Molecular Mechanisms of Learning and Memory
BOX 25.1 PATH OF DISCOVERY: What Attracted Me to the Study of Learning and Memory in Aplysia? by Eric Kandel

BOX 25.2 BRAIN FOOD: Synaptic Plasticity: Timing Is Everything
BOX 25.3 PATH OF DISCOVERY: Memories of Memory, by Leon Cooper
BOX 25.4 BRAIN FOOD: The Wide World of Long-Term Synaptic Depression
An important first step in understanding the neurobiology of memory is identifying where different types of information are stored. As we saw in Chapter 24, basic neuroscientific research is beginning to answer this question. However, an equally important question concerns how information is stored. As Hebb pointed out, memories can result from subtle alterations in synapses, and these alterations can be widely distributed in the brain. This insight helps narrow the search for a physical basis of memory, synaptic modifications, but it also raises a dilemma. The synaptic modifications that underlie memory may be too small and too widely distributed to be observed and studied experimentally.
These considerations inspired some researchers, led by Eric Kandel of Columbia University, to study the nervous systems of simple invertebrate animals for insights into the molecular mechanisms of memory. Through the history of neuroscience, researchers have used a large menagerie of invertebrate creatures for neurobiological experiments. You are already familiar with the squid and the contribution of its giant axon and giant synapse to our understanding of cellular neurophysiology (see Chapters 4 and 5). Other experimental invertebrates are lobsters, crayfish, cockroaches, flies, bees, leeches, and nematode worms. The reason for using them is that invertebrates have some important experimental advantages, including small nervous systems with large neurons, known and reproducible connections between neurons, and simple genetics.
Invertebrates can be particularly useful for analyzing the neural basis of behavior. Although the behavioral repertoire of the average invertebrate is rather limited, many invertebrate species exhibit some of the simple forms of learning that were introduced in the last chapter. One species in particular has been used to study the neurobiology of learning, the sea snail Aplysia californica. Kandel shared the 2000 Nobel Prize in Physiology or Medicine for his seminal contributions to the understanding of memory mechanisms in this creature. Invertebrate research has clearly shown that Hebb was right: Memories can reside in synaptic alterations. Moreover, it has been possible to identify some of the molecular mechanisms that lead to this synaptic plasticity. Although nonsynaptic changes have also been found that may account for some types of memory, invertebrate research leaves little doubt that the synapse is an important site of information storage.
The past several decades have seen rapid advances in understanding how our own brains form memories. This progress has come from the study of neural activity in regions of the mammalian brain associated with different types of memory. Insight from theoretical analysis of neural networks helped focus attention on modifications most likely to store information, and new technologies have made feasible the detection of candidate mechanisms. One fruitful approach has been the use of electrical brain stimulation to produce measurable synaptic alterations whose mechanisms can be studied. Researchers could then ask if these same mechanisms contribute to natural memory formation. One of the interesting conclusions of this research is that the mechanisms of activity-dependent synaptic plasticity and memory formation in the adult brain have much in common with those operating during development for wiring the brain.
There is a growing sense of optimism among neuroscientists that we may soon know the physical basis of learning and memory. This investigation has benefited from the combined approaches of researchers in disciplines ranging from psychology to molecular biology. In this chapter, we’ll take a look at some of their discoveries.

It is useful to consider learning and memory as occurring in two stages: (1) the acquisition of a short-term memory and (2) the consolidation of a long-term memory (Figure 25.1). In this context, memory acquisition (learning) occurs by a physical modification of the brain caused by incoming sensory information. This is different from working memory introduced in Chapter 24, which is vulnerable to erasure by distraction and has a very limited capacity (think about holding a phone number in mind). Working memory can be achieved by keeping neural activity going with continuous rehearsal, and does not require any lasting physical change in the brain. In contrast, short-term memory survives distraction, has a large capacity, and can last minutes to hours with no conscious effort. Do you remember what you had for breakfast this morning or for dinner last night? These memories persist for some time without rehearsal but are considered to be “short term” because they will be forgotten unless they are consolidated into long-term memory. Thus, you probably do not readily remember what you had for dinner two weeks ago on Tuesday because the brain changes that encoded this information have since faded away.

FIGURE 25.1 The flow of sensory information into long-term memory. The first step is memory acquisition, by which experiences are encoded by synaptic modifications. The second step is memory consolidation, by which temporary synaptic changes are made permanent. Description
Memory consolidation, introduced in Chapter 24, is the process by which some experiences, held temporarily by transient modifications of neurons, are selected for permanent storage in long-term memory. Perhaps last Tuesday’s dinner coincided with an emotionally charged event, like a first date with the love of your life. In that case, it would not be surprising if every detail of the evening was etched into your long-term memory. This example makes the point that not all memories are created equal. The brain has mechanisms that ensure that some experiences are retained while others are lost.
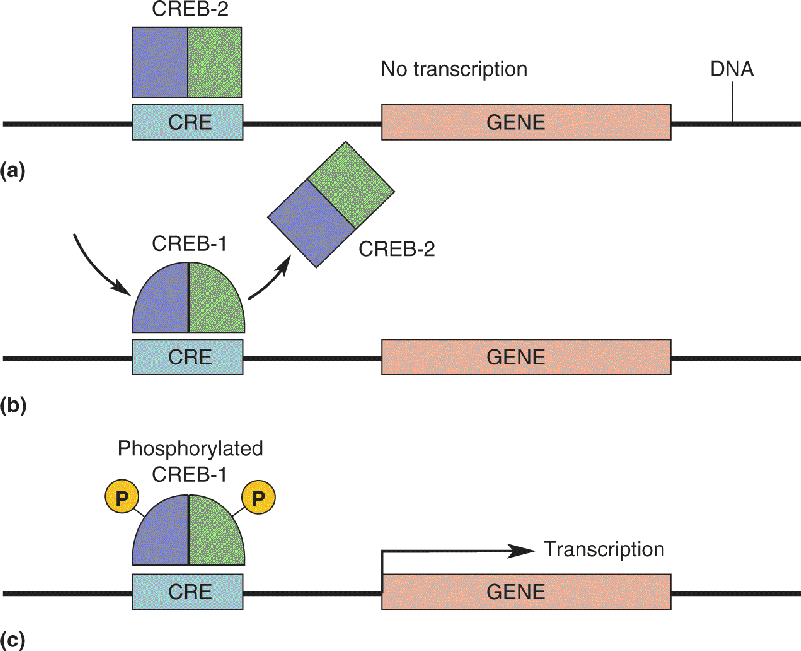
We will divide our discussion of memory mechanisms into those responsible for the initial acquisition of short-term memory and those acting to convert a temporary change into a permanent one. We’ll see that acquisition occurs by modifying synaptic transmission between neurons and that synaptic consolidation requires, in addition, new gene expression and protein synthesis.
“While it might appear that I am doing nothing, at the cellular level I am really quite busy.” We don’t know to whom this statement should be attributed, but it certainly applies to memory. In the last chapter we discussed different types of memories and where they are stored. For example, we learned that declarative memories (facts, events, places, faces) ultimately reside in the cerebral cortex. However, when it comes to information storage, no neuron is spared. Virtually every neuron in the nervous system can form a memory of recent patterns of activity. By the same token, countless molecular mechanisms participate in information storage of various kinds, so our coverage here will necessarily be selective. As a general example, let’s consider what happens in the cerebral cortex when a novel experience becomes familiar (Figure 25.2).

FIGURE 25.2 Famous people who may be familiar to you. What happened in your brain when you first saw photos or video of these people and formed a memory?
We and other primates are experts at using vision to recognize and discriminate differences between familiar objects and individuals. Where is this information stored? According to Hebb, if an engram is based on information from only one sensory modality, it should be possible to localize it within the regions of cortex that serve this modality (see Chapter 24). For example, if the engram relies only on visual information, then we would expect it to reside within the visual cortex. Studies of visual discrimination in monkeys are consistent with this proposal.
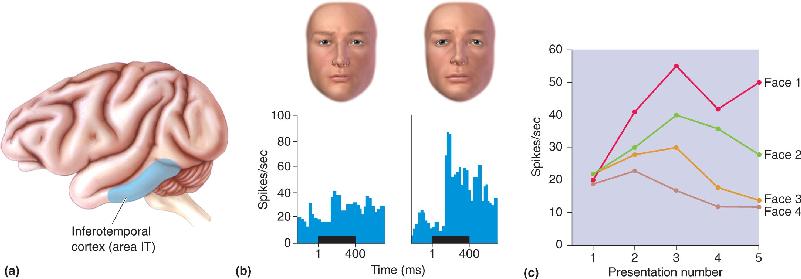
Macaque monkeys can be trained to discriminate images of objects and associate them with a food reward. However, they lose this ability when lesions are made in the inferotemporal cortex. This region contains area IT (Figure 25.3a) which we learned in Chapter 10 is part of the “ventral stream,” a series of higher order visual areas concerned with visual perception. After lesions of inferotemporal cortex, monkeys appear to be unable to recognize familiar objects, even though basic visual capacities remain intact. IT therefore appears to be both a visual area and an area involved in memory storage. This conclusion is further supported by a fascinating clinical condition called prospagnosia, a selective amnesia for familiar faces (including one’s own) that can result from damage to the inferotemporal cortex in humans.

FIGURE 25.3 Responses to faces in the inferotemporal cortex. (a) The location of area IT in the inferior temporal lobe of a macaque monkey. (b) IT neurons respond to faces, and these responses can be highly selective. The histograms show the response of a neuron in area IT to slightly different images of human faces. The horizontal bar under each histogram indicates when the stimulus was presented. (c) Changing responses of a cell as novel faces become familiar. When the four faces are presented for the first time, there is a moderate response to each. With subsequent presentations, the cell becomes more responsive to faces 1 and 2 and less responsive to faces 3 and 4. Acquisition of face selectivity correlates with the ability of the animal to recognize and distinguish among these faces. (Sources: Part b, adapted from Leopold et al., 2006, Fig. 6; part c, adapted from Rolls et al., 1989, Fig. 1.) Description
Like most cortical neurons, IT neurons typically show the property of stimulus selectivity; that is, they respond with a barrage of action potentials to the presentation of some but not all stimuli. As we learned back in Chapter 10, IT neurons have the distinction of responding to complex images and shapes that can include familiar faces. In a typical experiment, an electrode is used to record from an IT neuron in an alert monkey. When shown a series of images of familiar faces (other monkeys in the colony or the experimenters), the neuron responds vigorously to some but not all images: The neuron shows stimulus (face) selectivity (Figure 25.3b).
Now, what happens to an IT neuron as a visual recognition memory is formed, when a novel set of faces becomes familiar? The first time new faces are seen, the cell responds at about the same moderate level to all of them: There are responses but no selectivity (Figure 25.3c, presentation 1). However, with repeated presentations, the responses change and selectivity emerges. The response of the neuron grows to some faces and diminishes to others. With continued presentation of the same group of faces, the response of the neuron becomes more stable and more selective (Figure 25.3c, presentations 4 and 5). Other nearby neurons in IT show similar changes, but their responses grow and diminish to different faces. Are we observing the birth of a memory trace? There are good reasons to think so. Shifts in the selectivity of cortical neurons are a very common cellular correlate of memories formed in other modalities (audition, somatic sensation, etc.) as well.
Distributed Memory Storage. Analysis of a simple “neural network” model helps illustrate what is behind an experience-dependent shift in neuronal selectivity. Consider the network of connected neurons depicted in Figure 25.4. Three stimuli (say, the faces of Mark, Barry, and Mike) are conveyed by separate inputs to three postsynaptic cortical neurons (call them A, B, and C). Initially, in our first experience of these gentlemen, we find that neurons A, B, and C respond moderately to Mark, Barry, and Mike. There is no selectivity, and there is no neural response that could be used to distinguish one face from another. However, after repeated exposure to Mark, Barry, and Mike, the neurons of the network acquire selectivity; although all neurons respond to all faces, neuron A responds best to Mark, neuron B responds best to Barry, and neuron C responds best to Mike. This transformation of the responses to (now) familiar faces occurred by adjustments in the strength or “weights” of the three synaptic inputs converging on the cortical neurons.

FIGURE 25.4 A model of a distributed memory. (a) In this simple neural network, three inputs conveying information about the appearance of three faces (Mark, Barry, and Mike) synapse on three cortical neurons: cells A, B, vand C. (b) Before learning to recognize these faces, every neuron in the network responds moderately to every face. There is no selectivity for one face over another. (c) After learning, the neurons display a face preference. Cell A prefers Mark, cell B prefers Barry, and cell C prefers Mike. Notice that comparing the relative responses of all three neurons can determine which face is being viewed. For example, Mark evokes a strong response in A, a moderate response in B, and a weak response in C. Even if neuron A were to die, Mark would continue to be represented by a specific pattern of activity in neurons B and C. Description
Where is the “memory” in this network? Put another way, how does output from our three cortical neurons uniquely represent Mark, Barry, or Mike? The answer is that after learning, there is a unique pattern or ratio of activity in the three neurons for each face. Mark is represented by high activity in neuron A, moderate activity in neuron B, and weak activity in neuron C. We call this a distributed memory. By analogy, consider how a color is represented in the visual system, not solely by the output of any one type of cone photoreceptor, but by a comparison of the activity in all the three types of cone photoreceptor (Chapter 9).
To understand the advantages of this type of memory storage, let’s consider an alternative in which the memory were encoded solely by neuron A: When it is active, Mark would be recalled. After learning, neuron A would become a “Mark detector” that certainly could store the memory of Mark, but what would happen if neuron A died from a bump on the head or some other mishap of normal life? Poof, there goes Mark. The distributed memory avoids this problem because no single neuron represents Mark; he is represented by the pattern of activity across all neurons in the cortical network. If neuron A met an early demise, there is still a unique pattern or ratio of activity in neurons B and C that can represent Mark. The more neurons there are in the network, the more unique memories can be stored and the more resistant the memories are to damage to individual neurons. This is a good thing, because although they are numerous, neurons in the brain die every day.
Using artificial neural network models created in the laboratory with a computer, researchers can ask what happens when neurons in the network are gradually removed. The answer is that memories show what is called graceful degradation. Instead of a catastrophic loss of any one memory, representations tend to blend together as neurons are lost, such that one memory gets confused with another. This type of memory loss is similar to what often happens in old age or following the death of a large number of neurons due to a brain disease.
Neural network models can reproduce the experimental observations of experience-dependent shifts in neuronal selectivity, thereby yielding insights into how memory is stored. As we have said, one such insight is that memories are distributed and show graceful degradation in response to a loss of neurons. Another key insight is that the physical change that leads to memory can be the modification of synaptic weight that changes the input–output relationships of neurons. Synapses store memories.
The notion of a synaptic basis for memory received strong experimental support from Eric Kandel’s studies of the marine snail Aplysia. Kandel and colleagues were able to show that simple forms of learning, such as habituation and sensitization, were accompanied by changes in the strength of synaptic transmission between sensory neurons and motor neurons. Moreover, they were able to dissect many of the molecular mechanisms that underlie these changes. These studies provided a strong foundation for subsequent analysis of synaptic modification in the mammalian brain (Box 25.1).
What Attracted Me to the Study of Learning and Memory in Aplysia?
There was little in my early life to indicate that the biology of mind would become the passion of my academic career. In fact, there was little to suggest that I would have an academic career. Rather, my early life was shaped in large part by the traumatic events that occurred in the place of my birth: Vienna, Austria.
I was born in November 1929. In March 1938, when I was eight years old, Hitler entered Austria and was received by the Viennese with enormous enthusiasm. Within hours, that enthusiasm turned into an almost indescribable outburst of anti-Semitic violence. After a humiliating and frightening year, my older brother, Ludwig, and I were able to leave Vienna in April 1939. The two of us crossed the Atlantic by ourselves to live with our grandparents in New York. Our parents joined us six months later.
The spectacle of Vienna under the Nazis presented me for the first time with the dark side of human behavior. How is one to understand the sudden viciousness of so many people? How could a highly cultivated society listen to Haydn, Mozart, and Beethoven one day and the next day embrace the brutality of Kristallnacht? This question still haunted and fascinated me while in college at Harvard, where I majored in twentieth-century history and literature. I wrote my honors dissertation on the attitude of three German writers toward National Socialism, and I intended to do graduate work in modern European intellectual history. But at the end of my junior year, I decided that to obtain insights into the human mind and its capability for good and evil, it would be better to become a psychoanalyst rather than an intellectual historian.
I entered medical school in the fall of 1952, dedicated to becoming a psychoanalyst. While in medical school I loved the clinical work but had no particular interest in basic science. In my senior year, however, I decided that perhaps even a New York psychoanalyst should know something about the brain, so I took an elective at Columbia University with the neurophysiologist Harry Grundfest.
In Grundfest’s laboratory I was astonished to discover that science in the laboratory is dramatically different from taking courses and reading books.
Knowing of my interest in behavior, Grundfest suggested that I set up an electrophysical system to record from the large axon of the crayfish, which controls the animal’s tail and thus its escape from predators. I learned how to manufacture glass microelectrodes for insertion into individual nerve cells of crayfish and how to obtain and interpret electrical recordings from them. It was in the course of those experiments, which were almost laboratory exercises, since I was not exploring new ground scientifically or conceptually, that I first began to feel the excitement of working on my own. Whenever I penetrated a cell, I, too, could hear the crack of an action potential. I am not fond of the sound of gunshots, but I found the “bang! bang! bang!” of action potentials intoxicating. The idea that I had successfully impaled a cell and was actually listening in on the brain of the crayfish as it conveyed messages seemed marvelously intimate. I was becoming a true psychoanalyst: I was listening to the deep, hidden thoughts of my crayfish!
Had I not been exposed to the excitement of actually doing research, of carrying out experiments to discover something new, I would have ended up with a very different career and, I presume, a very different life.
I began to realize that what makes science so distinctive is not just the experiments themselves but also the social context, the sense of equality between student and teacher, and the open, ongoing, and brutally frank exchange of ideas and criticism.
Based on my six-month stay in his laboratory, Grundfest nominated me for a research position at the National Institutes of Health. I arrived at the NIH in July 1957, just after Brenda Milner published her classic research showing that complex memories—for people, places, and objects—are localized in the hippocampus. I realized that the problems of memory storage, once the exclusive province of psychologists and psychoanalysts, were now approachable with the methods of cell biology. What are the cellular mechanisms for that storage? I wondered. No one knew anything about the nerve cells of the hippocampus at that time. I thought perhaps the nerve cells that participate in memory storage would have novel properties that would speak to me of memory!
Together with Alden Spencer, a young colleague from NIH, I set out to study the properties of hippocampal nerve cells. We were the first scientists in the world to record signals from those cells. Our work showed, surprisingly, that these cells from the region of the brain that encodes our dearest memories function pretty much the same way as other nerve cells in the brain. I now realized that these cells did not speak to us about memory. We had climbed Mt. Everest, but we had no view.
I further realized that to explore memory I would need to study not nerve cells per se, but nerve cells during a learning experience that leads to the formation of a memory. This was too difficult to do in a complex structure like the hippocampus: In the late 1950s, we did not even know what sensory input affected hippocampal cells. Alden and I tried visual, tactile, and auditory inputs, all without effect. I became convinced that to succeed in bringing the power of cell biology to bear on the study of learning and memory, I would first have to take a very different approach, a reductionist approach. My first step had to be to study not the most complex case but the simplest case of memory storage—and to study it in the simplest, most tractable experimental animal available.
While a reductionist strategy was within the realm of traditional biology, most investigators were reluctant to apply it to mental processes such as learning and memory. From the outset it seemed to me that the mechanisms of memory storage are so important for survival that they must have been conserved through evolution. Moreover, a molecular analysis of learning, no matter how simple the animal or the task, was likely to reveal those mechanisms.
I needed to develop an experimental system in which a simple reflex behavior, controlled by a small number of large, accessible nerve cells, could be modified by a simple form of learning like classical conditioning. Only then could I relate the animal’s overt learned behavior to cellular and molecular events occurring in the neurons that control the behavior.
After looking at crayfish, lobsters, worms, and flies, I settled on the marine snail Aplysia, which has extremely large nerve cells that are conducive to recording. One of the two people in the world working on Aplysia at that time was Ladislav Tauc, so I spent 1962–1963 in Paris with him, and I have worked on Aplysia ever since.
In the early 1960s we had no frame of reference for studying the biological basis of memory formation and storage. Two conflicting theories prevailed. One was the aggregate field approach, which assumed that information is stored in the bioelectric field generated by the aggregate activity of many neurons. The other was the cellular connectionist approach, which derived from Santiago Ramón y Cajal’s idea that memory is stored as an anatomical change in the strength of synaptic connections between nerve cells (Cajal, 1894). In 1948 Jerzy Konorski renamed Cajal’s concept “synaptic plasticity” (Konorski, 1948).
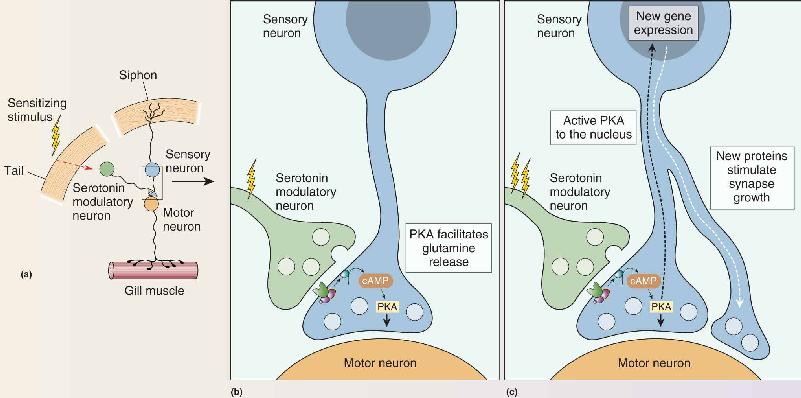
In my studies of Aplysia my work focused on the cellular substrates of the gill-withdrawal reflex that occurs when the siphon of the animal is touched (Figure A). This reflex undergoes sensitization (a simple form of learning) when a noxious stimulus is applied to the tail of the animal. I found that short-term memory results from a transient strengthening of preexisting synaptic connections, due to the modification of preexisting proteins, whereas long-term memory results from a persistent strengthening of synaptic connections brought about by alterations in gene expression, the synthesis of new proteins, and the growth of new synaptic connections. I discovered that the transient strengthening results in an increase in the amount of transmitter released by the sensory neuron onto the motor neuron that controls the gill musculature. This increase is produced by activation by the tail stimulus of serotonergic modulatory neurons (Figure B part a). Serotonin increases the strength of the synapse between sensory and motor neurons by increasing the concentration of cAMP, an intracellular signaling molecule in sensory neurons that activates protein kinase A (PKA). When we similarly simply injected cAMP directly into the sensory neuron, it resulted in an increase in the release of the transmitter (glutamate) into the synaptic cleft, thus temporarily strengthening the connection with the motor neuron (Figure B part b).

Figure A The gill-withdrawal reflex in Aplysia. (a) The mantle is held aside to show the gill in its normal position. (b) The gill retracts when water is sprayed on the siphon. Description

Figure B A mechanism for sensitization of the gill-withdrawal reflex. (a) A simple wiring diagram showing the minimal circuitry for sensitization of the gill withdrawal reflex. A noxious stimulus to the tail activates serotonergic modulatory neurons that influence synaptic transmission at the sensory–motor synapse. (b) Serotonin stimulates a rise in cAMP and activation of PKA in the sensory nerve terminal, causing an increase in the amount of glutamate released when the siphon is touched. (c) Repeated activation of the serotonergic modulatory neurons causes long-term sensitization, requiring new nuclear gene expression and protein synthesis. Description
Beginning in 1980, the insights and methods of molecular biology enabled us to identify common mechanisms of short-term memory in different animals and to explore how short-term memory is converted to long-term memory. We found that, following long-term sensitization, PKA moves into the nucleus and activates gene expression, leading to the synthesis of new proteins and a twofold increase in the number of synaptic connections made by Aplysia’s sensory neurons (Figure B part c). Moreover, the dendrites of the motor neurons, which receive the signals from the sensory neurons, grow and remodel to accommodate the additional sensory input.
Together, these early cellular studies of simple behaviors provided direct evidence supporting Cajal’s suggestion that synaptic connections between neurons are not immutable; they can be modified in learning, and those anatomical modifications are likely to subserve memory storage. In the gill-withdrawal reflex of Aplysia, changes in synaptic strength occur not only in the connections between sensory and motor neurons but also in the connections between sensory neurons and interneurons. Thus, even in a simple reflex, memory appears to be distributed among multiple sites. Studies showed further that a single synaptic connection is capable of being modified in opposite ways by different forms of learning and for different periods of time, paralleling the different stages of memory.
By 1980 my progress on Aplysia had been so heartening that I summoned up the courage to return to the hippocampus. There I found, much as Charles Darwin might have predicted, that once nature finds a solution that works, it tends to hold on to it. In other words, the same general principles that govern short- and long-term memory storage in simple animals also apply to complex ones.
Cajal SR. 1894. The Croonian Lecture: la fine structure des centres nerveux. Proceedings of the Royal Society, London 55:344–468.
Konorski J. 1948. Conditioned Reflexes and Neuron Organization. Cambridge, MA: University Press.
Consideration of neural network models, such as the one shown in Figure 25.4, indicates that both increases and decreases in synaptic weights can shift neuronal selectivity and store information. We begin our discussion of how this synaptic plasticity occurs with long-term potentiation (LTP), originally discovered in the hippocampus, a brain region critical for memory formation. (LTP was also discussed in the context of brain development in Chapter 23.)
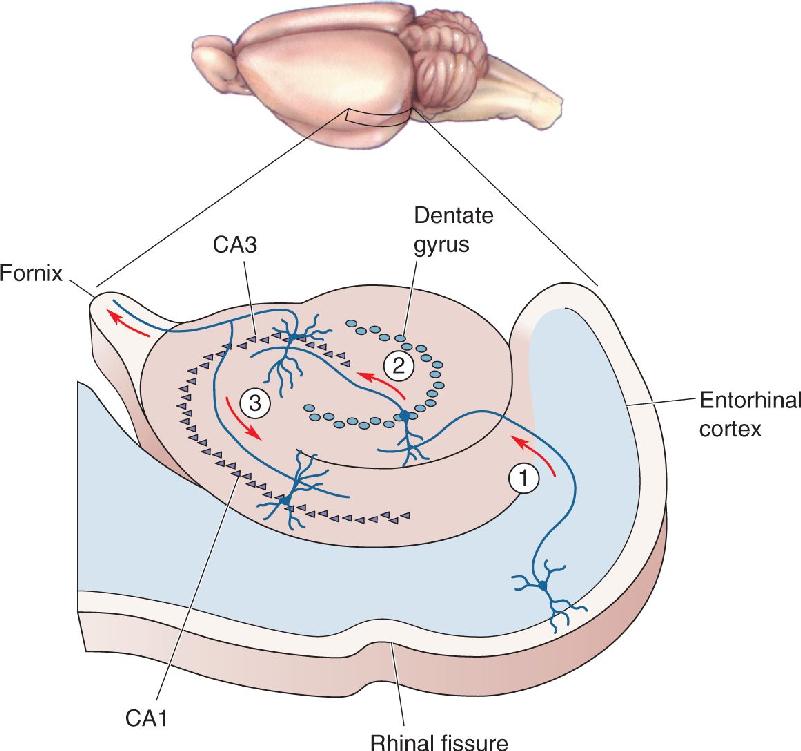
Anatomy of the Hippocampus. The hippocampus consists of two thin sheets of neurons folded onto each other. One sheet is called the dentate gyrus, and the other Ammon’s horn. Of the four divisions of Ammon’s horn, we will focus on two: CA3 and CA1 (CA stands for cornu Ammonis, Latin for “Ammon’s horn”).
Recall from Chapter 24 that a major input to the hippocampus is the entorhinal cortex. The entorhinal cortex sends information to the hippocampus by way of a bundle of axons called the perforant path. Perforant path axons synapse on neurons of the dentate gyrus. Dentate gyrus neurons give rise to axons (called mossy fibers) that synapse on cells in CA3. The CA3 cells give rise to axons that branch. One branch leaves the hippocampus via the fornix. The other branch, called the Schaffer collateral, forms synapses on the neurons of CA1. These connections, summarized in Figure 25.5, are sometimes called the trisynaptic circuit, because three sets of synaptic connections are involved:

FIGURE 25.5 Some microcircuits of the hippocampus. ① Information flows from the entorhinal cortex via the perforant path to the dentate gyrus. ② The dentate gyrus granule cells emit axons called mossy fibers that synapse upon pyramidal neurons in area CA3. ③ Axons from the CA3 neurons, called the Schaffer collaterals, synapse upon pyramidal neurons in area CA1. Description
- Entorhinal cortex → dentate gyrus (perforant path) synapses
Because of its very simple architecture and organization, the hippocampus is an ideal place to study synaptic transmission in the mammalian brain. In the late 1960s, researchers discovered that the hippocampus could actually be removed from the brain (usually in experimental animals) and cut up like a loaf of bread, and that the resulting slices could be kept alive in vitro for many hours. In such a brain slice preparation, fiber tracts can be stimulated electrically and synaptic responses recorded. Because cells in the slice can be observed, stimulating and recording electrodes can be positioned with the precision previously available only in invertebrate preparations. This brain slice preparation has greatly facilitated the study of LTP.
Properties of LTP in CA1. In 1973, an important discovery was made in the hippocampus by Timothy Bliss and Terje Lømo, working together in Norway. They found that brief, high-frequency electrical stimulation of the perforant path synapses on the neurons of the dentate gyrus produced LTP. It was subsequently shown that most excitatory (and many inhibitory) synapses support LTP, and that the mechanisms can vary from one synapse type to another. However, the most sophisticated understanding of LTP has come from studying the Schaffer collateral synapses on the CA1 pyramidal neurons in brain slice preparations. This will be our focus.
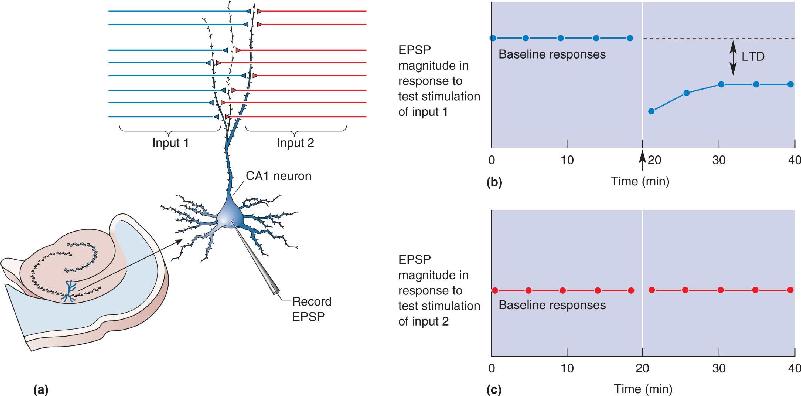
In a typical experiment, the effectiveness of the Schaffer collateral synapse is monitored by giving a bundle of presynaptic axons a brief electrical stimulus, then measuring the size of the resulting EPSP in a postsynaptic CA1 neuron (Figure 25.6). Usually such a test stimulation is given every minute or so for 15–30 minutes to ensure that the baseline response is stable. Next, to induce LTP, the same axons are given a tetanus, a brief burst of high-frequency stimulation (typically 50–100 stimuli at a rate of 100/sec). Usually this tetanus induces LTP, and subsequent test stimulation evokes an EPSP that is much greater than it was during the initial baseline period. In other words, the tetanus has caused a modification of the stimulated synapses so they are more effective. Other synaptic inputs onto the same neuron that did not receive tetanic stimulation do not show LTP. This property, that only the active inputs show the synaptic plasticity, is called input specificity.

FIGURE 25.6 Long-term potentiation in CA1. (a) The response of a CA1 neuron is monitored as two inputs are alternately stimulated. LTP is induced in input 1 by giving this input a tetanus. (b) The graph shows a record of the experiment. The tetanus to input 1 (arrow) yields a potentiated response to stimulation of this input. (c) LTP is input-specific, so there is no change in the response to input 2 after a tetanus to input 1. Description
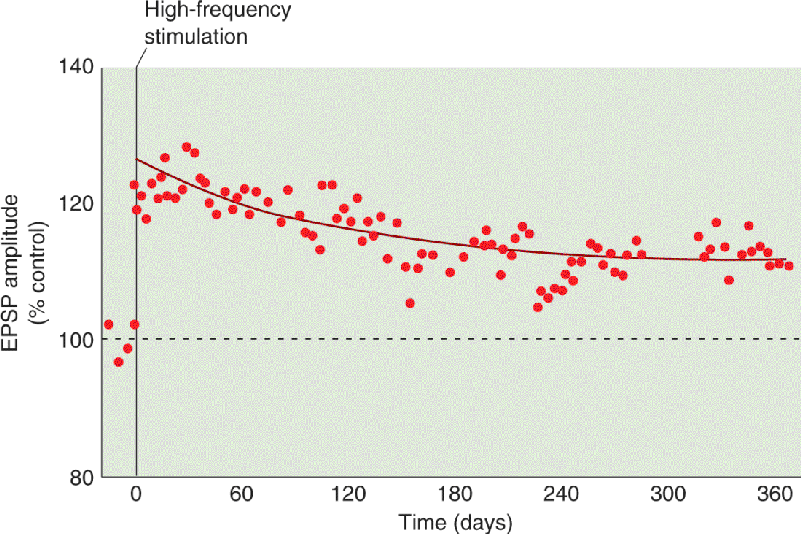
One remarkable feature of this plasticity is that it can be induced by a brief tetanus, lasting less than a second, consisting of stimulation at frequencies well within the range of normal axon firing. A second remarkable feature of LTP is its longevity. LTP induced in CA1 of awake animals can last many weeks, possibly even a lifetime (Figure 25.7). No wonder this form of synaptic plasticity has attracted interest as a candidate mechanism for declarative memory.

FIGURE 25.7 LTP can last a long, long time. In this experiment, LTP was induced with tetanic stimulation using electrodes implanted into the hippocampus of an awake rat. Each data point is the amplitude of the EPSP evoked with electrical stimulation of the synapses that had been tetanized. The LTP was still evident a year later. (Source: Adapted from Abraham et al., 2002.) Description
Subsequent research has shown that high-frequency stimulation is not an absolute requirement for LTP. Rather, what is required is that synapses be active at the same time that the postsynaptic CA1 neuron is strongly depolarized. In order to achieve the necessary depolarization with a tetanus, (1) synapses must be stimulated at frequencies high enough to cause temporal summation of the EPSPs, and (2) enough synapses must be active simultaneously to cause significant spatial summation of EPSPs. This second requirement is called cooperativity, because coactive synapses must cooperate to produce enough depolarization to cause LTP.
Consider for a moment how the cooperativity property of hippocampal LTP could be used to form associations. Imagine a hippocampal neuron receiving synaptic inputs from three sources: I, II, and III. Initially, no single input is strong enough to evoke an action potential in the postsynaptic neuron. Now imagine that inputs I and II repeatedly fire at the same time. Because of spatial summation, inputs I and II are now capable of firing the postsynaptic neuron and of causing LTP. Only the active synapses will be potentiated, and these, of course, are those belonging to inputs I and II. Now, because of potentiation of their synapses, either input I or input II can fire the postsynaptic neuron (but not input III). Thus, LTP has caused an association of inputs I and II. In this way, the sight of a duck could be associated with the quack of a duck (they often occur at the same time), but never with the bark of a dog.
Speaking of associations, remember the idea of a Hebb synapse, introduced in Chapter 23, to account for aspects of visual development? LTP in CA1 is Hebbian: Inputs that fire together wire together.
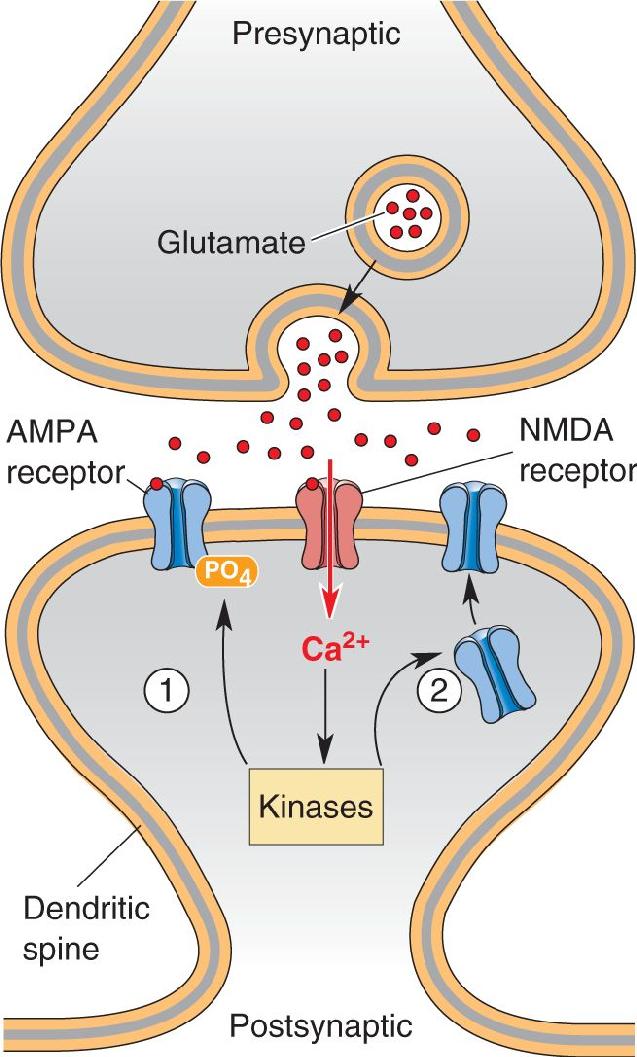
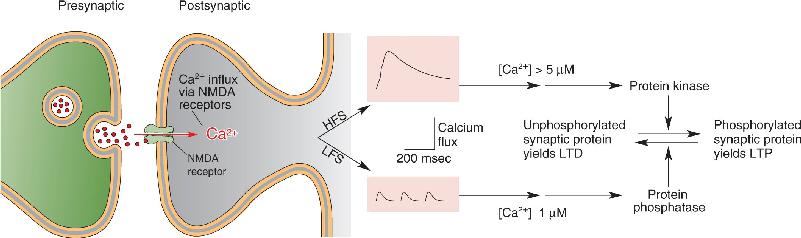
Mechanisms of LTP in CA1. Excitatory synaptic transmission in the hippocampus is mediated by glutamate receptors. Na+ ions passing through the AMPA subclass of glutamate receptor cause the EPSP at the Schaffer collateral–CA1 pyramidal cell synapse. However, CA1 neurons also have postsynaptic NMDA receptors. Recall that these glutamate receptors have the unusual property that they conduct Ca2+ ions, but only when glutamate binds and the postsynaptic membrane is depolarized enough to displace Mg2+ ions that clog the channel (Figure 25.8). Thus, Ca2+ entry through the NMDA receptor specifically signals when presynaptic and postsynaptic elements are active at the same time (Box 25.2).

FIGURE 25.8 NMDA receptors activated by simultaneous presynaptic and postsynaptic activity. (a) Presynaptic activation causes the release of glutamate, which acts on postsynaptic AMPA receptors and NMDA receptors. At the negative resting membrane potential, the NMDA receptors pass little ionic current because they are blocked with Mg2+. (b) When glutamate release coincides with depolarization sufficient to displace the Mg2+, then Ca2+ enters the postsynaptic neuron via the NMDA receptor. Description
When enough synapses are active at the same time, the postsynaptic neuron will be depolarized sufficiently to fire an action potential. Donald Hebb proposed that each individual synapse grows a little stronger when it successfully participates in the firing of the postsynaptic neuron. The phenomenon of LTP comes close to satisfying Hebb’s ideal. The synapse gets stronger when the glutamate released by the presynaptic terminal binds to postsynaptic NMDA receptors and the postsynaptic membrane is depolarized strongly enough to displace Mg2+ from the NMDA receptor channel.
Is there a role for postsynaptic action potentials in this “strong” depolarization? The first evidence that appropriate timing of a postsynaptic action potential might be important for LTP was obtained in the early 1980s by William Levy and Oswald Steward at the University of Virginia. They found that LTP occurred if a postsynaptic action potential occurred simultaneously with, or slightly after, presynaptic release of glutamate. However, action potentials are generated in the soma in response to depolarization of the membrane beyond threshold. Because this happens far away from the synapses located out on the dendritic tree, it was assumed for a time that the actual occurrence of the spike was not important for the mechanism of synaptic potentiation. The important thing was the strong depolarization in the dendrite, due to summed synaptic currents, which, coincidentally, was also usually sufficient to evoke a postsynaptic action potential.
While it remains true that the key is strong postsynaptic depolarization, researchers took another look at the role of the postsynaptic spike in LTP. This new attention resulted from the discovery that action potentials generated in the soma can actually “back-propagate” into the dendrites of some cells. Thus, Henry Markram, Bert Sakmann, and their colleagues at the Max Planck Institute investigated what happens when a postsynaptic spike is generated (via a microelectrode) at various time intervals before or after an EPSP. Remarkably, they found that if a postsynaptic action potential follows the EPSP within about 50 msec, the synapse potentiates. Nothing happens in response to the spike or the EPSP alone; LTP results specifically from the precise timing of EPSP and spike, just as Hebb suggested! In addition, the timing requirements for LTP in these studies agreed very well with those originally reported by Levy and Steward. This is an example of what is now referred to as spike timing–dependent plasticity.
What accounts for the LTP-promoting effect of a back-propagating action potential? The answer, of course, is strong depolarization. NMDA receptors have a very high affinity for glutamate, so the transmitter remains bound to the receptor for many tens of milliseconds. However, this bound glutamate does nothing if the postsynaptic membrane is not depolarized strongly, because the channel is clogged with Mg2+. The timely occurrence of the action potential is sufficient to awaken these dormant channels by ejecting the Mg2+. Then, as long as glutamate is still bound to the receptor, Ca2+ will enter the cell and trigger the mechanism of LTP.
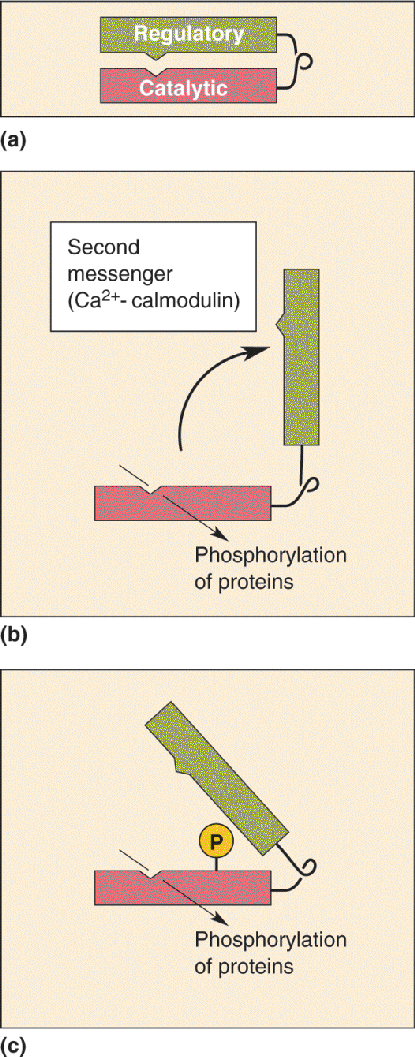
Considerable evidence now links this rise in postsynaptic [Ca2+]i to the induction of LTP. For example, LTP induction is prevented if NMDA receptors are pharmacologically inhibited, or if rises in postsynaptic [Ca2+]i are prevented by the injection of a Ca2+ chelator into the postsynaptic neuron. The rise in [Ca2+]i activates two protein kinases: protein kinase C and calcium-calmodulin-dependent protein kinase II, also known as CaMKII (pronounced “cam-K-two”). Recall from Chapters 5 and 6 that protein kinases regulate other proteins by phosphorylating (attaching phosphate groups to) them.
Following the rise in postsynaptic [Ca2+]i and the activation of the kinases, the molecular trail that leads to a potentiated synapse gets harder to follow. Current research suggests that this trail may actually branch (Figure 25.9). One path appears to lead toward an increased effectiveness of existing postsynaptic AMPA receptors by way of phosphorylation. Phosphorylation of the AMPA receptor, by either protein kinase C or CaMKII, leads to a change in the protein that increases the ionic conductance of the channel. The other path leads to the insertion of entirely new AMPA receptors into the postsynaptic membrane. According to a current model, vesicular organelles studded with AMPA receptors lie in wait near the postsynaptic membrane. In response to CaMKII activation, the vesicle membrane fuses with the postsynaptic membrane, and the new AMPA receptors are thereby delivered to the synapse. This addition of new membrane causes the spines to swell (Figure 25.10).

FIGURE 25.9 Routes for the expression of LTP in CA1. Ca2+ entering through the NMDA receptor activates protein kinases. This can cause LTP by ① changing the effectiveness of existing postsynaptic AMPA receptors or ② stimulating the insertion of new AMPA receptors. Description

FIGURE 25.10 The growth of spines following LTP. A segment of dendrite was filled with a fluorescent dye and imaged in living tissue using a special microscope. After LTP, the spines grew and sometimes sprouted to accommodate new synapses. Each frame is a snapshot of the dendrite at a different time, indicated in the upper-right corner (in minutes). At the time marked 0 min, the yellow dot indicates that this spine was repetitively activated with glutamate to induce LTP. After LTP, the spine grew to accommodate more AMPA receptors. (Photos courtesy of Dr. Miquel Bosch, Massachusetts Institute of Technology.)
Evidence also indicates that synaptic structure changes following LTP. In particular, postsynaptic dendritic spines appear to bud and form new synaptic contacts with axons. Thus, following LTP, a single axon can make multiple synapses on the same postsynaptic neuron, which is not the normal pattern in CA1. This sprouting of synapses not only increases the responsive postsynaptic surface but also increases the probability that an action potential in the axon will trigger presynaptic glutamate release.
We have seen from our simple neural network model in Figure 25.4 that information can be stored as both decreases and increases in synaptic effectiveness. Recall Hebb’s theory that a synapse grows stronger, or potentiates, when the activity of that synapse correlates with the strong activation of the postsynaptic neuron by other converging inputs. An extension of Hebb’s theory, designed to account for bidirectional (up and down) regulation of synaptic strength, is called the BCM theory, named for its authors, Elie Bienenstock, Leon Cooper, and Paul Munro, working at Brown University.
After sharing the 1972 Nobel Prize in Physics for his development of a theory of superconductivity, Cooper became interested in the problem of memory storage by large networks of neurons (Box 25.3). He and his students Bienenstock and Munro recognized that experience-dependent changes in neuronal stimulus selectivity reflect the synaptic modifications that store memory in neural networks, and they devised a synaptic “learning rule” to account for how synapses potentiate and depress as the sensory environment is changed. A key assumption of the BCM theory, published in 1982, is that synapses will undergo synaptic weakening instead of LTP when they are active at the same time the postsynaptic cell is only weakly depolarized by other inputs. This idea inspired a search for long-term synaptic depression in hippocampal area CA1, using stimuli that were designed to evoke a modest postsynaptic response. In 1992 Serena Dudek and Mark Bear, working together at Brown University, showed that tetanic stimulation of the Schaffer collaterals at low frequencies (1–5 Hz) indeed produces synaptic weakening (Figure 25.11). Because it occurs only at the stimulated synapses, it is often referred to as homosynaptic long-term depression (LTD).

FIGURE 25.11 Homosynaptic LTD in the hippocampus. (a) The response of a CA1 neuron is monitored as two inputs are alternately stimulated. LTD is induced in input 1 by giving this input a 1 Hz tetanus. (b) The graph shows a record of the experiment. The low-frequency tetanus to input 1 (arrow) yields a depressed response to stimulation of this input. (c) LTD is input-specific, so there is no change in the response to input 2 after a tetanus to input 1. Description
I have been asked many times: “What led you from physics to neuroscience?” The best I can come up with is to repeat Humphrey Bogart’s response to Claude Rains in Casablanca: “I was misinformed.” After the publication of our theory on superconductivity, I worked on other “many-electron” problems. I came to believe that the mathematical techniques used there could be applied to “many-neuron” problems. While some of them did prove useful, for the most part they were irrelevant. But what was most useful, perhaps, was my conviction that theory, essential in the physical sciences, is essential in neuroscience as well.
So when I strayed from the lofty realm of theoretical physics to the earthy problem of the brain, my first effort was to attempt to construct networks of neurons that would display some of the qualitative features associated with what I called animal memory. Initial success was the seduction that lured me further into this exotic domain. It is this journey that I will describe.
We realized in the early 1970s that networks of neurons can form distributed representations of the world that are “associative” (recollection of one memory can lead to the recollection of another linked to it by experience) and “content addressable” (memories are accessed by content rather than by a physical address in the network). Such representations are resistant to the loss of individual neurons and synapses and thus provide a candidate substrate for memory storage in the animal brain. But how can these representations be constructed in networks of neurons? That is, how can the strengths of the vast numbers of synapses that make up neuronal networks be adjusted to obtain a mapping that corresponds to an appropriate memory?
This might result if synaptic modification (or learning) follows the famous Hebbian rule. But Hebbian synaptic modification needs stabilization. Elie Bienenstock, Paul Munro, and I therefore proposed in 1982 a form of bidirectional synaptic modification that combines Hebbian modification—synaptic potentiation that occurs when pre- and postsynaptic neurons are both strongly activated—with “anti-Hebbian” synaptic weakening that occurs when presynaptic activity happens in the absence of a strong postsynaptic response. We further proposed that the critical level of postsynaptic response at which the polarity of synaptic modification changes from weakening to strengthening, called the “modification threshold,” varies according to the history of postsynaptic cell activity. Together, these assumptions lead to stabilization and various other desirable properties. The resulting theory has become known as BCM synaptic modification.
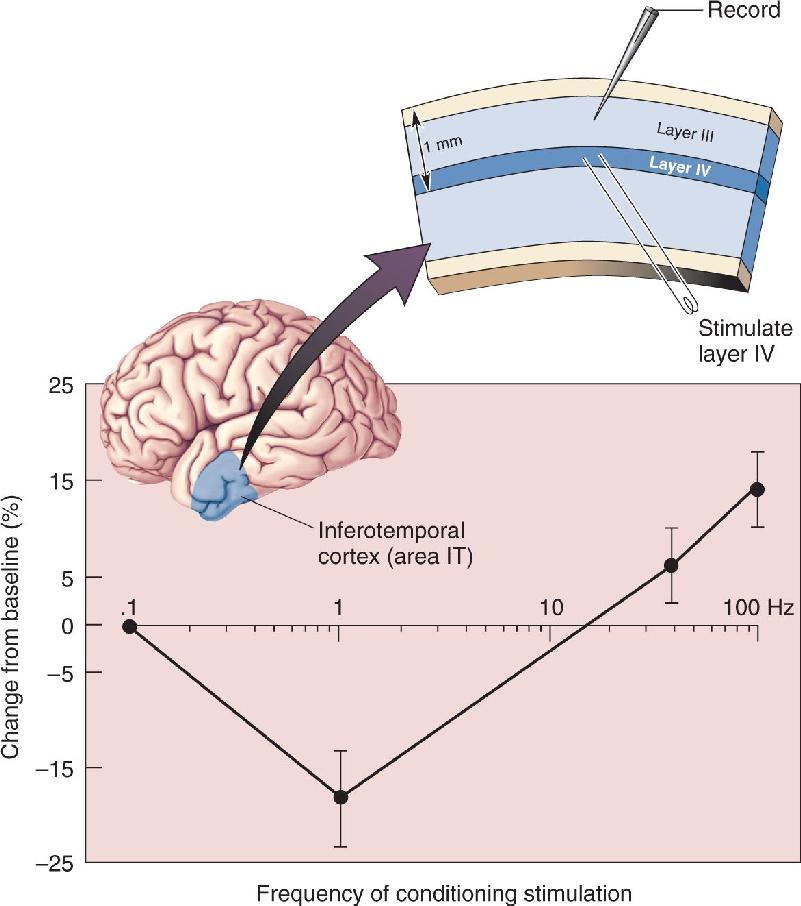
In the late 1980s I began a long and very fruitful collaboration with Mark Bear who was then also at Brown. Mark and his students performed experiments to test the validity of BCM assumptions at excitatory glutamatergic synapses in the cerebral cortex. The shape of the BCM synaptic modification function was first confirmed by Dudek and Bear (1992) in hippocampus, and Kirkwood and Bear (1994) showed that the result was the same in visual cortex. Since then, similar findings have been confirmed in many different regions of neocortex in many species of both young and old animals. Of particular interest are data showing that the same principles of synaptic plasticity apply in the human inferotemporal cortex, a region believed to be a repository of visual memories. Together, the data support the idea that very similar principles guide synaptic plasticity in many species in widely different regions of the brain.
According to BCM, the modification threshold, θm, must vary depending on the history of postsynaptic cortical activity. An experimental test of this hypothesis was first reported by Kirkwood, Marc Rioult, and Bear (1996). They compared the synaptic modification function in the visual cortex of normal animals with that in the visual cortex of animals reared in complete darkness and found a shift of this function in accordance with the theoretical postulate. Elizabeth Quinlan, Ben Philpot, and Bear, in collaboration with Richard Huganir at Johns Hopkins School of Medicine, went on to show in 1999 that the ratio of two distinct subunits of the cortical NMDA receptor is set according to the activation history of the cortex, providing a potential mechanism for the sliding modification threshold.
Consequences of BCM synaptic modification in networks modeled after visual cortex have been shown by my students Nathan Intrator, Harel Shouval, Brian Blais, and many others, using analysis and simulations, to be in agreement with experimental observations on the shifts in neuronal selectivity that have been observed by rearing animals in various visual environments. Thus, the BCM theory provides a bridge between the molecular mechanisms of synaptic modification and the systems-level properties of distributed information storage.
Given the level of skepticism displayed when synaptic modification ideas were discussed 40 years ago, I think it is reasonable to say that we have made considerable progress. Our initial aim to build a theoretical structure relevant to a fundamental brain process that was sufficiently concrete so that it could be tested by experiment has been accomplished. It is particularly gratifying that theory has inspired experiments that, in addition to confirming the various postulates and predictions of our theory, have led to the discovery of new phenomena such as homosynaptic long-term depression and metaplasticity. Possibly most important, we have an excellent example of the fruitful interaction of theory with experiment in neuroscience.
Bienenstock EL, Cooper LN, Munro PW. 1982. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. Journal of Neuroscience 2:32–48.
Blais B, Cooper LN, Shouval H. 2000. Formation of direction selectivity in natural scene environments. Neural Computation 12:1057–1066.
Blais BS, Intrator N, Shouval HZ, Cooper LN. 1998. Receptive field formation in natural scene environments: comparison of single-cell learning rules. Neural Computation 10:1797–1813.
Dudek SM, Bear MF. 1992. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proceedings of the National Academy of Sciences USA 89:4363–4367.
Kirkwood A, Bear MF. 1994. Homosynaptic long-term depression in the visual cortex. Journal of Neuroscience 14:3404–3412.
Kirkwood A, Rioult MC, Bear MF. 1996. Experience-dependent modification of synaptic plasticity in visual cortex. Nature 381:526–528.
Quinlan EM, Philpot BD, Huganir RL, Bear MF. 1999. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nature Neuroscience 2:352–357.
Shouval H, Intrator N, Cooper LN. 1997. BCM network develops orientation selectivity and ocular dominance in natural scene environment. Vision Research 37:3339–3342.
It has now been established experimentally that bidirectional plasticity of many cortical synapses is indeed governed by two simple rules:
- Synaptic transmission occurring at the same time as strong depolarization of the postsynaptic neuron causes LTP of the active synapses.
- Synaptic transmission occurring at the same time as weak or modest depolarization of the postsynaptic neuron causes LTD of the active synapses.
Although these rules apply to many cortical synapses, it is important to appreciate that LTD is a widespread form of synaptic plasticity. The properties and mechanisms of LTD vary from one synapse type to the next (Box 25.4). At some synapses, the timing of pre- and postsynaptic actions potentials is a key variable. As discussed in Box 25.2, LTP can result when the EPSP caused by synaptic glutamate release precedes an action potential in the postsynaptic neuron; this is an example of spike timing–dependent plasticity. At many of these same synapses, LTD can result instead when the EPSP caused by glutamate release follows a postsynaptic action potential (Figure 25.12).

FIGURE 25.12 Spike timing–dependent plasticity. When postsynaptic spikes consistently follow the EPSPs induced by presynaptic spiking, the synapse grows stronger. However, when the postsynaptic spikes consistently precede the EPSPs, the synapse grows weaker. This graph relates the change in synaptic strength to the relative timing difference. Description
We saw in Chapter 14 that the cerebellum is important for learning and remembering motor skills. The unusual circuitry of the cerebellar cortex suggested to David Marr at the University of Cambridge how this learning might occur. The output of the cerebellar cortex arises from large neurons called Purkinje cells, and these cells receive two converging inputs. Each Purkinje cell receives input from a single climbing fiber that arises from a nucleus in the medulla called the inferior olive. The climbing fiber synapses are very powerful and always cause the Purkinje cell to fire action potentials. Parallel fibers arising from cerebellar granule cells provide the second input, and the organization is very different. Each Purkinje cell receives weak parallel fiber synapses from as many as 100,000 different granule cells. Marr proposed that this unusual convergence of parallel and climbing fiber inputs onto Purkinje cell dendrites serves motor learning. He proposed that (1) the climbing fiber input carries error signals indicating a movement has failed to meet expectations, and (2) corrections are made by adjusting the effectiveness of the parallel fiber inputs to the Purkinje cell. The theory was modified by James Albus at the Goddard Space Flight Center in Greenbelt, Maryland, to explicitly predict LTD of the parallel fiber synapse if it is active at the same time as the climbing fiber input to the postsynaptic Purkinje cell.
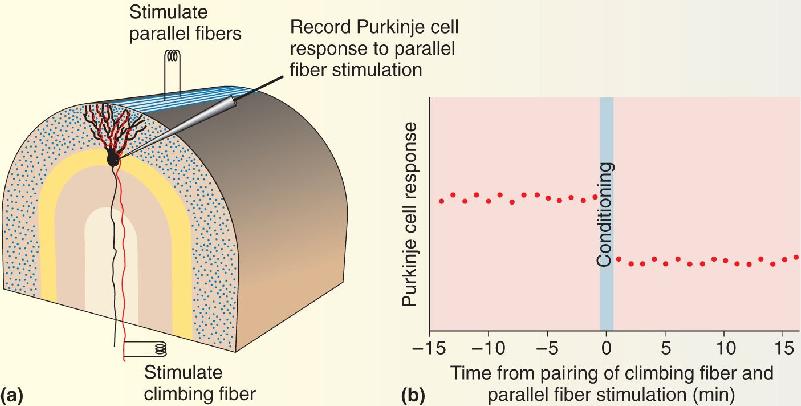
Masao Ito and his colleagues at the University of Tokyo tested this idea by pairing electrical stimulation of the climbing fibers with stimulation of the parallel fibers. Remarkably, they found that after this pairing procedure, activation of the parallel fibers alone resulted in a smaller postsynaptic response in the Purkinje cell (Figure A). It is now understood that the requirements for induction of this form of LTD are a large surge in postsynaptic [Ca2+] caused by activation of the climbing fiber, coincident with activation of metabotropic glutamate receptor 1 (mGluR1) by the parallel fibers. This conjunction triggers the internalization of AMPA receptors and the depression of synaptic transmission at the parallel fiber synapses. A mechanistically similar form of mGluR-dependent LTD was later discovered in the hippocampus, although it does not require a Ca2+ surge.

Figure A Cerebellar LTD. (a) The experimental arrangement for demonstrating LTD. The magnitude of the Purkinje cell response to stimulation of a “beam” of parallel fibers is monitored. Conditioning involves pairing parallel fiber stimulation with climbing fiber stimulation. (b) A graph of an experiment performed in this way. After the pairing, LTD of the response to parallel fiber stimulation results. Description
At other synapses in the brain, activation of mGluRs triggers LTD by a different mechanism altogether. For example, in the nucleus accumbens, activation of postsynaptic mGluR5 stimulates the synthesis of endocannabinoids, which travel retrogradely to the presynaptic terminal and cause a persistent depression of glutamate release. (Endocannabinoids were introduced in Chapter 6; see Box 6.2.)
Yet another LTD variant has been observed in the neocortex. Endocannabinoids in some neocortical pyramidal neurons are released in response to dendritic action potentials. If these endocannabinoids impinge on glutamatergic axon terminals at the same time they are releasing glutamate, then these synapses will be depressed. This mechanism yields a timing requirement on LTD, such that it is induced when a postsynaptic spike (causing the release of endocannabinoids) precedes the presynaptic spike by a few tens of milliseconds.
Each mechanism for LTD imposes different rules on the patterns of activity that yield synaptic plasticity. We can speculate that these have evolved to optimize the contribution of synaptic plasticity to the functions of different brain circuits.
As is the case for LTP, we know most about the mechanism of homosynaptic LTD in area CA1 of the hippocampus, so this will be our focus.
Mechanisms of LTD in CA1. At the Schaffer collateral–CA1 synapse, two distinct forms of homosynaptic LTD have been described. The first one to be discovered depends on activation of the NMDA receptor. The second form, discovered a few years later, requires activation of G-protein coupled metabotropic glutamate receptors (mGluRs). Here we will focus on NMDA receptor-dependent LTD.
Because NMDA receptors admit Ca2+ to the postsynaptic neuron, it came as no surprise that a rise in postsynaptic [Ca2+] is necessary to trigger LTD. But how can the same signal, Ca2+ entry through the NMDA receptor, trigger both LTP and LTD? The key difference lies in the level of NMDA receptor activation (Figure 25.13). When the postsynaptic neuron is only weakly depolarized, the partial blocking of the NMDA receptor channels by Mg2+ prevents all but a trickle of Ca2+ into the postsynaptic neuron. On the other hand, when the postsynaptic neuron is strongly depolarized, the Mg2+ block is displaced entirely, and Ca2+ floods into the postsynaptic neuron. These different types of Ca2+ response selectively activate different types of enzymes. Instead of the kinases that are activated by high [Ca2+]i, modest and prolonged elevations in [Ca2+]i activate protein phosphatases, enzymes that pluck phosphate groups off proteins. Therefore, if LTP is putting phosphate groups on, LTD apparently is taking them off. Indeed, biochemical evidence now indicates that AMPA receptors are dephosphorylated in response to stimulation that induces LTD (Figure 25.14). Moreover, the induction of hippocampal LTD can also be associated with the internalization of AMPA receptors at the synapse. Thus, LTP and LTD appear to reflect the bidirectional regulation of both the phosphorylation and the number of postsynaptic AMPA receptors.

FIGURE 25.13 NMDA receptor activation and bidirectional synaptic plasticity. The long-term change in synaptic transmission is graphed as a function of the level of NMDA receptor activation during conditioning stimulation. The level of NMDA receptor activation at which the polarity of synaptic modification switches from LTD to LTP is called the modification threshold. Description

FIGURE 25.14 A model for how Ca2+ can trigger both LTP and LTD in the hippocampus. High-frequency stimulation (HFS) yields LTP by causing a large elevation of [Ca2+]. Low-frequency stimulation (LFS) yields LTD by causing a smaller elevation of [Ca2+]. (Source: Adapted from Bear and Malenka, 1994, Fig. 1.) Description
Glutamate Receptor Trafficking. Besides revealing much about the likely synaptic basis for learning and memory, studies of LTP and LTD have led to a much deeper understanding of how synaptic transmission is maintained in the brain. Current research suggests that AMPA receptors in the postsynaptic membrane are continually being added and removed even in the absence of synaptic activity. Researchers estimate that half the synaptic AMPA receptors are replaced every 15 minutes! However, despite this remarkable turnover, synaptic transmission will remain stable as long as one receptor is added whenever one receptor is removed. LTP and LTD disrupt this equilibrium, leading to a net increase or decrease in the capacity of the synaptic membrane for AMPA receptors.
The capacity of the postsynaptic membrane is determined by the size of a scaffold of what has been termed slot proteins. Imagine the scaffold is like an egg carton, and the slot proteins form each of the egg cups. AMPA receptors are the eggs that fill the carton. As long as the size of the carton doesn’t change, synaptic transmission is stable even if the individual eggs are continually replaced (Figure 25.15).

FIGURE 25.15 An egg carton model of AMPA receptor trafficking at the synapse. Each egg represents an AMPA receptor, and the carton represents the capacity of the synapse for receptors, which might be determined by the amount of PSD-95. (a) The initial steady state. Each AMPA receptor that is removed is replaced with a new receptor. (b) LTP. More PSD-95 is added, increasing the synaptic capacity for AMPA receptors. The new receptors (blue) contain the GluR1 subunit. (c) The new steady state. Over time, ongoing turnover of receptors replaces those with GluR1. (d) LTD. Some PSD-95 is destroyed, decreasing the synaptic capacity for AMPA receptors. (e) The new steady state following LTD. Description
Stable LTP requires increasing the size of the carton and providing new eggs. The details of how this occurs at a molecular level remain an active area of research, and today’s conclusions may be overturned by tomorrow’s experiments. However, there is evidence that a protein called PSD-95 (a postsynaptic density protein with a molecular weight of 95 kilodaltons) may comprise the egg carton. Increasing the expression of PSD-95 in neurons increases the synaptic capacity for AMPA receptors. In addition, there is evidence that the new eggs may be AMPA receptors that contain a distinctive subunit called GluR1. LTP can selectively increase in the number of GluR1-containing AMPA receptors in the membrane. Over time, these receptors are replaced by those that lack GluR1. By analogy, imagine the neuron contains a stash of blue Easter eggs that can be delivered to the carton in response to LTP-inducing stimulation. Over time, the blue eggs are replaced by uncolored eggs. But because the size of the carton is increased, there continues to be a net increase in the number of eggs.
Conversely, stable LTD requires reducing the size of the egg carton, which reduces the capacity for eggs. Indeed, research has shown that LTD-inducing stimulation leads to both destruction of PSD-95 and a net loss of AMPA receptors from the postsynaptic membrane.
LTP and LTD have attracted a lot of interest because theoretical work shows that these mechanisms of synaptic plasticity can contribute to the formation of declarative memories. Recent research indicates that the types of NMDA receptor-dependent synaptic plasticity that have been characterized in the hippocampus also occur throughout the neocortex, including area IT where memories of familiar faces are created (Figure 25.16). It appears that plasticity at many synapses in the cerebral cortex may be governed by similar rules and might use similar mechanisms. (But remember, there are many exceptions to these “rules,” and they do not apply to all synapses, even within a single structure.)

FIGURE 25.16 Bidirectional synaptic modifications in human area IT. Slices of human temporal cortex, removed during the course of surgery to gain access to deeper structures, were maintained in vitro. Synaptic responses were monitored following various types of tetanic stimulation. As in rat CA1, stimulation of 1 Hz produced LTD, while 100 Hz stimulation produced LTP. (Source: Adapted from Chen et al., 1996.) Description
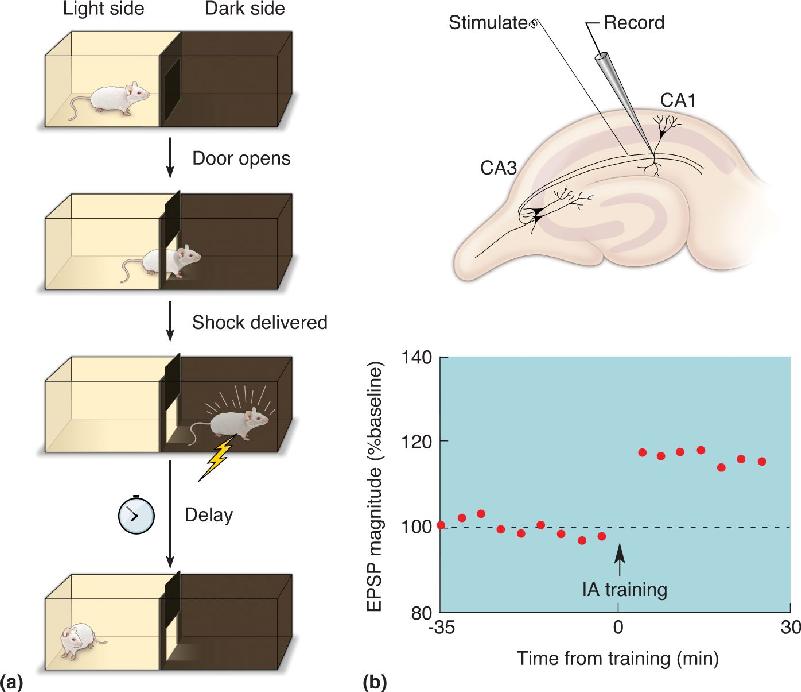
LTP and LTD are clearly appealing models, but what evidence links them to memory? So far, all we’ve described is a possible neural basis for a memory of having one’s brain electrically stimulated! One approach has been to insert stimulating and recording electrodes in the hippocampus and use these to monitor the state of synaptic transmission during learning. Because of the distributed nature of memory, success with this approach required the use of a particularly robust type of learning called inhibitory avoidance. In this experiment, a rat learns to associate a place (the dark side of a box) with an aversive experience (a foot shock) (Figure 25.17a). Animals of all types (from flies to humans) will learn to avoid the place they received the shock after only one trial (depending, of course, on the strength of the shock). This type of learning is not subtle, and neither are the patterns of hippocampal activation it produces. The widespread activation of the hippocampus after inhibitory avoidance training gave researchers the opportunity to detect changes in synaptic transmission at Schaffer collateral–CA1 synapses, and voilà!—LTP was observed (Figure 25.17b). In other experiments, exposing animals to a novel environment without a foot shock caused LTD instead. These experiments tell us that learning does indeed induce LTP and LTD at hippocampal synapses.

FIGURE 25.17 LTP in CA1 induced by learning. In this experiment, electrodes were implanted in the rat’s hippocampus to monitor the strength of synaptic transmission before and after inhibitory avoidance training. (a) The rat is placed in a box divided by a closed door that separates the light side from the dark side. When the door is opened, the rat scurries into the dark side to avoid the light. On the dark side, a foot shock is delivered to the rat. To test for the creation of a memory trace, one can measure the time it takes for the rat to re-enter the dark side at various time points after the initial experience. (b) Recordings of synaptic transmission in area CA1 showed evidence of LTP when this type of memory was formed. (Source: Adapted from Whitlock et al., 2006.) Description
Another approach has been to see whether the molecules involved in LTP and LTD are also involved in learning and memory. For example, both forms of synaptic plasticity may require activation of the NMDA receptors. To assess the possible role of hippocampal NMDA receptors in learning, researchers injected an NMDA receptor blocker into the hippocampus of rats undergoing inhibitory avoidance training. This treatment prevented formation of a memory of the aversive experience. These experiments built on pioneering studies performed by Richard Morris in the late 1980s at the University of Edinburgh, in which NMDA receptor blockers were infused into the hippocampus of rats while they were being trained in a water maze (see Figure 24.20). Unlike normal animals, these rats failed to learn the rules of the game or the location of the escape platform. This finding provided the first evidence that NMDA-receptor-dependent processes play a role in memory.
A revolutionary new approach to the molecular basis of learning and memory was introduced by Susumu Tonegawa at the Massachusetts Institute of Technology. Tonegawa, who switched to neuroscience after winning the 1987 Nobel Prize for his research in immunology, recognized that molecules and behavior could be connected by manipulating the genes of experimental animals. This approach had already been tried with success in simple organisms like fruit flies (Box 25.5), but not in mammals. In their first experiment in mice, Tonegawa, Alcino Silva, and their colleagues “knocked out” (deleted) the gene for one subunit (α) of CaMKII, and found parallel deficits in hippocampal LTP and memory. Since then, many genes have been manipulated in mice, with the aim of assessing the role of LTP and LTD mechanisms in learning. LTP, LTD, and learning clearly have many common requirements.
Of the several hundred thousand proteins manufactured by a neuron, some may be more important than others when it comes to learning. It is even possible that some proteins are uniquely involved in learning and memory. Needless to say, we could gain considerable insight about the molecular basis of learning and learning disorders if such hypothetical “memory molecules” could be identified.
Recall that each protein molecule is the readout of a gene. One way to identify a “memory protein” is to delete genes one at a time and see if specific learning deficits result. This is precisely the strategy that Seymour Benzer, Yadin Dudai, and their colleagues at the California Institute of Technology tried using the fruit fly Drosophila melanogaster. Drosophila has long been a favorite species of geneticists, but one might reasonably question to what extent a fruit fly learns. Fortunately, Drosophila can perform the same tricks that other invertebrate species like Aplysia have mastered. For example, fruit flies can learn that a particular odor predicts a shock. They demonstrate this memory after training by flying away when the odor is presented. The strategy is to produce mutant flies by exposing them to chemicals or X-rays. They are then bred and their offspring are screened for behavioral deficits. The first mutant displaying a fairly specific learning deficit was described in 1976 and called Dunce. Other memory-deficient mutants were later described and given vegetable names, such as Rutabaga and Cabbage. The next challenge was to identify exactly which proteins had been deleted. It turned out that all three of these memory mutants lacked particular enzymes in intracellular signaling pathways.
In these early Drosophila studies, the mutations that were induced occurred at random, followed by extensive screening, first to find a learning deficit and then to determine exactly which gene was missing. More recently, however, genetic engineering techniques have made it possible to make very specific deletions of known genes, not only in Drosophila but also in mammals. Thus, for example, in 1992 Susumu Tonegawa, Alcino Silva, and their colleagues at the Massachusetts Institute of Technology were able to isolate and delete one subunit (α) of the calcium-calmodulin-dependent protein kinase II in mice. Experiments had already suggested that this enzyme is critical for the induction of long-term potentiation. Sure enough, these mice had a clear deficit in LTP in the hippocampus and the neocortex. And, when tested in the Morris water maze, they were found to have a severe memory deficit. Thus, these mice were memory mutants, just like their distant cousins Dunce, Rutabaga, and Cabbage.
Are we to conclude that the missing proteins in these mutants are the elusive “memory molecules”? No. All the mutants displayed other behavioral deficits in addition to memory. We can only conclude, at present, that animals growing up without these proteins are unusually poor learners. However, the studies do underscore the critical importance of specific second messenger pathways in translating a fleeting experience into a lasting memory.
Despite the power of this genetic approach, it has some serious limitations. Loss of a function, like LTP or learning, might be a secondary consequence of developmental abnormalities caused by growing up without a particular protein. Moreover, since the protein is missing in all cells that normally express it, pinpointing where and how a molecule contributes to learning can be difficult. For these reasons, researchers have attempted to devise ways to restrict their genetic manipulations to specific times and specific locations. In one interesting example of this approach, Tonegawa and his colleagues found a way to restrict the genetic deletion of NMDA receptors to the CA1 region in mice, starting at about 3 weeks of age. These animals showed a striking deficit in LTP, LTD, and water maze performance, thus revealing an essential role for CA1 NMDA receptors in this type of learning.
If too little hippocampal NMDA receptor activation is bad for learning and memory, what would happen if we boosted the number of NMDA receptors? Amazingly, animals engineered to produce more than the normal number of NMDA receptors show enhanced learning ability in some tasks. Taken together, the pharmacological and genetic studies show that hippocampal NMDA receptors play a key role not only in synaptic modification, such as LTP and LTD, but also in learning and memory.
Synaptic plasticity is widespread in the brain, and analysis by theoretical neuroscientists has revealed that this can present a problem. To illustrate, let’s consider Hebbian synaptic strengthening. Synapses potentiate when they are active at the same time as their postsynaptic target neuron. As they undergo LTP, these synapses exert more influence on the postsynaptic cell, making it more likely to respond and thereby causing further potentiation of all synapses that are active at the same time. Computer simulations show that eventually all synapses on the neuron will potentiate, and stimulus selectivity (and memory) will be lost. A similar problem can arise with synaptic weakening: By reducing postsynaptic activity, LTD makes synapses more likely to be weakened until they eventually disappear altogether. Unchecked synaptic plasticity can therefore lead to unstable neuronal responses. As we learned in Chapter 15, homeostasis is the term used to describe regulatory processes that maintain the internal environment of the body within a narrow physiological range. There must be homeostatic mechanisms that provide stability and keep synaptic weights within a useful dynamic range. We will discuss two such mechanisms here.
Metaplasticity. Consider again the graph in Figure 25.13. It shows that weak NMDA receptor activation causes LTD and strong NMDA receptor activation causes LTP. At some level of moderate NMDA receptor activation, between that required for LTD and for LTP, there is no net change. This value is called the synaptic modification threshold. The BCM theory proposed that the value of the modification threshold adjusts depending on the history of integrated postsynaptic activity. Therefore, when activity rises, due perhaps to too much LTP, the modification threshold slides up, making LTP more difficult to produce. Conversely, if activity levels fall, due perhaps to too much LTD, the modification threshold slides down, making LTD less likely and LTP easier to produce. This general concept, that the rules of synaptic plasticity change depending on the history of synaptic or cellular activity, is called metaplasticity. Computer simulations show that ongoing adjustments of the value of the modification threshold ensure that synaptic modifications are constrained to maintain neuronal stimulus selectivity and memory.
Research inspired by the BCM theory has confirmed the existence of metaplasticity. Although many different mechanisms contribute to the sliding modification threshold, one appears to be adjustments in the molecular composition of NMDA receptors themselves. NMDA receptors are composed of four subunits: two NR1 subunits and two NR2 subunits. At many synapses in the cerebral cortex, two types of NR2 subunits are used to construct the receptor: NR2A and NR2B. The ratio of NR2A to NR2B subunits determines the properties of the receptor, including how much Ca2+ can pass and what intracellular enzymes are activated. LTP is favored when more NR2B-containing receptors are expressed at the synapse, whereas LTD is favored when more NR2A-containing receptors are expressed. The ratio of NR2A- to NR2B-containing receptors depends in part on the relative abundance of these proteins in the neuron. Research has shown that after a period of high cortical activity, NR2A levels increase and NR2B levels decrease, promoting LTD over LTP. On the other hand, NR2B levels increase and NR2A levels decrease after a period of low cortical activity, promoting LTP over LTD (Figure 25.18). These changes in NMDA receptor subunit composition occur relatively slowly, over the course of hours, presumably because they depend on the synthesis of new protein subunits.

FIGURE 25.18 The sliding modification threshold. Experiments in which cortical activity is reduced for several days reveal a shift in the curve that relates stimulation frequency to LTD and LTP. Lowering activity favors LTP over LTD, whereas raising activity favors LTD over LTP. This shift is accounted for in part by changes in the subunit composition of NMDA receptors. NMDA receptors with more NR2B admit more Ca2+. (Source: Adapted from Bear, 2003.) Description
Synaptic Scaling. In a classic series of experiments dating back to the 1930s, the eminent physiologist Walter Cannon (introduced in Chapter 18) showed that cutting the nerve to a muscle leads to an increase in the electrical excitability and sensitivity of the muscle to ACh, the neurotransmitter of the neuromuscular junction. This phenomenon, called denervation supersensitivity, was later shown to be a widespread response of neurons to the loss of synaptic input. Denervation is not necessary to induce supersensitivity, however. A similar response occurs if the neurotransmitter receptors are blocked pharmacologically or if muscles or neurons are electrically silenced with tetrodotoxin (TTX). Cannon suggested this was likely to represent a homeostatic response of excitable cells to the loss of input.
An analogous phenomenon occurs in cortical neurons after manipulations of overall synaptic input. When cortical neurons are silenced with TTX, their electrical excitability increases, as does the strength of excitatory synapses that impinge on them. But what does this gross adjustment of overall synaptic strength do to the carefully tuned patterns of synaptic weights that have stored memories? Gina Turrigiano and her colleagues at Brandeis University discovered that relative differences in the strengths of synapses on a neuron are unchanged, even as the absolute levels go up or down; that is, the neuron adjusts by multiplying (or dividing) the values of all synaptic weights by the same number. This adjustment of absolute synaptic effectiveness that preserves the relative distribution of synaptic weights is called synaptic scaling.
As with metaplasticity, multiple mechanisms contribute to synaptic scaling. One appears to use Ca2+ entering the soma through voltage-gated Ca2+ channels and activation of calcium-calmodulin-dependent kinase IV (CaMKIV, a close relative of CaMKII) to regulate gene expression. A period of elevated activity increases CaMKIV-dependent gene expression, whereas a period of inactivity decreases it. The ultimate consequence of these changes in gene expression is the cellwide removal or insertion of glutamate receptors at synapses (both NMDA receptors and AMPA receptors). Like metaplasticity, scaling occurs over a longer time period (hours to days) than does the induction of LTP or LTD (seconds to minutes). This time is necessary for the synthesis (or degradation) of proteins required to adjust the strengths of the thousands of synapses impinging on the neuron.
With metaplasticity and scaling, the neuron keeps a lid on the roiling boil of ongoing synaptic plasticity. When activity is too high for too long, these mechanisms kick in to promote LTD and scale down synaptic weights. When activity is too low, they promote LTP and scale up synaptic weights. Proper neuronal function, experience-dependent shifts in selectivity, and learning and memory all require the appropriate balance of synaptic change and stability.
We have seen that memory can result from experience-dependent alterations in synaptic transmission. In most examples of synaptic plasticity, transmission is initially modified as a result of changing the number of phosphate groups that are attached to proteins in the synaptic membrane. In the case of LTD and LTP, this occurs at the postsynaptic AMPA receptor and at proteins regulating AMPA receptor number at the synapse.
Adding phosphate groups to a protein can change synaptic effectiveness and form a memory, but only as long as the phosphate groups remain attached to that protein. Phosphorylation as a long-term memory consolidation mechanism is problematic for two reasons:
- Phosphorylation of a protein is not permanent. Over time, the phosphate groups are removed, thereby erasing the memory.
- Protein molecules themselves are not permanent. Most proteins in the brain have a lifespan of less than 2 weeks and are undergoing a continual process of replacement. Memories tied to changes in individual protein molecules would not be expected to survive this rate of molecular turnover.
Thus, we need to consider mechanisms that might convert what initially is a change in synaptic protein phosphorylation to a form that can last a lifetime.
Phosphorylation of synaptic proteins, and memory, could be maintained if the kinases, the enzymes that attach phosphate groups to proteins, were made to stay “on” all the time. Normally the kinases are tightly regulated and are “on” only in the presence of a second messenger. But what if learning changed these kinases so they no longer required the second messenger? The relevant synaptic proteins would remain phosphorylated all the time.
Recent evidence suggests that some kinases can become independent of their second messengers. Let’s consider as examples the changes that occur in two protein kinases during LTP in the hippocampus.
CaMKII. Recall that the entry of Ca2+ into the postsynaptic cell and the activation of CaMKII are required for the induction of LTP in CA1. Research has shown that CaMKII stays “on” long after [Ca2+]i has fallen back to a low level.
CaMKII consists of ten subunits arranged in a rosette pattern. Each subunit catalyzes the phosphorylation of substrate proteins in response to a rise in Ca2+-calmodulin. How might CaMKII be switched permanently on? The answer requires some knowledge of how this enzyme is normally regulated (Figure 25.19). Each subunit is built like a pocket knife, with two parts connected by a hinge. One part, the catalytic region, performs the phosphorylation reaction. The other part is called the regulatory region. Normally, in the absence of the appropriate second messenger, the knife is closed and the catalytic region is covered by the regulatory region. This keeps the enzyme “off.” The normal action of the second messenger (Ca2+-calmodulin) is to pry the knife open, but only as long as the messenger is present. When the messenger is removed, the molecule usually snaps shut, and the kinase turns off again. After LTP, however, it appears that the knife fails to close completely in the α subunits of CaMKII. The exposed catalytic region continues to phosphorylate CaMKII substrates.

FIGURE 25.19 The regulation of CaMKII. (a) The hingelike subunit of CaMKII is normally “off” when the catalytic region is covered by the regulatory region. (b) The hinge opens upon activation of the molecule by Ca2+-bound calmodulin, freeing the catalytic region to add phosphate groups to other proteins. (c) A large elevation of Ca2+ can cause phosphorylation (P) of one subunit by another (autophosphorylation), which enables the catalytic region to stay “on” permanently. Description
How is the hinge of the protein kinase molecule kept open? The answer lies in the fact that CaMKII is an autophosphorylating protein kinase; each subunit within the CaMKII molecule can be phosphorylated by a neighboring subunit. The consequence of subunit phosphorylation is that the hinge stays open. If the initial activation of CaMKII by Ca2+-calmodulin is sufficiently strong, autophosphorylation will occur at a faster rate than dephosphorylation, and the molecule will be switched on. Persistent activity of CaMKII could contribute to the maintenance of synaptic potentiation, for example, by keeping the postsynaptic AMPA receptors phosphorylated. The general idea that an autophosphorylating kinase could store information at the synapse, initially proposed by John Lisman at Brandeis University, is called the molecular switch hypothesis.
Protein Kinase M Zeta. Recent research has implicated a new player in the maintenance of LTP and certain forms of memory, protein kinase M zeta (PKMς). Excitement about the role of this protein kinase originates with the work of Todd Sacktor at the State University of New York Downstate Medical Center. Sacktor and his colleagues showed that an intracerebral injection of a small peptide called ZIP, designed to specifically inhibit PKMς, can erase LTP and memories established many days before the injection. Simply put, ZIP zaps memories. This intriguing finding suggests that persistent PKMς activity maintains changes in synaptic strength by continuing to phosphorylate its substrates. ZIP, by temporarily inhibiting the kinase, allows these substrates to become dephosphorylated, thus erasing the memory trace.
How does PKMς become persistently active in response to synaptic activity? A current model suggests that the mRNA for this kinase exists at synapses but is normally not translated into protein. Strong synaptic activation, and the corresponding rise in [Ca2+], trigger a burst of synaptic protein synthesis and the birth of new PKMς molecules. PKMς phosphorylates synaptic proteins involved in regulating AMPA receptor number and, in addition, proteins involved in the regulation of mRNA translation at the synapse. By switching translation on in the absence of elevated [Ca2+], PKMς levels can be replenished in the face of ongoing degradation of the kinase molecules.
The erasure of memory by ZIP has been reproduced by several laboratories, but at this point it is unclear if it acts specifically by inhibiting PKMς. It is certain, however, that unlocking the mystery of how ZIP works will reap great rewards in our understanding of memory mechanisms.
The idea that protein synthesis is important for memory consolidation is not new. It has been investigated extensively since the introduction of drugs in the 1960s that selectively inhibit the assembly of protein from messenger RNA. Protein synthesis inhibitors can be injected into the brains of experimental animals as they are trained to perform a task, and deficits in learning and memory can be assessed. These studies reveal that if brain protein synthesis is inhibited at the time of training, the animals learn normally but fail to remember when tested days later. A deficit in long-term memory is also often observed if the inhibitors are injected shortly after training. However, the memories become increasingly resistant to the inhibition of protein synthesis as the interval between the training and the injection of inhibitor is increased. These findings indicate a requirement for new protein synthesis during the period of memory consolidation, when short-term memories are converted into long-term ones.
Consider the example of inhibitory avoidance memory (see Figure 25.17a). As we discussed, the memory is created in a single trial and can be measured by how much a rat avoids the location where it received a foot shock (usually the dark side of a box with two rooms separated by a door). Normally, this memory is very stable and lasts a long time (days to weeks depending on the strength of the shock). If the animal is injected with a protein synthesis inhibitor shortly before the training, learning occurs normally, as measured by the animal’s immediate avoidance of the dark side. However, this memory fades within a day without new protein synthesis. Similarly, inhibiting protein synthesis at the time of a tetanus has no effect on the induction of LTP in the hippocampus, but instead of lasting days to months, the synaptic potentiation gradually disappears over a few hours.
Synaptic Tagging and Capture. From what we have learned so far, memory formation appears initially to involve the rapid modification of existing synaptic proteins. These modifications, perhaps with the help of persistently active kinases, work against the factors that would erase a memory (such as molecular turnover). It’s a losing battle, unless a new protein arrives at the modified synapse and converts the temporary change in the synapse to a more permanent one. But how do the proteins required for consolidating synaptic changes and memories find the modified synapses? An answer is suggested by a clever series of experiments performed collaboratively in the late 1990s by Julietta Frey in Magdeburg, Germany, and Richard Morris in Edinburgh, Scotland.
Frey had shown previously that LTP caused by “weak” tetanic stimulation, which activates briefly only a small number of synapses, decays back to baseline over the course of an hour or two because it fails to trigger protein synthesis. On the other hand, repeated episodes of “strong” stimulation, recruiting a larger number of synapses, induces long-lasting LTP because it stimulates new protein synthesis (Figure 25.20a,b). Frey and Morris asked if the newly synthesized proteins acted only on the synapses whose activity triggered their synthesis. They found that the wave of protein synthesis triggered by strong stimulation of one synaptic input to a hippocampal neuron would also consolidate the LTP caused by the weak stimulation of a second input (Figure 25.20c). It appeared that the weak stimulation endows synapses with a tag that enables them to capture the newly synthesized proteins that consolidate LTP. By varying the interval between the weak and strong stimulation of the two inputs, Frey and Morris were able to determine that the tag lasts about 2 hours. In this way, a trivial event that would otherwise be forgotten, like last Tuesday’s dinner, might be seared into long-term memory if it occurs within 2 hours of a momentous event that triggers a wave of new protein synthesis, like the first kiss of the love of your life. The molecular mechanism for synaptic tagging has not been fully elucidated, but it should come as no surprise that it is believed to involve phosphorylation of synaptic proteins by various kinases, including CaMKII and PKMς.

FIGURE 25.20 Synaptic tagging and capture. (a) The persistence of LTP depends on whether synaptic stimulation is strong enough to trigger protein synthesis in the postsynaptic neuron. Weak stimulation induces LTP of the EPSP that rapidly decays. Strong stimulation induces LTP and stimulates the protein synthesis that converts a temporary synaptic change into a long-lasting one. (b) Two inputs to the same neuron are stimulated in alternation. Weak stimulation of input 1 induces LTP that would normally fade, but if it is followed an hour later by strong stimulation of input 2, the wave of new proteins can be captured by synapses tagged by the weak stimulation of input 1. The timely arrival of new protein converts the short-lasting LTP into long-lasting LTP. Description
CREB and Memory. What regulates the protein synthesis that is required for memory consolidation? Recall that the very first step in protein synthesis is the generation of an mRNA transcript of a gene (see Figure 2.9). This process of gene expression is regulated by transcription factors in the nucleus. One transcription factor is called the cyclic AMP response element binding protein (CREB). CREB is a protein that binds to specific segments of DNA, called cyclic AMP response elements (CREs), and functions to regulate the expression of neighboring genes (Figure 25.21). There are two forms of CREB: CREB-2 represses gene expression when it binds to the CRE; CREB-1 activates transcription, but only when it is phosphorylated by protein kinase A. In a seminal study published in 1994, Tim Tully and Jerry Yin at Cold Spring Harbor Laboratory showed that CREB regulates the gene expression required for memory consolidation in the fruit fly Drosophila melanogaster (see Box 25.5).

FIGURE 25.21 The regulation of gene expression by CREB. Shown here is a piece of DNA containing a gene whose expression is regulated by the interaction of a CREB protein with a CRE on the DNA. (a) CREB-2 functions as a repressor of gene expression. (b) CREB-1, an activator of gene expression, can displace CREB-2. (c) When CREB-1 is phosphorylated by protein kinase A (and other kinases), transcription can ensue. Description
In their first series of experiments, Tully and Yin bred Drosophila that would make extra copies of the fly’s version of CREB-2 (called dCREBb) when the animal was warmed up (a miracle of fly genetic engineering that is not possible in mammals). This manipulation repressed all gene expression that is regulated by the CREs, and also blocked memory consolidation in a simple memory task. Thus, CREB-regulated gene expression is critical for memory consolidation in fruit flies. More interesting, however, is what they found when they generated flies that could make extra copies of fly CREB-1 (called dCREBa). Now, tasks that would take normal flies many trials to learn could be remembered after a single training trial. These mutant flies had a “photographic” memory! And these results are not peculiar to flies; CREB has been implicated in regulating the consolidation of sensitization in Aplysia, as well as long-term potentiation and spatial memory in mice.
As we have discussed, not all experiences are remembered equally. Some are seared permanently into our memories. Others remain with us only briefly and then fade away. The modulation of gene expression by CREB provides a molecular mechanism that can control the strength of a memory.
A failure of memories to consolidate is a feature of numerous brain disorders as well as the aging process. The recent understanding of how consolidation is regulated has spawned a new industry focused on developing memory-enhancing drugs. Such drugs could greatly improve the quality of life for individuals with neurological diseases, such as Alzheimer’s. However, there is also the allure of performance enhancement in healthy people. By analogy, consider the widespread use of such drugs as Viagra introduced to treat male erectile dysfunction. “Viagra for the brain,” as such memory enhancers have been whimsically called, continues to be pursued. Although compounds have been discovered that boost memory consolidation, their side effects have thus far prevented their development into drugs. As was the case for drugs that enhance athletic performance, the ethics of using memory boosters in the absence of a clear medical justification are certain to be hotly debated.
Structural Plasticity and Memory. How does the synapse make use of the timely occurrence of gene expression and the arrival of a new protein? One possibility is that newly synthesized proteins (such as PKMς) switch local synaptic protein synthesis on in order to maintain a synaptic change. However, to account for the fact that blocking protein synthesis fails to disrupt memories that have already been consolidated, we would have to further posit that these newly synthesized proteins have a lifespan that is long enough to survive the temporary inhibition of protein synthesis.
Another possibility is that the lasting imprint of new protein synthesis is the construction (or demolition) of synapses. Work on the invertebrate Aplysia has shown that some types of long-term (but not short-term) memory can cause a doubling of the number of synapses made by some neurons!
Do similar structural changes occur in the mammalian nervous system after learning? This is a difficult problem to solve experimentally because of the complexity of the mammalian brain and the distributed nature of memory. One approach has been to compare brain structure in animals that have had ample opportunity to learn with that of animals that have had little chance to learn. For example, putting a laboratory rat in a “complex” environment filled with toys and playmates (other rats) has been shown to increase the number of synapses per neuron in the occipital cortex by about 25%. Very recently, advances in microscopy and methods for cell labeling (see Box 2.1) have made it possible for researchers to image the same neuron in living mice over the course of many days. Altering the visual or tactile environment stimulates the formation of new dendritic spines, important sites of excitatory synaptic transmission, in the visual and somatosensory cortex, respectively (Figure 25.22). As exposure to this new environment is increased, these new synapses grow larger and other synapses on the same dendrites are eliminated, as would be expected if the mechanisms of LTP and LTD are at work to encode a memory. The new spines may shrink if the animal is returned to the original environment but do not disappear, consistent with the interpretation that these persistent spines contribute to a long-term memory of the altered environment.

FIGURE 25.22 Synaptic remodeling in the cerebral cortex during learning and memory. This illustration summarizes some of the structural changes that have been observed in the neocortex when mice are exposed to new sensory environments that are encoded as memory. (Source: Adapted from Hofer and Bonhoeffer, 2010, Fig. 1.) Description
It is important to recognize that there are limits to structural plasticity in the adult brain. As we discussed in Chapter 23, large changes in brain circuitry are generally confined to critical periods of early life. The growth and retraction of most axons in the adult central nervous system (CNS) are restricted to no more than a few tens of micrometers. But it is now very clear that the end of a critical period does not necessarily signify an end to changes in the structure of axon terminals or the effectiveness of their synapses.
Learning and memory can occur at synapses. Regardless of the species, brain location, and memory type, many of the underlying mechanisms appear to be universal. Events are represented first as changes in the electrical activity of the brain, then as intracellular second messengers, and next as modifications of existing synaptic proteins. These temporary changes are converted to permanent ones, and long-term memories, by altering the structure of the synapse. In many forms of memory, this involves new protein synthesis and the assembly of new microcircuits. In other forms of memory, existing circuits may be disassembled. In either case, learning requires many of the same mechanisms that were used to refine brain circuitry during development.
One universal feature is the involvement of Ca2+. Clearly, calcium does far more than build strong bones and teeth. Not only is it critical for neurotransmitter secretion and muscle contraction, but it is also involved in nearly every form of synaptic plasticity. Because it is a charge-carrying ion on the one hand and a potent second messenger substance on the other, Ca2+ has a unique ability to directly couple electrical activity with long-term changes in the brain.
Can basic neuroscience research take us from ions to intelligence? From calcium to cognition? If you remember what you have learned in this textbook, and if synaptic plasticity truly is the basis of declarative memory, it would appear that the answer is yes.
cyclic AMP response element binding protein (CREB) (p. 894)
1. What is the most common cellular correlate of memory formation in the cerebral cortex? What does this say about how memories are stored?
2. How does one account for the graceful degradation of memory as neurons die over the lifespan?
4. Sketch the trisynaptic circuit of the hippocampus.
5. How can the mechanisms of LTP serve associative memory?
6. What property of the NMDA receptor makes it well suited to detect coincident presynaptic and postsynaptic activity? How could Ca2+ entering through the NMDA receptor possibly trigger both LTP and LTD in CA1 and the neocortex?
7. Compare and contrast metaplasticity and synaptic scaling.
8. In H.M and R.B. (see Chapter 24), destruction of the hippocampus appears to have impaired the mechanism that “fixes” new memories in the neocortex. Propose a mechanism involving CREB explaining why this might be true.
Abraham WC, Robins A. 2005. Memory retention: the synaptic stability versus plasticity dilemma. Trends in Neuroscience 28:73–78.
Bear MF. 1996. A synaptic basis for memory storage in the neocortex. Proceedings of the National Academy of Sciences USA 93:13453–13459.
Cooper LN, Bear MF. 2012. The BCM theory of synapse modification at 30: interaction of theory and experiment. Nature Reviews Neuroscience 13:798–810.
Kandel ER. 2006. In Search of Memory: The Emergence of a New Science of Mind. New York: Norton.
Kessels HW, Malinow R. 2009. Synaptic AMPA receptor plasticity and behavior. Neuron 61:340–350.
Malenka RC, Bear MF. 2004. LTP and LTD: an embarrassment of riches. Neuron 44:5–21.
Additional figures