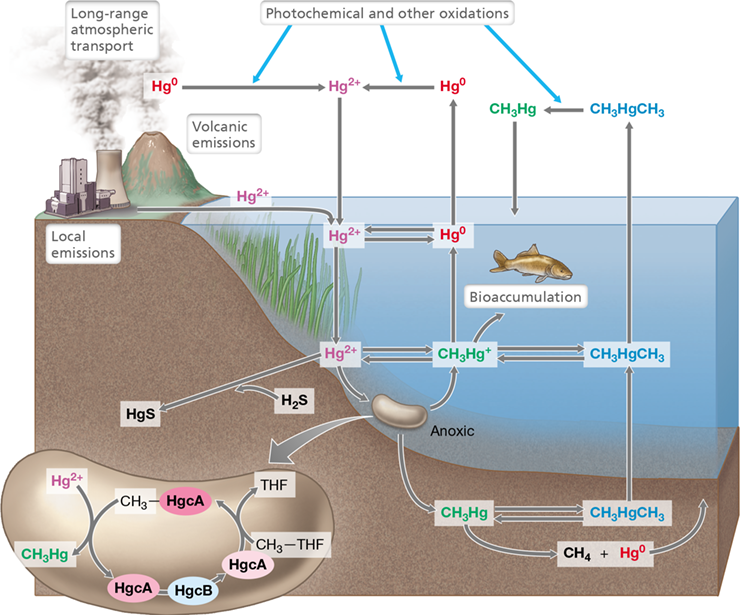
Figure 21.22 Biogeochemical cycling of mercury.

The major reservoirs of mercury are water and sediments. Mercury in water can be concentrated in animal tissues; it can be precipitated as HgS from sediments. The volatile forms of mercury are Hg0 and CH3HgCH3. The enlarged bacterial cell shows the enzyme system responsible for mercury methylation by sulfate-reducing bacteria. Common forms of mercury are shown in different colors. THF, tetrahydrofolate. HgcA and HgcB are proteins that function to methylate mercury.
Microbial Redox Cycle for Mercury
Mercuric ion readily adsorbs to particulate matter, and when deposited in anoxic environments, such as lake or marine sediments, Hg2+ can be metabolized from there by anaerobic microbes. Microbial activity methylates Hg, yielding methylmercury, CH3Hg+ (Figure 21.22), and this can be further methylated to form dimethylmercury, Hg(CH3)2. Methylmercury formation is associated with the activities of sulfate-reducing and iron-reducing Bacteria and methanogenic Archaea; very few aerobes have been shown to methylate mercury. In contrast to methylmercury, the formation of dimethylmercury is likely not due to microbes but instead to abiotic processes.
Methylmercury and dimethylmercury are readily absorbed through the skin of animals and are potent neurotoxins. Dimethylmercury is an extremely toxic neurotoxin and only a few microliters of liquid is sufficient to kill a human. Symptoms are slow to develop but unstoppable as this uncharged form crosses the blood–brain barrier. In addition, CH3Hg+ is soluble and thus easily concentrated in the food chain, primarily in the muscle tissues of fish, and its concentration increases with each trophic level (a process called biomagnification), causing a threat to humans whose diets rely heavily on fish. Methylmercury and Hg2+ are about 100 times more toxic than Hg0, and their accumulation in the aquatic food chain is particularly acute in freshwater lakes and marine coastal waters where enhanced levels of CH3Hg+ have been detected in fish caught for human consumption. In addition to their neurotoxicity, mercuric compounds also cause liver and kidney damage in humans and other animals, and methylmercury is a suspected human carcinogen.
Mastering Microbiology
Art Activity: Figure 21.17 Biogeochemical cycling of mercury
Biochemistry of Mercury Methylation
Methylation of Hg2+ by sulfate-reducing bacteria requires two genes, hgcA (encoding a methyltransferase corrinoid protein) and hgcB, encoding a [4Fe-4S] ferredoxin-containing protein (Figure 21.22). In the reaction sequence (Figure 21.22), a methyl group is transferred from methylated HgcA to inorganic Hg2+. HgcB then regenerates the reduced form of HgcA that accepts a methyl group from the coenzyme methyltetrahydrofolate (Figure 21.22). Identification of hgcA and hgcB has fueled surveys of their distribution in genomic and metagenomic sequences and has revealed that genes for mercury methylation are widely distributed in species of Bacteria and methanogenic Archaea. The discovery of mercury methylation genes paves the way for developing genetic tools for surveying environments for their potential to methylate mercury and should contribute to our understanding of the physiological significance of methylation. It also allows for the identification of environmental variables that control the expression of mercury methylation genes and insight into the observation that mercury methylation also occurs in oxic marine surface waters.
Several other microbial transformations of mercury occur in anoxic sediments, including reactions catalyzed by sulfate-reducing bacteria (H2S+Hg2+→HgS) and methanogens (CH3Hg+→CH4+Hg0) (Figure 21.22). The solubility of mercuric sulfide (HgS) is very low, so in sulfidic sediments, most Hg exists as HgS. But upon aeration, HgS can be oxidized to Hg2+ and SO4 2− by sulfide-oxidizing bacteria (Sections 14.7 and 15.12), and the Hg2+ is eventually converted to CH3Hg+. However, it is not the Hg in HgS that is oxidized here, but instead the sulfide, probably by organisms related to Acidithiobacillus or other sulfur-oxidizing bacteria (Section 15.12).
Mercury Resistance
At sufficiently high concentrations, Hg2+ and CH3Hg+ can be toxic to microorganisms as well as to macroorganisms. However, several gram-positive and gram-negative bacteria convert toxic forms of Hg to nontoxic or less toxic forms. These mercury-resistant bacteria employ the enzyme organomercury lyase to degrade the highly toxic CH3Hg+ to Hg2+ and methane (CH4), and the NADPH (or NADH)-linked enzyme mercuric reductase to reduce Hg2+ to Hg0, which is volatile and thus mobile (Figure 21.23).
Figure 21.23 Mechanism of mercury transformations and resistance.

(a) The mer operon. MerR can function as either a repressor (in the absence of Hg2+) or a transcriptional activator (in the presence of Hg2+). (b) Transport and reduction of Hg2+ and CH3Hg+; the Hg2+ is bound by cysteine residues in the MerP and MerT proteins. MerA is the enzyme mercuric reductase and MerB is organomercury lyase.
In many mercury-resistant bacteria, which are predominantly aerobes, genes encoding Hg resistance (mer genes) reside on plasmids or transposons (Sections 6.2 and 9.11). Mer genes are arranged in an operon under control of the regulatory protein MerR, which can function as either a repressor or an activator of transcription (Section 7.3), depending on Hg availability. In the absence of Hg2+, MerR functions as a repressor and binds to the operator region of the mer operon, thus preventing transcription of the structural genes, merTPABD. However, when Hg2+ is present, it forms a complex with MerR, which then binds to the mer operon and functions as an activator of transcription of mer structural genes (Figure 21.23).
MerP is a periplasmic mercuric ion–binding protein; it binds Hg2+ and transfers it to the membrane transport protein MerT, which interacts with mercuric reductase (MerA) to reduce Hg2+ to Hg0 (Figure 21.23b). Thus, Hg2+ is not released into the cytoplasm and the final result is the release of Hg0 from the cell. Mercuric ion produced from the activity of MerB is trapped by MerT and reduced by MerA, again releasing Hg0 (Figure 21.23b). In this way, Hg2+ and CH3Hg+ are converted to the relatively nontoxic Hg0, which is nonpolar and exits the cell through the cytoplasmic membrane.
Check Your Understanding
What forms of mercury are most toxic to organisms?
21.9 Human Impacts on the Carbon and Nitrogen Cycles
21.9 Human Impacts on the Carbon and Nitrogen Cycles
21.9 Human Impacts on the Carbon and Nitrogen Cycles
Human activities have major effects on the carbon and nitrogen cycles, and these effects have significance for the health of our planet in general. The period of marked human influence on these nutrient cycles began with the Industrial Revolution in the 1800s and is informally termed the Anthropocene, a new geological epoch. Although the greatest human impacts have been on the release of CO2 through the burning of fossil fuels (oil, gas, and coal) and from extensive and ongoing deforestation, human activity has also profoundly affected the nitrogen cycle. We discussed earlier the close coupling of the carbon and nitrogen cycles (Section 21.1), and here we consider some of the projected biogeochemical consequences of human alteration of these two critical nutrient cycles.
CO2, Other Trace Gases, and Global Warming
Atmospheric CO2 levels have increased approximately 40% since the beginning of the Industrial Revolution and are now higher than at any time in the last 800,000 years. In mid-2019, atmospheric levels of CO2 reached 415 parts per million—its highest level in human history (Figure 21.24). Carbon dioxide is one of several trace gases (primarily water vapor, CO2, CH4, and N2O) that comprise less than 0.5% of the atmosphere but contribute significantly to terrestrial and atmospheric warming due to the greenhouse effect, the ability of these gases to trap the infrared radiation emitted by Earth. Atmospheric concentrations of all of these trace gases are rapidly increasing as a consequence of human activities (Figure 21.24). The change in global warming potential resulting from the addition of these gases to the atmosphere is expressed as radiative forcing, defined as the difference between sunlight energy absorbed by Earth and energy radiated back to space, measured in watts per square meter of Earth’s surface (W m−2; Figure 21.24d). Radiative forcing is used to calculate an Annual Greenhouse Gas Index (AGGI), defined as the ratio of the total direct radiative forcing due to long-lived greenhouse gases for a given year to that which was present in 1990 (the baseline year for the Kyoto Protocol for controlling greenhouse gas emissions).
Figure 21.24 Past 40-year increases in greenhouse gases and associated radiative forcing.

Global average values for (a) CO2, (b) CH4, (c) N2O. Increases in CO2 have averaged 1.4 ppm per year before 1995 and 2 ppm per year thereafter. (d) Radiative forcing from chlorofluorocarbons (CFCs) is now stable or declining as a result of the Montreal Protocol banning production of substances that deplete the ozone layer. Greenhouse gas measurements are continuously collected by the National Oceanic and Atmospheric Association on Mauna Loa, Hawaii.
In 2018, the AGGI was 1.43, an increase in radiative forcing of 43% since 1990. Climate models incorporate both the radiative forcing values and the atmospheric lifetimes of the major greenhouse gases. Although methane and N2O have greater radiative forcing per unit molecule than CO2 (58- and 206-fold, respectively), they are at much lower concentrations and therefore contribute less to warming (Figure 21.24d). CO2 is the major contributor to the AGGI in terms of both amount and rate of increase. The atmospheric lifetime of a molecule of N2O is around 150 years. In contrast, there is more uncertainty about the atmospheric lifetimes of methane and CO2, now estimated at about 10 and 120 years, respectively. The long lifetimes of N2O and CO2 mean that once these gases are added to the atmosphere, they will impact Earth’s climate for centuries. In contrast, the much shorter lifetime of methane indicates that any reductions in emissions would have a much more immediate impact on warming.
Also shown in Figure 21.24d is radiative forcing attributed to chlorofluorocarbons (CFCs). There is no natural source of CFCs. These compounds were chemically synthesized for use as refrigerants, aerosol propellants, and cleaning solvents. Following the discovery that they were destroying stratospheric ozone, their manufacture was effectively suspended through an international agreement, the Montreal Protocol on substances that deplete the ozone layer, which entered into force on January 1, 1989. Although the levels of major CFCs now appear to be declining, their long atmospheric lifetimes mean they will remain in the atmosphere for over 100 years. Unfortunately, analyses have identified a 25% increase in CFC release originating in eastern Asia since 2012, pointing to the difficulties in enforcing international Earth-benefiting agreements.
CO2, and Its Effects on Aquatic Microbial Ecosystems
The increase in atmospheric CO2 concentration, measured across a global network of sampling stations (Figure 21.24a), is currently about 2 parts per million per year. This increase would be much more rapid were it not for the high solubility of CO2 in water, which produces carbonic acid (H2CO3); much anthropogenic CO2 thus dissolves in the oceans (Figures 21.1 and 21.20c). The surface waters of the oceans have taken up an estimated 500 billion tons of CO2 from the atmosphere out of a total of 1300 billion tons of total anthropogenic emissions, thus modulating the greenhouse effect somewhat. The increase in average Earth air temperature, estimated to have increased 0.75 °C in the twentieth century and projected to increase by anywhere from 1.1 to 6.4 °C in the twenty-first century, would also have been more rapid without the heat absorption of the oceans. Since three orders of magnitude more energy is required to raise the temperature of a cubic meter of water than a cubic meter of air, it can be calculated that over 80% of the heat retained on Earth as a result of the greenhouse effect thus far has actually entered the ocean.
Although there is considerable uncertainty about the consequences of ocean warming and CO2 consumption on Earth’s biological systems, there is agreement on how these changes will affect biogeochemistry. Warmer ocean surface waters are more buoyant because of their lower density than are deeper waters. Thus, as occurs seasonally in lakes (Section 20.9), the oceans will become more stratified with future global warming. Stratification tends to slow the transfer of nutrients from deeper waters that are needed to nourish phototrophic microbes at the base of the food web in surface waters. This in turn reduces ocean productivity and export of a portion of that production to the deeper ocean through sedimentation (the biological pump, Figure 21.1).
Ocean warming is also contributing to the expansion of oxygen minimum zones (OMZs), regions of naturally occurring low O2 concentration in subsurface waters between 100 and 1000 m in depth (Section 20.10). OMZs are a consequence of both the reduced solubility of O2 in warmer water and the increasing stratification associated with surface warming, which reduces mixing of surface and subsurface waters. Animals will be excluded from the expanding OMZs in contrast to the enhancement of anaerobic microbial processes, such as denitrification and anammox, that directly influence the nitrogen cycle and production of the greenhouse gas N2O.
Acidification of the ocean resulting from the ongoing dissolution of anthropogenic CO2 has reduced ocean pH by 0.1 pH units since the beginning of the Industrial Revolution and may further reduce the pH by 0.3–0.4 units by the year 2100. The ongoing reduction in carbonate (CO3 2−) concentration, a consequence of increasing acidification, is expected to be detrimental to marine calcifiers (organisms synthesizing CaCO3 shells or skeletons, Figure 21.20). Since the concentration of Ca in seawater is relatively constant, continued reduction in CO3 2− will ultimately reach a point where the dissolution of existing CaCO3 is chemically favored, ultimately releasing more dissolved CO2 (Figure 21.20), which reduces the capacity of the oceans to absorb more atmospheric CO2.
Although the biological response to ocean acidification is unknown, it is likely that coral reef ecosystems, a major component of the marine biosphere (Section 23.13), will cease to occur naturally on Earth if CO2 emissions continue at their present rate (Figure 21.24a and see the chapter opener on page 693). Calcification in foraminifera (Section 18.6) will likely be impaired significantly by ocean acidification, as will calcification in coccolithophores (Figure 21.20; Section 18.7). Over periods of a century or so, the invasion of anthropogenic CO2 into the deep ocean will ultimately result in a significant reduction in the levels of CaCO3 sequestered there, and this will likely affect the carbon cycle in major but as yet unpredictable ways.
Methane and Global Warming: Wetlands, Permafrost, and Sea Ice
Increases in atmospheric methane have contributed to about one-fifth of the increase in radiative forcing since 1750. There are many natural sources of methane as well as several sources linked to human activities (Figure 21.25). Major natural sources of methane are wetlands (including melting permafrost), ruminants, and rice cultivation. Analysis of the stable isotopic composition of new atmospheric methane (Section 19.10) suggests that the uptick since 2007 is primarily fueled by emissions from tropical wetlands, the world’s largest natural source of atmospheric methane (Figure 21.25). On the human side, fossil fuel and biomass burning make up nearly a quarter of all methane emissions, and the steadily increasing numbers of domesticated ruminants used as a food source constitute a major source of methane as well.
Figure 21.25 Major sources of global methane emissions.

Total methane emissions are 500–600 million metric tons per year.
Ongoing planetary warming will also increase atmospheric inputs of methane by destabilizing coastal methane hydrates (Figure 21.4) and by the melting of permafrost. That is, there is positive climate feedback linked to methane release. As Earth warms in response to the accumulation of greenhouse gases, attention has focused on high-latitude regions where temperatures are rising twice as fast as the global average. As temperatures rise, normally frozen ground (the permafrost subsurface layer in tundra) thaws, making vast stores of organic carbon available for microbial decomposition. Accumulated over thousands of years from past plant and animal life, the pool of carbon in the permafrost zone is estimated to be 1300–1600 billion tons. This is about 50% of global soil carbon and twice as much carbon as is currently in the atmosphere.
Predicting the rate and extent of microbial conversion of permafrost organic carbon to greenhouse gases (primarily CO2 and CH4) is essential in developing future climate scenarios. It is estimated that 5–15% of permafrost carbon will decompose in this century, releasing primarily CO2 at a rate unlikely to cause abrupt climate change in the near future (a portion of the methane will be oxidized to CO2 by methanotrophs). However, thawing permafrost will impact global climate for centuries, with current models estimating that about 60% of total permafrost carbon emissions will occur past the year 2100. Predicting the end result of such releases on the habitability of Earth is impossible at this point except to say that microbial responses to continued warming will unquestionably occur and will have consequences for all life on Earth.
An additional positive climate feedback is associated with the rapid loss of summer sea ice in the Arctic Ocean, reduced by more than 11% since 1979. Instead of reflecting back most sunlight as ice does, the sea ice melt has opened a new expanse of dark open water to absorb the sun’s energy. This extra energy input, and associated flux of moisture and heat to the Arctic atmosphere, is contributing to a strong local positive feedback called the Arctic amplification. As a result, surface temperatures in the Arctic are rising twice as fast as at lower latitudes and should increase releases of methane from hydrates and of methane and CO2 from permafrost accordingly. The enormous loads of organic carbon in the Arctic place a special focus on this region as an area where microbial activities will continue to surge significantly and accelerate global warming.
Anthropogenic Effects on the Nitrogen Cycle
Anthropogenic impacts on the microbial ecology of the nitrogen cycle are nearly as profound as those on the carbon cycle. The yearly industrial production of nitrogenous fertilizers through the Haber–Bosch process, which combines N2+H2 to form NH3 under high temperature and pressure, is now comparable to the amount of fixed nitrogen entering the biosphere through biological nitrogen fixation, a key link in the nitrogen cycle (Section 21.3). Most of the industrially produced N is used in agriculture, and a significant fraction runs off to the oceans and contributes to coastal eutrophication (Section 20.10). Large amounts are also lost as gaseous nitrogen compounds (N2, N2O, and NO), primarily from nitrification of NH3 and denitrification of NO3 − (Section 21.3). N2O emission is now increasing at a rate of 0.2–0.3% per year (Figure 21.24c). Agriculture—including the microbial breakdown of manure and urine—contributes about 80% of N2O emissions in the United States. Lesser amounts come from motor vehicle emissions and industrial production of fertilizers and nitrogen-based polymers.
Transport of N from industrial and agricultural centers through the atmosphere fertilizes both terrestrial and marine systems and could cause one of at least two effects. On the one hand, if deposition suppresses microbial nitrogen fixation, this would to some degree mitigate the fertilization effect. On the other hand, a greater supply of both CO2 and iron (caused by greater deposition of dust from areas of increasing desertification, Sections 20.6 and 20.7) along with increased N depositions could enhance primary production, since iron is also often a limiting nutrient. Either way, major effects on the carbon cycle should be expected from human inputs in the nitrogen cycle.
Although changes in Earth’s biosphere from human intervention in microbial nutrient cycles are a certainty, precisely what these changes will be is less clear. However, because major nutrient cycles are closely coupled (Section 21.1 and Figure 21.6), it is likely that the significant changes already in play in the carbon and nitrogen cycles will have feedback effects on other nutrient cycles as well. Collectively, these events could upset the balance and interrelationships of Earth’s nutrient cycles in general—cycles driven by the activities of microbial communities keenly attuned to their environments—and have significant (and likely negative) consequences for plants, animals, and humans.
Check Your Understanding
What is the greenhouse effect, and what causes it?
What is the fate of most nitrogen used in agricultural applications?
Why are the OMZs expanding, and what are the likely impacts on nutrient cycles?
Explore the Microbial World Solving the Marine Methane Paradox
Explore the Microbial World Solving the Marine Methane Paradox
Solving the Marine Methane Paradox
An anomaly in the concentration of dissolved methane in oxygenated ocean surface waters was first observed in the latter half of the last century. A region from the surface to approximately 300 meters depth is supersaturated in methane relative to the concentration expected if the ocean was in equilibrium with atmospheric methane (**Figure 1*a***). About 75% of the oceanic flux of methane to the atmosphere derives from this source (other sources being melting methane hydrates and methane generated in marine sediments; see Figures 21.3, 21.4, and 21.5). Because methane is a major greenhouse gas (Section 21.9), identifying its marine origins has been considered essential for predicting how changes in ocean temperatures might exacerbate climate change.
Figure 1 The marine methane paradox.

(a) Representative profile of dissolved methane in surface waters of the Atlantic Ocean showing supersaturation of methane in the upper 300 meters. (b) The pathway for methylphosphonic acid biosynthesis, first identified in the marine ammonia-oxidizing archaeon Nitrosopumilus maritimus (scanning electron micrograph). Phosphoenolpyruvic acid (PEP) is an intermediate in glycolysis (Section 3.6 and Figure 3.11). Methylphosphonic acid synthase (MpnS) generates the methylphosphonic acid that is subsequently cleaved to methane and inorganic phosphate by the enzyme C–P lyase present in many marine bacteria. The enzyme phosphoenolpyruvic acid mutase (PepM) catalyzes the first step of all known phosphonic acid biosynthesis pathways, and the presence of the pepM gene is diagnostic for organisms having the capacity for phosphonic acid biosynthesis. (c) Structures of the phosphonate antibiotic fosfomycin and the phosphonate herbicide glyphosate.
An initial clue came from the light isotopic composition of carbon in methane from marine surface waters, a result consistent with a microbial source (Section 19.10). However, this presented a conundrum—only obligately anaerobic methanogens are known to produce methane (Section 17.2), and marine surface waters are highly oxic. Thus, at first glance, a microbial source for this methane did not make sense. However, a solution to this paradox ultimately came from studies of the marine phosphorus cycle.
Dissolved phosphorus, often a limiting nutrient controlling primary production in the ocean, is present in both inorganic (typically as phosphate) and organic forms. About 10% of the dissolved organic phosphorus is in the form of phosphonates, compounds characterized by a C–P bond (Figure 1b, c). About 10% of marine microbes are capable of phosphonate biosynthesis; however, not all marine organisms are capable of using phosphonate. Cleavage of the C–P bond, required for release of inorganic phosphate, requires an enzyme called C–P lyase. Since phosphonates are abundant in upper ocean waters and the C–P bond can be cleaved by many marine microbes, it was hypothesized that methylphosphonate, or the protonated form methylphosphonic acid (Figure 1b), could be the source of the surface water methane observed.
Subsequent experiments showed that several aerobic marine bacteria could cleave methylphosphonate and use the released phosphate to support growth. However, this brought up another question: What was the source of the methylphosphonic acid? This riddle was solved when a pathway for its biosynthesis was discovered in the marine ammonia-oxidizing archaeon Nitrosopumilus maritimus1 (Figure 1b). Identification of the biochemical pathway and supporting genes from this organism facilitated surveys of other marine microbes and marine metagenomes for similar genes. These studies revealed that the pathway for methylphosphonic acid biosynthesis is widely distributed in marine bacteria, including the abundant chemotrophic bacterium Pelagibacter and phototroph Prochlorococcus (Sections 20.11 and 20.12).
Quantitative studies have confirmed that levels of methylphosphonate in the marine pool of dissolved organic phosphorus can account for the marine methane anomaly, solving the methane paradox and identifying an important new process in the cycling of carbon and phosphorus in the ocean.2 But in addition to marine sources of phosphonates, a few phosphonates are of medical or agricultural significance, including the antibiotic fosfomycin—a potent clinical weapon for treating urinary tract infections—and the widely used herbicide glyphosate (marketed as Roundup®) (Figure 1c). Glyphosate can be catabolized as a sole source of carbon, nitrogen, and phosphate by several different bacteria. However, because of the large amounts of this herbicide used agriculturally, microbial ecological studies of glyphosate biodegradation is an area in need of much further research if we are to understand the fate of terrestrial phosphonates as well as we now do marine phosphonates.
1 Metcalf et al. 2012. Synthesis of methylphosphonic acid by marine microbes: a source for methane in the aerobic ocean. Science 337 : 1104.
2 Repeta et al. 2016. Marine methane paradox explained by bacterial degradation of dissolved organic matter. Nat. Geosci. 9 : 884.
Chapter Review
Go to Mastering Microbiology for videos, animations, practice tests, and more.
I Carbon, Nitrogen, and Sulfur Cycles
21.1 The oxygen and carbon cycles are interconnected through the complementary activities of autotrophic and heterotrophic organisms. Microbial decomposition is the single largest source of CO2 released to the atmosphere.
Q What are methane hydrates, and why are these deposits of concern to climate scientists?
21.2 Under anoxic conditions, organic matter is degraded to CH4 and CO2. Methane is formed primarily from acetate and the reduction of CO2 by H2. The H2 and acetate are supplied by syntrophic bacteria that depend on H2 consumption by a partner as the basis of their energetics.
**Q How can organisms such as Syntrophobacter and Syntrophomonas grow when their metabolism is based on thermodynamically unfavorable reactions? How does coculture of these syntrophs with certain other bacteria allow them to grow?**
21.3 The principal form of nitrogen on Earth is N2, which can be used as a N source only by nitrogen-fixing bacteria. Ammonia produced by nitrogen fixation or by ammonification can be assimilated into organic matter or oxidized to NO3 −. Denitrification and anammox cause major losses of fixed nitrogen from the biosphere.
Q Compare and contrast the processes of nitrification and denitrification in terms of the organisms involved, the environmental conditions that favor each process, and the changes in nutrient availability that accompany each process.
21.4 Bacteria play major roles in both the oxidative and reductive sides of the sulfur cycle. Sulfur- and sulfide-oxidizing bacteria produce SO4 2−, whereas sulfate-reducing bacteria consume SO4 2−, producing H2S. Because sulfide is toxic and reacts with various metals, SO4 2− reduction is an important biogeochemical process. Dimethyl sulfide is the major organic sulfur compound of ecological significance in nature.
Q If sulfur chemolithotrophs had never evolved, would there be a problem in the microbial cycling of sulfur compounds? What impact would coastal marine eutrophication have on microbial sulfur transformations?
II Other Nutrient Cycles
21.5 Iron and manganese exist naturally in two oxidation states: Fe2+/Fe3+ and Mn2+/Mn4+, and typically as insoluble minerals. Bacteria reduce the oxidized metals in anoxic environments, having evolved mechanisms for exporting electrons to the insoluble electron acceptors outside the cell to carry out the anaerobic respiration.
**Q Compare the systems employed by Geobacter and Shewanella for reducing metals outside the cell.**
21.6 The reduced forms of iron and manganese can be oxidized by a wide variety of bacteria. Ferrous iron is soluble at acidic pH, but at neutral pH, Fe2+ and Mn2+ are only soluble under anoxic conditions, and thus organisms that oxidize these metals must live at anoxic–oxic interfaces.
**Q Contrast the habitats of the iron-oxidizing bacteria Acidithiobacillus and Gallionella.**
21.7 P, Ca, and Si are elements cycled by microbial activities, primarily in aquatic environments. Calcium and silicon play important roles in the biogeochemistry of the oceans as components of the exoskeletons of coccolithophores and diatoms, respectively.
Q In what ways are Ca and Si cycling in ocean waters similar, and in what ways do they differ? How do the calcium and silicon cycles couple to the carbon cycle?
III Humans and Nutrient Cycling
21.8 A major toxic form of Hg in nature is CH3Hg+, which can yield Hg2+ that is reduced by bacteria to Hg0. Genes conferring resistance to the toxicity of Hg, such as those that encode enzymes that can detoxify or pump out the metal, often reside on plasmids or transposons.
**Q How are Hg2+ and CH3Hg+ detoxified by the mer system?**
21.9 Anthropogenic inputs of CO2 and reactive nitrogen are impacting major nutrient cycles. Although some consequences are reasonably well understood, including expansion of oxygen minimum zones (OMZs) and impaired growth of calcareous organisms, the long-term changes to the nutrient cycles that sustain Earth’s biosphere remain unclear.
Q Discuss two different ways that microorganisms contribute to positive feedbacks in climate change.
Application Questions
Compare and contrast the carbon, sulfur, and nitrogen cycles in terms of the physiologies of the organisms that participate in each cycle. Which physiologies are part of one cycle but not another?
14C-labeled cellulose is added to a vial containing some anoxic freshwater lake sediments and sealed under anoxic conditions. A few hours later, 14CH4 appears in the vial. Discuss what has happened to yield such a result.
Carbon can be sequestered in the ocean in a variety of forms. Discuss the different forms, their biological sources, and how a warming Earth will influence them.
Chapter Glossary
anaerobic respiration in which NO3 − or NO2 − is reduced to nitrogen gases, primarily N2 Global warming
the warming of the atmosphere and oceans attributed to anthropogenic release of greenhouse gases, primarily CO2, that trap infrared radiation emitted by Earth, ultimately leading to major changes in climate Humus
dead organic matter, some of which functions as electron shuttles for the microbial reduction of metal oxides Radiative forcing
the difference between sunlight energy absorbed by Earth and energy radiated back to space Syntrophy
a process whereby two or more microorganisms cooperate to degrade a substance neither can degrade alone