Spinal Control of Movement
Graded Control of Muscle Contraction by Alpha Motor Neurons
BOX 13.1 OF SPECIAL INTEREST: ALS: Glutamate, Genes, and Gehrig
BOX 13.3 OF SPECIAL INTEREST: Duchenne Muscular Dystrophy
BOX 13.4 PATH OF DISCOVERY: Nerve Regeneration Does Not Ensure Full Recovery, by Timothy C. Cope
The Generation of Spinal Motor Programs for Walking
We are now ready to turn our attention to the system that actually gives rise to behavior. The motor system consists of all our muscles and the neurons that control them. The importance of the motor system was summarized by the pioneering English neurophysiologist Charles Sherrington in the Linacre lecture of 1924: “To move things is all that mankind can do . . . for such the sole executant is muscle, whether in whispering a syllable or in felling a forest.” A moment’s thought will convince you that the motor system is also incredibly complex. Behavior requires the coordinated action of various combinations of almost 700 muscles in a changing and often unpredictable environment.
Have you heard the expression “running around like a chicken with its head cut off”? It comes from the observation that some complex patterns of behavior (running around a barnyard, at least briefly) can be generated without the participation of the brain. There is a considerable amount of circuitry within the spinal cord for the coordinated control of movements, particularly stereotyped (repetitive) ones such as those associated with locomotion. This point was established early in this century by Sherrington and his English contemporary Thomas Graham Brown, who showed that rhythmic movements could be elicited in the hind legs of cats and dogs long after their spinal cords had been severed from the rest of the central nervous system. Today’s view is that the spinal cord contains certain motor programs for the generation of coordinated movements, and that these programs are accessed, executed, and modified by descending commands from the brain. Thus, motor control can be divided into two parts: (1) the spinal cord’s command and control of coordinated muscle contraction, and (2) the brain’s command and control of the motor programs in the spinal cord.
In this chapter, we will explore the peripheral somatic motor system: the joints, skeletal muscles, and spinal motor neurons and interneurons, and how they communicate with each other. In Chapter 14, we will take a look at how the brain influences the activity of the spinal cord.
Based on their appearance under the microscope, the muscles in the body can be described according to two broad categories: striated and smooth. But they are also distinct in other ways. Smooth muscle lines the digestive tract, arteries, and related structures and is innervated by nerve fibers from the autonomic nervous system (see Chapter 15). Smooth muscle plays a role in peristalsis (the movement of material through the intestines) and the control of blood pressure and blood flow. There are two types of striated muscle: cardiac and skeletal. Cardiac muscle is heart muscle, which contracts rhythmically even in the absence of any innervation. Innervation of the heart from the autonomic nervous system (ANS) functions to accelerate or slow down the heart rate. (Recall Otto Loewi’s experiment in Chapter 5.)
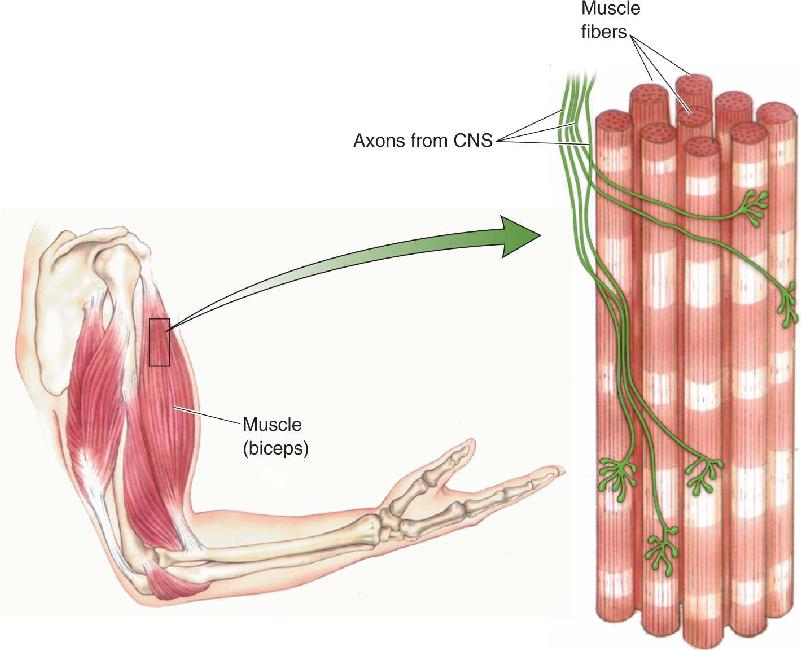
Skeletal muscle constitutes the bulk of the muscle mass of the body and functions to move bones around joints, to move the eyes within the head, to inhale and exhale, to control facial expression, and to produce speech. Each skeletal muscle is enclosed in a connective tissue sheath that, at the ends of the muscle, forms the tendons. Within each muscle are hundreds of muscle fibers, the cells of skeletal muscle, and each fiber is innervated by a single axon branch from the central nervous system (CNS) (Figure 13.1). Because skeletal muscle is derived embryologically from 33 paired somites (see Chapter 7), these muscles and the parts of the nervous system that control them are collectively called the somatic motor system. We focus our attention on this system here because it is under voluntary control and it generates behavior. (The visceral motor system of the ANS will be discussed in Chapter 15.)

FIGURE 13.1 The structure of skeletal muscle. Each muscle fiber is innervated by a single axon.
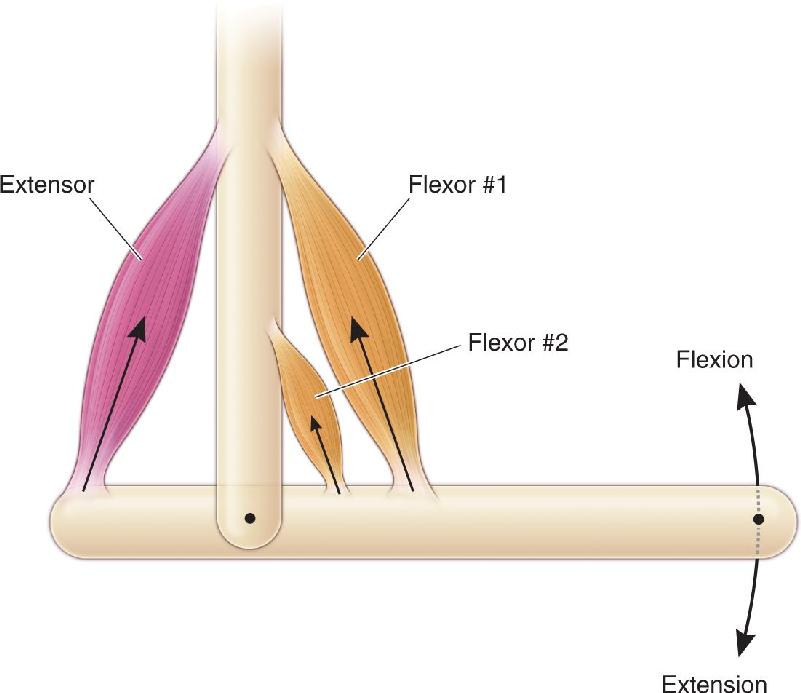
Consider the elbow joint (Figure 13.2). This joint is formed where the humerus, the upper arm bone, is bound by fibrous ligaments to the radius and ulna, the bones of the lower arm. The joint functions like a hinge on a pocket knife. Movement in the direction that closes the knife is called flexion, and movement in the direction that opens the knife is called extension. Note that muscles only pull on a joint; they cannot push. The major muscle that causes flexion is the brachialis, whose tendons insert into the humerus at one end and into the ulna at the other. Two other muscles cause flexion at this joint, the biceps brachii and the coracobrachialis (which lies under the biceps). Together, these muscles are called flexors of the elbow joint, and, because the three muscles all work together, they are called synergists of one another. The two synergistic muscles that cause extension of the elbow joint are the triceps brachii and the anconeus; these two muscles are called extensors. Because the flexors and extensors pull on the joint in opposite directions, they are called antagonists to one another. The relationships between these muscles and bones, and the forces and movements they generate, are shown schematically in Figure 13.3. Even the simple flexion of the elbow joint requires the coordinated contraction of the synergistic flexor muscles and the relaxation of the antagonistic extensor muscles. Relaxing the antagonists allows movements to be faster and more efficient because the muscles are not working against one another.
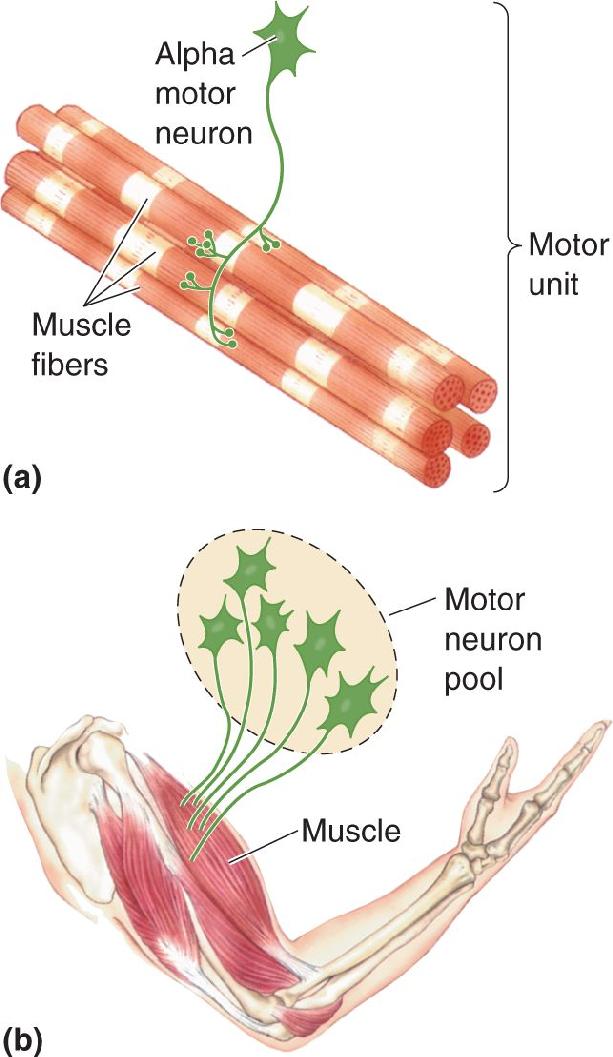

FIGURE 13.2 Major muscles of the elbow joint. The biceps and triceps are antagonistic muscles. Contraction of the biceps causes flexion of the elbow, and contraction of the triceps causes extension. Description

FIGURE 13.3 How contracting muscles flex or extend a joint. Contractions of the flexors pull the right end of the bone upward (flexion). Contraction of the extensor pulls the left end of the bone upward, causing the right end to pivot downward (extension). Flexor #1 and flexor #2 are synergists. Flexors #1 and #2 are antagonist muscles of the extensor.
Other terms to note about somatic musculature refer to the location of the joints they act on. The muscles that are responsible for movements of the trunk are called axial muscles; those that move the shoulder, elbow, pelvis, and knee are called proximal (or girdle) muscles; and those that move the hands, feet, and digits (fingers and toes) are called distal muscles. The axial musculature is very important for maintaining posture, the proximal musculature is critical for locomotion, and the distal musculature, particularly of the hands, is specialized for the manipulation of objects.
The somatic musculature is innervated by the somatic motor neurons in the ventral horn of the spinal cord (Figure 13.4). These cells are sometimes called lower motor neurons to distinguish them from the higher order upper motor neurons of the brain that supply input to the spinal cord. Remember, only the lower motor neurons directly command muscle contraction. Sherrington called these neurons the final common pathway for the control of behavior.

FIGURE 13.4 Muscle innervation by lower motor neurons. The ventral horn of the spinal cord contains motor neurons that innervate skeletal muscle fibers. Description
The axons of lower motor neurons bundle together to form ventral roots; each ventral root joins with a dorsal root to form a spinal nerve that exits the cord through the notches between vertebrae. Recall from Chapter 12 that there are as many spinal nerves as there are notches between vertebrae; in humans, this adds up to 30 on each side. Because they contain sensory and motor fibers, they are called mixed spinal nerves. The motor neurons that provide fibers to one spinal nerve are said to belong to a spinal segment, named for the vertebra where the nerve originates. The segments are cervical (C) 1–8, thoracic (T) 1–12, lumbar (L) 1–5, and sacral (S) 1–5 (see Figure 12.11).
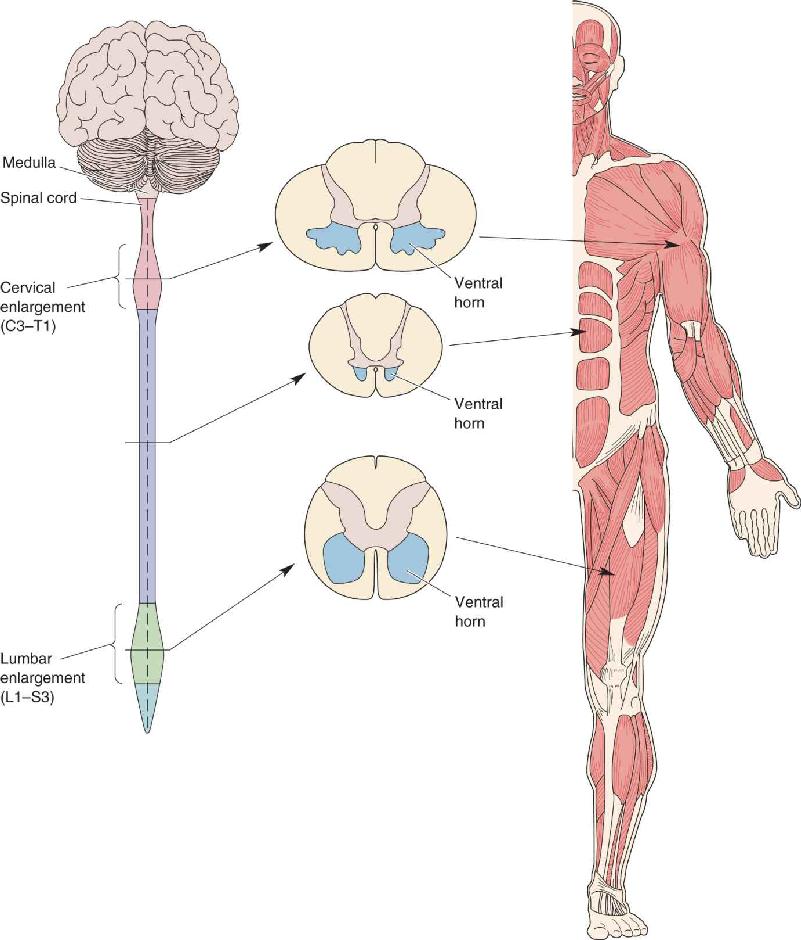
Skeletal muscles are not distributed evenly throughout the body, nor are lower motor neurons distributed evenly within the spinal cord. For example, innervation of the more than 50 muscles of the arm originates entirely from spinal segments C3–Tl. Thus, in this region of the spinal cord, the dorsal and ventral horns appear swollen to accommodate the large number of spinal interneurons and motor neurons that control the arm musculature (Figure 13.5). Similarly, spinal segments Ll–S3 have swollen dorsal and ventral horns because this is where the neurons controlling the leg musculature reside. Thus, we can see that the motor neurons that innervate distal and proximal musculature are found mainly in the cervical and lumbar–sacral segments of the spinal cord, whereas those innervating axial musculature are found at all levels.

FIGURE 13.5 The distribution of motor neurons in the spinal cord. The cervical enlargement of the spinal cord contains the motor neurons that innervate the arm muscles. The lumbar enlargement contains the neurons that innervate the muscles of the leg. Description
The lower motor neurons are also distributed within the ventral horn at each spinal segment in a predictable way, depending on their function. The cells innervating the axial muscles are medial to those innervating the distal muscles, and the cells innervating flexors are dorsal to those innervating extensors (Figure 13.6).

FIGURE 13.6 The distribution of lower motor neurons in the ventral horn. Motor neurons controlling flexors lie dorsal to those controlling extensors. Motor neurons controlling axial muscles lie medial to those controlling distal muscles. Description
There are two categories of lower motor neurons of the spinal cord: alpha motor neurons and gamma motor neurons (the latter are discussed later in the chapter). Alpha motor neurons directly trigger the generation of force by muscles. One alpha motor neuron and all the muscle fibers it innervates collectively make up the elementary component of motor control; Sherrington called it the motor unit. Muscle contraction results from the individual and combined actions of motor units. The collection of alpha motor neurons that innervates a single muscle (e.g., the biceps brachii) is called a motor neuron pool (Figure 13.7).

FIGURE 13.7 A motor unit and motor neuron pool. (a) A motor unit is an alpha motor neuron and all the muscle fibers it innervates. (b) A motor neuron pool is all the alpha motor neurons that innervate one muscle. Description
Graded Control of Muscle Contraction by Alpha Motor Neurons. It is important to exert just the right amount of force during movements: Too much, and you’ll crush the egg you just picked up, while also wasting metabolic energy. Too little, and you may lose the swim race. Most of the movements we make, such as walking, talking, and writing, require only weak muscle contractions. Now and then we need to jog, hop, or lift a pile of books, and stronger contractions are necessary. We reserve the maximal contraction force of our muscles for rare events, such as competitive sprinting or scrambling up a tree to escape a charging bear. The nervous system uses several mechanisms to control the force of muscle contraction in a finely graded fashion.
The first way the CNS controls muscle contraction is by varying the firing rate of motor neurons. An alpha motor neuron communicates with a muscle fiber by releasing the neurotransmitter acetylcholine (ACh) at the neuromuscular junction, the specialized synapse between a nerve and a skeletal muscle (see Chapter 5). Because of the high reliability of neuromuscular transmission, the ACh released in response to one presynaptic action potential causes an excitatory postsynaptic potential (EPSP) in the muscle fiber (sometimes also called an end-plate potential) large enough to trigger one postsynaptic action potential. By mechanisms we will discuss in a moment, a postsynaptic action potential causes a twitch—a rapid sequence of contraction and relaxation—in the muscle fiber. A sustained contraction requires a continual barrage of action potentials. High-frequency presynaptic activity causes temporal summation of the postsynaptic responses, as it does for other types of synaptic transmission. Twitch summation increases the tension in the muscle fibers and smoothes the contraction (Figure 13.8). The rate of firing of motor units is therefore one important way the CNS grades muscle contraction.

FIGURE 13.8 From muscle twitch to sustained contraction. (a) A single action potential in an alpha motor neuron causes the muscle fiber to twitch. (b) The summation of twitches causes a sustained contraction as the number and frequency of incoming action potentials increase. Description
A second way the CNS grades muscle contraction is by recruiting additional synergistic motor units. The extra tension provided by the recruitment of an active motor unit depends on how many muscle fibers are in that unit. In the antigravity muscles of the leg (muscles that oppose the force of gravity when standing upright), each motor unit tends to be quite large, with an innervation ratio of over 1000 muscle fibers per single alpha motor neuron. In contrast, the smaller muscles that control the movement of the fingers and the rotation of the eyes are characterized by much smaller innervation ratios, as few as three muscle fibers per alpha motor neuron. In general, muscles with a large number of small motor units can be more finely controlled by the CNS.
Most muscles have a range of motor unit sizes, and these motor units are usually recruited in the order of smallest first, largest last. This orderly recruitment explains why finer control is possible when muscles are under light loads than when they are under greater loads. Small motor units have small alpha motor neurons, and large motor units have large alpha motor neurons. Thus, one way orderly recruitment occurs is that small neurons, as a consequence of the geometry and physiology of their soma and dendrites, are more easily excited by signals descending from the brain. The idea that the orderly recruitment of motor neurons is due to variations in alpha motor neuron size is called the size principle, first proposed in the late 1950s by Harvard University neurophysiologist Elwood Henneman.
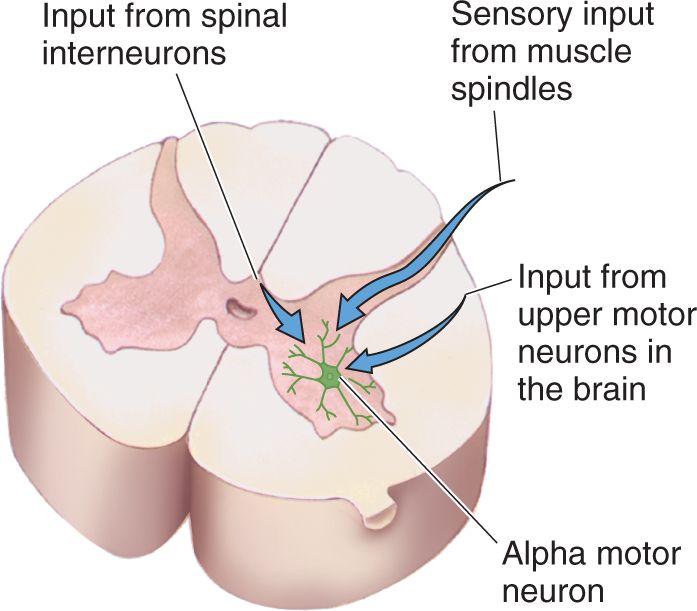
Inputs to Alpha Motor Neurons. Alpha motor neurons excite skeletal muscles. Therefore, to understand the control of muscles, we must understand what regulates motor neurons. Lower motor neurons are controlled by synaptic inputs in the ventral horn. There are only three major sources of input to an alpha motor neuron, as shown in Figure 13.9. The first source is dorsal root ganglion cells with axons that innervate a specialized sensory apparatus embedded within the muscle known as a muscle spindle. As we shall see, this input provides feedback about muscle length. The second source of input to an alpha motor neuron derives from upper motor neurons in the motor cortex and brain stem. This input is important for the initiation and control of voluntary movement and will be discussed in more detail in Chapter 14. The third and largest input to an alpha motor neuron derives from interneurons in the spinal cord. This input may be excitatory or inhibitory and is part of the circuitry that generates the spinal motor programs.

FIGURE 13.9 An alpha motor neuron and its three sources of input. Description
If you have ever eaten different parts of chicken, you know that not all muscle is the same; the leg has dark meat and the breast and wing have white meat. The different appearance, and taste, of the various muscles are due to the biochemistry of the constituent muscle fibers. The red (dark) muscle fibers are characterized by a large number of mitochondria and enzymes specialized for oxidative energy metabolism. These, sometimes called slow (S) fibers, are relatively slow to contract but can sustain contraction for a long time without fatigue. They are typically found in the antigravity muscles of the leg and torso and in the flight muscles of birds that fly (as opposed to domesticated chickens). In contrast, the pale (white) muscle fibers contain fewer mitochondria and rely mainly on anaerobic (without oxygen) metabolism. These fibers, sometimes called fast (F) fibers, contract rapidly and powerfully, but they also fatigue more quickly than slow fibers. They are typical of muscles involved in escape reflexes; for example, the jumping muscles of frogs and rabbits. In humans, the arm muscles contain a large number of white fibers. Fast fibers can be further divided into two subtypes: Fatigue-resistant (FR) fibers generate moderately strong and fast contractions and are relatively resistant to fatigue; fast fatigable (FF) fibers generate the strongest, fastest contractions but are quickly exhausted when stimulated at high frequency for long periods.
Even though all three types of muscle fibers can (and usually do) coexist in a given muscle, each motor unit contains muscle fibers of only a single type. Thus, there is one type of slow motor unit that contains only slowly fatiguing red fibers, and there are two types of fast motor units, each containing either FR or FF white fibers (Figure 13.10). Just as the muscle fibers of the three types of units differ, so do many of the properties of alpha motor neurons. For example, the motor neurons of the FF units are generally the biggest and have the largest diameter, fastest conducting axons; FR units have motor neurons and axons intermediate in size; and slow units have small-diameter, slowly conducting axons. The firing properties of the three types of motor neuron also differ. Fast (FF) motor neurons tend to generate occasional high-frequency bursts of action potentials (30–60 impulses per second), whereas slow motor neurons are characterized by relatively steady, low-frequency activity (10–20 impulses per second).

FIGURE 13.10 Three types of motor units and their contractile properties. (a) A single action potential triggers contraction strengths of different force and time-course in each of the three types of motor units. (b) Repeated trains of action potentials at 40 Hz over many minutes lead to different rates of fatigue in the three types of motor units. (Source: Adapted from Burke et al., 1973.) Description
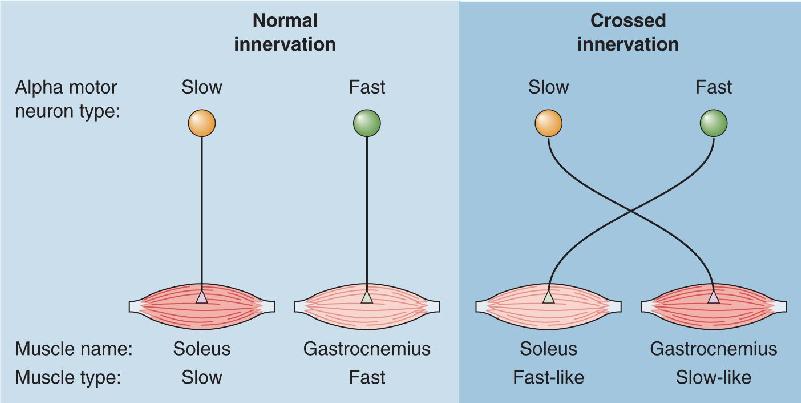
Neuromuscular Matchmaking. The precise matching of particular motor neurons to particular muscle fibers raises an interesting question. Since we’ve been talking about chickens, let’s pose the question this way: Which came first, the muscle fiber or the motor neuron? Perhaps during early embryonic development, there is a matching of the appropriate axons with the appropriate muscle fibers. Alternatively, we could imagine that the properties of the muscle are determined solely by the type of innervation it gets. If it receives a synaptic contact from a fast motor neuron, it becomes a fast fiber and vice versa for slow units.
John Eccles and his colleagues, working at the Australian National University, addressed this question with an experiment in which the normal innervation of a fast muscle was removed and replaced with a nerve that normally innervated a slow muscle (Figure 13.11). This procedure resulted in the muscle’s acquiring slow properties, including not only the type of contraction (slow, fatigue-resistant) but also a switch in much of its underlying biochemistry. This change is referred to as a switch of muscle phenotype—its physical characteristics—because the types of proteins expressed by the muscle were altered by the new innervation. Work by Terje Lømo and his colleagues in Norway suggests that this switch in muscle phenotype can be induced simply by changing the activity in the motor neuron from a fast pattern (occasional bursts at 30–60 spikes per second) to a slow pattern (steady activity at 10–20 spikes per second). These findings are particularly interesting because they raise the possibility that neurons switch phenotype as a consequence of synaptic activity (experience), and that this may be a basis for learning and memory (see Chapters 24 and 25).

FIGURE 13.11 A crossed-innervation experiment. Forcing slow motor neurons to innervate a fast muscle causes the muscle to switch to assume slow properties. Description
Besides the alterations imposed by patterns of motor neuron activity, simply varying the absolute amount of activity can also change muscle fibers. A long-term consequence of increased activity (especially due to isometric exercise) is hypertrophy, or exaggerated growth, of the muscle fibers as seen in bodybuilders. Conversely, prolonged inactivity leads to atrophy, or degeneration, of muscle fibers, which can happen when joints are immobilized in a cast following an injury. Clearly, there is an intimate relationship between the lower motor neuron and the muscle fibers it innervates (Box 13.1).
Amyotrophic lateral sclerosis (ALS) is a particularly cruel disease that was first described in 1869 by the French neurologist Jean-Martin Charcot. The initial signs of the disease are muscle weakness and atrophy. Usually over the course of 1–5 years, all voluntary movement is lost; walking, speaking, swallowing, and breathing gradually deteriorate. Death is usually caused by failure of the respiratory muscles. Because the disease often has no effect on sensations, intellect, or cognitive function, patients are left to watch their bodies slowly waste away, keenly aware of what is happening. The disease is relatively rare, afflicting one out of approximately every 20,000 individuals. Still, an estimated 30,000 Americans are currently diagnosed with ALS. Its most famous victim was Lou Gehrig, a star baseball player with the New York Yankees, who died of ALS in 1936. In the United States, ALS is often called Lou Gehrig’s disease.
Muscle weakness and paralysis are characteristics of motor unit damage. Indeed, the main pathology associated with ALS is the degeneration of the large alpha motor neurons. The large neurons of the motor cortex that innervate alpha motor neurons are also affected, but, curiously, other neurons in the CNS are generally spared. The selective damage to motor neurons explains the selective loss of motor functions in ALS patients.
There appear to be many causes of ALS, most of them still unknown. One suspected cause is excitotoxicity. As we learned in Chapter 6, overstimulation by the excitatory neurotransmitter glutamate and closely related amino acids can cause the death of otherwise normal neurons (see Box 6.4). Many ALS patients have elevated levels of glutamate in their cerebrospinal fluid. Excitotoxicity has been implicated in the unusually high incidence of ALS on the island of Guam that occurred before World War II. It has been suggested that one environmental cause in Guam may have been the ingestion of cycad nuts, which contain an excitotoxic amino acid. In addition, research indicates that a glutamate transporter may be defective in ALS, thereby prolonging the exposure of active neurons to extracellular glutamate. Thus, the first drug approved by the U.S. Food and Drug Administration for the treatment of ALS was riluzole, a blocker of glutamate release. The drug treatment can slow the disease by only a few months, however, and unfortunately, the long-term outcome is the same.
Only 10% of ALS cases are obviously inherited, and screens for defective genes have pointed to several mutations that can lead to ALS. The first mutation, discovered in 1993, leads to defects in the enzyme superoxide dismutase. A toxic by-product of cellular metabolism is the negatively charged molecule O2–, called the superoxide radical. Superoxide radicals are extremely reactive and can inflict irreversible cellular damage. Superoxide dismutase is a key enzyme that causes superoxide radicals to lose their extra electrons, converting them back to oxygen. Thus, the loss of superoxide dismutase would lead to a buildup of superoxide radicals and cellular damage, particularly in cells that are metabolically very active. The death of motor neurons seems to depend on the actions of glial cells that surround them.
More recent research has identified mutations of about 15 more genes that can cause inherited forms of ALS. They affect a surprisingly wide variety of basic cellular processes. Some mutations cause defects in proteins that normally bind and regulate RNA during transcription. Others affect proteins involved in the trafficking of vesicles, protein secretion, cell division, ATP production, or the dynamics of the cytoskeleton. Genome-wide association studies, which examine a large number of gene variations to reveal which are associated with a disease, suggest that the coincidence of two mutations of distinctly different genes can also cause ALS. The picture that is emerging is that ALS can have many distinct causes; it is really a group of diseases that happen to share similar clinical characteristics.
There is still much to be learned about selective motor neuron loss in ALS. What we know so far has led to new ideas for possible treatments, including the use of neuronal stem cells to replace lost neurons and glia, and genetics-based strategies to suppress the effects of mutations. Translating these ideas into effective treatments for ALS patients is an exciting but still distant possibility.
Muscle contraction is initiated by the release of acetylcholine (ACh) from the axon terminals of alpha motor neurons, as we said. ACh produces a large EPSP in the postsynaptic membrane due to the activation of nicotinic ACh receptors. Because the membrane of the muscle cell contains voltage-gated sodium channels, this EPSP is sufficient to evoke an action potential in the muscle fiber (but see Box 13.2). By the process of excitation–contraction coupling, this action potential (the excitation) triggers the release of Ca2+ from an organelle inside the muscle fiber, which leads to contraction of the fiber. Relaxation occurs when the Ca2+ levels are lowered by reuptake into the organelle. To understand this process, we must take a closer look at the muscle fiber.
The neuromuscular junction is an exceptionally reliable synapse. A presynaptic action potential causes the contents of hundreds of synaptic vesicles to be released into the synaptic cleft. The liberated ACh molecules act at densely packed nicotinic receptors in the postsynaptic membrane, and the resulting EPSP is many times larger than what is necessary to trigger an action potential, and twitch, in the muscle fiber—normally, that is.
In a clinical condition called myasthenia gravis, the ACh released is far less effective, and neuromuscular transmission often fails. The name is derived from the Greek for “severe muscle weakness.” The disorder is characterized by weakness and fatigability of voluntary muscles, typically including the muscles of facial expression, and it can be fatal if respiration is compromised. The disease strikes roughly one in 10,000 people of all ages and ethnic groups. An unusual feature of myasthenia gravis is that the severity of the muscle weakness fluctuates, even over the course of a single day.
Myasthenia gravis is an autoimmune disease. In 1973, Jim Patrick and Jon Lindstrom, working at the Salk Institute in California, discovered that rabbits injected with purified nicotinic ACh receptors generated antibodies to their own ACh receptors and contracted a rabbit version of myasthenia gravis. For reasons we don’t understand, the immune systems of most myasthenia-afflicted humans generate antibodies against their own nicotinic ACh receptors. The antibodies bind to the receptors, interfering with the normal actions of ACh at the neuromuscular junctions. In addition, the binding of antibodies to the receptors leads to secondary, degenerative changes in the structure of the neuromuscular junctions that also make transmission much less efficient.
An effective treatment for myasthenia gravis is the administration of drugs that inhibit the enzyme acetylcholinesterase (AChE). Recall from Chapters 5 and 6 that AChE breaks down ACh in the synaptic cleft. In low doses, AChE inhibitors can strengthen neuromuscular transmission by prolonging the life of released ACh. But the drugs are imperfect and the therapeutic window is narrow. As we saw in Box 5.5, too much ACh in the cleft leads to desensitization of the receptors and a block of neuromuscular transmission. Different muscles may respond differently to the same drug dose. The increased levels of ACh can also affect the ANS, leading to side effects such as nausea, vomiting, abdominal cramps, diarrhea, and bronchial secretions. Another common treatment for myasthenia gravis involves suppression of the immune system, either with drugs or by surgical removal of the thymus gland.
With careful and continual medical treatment, the long-term prognosis is good and life expectancy is normal for patients with this disease of the neuromuscular junction.
The structure of a muscle fiber is shown in Figure 13.12. Muscle fibers are formed early in fetal development by the fusion of muscle precursor cells, or myoblasts, which are derived from the mesoderm (see Chapter 7). This fusion leaves each cell with more than one cell nucleus, so individual muscle cells are said to be multinucleated. The fusion elongates the cells (hence the name fiber), and fibers can range from 1 to 500 mm in length. Muscle fibers are enclosed by an excitable cell membrane called the sarcolemma.

FIGURE 13.12 The structure of a muscle fiber. T tubules conduct electrical activity from the surface membrane into the depths of the muscle fiber. Description
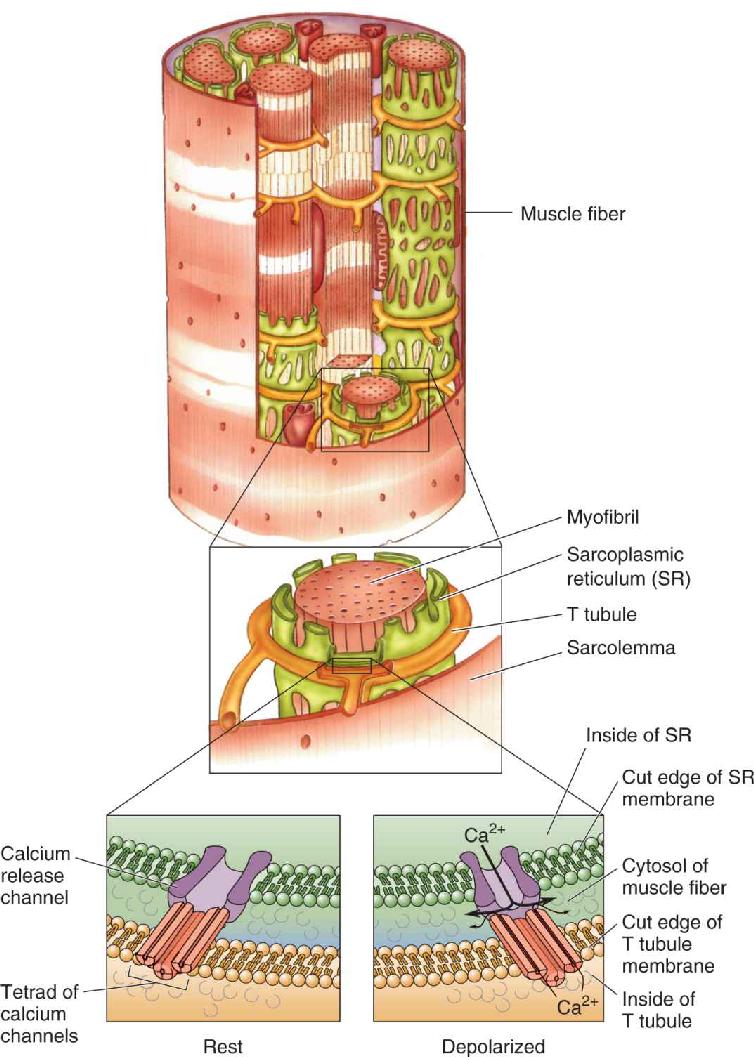
Within the muscle fiber are a number of cylindrical structures called myofibrils, which contract in response to an action potential sweeping down the sarcolemma. Myofibrils are surrounded by the sarcoplasmic reticulum (SR), an extensive intracellular sac that stores Ca2+ (similar in appearance to the smooth endoplasmic reticulum of neurons; see Chapter 2). Action potentials sweeping along the sarcolemma gain access to the sarcoplasmic reticulum deep inside the fiber by way of a network of tunnels called T tubules (T for transverse). These are like inside-out axons; the interior of each T tubule is continuous with the extracellular fluid.
Where the T tubule comes in close apposition to the SR, there is a specialized coupling of the proteins in the two membranes. A voltage-sensitive cluster of four calcium channels, called a tetrad, in the T tubule membrane is linked to a calcium release channel in the SR. As illustrated in Figure 13.13, the arrival of an action potential in the T tubule membrane causes a conformational change in the voltage-sensitive tetrad of channels, which opens the calcium release channel in the SR membrane. Some Ca2+ flows through the tetrad channels, and even more Ca2+ flows through the calcium-release channel, and the resulting increase in free Ca2+ within the cytosol causes the myofibril to contract.

FIGURE 13.13 The release of Ca2+ from the sarcoplasmic reticulum. Depolarization of the T tubule membrane causes conformational changes in proteins that are linked to calcium channels in the SR, releasing stored Ca2+ into the cytosol of the muscle fiber. Description
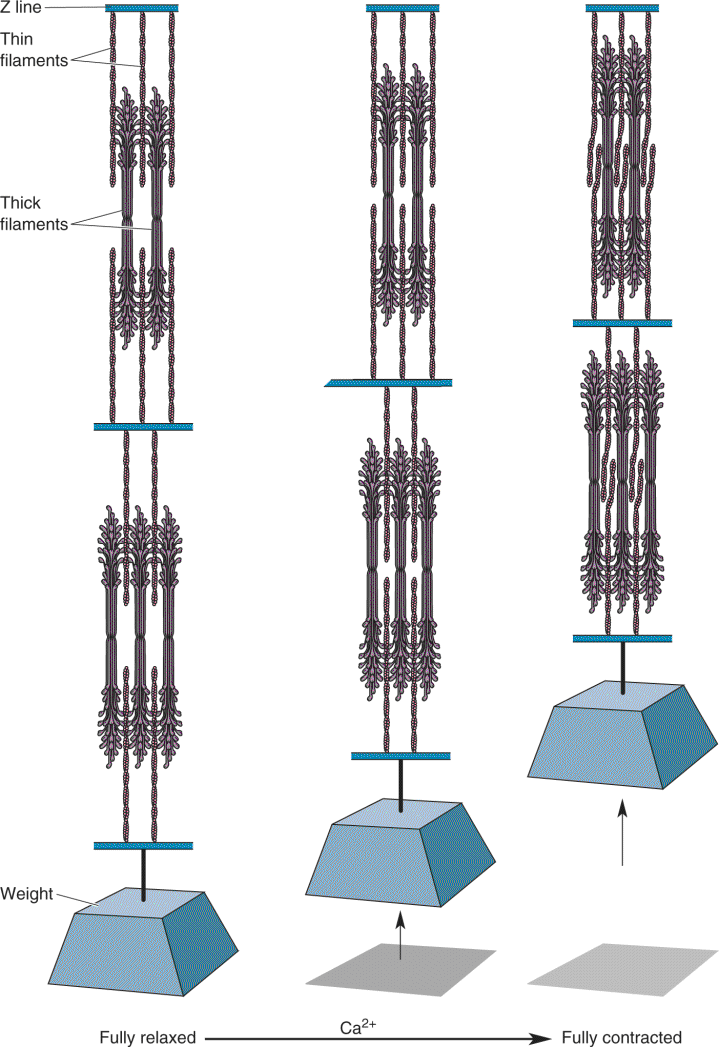
A closer look at the myofibril reveals how Ca2+ triggers contraction (Figure 13.14). The myofibril is divided into segments by disks called Z lines (named for their appearance when viewed from the side). A segment composed of two Z lines and the myofibril in between is called a sarcomere. Anchored to each side of the Z lines is a series of bristles called thin filaments. The thin filaments from adjacent Z lines face one another but do not come in contact. Between and among the two sets of thin filaments are a series of fibers called thick filaments. Muscle contraction occurs when the thin filaments slide along the thick filaments, bringing adjacent Z lines toward one another. In other words, the sarcomere becomes shorter in length. This sliding-filament model of sarcomere shortening is shown in Figure 13.15.


FIGURE 13.15 The sliding-filament model of muscle contraction. Myofibrils shorten when the thin filaments slide toward one another on the thick filaments. Description
The sliding of the filaments with respect to one another occurs because of the interaction between the major thick filament protein, myosin, and the major thin filament protein, actin. The exposed “heads” of the myosin molecules bind actin molecules and then undergo a conformational change that causes them to pivot (Figure 13.16). This pivoting causes the thick filament to move with respect to the thin filament. Adenosine triphosphate (ATP) then binds to the myosin heads and the heads disengage and “uncock” so that the process can repeat itself. Repeating this cycle enables the myosin heads to “walk” along the actin filament.

FIGURE 13.16 The molecular basis of muscle contraction. The binding of Ca2+ to troponin shifts tropomyosin and allows the myosin heads to bind to the actin filament. Then the myosin heads pivot, causing the filaments to slide with respect to one another. Description
When the muscle is at rest, myosin cannot interact with actin because the myosin attachment sites on the actin molecule are covered by a complex of two proteins: tropomyosin and troponin. Ca2+ initiates muscle contraction by binding to troponin and causing tropomyosin to shift its position, thereby exposing the sites where myosin binds to actin. Contraction continues as long as Ca2+ and ATP are available; relaxation occurs when the Ca2+ is sequestered by the SR. The reuptake of Ca2+ by the SR depends on the action of a calcium pump and hence also requires ATP.
We can summarize the steps of excitation–contraction coupling as follows:
- An action potential occurs in an alpha motor neuron axon.
- ACh is released by the axon terminal of the alpha motor neuron at the neuromuscular junction.
- Nicotinic receptor channels in the sarcolemma open, and the postsynaptic sarcolemma depolarizes (EPSP).
- Voltage-gated sodium channels in the sarcolemma open and an action potential is generated in the muscle fiber, which sweeps down the sarcolemma and into the T tubules.
- Depolarization of the T tubules causes Ca2+ release from the SR.
- Tropomyosin shifts position and myosin binding sites on actin are exposed.
- An ATP binds to each myosin head and it disengages from actin.
- The cycle continues as long as Ca2+ and ATP are present.
- As EPSPs end, the sarcolemma and T tubules return to their resting potentials.
- Ca2+ is sequestered by the SR by an ATP-driven pump.
- Myosin binding sites on actin are covered by tropomyosin.
You can now understand why death causes stiffening of the muscles, a condition known as rigor mortis. Starving the muscle cells of ATP prevents the detachment of the myosin heads and leaves the myosin attachment sites on the actin filaments exposed for binding. The end result is the formation of permanent attachments between the thick and thin filaments.
Since the proposal of the sliding-filament model in 1954 by English physiologists Hugh Huxley, Andrew Huxley, and their colleagues, there has been a tremendous amount of progress in identifying the detailed molecular mechanisms of excitation–contraction coupling in muscle. This progress has resulted from a multidisciplinary approach to the problem, with critical contributions made by the use of electron microscopy as well as biochemical, biophysical, and genetic methods. The application of molecular genetic techniques also has added important new information to our understanding of muscle function, in both health and disease (Box 13.3).
Muscular dystrophy is a group of inherited disorders, all of which are characterized by progressive weakness and deterioration of muscle. The most common type, Duchenne muscular dystrophy, afflicts about one in 3500 boys before adolescence. The disease is first detected as a weakness of the legs and usually puts its victims in wheelchairs by the time they reach age 12. The disease continues to progress, and afflicted males typically do not survive past the age of 30.
The characteristic hereditary pattern of this disease, which afflicts only males but is passed on from their mothers, led to a search for a defective gene on the X chromosome. Major breakthroughs came in the late 1980s when the defective region of the X chromosome was identified. Researchers discovered that this region contains the gene that codes for a cytoskeletal protein dystrophin. The dystrophin gene is enormous—2.6 million base pairs—and its size makes it unusually vulnerable to mutations. Boys with Duchenne muscular dystrophy have an entirely dysfunctional dystrophin gene: They cannot produce the mRNA encoding dystrophin. A milder form of the disease, called Becker muscular dystrophy, is associated with an altered mRNA encoding a portion of the dystrophin protein.
Dystrophin is a large protein that helps to link the muscle cytoskeleton, lying just under the sarcolemma, to the extracellular matrix. The protein also seems to be important for helping muscle deal with oxidative stress. Dystrophin must not be strictly required for muscle contraction because movements in afflicted boys appear to be normal during their first few years of life. The absence of dystrophin may lead to secondary changes in the contractile apparatus, eventually resulting in muscle degeneration. It is interesting to note that dystrophin is also concentrated in axon terminals in the brain, where it might contribute to excitation-secretion coupling.
Intensive efforts are underway to find a strategy for treating, or even curing, Duchenne muscular dystrophy with some form of gene therapy. One long-standing idea is to introduce an artificial gene that essentially repairs the patient’s defective dystrophin gene or mimics a normal dystrophin gene. A big challenge, as with most attempts at gene therapy, has been to get the artificial gene into dystrophic muscle cells safely and effectively. Specially engineered forms of viruses that carry the gene, infect muscle cells, and induce the cells to express dystrophin are often used. Another approach is to transplant stem cells—immature cells that can grow and differentiate into mature, normal muscle cells that express dystrophin—into dystrophic muscles. Stem cell therapy has been very promising when tested in mouse models of muscular dystrophy. Yet, another strategy is to test small molecules that might minimize muscle degeneration, promote muscle regeneration, mitigate encoding problems by mutant dystrophin genes, or promote the production of other muscle proteins that can substitute for dystrophin.
There is no cure for Duchenne muscular dystrophy as yet, but some of the new treatment strategies have shown promise in clinical trials. It is exciting to think that a devastating genetic disease such as Duchenne muscular dystrophy might soon be treatable.
We’ve traced the action potentials sweeping down the axon of the alpha motor neuron and seen how this causes contraction of the muscle fibers in the motor unit. Now let’s explore how the activity of the motor neuron is itself controlled. We begin with a discussion of the first source of synaptic input to the alpha motor neuron introduced earlier—sensory feedback from the muscles themselves.
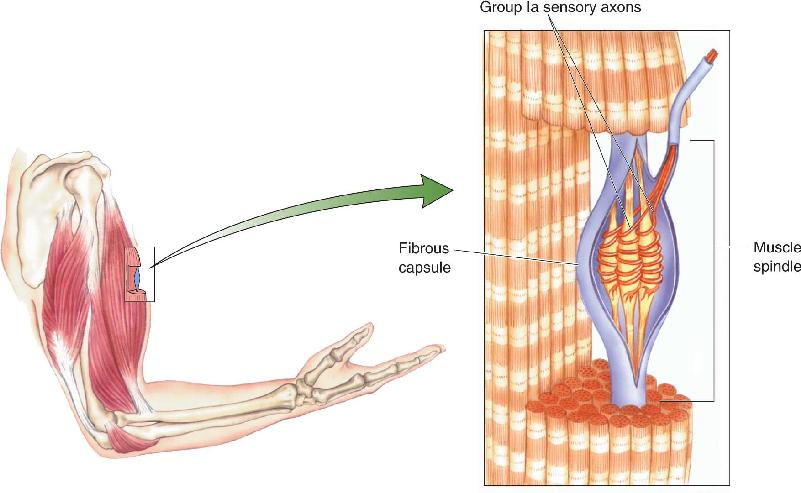
As we mentioned already, deep within most skeletal muscles are specialized structures called muscle spindles (Figure 13.17). A muscle spindle, also called a stretch receptor, consists of several types of specialized skeletal muscle fibers contained in a fibrous capsule. The middle third of the capsule is swollen, giving the structure the shape for which it is named. In this middle (equatorial) region, group Ia sensory axons wrap around the muscle fibers of the spindle. The spindles and their associated Ia axons, specialized for the detection of changes in muscle length (stretch), are examples of proprioceptors. These receptors are a component of the somatic sensory system that is specialized for “body sense,” or proprioception (from the Latin for “one’s own”), which informs us about how our body is positioned and moving in space.

FIGURE 13.17 A muscle spindle and its sensory innervation.
Recall from Chapter 12 that group I axons are the thickest myelinated axons in the body, meaning that they conduct action potentials very rapidly. Within this group, Ia axons are the largest and fastest. Ia axons enter the spinal cord via the dorsal roots, branch repeatedly, and form excitatory synapses upon both interneurons and alpha motor neurons of the ventral horns. The Ia inputs are also very powerful. Neurophysiologist Lorne Mendell, working at Harvard with Henneman, was able to show that a single Ia axon synapses on virtually every alpha motor neuron in the pool innervating the same muscle that contains the spindle.
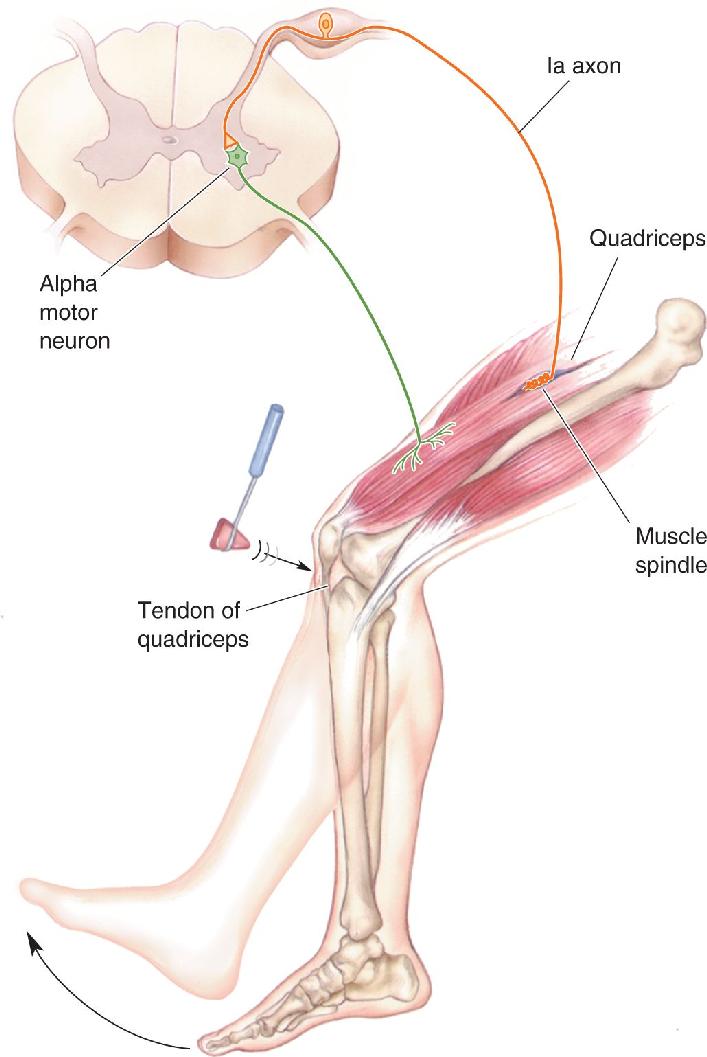
The Stretch Reflex. The function of this sensory input to the spinal cord was first shown by Sherrington, who noted that when a muscle is pulled on, it tends to pull back (contract). The fact that this stretch reflex, sometimes called the myotatic reflex (myo from the Greek for “muscle,” tatic from the Greek for “stretch”), involves sensory feedback from the muscle was shown by cutting the dorsal roots. Even though the alpha motor neurons were left intact, this procedure eliminated the stretch reflex and caused a loss of muscle tone. Sherrington deduced that the motor neurons must receive a continual synaptic input from the muscles. Later work showed that the discharge of Ia sensory axons is closely related to the length of the muscle. As the muscle is stretched, the discharge rate goes up; as the muscle is shortened and goes slack, the discharge rate goes down.
The Ia axon and the alpha motor neurons on which it synapses constitute the monosynaptic stretch reflex arc — “monosynaptic” because only one synapse separates the primary sensory input from the motor neuron output. Figure 13.18 shows how this reflex arc serves as an antigravity feedback loop. When a weight is placed on a muscle and the muscle starts to lengthen, the muscle spindles are stretched. The stretching of the equatorial region of the spindle leads to depolarization of the Ia axon endings due to the opening of mechanosensitive ion channels (see Chapter 12). The resulting increased action potential discharge of the Ia axons synaptically depolarizes the alpha motor neurons, which respond by increasing their action potential frequency. This causes the muscle to contract, thereby shortening it.

FIGURE 13.18 The stretch reflex. This illustration shows the response of an Ia axon and a motor neuron to the sudden addition of weight that stretches the muscle. Description
The knee-jerk reflex is one example of the stretch reflex. When your doctor taps the tendon beneath your kneecap, the tendon very briefly stretches the quadriceps muscle of your thigh, which then reflexively contracts and causes your leg to extend (Figure 13.19). The knee-jerk reflex tests the intactness of the nerves and muscles in this reflex arc. Stretch reflexes can also be elicited by stretching muscles of the arm, the ankle, and the jaw.

Peripheral sensory and motor nerves are vulnerable to various type of injury. As we learned earlier, the cut axons of peripheral nerves can often regenerate and reestablish their connections with muscle (see Figure 13.11). Are regenerated axons and synapses as effective as normal axons and synapses? This question has been studied carefully in the neural circuits of the stretch reflex (Box 13.4).
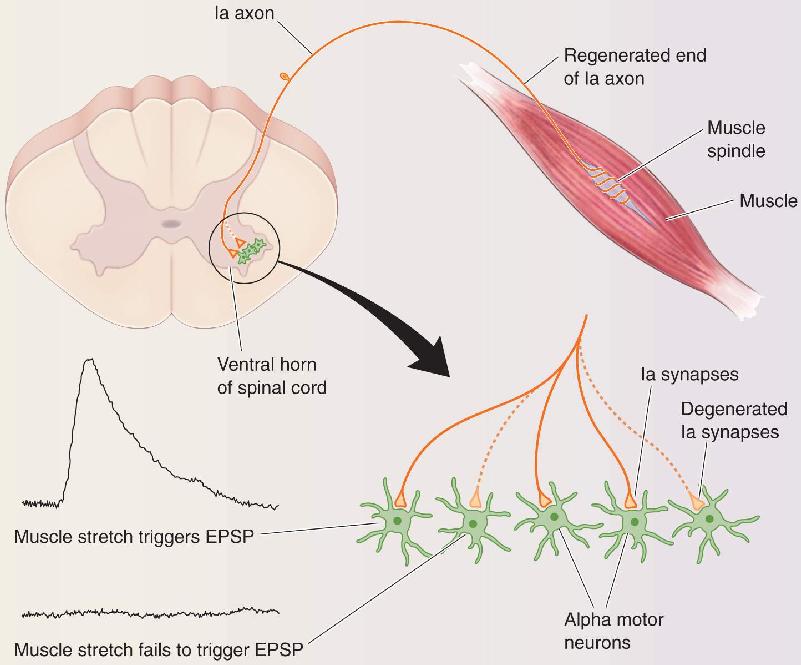
Tapping a muscle’s tendon can cause the muscle to contract and a leg to jerk. The monosynaptic circuit underlying this stretch reflex is shown in Figure 13.19. You will not be surprised to hear that cutting the sensory and motor nerves interrupts the reflex. Peripheral nerves can often regrow, however. What would you expect after the cut axons grow back into the muscle? The surprising answer is that the stretch reflex does not recover, even though voluntary contractions regain substantial force. It would seem easy to figure out why. Every element of this circuit is accessible to measurements, including the firing patterns of group Ia axons that encode muscle length, firing patterns of the motor neurons, the force of contraction produced by the stretched muscle and its synergists, and even the excitatory postsynaptic potentials (EPSPs) produced by the synapses between Ia axons and motor neurons in the spinal cord. Studies of this problem have fascinated me for more than 20 years. They have given me a rare opportunity to understand how a neural circuit can generate normal behavior, how a circuit responds to injury, and what factors limit the nervous system’s capacity to recover from injury.
The recovery problem was most likely on the sensory side of the circuit. It couldn’t be a deficit in the motor neurons or muscle or their restored connections because the muscle contracted normally during reflexes triggered by sensory stimuli other than stretch. At first, the most likely alternative hypothesis seemed to be that regenerating sensory axons end up reconnecting with the wrong sensory receptor organs in the periphery. It was well known that sensory axons in severed nerves reconnect somewhat indiscriminately with their targets, meaning that a reduced number of Ia axons would be available after regeneration to detect muscle stretch and to excite motoneurons. Even so, a sizable fraction of Ia axons do manage to reinnervate their normal targets; Lorne Mendell and his coworkers found that nearly 40% of regenerating Ia axons reconnected with muscle spindles. Even if the total excitation from fewer Ia axons were too weak to excite motoneurons to fire during muscle stretch, we would still expect that the action potentials in the reconnected Ia axons would provide a boost to the force of ongoing muscle contraction. In the laboratory, however, Brian Clark and I found no detectable firing modulation of motor units by stretching self-reinnervated muscles. Our colleague Richard Nichols, using different methods, corroborated our findings. The results were clear and puzzling: Muscle stretch utterly fails to recruit motor neurons after recovery from nerve cuts.
So what defect accounts for the nearly complete and persistent absence of stretch reflexes following injury? A key result emerged from our studies of EPSPs recorded from motor neurons during natural stretch of the muscle. The Ia synaptic potentials were weaker, of course, in part because roughly half of the Ia axons were not responding appropriately to muscle stretch. In addition, Edyta Bichler and Katie Bullinger in our lab made the prescient observation that these diminished EPSPs could be found only in about half of the motor neurons studied, while the other half showed no EPSPs at all (Figure A). Normally, Ia axons produce an EPSP in every motor neuron that innervates the same muscle. These observations highlighted a key shortcoming of Ia sensory neurons that regenerate their damaged peripheral axons: While some reconnect with muscle spindle receptors in the muscle, they also disconnect from many motor neurons in the spinal cord.

Recently, a structural explanation for the loss of the stretch reflex was provided by Francisco Alvarez and his laboratory in collaboration with ours: A probe that allows microscopic identification of Ia synaptic terminals revealed the loss of more than 70% of Ia synapses on the proximal dendrites of motor neurons. We also found that regenerating Ia axons actually retract their branches from the areas where motor neuron cell bodies and dendrites reside. Synaptic loss and axonal retraction in the spinal cord occur despite successful regeneration of the injured Ia axons’ branches in the muscle.
What is the value of these findings? The circuits of the stretch reflex play an important role during normal movement by sensing the configuration of the body and limbs and regulating the way they respond to mechanical disturbances. The reorganization of spinal circuits after nerve injury helps us to understand why some movement disorders persist despite the regeneration of axons. Our findings may also apply to circuits beyond the spinal cord. For example, we might consider whether there are similar changes to synapses on corticospinal neurons after disruption of descending motor tracts; this could have implications for therapeutic strategies for dealing with spinal cord injury. Our findings so far strongly motivate us to move forward and learn more about the biological processes that underlie the degeneration of neurons.
Many people participated in these studies, including undergraduate, graduate, and post-doctoral students. Our progress rested on the diverse expertise and hard work of these collaborators. I believe that a team approach is an absolute necessity for tackling the extraordinarily complex functions and malfunctions of the central nervous system. Working collaboratively exposes us to new ideas and promotes our growth as scientists.
Bullinger KL, Nardelli P, Pinter MJ, Alvarez FJ, Cope TC. 2011. Permanent central synaptic disconnection of proprioceptors after nerve injury and regeneration. II. Loss of functional connectivity with motoneurons. Journal of Neurophysiology 106:2471–2485.
Haftel VK, Bichler EK, Wang QB, Prather JF, Pinter MJ, Cope TC. 2005. Central suppression of regenerated proprioceptive afferents. Journal of Neuroscience 25:4733–4742.
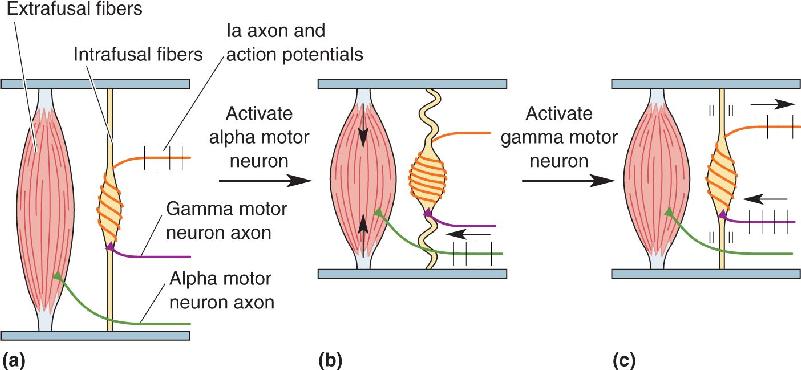
The muscle spindle contains modified skeletal muscle fibers within its fibrous capsule. These muscle fibers are called intrafusal fibers, to distinguish them from the more numerous extrafusal fibers that lie outside the spindle and form the bulk of the muscle. An important difference between the two types of muscle fibers is that only extrafusal fibers are innervated by alpha motor neurons. Intrafusal fibers receive their motor innervation by another type of lower motor neuron called a gamma motor neuron (Figure 13.20).

FIGURE 13.20 Alpha motor neurons, gamma motor neurons, and the muscle fibers they innervate.
Imagine a situation in which muscle contraction is commanded by an upper motor neuron. The alpha motor neurons respond, the extrafusal fibers contract, and the muscle shortens. The response of the muscle spindles is shown in Figure 13.21. If they were to become slack, the Ia axons would become silent and the spindle would go “off the air,” no longer providing information about muscle length. This does not happen, however, because the gamma motor neurons are also activated. Gamma motor neurons innervate the intrafusal muscle fiber at the two ends of the muscle spindle. Activation of these fibers causes a contraction of the two poles of the muscle spindle, thereby pulling on the noncontractile equatorial region and keeping the Ia axons active. Notice that the activation of alpha and gamma motor neurons has opposite effects on Ia output; alpha activation alone decreases Ia activity, while gamma activation alone increases Ia activity.

FIGURE 13.21 The function of gamma motor neurons. (a) Activation of alpha motor neurons causes the extrafusal muscle fibers to shorten. (b) If the muscle spindle were to become slack, it would go “off the air” and no longer report the length of the muscle. (c) Activation of gamma motor neurons causes the poles of the spindle to contract, keeping it active and “on the air.” Description
Recall from our discussion earlier that the monosynaptic stretch reflex arc can be viewed as a feedback loop. The principles of feedback control systems are that a set point is determined (in this case, the desired muscle length), deviations from the set point are detected by a sensor (the Ia axon endings), and deviations are compensated for by an effector system (alpha motor neurons and extrafusal muscle fibers), returning the system to the set point. Changing the activity of the gamma motor neurons changes the set point of the stretch feedback loop. This circuit, gamma motor neuron → intrafusal muscle fiber → Ia afferent axon → alpha motor neuron → extrafusal muscle fibers, is sometimes called the gamma loop.
During most normal movements, alpha and gamma motor neurons are simultaneously activated by descending commands from the brain. By regulating the set point of the stretch feedback loop, the gamma loop provides additional control of alpha motor neurons and muscle contraction.
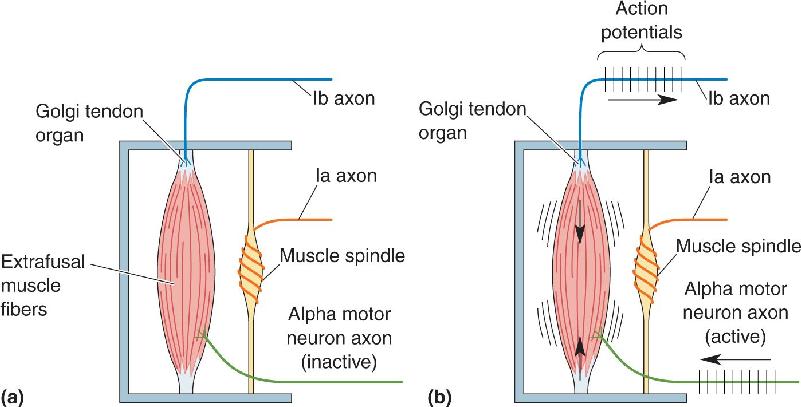
Muscle spindles are not the only source of proprioceptive inputs from the muscles. Another sensor of skeletal muscle is the Golgi tendon organ, which acts like a very sensitive strain gauge; that is, it monitors muscle tension, or the force of contraction. Golgi tendon organs are about 1 mm long and 0.1 mm wide. They are located at the junction of the muscle and the tendon and are innervated by group Ib sensory axons, which are slightly smaller than the Ia axons innervating the muscle spindles. Within the Golgi tendon organ, thin branches of the Ib axon entwine among the coils of collagen fibrils (Figure 13.22). When the muscle contracts, the tension on the collagen fibrils increases. As the fibrils straighten and squeeze the Ib axons, their mechanosensitive ion channels are activated and action potentials can be triggered.

It is important to note that while spindles are situated in parallel with the muscle fibers, Golgi tendon organs are situated in series (Figure 13.23). This different anatomical arrangement helps to determine the types of information these two sensors provide the spinal cord: Ia activity from the spindle encodes muscle length information, while Ib activity from the Golgi tendon organ encodes muscle tension information.

FIGURE 13.23 The organization of muscle proprioceptors. (a) Muscle spindles are arranged parallel to the extrafusal fibers; Golgi tendon organs lie in series, between the muscle fibers and their points of attachment. (b) Golgi tendon organs respond to increased tension on the muscle and transmit this information to the spinal cord via group Ib sensory axons. Because the activated muscle does not change length, the Ia axons remain silent in this example. Description
The Ib axons enter the spinal cord, branch repeatedly, and synapse on special interneurons called Ib inhibitory interneurons in the ventral horn. Ib interneurons also receive inputs from other sensory receptors and from descending pathways. Some of the Ib interneurons form inhibitory connections with the alpha motor neurons innervating the same muscle (Figure 13.24). This is the basis for another spinal reflex. In extreme circumstances, this Ib reflex arc may protect the muscle from being overloaded. However, its normal function is to regulate muscle tension within an optimal range. As muscle tension increases, the inhibition of the alpha motor neuron slows muscle contraction; as muscle tension falls, the inhibition of the alpha motor neuron is reduced, and muscle contraction increases. This type of proprioceptive feedback is thought to be particularly important for the proper execution of fine motor acts, such as the manipulation of fragile objects with the hands, which require a steady, but not too powerful, grip.

FIGURE 13.24 Golgi tendon organ circuit. The Ib axon of the Golgi tendon organ excites an inhibitory interneuron, which inhibits the alpha motor neurons of the same muscle.
Proprioception from the Joints. We have focused on the proprioceptors that are involved in reflex control of the spinal motor neurons. However, besides muscle spindles and Golgi tendon organs, a variety of proprioceptive axons are present in the connective tissues of joints, especially within the fibrous tissue surrounding the joints (joint capsules) and ligaments. These mechanosensitive axons respond to changes in the angle, direction, and velocity of movement in a joint. Most are rapidly adapting, meaning that sensory information about a moving joint is plentiful, but nerves encoding the resting position of a joint are few. We are, nevertheless, quite good at judging the position of a joint, even with our eyes closed. It seems that information from joint receptors, muscle spindles, and Golgi tendon organs, and probably from receptors in the skin, is combined within the CNS to estimate joint angle. Removing one source of information can be compensated for by the use of the other sources. When an arthritic hip is replaced with a steel and plastic one, patients can still tell the angle between their thigh and their pelvis, despite the fact that all their hip joint mechanoreceptors are sitting in a jar of formaldehyde.
The actions of Ib inputs from Golgi tendon organs on alpha motor neurons are entirely polysynaptic; they are all mediated by intervening spinal interneurons. Indeed, most of the input to the alpha motor neurons comes from interneurons of the spinal cord. Spinal interneurons receive synaptic input from primary sensory axons, descending axons from the brain, and collaterals of lower motor neuron axons. The interneurons are themselves networked together in a way that allows coordinated motor programs to be generated in response to their many inputs.
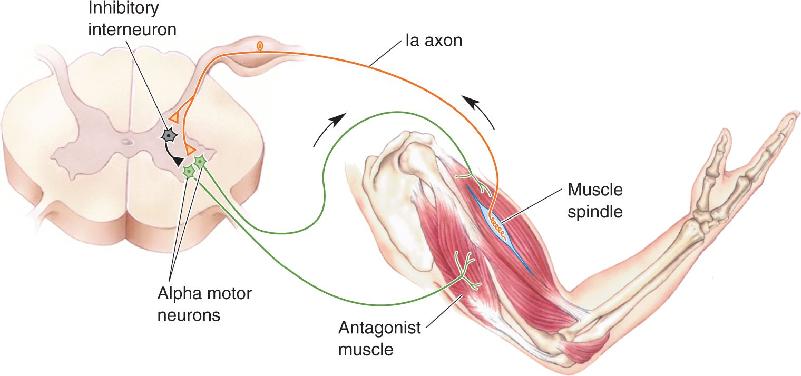
Inhibitory Input. Interneurons play a critical role in the proper execution of even the simplest reflexes. Consider the stretch reflex, for example. Compensation for the lengthening of one set of muscles, such as the flexors of the elbow, involves contraction of the flexors via the stretch reflex but also requires relaxation of the antagonist muscles, the extensors. This process is called reciprocal inhibition, the contraction of one set of muscles accompanied by the relaxation of their antagonist muscles. The importance of this is obvious; imagine how hard it would be to lift something by contracting your biceps if its antagonist muscles (e.g., your triceps) were constantly opposing you. In the case of the stretch reflex, reciprocal inhibition occurs because collaterals of the Ia axons synapse on inhibitory spinal interneurons that contact the alpha motor neurons supplying the antagonist muscles (Figure 13.25).

FIGURE 13.25 Reciprocal inhibition of flexors and extensors of the same joint. Description
Reciprocal inhibition is also used by descending pathways from the brain to overcome the powerful stretch reflex. Consider a situation in which the flexors of the elbow are voluntarily commanded to contract. You might expect the resulting stretch of the antagonist extensor muscles to activate their stretch reflex arc, which would strongly resist flexion of the joint. However, the descending pathways that activate the alpha motor neurons controlling the flexors also activate interneurons, which inhibit the alpha motor neurons that supply the antagonist muscles.
Excitatory Input. Not all interneurons are inhibitory. An example of a reflex mediated in part by excitatory interneurons is the flexor reflex, sometimes called the flexor withdrawal reflex (Figure 13.26). This is a complex, polysynaptic reflex arc used to withdraw a limb from an aversive stimulus (such as the withdrawal of your foot from the thumbtack in Chapter 3). The flexor reflex is remarkably specific. The speed of withdrawal depends on how painful the stimulus is. The direction of withdrawal depends on the location of the stimulus; for example, hot stimuli applied to your palm and to the back of your hand trigger withdrawals in opposite directions (as you would hope!).

FIGURE 13.26 Circuitry of the flexor withdrawal reflex. Description
The flexor reflex is far slower than the stretch reflex, indicating that a number of interneurons intervene between the sensory stimulus and the coordinated motor act. The flexor reflex is activated by the small, myelinated Aδ nociceptive axons that trigger pain (see Chapter 12). The nociceptive axons entering the spinal cord branch profusely and activate interneurons in several different spinal segments. These cells eventually excite the alpha motor neurons that control all the flexor muscles of the affected limb (and, needless to say, inhibitory interneurons are also recruited to inhibit the alpha motor neurons that control the extensors).
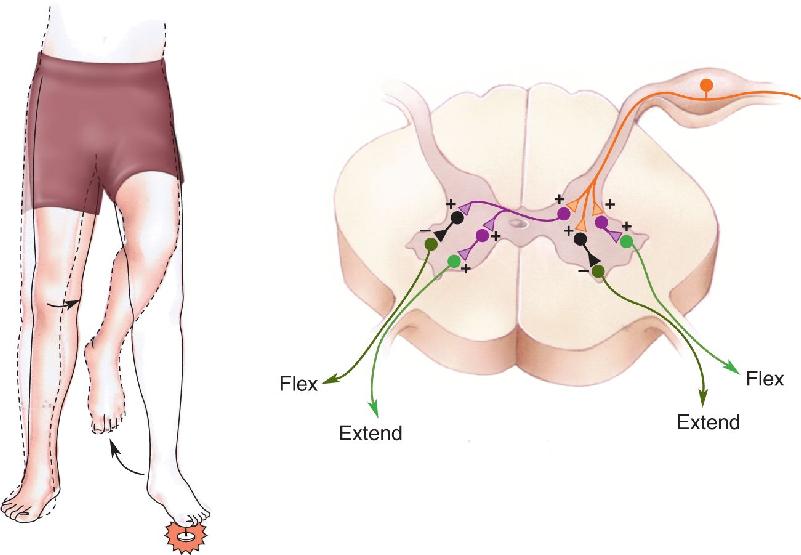
You’re walking around barefoot, and you step on a tack. Thanks to the flexor reflex, you reflexively yank your foot up. But where would that leave the rest of your body if nothing else happened? Falling to the floor, most likely. Luckily, an additional component of the reflex is recruited: the activation of extensor muscles and the inhibition of flexors on the opposite side. This is called the crossed-extensor reflex, and it is used to compensate for the extra load imposed by limb withdrawal on the antigravity extensor muscles of the opposite leg (Figure 13.27). Notice that this is another example of reciprocal inhibition, but in this case, activation of the flexors on one side of the spinal cord is accompanied by inhibition of the flexors on the opposite side.

FIGURE 13.27 Circuitry of the crossed-extensor reflex. Description
The Generation of Spinal Motor Programs for Walking
The crossed-extensor reflex, in which one limb extends as the other limb flexes, seems to provide a building block for locomotion. When you walk, you alternately withdraw and extend your two legs. All that’s lacking is a mechanism to coordinate the timing. In principle, this could be a series of descending commands from upper motor neurons. However, as we already suspected from our consideration of headless chicken behavior, it seems likely that this control is exerted from within the spinal cord. Indeed, a complete transection of a cat’s spinal cord at the mid-thoracic level leaves the hind limbs capable of generating coordinated walking movements. The circuit for the coordinated control of walking must reside, therefore, within the spinal cord. In general, circuits that give rise to rhythmic motor activity are called central pattern generators.
How do neural circuits generate rhythmic patterns of activity? Different circuits use different mechanisms. However, the simplest pattern generators are individual neurons whose membrane properties endow them with pacemaker properties. An interesting example comes from the work of Sten Grillner and his colleagues in Stockholm, Sweden. Based on the assumption that the spinal central pattern generators for locomotion in different species are variations on a plan that was established in a common ancestor, Grillner focused on the mechanism for swimming in the lamprey, a jawless fish that has evolved slowly over the course of the past 450 million years. Lampreys swim by undulating their elongated bodies. They lack limbs and even pairs of fins, but the coordinated rhythmic contractions of their body muscles during swimming closely resemble the contraction patterns necessary for terrestrial animals to walk.
The lamprey spinal cord can be dissected and kept alive in vitro for several days. Electrical stimulation of the stumps of axons descending from the brain can generate alternating rhythmic activity in the spinal cord, mimicking that which occurs during swimming. In an important series of experiments, Grillner showed that the activation of NMDA receptors on spinal interneurons was sufficient to generate this locomotor activity.
Recall from Chapter 6 that NMDA receptors are glutamate-gated ion channels with two peculiar properties: (1) They allow more current to flow into the cell when the postsynaptic membrane is depolarized, and (2) they admit Ca2+ as well as Na+ into the cell. In addition to NMDA receptors, spinal interneurons possess calcium-activated potassium channels. Now imagine the cycle that is initiated when NMDA receptors are activated by glutamate (Figure 13.28):

FIGURE 13.28 Rhythmic activity in a spinal interneuron. Some neurons respond to the activation of NMDA receptors with rhythmic depolarization. (a) In the resting state, the NMDA receptor channels and the calcium-activated potassium channels are closed. (b) Glutamate causes the NMDA receptors to open, the cell membrane to depolarize, and Ca2+ to enter the cell. (c) The rise in intracellular [Ca2+] causes the Ca2+-activated potassium channels to open. Potassium ions leave the neuron, hyperpolarizing the membrane. The hyperpolarization allows Mg2+ to enter and clog the NMDA channel, arresting the flow of Ca2+. (d) As [Ca2+] falls, the potassium channels close, resetting the membrane for another oscillation. (Source: Adapted from Wallen and Grillner, 1987.) Description
- Na+ and Ca2+ flow into the cell through the NMDA receptors.
- The membrane depolarizes, and the cycle repeats.
It is easy to imagine how intrinsic pacemaker activity in spinal interneurons might act as the primary rhythmic driving force for sets of motor neurons that in turn command cyclic behaviors like walking. However, pacemaker neurons are not solely responsible for generating rhythms in vertebrates. They are embedded within interconnected circuits, and it is the combination of intrinsic pacemaker properties and synaptic interconnections that produces rhythm.
An example of a possible pattern-generating circuit for walking is shown in Figure 13.29. According to this scheme, walking is initiated when a steady input excites two interneurons that connect to the motor neurons controlling the flexors and extensors, respectively. The interneurons respond to a continuous input by generating bursts of outputs (see Figure 13.28). The activity of these two interneurons alternates because they inhibit each other via another set of interneurons, which are inhibitory. Thus, a burst of activity in one interneuron strongly inhibits the other, and vice versa. Then, using the spinal cord circuitry of the crossed-extensor reflex (or a similar circuit), the movements of the opposite limb could be coordinated so that flexion on one side is accompanied by extension on the other. The addition of more interneuronal connections between the lumbar and cervical spinal segments could account for the swinging of the arms that accompanies walking, or the coordination of forelimbs and hind limbs in four-legged animals.

FIGURE 13.29 A possible circuit for rhythmic alternating activity. Description
Work on many vertebrate species, from lampreys to humans, has shown that locomotor activity in the spinal cord and its coordination depend on multiple mechanisms. Such complexity is not surprising when we consider the demands on the system—for example, the adjustments necessary when one foot strikes an obstacle while walking, or the changes in output that are necessary to walk forward or backward, or to go from walking, to jogging, to running, to jumping.
We can draw several conclusions from the preceding discussion of the spinal control of movement. First, a great deal has been learned about movement and its spinal control by working at different levels of analysis, ranging from biochemistry and genetics to biophysics and behavior. Indeed, a complete understanding, whether of excitation–contraction coupling or central pattern generation, requires knowledge derived from every approach. Second, sensation and movement are inextricably linked even at the lowest levels of the neural motor system. The normal function of the alpha motor neuron depends on direct feedback from the muscles themselves and indirect information from the tendons, joints, and skin. Third, the spinal cord contains an intricate network of circuits for the control of movement; it is far more than just a conduit for somatic sensory and motor information.
Evidently, coordinated and complex patterns of activity in these spinal circuits can be driven by relatively crude descending signals. This leaves the question of precisely what the upper motor neurons contribute to motor control—the subject of the next chapter.
1. What did Sherrington call the “final common pathway,” and why?
2. Define, in one sentence, motor unit. How does it differ from motor neuron pool?
3. Which is recruited first, a fast motor unit or a slow motor unit? Why?
5. Your doctor taps the tendon beneath your kneecap and your leg extends. What is the neural basis of this reflex? What is it called?
7. Lenny, a character in Steinbeck’s classic book Of Mice and Men, loved rabbits, but when he hugged them, they were crushed to death. Which type of proprioceptive input might Lenny have been lacking?
Kernell D. 2006. The Motoneurone and its Muscle Fibres. New York: Oxford University Press.
Lieber RL. 2002. Skeletal Muscle Structure, Function, and Plasticity, 2nd ed. Baltimore: Lippincott, Williams & Wilkins.
Poppele R, Bosco G. 2003. Sophisticated spinal contributions to motor control. Trends in Neurosciences 26:269–276.
Schouenborg J, Kiehn O, eds. 2001. The Segerfalk symposium on principles of spinal cord function, plasticity, and repair. Brain Research Reviews 40:1–329.
Stein PSG, Grillner S, Selverston AI, Stuart DG, eds. 1999. Neurons, Networks, and Motor Behavior. Cambridge, MA: MIT Press.
Windhorst U. 2007. Muscle proprioceptive feedback and spinal networks. Brain Research Bulletin 73:155–202.
Additional figures