Mental Illness
The Promise and Challenge of Molecular Medicine in Psychiatry

BOX 22.1 OF SPECIAL INTEREST: Agoraphobia with Panic Attacks
Other Disorders Characterized by Increased Anxiety
Regulation of the HPA Axis by the Amygdala and Hippocampus
BOX 22.2 OF SPECIAL INTEREST: A Magical Orange Grove in a Nightmare
BOX 22.3 PATH OF DISCOVERY: Tuning Depression Circuits, by Helen Mayberg
Neurology is a branch of medicine concerned with the diagnosis and treatment of nervous system disorders. We have discussed many neurological disorders in this book, ranging from multiple sclerosis to aphasia. While they are significant and fascinating in their own right, neurological disorders also help illustrate the role of physiological processes in normal brain function, such as the importance of myelin for action potential conduction and the role of the frontal lobe in language.
Psychiatry, on the other hand, has a different focus. This branch of medicine is concerned with the diagnosis and treatment of disorders that affect the mind, or psyche. (In Greek mythology, the beautiful young woman Psyche was the personification of the human soul.) Aspects of brain function that are disturbed by mental illness—our fears, moods, and thoughts—were once considered beyond the reach of neuroscience. But, as we saw in the earlier chapters of Part III, many higher brain functions have begun to yield their secrets. Today, there is hope that neuroscience will also solve the riddle of mental illness.
In this chapter, we will discuss some of the most severe and prevalent psychiatric disorders: anxiety disorders, affective disorders, and schizophrenia. Once again, we will see that a great deal can be learned about the nervous system by studying what happens when things go wrong.
Human behavior is the product of brain activity, and the brain is the product of two interacting factors: heredity and environment. Obviously, one important determinant of your individualism is your complement of DNA, which, unless you have an identical twin, is unique. This means that physically your brain, like your fingerprints, is different from all others. A second factor that makes your brain unique is your history of personal experience. Experiences can include trauma and disease, but, as we saw in the case of somatosensory map plasticity (see Chapter 12), the sensory environment itself can leave a permanent mark on the brain. (We’ll return to this theme in Part IV of this book when we discuss development, learning, and memory.) Thus, despite the gross physical similarities you might share with a genetic twin, at a fine scale, neither your brains nor your behaviors are identical. To complicate matters further, variations in genetic makeup and past experience make the brain differentially susceptible to modification by subsequent experiences. These genetic and experiential variations, all ultimately expressed as physical changes in the brain, give rise to the full range of behaviors exhibited by the human population.
Health and illness are two points along a continuum of bodily function, and the same can be said for mental health and mental illness. While we all have our odd characteristics, an individual is said to be “mentally ill” at the point when the person has a diagnosable disorder of thought, mood, or behavior that causes distress or impaired functioning. An unfortunate legacy of our past ignorance about brain function is the common distinction drawn between “physical” and “mental” health. The philosophical roots of this distinction can be traced to Descartes’s proposed separation of body and mind (see Chapter 1). Disorders of the body (which, for Descartes, included the brain) had an organic basis and were the concern of physicians and medicine. Disorders of the mind, on the other hand, were considered spiritual or moral and were the concern of clergymen and religion. The fact that most disorders of mood, thought, and behavior have, until very recently, remained resistant to biological explanations or treatments has reinforced this dichotomy.
An important advance in the secularization of mental illness was the emergence of the medical discipline of psychiatry, devoted to treating disorders of human behavior. The Austrian neurologist and psychiatrist Sigmund Freud (1856–1939) had an enormous impact on the new field, especially in the United States (Figure 22.1). Freud’s theory of psychoanalysis is based on two major assumptions that (1) much of mental life is unconscious (beyond awareness), and (2) past experiences, particularly in childhood, shape how a person will feel and respond throughout life. According to Freud, mental illness results when the unconscious and conscious elements of the psyche come into conflict. The way to resolve the conflict, and to treat the illness, is to help the patient unearth the hidden secrets of the unconscious. Often, these dark secrets are related to incidents (e.g., physical, mental, or sexual abuse) that occurred during childhood and were suppressed from consciousness.
FIGURE 22.1 Sigmund Freud. Freud proposed psychoanalytic theories of mental illness.
A different theory of personality, championed by Harvard University psychologist B. F. Skinner (1904–1990), is based on the assumption that many behaviors are learned responses to the environment. Behaviorism rejects the notions of underlying conflicts and the unconscious and focuses instead on observable behaviors and their control by the environment. In Chapter 16, we learned about some of the forces that motivate behavior. The probability of a type of behavior increases when it satisfies a craving or produces a pleasurable sensation (positive reinforcement), and it decreases when the consequences are deemed unpleasant or unsatisfactory (negative reinforcement). According to this theory, mental disorders may represent maladaptive behaviors that are learned. Treatment consists of active attempts to “unlearn” through behavior modification, either by introducing new types of behavioral reinforcement or by providing an opportunity to observe and recognize behavioral responses that are appropriate.
Such “psychosocial” approaches to treating mental illness have a sound neurobiological basis. The brain is structurally modified through learning and early experience, and these modifications will alter behavioral responses. Treatment relies on psychotherapy, the use of verbal communication to help the patient. Of course, “talk therapy” is not appropriate for all mental disorders, any more than a particular antibiotic is appropriate for all infections. However, until the revolution in biological psychiatry, variations in psychotherapy were the only tools available to psychiatrists. Moreover, despite the shift in “blame” away from one’s moral character and toward early childhood experience, psychotherapy contributed to the stigmatizing notion that mental illness (in contrast to physical illness) could be overcome by willpower alone. Freud himself recognized the shortcomings of psychotherapy, stating that the “deficiencies in our [the psychoanalytic] description would probably vanish if we were already in a position to replace the psychological terms by physiological or chemical ones” (1920, p. 54). Now, nearly a century later, neuroscience has advanced to a point where this goal seems attainable.
A spectacular success in the early biological diagnosis and treatment of mental illness actually occurred during Freud’s time. A major psychiatric disorder at the turn of the twentieth century was called general paresis of the insane, afflicting 10–15% of all institutionalized psychiatric patients. The disorder had a progressive course, starting with symptoms of mania—excitement, euphoria, and grandiose delusions—and evolved to cognitive deterioration and, ultimately, paralysis and death. Initially blamed on psychological factors, the cause was eventually traced to infection of the brain with Treponema pallidum, the microorganism that causes syphilis. Once the cause was known, increasingly effective treatments quickly followed. By 1910, German microbiologist Paul Ehrlich had established that the drug arsphenamine could act as a “magic bullet,” killing the T. pallidum in the blood without damaging its human host. Eventually, the antibiotic penicillin (discovered in 1928 by British microbiologist Alexander Fleming) was found to be so effective in killing the microorganism that established brain infections could be completely eradicated. Thus, when penicillin became widely available by the end of World War II, a major psychiatric disorder was virtually eliminated.
A number of other mental illnesses can be traced directly to biological causes. For example, a dietary deficiency in niacin (a B vitamin) can cause agitation, impaired reasoning, and depression. The penetration of HIV (the AIDS virus) into the brain causes progressive cognitive and behavioral impairments. A form of obsessive-compulsive disorder (discussed later) has been linked to an autoimmune response triggered by streptococcal pharyngitis (strep throat) in children. Understanding the causes of these diseases will lead to treatments and, ultimately, to cures of the associated mental disorders.
The Promise and Challenge of Molecular Medicine in Psychiatry. Of course, serious mental disorders also occur in well-nourished and infection-free individuals. Although the causes in most cases remain to be determined, it is safe to say that the roots of these disorders lie in altered brain anatomy, chemistry, and function. An exciting new way to understand brain malfunction has been unleashed by knowledge of the human genome. As in other complex diseases like cancer, gene mutations can cause or confer risk for psychiatric disease, and major efforts are now well underway to identify these genes. The approach of using genetic information to develop a treatment is sometimes referred to as molecular medicine.
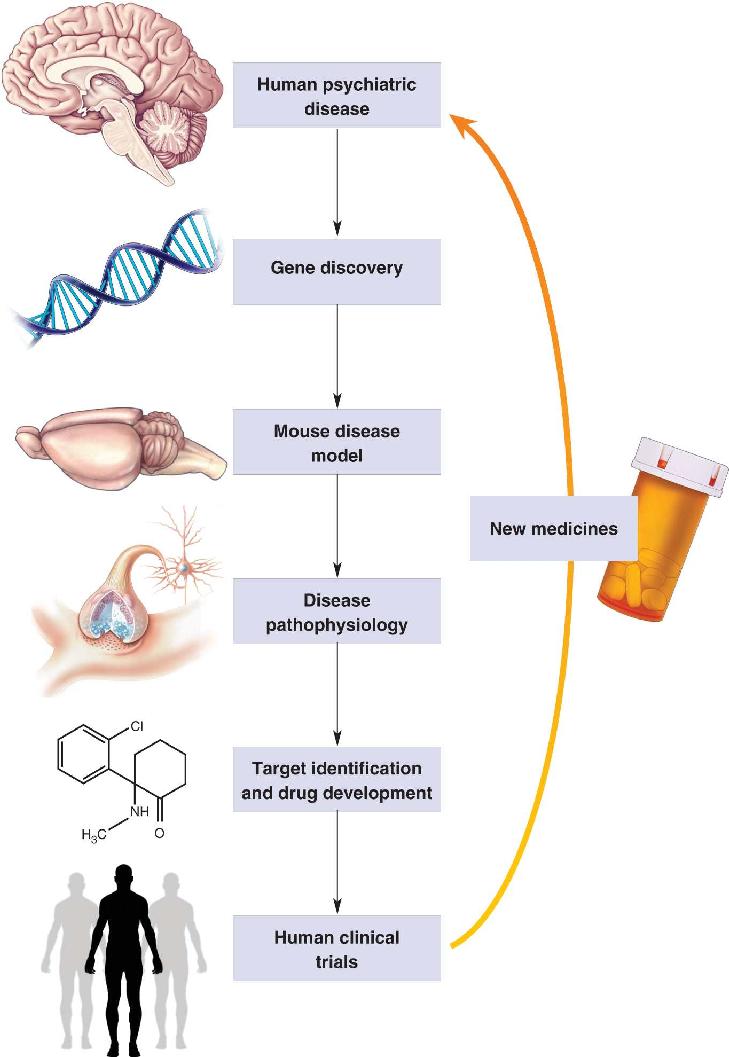
A path from gene to treatment is illustrated in Figure 22.2. Searching the DNA of individuals with a psychiatric disease may reveal causative gene mutations that can be reproduced in genetically engineered mice. By comparing the neurobiology of these animals with normal, “wild type” mice, researchers can determine how the brain functions differently in association with these mutations. Discovery of an abnormal physiological condition, or pathophysiology, may suggest biological processes that can be targeted with drug therapy—for example, too much or too little of a neurotransmitter. If drug candidates succeed in human clinical trials, then new therapeutics can be introduced to treat the disease.

FIGURE 22.2 Molecular medicine. A path from genes to treatments for psychiatric disorders. Description
Despite the enormous promise of molecular medicine, brain diseases present some unique challenges. First, mental disorders are diagnosed by clinicians based on how they appear or are described by the patient (signs and symptoms), not by knowledge of their underlying cause (etiology). It is now understood that the same diagnosis may arise from many causes, so no single treatment approach is likely to succeed in all patients, thus complicating clinical trials. Second, not all mental illnesses have a clear genetic basis, and for those that do, a large number of genes have been implicated. In some cases, it appears that pathophysiology may be caused by inheritance of numerous small mutations in many different genes. In these cases, although no single mutation has much effect, together they greatly increase the risk for a mental illness (metaphorically, death by a thousand small knife cuts). In other cases, duplication or deletion of a gene or segment of genes, called gene copy number variants, might be the single cause of the diagnosis. Although each specific variant occurs rarely in the human population, variation in many different segments of DNA can result in the same diagnosis (by analogy, death by a gunshot wound; although the end result is the same, each fatal wound can uniquely affect a different part of the body). This genetic complexity interferes with development of broadly useful animal models.
A radical new approach to overcome these challenges is to study the pathophysiology of neurons from individual patients. Don’t worry; this does not entail a brain biopsy! Rather, the approach takes advantage of the recent discovery that if skin cells scraped from a patient are treated with the correct mixture of chemicals, they can be transformed into what are called induced pluripotent stem cells, or simply iPSCs. Treatment with another mixture of chemicals can then cause these cells to differentiate into neurons that can be kept alive in a culture dish. These neurons can then be compared with those from healthy people to determine their pathophysiology. However, the major challenge with this approach is that the brain is far more complicated than a single neuron. The brain is composed of myriad cell types that are richly interconnected, and gene mutations manifest differently in different types of neurons. Treatment for pathophysiology of a neuron may not be appropriate for the pathophysiology of a brain.
Despite these sobering reminders that brains and brain diseases are extraordinarily complex, there is much optimism in the field that the challenges can and soon will be overcome. Now let’s explore the major psychiatric disorders and see how neuroscience has already both provided insight about their possible causes and contributed to their treatment.
Fear is an adaptive response to threatening situations. As we learned in Chapter 18, fear is expressed by the autonomic fight-or-flight response, mediated by the sympathetic division of the autonomic nervous system (ANS) (see Chapter 15). Many fears are innate and species-specific. A mouse does not need to be taught to fear a cat. But fear is also learned. One touch is usually all it takes to cause a horse to fear an electric fence. The adaptive value of fear is obvious. As the old aviation saying goes, “There are old pilots, and there are bold pilots, but there are no old bold pilots.” But fear is not an appropriate or adaptive response in all circumstances. An inappropriate expression of fear characterizes anxiety disorders, the most common of psychiatric disorders.
It has been estimated that in any given year, over 15% of Americans will suffer from one of the recognized anxiety disorders listed in Table 22.1. Although they differ in terms of the real or imagined stimuli that evoke the anxiety, and the behavioral responses the individual uses to attempt to reduce it, these disorders have in common the pathological expression of fear.
Frequent panic attacks consisting of discrete periods with the sudden onset of intense apprehension, fearfulness, or terror, often associated with feelings of impending doom
Anxiety about, or the avoidance of, places or situations from which escape might be difficult or embarrassing, or in which help may not be available in the event of a panic attack
At least 6 months of persistent and excessive anxiety and worry
Clinically significant anxiety provoked by exposure to a specific feared object or situation, often leading to avoidance behavior
Clinically significant anxiety provoked by exposure to certain types of social or performance situations, often leading to avoidance behavior
Source: Adapted from American Psychiatric Association, 2013.
Panic Disorder. Panic attacks are sudden feelings of intense terror that occur without warning. The symptoms include palpitations, sweating, trembling, shortness of breath, chest pain, nausea, dizziness, tingling sensations, and chills or blushing. Most people report an overwhelming fear that they are dying or “going crazy,” and flee from the place where the attack begins, often seeking emergency medical assistance. The attacks are short-lived, however, usually lasting less than 30 minutes. Panic attacks can occur in response to specific stimuli. They may be a feature of a number of anxiety disorders, but they can also occur spontaneously.
The condition that psychiatrists call panic disorder is characterized by recurring, seemingly unprovoked panic attacks and a persistent worry about having further attacks. About 2% of the population suffers from panic disorder, which is twice as common in women as in men. The onset of the disorder is most common after adolescence but before the age of 50 years. Half the individuals who have panic disorder will also suffer from major depression (see below), and 25% of them will become alcoholic or develop substance-abuse problems.
Agoraphobia. Severe anxiety about being in situations where escape might be difficult or embarrassing is characteristic of agoraphobia (from the Greek, for “fear of an open marketplace”). The anxiety leads to avoidance of situations irrationally perceived as threatening, such as being alone outside the home, in a crowd of people, in a car or airplane, or on a bridge or elevator. Agoraphobia is often an adverse outcome of panic disorder, as the situation in Box 22.1 describes. About 5% of the population is agoraphobic, with the incidence among women being twice that of men.
To appreciate the distress and disruption caused by anxiety disorders, consider the following case history from Nancy C. Andreasen’s book, The Broken Brain.
Greg Miller is a 27-year-old unmarried computer programmer. When asked about his main problem, he replied, “I am afraid to leave my house or drive my car.”
The patient’s problems began approximately one year ago. At that time he was driving across the bridge that he must traverse every day in order to go to work. While driving in the midst of the whizzing six-lane traffic, he began to think (as he often did) about how awful it would be to have an accident on that bridge. His small, vulnerable VW convertible could be crumpled like an aluminum beer can, and he could die a bloody, painful death or be crippled for life. His car could even hurtle over the side of the bridge and plunge into the river.
As he thought about these possibilities, he began to feel increasingly tense and anxious. He glanced back and forth at the cars on either side of him and became frightened that he might run into one of them. Then he experienced an overwhelming rush of fear and panic. His heart started pounding and he felt as if he were going to suffocate. He began to take deeper and deeper breaths, but this only increased his sense of suffocation. His chest felt tight and he wondered if he might be about to die of a heart attack. He certainly felt that something dreadful was going to happen to him quite soon. He stopped his car in the far right lane in order to try to regain control of his body and his feelings. Traffic piled up behind with many honking horns, and drivers pulled around him yelling obscenities. On top of his terror, he experienced mortification. After about three minutes, the feeling of panic slowly subsided, and he was able to proceed across the bridge and go to work. During the remainder of the day, however, he worried constantly about whether or not he would be able to make the return trip home across the bridge without a recurrence of the same crippling fear.
He managed to do so that day, but during the next several weeks he would begin to experience anxiety as he approached the bridge, and on three or four occasions he had a recurrence of the crippling attack of panic. The panic attacks began to occur more frequently so that he had them daily. By this time he was overwhelmed with fear and began to stay home from work, calling in sick each day. He knew that his main symptom was an irrational fear of driving across the bridge, but he suspected that he might also have some type of heart problem. He saw his family doctor, who found no evidence of any serious medical illness, and who told him that his main problem was excessive anxiety. The physician prescribed a tranquilizer for him and told him to try to return to work.
For the next six months, Greg struggled with his fear of driving across the bridge. He was usually unsuccessful and continued to miss a great deal of work. Finally, he was put on disability for a few months and told by the company doctor to seek psychiatric treatment. Greg was reluctant and embarrassed to do this, and instead he stayed home most of the time, reading books, listening to records, playing chess on his Apple computer, and doing various “handy-man” chores around the house. As long as he stayed home, he had few problems with anxiety or the dreadful attacks of panic. But when he tried to drive his car, even to the nearby shopping center, he would sometimes have panic attacks. Consequently, he found himself staying home nearly all the time and soon became essentially housebound. (Andreasen, 1984, pp. 65–66)
Other Disorders Characterized by Increased Anxiety
Several disorders that are no longer classified by the American Psychiatric Association as “anxiety disorders” are nevertheless characterized by increased anxiety. Two of the most prevalent are post-traumatic stress disorder and obsessive-compulsive disorder.
Post-Traumatic Stress Disorder. To a pathologist, trauma refers to a wound caused by sudden violence. In the realm of psychiatry, the term refers to the psychological wounds of experiencing or witnessing a shocking event or events. A long-lasting consequence can be post-traumatic stress disorder, or PTSD. The symptoms of PTSD can include increased anxiety, intrusive memories, dreams or flashbacks of the traumatic experiences, irritability, and emotional numbness. PTSD affects 3.5% of the adult population in the United States.
Obsessive-Compulsive Disorder. People with obsessive-compulsive disorder (OCD) have obsessions, which are recurrent, intrusive thoughts, images, ideas, or impulses that the person perceives as being inappropriate, grotesque, or forbidden. Common themes are thoughts of contamination with germs or body fluids, thoughts that the sufferer has unknowingly caused harm to someone, and violent or sexual impulses. These thoughts are recognized by the affected individual as being foreign, and they evoke considerable anxiety. People with OCD also have compulsions, which are repetitive behaviors or mental acts that are performed to reduce the anxiety associated with obsessions. Examples are repeated hand-washing, counting, and checking to make sure that something is not out of place. OCD affects over 2% of the population, with an equal incidence among men and women. The disorder usually appears in young adult life, and the symptoms fluctuate in response to stress levels.
A genetic predisposition has been established for many anxiety disorders. Other anxiety disorders appear to be rooted more in the occurrence of stressful life events.
Fear is normally evoked by a threatening stimulus, called a stressor, and it is manifest by a response known as the stress response. As mentioned previously, the stimulus–response relationship can be strengthened by experience (recall the horse and the electric fence), but it can also be weakened. Consider, for example, an expert skier who no longer views a precipitous drop as fearful. A healthy person regulates the stress response through learning. The hallmark of anxiety disorders is the occurrence of an inappropriate stress response either when a stressor is not present or when it is not immediately threatening. Thus, a key to understanding anxiety is to understand how the stress response is regulated by the brain.
The Stress Response. The stress response is the coordinated reaction to threatening stimuli. It is characterized by the following:
- Activation of the sympathetic division of the ANS
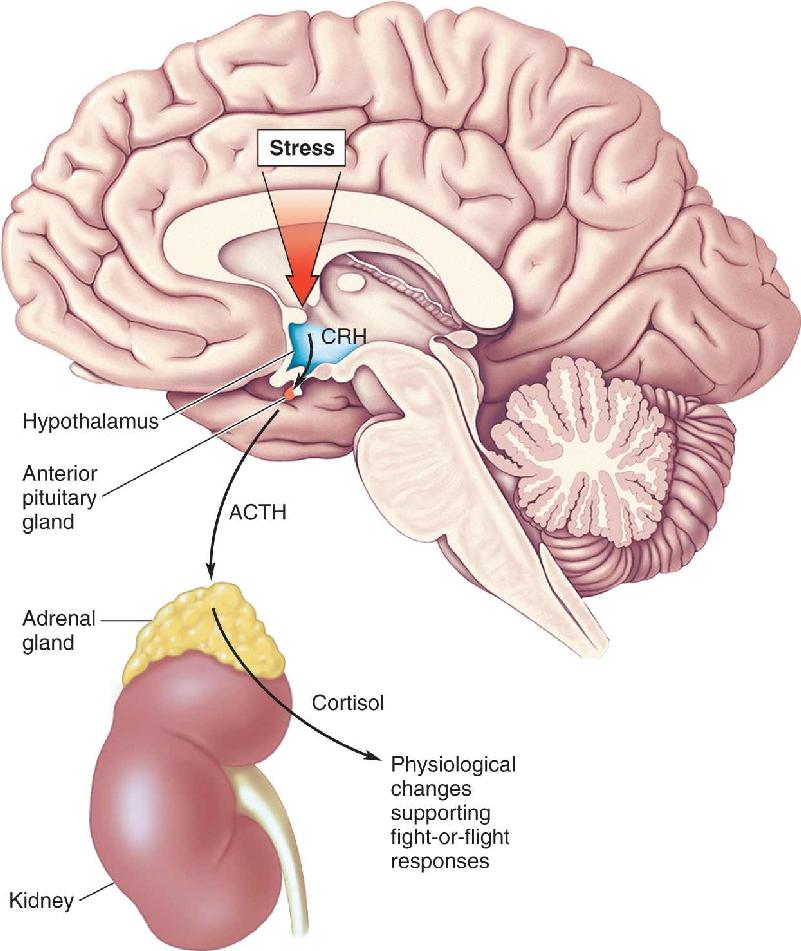
It should come as no surprise that the hypothalamus is centrally involved in orchestrating appropriate humoral, visceromotor, and somatic motor responses (see Chapters 15 and 16). To get an idea of how this response is regulated, let’s focus on the humoral response, which is mediated by the hypothalamic-pituitary-adrenal (HPA) axis (Figure 22.3).

FIGURE 22.3 The hypothalamic-pituitary-adrenal axis. The HPA axis regulates the secretion of cortisol from the adrenal gland in response to stress. CRH is the chemical messenger between the paraventricular nucleus of the hypothalamus and the anterior pituitary gland. ACTH released by the pituitary gland travels in the bloodstream to the adrenal gland lying atop the kidney, where it stimulates cortisol release. Cortisol contributes to the body’s physiological response to stress. Description
As we learned in Chapter 15, the hormone cortisol (a glucocorticoid) is released from the adrenal cortex in response to an elevation in the blood level of adrenocorticotropic hormone (ACTH). ACTH is released by the anterior pituitary gland in response to corticotropin-releasing hormone (CRH). CRH is released into the blood of the portal circulation by parvocellular neurosecretory neurons in the paraventricular nucleus of the hypothalamus. Thus, this arm of the stress response can be traced back to activation of the CRH-containing neurons of the hypothalamus. Much can be learned about anxiety disorders by understanding how the activity of these neurons is regulated. For example, when CRH is overexpressed in genetically engineered mice, the animals display increased anxiety-like behaviors. When the receptors for CRH are genetically eliminated from mice, they have less anxiety-like behavior than normal mice.
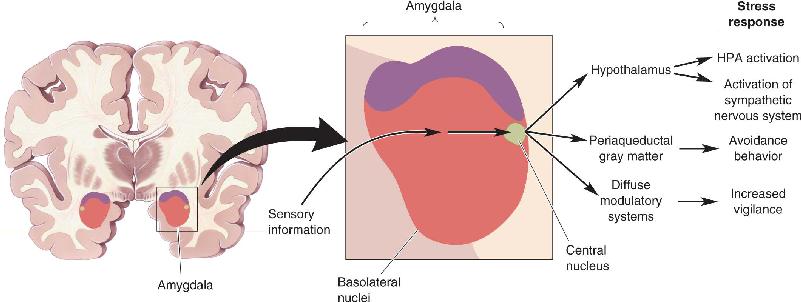
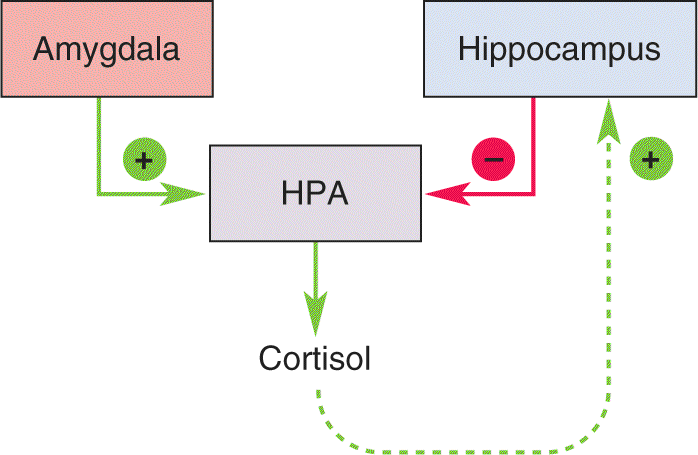
Regulation of the HPA Axis by the Amygdala and Hippocampus. The CRH neurons of the hypothalamus are regulated by two structures that were introduced in earlier chapters: the amygdala and the hippocampus (Figure 22.4). As we learned in Chapter 18, the amygdala is critical to fear responses. Sensory information enters the basolateral amygdala, where it is processed and relayed to neurons in the central nucleus. When the central nucleus of the amygdala becomes active, the stress response ensues (Figure 22.5). Inappropriate activation of the amygdala, as measured using fMRI (see Box 7.3), has been associated with some anxiety disorders. Downstream from the amygdala is a collection of neurons called the bed nucleus of the stria terminalis. The bed nucleus neurons activate the HPA axis and the stress response.

FIGURE 22.4 The location of the amygdala and hippocampus.

FIGURE 22.5 Control of the stress response by the amygdala. The amygdala receives ascending sensory information from the thalamus as well as descending inputs from the neocortex. This information is integrated by the basolateral nuclei and is relayed to the central nucleus. Activation of the central nucleus leads to the stress response.
The HPA axis is also regulated by the hippocampus. However, hippocampal activation suppresses, rather than stimulates, CRH release. The hippocampus contains numerous glucocorticoid receptors that respond to the cortisol released from the adrenal gland in response to HPA system activation. Thus, the hippocampus normally participates in the feedback regulation of the HPA axis by inhibiting CRH release (and the subsequent release of ACTH and cortisol) when circulating cortisol levels get too high. Continuous exposure to cortisol, such as during periods of chronic stress, can cause hippocampal neurons to wither and die in experimental animals (see Box 15.1). This degeneration of the hippocampus may set off a vicious cycle, in which the stress response becomes more pronounced, leading to even greater cortisol release and more hippocampal damage. Human brain imaging studies have shown a decrease in the volume of the hippocampus in some people suffering from PTSD.
To summarize, the amygdala and the hippocampus regulate the HPA axis and the stress response in a push–pull fashion (Figure 22.6). Anxiety disorders have been related to both hyperactivity of the amygdala and diminished activity of the hippocampus. It is important to keep in mind, however, that the amygdala and hippocampus both receive highly processed information from the neocortex. Indeed, another consistent finding in humans with anxiety disorders has been elevated activity of the prefrontal cortex.

FIGURE 22.6 Push–pull regulation of the HPA axis by the amygdala and hippocampus. Amygdala activation stimulates the HPA system and the stress response (green lines). Hippocampal activation, on the other hand, suppresses the HPA system (red line). Because the hippocampus has glucocorticoid receptors that are sensitive to circulating cortisol, it is important in the feedback regulation of the HPA axis in preventing excessive cortisol release.
Several treatments are available for anxiety disorders. In many cases, patients respond well to psychotherapy and counseling; in other cases, specific medications are more effective.
Psychotherapy. We have seen that there is a strong learning component to fear, so it should not be surprising that psychotherapy can be an effective treatment for many of the anxiety disorders. The therapist gradually increases the exposure of the patient to the stimuli that produce anxiety, reinforcing the notion that the stimuli are not dangerous. At the neurobiological level, the aim of the psychotherapy is to alter connections in the brain such that the real or imagined stimuli no longer evoke the stress response.
Anxiolytic Medications. Medications that reduce anxiety, described as anxiolytic drugs, act by altering chemical synaptic transmission in the brain. The major classes of drugs currently used in the treatment of anxiety disorders are benzodiazepines and serotonin-selective reuptake inhibitors.
Recall that GABA is an important inhibitory neurotransmitter in the brain. GABAA receptors are GABA-gated chloride channels that mediate fast inhibitory postsynaptic potentials (see Chapter 6). The proper action of GABA is critical to the proper functioning of the brain: Too much inhibition results in coma, and too little results in seizures. In addition to its GABA-binding site, the GABAA receptor contains sites where chemicals can act to powerfully modulate channel function. Benzodiazepines bind to one of these sites and act to make GABA much more effective in opening the channel and producing inhibition (Figure 22.7). The site on the receptor that binds benzodiazepines is believed to be used normally by a naturally occurring brain chemical, although the identity of the endogenous molecule has not been established.

FIGURE 22.7 The action of benzodiazepine. Benzodiazepines bind to a site on the GABAA receptor that makes it much more responsive to GABA, the major inhibitory neurotransmitter in the forebrain. A different site can bind ethanol and also make the receptor more responsive to GABA. Description
Benzodiazepines, of which Valium (diazepam) is perhaps the most well known, are highly effective treatments for acute anxiety. Indeed, virtually all drugs that stimulate GABA actions are anxiolytic, including the active ingredient in alcoholic beverages, ethanol. A reduction in anxiety is likely to explain, at least in part, the widespread social use of alcohol. The anxiolytic effects of alcohol are also an obvious reason that anxiety disorders and alcohol abuse often go hand-in-hand.
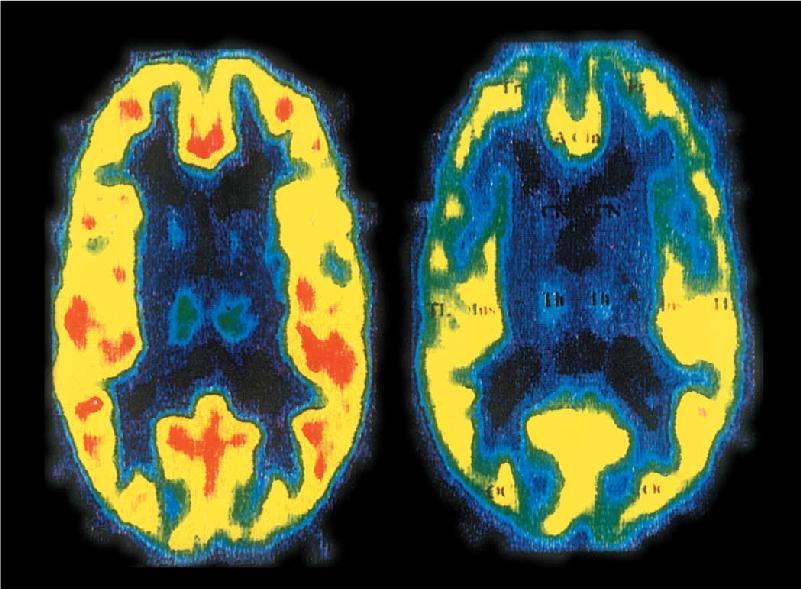
We may infer that the calming actions of benzodiazepines are due to the suppression of activity in the brain circuits used in the stress response. Benzodiazepine treatment might be required to restore normal function to these circuits. Indeed, a study of patients with panic disorder using positron emission tomography (PET) imaging (see Box 7.3) demonstrated that the number of benzodiazepine binding sites was reduced in regions of the frontal cortex that show hyperactive responsiveness during anxiety (Figure 22.8). These results are promising not only because they might reveal the sites of benzodiazepine action in the brain but also because they suggest that an alteration in the endogenous regulation of GABA receptors is a cause of the anxiety disorder.

FIGURE 22.8 Diminished binding of radioactive benzodiazepine in a patient with panic disorder. PET scans in the horizontal plane of the brain of a healthy person (left) and the brain of a person suffering from panic disorder (right). The color-coding indicates the number of benzodiazepine binding sites in the brain (hot colors indicate more; cool colors indicate fewer). The frontal cortex, at the top of the scan, shows many fewer binding sites in the individual with panic disorder. (Source: Nutt DJ, Malizia AL. 2001. New insights into the role of the GABAA-benzodiazepine receptor in psychiatric disorder. The British Journal of Psychiatry 179:390–396.)
Serotonin-selective reuptake inhibitors (SSRIs) are widely used in the treatment of mood disorders, as we will discuss in a moment. However, SSRIs are also highly effective for treating other psychiatric disorders, notably including OCD. Recall that serotonin is released throughout the brain by a diffuse modulatory system originating in the raphe nuclei of the brain stem (see Figure 15.13). The actions of serotonin are mediated by G-protein-coupled receptors and are terminated by reuptake, via serotonin transporter proteins, into the axon terminal. Thus, as the name implies, SSRIs act to prolong the actions of released serotonin at their receptors by inhibiting reuptake. In a recent study, the presence in some families of a rare mutation in the serotonin transporter gene was associated with a high incidence of OCD, further implicating serotonin in the origins of this disease.
Unlike the benzodiazepines, however, the anxiolytic actions of the SSRIs are not immediate. Therapeutic effects develop slowly, over a period of weeks, in response to regular daily dosing. This finding indicates that the immediate rise in extracellular serotonin caused by the SSRI is not responsible for the anxiolytic effect. Rather, the effect appears to be due to an adaptation of the nervous system to chronically elevated brain serotonin, via some structural or functional change that is not understood. We will return to a discussion of the actions of SSRIs when we discuss depression. However, it is very interesting in the context of anxiety disorders that one adaptive response to SSRIs is an increase in the glucocorticoid receptors in the hippocampus. SSRIs might act to dampen anxiety by enhancing the feedback regulation of the CRH neurons in the hypothalamus (see Figure 22.6).
Although benzodiazepines and SSRIs have proven to be effective in treating a wide variety of anxiety disorders, novel drugs are now being developed based on our new understanding of the stress response. One promising drug target is the receptors for CRH. Not only is CRH used by hypothalamic neurons to control ACTH release from the pituitary but it is also used as a neurotransmitter in some of the central circuits involved in the stress response. For example, some neurons of the central nucleus of the amygdala contain CRH, and injections of CRH into the brain can produce the full-blown stress response and signs of anxiety. Thus, there is hope that antagonists of CRH receptors will be useful for the treatment of some anxiety disorders.
Affect is the medical term for emotional state or mood; affective disorders are disorders of mood. In a given year, over 9% of the population will suffer from one of the mood disorders.
An occasional, brief feeling of low mood—getting “the blues”—is a common response to life’s events, such as suffering a loss or disappointment, and we would not call this a disorder. However, the affective disorder that psychiatrists and psychologists call depression is something more prolonged and much more severe, characterized by a feeling that one’s emotional state is no longer under one’s control. It can occur suddenly, often without obvious external cause, and if left untreated, it usually lasts 4–12 months.
Depression is a serious disease. It is a main precipitating cause of suicide, which claims more than 38,000 lives each year in the United States. Depression is also widespread. Perhaps as many as 20% of the population will suffer a major, incapacitating episode of depression during their lifetime. In a subset of patients with bipolar disorder, bouts of depression are punctuated with emotional highs that can also be highly disruptive.
Major Depression. The mental illness known as major depression is the most common mood disorder, affecting 6% of the population every year. The cardinal symptoms are lowered mood and decreased interest or pleasure in all activities. For a diagnosis of major depression, these symptoms must be present every day for a period of at least 2 weeks and not be obviously related to bereavement. Other symptoms also occur, including:
Episodes of major depression usually don’t last longer than 2 years, although the disease has a chronic, unremitting course in about 17% of patients. Without treatment, however, depression will recur in 50% of cases, and after three or more episodes, the odds of recurrence increase to over 90%. Another expression of depression, afflicting 2% of the adult population, is called dysthymia. Although milder than major depression, dysthymia has a chronic, “smoldering” course, and it seldom disappears spontaneously. Major depression and dysthymia are twice as common in women as men.
Bipolar Disorder. Like major depression, bipolar disorder is a recurrent mood disorder. It consists of repeated episodes of mania, or mixed episodes of mania and depression, and therefore is also called manic-depressive disorder. Mania (derived from a French word meaning “crazed” or “frenzied”) is a distinct period of abnormally and persistently elevated, expansive, or irritable mood. During the manic phase, other common symptoms include:
- Increased talkativeness or feelings of pressure to keep talking
- Flight of ideas, or a subjective experience that thoughts are racing
Another symptom is impaired judgment. Spending sprees, offensive or disinhibited behavior, promiscuity, or other reckless behaviors are common.
According to current diagnostic criteria, there are two types of bipolar disorder. Type I bipolar disorder is characterized by the manic episodes just described (with or without incidents of major depression), and occurs in about 1% of the population, equally among men and women. Type II bipolar disorder, affecting about 0.6% of the population, is characterized by hypomania, a milder form of mania that is not associated with marked impairments in judgment or performance. Indeed, hypomania in some may take the form of a marked increase in efficiency, accomplishment, or creativity (Box 22.2). However, type II bipolar disorder is also always associated with episodes of major depression. When hypomania alternates with periods of depression that are not severe enough to warrant the description “major” (i.e., fewer symptoms and shorter duration), the disorder is called cyclothymia.
Winston Churchill called it his “black dog.”1 The writer F. Scott Fitzgerald often found himself “hating the night when I couldn’t sleep and hating the day because it went toward night.”2 It was the most “terrible of all the evils of existence” for the composer Hector Berlioz.3 They were speaking of their lifelong bouts with depression. From the Scottish poet Robert Burns to the American grunge rocker Kurt Cobain, extraordinarily creative people have suffered inordinately from affective disorders. Biographical studies of accomplished artists have been consistent and alarming; their estimated rates of major depression are about 10 times higher than in the general population, and their rates of bipolar disorder may be up to 30 times higher.
Many artists have eloquently described their misfortunes. But can mood disorders actually reinforce great talent and creative productivity? Certainly, most people with mood disorders are not artistic or unusually imaginative, and most artists are not manic-depressive. However, artists with bipolar disorders can sometimes draw vigor and inspiration from their condition. Edgar Allan Poe wrote of his cycles of depression and mania, “I am excessively slothful, and wonderfully industrious—by fits.”4 The poet Michael Drayton mused about “that fine madness . . . which rightly should possess a poet’s brain.”5 Studies have suggested that hypomania can heighten certain cognitive processes, increase original and idiosyncratic thought, and even enhance linguistic skills. Manic states can also reduce the need for sleep, foster intense and obsessive concentration, create unmitigated self-confidence, and eliminate concern for social norms—just what you need, perhaps, to push the envelope of artistic creativity.
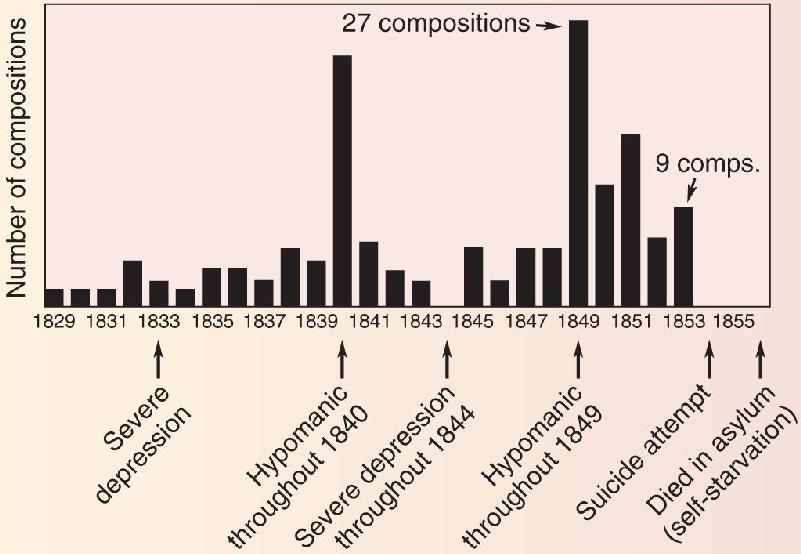
The poet’s madness is much more often a scourge than an inspiration. For Robert Lowell, manic experiences were “a magical orange grove in a nightmare.”6 Virginia Woolf’s husband described how “she talked almost without stopping for two or three days, paying no attention to anyone in the room or anything said to her.”7 It is hard to overstate the depths of melancholy that can accompany major depression. The suicide rate among accomplished poets is said to be 5 to 18 times higher than in the general population. Poet John Keats once wrote desperately, “I am in that temper that if I were under water I would scarcely kick to come to the top.”8 But when Keats’s mood pitched the other way, he wrote most of his best poetry during a 9-month period in 1819, before dying of tuberculosis at age 25. Figure A shows how Robert Schumann’s wildly fluctuating output of musical compositions coincided with the oscillations of his manic-depressive episodes.

Figure A Schumann’s output of musical composition. (Source: Adapted from Slater and Meyer, 1959). Description
The psychiatrist Kay Redfield Jamison has suggested that “depression is a view of the world through a dark glass, and mania is that seen through a kaleidoscope—often brilliant but fractured.”9 Today, we are lucky to have effective treatments for both conditions, for the dark glass and the kaleidoscope carry a heavy price.
1 Quoted in Ludwig AM. 1995. The Price of Greatness: Resolving the Creativity and Madness Controversy. New York: Guilford Press, p. 174.
2 F. Scott Fitzgerald. 1956. The Crack-Up. In The Crack-Up and Other Stories. New York: New Directions, pp. 69–75.
3 Hector Berlioz. 1970. The Memoirs of Hector Berlioz, trans. David Cairns. St. Albans, England: Granada, p. 142.
4 Edgar Allan Poe. 1948. Letter to James Russell Lowell, June 2, 1844. In The Letters of Edgar Allan Poe, Vol. 1, ed. John Wand Ostrom. Cambridge, MA: Harvard University Press, p. 256.
5 Michael Drayton. 1753. “To my dearly beloved Friend, Henry Reynolds, Esq.; of Poets and Poesy,” lines 109–110, The Works of Michael Drayton, Esq., vol. 4, London: W. Reeve.
6 Ian Hamilton. 1982. Robert Lowell: A Biography. New York: Random House, p. 228.
7 Leonard Woolf. 1964. Beginning Again: An Autobiography of the Years 1911 to 1918. New York: Harcourt Brace, pp. 172–173.
8 Quoted by Kay Jamison in a presentation at the Depression and Related Affective Disorders Association/Johns Hopkins Symposium, Baltimore, Maryland, April 1997.
9 Jamison KR. Manic-depressive illness and creativity. Scientific American 272: 62–67.
Like most other mental illnesses, affective disorders reflect the altered functioning of many parts of the brain at the same time. How else can we explain the coexistence of symptoms ranging from eating and sleeping disorders to a loss of the ability to concentrate? For this reason, research has focused on the role of the diffuse modulatory systems, with their wide reach and diverse effects. However, in the last few years, disruption of the HPA system and related cortical areas has also been implicated as playing an important role in depression. Let’s take a closer look at the neurobiology of mood disorders.
The Monoamine Hypothesis. The first real indication that depression might result from a problem with the central diffuse modulatory systems came in the 1960s. A drug called reserpine, introduced to control high blood pressure, caused severe depression in about 20% of cases. Reserpine depletes central catecholamines and serotonin by interfering with their loading into synaptic vesicles. Another class of drugs that were introduced to treat tuberculosis caused a marked mood elevation. These drugs inhibit monoamine oxidase (MAO), the enzyme that destroys catecholamines and serotonin. Another piece of the puzzle fell into place when neuroscientists recognized that the drug imipramine, introduced some years earlier as an antidepressant, inhibits the reuptake of released serotonin and norepinephrine, thus promoting their action in the synaptic cleft. As a result of these observations, researchers developed the hypothesis that mood is closely tied to the levels of released “monoamine” neurotransmitters—norepinephrine and/or serotonin—in the brain. According to this idea, called the monoamine hypothesis of mood disorders, depression is a consequence of a deficit in one of these diffuse modulatory systems (Figure 22.9). Indeed, as we will see in a moment, many of the modern drug treatments for depression have in common enhanced neurotransmission at central serotonergic and/or noradrenergic synapses.

FIGURE 22.9 The diffuse modulatory systems implicated in affective disorders. The norepinephrine and serotonin systems, introduced in Chapter 15, are characterized by the broad reach of their axonal projections. Description
A direct correlation between mood and modulator, however, is too simplistic. Perhaps the most striking problem is the clinical finding that the antidepressant action of all of these drugs takes several weeks to develop, even though they have almost immediate effects on transmission at the modulatory synapses. Another concern is that other drugs that raise NE levels in the synaptic cleft, like cocaine, are not effective as antidepressants. A new hypothesis is that the effective drugs promote long-term adaptive changes in the brain, involving alterations in gene expression, which alleviate the depression. One adaptation occurs in the HPA axis, which, as we’ll discuss next, has also been implicated in mood disorders.
The Diathesis–Stress Hypothesis. Evidence clearly indicates that mood disorders run in families and that our genes predispose us to this type of mental illness. The medical term for a predisposition for a certain disease is diathesis. However, researchers have also established that early childhood abuse or neglect and other stresses of life are important risk factors in the development of mood disorders in adults. According to the diathesis–stress hypothesis of affective disorders, the HPA axis is the main site where genetic and environmental influences converge to cause mood disorders.
As we have seen, exaggerated activity of the HPA system is associated with anxiety disorders. However, anxiety and depression often coexist (in fact, this “comorbidity” is the rule rather than the exception). Indeed, one of the most robust findings in all of biological psychiatry is hyperactivity of the HPA axis in severely depressed patients: Blood cortisol levels are elevated, as is the concentration of CRH in the cerebrospinal fluid. Could this hyperactive HPA system, and the resulting deleterious effects on brain function, be the cause of depression? Animal studies are highly suggestive. Injected CRH into the brains of animals produces behavioral effects that are similar to those of major depression: insomnia, decreased appetite, decreased interest in sex, and, of course, an increased behavioral expression of anxiety.
Recall that the activation of the hippocampal glucocorticoid receptors by cortisol normally leads to feedback inhibition of the HPA axis (see Figure 22.6). In depressed patients, this feedback is disrupted, explaining why HPA function is hyperactive. A molecular basis for the diminished hippocampal response to cortisol is a decreased number of glucocorticoid receptors. What regulates glucocorticoid receptor number? In a fascinating parallel with the factors implicated in mood disorders, the answer is genes, monoamines, and early childhood experience.
Glucocorticoid receptors, like all proteins, are a product of gene expression. In rats, it has been shown that the amount of glucocorticoid receptor gene expression is regulated by early sensory experience. Rats that received a lot of maternal care as pups express more glucocorticoid receptors in their hippocampus, less CRH in their hypothalamus, and reduced anxiety as adults. The maternal influence can be replaced by increasing the tactile stimulation of the pups. Tactile stimulation activates the ascending serotonergic inputs to the hippocampus, and the serotonin triggers a long-lasting increase in the expression of the glucocorticoid receptor gene. More glucocorticoid receptors equip the animal to respond to stressors as adults. However, the beneficial effect of experience is restricted to a critical period of early postnatal life; stimulation of the rats as adults does not have the same effect. Childhood abuse and neglect, in addition to genetic factors, are known to put people at risk for developing mood and anxiety disorders, and these animal findings suggest one cause. Elevations in brain CRH, and decreased feedback inhibition of the HPA system, may make the brain especially vulnerable to depression.
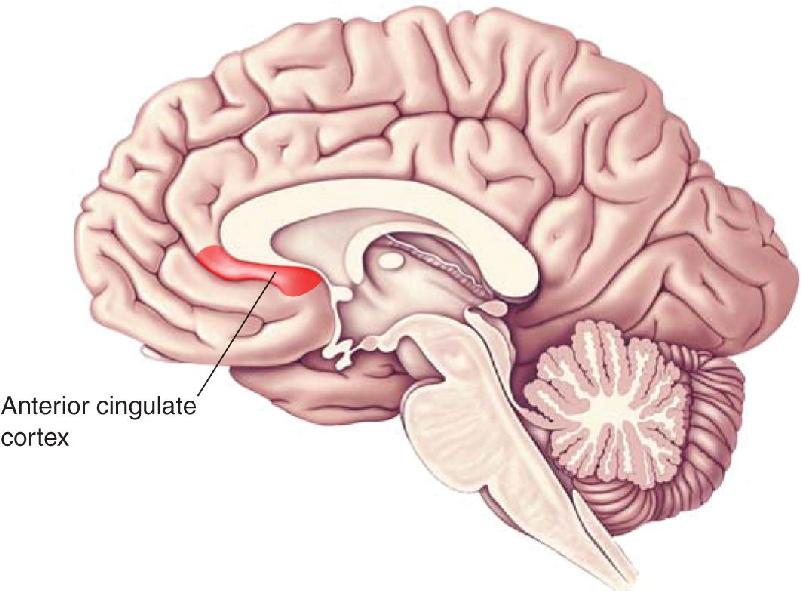
Anterior Cingulate Cortex Dysfunction. Functional brain imaging studies have consistently found increased resting-state metabolic activity in the anterior cingulate cortex of depressed patients (Figure 22.10). This region of the brain is considered to be a “node” in an extensive network of interconnected structures that include other regions of the frontal cortex, hippocampus, amygdala, hypothalamus, and brain stem. The hypothesis that anterior cingulate cortex dysfunction contributes to the symptoms of major depression is supported by a number of findings, including studies that have shown that activity here is increased by autobiographical recall of a sad event and is decreased following successful medical treatment for depression. Based on these findings, the anterior cingulate cortex is considered to be an important link between an internally generated emotional state and the HPA.

FIGURE 22.10 The anterior cingulate cortex. Activity in this region assessed by PET or fMRI imaging is increased in patients suffering from major depression and reduced by successful treatments.
Mood disorders are very common, and the burden they impose on human health, happiness, and productivity is enormous. Fortunately, a number of helpful treatments are available.
Electroconvulsive Therapy. It might surprise you to learn that one of the most effective treatments for depression and mania involves inducing seizure activity in the temporal lobes. In electroconvulsive therapy (ECT), electrical currents are passed between two electrodes placed on the scalp. Localized electrical stimulation triggers seizure discharges in the brain, but the patient is given anesthesia and muscle relaxants to prevent violent movements during treatment. An advantage of ECT is that relief can occur quickly, sometimes after the first treatment session. This attribute of ECT is especially important in cases where suicide risk is high. An adverse effect of ECT, however, is memory loss. As we will see in Chapter 24, temporal lobe structures (including the hippocampus) play a vital role in memory. ECT usually disrupts memories for events that occurred before treatment, and this can extend back as far as 6 months. In addition, ECT can temporarily impair the storage of new information.
The mechanism by which ECT relieves depression is unknown. However, as mentioned earlier, one temporal lobe structure affected by ECT is the hippocampus, which we have seen is involved in regulating CRH and the HPA axis.
Psychotherapy. Psychotherapy can be effective in treating mild to moderate cases of depression. The main goal of psychotherapy is to help depressed patients overcome negative views of themselves and their future. The neurobiological basis of the treatment has not been established, although we can infer that it relates to establishing cognitive, neocortical control over the activity patterns in disturbed circuits.
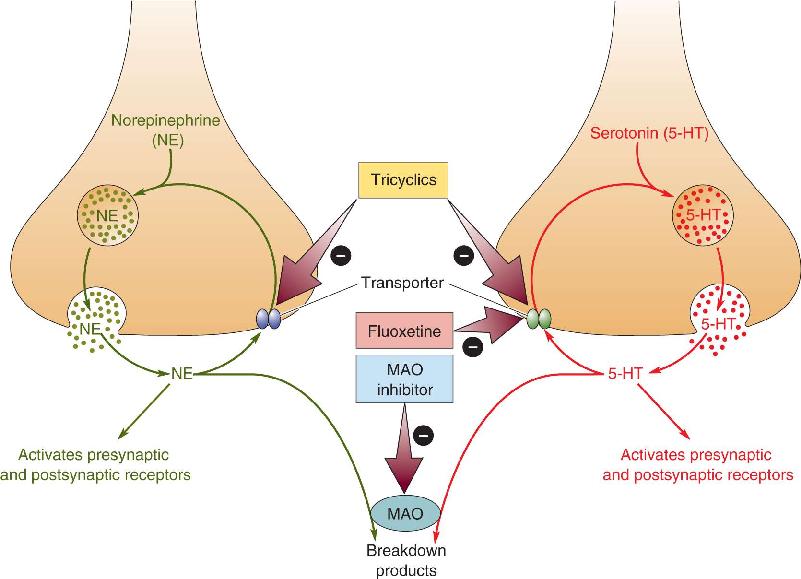
Antidepressants. A number of highly effective pharmacological treatments are available for mood disorders. Antidepressant drugs include (1) tricyclic compounds (named for their chemical structure), such as imipramine, which among other actions, block the reuptake of both norepinephrine and serotonin by transporters; (2) SSRIs, such as fluoxetine, which act only on serotonin terminals; (3) NE- and 5-HT-selective reuptake inhibitors, such as venlafaxine; and (4) MAO inhibitors, such as phenelzine, which reduce the enzymatic degradation of serotonin and norepinephrine (Figure 22.11). All of these drugs elevate the levels of monoamine neurotransmitters in the brain; however, as mentioned, their therapeutic actions take weeks to develop.

FIGURE 22.11 Antidepressant drugs and the biochemical life cycles of norepinephrine and serotonin. MAO inhibitors, tricyclics, and SSRIs are used as antidepressants. MAO inhibitors enhance the actions of NE and 5-HT by preventing their enzymatic destruction. Tricyclics enhance NE and 5-HT action by blocking uptake. SSRIs act the same way but are selective for serotonin. Description
The adaptive response in the brain for the clinical effectiveness of these drugs has not been established with certainty. Nonetheless, an intriguing finding is that clinically effective treatment with antidepressants dampens the hyperactivity of the HPA system and the anterior cingulate cortex in humans. Animal studies suggest that this effect may be due, in part, to increased glucocorticoid receptor expression in the hippocampus, which occurs in response to a long-term elevation in serotonin. Recall that CRH plays a crucial role in the stress response of the HPA axis. New drugs that act as CRH receptor antagonists are currently under development and testing. Recent research has also shown that prolonged treatment with SSRIs increases neurogenesis, the proliferation of new neurons, in the hippocampus. (Neurogenesis is discussed further in Chapter 23.) Remarkably, this proliferation may be important for the beneficial behavioral effects of SSRIs, presumably in part by boosting the control of the HPA by the hippocampus.
The long delay between treatment onset and antidepressant effect presents not only a scientific mystery but also a clinical challenge. Patients can feel discouraged when their expectations for improvement are not met, and this can temporarily exacerbate the depression. This is a serious limitation, particularly in cases where there is a high risk of suicide. Thus, the search is on for rapidly acting antidepressant medications that do not require weeks of treatment to become effective. The hope for attaining this goal has been fueled by recent findings that a single intravenous dose of the anesthetic drug ketamine can rapidly alleviate symptoms of depression for several days. Although these findings support the concept of a rapidly acting antidepressant, ketamine itself is not a practical treatment for depression. As we will discuss later in the context of schizophrenia, ketamine can cause severe psychotic episodes that require hospitalization. It is only after the drug is eliminated from the body, and psychotic symptoms diminish, that antidepressant effects are observed. Thus, as with other medical treatments for depression, the therapeutic effect is apparently caused by some adaptive response to the drug. However, in the case of ketamine, this adaptation occurs much more rapidly than with the other antidepressants used in clinical practice today.
Lithium. By now, you probably have formed the (correct) impression that, until recently, most treatments for psychiatric disorders were discovered virtually by chance. For example, ECT was introduced initially in the 1930s as a treatment of last resort for psychotic behavior based on the mistaken belief that epilepsy and schizophrenia could not coexist in the same person. Only later was it shown to be an effective treatment for major depression for reasons that still remain unknown.
“Enlightened serendipity” was again at work in the discovery of a highly effective treatment for bipolar disorder. Working in the 1940s, Australian psychiatrist John Cade was searching for psychoactive substances in the urine of manic patients. He injected guinea pigs with urine or urinary constituents and observed their behavioral effects. Cade wanted to test the effect of uric acid, but he had difficulty getting it into solution. Instead, he used lithium urate because it dissolved easily and was readily available in the pharmacy. He observed, quite unexpectedly, that this treatment calmed the guinea pigs (he had predicted the opposite effect). Because other lithium salts also produced this behavioral effect, he concluded that it was the lithium, not a constituent of urine, that was responsible. He went on to test lithium treatment on patients with mania, and, amazingly, it worked. Subsequent studies showed that lithium is highly effective in stabilizing the mood of patients with bipolar disorder, by preventing not only the recurrence of mania but also the episodes of depression (Figure 22.12).

FIGURE 22.12 The mood-stabilizing effect of lithium treatment in five patients. (Source: Adapted from Barondes, 1993, p. 139.)
Lithium affects neurons in many ways. In solution, it is a monovalent cation that passes freely through neuronal sodium channels. Inside the neuron, lithium prevents the normal turnover of phosphatidyl inositol (PIP2), a precursor for important second messenger molecules that are generated in response to activation of some G-protein-coupled neurotransmitter receptors (see Chapter 6). Lithium also interferes with the actions of adenylyl cyclase, essential for the generation of the second messenger cyclic-AMP, and glycogen synthase kinase, a critical enzyme in cellular energy metabolism. Why lithium is such an effective treatment for bipolar disorder, however, remains completely unknown. Like other antidepressants, the therapeutic effects of lithium require long-term use. The answer, again, appears to lie in an adaptive change in the central nervous system (CNS), but the nature of this change remains to be determined.
Deep Brain Stimulation. In a substantial fraction of patients, severe depression fails to respond to ECT, medicine, or talk therapy. In such cases, more drastic measures are called for, and one entails undergoing a surgical procedure in which an electrode is implanted deep in the brain. This approach to treat depression was pioneered by Helen Mayberg, a neurologist at Emory University (Box 22.3). Recall that activity in the anterior cingulate cortex is increased by sadness and decreased by successful treatment with standard antidepressant medications. The observation that activity in this region fails to decrease in patients with unrelenting, treatment-resistant depression inspired Mayberg to contemplate using direct brain stimulation to modulate activity here. Although it seems counterintuitive, electrical stimulation can actually decrease activity in brain circuits that are chronically overactive (the reasons remain unclear but likely include recruitment of inhibitory neurons). Indeed, Mayberg and a team of neurosurgeons at the University of Toronto found that electrical stimulation of a circumscribed region of the anterior cingulate cortex, comprising Brodmann’s area 25, could produce immediate relief from depression.
It was never my plan to study depression. I am a neurologist, and depression was generally considered to be beyond the purview of my medical discipline. While many patients with neurological disorders develop depression, it was often seen as a nonspecific response to a distressing diagnosis (stroke, Parkinson’s disease, Alzheimer’s disease, and the like). Furthermore, the notion that a global change like depression could be localized to specific brain regions, the way a language deficit might be traced to disruption of specific parts of the frontal or temporal lobes, was not intuitive. For the most part, strategies to study and treat depression in neurological patients mirrored those in patients with depression without identified neurological disease—focusing on brain chemistry—that is, until the early 1990s when advances in neuroimaging changed the playing field.
By 2001, we had learned a lot about the functional neuroanatomy of depression. Using positron emission tomography and functional magnetic resonance imaging, we had identified activity patterns that subdivided depressed patients by their symptom clusters. We also studied changes that distinguished the response to antidepressant drugs from that of psychotherapy and identified baseline patterns that might guide treatment selection for each treatment. A brain wiring diagram of depression was emerging.
It was around this time that we had the opportunity to directly examine the role of the subcallosal cingulate region (Brodmann’s area 25) in our evolving depression circuit (Figure A). We had converging evidence of common changes in this region across a wide variety of effective antidepressant treatments. We also knew that failure to effect changes in this region were associated with treatment nonresponse. We hypothesized that relief from major depression could be achieved with focal brain stimulation using a well-established neurosurgical technique used to treat Parkinson’s disease—deep brain stimulation (DBS). Inserting electrodes into our intended target, the subcallosal cingulate white matter, was not technically more difficult or of higher risk than the basal ganglia implantations used for Parkinson’s disease, or so said the surgeon. We became convinced it should be attempted, but what patient would be appropriate for such a procedure?

Figure A Abnormal activity in the anterior cingulate cortex and the use of DBS to correct it. Upper left: PET scan of a depressed patient demonstrating increased blood flow—indicating hyperactivity—in the subcallosal cingulate cortex (red). DBS quiets this region. Upper right: Diffusion-weighted MRI scan used prior to surgery to identify the intersection of three white matter bundles passing through the subcallosal cingulate region thus defining the optimal location to implant the DBS electrode. Lower left: Structural MRI scan used in the operating room to plan and verify the targeted location of the implanted DBS electrode. Lower right: Postsurgical skull X-ray showing the actual implanted DBS electrodes. (Source: Courtesy of Dr. Helen Mayberg.)
Treatment-resistant depression is a dire condition defined by failure to respond to multiple available antidepressant treatments including electroconvulsive therapy. What I had not appreciated in my years of studying depression was the banality of our definitions and rating scales, as they failed to capture the degree of suffering experienced by patients in what can only be described as a malignant condition, a pervasive state of sustained mental pain and physical immobility with no “off switch.”
I can still remember that first case on the morning of May 23, 2003. We were prepared technically: where to implant, what side effects to watch for. But otherwise, we had no expectations. How could there be when what you are doing is something that has never been done before? Our patient was awake (DBS electrodes are implanted using local anesthesia), and it was easy enough to monitor the obvious—discomfort, pain, general distress. The primary goal was to get the electrodes implanted and then turn them on and make sure nothing bad happened. Our mindset going in was that the real work would come later as we tested various stimulation parameters to achieve clinical effects—a process we thought would take weeks, like other antidepressant treatments.
The plan was to observe, keep the patient safe, and if something didn’t seem right, turn it off. So we weren’t expecting it when the patient’s mood abruptly lifted during testing of the second contact on the left electrode. As the current was turned up, the patient suddenly asked if we had done something different. She felt calm, with a lightness and serenity she hadn’t felt in a long time. I was looking right at her on the nonsterile side of the surgical table. Her eyes were wider, looking around; her speech was noticeably louder and less halting; and she was more engaged with the room and with me. It was as if we had hit a spot and literally turned her “negative” feeling off, releasing the rest of her brain to go about doing whatever it wanted to do. And then we turned down the current back to zero; the relief faded and the void returned. That moment changed everything I knew about depression and how to study it.
Recall that during most neurosurgical procedures, the patient remains awake, which is possible because there are no pain receptors in the brain. Thus, the patients in Mayberg’s study could report the effect of stimulation during the operation. They described a “sudden calmness” or “lightness” and “disappearance of the void” when the stimulator was turned on. These patients were discharged from the hospital with the implanted electrodes connected to a battery-operated stimulator that continuously applied electric pulses. The majority of patients experienced continued relief from their depression.
These findings have generated considerable excitement in the field but are still considered to be preliminary. Additional studies are underway to confirm these initial results. Obviously, brain surgery is always considered a treatment of last resort.
Although their severity might be hard to fully comprehend, we all have some idea of what mood and anxiety disorders are like because they are extremes in the spectrum of brain states that are part of normal experience. The same cannot be said for schizophrenia. This severe mental disorder distorts thoughts and perceptions in ways that healthy people find difficult to understand. Schizophrenia is a major public health problem, affecting 1% of the adult population. Over 2 million people suffer from the disorder in the United States alone.
Schizophrenia is characterized by a loss of contact with reality and a disruption of thought, perception, mood, and movement. The disorder typically becomes apparent during adolescence or early adulthood and usually persists for life. The name, introduced in 1911 by Swiss psychiatrist Eugen Bleuler, roughly means “divided mind,” because of his observation that many patients seemed to oscillate between normal and abnormal states. However, there are many variations in the manifestations of schizophrenia, including those that show a steadily deteriorating course. Indeed, it is still not clear whether what is called schizophrenia is a single disease or several.
The symptoms of schizophrenia fall into two categories: positive and negative. Positive symptoms reflect the presence of abnormal thoughts and behaviors, such as:
Negative symptoms reflect the absence of responses that are normally present. These symptoms include:
Individuals affected by schizophrenia often have delusions organized around a theme; for example, they may believe that powerful adversaries are out to get them. These are often accompanied by auditory hallucinations (such as hearing imaginary voices) related to the same delusional theme. There can also be a lack of emotional expression (called a “flat affect”), coupled with disorganized behavior and incoherent speech. Speech may be accompanied by silliness and laughter that appear to have no relation to what is being said. In some cases, schizophrenia is accompanied by peculiarities of voluntary movement, such as immobility and stupor (catatonia), bizarre posturing and grimacing, and senseless, parrot-like repetition of words or phrases.
Understanding the neurobiological basis for schizophrenia represents one of the greatest challenges of neuroscience because the disorder affects many of the characteristics that make us human: thought, perception, self-awareness. Although considerable progress has been made, we still have much more to learn.
Genes and the Environment. Schizophrenia runs in families. As shown in Figure 22.13, the likelihood of having the disorder varies in relation to the number of genes that are shared with an affected family member. If your identical twin has schizophrenia, the probability is about 50% that you will also have it. The chances you will have the disease decline as the number of genes you share with an affected family member decreases. These findings argue that schizophrenia is primarily a genetic disorder. Recently, researchers have identified several specific genes that seem to increase susceptibility to schizophrenia. Nearly all of these genes have important roles in synaptic transmission, its plasticity, or the growth of synapses.

FIGURE 22.13 The familial nature of schizophrenia. The risk of developing schizophrenia increases with the number of shared genes, suggesting a genetic basis for the disease. (Source: Adapted from Gottesman, 1991, p. 96.) Description
Remember, however, that identical twins have exactly the same genes. So why, in 50% of cases, is one sibling spared when the other has schizophrenia? The answer must lie in the environment. In other words, faulty genes seem to make some people vulnerable to environmental factors that cause schizophrenia. Although the symptoms may not appear until a person reaches his or her twenties, considerable evidence indicates that the biological changes causing the condition begin early in development, perhaps prenatally. Viral infections during fetal and infant development have been implicated as contributing causes, as has poor maternal nutrition. In addition, environmental stresses throughout life are known to exacerbate the course of the disorder. A number of studies have suggested that use of marijuana increases the risk for developing schizophrenia in genetically vulnerable adolescents.
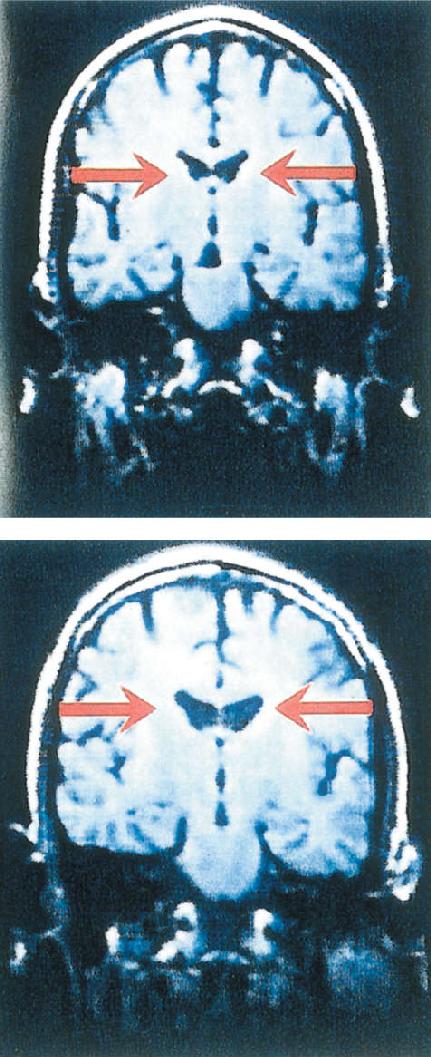
Schizophrenia is associated with physical changes in the brain. An interesting example appears in Figure 22.14. The figure shows brain scans of identical twins, one with schizophrenia and one without. Normally, the structures of the brains of identical twins are nearly identical. However, in this case, the brain of the schizophrenic sibling shows enlarged lateral ventricles, which reflects the shrinkage of brain tissue around them. This difference is consistent when large numbers of people are sampled; the brains of schizophrenics have, on average, a significantly larger ventricle-to-brain-size ratio than people who do not have the disorder.

FIGURE 22.14 Enlarged lateral ventricles in schizophrenia. These MRI scans are from the brains of identical twins. The sibling on the top was normal; the one on the bottom was diagnosed with schizophrenia. Notice the enlarged lateral ventricles in the schizophrenic sibling, indicating a loss of brain tissue. (Source: Barondes, 1993, p. 153.)
Such pronounced structural changes are not always apparent in the brains of schizophrenics, however. Important physical changes in their brains also occur in the microscopic structure and function of cortical connections. For example, schizophrenics often have defects in the myelin sheaths surrounding axons in their cerebral cortex, although it is not clear whether this is a cause or consequence of the disease. Another common finding in schizophrenia is reduced cortical thickness and abnormal neuronal lamination (Figure 22.15). Changes in synapses and several neurotransmitter systems have also been implicated. As we’ll see next, particular attention has focused on alterations in chemical synaptic transmission mediated by dopamine and glutamate.

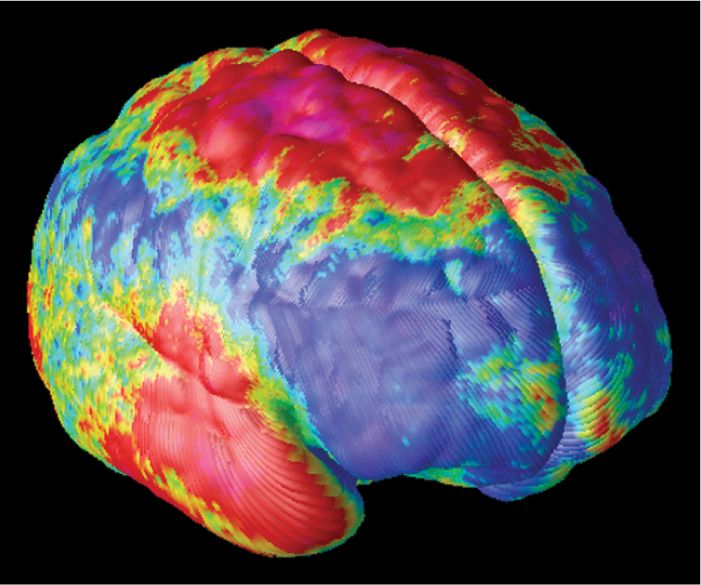
FIGURE 22.15 Loss of cortical gray matter in schizophrenics during adolescence.. The brains of 12 patients with early-onset schizophrenia were imaged repeatedly over the course of 5 years, between the ages of 13 to 18. This image shows the average annual change in the thickness of their cortical gray matter, with red colors indicating regions of greatest loss and blue indicating no change. Severe loss (up to 5% annually) is observed in parietal, motor and anterior temporal cortex. (Source: Thompson et al., 2001, Figure 1, with permission.)
The Dopamine Hypothesis. Recall that dopamine is the neurotransmitter used by another of the diffuse modulatory systems (Figure 22.16). A link between the mesocorticolimbic dopamine system and schizophrenia has been made on the basis of two main observations. The first relates to the effects of amphetamine in otherwise healthy people. Remember from our discussion in Chapter 15 that amphetamine enhances neurotransmission at catecholamine-utilizing synapses and causes the release of dopamine. Amphetamine’s normal stimulant action bears little resemblance to schizophrenia. However, because of its addictive properties, users of amphetamines often risk taking more and more to satisfy their cravings. The resulting overdose can lead to a psychotic episode with positive symptoms that are virtually indistinguishable from those of schizophrenia. This suggests that psychosis is somehow related to too much catecholamine in the brain.

FIGURE 22.16 The dopaminergic diffuse modulatory systems of the brain. The mesocorticolimbic dopamine system, which arises in the ventral tegmental area, has been implicated in the cause of schizophrenia. A second dopaminergic system arising from the substantia nigra is involved in the control of voluntary movement by the striatum. Description
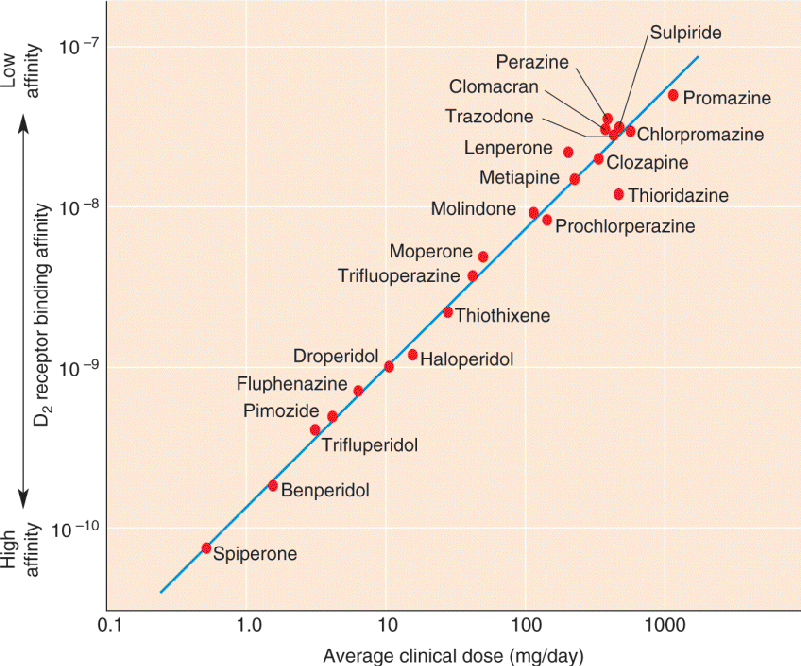
A second reason to associate dopamine with schizophrenia relates to the CNS effects of drugs that are effective in reducing the positive symptoms of the disorder. In the 1950s, researchers discovered that the drug chlorpromazine, initially developed as an antihistamine, could prevent the positive symptoms in schizophrenia. Chlorpromazine and other related antipsychotic drugs, collectively called neuroleptic drugs, were later found to be potent blockers of dopamine receptors, specifically the D2 receptor. When a large number of neuroleptics are examined, the correlation between the dosage effective for controlling schizophrenia and their ability to bind to D2 receptors is impressive (Figure 22.17). Indeed, these same drugs are effective in the treatment of amphetamine and cocaine psychoses. According to the dopamine hypothesis of schizophrenia, psychotic episodes in schizophrenia are triggered specifically by the activation of dopamine receptors.

FIGURE 22.17 Neuroleptics and D2 receptors. The neuroleptic dosages effective in controlling schizophrenia correlate well with the binding affinities of the drugs for D2 receptors. The units on the Y axis are the molar concentrations of drug that inhibit half of the D2 receptors in the brain. Higher affinity drugs block the receptors at lower concentrations. (Source: Adapted from Seeman, 1980.) Description
Despite the tantalizing link between the positive symptoms of schizophrenia and dopamine, there seems to be more to the disorder than an overactive dopamine system. One indication is that newly developed antipsychotic drugs, like clozapine, have little effect on D2 receptors. These drugs are called atypical neuroleptics, indicating that they act in a novel way. The mechanism by which these compounds exert their neuroleptic effect has not been established with certainty, but an interaction with serotonin receptors is suspected.
The Glutamate Hypothesis. Another indication that there is more to schizophrenia than dopamine comes from the behavioral effects of phencyclidine (PCP) and ketamine. These drugs were introduced in the 1950s as anesthetics. However, many patients experienced adverse side effects, sometimes lasting for days, which included hallucinations and paranoia. Although it is no longer used clinically, PCP is now a common illegal drug of abuse, known by users as “angel dust” or “hog.” Ketamine, which is still used in veterinary medicine, has also made it to the street where it is referred to as “special K” or “vitamin K.” PCP and ketamine intoxication cause many of the symptoms of schizophrenia, both positive and negative. However, neither drug has an effect on dopaminergic transmission; they affect synapses that use glutamate as a neurotransmitter.
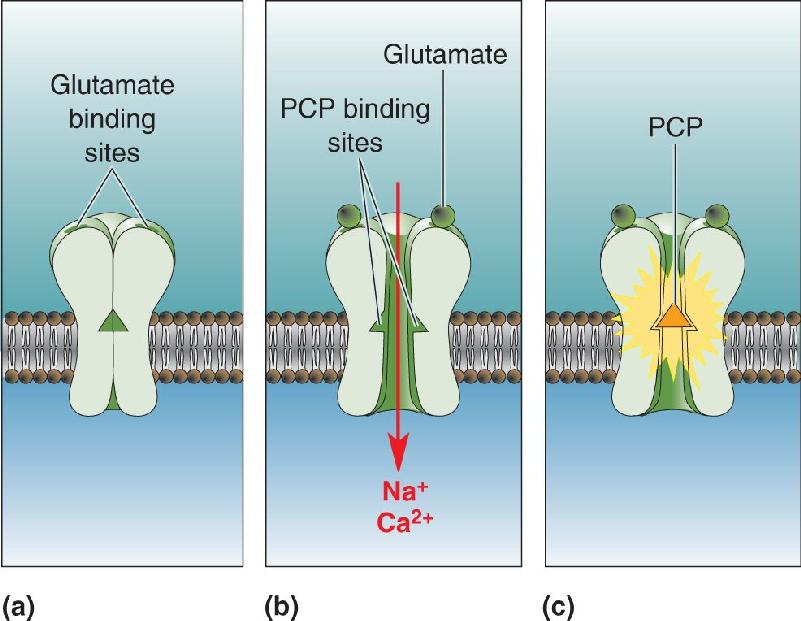
Recall from Chapter 6 that glutamate is the main fast excitatory neurotransmitter in the brain, and that NMDA receptors are one subtype of glutamate receptor. PCP and ketamine act by inhibiting NMDA receptors (Figure 22.18). Thus, according to the glutamate hypothesis of schizophrenia, the disorder reflects diminished activation of NMDA receptors in the brain.

FIGURE 22.18 Blocking of the NMDA receptor by PCP. NMDA receptors are glutamate-gated ion channels. (a) In the absence of glutamate, the channel is closed. (b) In the presence of glutamate, the channel is open, exposing PCP binding sites. (c) The channel is blocked when PCP enters and binds. The blockade of brain NMDA receptors by PCP produces effects on behavior that resemble the symptoms of schizophrenia. Description
In order to study the neurobiology of schizophrenia, neuroscientists have attempted to establish animal models of the disorder. Low doses of PCP administered chronically to rats produce changes in brain biochemistry and behavior that resemble those in schizophrenic patients. Mice that have been genetically engineered to express fewer NMDA receptors also display some schizophrenia-like behaviors, including repetitive movements, agitation, and altered social interactions with other mice (Figure 22.19). Of course, we don’t know whether the mutant mice feel paranoid or hear imaginary voices. But it is significant that the observable behavioral abnormalities can be lessened by treating the mice with either conventional or atypical neuroleptic drugs.

FIGURE 22.19 Social withdrawal in mutant mice with reduced numbers of NMDA receptors. The mice on the left have a normal number of NMDA receptors. The photographs were taken 30 minutes apart over 2 hours to monitor social behavior. These mice tended to nest together. The mice on the right have been genetically altered to express fewer NMDA receptors. Notice that these mice tended to avoid social contact with one another. (Source: Mohn et al., 1999, p. 432.)
While all drugs that inhibit NMDA receptors will impair memory and cognition, not all reproduce the positive symptoms of schizophrenia in humans. The key difference lies in the mechanism of action. PCP and ketamine do not interfere with the binding of glutamate to the receptor like other NMDA receptor inhibitors. Instead, they act by entering the channel and clogging the pore. Consequently, blockade by PCP and ketamine is possible only when the receptors are active and the channels are open. This feature has led researchers to wonder if the psychomimetic effects of these drugs are mediated by a select population of neurons with high ongoing activity and tonic NMDA receptor activation. One such population is comprised of cortical GABAergic neurons in the cerebral cortex. Inhibition of the NMDA receptors on these neurons might distort thinking and the processing of sensory information. Notably, postmortem examination of the brains of individuals with schizophrenia has found the cortex to be deficient in many interneurons.
The treatment of schizophrenia consists of drug therapy combined with psychosocial support. As mentioned earlier, the conventional neuroleptics, such as chlorpromazine and haloperidol, act at D2 receptors. These drugs reduce the positive symptoms of schizophrenia in the majority of patients. Unfortunately, the drugs also have numerous side effects related to their actions on the dopaminergic input to the striatum that arises from the substantia nigra (see Chapter 14). Not surprisingly, the effects of blocking dopamine receptors in the striatum resemble the symptoms of Parkinson’s disease, including rigidity, tremor, and difficulty initiating movements. Chronic treatment with conventional neuroleptics also can result in the emergence of tardive dyskinesia, which is characterized by involuntary movements of the lips and jaw. Many of these side effects are avoided by using atypical neuroleptics, such as clozapine and risperidone, because they do not act directly on the dopamine receptors in the striatum. These medications are also more effective against the negative symptoms of schizophrenia.
The newest focus of drug research is the NMDA receptor. Investigators hope that increasing NMDA receptor responsiveness in the brain, perhaps in combination with decreasing D2 receptor activation, will further alleviate the symptoms of schizophrenia.
Neuroscience has had a huge impact on psychiatry. Mental illness is now recognized as the consequence of pathologic modifications of the brain, and psychiatric treatments today are focused on correcting these changes. Just as importantly, neuroscience has changed how society views people who suffer from mental illness. Suspicion about mentally ill people is slowly giving way to compassion. Mental illnesses today are recognized as diseases of the body, just like hypertension or diabetes.
Despite remarkable progress in treating psychiatric disorders, we have a very incomplete understanding of how current treatments work their magic on the brain. In the case of drug therapy, we know with great precision about how chemical synaptic transmission is affected. But we do not know why, in many cases, the therapeutic effect of a drug takes weeks to emerge. Even less is known about how psychosocial treatments act on the brain. In general, the answer seems to lie in adaptive changes that occur in the brain in response to treatment.
We also do not know the causes of most mental disorders. It is clear that our genes either put us at risk or protect us. However, the environment also plays an important role. Environmental stresses before birth may contribute to schizophrenia, and those after birth may precipitate depression. Not all environmental effects are bad, however. Appropriate sensory stimulation, especially in early childhood, can apparently produce adaptive changes that help protect us from developing mental illnesses later in life.
Psychiatric disorders and their treatment illustrate that our brains and behaviors are influenced by past experience, whether it is exposure to inescapable stress or to pharmacologically elevated levels of serotonin. Of course, much more subtle sensory experiences also leave their mark on the brain. In Part IV, we will explore how sensory experiences modify the brain during development and during learning.
hypothalamic-pituitary-adrenal (HPA) axis (p. 759)
serotonin-selective reuptake inhibitor (SSRI) (p. 762)
monoamine hypothesis of affective disorders (p. 766)
diathesis–stress hypothesis of affective disorders (p. 766)
1. How and where in the brain do benzodiazepines act to reduce anxiety?
2. Depression is often accompanied by bulimia nervosa, which is characterized by frequent eating binges followed by purging. Where does the regulation of mood and appetite converge in the brain?
3. Snuggling with your mom as a baby might help you cope with stress better as an adult. Why?
4. What three types of drugs are used to treat depression? What do they have in common?
5. Psychiatrists often refer to the dopamine theory of schizophrenia. Why do they believe dopamine is linked to schizophrenia? Why must we be cautious about accepting a simple correlation between schizophrenia and too much dopamine?
American Psychiatric Association. 2013. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Arlington, VA: American Psychiatric Association.
Andreasen NC. 2004. Brave New Brain: Conquering Mental Illness in the Era of the Genome. New York: Oxford University Press.
Charney DS, Nestler EJ, eds. 2004. Neurobiology of Mental Illness, 2nd ed. New York: Oxford University Press.
Harrison PJ, Weinberger DR. 2005. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Molecular Psychiatry 10:40–68.
Holtzheimer PE, Mayberg HS. 2011. Deep brain stimulation for psychiatric disorders. Annual Review of Neuroscience 34:289–307.
Insel TR. 2012. Next generation treatments for psychiatric disorders. Science Translational Medicine 4:1–9.
Additional figures