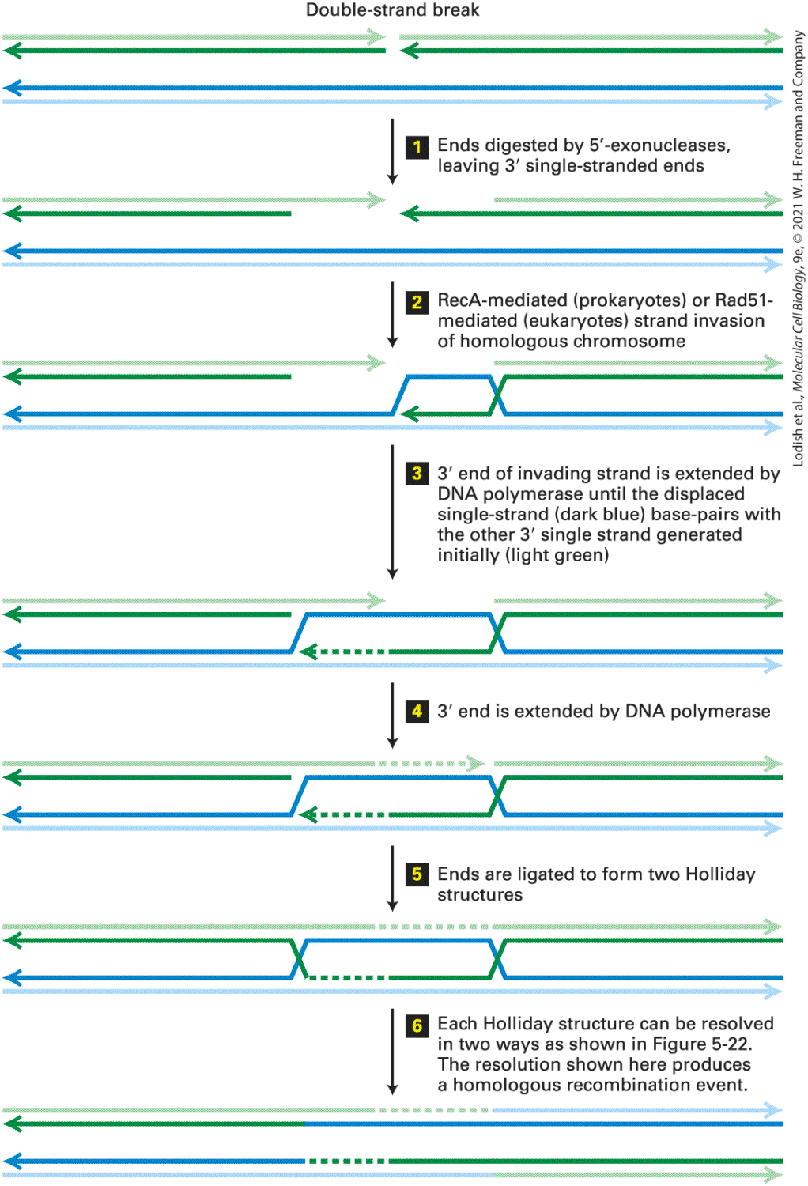
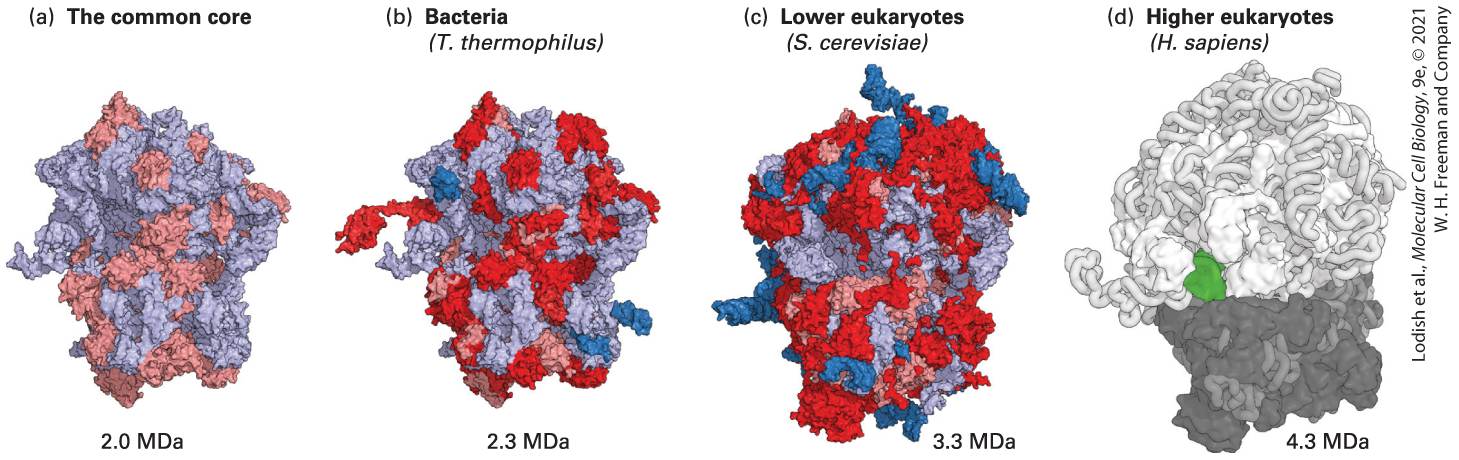
Introduction
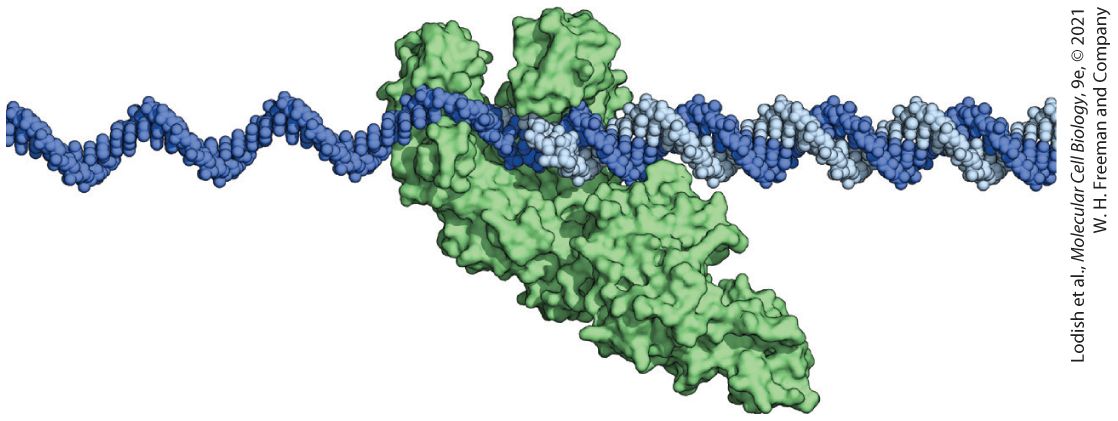
Chapter 5 Fundamental Molecular Genetic Mechanisms Model of DNA Polymerase I from Escherichia coli in the process of replicating DNA. The universal mechanism for copying information in DNA is replication by DNA polymerase, a remarkable copying and proofreading machine. The E. coli DNA polymerase I shown here can synthesize a complementary copy of the template strand at a rate of about 500 bases per second, making a mistake only once in every bases.
5.4 Transcription of Protein-Coding Genes and Formation of mRNA
5.7 Viruses: Parasites of the Cellular Genetic System One of the foundational principles of cell biology is that the cells that make up a multicellular organism are autonomous — each cell carries its own set of genetic instructions for making all of the specific components of cellular structures, cellular catalysts, and molecular machines that make up a functioning cell of a given type. As was described in Chapter 3, the vast majority of these active cellular components are proteins. The specific function of each individual protein is dictated by its sequence of amino acids, the instructions for which are ultimately encoded in the sequence of nucleotides in deoxyribonucleic acid (DNA), the biological information-carrying molecule. One copy of DNA of the human genome encompassing all 23 chromosomes is about nucleotides long and encodes about 20,000 different proteins. In physical terms, one copy of the genome adds up to a thread of DNA that is about one meter in total length, but that is so fine that two complete copies are compacted into the nucleus of each of the approximately cells in our body. This information, under the control of a complex regulatory network, is then read out to
express the appropriate sets of proteins for each of the specific cell types in the body. Thus the basic principle of autonomy of cellular information implies a system of stored and expressed genetic information that is so vast as to defy comprehension. Yet the general rules, as they are now well understood, for how biological information is managed are in fact quite straightforward and are the same in virtually all forms of life. In this chapter, we describe the basic principles for storage, replication, and expression of biological information. A full understanding of this subject not only requires an understanding of the structure and biochemical properties of the molecules involved, but also requires the application of biological information theory. Let us first see how information theory can be used to compute the information-carrying capacity of DNA and to compare this to the memory capacity of a computer. Whether in a digital computer or in a biological system, the smallest unit of information is a bit, which corresponds to a unit that can have one of two values — in a digital computer these values correspond to 1 and 0. DNA is made up of two complementary strands that can have one of four possible bases (A, G, C, or T) at each position. The information in n bits can specify possible values; thus each base can be specified by two bits and is therefore said to contain two bits of information. The two strands are base paired with one another according to the principles of base pairing, such that A always pairs with T and G always pairs with C. Because of the fixed rules for base pairing, the sequence of the second complementary strand of DNA is determined by the first and does not add to the information carrying capacity of DNA, but it does add redundancy of information which, as we will see, allows DNA
to be readily copied and for damage in DNA to be repaired. Since the human genome contains a total of base pairs, the total information capacity is bits, which corresponds to about bytes of information ( , which is the unit that is used to measure computer memory). A single copy of the human genome weighs only grams and thus on a weight basis, DNA has enormous capacity to store information — in theory, the same information contained in a million one-terabyte laptop computer hard drives could be stored in a mass of DNA the size of a grain of sand. When cells divide, each daughter cell must receive its own exact copy of the information contained in DNA. DNA is copied or replicated by separation of the two complementary strands of a DNA molecule and then enzymes known as DNA polymerases use each strand as a template for the synthesis of two identical copies of the parent molecule by following the rules of base pairing. From the perspective of an information-copying reaction, we are interested in both the rate at which the genome can be copied and the fidelity of the reaction, which is related to how frequently errors are produced during copying. From fundamental chemical principles, fidelity is dictated by the difference in energy between a correct and an incorrect base pair in the replication reaction. A typical DNA polymerase incorporates one nucleotide every 2 milliseconds. In this timeframe the theoretical rate of errors in replication would be one mistake in every bases, which is the measured fidelity of replication catalyzed by the simplest DNA polymerases. However, if the human genome were replicated with such a low-fidelity polymerase, each parent would pass on about one million new mutations to each of their children.
The measured fidelity of DNA replication in humans corresponds to one new mutation in bases per generation. In this chapter, we will see how elaborate proofreading and repair processes have evolved to achieve this 1000-fold enhancement in the fidelity in the replication and transmission of information in DNA. Gene expression is the mechanism by which the information in the DNA sequence of a gene is used as the instructions for production of a functional protein or RNA molecule. For protein-coding genes, the flow of information within cells follows the outline which is known as the central dogma of molecular biology. Before any of the details of how information flows from DNA to proteins were understood, molecular biologists used the basic principles of information theory to deduce how a DNA sequence comprised of four letters (A, G, C, and T) could encode a protein sequence made up of 20 different amino acids. In theory, at least five bits of information (which could give 32 different possibilities) would be required to specify one of 20 amino acids. For a simple code in which blocks of nucleotides in DNA would code for each amino acid, a block size of a least three nucleotides would be required. We now know that the nucleotide sequences of genes are in fact read three nucleotides at a time in blocks known as codons. Working out the universal genetic code for which amino acid is coded by each of the 64 possible codons was a monumental achievement of twentieth-century science. The genetic code table is the central component of all of the algorithms that are used to analyze new genomic sequences to deduce the protein sequences that they encode.
The information in the nucleotide sequence of DNA is not translated directly into an amino acid sequence; instead, through the process of transcription, the information stored in DNA is copied into a messenger ribonucleic acid (mRNA). Like DNA, RNA carries information in the form of a sequence of four nucleotides, and the process of transcription is similar to replication in that the rules of base pairing are used to synthesize an mRNA of complementary sequence from a DNA template. RNA is generally single stranded and RNA is chemically less stable than DNA. Thus mRNA serves as a temporary copy of the information in a gene that allows the expression of each gene sequence to be controlled independently. The nucleotide sequence of an mRNA molecule contains information that specifies the correct order of amino acids during the synthesis of a protein. In this process, known as translation, the nucleotide sequence of an mRNA molecule is read by a second type of RNA called transfer RNA (tRNA). The reading occurs by the base pairing of each of the three nucleotide codons of the mRNA with a complementary anti-codon triplet in the tRNA. The accurate, stepwise assembly of amino acids into proteins takes place in the context of a remarkable macromolecular machine, the ribosome, which is composed of a third type of RNA, ribosomal RNA (rRNA), and associated proteins. As the correct amino acids are brought into sequence by tRNAs, they are linked by peptide bonds to make proteins. Another type of information that is encoded in DNA is represented by the control sequences that regulate gene expression and determine which
genes are on and which are off in each of the cell types in the body. Typically, gene expression is regulated at three possible control points: transcription initiation, mRNA stability, or initiation of translation. Regulation of transcription initiation by the control sequences at a gene promoter and the protein transcription factors that interact with them are discussed in Chapter 9. Other types of control sequences that govern the rate at which a given mRNA will be translated or the rate at which the mRNA will be degraded are discussed in Chapter 10. In this chapter, we first review the chemical and structural properties of DNA that make this molecule ideally suited as the carrier of genetic information in the cell. We next consider the molecular problems involved in DNA replication and the complex cellular machinery that ensures accurate copying of the genetic material. The next section describes how damage to DNA is repaired and how regions of different DNA molecules are exchanged in the process of recombination to generate new combinations of traits in the individual organisms of a species. In the next several sections, we discuss the basic processes summarized in
Figure 5-1: transcription of DNA into mRNA precursors and processing of these precursors to make functional mRNA molecules. After outlining the functions of mRNA, tRNA, and rRNA in protein synthesis, we present a detailed description of these and other components in the biochemical steps of translation.
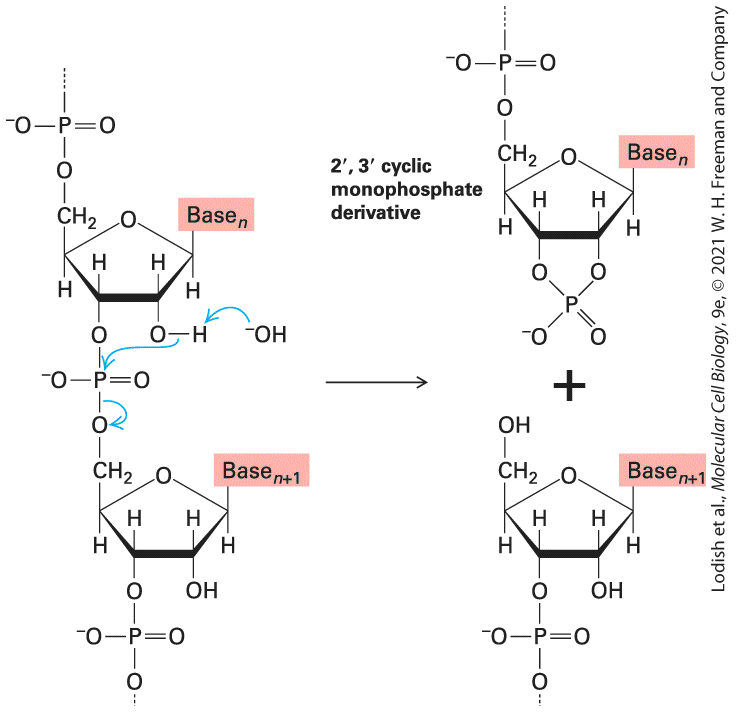
FIGURE 5-1 Overview of four basic molecular genetic processes. In this chapter, we cover the three processes that lead to production of proteins ( 1 – 3 ) and the process for replicating DNA ( 4 ). During transcription of a protein-coding gene by RNA polymerase ( 1 ), the four-base DNA code specifying the amino acid sequence of a protein is copied, or transcribed, into a precursor messenger RNA (pre-mRNA) by the polymerization of ribonucleoside triphosphate monomers (rNTPs). Removal of noncoding sequences and other modifications to the pre-mRNA ( 2 ), collectively known as RNA processing, produce a functional mRNA, which is transported to the cytoplasm. During translation ( 3 ), the four-base code of the mRNA is decoded into the 20–amino acid language of proteins. Transfer tRNAs (tRNAs) are the adaptors that read a codon of three nucleotides on the mRNA. In preparation for translation, tRNAs are charged with the correct amino acid by enzymes known as aminoacyl-tRNA synthetases. Ribosomes, the macromolecular machines that translate the mRNA code, are composed of two subunits assembled in the nucleolus from ribosomal RNAs (rRNAs) and multiple proteins (left). After transport to the cytoplasm, ribosomal subunits associate with an mRNA and carry out protein synthesis with the help of charged tRNAs and translation factor proteins. During DNA replication ( 4 ), which occurs only in cells preparing to divide, deoxyribonucleoside triphosphate monomers (dNTPs) are
polymerized to yield two identical copies of each chromosomal DNA molecule. Each daughter cell receives one of the identical copies. The final section of the chapter presents basic information about viruses — parasites that exploit the cellular machinery for DNA replication, transcription, and protein synthesis. In addition to being significant pathogens, viruses are important models for studying these cellular mechanisms of macromolecular synthesis and other cellular processes. Viruses continue to teach us important lessons in molecular cell biology today and have been adapted as experimental tools for introducing genes into cells, tools that are currently being tested for their effectiveness in human gene therapy.
Native DNA Is a Double Helix of Complementary Antiparallel Strands
5.1 The Double-Helical Structure of DNA DNA and RNA have very similar primary structures: both are linear polymers composed of only four different nucleotides. Recall from
Chapter 2 that all nucleotides consist of an organic base linked to a fivecarbon sugar that has a phosphate group attached to the carbon. Though chemically similar, DNA and RNA polymers in cells have quite different three-dimensional structures. Cellular DNA exists as two long, perfectly complementary, base-paired strands that can be as long as several hundred million nucleotides. Cellular RNAs usually exist as much shorter molecules (RNAs range in length from about 20 to thousands of nucleotides); they are typically single-stranded molecules that may have limited regions that form self-complementary base pairs. As we will see, the relative chemical stability of DNA and its unique redundant complementary structure make DNA an ideal molecule for the stable storage of genetic information. Native DNA Is a Double Helix of Complementary Antiparallel Strands The four nucleotides of DNA contain the purine bases adenine (A) and guanine (G) and the pyrimidine bases cytosine (C) and thymine (T) (see
Figure 2-17). A single DNA strand has a backbone composed of repeating
pentose-phosphate units from which the purine and pyrimidine bases extend as side groups. (Note that the single-letter abbreviations for these bases are also commonly used to denote all the nucleotides in nucleic acid polymers.) Like a polypeptide, a nucleic acid strand has an end-to-end chemical orientation: the end has a hydroxyl or phosphate group on the carbon of its terminal sugar; the end usually has a hydroxyl group on the carbon of its terminal sugar (Figure 5-2). This directionality, plus the fact that synthesis always proceeds to , has given rise to the convention that polynucleotide sequences are written and read in the direction (from left to right); for example, the sequence ATG is assumed to be ATG . The chemical linkage between adjacent nucleotides, commonly called a phosphodiester bond, actually consists of two phosphoester bonds, one on the side of the phosphate and another on the side.
FIGURE 5-2 Chemical directionality of a nucleic acid strand. Shown here are alternative representations of a single strand of DNA containing only three bases: cytosine (C), adenine (A), and guanine (G). (a) The chemical structure shows a hydroxyl group at the end and a phosphate group at the end. Note also that two phosphoester bonds link adjacent nucleotides; this two-bond linkage is commonly referred to as a phosphodiester bond. (b) In the stick diagram (top), the sugars are indicated as vertical lines and the phosphodiester bonds as slanting lines; the bases are denoted by their single-letter abbreviations. In the
simplest representation (bottom), only the bases are indicated. By convention, a polynucleotide sequence is always written in the direction (left to right) unless otherwise indicated. Description The illustration labeled A shows the vertical arrangement of single-stranded D N A. The 3 prime end of the D N A at the bottom contains guanine with a hydroxyl group, followed by adenine in the middle, and cytosine at the top connected to a phosphate group in the 5 prime end of the D N A. A phosphodiester bond links each nucleotide together. The illustration labeled B shows the stick diagram of C (cytosine), A (adenine), and G (guanine) connected by a phosphodiester bond with phosphate group in the 5 prime end and hydroxyl group in the 3 prime end. Below the stick diagram, a simple representation of the same structure as above reads, 5 prime C dash A dash G 3 prime. An understanding of how genetic information might be stored in the nucleotide sequence of DNA began to take shape in 1953 when James D. Watson and Francis H. C. Crick proposed that DNA has a double-helical structure. Their proposal was based on analysis of x-ray diffraction patterns of DNA fibers generated by Rosalind Franklin and Maurice Wilkins, which showed that the structure was helical, and analyses of the base composition of DNA from multiple organisms by Erwin Chargaff and colleagues. Chargaff’s studies revealed that while the base composition of DNA (percentages of A, T, G, and C) varies greatly between distantly related organisms, the percentage of A always equals the percentage of T, and the percentage of G always equals the percentage of C, in all organisms. Based on these discoveries and the structures of the four nucleotides, Watson and Crick performed careful molecular model building, proposing a double helix, with A always hydrogen-bonded to T
and G always hydrogen-bonded to C along the central axis of the double helix, as the structure of DNA. The most telling feature of the Watson and Crick model was that each of the four possible base pairs fit within the helical backbones in precisely the same manner. Thus any arbitrary sequence of nucleotides can be accommodated by the same double-helical structure. This immediately lead to the understanding that DNA functions as an informational molecule by carrying genetic information as a sequence of the four letters A, G, C, and T, just as a digital computer memory contains information encoded as a sequence of 1s and 0s. The extraordinary capacity of DNA to carry information stems from the fact that each nucleotide base is only about 50 atoms yet in the context of a DNA sequence can carry two bits of information — while even the most advanced computer memories require many thousands of atoms to store one bit of information. DNA consists of two associated polynucleotide strands that wind together to form a double helix. The two sugar-phosphate backbones are on the outside of the double helix, and the bases project into the interior. The adjoining bases in each strand stack on top of one another in parallel planes (Figure 5-3a). The orientation of the two strands is antiparallel; that is, their directions are opposite. The strands are held in precise register by formation of base pairs between the two strands: A is paired with T through two hydrogen bonds; G is paired with C through three hydrogen bonds (Figure 5-3b). This base-pair complementarity is a consequence of the size, shape, and chemical composition of the bases. The presence of thousands of such hydrogen bonds in a DNA molecule
contributes greatly to the stability of the double helix. Hydrophobic and van der Waals interactions between the stacked adjacent base pairs further stabilize the double-helical structure.
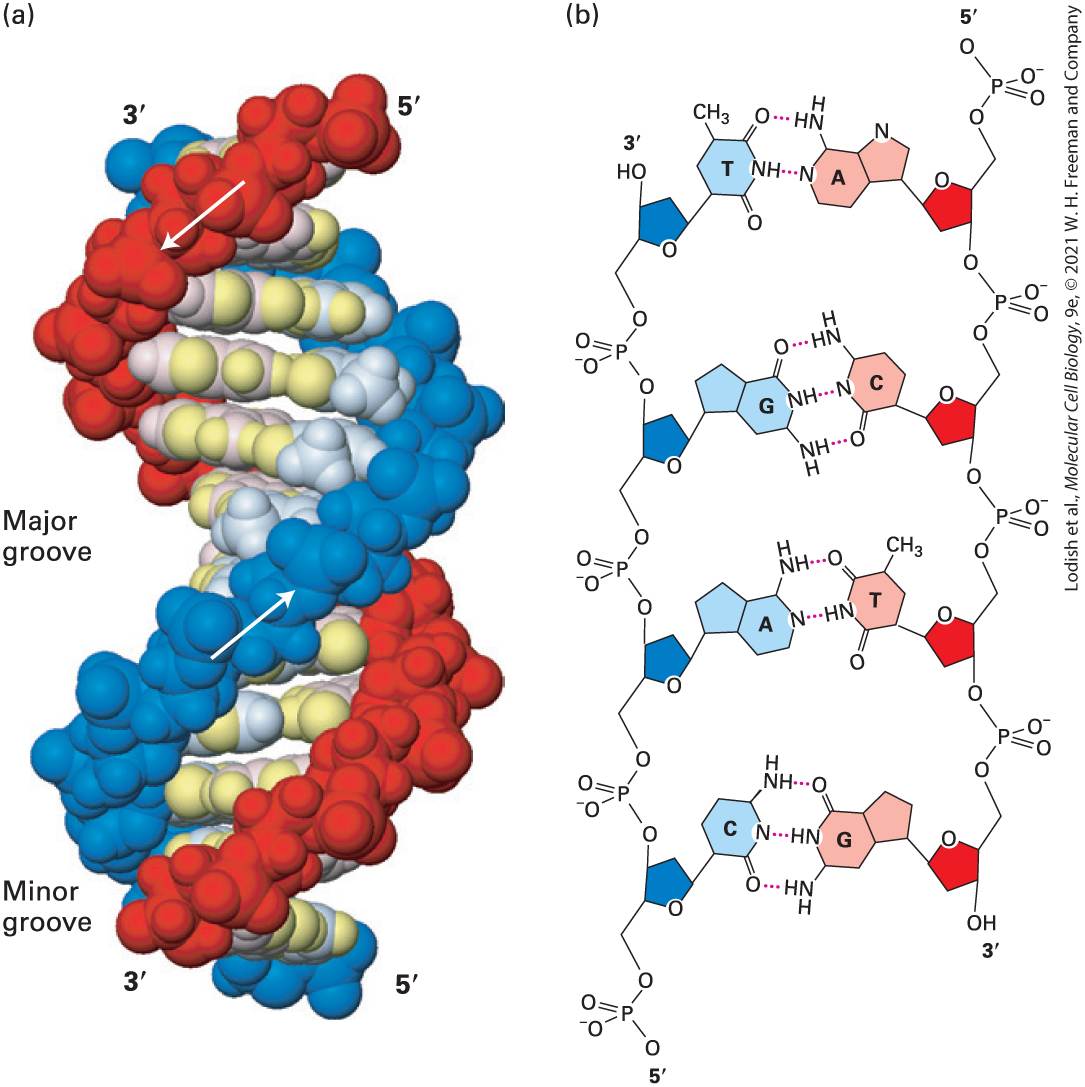
FIGURE 5-3 The DNA double helix. (a) Space-filling model of B DNA, the most common form of DNA in cells. The bases (light shades) project inward from the sugar-phosphate backbones (dark red and blue) of each strand, but their edges are accessible through major and minor grooves. Arrows indicate the direction of each strand. Hydrogen bonds between the bases are in the center of the structure. The major and minor grooves are lined
by potential hydrogen bond donors and acceptors (highlighted in yellow). (b) Chemical structure of DNA double helix. This extended schematic shows the two sugar-phosphate backbones and hydrogen bonding between the Watson-Crick base pairs, A·T and G·C. See R. E. Dickerson, 1983, Sci. Am. 249:94. [Part (a) Data from R. Wing et al., 1980, Nature 287:755, PDB ID 1bna.] Description In the illustration labeled A, Two arrows point in opposite directions along the length of the two strands of the helix. An arrow in the red strand points downward from 5 prime to 3 prime and another arrow in the blue strand point upward from 5 prime to 3 prime. The bases and sugar-phosphate backbones in the D N A are represented in colored beads. Grooves formed because of the spiral shape of the ribbon-like strands are labeled Minor and major. Minor is the smaller-sized groove, and major is the largersized groove. In the illustration labeled B, Adenine from one strand is paired with Thymine from the other strand, and Guanine from one strand is paired with Cytosine from the other strand at the center of the double helix. Adenine’s and Thymine’s have two hydrogen bonds between them. Guanine’s and Cytosine’s have three hydrogen bonds between them. The nitrogenous bases attach to the sugar and phosphate groups along the length of each D N A strand. In natural DNA, A always hydrogen-bonds with T and G with C, forming A·T and G·C base pairs as shown in Figure 5-4. These associations, always between a larger purine and a smaller pyrimidine, are often called WatsonCrick base pairs. Two polynucleotide strands, or regions thereof, in which all the nucleotides form such base pairs are said to be complementary. However, in theory and in synthetic DNAs, other base pairs can form. For example, guanine (a purine) could theoretically form hydrogen bonds with thymine (a pyrimidine) within the space available in the helix. Similarly, pairing between adenosine and cytosine could also be accommodated. However, as shown in Figure 5-4, nonstandard G·T and A·C base pairs
deviate sufficiently from the precise geometry of natural base pairs to be excluded from double-stranded DNA by the DNA-copying enzyme, as described later in this chapter.
FIGURE 5-4 (a) The structure of the standard A·T and G·C base pairs showing the angle and distance between the bonds connecting the bases to the ribose-phosphate backbone. Because the geometry of the base pairs is almost identical, both of these base pairs are accommodated in the double-helical structure of DNA. (b) Nonstandard base pairs, such as G·T and A·C, have very different bond geometries and are thus excluded from natural DNA by the copying mechanism of DNA polymerase. Description The illustration labeled A shows the chemical structure of two nucleotide base pairs of a D N A strand – Thymine and adenine (T A), cytosine and guanine (C G). A bidirectional pink arrow below the chemical structures of T-A and C-G indicates the
specific distance and angle between bonds. T-A bond matches both the distance and angle of the pink line and the C-G angle is slightly steeper. The illustration labeled B shows the chemical structure of two nucleotide base pairs of a D N A strand – Cytosine and adenine (C A), thymine and guanine (T G). A bidirectional pink arrow below the chemical structure of C-A and T-G indicates the specific distance and angle between bonds. Both C A and T G are tilted to the opposite angle with the same distance. Almost all DNA in cells takes the form of a right-handed helix. The x-ray diffraction pattern of DNA indicates that the stacked bases are regularly spaced 0.34-nm apart along the helix axis. The helix makes a complete turn every to , depending on the sequence; thus there are about 10–10.5 base pairs per turn. This helical form, referred to as the B form of DNA, is the normal form present in most DNA stretches in cells. On the outside of the helix, the spaces between the intertwined strands form two helical grooves of different widths, described as the major groove and the minor groove (see Figure 5-3a). As a consequence, the atoms on the edges of each base within these grooves are accessible from outside the helix, forming two types of binding surfaces. DNA-binding proteins can read the sequence of bases in duplex DNA by contacting atoms in either the major or the minor grooves. Although the atoms facing the surfaces of the major and minor grooves differ between the four bases, the overall structure of the intertwined strands of DNA is uniform regardless of the base sequence. Any irregularities in the DNA caused by chemical damage or mispairing of bases will disrupt this uniform structure and, as we will see in Section 5.3, will allow DNA repair enzymes to identify and act on the site of the damage.
Important modifications in the structure of standard B-form DNA come about as a result of protein binding to specific DNA sequences. Although the multitude of hydrogen and hydrophobic bonds between the bases provides stability to DNA, the double helix is flexible about its long axis. Unlike the α helix in proteins (see Figure 3-4), it has no hydrogen bonds parallel to the axis of the helix. This property allows DNA to bend when complexed with a DNA-binding protein, such as the transcription factor TBP (Figure 5-5). Bending of DNA is also critical to the dense packing of DNA in chromatin, the protein-DNA complex in which nuclear DNA occurs in eukaryotic cells (see Chapter 8).
FIGURE 5-5 Interaction with a protein such as TBP can bend DNA. The conserved C-terminal domain of the TATA box–binding protein (TBP) binds to the minor groove of specific DNA sequences rich in A and T, untwisting and sharply bending the double helix. Transcription of most eukaryotic genes requires participation of TBP. [Data from D. B. Nikolov and S. K. Burley, 1997, Proc. Nat’l. Acad. Sci. USA 94:15, PDB ID 1cdw.] Why did DNA, rather than RNA, evolve to be the carrier of genetic information in cells? The hydrogen at the position in the deoxyribose of DNA makes it a far more stable molecule than RNA, which instead has a
hydroxyl group at the position of ribose (see Figure 2-16). The - hydroxyl groups in RNA participate in the slow, -catalyzed hydrolysis of phosphodiester bonds at neutral (as shown in Figure 5-6). The absence of -hydroxyl groups in DNA prevents this process. Therefore, the presence of deoxyribose in DNA makes it a more stable molecule — a characteristic that is critical to its function in the long-term storage of genetic information.
DNA Can Undergo Reversible Strand Separation
FIGURE 5-6 Spontaneous hydrolysis of RNA catalyzed by the -hydroxyl group. The -hydroxyl group in RNA can act as a nucleophile, attacking the phosphodiester bond. The , cyclic monophosphate derivative is further hydrolyzed to a mixture of and monophosphates. Importantly, this facile mechanism of phosphodiester bond hydrolysis cannot occur in DNA, which lacks -hydroxyl groups. Description The illustration shows the hydrolysis of R N A to 2 prime 3 prime cyclic monophosphate derivative. The hydroxyl group in the second position of the ribose sugar acts on the phosphodiester bond connected to two bases labeled base subscript n at the top and base subscript n plus 1 at the bottom. Phosphodiester bond is attached to C 5 of base subscript n and C 3 of base subscript n plus 1. The nucleophilic activity of 2 prime hydroxyl group hydrolyzes the phosphodiester bond between the bases of R N A and forms 2 prime 3 prime cyclic monophosphate derivative. 2 prime 3 prime cyclic monophosphate derivative contains two ribose sugars. The first ribose sugar is with base subscript n attaches to two phosphate groups at C 2, C 3 and C 5, and the second ribose sugar with base subscript n plus 1 has a hydroxyl group at C 5 and phosphate group at C 3. DNA Can Undergo Reversible Strand Separation During replication and transcription of DNA, the strands of the double helix must separate to allow the internal edges of the bases to pair with the bases of the nucleotides being polymerized into new complementary polynucleotide chains. In later sections, we describe the cellular mechanisms that separate and subsequently reassociate DNA strands during replication and transcription. Here we discuss the fundamental
factors that influence the separation and reassociation of DNA strands. These properties of DNA were elucidated by in vitro experiments. The unwinding and separation of DNA strands, referred to as denaturation, or melting, can be induced experimentally by increasing the temperature of a solution of DNA. As the thermal energy increases, the resulting increase in molecular motion eventually breaks the hydrogen bonds and other forces that stabilize the double helix. The strands then separate, driven apart by the electrostatic repulsion of the negatively charged deoxyribose-phosphate backbones of the two strands. Near the denaturation temperature, a small increase in temperature causes a rapid, nearly simultaneous loss of the multiple weak interactions holding the strands together along the entire length of the DNA molecules (Figure 57a). The stacked base pairs in duplex DNA absorb less ultraviolet (UV) light than the unstacked bases in single-stranded DNA, and this change leads to an abrupt increase in the absorption of UV light. This phenomenon, known as hyperchromicity, is useful for monitoring DNA denaturation.
EXPERIMENTAL FIGURE 5-7 G·C content of DNA affects melting temperature. The temperature at which DNA denatures increases with the proportion of G·C pairs. (a) Melting of double-stranded DNA can be monitored by its absorption of UV light at . As regions of double-stranded DNA unpair, the absorption of light by those regions increases almost twofold. The temperature at which half the bases in a double-stranded DNA sample have denatured is denoted (for “temperature of melting”). (b) The is a function of the G·C content of the DNA; the greater the percentage, the higher the . Description In the graph labeled A, the vertical axis plots fraction single-stranded ranging from 0.5 to 1.0 in increments of 0.25. The horizontal axis plots temperature in degree Celsius ranging from 75 to 90 in increments of 5. In 40 percent G C pairs, the purple line starts at (75, 0.55), increases at (80, 0.55), peaks at (85, 1.0), and ends at (90, 1.0). A vertical and horizontal dotted line at (82.5, 0.75) represents the melting point (Big T subscript small m). In 50 percent G C pairs, the blue line starts at (75, 0.55), increases at (82.5, 0.55), peaks at (87, 1.0), and ends at (90, 1.0). The double-stranded D N A is labeled at the starting point of the graph and the single-stranded D N A is labeled at the peaks of the graph In the graph labeled B, the vertical axis plots percentage of G C pairs ranging from 0 to 100 in increments of 20. The horizontal axis plots melting temperature in degree Celsius ranging from 0 to 110 in varying increments. A linear graph starts at (70, 0) and ends at (110, 100). The melting temperature at which DNA strands separate depends on several factors. Molecules that contain a greater proportion of G·C pairs require higher temperatures to denature because the three hydrogen bonds in G·C pairs make these base pairs more stable than A·T pairs, which have only two hydrogen bonds. Indeed, the percentage of G·C base pairs in a DNA sample can be estimated from its (Figure 5-7b). The ion concentration of the solution also influences the because the negatively
DNA Molecules Can Acquire Torsional Stress
charged phosphate groups in the two strands are shielded by positively charged ions. When the ion concentration is low, this shielding is decreased, thus increasing the repulsive forces between the strands and reducing the . Agents that destabilize hydrogen bonds, such as formamide or urea, also lower the . Finally, extremes of denature DNA at low temperatures. At low (acid) , the bases become protonated and thus positively charged, repelling each other. At high (alkaline) , the bases lose protons and become negatively charged, again repelling each other because of their similar charges. In cells, and temperature are, for the most part, maintained at a constant level. These features of DNA denaturation are most useful for manipulating DNA in a laboratory setting. The single-stranded DNA molecules that result from denaturation form random coils without an organized structure. Lowering the temperature, increasing the ion concentration, or neutralizing the causes two complementary strands to reassociate into a perfect double helix. The extent of such renaturation is dependent on time, the DNA concentration, and the ion concentration. Two DNA strands that are not related in sequence will remain as random coils and will not renature, but they will not inhibit complementary DNA partner strands from finding each other and renaturing. Denaturation and renaturation of DNA are the basis of nucleic acid hybridization, a powerful technique used to study the relatedness of two DNA samples and to detect and isolate specific DNA molecules in a mixture containing numerous different DNA sequences (see Chapter 6).
DNA Molecules Can Acquire Torsional Stress Many bacterial genomic DNAs and many viral DNAs are circular molecules. Circular DNA molecules also occur in mitochondria, which are present in almost all eukaryotic cells, and in chloroplasts, which are present in plants and some unicellular eukaryotes. Although eukaryotic nuclear DNA is linear, long loops of DNA are fixed in place within chromosomes (see Chapter 8). Each of the two strands in a circular DNA molecule or in a fixed loop of a eukaryotic chromosome forms a closed structure without free ends and is therefore subject to torsional stress. When DNA is either overwound or underwound, meaning that it has more or fewer helical turns than B-form linear DNA of the same length, the torsional stress thus produced will be relieved by the DNA molecule twisting back on itself forming supercoils (Figure 5-8a). A similar topological interconversion between turns and supercoils can be demonstrated by the twists that form in a rubber band after being rolled between your fingertips. As we will see in the next section, when DNA replicates, the two strands must unwind; the remaining portion of the molecule would gain one supercoil for the unwinding of every complete turn of the strands in the double helix, which occurs every 10 base pairs. Since a typical replication fork moves at a rate of about 500 bases per second, supercoils would be introduced into the DNA ahead of the moving replication fork at a rate of 50 per second. To relieve the torsional stress that would be caused by supercoils developed during replication, all cells
contain topoisomerase I. This enzyme binds to DNA at random sites and breaks a phosphodiester bond in one strand. Such a one-strand break in DNA is called a nick. The broken end then winds around the uncut strand, leading to loss of supercoils (Figure 5-8b). Finally, the same enzyme joins (ligates) the two ends of the broken strand. Another type of enzyme, topoisomerase II, relieves torsional stress in DNA by making breaks in both strands of a double-stranded DNA and then religating them. EXPERIMENTAL FIGURE 5-8 Topoisomerase I relieves torsional stress on DNA. (a) Electron micrograph of SV40 viral DNA. When the circular DNA of the SV40 virus is isolated and separated from its associated protein, the DNA duplex is underwound and assumes the supercoiled configuration. (b) If a supercoiled DNA is nicked (i.e., one strand cleaved), the strands can rewind, leading to loss of a supercoil. Topoisomerase I catalyzes this reaction and also reseals the broken ends. All the supercoils in isolated SV40 DNA can
be removed by the sequential action of this enzyme, producing the relaxed-circle conformation. For clarity, the shapes of the molecules at the bottom have been simplified. KEY CONCEPTS OF SECTION 5.1 The Double-Helical Structure of DNA Deoxyribonucleic acid (DNA), the genetic material, is made up of two long, unbranched polymers of nucleotides. A nucleotide consists of a phosphorylated pentose linked to either a purine-adenine (A) or guanine (G), or a pyrimidine-cytosine (C) or thymine (T). Adjacent nucleotides in a polynucleotide are linked by phosphodiester bonds. The entire strand has a chemical directionality with and ends (see Figure 5-2). Natural DNA contains two complementary antiparallel polynucleotide strands wound together into a regular right-handed double helix with the bases on the inside and the two sugar-phosphate backbones on the outside (see Figure 5-3). The double-stranded structure of DNA is significantly stabilized by noncovalent bonds consisting of hydrogen bonds between pairs of bases on opposite strands and by hydrophobic interactions between adjacent base pairs stacked perpendicular to the helix axis. The standard Watson-Crick base pairs — G·C and A·T — have nearly identical dimensions and are the only base pairs accommodated by the native double-helical structure. Binding of protein to DNA can deform its helical structure, causing local bending or unwinding of the DNA molecule. Heat causes the DNA strands to separate (denature). The melting temperature of DNA increases with the percentage of G·C base pairs. Under suitable conditions, separated complementary nucleic acid strands will renature. Overwinding or underwinding of DNA causes long DNA molecules to twist on themselves, forming supercoils (see Figure 5-7). Unwinding of chromosomal DNA during replication would generate a large number of supercoils except that enzymes called topoisomerases relieve this torsional stress by removing supercoils from DNA molecules.
5.2 DNA Replication
5.2 DNA Replication In a human cell, the genetic information encoded in DNA resides in 23 pairs of chromosomes, with each chromosome consisting of a doublestranded helical molecule hundreds of millions of nucleotides long. When cells divide, each of the two daughter cells must contain exactly the same genetic information as the parent cell. Thus every cell division must be accompanied by the exact copying of the DNA sequence of each chromosome by a process known as DNA replication. A key feature of the double-helical DNA structure that was immediately apparent to Watson and Crick was that the invariant rules for base pairing (A with T and G with C) mean that each strand of a DNA polymer carries the same information in complementary form. The implication was that DNA replication could be achieved by separating the two strands of the DNA polymer and then using each strand as a template in the synthesis of new, complementary strands, thus producing two duplicate, double-stranded copies of the original DNA molecule. An understanding of the biochemical mechanism for copying of DNA came from the isolation of the first template-directed DNA polymerase enzyme from extracts of Escherichia coli cells by Arthur Kornberg and colleagues in 1956. Although we now know that the isolated enzyme known as DNA polymerase I is involved in DNA repair and not the replication of the E. coli chromosome, all template-directed DNA
DNA Polymerases Require a Template and a Primer to Replicate DNA
polymerases operate by the same fundamental rules. The lessons learned from studying DNA polymerase I have proved to be universal. DNA Polymerases Require a Template and a Primer to Replicate DNA All DNA polymerases have the same requirements for synthesis of a new DNA strand: i. A single-stranded DNA template. ii. A DNA primer base paired with the template, and with a free hydroxyl group at the end of the primer to accept a new nucleotide. iii. A source of deoxyribonucleoside -triphosphate (dNTPs) precursors. With a primer base-paired to the template strand, a DNA polymerase will add deoxyribonucleotides to the free hydroxyl group at the end of the
primer, as directed by the sequence of the template strand. The energy for this reaction comes from the release of diphosphate when a phosphoester bond forms between the oxygen and the α phosphate of the appropriate dNTP. The equilibrium for the reaction is driven further toward chain elongation by pyrophosphatase, an enzyme that catalyzes cleavage of the released into two molecules of inorganic phosphate (Figure 5-9). After the addition of a nucleotide, a new primer end will be available to accept the next nucleotide. Polymerization thus proceeds in the direction and will continue until the growing end of the primer reaches the end of the template strand.
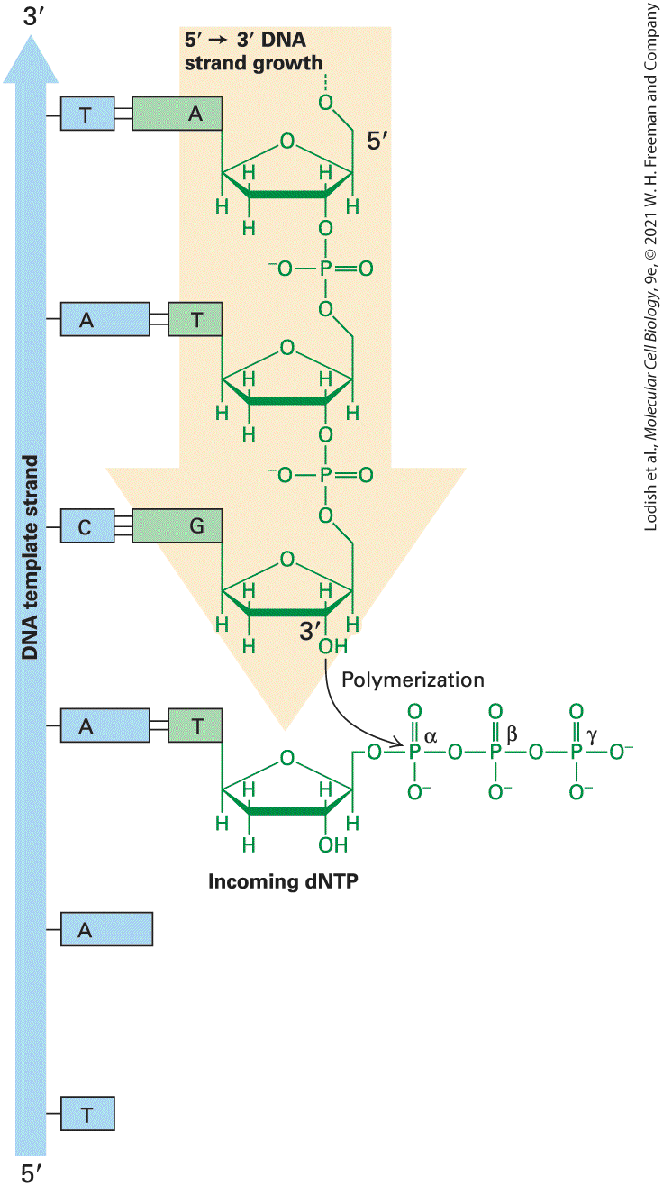
FIGURE 5-9 DNA is synthesized from dNTP precursors. The three requirements for DNA synthesis are a primer strand with a free terminus, a template strand that is base paired to the primer, and a source of deoxyribonucleoside triphosphates (dNTPs). DNA polymerase will act on this substrate to add a new dNTP at the end of the primer strand as specified by base pairing between the added base and the template DNA strand. DNA polymerase catalyzes formation of a phosphodiester bond between the oxygen of the primer strand and the α phosphate of a correctly base-paired dNTP. New DNA strands are always synthesized in the direction and are opposite in polarity to their template DNA strands. Description The illustration on the left shows a vertical D N A template strand from 5 prime to 3 prime direction (bottom to top) with Thymine, Adenine, Adenine, Cytosine, Adenine, and Thymine bases. The complementary strand on the right from 5 prime to 3 prime direction (top to bottom) has Adenine, Thymine, Guanine, and Thymine bases complementary to D N A template strand. The hydrogen bonds between adenine and thymine are two and guanine and cytosine are three. The incoming d N T P's binds to the complementary base of the template D N A strand and polymerization occurs between the 3 prime oxygen of the newly synthesized strand and the alpha phosphate of the d N T P's. The d N T P's contains three phosphate groups, alpha, beta, and gamma. The faithful replication of chromosomal DNA is crucial for life, and DNA polymerases have evolved to replicate DNA with extraordinary speed and fidelity. A typical DNA polymerase can add a single nucleotide to a growing chain in about 2 milliseconds, but this does not give much time for the polymerase to discriminate between a correctly and an incorrectly base-paired nucleotide. If the fidelity of polymerization was based only on the difference in energy between the correctly and incorrectly hydrogenbonded nucleotide pair, the polymerase would make at least one mistake
every 50 nucleotides. A much higher degree of fidelity is achieved by the active site of the polymerase only accommodating the base pairs with the exact geometry of a normal Watson-Crick base pair while rejecting nonstandard base-pair geometries even if they are only slightly different (see Figure 5-4). By using this type of geometric selection, most polymerases can achieve a fidelity of initial nucleotide incorporation that would produce error rates of about 1 incorrect nucleotide per (10,000) polymerized nucleotides. The fidelity of DNA replication in most organisms is about 1 mistake in (one billion) nucleotides incorporated into a growing strand, which is much greater than can be explained by the maximum selectivity of base pairing. This remarkable accuracy is largely due to proofreading by DNA polymerases. Proofreading depends on the exonuclease activity of some DNA polymerases. When an incorrect base is incorporated during DNA synthesis, base pairing between the nucleotide of the nascent strand and the template strand does not occur. As a result, the polymerase pauses, then transfers the end of the growing chain to its exonuclease site, where the incorrect mispaired base is removed (Figure 5-10). The end is then transferred back to the polymerase site, where this region is copied correctly. Most DNA polymerases require a high degree of fidelity and thus have proofreading. For example, all three E. coli DNA polymerases have proofreading exonuclease activity.
FIGURE 5-10 Proofreading by DNA polymerase. All DNA polymerases have a similar three-dimensional structure, which resembles a half-opened right hand. The “fingers” bind the single-stranded segment of the template strand, and the polymerase catalytic activity (Pol) lies in the junction between the fingers and “palm.” As long as the correct nucleotides are added to the end of the growing strand, it remains in the polymerase site. Incorporation of an incorrect base at the end results in a substrate that does not satisfy the requirements for polymerization of the next nucleotide and the polymerase pauses. Because the end of the primer strand is not stably base-paired to the template, it is free to move to the exonuclease site (Exo) about away, where the mispaired base is removed. Subsequently, the new end, now freed of the mispaired nucleotide, can base pair properly with the template and polymerization can resume. See C. M. Joyce and T. T. Steitz, 1995, J. Bacteriol. 177:6321, and S. Bell and T. Baker, 1998, Cell 92:295. Description The illustrations depict the process of D N A proofreading. The D N A polymerases are illustrated to show how they resemble a hand with fingers, a palm, and a thumb. The illustration on the left shows the finger part on the left, labeled Pol (polymerase site), and the lower palm area labeled, Exo (exonuclease site). The double-stranded D N A with a template strand and growing strand passes through the palm in the hand like structure. The growing strand ends in the Pol site and the template strand remains adjacent to the pol site. The illustration on the right shows the same structure and labels but the growing strand ends in the Exo site and the template strand remains adjacent to the pol site.
Duplex DNA Is Unwound, and Daughter Strands Are Formed at the DNA Replication Fork
Duplex DNA Is Unwound, and Daughter Strands Are Formed at the DNA Replication Fork In order for duplex DNA to function as a template during replication, the two intertwined strands must be unwound, or melted, to make their bases available for pairing with the bases of the dNTPs that are polymerized into the newly synthesized daughter strands. This unwinding of the parent DNA strands is performed by enzymes called helicases. Unwinding begins at segments in a DNA molecule called replication origins, or simply origins. The nucleotide sequences of origins from different organisms vary greatly, although they usually contain AT-rich sequences. DNA polymerases cannot initiate replication de novo because, as we have seen, all of these polymerases require a free primer terminus. However, RNA polymerases that have a similar mechanism to DNA polymerases do have the capability of initiating new strand synthesis from a template without a preexisting primer (see Figure 5-23a). Once helicases have unwound the parent DNA at an origin, a specialized RNA polymerase called primase forms a short (∼12-nucleotide) RNA primer complementary to the unwound template strands. The primer, still base-paired to its complementary DNA strand, is then elongated by DNA polymerase α for another 25 nucleotides or so, forming a primer made of RNA at the end and DNA at the end. This primer is further extended by DNA polymerase δ, thereby forming a new daughter strand.
The DNA region at which all these proteins come together to carry out the synthesis of daughter strands is called the replication fork. As replication proceeds, the replication fork and the associated proteins move away from the origin. As noted earlier, local unwinding of duplex DNA produces torsional stress, which is relieved by topoisomerase I. In order for DNA polymerases to move along and copy a duplex DNA, helicase must sequentially unwind the duplex and topoisomerase must remove the supercoils that form. A major complication in the operation of a DNA replication fork arises from two properties of DNA: the two strands of the parent DNA duplex are antiparallel, and DNA polymerases (like RNA polymerases) can add nucleotides to the growing daughter strands only in the direction. Synthesis of one daughter strand, called the leading strand, can proceed continuously from a single RNA primer in the direction, the same direction as the movement of the replication fork (Figure 5-11). The problem comes in the synthesis of the other daughter strand, called the lagging strand.
FIGURE 5-11 Leading-strand and lagging-strand DNA synthesis. Nucleotides are added by a DNA polymerase to each growing daughter strand in the direction (indicated by arrowheads). The leading strand is synthesized continuously from a single RNA primer (red) at its end. The lagging strand is synthesized discontinuously from multiple RNA primers that are formed periodically as each new region of the parent duplex is unwound. Elongation of these primers initially produces Okazaki fragments. As each growing fragment approaches the previous primer, that primer is removed and the fragments are ligated. Repetition of this process eventually results in synthesis of the entire lagging strand. Description The illustration shows D N A replication fork with an arrow pointing toward the left labeled direction of the replication fork. A continuous arrow complementary to the 3 prime to 5 prime of parental D N A strand (light blue) is the leading strand (Green) and three short discontinuous arrows complementary to the 5 prime to 3 prime parental D N A strand (Blue) is the lagging strand (Light green). A single R N A primer (red) is in the 5 prime of the leading strand and three R N A primers are in the 5 prime of the lagging strand. An arrow combining the R N A primer and short sequences of D N A nucleotides are labeled as Okazaki fragment.
A DNA Replication Fork Advances by Cooperation of Multiple Proteins
Because growth of the lagging strand must occur in the direction, copying of this strand must proceed in the opposite direction from the movement of the replication fork. A cell accomplishes this feat by synthesizing a new primer every 100 to 200 nucleotides on that template strand as more of the strand is exposed by unwinding. Each of these primers, base-paired to the template strand, is elongated in the direction, forming discontinuous segments named Okazaki fragments after their discoverer, Reiji Okazaki (see Figure 5-11). The RNA primer of each Okazaki fragment is removed and replaced by DNA chain growth from the neighboring Okazaki fragment; and an enzyme called DNA ligase joins the adjacent fragments. A DNA Replication Fork Advances by Cooperation of Multiple Proteins Detailed understanding of the eukaryotic proteins that participate in DNA replication initially came from studies with small viral DNAs, particularly the circular genome of SV40, a virus which infects monkeys. Virusinfected cells replicate large numbers of the simple viral genome in a short period of time, which makes them an ideal model system for studying basic aspects of DNA replication. Because simple viruses such as SV40 depend largely on the DNA replication machinery of their host cells (in this case monkey cells), they offer a unique opportunity to study the replication of multiple identical, small DNA molecules by cellular proteins. Figure 5-12 depicts the multiple proteins that coordinate the copying of SV40 DNA at a replication fork. The assembled proteins at a
replication fork further illustrate the concept of molecular machines introduced in Chapter 3. These multicomponent complexes permit the cell to carry out an ordered sequence of events that accomplishes essential cell functions.
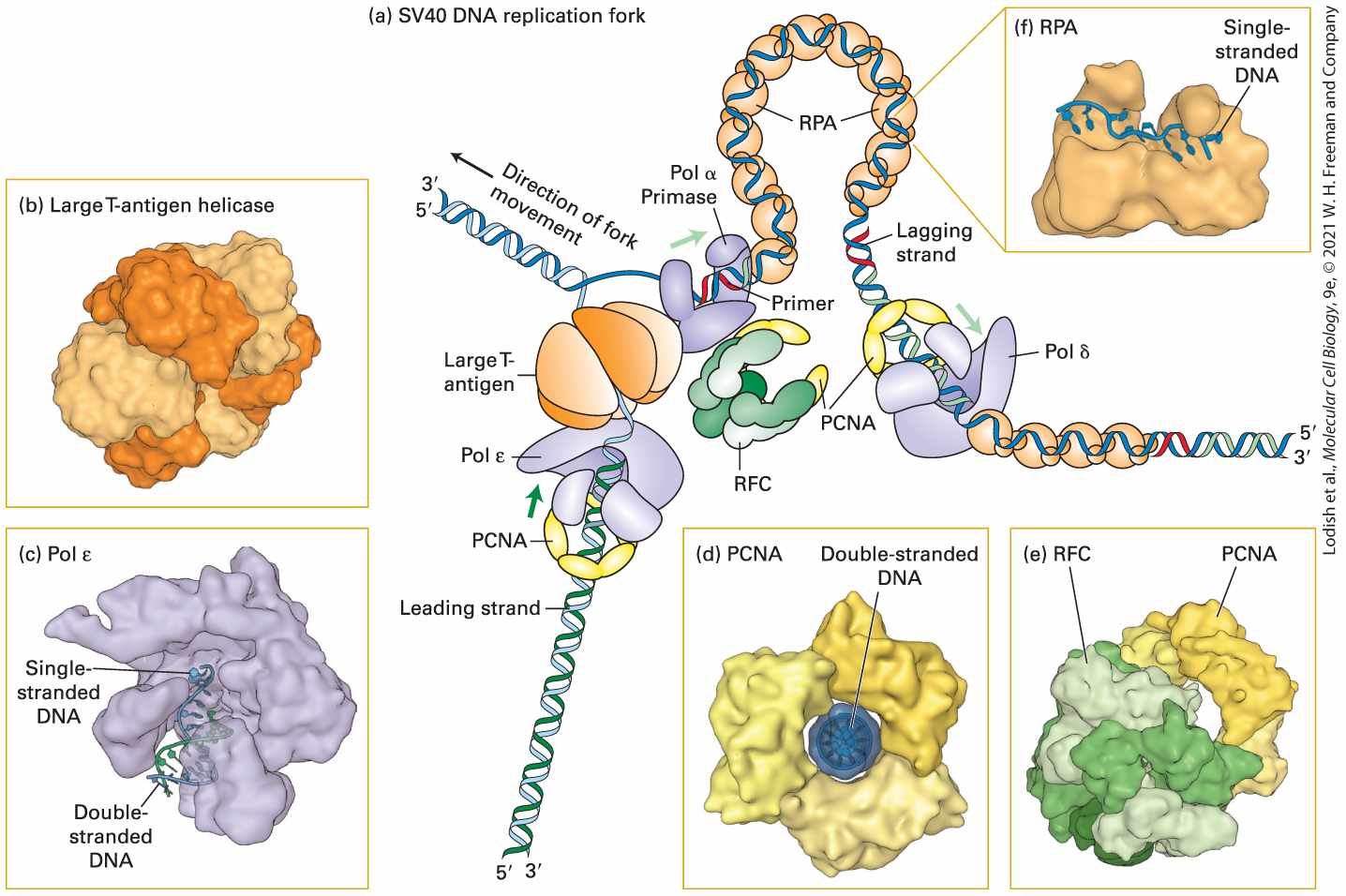
FIGURE 5-12 Model of an SV40 DNA replication fork. (a) A hexamer of large T-antigen, a viral protein, functions as a helicase to unwind the parent DNA strands. The leading strand is extended by DNA polymerase ε (Pol ε) up to the replication fork. Pol ε is bound to a ring of PCNA that surrounds the daughter double-stranded DNA so that the Pol ε–PCNA complex remains stably associated with the replication fork. The single-stranded region of the lagging-strand template generated by T-antigen helicase is bound by multiple copies of the heterotrimeric protein RPA. Primers for lagging-strand synthesis (red, RNA; light green, DNA) are synthesized by a complex of primase and DNA polymerase α (Pol α). The end of each primer synthesized by Pol α–primase is then bound by a PCNA–Pol δ complex, which extends the primer and synthesizes most of each Okazaki fragment. (b) The helicase domain of SV40 T-antigen forms a hexameric replicative helicase. Subunits are shown in alternating light and dark orange. (c) Model of DNA polymerase ε extending the -end of
the leading strand. (d) The three subunits of PCNA, shown in different shades of yellow, form a circular structure with a central hole through which daughter double-stranded DNA passes. (e) RFC, the pentameric “clamp-loader” (monomers shown in different shades of green) is shown bound to a circular trimer PCNA before the PCNA clamp is opened. (f) The large subunit of RPA contains two domains that bind single-stranded DNA. Note that the single DNA strand is extended, with the bases oriented in an optimal conformation for replication by Pol δ. See M. O’Donnell, L. Langston, and B. Stillman, 2013, Cold Spring Harbor Perspect. Biol. 5:a010108. [Part (b) Data from D. Li et al., 2003, Nature 423:512, PDB ID 1n25. Part (c) Data from M. Hogg et al., 2014, Nat. Struct. Mol. Biol., 21:49, PDB ID 4m8o. Part (d) Data from J. M. Gulbis et al., 1996, Cell 87:297, PDB ID 1axc. Part (e) Data from G. D. Bowman, M. O’Donnell, and J. Kuriyan, 2004, Nature 429:724, PDB ID 1sxj. Part (f) Data from A. Bochkarev et al., 1997, Nature 385:176, PDB ID 1jmc.] Description The main (a) illustration is labeled S V 40 D N A Replication Fork. It shows a drawing of ribbons of D N A flowing through 5 different proteins. At the bottom left, the leading strand of D N A moves through P C N A, Pol epsilon, Large T-Antigen proteins. At the same time, on the right, another genetic strand goes through a different Pol delta, then P C N A, then R P A, to a Pol alpha primase before meeting the leading strand. An arrow where they meet is labeled direction of fork movement. (b) is a close up inset drawing of Large T-antigen helicase (c) is a close up inset of Pol epsilon, with labels for single strand D N A and double strand D N A shown in the lower middle of the protein. (d) is an inset of P C N A that is surrounding a purple circle of double stranded D N A. (e) is R F C, which is attached at the bottom left to a P C N A protein. (f) is R P A with a label showing single stranded D N A moving across the top part of this protein. The molecular machine that replicates SV40 DNA contains only one viral protein; all other proteins involved in SV40 DNA replication are provided by the host cell. This viral protein, large T-antigen, forms a hexameric replicative helicase, a protein that uses energy from ATP hydrolysis to
unwind the parent strands at a replication fork. Primers for the leading and lagging daughter strands are synthesized by a complex of primase, which synthesizes a short RNA primer (∼12 nucleotides), and DNA polymerase α (Pol α), which extends the RNA primer with deoxyribonucleotides for another 25 nucleotides or so, forming a mixed RNA-DNA primer. The short RNA-DNA primer is extended by the high fidelity DNA polymerase δ (Pol δ), which possess a proofreading mechanism based on exonuclease activity. During the replication of cellular DNA, Pol δ synthesizes lagging-strand DNA, while a second high-fidelity polymerase, DNA polymerase ε (Pol ε), synthesizes most of the length of the leading strand. Pol δ and Pol ε each form a complex with PCNA (proliferating cell nuclear antigen), which displaces the primase–Pol α complex following primer synthesis. As illustrated in Figure 5-12d, PCNA is a homotrimeric protein that has a central hole through which the daughter duplex DNA passes, thereby preventing the PCNA–Pol δ and PCNA–Pol ε complexes from dissociating from the template. As such, PCNA is known as a sliding clamp that enables Pol δ and Pol ε to remain stably associated with a single template strand for thousands of nucleotides. A pentameric protein called RFC (replication factor C) functions to open the PCNA ring so that it can encircle the short region of double-stranded DNA synthesized by Pol α. Consequently, RFC is called a clamp loader. After parent DNA is separated into single-stranded templates at the replication fork, the leading strand is extended by Pol ε, which can extend the growing strand up to the replication fork. The single-stranded template for lagging-strand synthesis is bound by multiple copies of RPA
DNA Replication Occurs Bidirectionally from Each Origin
(replication protein A), a heterotrimeric protein (Figure 5-12c). Binding of RPA maintains the template in a uniform conformation that is optimal for copying by Pol δ. Bound RPA proteins are dislodged from the parent strand by Pol δ as it synthesizes the complementary strand base-paired with the parent strand. Several other eukaryotic proteins that function in DNA replication are not depicted in Figure 5-12. For example, topoisomerase I associates with the parental double-stranded DNA ahead of the replicative helicase (i.e., to the left of large T-antigen in Figure 5-12) to remove torsional stress introduced by the unwinding of the parent strands (see Figure 5-7a). Ribonuclease H and FEN I remove the ribonucleotides at the ends of Okazaki fragments; these ribonucleotides are replaced by deoxyribonucleotides added by Pol δ as it extends the upstream Okazaki fragment. Successive Okazaki fragments are coupled by DNA ligase through standard phosphoester bonds. Other specialized DNA polymerases are involved in the repair of mismatches and lesions in DNA (see Section 5.3). DNA Replication Occurs Bidirectionally from Each Origin As indicated in Figures 5-11 and 5-12, both parent DNA strands that are exposed by local unwinding at a replication fork are copied into daughter strands. In theory, DNA replication from a single origin could involve one replication fork that moves in one direction. Alternatively, two replication
forks might assemble at a single origin and then move in opposite directions, leading to bidirectional growth of both daughter strands. We now know that all bacterial, archaeal, and eukaryotic cells employ a bidirectional mechanism of DNA replication. In the case of SV40 DNA, replication is initiated by the binding of two large T-antigen hexameric helicases to the single SV40 origin and the assembly of other proteins to form two replication forks. These forks then move away from the SV40 origin in opposite directions, and leading- and lagging-strand synthesis occurs at both forks. As shown in Figure 5-13, the left replication fork extends DNA synthesis in the leftward direction; similarly, the right replication fork extends DNA synthesis in the rightward direction.
FIGURE 5-13 Bidirectional mechanism of DNA replication. The left replication fork here is comparable to the replication fork diagrammed in Figure 5-29 (although that figure also shows proteins other than large T-antigen, which are not shown here). Top: Two large Tantigen hexameric helicases first bind at the replication origin in opposite orientations. Step 1 : Using energy provided by ATP hydrolysis, the helicases move in opposite directions, unwinding the parent DNA and generating single-stranded templates, which are bound by RPA proteins (not shown). Step 2 : Primase–Pol α complexes synthesize short primers (red) base-paired to each of the separated parent strands. Step 3 : PCNA-Rfc–Pol ε complexes replace the primase–Pol α complexes and extend the short primers, generating the leading strands (dark green) at each replication fork. Step 4 : The helicases further unwind the parent strands, and RPA proteins bind to the newly exposed single-stranded regions. Step 5 : PCNA-Rfc–Pol ε complexes extend the leading strands farther. Step 6 : Primase–Pol α complexes synthesize primers for lagging-strand synthesis at each replication fork. Step 7 : PCNA-Rfc–Pol δ complexes displace the primase–Pol α complexes and extend the lagging-strand Okazaki fragments (light green), which are eventually ligated to the ends of the leading strands. The position where ligation occurs is represented by a circle. Replication continues by further unwinding of the parent strands and synthesis of leading and lagging strands as in steps 4 – 7 . (Although depicted here as individual steps for clarity, unwinding and synthesis of leading and lagging strands occur concurrently.) Description The steps involved in the process of bidirectional mechanism of D N A replication are as follows: Step 1: Unwinding. Step 2: Leading strand primer synthesis. Step 3: Leading strand extension. Step 4: Unwinding. Step 5: Leading strand extension. Step 6: Lagging strand primer synthesis.
Step 7: Lagging strand extension. Unlike SV40 DNA, eukaryotic chromosomal DNA molecules contain multiple replication origins separated by tens to hundreds of kilobases. A six-subunit protein called ORC, for origin recognition complex, binds to each origin and associates with other proteins required to load cellular hexameric helicases composed of six homologous MCM (minichromosome maintenance) proteins. Two MCM helicases, oriented in opposite directions, separate the parent strands at an origin, and RPA proteins bind to the resulting single-stranded DNA. Synthesis of primers and subsequent steps in the replication of cellular DNA are thought to be analogous to those in SV40 DNA replication (see Figure 5-13). Cell division begins with the duplication of chromosomes; therefore, the initiation of DNA replication is usually the first step in the cell division cycle. Cellular DNA replication is initiated by activation of MCM helicase by a specific protein kinase (DDK), which in turn is regulated by S-phase cyclin-dependent kinases. Other cyclin-dependent kinases regulate additional aspects of cell proliferation, including the complex process of mitosis by which a eukaryotic cell divides into two daughter cells. Mitosis and another specialized type of cell division called meiosis, which generates haploid sperm and egg cells, is discussed in Chapter 6. We discuss the various regulatory mechanisms that determine the rate of cell division in Chapter 19. KEY CONCEPTS OF SECTION 5.2
DNA Replication DNA polymerases require, as a substrate, a template strand that is base paired to a primer strand with an available hydroxyl group. Newly synthesized DNA extends from the primer in the direction. The high fidelity of DNA replication comes from rigorous geometric constraints for only A·T and G·C base pairs at the polymerase active site, and by a proofreading exonuclease that removes incorrect base pairs. Replication begins at a sequence called an origin. Each eukaryotic chromosomal DNA molecule contains multiple replication origins. Replication initiates at an origin by unwinding of the DNA and initiation of new synthesis by an RNA polymerases known as primase. After initiation replication proceeds from a replication fork, at which one daughter strand (the leading strand) is elongated continuously. The other daughter strand (the lagging strand) is formed discontinuously (see Figure 5-11). Discontinuous replication of the lagging strand proceeds by formation of Okazaki fragments synthesized every 100 to 200 nucleotides. The ribonucleotide primers at the end of each Okazaki fragment are removed and replaced by elongation from the end of the next Okazaki fragment. Finally, adjacent Okazaki fragments are joined by DNA ligase. Helicases use energy from ATP hydrolysis to separate the parent (template) DNA strands, which are initially bound by multiple copies of a single-stranded DNAbinding protein, RPA. Primase synthesizes a short RNA primer, which remains basepaired to the template DNA. This primer is initially extended at the end by DNA polymerase α (Pol α), resulting in a short daughter strand made up of RNA and DNA. Most of the DNA in eukaryotic cells is synthesized by Pol δ and Pol ε, which take over from Pol α and continue elongation of the daughter strands in the direction. Pol δ synthesizes most of the length of the lagging strand, while Pol ε synthesizes the leading strand. Pol δ and Pol ε remain stably associated with the template by binding to PCNA, a trimeric protein that encircles the daughter duplex DNA, functioning as a sliding clamp. DNA replication generally occurs by a bidirectional mechanism in which two replication forks form at an origin and move in opposite directions, and both template strands are copied at each fork. MCM helicases initiate eukaryotic DNA replication in vivo at multiple origins spaced along chromosomal DNA. Synthesis of eukaryotic DNA is regulated by controls on the binding and activity of these helicases.
5.3 DNA Repair and Recombination
5.3 DNA Repair and Recombination Damage to DNA is unavoidable and arises in many ways. DNA damage can be caused by spontaneous cleavage of chemical bonds in DNA, by environmental agents such as ultraviolet and ionizing radiation, and by reaction with genotoxic chemicals that are by-products of normal cellular metabolism or occur in the environment. A change in the normal DNA sequence, called a mutation, can occur during replication when a DNA polymerase inserts the wrong nucleotide as it reads a damaged template. Mutations also occur at a low frequency as the result of copying errors introduced by DNA polymerases when they replicate an undamaged template. In humans, mutations that compromise the integrity of the genome can have multiple undesirable consequences. Mutations in the DNA in germ cells will be passed on to offspring, usually with deleterious effects. By contrast, mutations in somatic cells that affect genes that are normally involved in the careful regulation of cell division can cause uncontrolled cell division, eventually leading to tumor formation and cancer. It is thus not surprising that cells have evolved elaborate mechanisms to repair different kinds of damage to the integrity of DNA before the damage becomes fixed as a mutation that is passed to daughter cells.
Chemical and Radiation Damage to DNA Can Lead to Mutations
DNA repair mechanisms take advantage of the inherent redundancy of information contained in complementary strands of DNA and correct the damaged strand using information from the undamaged strand that is still intact. To accomplish this, most DNA repair processes follow the same general outline. All repair processes must first identify the site of the damage. This is usually done by scanning for disruptions to the regular structure of the DNA helix. Once a lesion is identified, most repair processes then selectively remove a portion of the damaged strand and copy the missing information from the intact complementary strand. In this section, we will review the mechanisms for repair of mismatches between normal bases, repair of chemically altered bases, and repair of broken ends of DNA. Chemical and Radiation Damage to DNA Can Lead to Mutations DNA is continually subjected to a barrage of damaging chemical reactions; estimates of the number of DNA-damage events in a single human cell range from to per day. Even if DNA were not exposed to damaging chemicals, certain aspects of DNA structure are inherently unstable. For example, the bond connecting a purine base to deoxyribose is prone to hydrolysis at a low rate under physiological conditions, leaving a sugar without an attached base. Thus coding information is lost on that strand, and this loss can lead to a mutation during DNA replication. Normal cellular reactions, including the movement of electrons along the electron-transport chain in mitochondria and lipid oxidation in
peroxisomes (see Chapter 12), produce several chemicals that react with and damage DNA, including hydroxyl radicals and superoxide . These chemicals can also cause chemical damage to DNA that can lead to mutations, including mutations that lead to cancers. Many spontaneous mutations are point mutations, which involve a change in a single base pair in the DNA sequence. If a point mutation falls within the protein-coding region of a gene, the mutation may change one codon to another, thus changing the amino acid sequence of the encoded protein. Another possibility is that the mutation may introduce a stop codon in the protein sequence, leading to early termination. Point mutations outside of the coding sequence can also interfere with gene function by, for example, altering the regulation of a gene’s transcription, as discussed in Chapter 8. One of the most frequent causes of point mutations is deamination of a cytosine (C) base, which converts it into a uracil (U) base. Another is deamination of the common modified base 5methylcytosine, which forms thymine when it is deaminated. If these alterations are not corrected before the DNA is replicated, the cell will use the strand containing U or T as a template to form a U·A or T·A base pair, thus creating a permanent change in the DNA sequence that is a mutation (Figure 5-14).
FIGURE 5-14 Deamination leads to point mutations. A spontaneous point mutation can be caused by deamination of 5-methylcytosine (C) to form thymine (T). If the resulting T·G base pair is not restored to the normal C·G base pair by base excision-repair mechanisms (step 1 ), it will lead to a permanent change in sequence (i.e., a mutation) following DNA replication (step 2 ). After one round of replication, one daughter DNA molecule will have the mutant T·A base pair and the other will have the wild-type C·G base pair. Description The illustration on the top shows the chemical structure of 5- methylcytosine (M e C) on the left. An arrow from M e C labeled deamination points to thymine. During this conversion, the amino group in the fourth position deaminated to form thymine. The illustration below shows a wild type D N A running in opposite directions contains M e C in one strand and guanine in another strand. The steps involved in the point mutation due to deamination are as follows: Step 1: Deamination. M e c is mutated to thymine. Step 2: Replication. Mutant D N A is formed with thymine in one strand (gray) and adenine in another strand (blue). Wild type D N A is formed with cytosine in one strand
Base Excision Repairs T-G Mismatches and Damaged Bases
(blue) and guanine in another strand (gray). High-Fidelity DNA Excision-Repair Systems Recognize and Repair Damage DNA excision-repair systems were first elucidated through a combination of genetic and biochemical studies in E. coli. Homologs of the key bacterial proteins exist in eukaryotes from yeasts to humans, indicating that these error-free mechanisms arose early in evolution to protect DNA integrity. Each of these systems functions in a similar manner: the damaged DNA strand is recognized, a segment of the damaged DNA strand is excised, and the gap is filled by DNA polymerase and ligase using the complementary DNA strand as a template. Base Excision Repairs T-G Mismatches and Damaged Bases In humans, the most common type of point mutation is a change from a C to a T, which is caused by deamination of 5-methyl C (see Figure 5-14). The conceptual problem with correcting a lesion in DNA caused by chemically normal bases that are mismatched is in determining which one is the normal and which one is the mutant DNA strand. But since a G·T mismatch is almost invariably caused by chemical conversion of C to U or
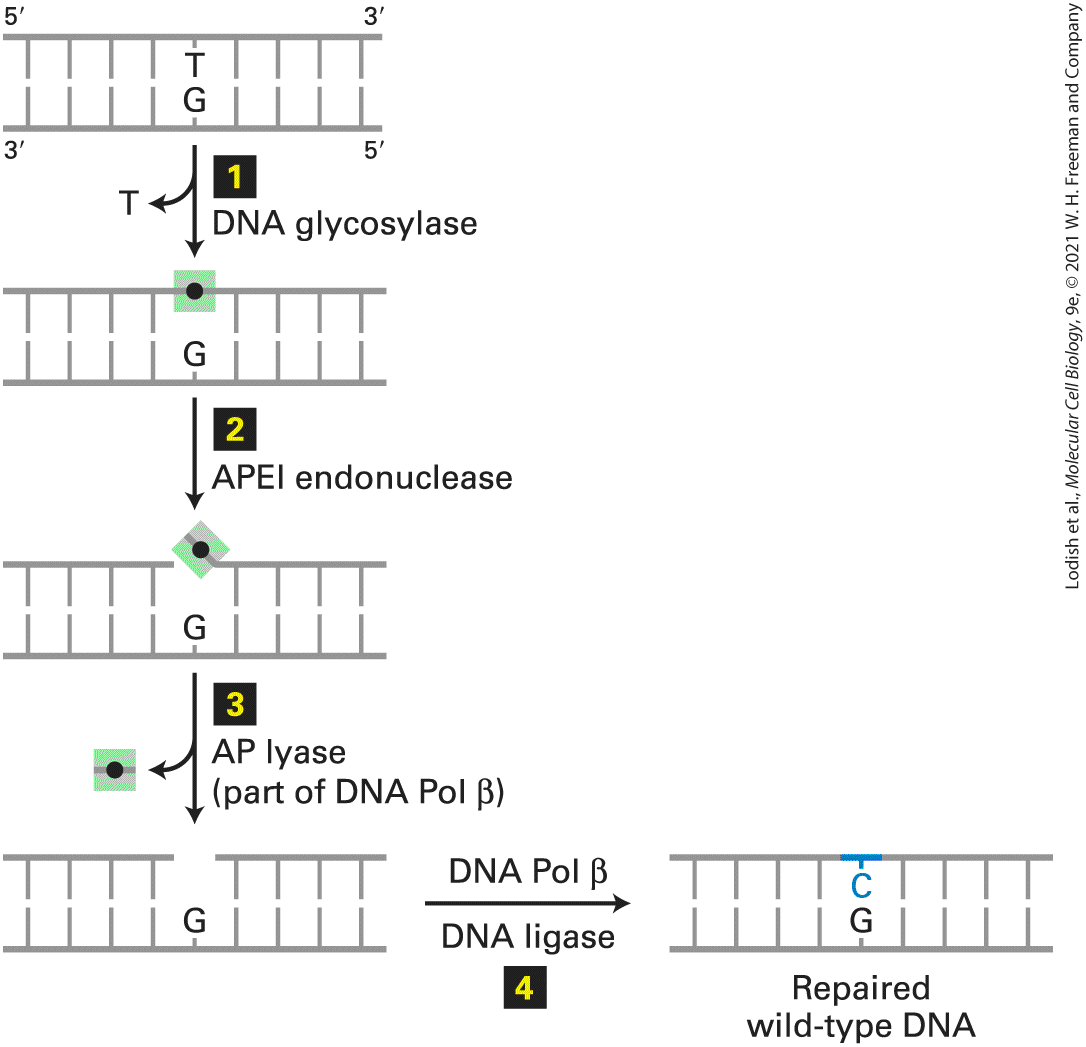
5-methyl C to T, the repair system evolved to selectively remove the T and replace it with a C. The G·T mismatch is recognized by a DNA glycosylase that flips the thymine base out of the helix and then hydrolyzes the bond that connects it to the sugar-phosphate DNA backbone. Following this initial incision, an endonuclease, APE1, cuts the DNA strand near the now abasic site. The deoxyribose phosphate lacking the base is then removed and replaced with a C by a specialized DNA polymerase that reads the G in the template strand (Figure 5-15).
FIGURE 5-15 Base excision repair of a T·G mismatch. A DNA glycosylase specific for G·T mismatches, which are usually formed by deamination of 5-methyl C (see Figure 5-34), flips the thymine base out of the helix and then cuts it away from the sugar-phosphate DNA backbone (step 1 ), leaving just the deoxyribose phosphate (black dot). An endonuclease specific for the resultant abasic site (apurinic endonuclease I, APE1) then cuts the DNA backbone (step 2 ), and the deoxyribose phosphate is removed by an endonuclease, apurinic lyase (AP lyase) associated with DNA polymerase β, a specialized DNA polymerase used in repair (step 3 ). The gap is then filled in by DNA Pol β and sealed by DNA ligase (step 4 , restoring the original G·C base pair. See O. Schärer, 2003, Angewandte Chemie 42:2946.
Description The illustration shows a double-stranded D N A with thymine in one strand mispaired with guanine in another strand. The steps involved in the excision of the thymine base are as follows: Step 1: Damage recognition and excision by D N A glycosylase. Step 2: Restriction of deoxyribose phosphate by A P E 1 endonuclease. Step 3: Excision of deoxyribose phosphate by A P lyase. Step 4: Gap repair by D N A polymerase beta and ligase. The base excision repair mechanism removes guanine and replaces it with cytosine. As mentioned earlier, this repair must take place prior to DNA replication because the incorrect base in this pair, T, occurs naturally in normal DNA. Consequently, it would be able to engage in normal Watson-Crick base pairing during replication, generating a stable point mutation that would no longer be recognized by repair mechanisms (see Figure 5-14, step 2 ). Human cells contain a battery of glycosylases, each of which is specific for a different set of chemically modified DNA bases. For example, one glycosylase removes 8-oxyguanine, an oxidized form of guanine, allowing its replacement by an undamaged G, and others remove bases modified by alkylating agents. The resulting abasic nucleotide is then replaced by the repair mechanism described above. A similar mechanism functions in the repair of lesions resulting from depurination, the loss of a guanine or adenine base from DNA caused by hydrolysis of the glycosylic bond between deoxyribose and the base. Depurination occurs spontaneously and is fairly common in mammals and birds because of their warm body
Mismatch Excision Repairs Other Mismatches and Small Insertions and Deletions
temperatures. The resulting abasic sites, if left unrepaired, generate mutations during DNA replication because they cannot specify the appropriate paired base. Mismatch Excision Repairs Other Mismatches and Small Insertions and Deletions Another process, also conserved in organisms from bacteria to humans, principally eliminates base-pair mismatches and insertions or deletions of one or a few nucleotides that are accidentally introduced by DNA polymerases during replication. As with base excision repair of a T in a TG mismatch, the conceptual problem with mismatch excision repair is determining which is the normal and which is the mutant DNA strand. In E. coli, this discrimination is achieved by preferentially correcting the base in the most recently synthesized strand of DNA which is the most likely to be incorrect. Sensing of the relative age of the two DNA strands takes advantage of a methylation reaction that occurs in the context of double-stranded DNA. Because methylation lags after DNA synthesis, the newly synthesized strand can be identified by it having relatively less methylation. Human cells are also able to discriminate the newly synthesized strand from the template strand immediately after DNA replication. However, this discrimination does not appear to be based on methylation of the DNA, but instead probably involves recognition of the end of the newly synthesized strand. Once identified, the mispaired segment of the daughter strand — the one with the replication error — is
excised and repaired to produce an exact complement of the template strand (Figure 5-16). In contrast to base excision repair, mismatch excision repair occurs after DNA replication.
FIGURE 5-16 Mismatch excision repair in human cells. The mismatch excision-repair pathway corrects errors introduced during replication. A complex of the MSH2 and MSH6 proteins (bacterial MutS homologs 1 and 6) binds to a mispaired segment of DNA in such a way as to distinguish between the template and the newly synthesized daughter strand (step 1 ). This binding triggers binding of MLH1 and PMS2 (both homologs of bacterial MutL). The resulting DNA-protein complex then binds an endonuclease that cuts the newly synthesized daughter strand. Next a DNA helicase unwinds the helix, and an exonuclease removes several nucleotides from the cut end of the daughter strand, including the mismatched base (step 2 ). Finally, as with base excision repair, the gap is filled in by a DNA polymerase (Pol δ, in this case) and sealed by DNA ligase (step 3 ). Description The illustration shows a double-stranded D N A with adenine in template strand mispaired with cytosine in newly synthesized daughter strand. The steps involved in the excision of the cytosine are as follows: Step 1: Damage recognition. Step 2: Unwinding and excision Step 3: Gap repair by D N A polymerase and ligase. The mismatch excision repair mechanism removes cytosine and replaces it with thymine in the daughter strand. Predisposition to a colon cancer known as hereditary nonpolyposis colorectal cancer results from an inherited loss-of-function mutation in one copy of either the MLH1 or the MSH2 gene. The MSH1 and MLH2 proteins are essential for DNA mismatch repair (see Figure 5-16). Cells with at least one functional copy of each of these genes exhibit normal mismatch repair. However, tumor cells frequently arise from individual cells that have experienced a random mutation in the second copy; when
Nucleotide Excision Repairs Chemical Adducts That Distort Normal DNA Shape
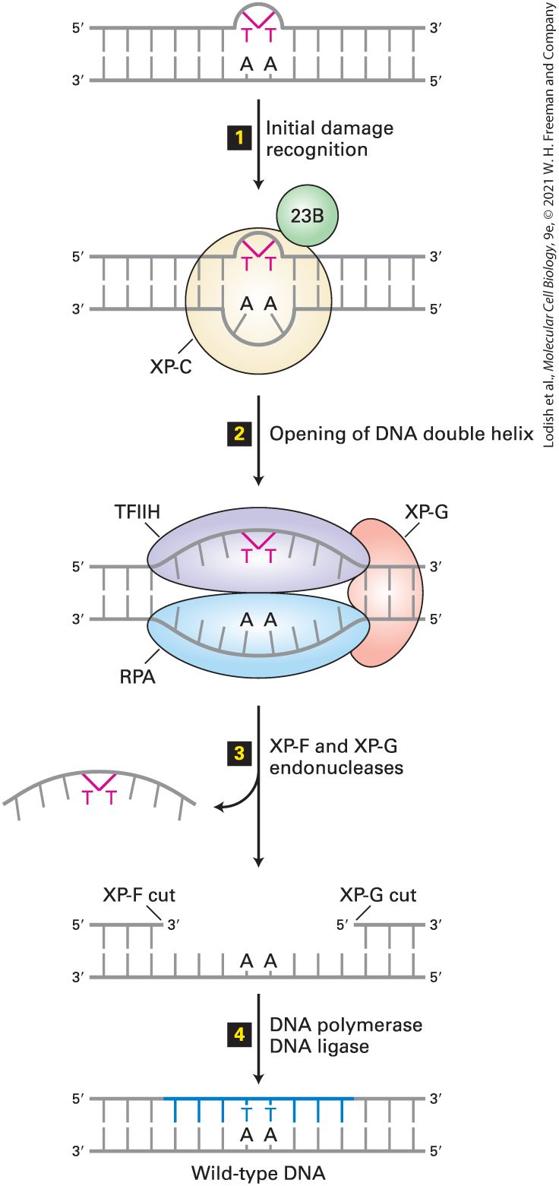
both copies of one gene have been inactivated, the mismatch repair system is no longer functional. Inactivating mutations in these genes are also common in noninherited forms of colon cancer. Nucleotide Excision Repairs Chemical Adducts That Distort Normal DNA Shape Cells use nucleotide excision repair to fix DNA regions containing chemically modified bases, often called chemical adducts, that distort the normal shape of DNA locally. A key to this type of repair is the ability of certain proteins to slide along the surface of a double-stranded DNA molecule looking for bulges or other irregularities in the shape of the double helix. For example, this mechanism repairs thymine-thymine dimers, a common type of chemical modification caused by UV light (Figure 5-17); these dimers interfere with both replication and transcription of DNA. Figure 5-18 illustrates how the nucleotide excisionrepair system repairs damaged DNA.
FIGURE 5-17 Formation of thymine-thymine dimers. (a) The most common type of DNA damage caused by UV irradiation is the formation of thymine-thymine dimers. (b) These lesions can be repaired by an excision-repair mechanism that recognizes the distortion they create in the shape of the DNA double helix. The red lines in (b) represent the UV-induced C — C bonds shown in (a). [Part (b) Data from K. McAteer et al., 1998, J. Mol Biol. 282:1013, PDB ID 1ttd.] Description The illustration labeled A shows two thymine residues attached to a phosphate molecule. The thymine residues on exposure to U V radiation undergoes a conformational change and forms a bond between C 5 and C 6. The illustration labeled B shows a 3 D model of the D N A double helix with a kink at the lower left section and a thymine-thymine dimer formation at the lower right section indicated in red.
FIGURE 5-18 Nucleotide excision repair in human cells. A DNA lesion that causes distortion of the double helix, such as a thymine-thymine dimer, is initially recognized by a complex of the XP-C (xeroderma pigmentosum C protein) and 23B proteins (step 1 ). This complex then recruits transcription factor TFIIH, whose helicase subunits, powered by ATP hydrolysis, partially unwind the double helix. XP-G and RPA proteins then bind to the complex and further unwind and stabilize the helix until a bubble of about 25 bases is formed (step 2 ). Then XP-G (now acting as an endonuclease) and XP-F, a second endonuclease, cut the damaged strand at points 24–32 bases apart on each side of the lesion (step 3 ). This releases the DNA fragment with the damaged bases, which is degraded to mononucleotides. Finally the gap is filled by DNA polymerase exactly as in DNA replication, and the remaining nick is sealed by DNA ligase (step 4 ). See J. Hoeijmakers, 2001, Nature 411:366, and O. Schärer, 2003, Angewandte Chemie 42:2946. Description The illustration shows a double-stranded D N A with thymine-thymine dimer in one strand and its complementary A A base pairs in another strand. The steps involved in the excision of the thymine-thymine dimer are as follows: Step 1: Damage recognition. Step 2: Unwinding. Step 3: Excision. Step 4: Ligation. Consider what happens to a thymine-thymine dimer not in the context of a double helix, but in a single strand of DNA at a replication fork. Such an unrepaired lesion of the template would not allow formation of a normal Watson-Crick base pair and the progress of a replicative polymerase would stall. In such events, progress of the replication fork is restored by
replacement of the high-fidelity replicative polymerase with a low-fidelity translesion polymerase. A replication fork stalled at an unrepaired thymine-thymine dimer on the template strand, produces a signal that leads to ubiquitylation of the sliding clamp PCNA (see Figure 5-13), which in turn triggers the replacement of a normal replicative polymerase such as Pol δ or Pol ε with the translesion polymerase, Pol η. Pol η has an active site that can accept distorted base pairs and lacks proofreading exonuclease activity. These features allow Pol η to form nonstandard base interactions, permitting replication past a thymine-thymine dimer on the template, but also render the polymerase prone to making errors during the replication of normal DNA. After replication has progressed past the lesion, the replication will eventually reset to the normal replicative polymerase. Ultimately, the process of replication past a lesion in the DNA will lead to a segment of DNA in the vicinity of the lesion that is relatively likely to contain mutations caused by errors in replication by translesion polymerase. This explains why mutagenesis with UV light causes a wide variety of base changes in the vicinity of T-T dinucleotides. Some 30 proteins are involved in the nucleotide excision-repair process, the first of which were identified through a study of the defects in DNA repair in cultured cells from individuals with xeroderma pigmentosum, a hereditary disease associated with a predisposition to cancer. Individuals with this disease frequently develop skin cancers called melanomas and squamous cell carcinomas if their skin is exposed to the
Two Systems Use Recombination to Repair Double-Strand Breaks in DNA
UV rays in sunlight. Mutations in any of at least seven different genes, called XP-A through XP-G, cause sensitivity to UV by inactivation of components of the nucleotide excision-repair system depicted in Figure 518. A second type of xeroderma pigmentosum mutation, which also causes sensitivity to UV rays, called XP-V, is in the Pol η gene and prevents replication past UV-induced lesions. Two Systems Use Recombination to Repair Double-Strand Breaks in DNA Ionizing radiation (e.g., x- and γ-radiation) and some anticancer drugs cause double-strand breaks in DNA. The failure to repair such a lesion would lead to the loss of the portion of the chromosome that was distal to the site of the break and in most cases would be lethal. Two systems have evolved to repair double-strand breaks: breaks can be precisely repaired using information from the intact homologous chromosome by a process of homologous recombination, discussed in the next section. Failing precise repair, double-strand breaks can be repaired by the error-prone mechanism of nonhomologous end joining (NHEJ). Error-Prone Repair by Nonhomolohous End Joining In the nonhomologous end joining pathway, the free broken ends of DNA are prevented from further degradation by binding to a complex of two proteins, Ku and DNA-PK. Two Ku and DNA-PK heterodimers bound to
DNA can then come together and, with the assistance of additional proteins including DNA ligase, join the ends of two DNA molecules (Figure 5-19). Since diffusion of DNA within the viscous nucleoplasm is fairly slow, the ends of a broken chromosome can often be rejoined before they diffuse apart. Even when correct ends are rejoined, the inevitable action of exonucleases will result in the loss of several base pairs at the joining point. If a deletion resulting from inexact end joining occurs within the coding sequence, the function of the affected gene will likely be lost.
FIGURE 5-19 Nonhomologous end joining. (a) When sister chromatids are not available to help repair double-strand breaks, nucleotide sequences are butted together that were not apposed in the unbroken DNA. These DNA ends are usually from the same chromosome locus and, when linked together, several base pairs are lost. Occasionally, ends from
different chromosomes are accidentally joined together. A complex of two proteins, Ku and DNA-dependent protein kinase (DNA-PK), binds to the ends of a double-strand break (step 1 ). After formation of a synapse, the ends are further processed by nucleases, resulting in removal of a few bases (step 2 ), and the two double-stranded molecules are ligated together (step 3 ). As a result, the double-strand break is repaired, but several base pairs at the site of the break are removed. See G. Chu, 1997, J. Biol. Chem. 272:24097; M. Lieber et al., 1997, Curr. Opin. Genet. Devel. 7:99; and D. van Gant et al., 2001, Nat. Rev. Genet. 2:196. (b) Structure of KU70/KU80 bound to a duplex DNA end. The complex is shown in a view down the DNA axis (left) and from the side (right). KU80 is light green, KU70 dark green, DNA blue. [Part (b) Data from J. R. Walker, R. A. Corpina, and J. Goldberg, 2001, Nature 412:607, PDB ID 1jey.] Description In the illustration labeled A, the steps involved in non-homologous end joining are as follows: Step 1: Attachment of D N A-dependent protein kinase and K U 70 and 80 heterodimer proteins to the ends of double-strand break. Step 2: Removal of unwanted bases and addition of proteins. Step 3: Ligation. The illustration labeled B shows the two different views of K U proteins. The end view on the left shows the base of the D N A and the side view on the right shows the D N A double helix after end joining. Occasionally, however, broken ends from different chromosomes or far apart on the same chromosome are joined together, leading to gross chromosomal rearrangements that can affect the functioning of genes. For example, incorrect joining could create a hybrid gene that codes for the N-terminal portion of one amino acid sequence fused to the C-terminal portion of a completely different amino acid sequence; or a chromosomal
Homologous Recombination Can Repair DNA Damage and Generate Genetic Diversity
rearrangement could bring the promoter of one gene close to the coding region of another gene, changing the level of expression or cell type in which that gene is expressed. Such translocations may generate chimeric genes that can have drastic effects on normal cell function, such as uncontrollable cell growth, which is the hallmark of cancer. Homologous Recombination Can Repair DNA Damage and Generate Genetic Diversity A variety of DNA lesions that are not repaired by the mechanisms discussed earlier can be repaired by mechanisms in which the damaged DNA sequence is copied from an undamaged copy of the same or a highly homologous sequence on the homologous chromosome in diploid organisms or the sister chromosome following DNA replication in haploid and diploid organisms. These mechanisms involve an exchange of strands between separate DNA molecules and hence are referred to as recombination. In addition to providing a mechanism for DNA repair, similar recombination mechanisms generate genetic diversity among the individuals of a species by causing the exchange of large regions of chromosomes between the maternal and paternal pair of homologous chromosomes during meiosis, the special type of cell division that generates germ cells (sperm and eggs) (see Figure 6-3). In fact, the exchange of regions of homologous chromosomes, called crossing over, is
required for proper segregation of chromosomes during the first meiotic cell division. Meiosis and the consequences of generating new combinations of maternal and paternal genes by recombination are discussed further in Chapter 6, and the mechanisms leading to proper segregation of chromosomes during meiosis are discussed in Chapter 19. Here we focus on the molecular mechanisms of DNA recombination, highlighting the exchange of DNA strands between two recombining DNA molecules. At one time, homologous recombination was thought to be a minor repair process in human cells. This view changed when it was realized that several human cancers are potentiated by inherited mutations in genes that are essential for homologous recombination repair. For example, some women with an inherited susceptibility to breast cancer have a mutation in one allele of either the BRCA1 or the BRCA2 gene, both of which encode proteins participating in this repair process. Loss or inactivation of the second allele inhibits the homologous recombination repair pathway and thus tends to induce cancer in mammary or ovarian epithelial cells. Yeasts can use homologous recombination to repair double-strand breaks induced by γ-irradiation. Isolation and analysis of radiation-sensitive (RAD) mutants that are deficient in this repair system facilitated the study of this process. Most of the yeast Rad proteins have homologs in the human genome, and the human and yeast proteins function in an essentially identical fashion. Repair of a Collapsed Replication Fork
An example of recombinational DNA repair is the repair of a replication fork that has come apart, known as a collapsed replication fork. If a break in the phosphodiester backbone (a nick) of one DNA strand is not repaired before a replication fork passes, the replicated portions of the daughter chromosomes become separated when the replication helicase reaches the nick in the parent strand because there are no covalent bonds between the two fragments of the parent strand on either side of the nick. This process is called replication fork collapse (Figure 5-20, step 1 ). If the break in the double-stranded daughter DNA molecule is not repaired, it is generally lethal to at least one daughter cell following cell division because of the loss of genetic information between the break and the end of the chromosome. The recombination process that repairs the resulting doublestrand break and regenerates a replication fork involves multiple enzymes and other proteins, only some of which are mentioned here.
FIGURE 5-20 Recombinational repair of a collapsed replication fork. Parent strands are light and dark blue. The leading daughter strand is dark green, and the lagging daughter strand light green. Diagonal lines in step 3 and beyond represent a single phosphodiester bond from the DNA strand of the corresponding color. Small black arrows following step 4 represent cleavage of the phosphodiester bonds at the crossover of DNA strands in the Holliday structure. See the text for a discussion. Description The seven steps involved in the repair mechanism are as follows: Step 1: Replication fork collapse. Step 2: Digestion by 5 prime exonucleases. Step 3: Rec A or Rad 51 mediated strand invasion. Step 4: Branch migration. Step 5: Strand cut at crossover. Step 6: Ligation. Step 7: Regeneration of replication fork. The first step in the repair of the double-strand break is exonucleolytic digestion of the strand whose end is at the broken end of DNA, leaving the strand with the end at the break single stranded (Figure 5-20, step 2 ). The lagging nascent strand (light green) base-paired to the unbroken parent strand (dark blue) is ligated to the unreplicated portion of the parent chromosome (light blue), as shown in Figure 5-20, step 2 . A protein required for the next step is RecA in bacteria, or the homologous Rad51 in Saccharomyces cerevisiae and other eukaryotes. Multiple RecA/Rad51 molecules bind to the single-stranded DNA and catalyze its hybridization
to a perfectly or nearly perfectly complementary sequence in another, homologous, double-stranded DNA molecule. The complementary strand of this target double-stranded DNA (dark blue) is displaced as a singlestranded loop of DNA over the region of hybridization to the invading strand (Figure 5-20, step 3 ). This RecA/Rad51–catalyzed invasion of a duplex DNA by a single-stranded complement of one of the strands is key to the recombination process. This process is called strand invasion, and because there is no change in the number of base pairs, it does not require an input of energy. Next the hybrid region between target DNA and the invading strand is extended away from the break by a process called branch migration (Figure 5-20, step 4 ). In this diagram, the diagonal lines represent only one phosphodiester bond. Molecular modeling and other studies show that the first base on either side of the branch is base-paired to a complementary nucleotide. As this branch migrates to the left, the number of base pairs remains constant; one new base pair formed with the invading strand (dark green) is matched by the loss of one base pair with the parent strand (blue). When the hybrid region extends beyond the end of the broken strand that was digested by the -exonuclease in step 2 (light blue), the singlestranded parent DNA thus generated (light blue) can base-pair with the complementary region of the other parent strand (dark blue), which becomes single-stranded as the branch migrates to the left (see Figure 520, step 4 ). The resulting structure is called a Holliday structure, after Robin Holliday, the geneticist who first proposed it as an intermediate in
genetic recombination. Again, the diagonal lines in the diagram following step 4 represent single phosphodiester bonds, and all bases in the Holliday structure are base-paired to complementary bases in the parent strands. Cleavage of the phosphodiester bonds that cross over from one parent strand to the other (step 5 ) and ligation of the and ends basepaired to the same parent strands (step 6 ) result in the generation of a structure similar to a replication fork. Rebinding of replication fork proteins results in extension of the leading strand past the point of the original strand break and re-initiation of lagging-strand synthesis (step 7 ), thus regenerating a replication fork. The overall process allows the ligated upper strand in the lower molecule following step 2 to serve as a template for extension of the leading strand in step 7 . Repair of a Double-Strand Break by Homologous Recombination A similar mechanism, called homologous recombination, can repair a double-strand break in a chromosome and can also exchange large segments of two double-stranded DNA molecules (Figure 5-21). First, the broken ends of the DNA molecule are digested by -exonucleases, leaving a single-stranded region of DNA with a end (step 1 ). RecA in bacteria or Rad51 in eukaryotes then catalyzes invasion of one of these ends into the homologous region of the homologous chromosome, as described above for repair of a collapsed replication fork (step 2 ). The end of the invading DNA strand is then extended by a DNA polymerase, displacing the parent strand as an enlarging single-stranded loop of DNA (dark blue)
(step 3 ). When the loop extends to a sequence that is complementary to the other broken and -exonuclease–digested end of DNA (the fragment on the left following step 1 ), the complementary sequences base-pair (diagram following step 3 ). This end is then extended by a DNA polymerase using the displaced single-stranded loop of parent DNA (dark blue) as a template (step 4 ).
FIGURE 5-21 Repair of a double-strand break by homologous recombination. For simplicity, each DNA double helix is represented by two parallel lines with the polarities of the strands indicated by arrowheads at their ends. The upper molecule has a doublestrand break. Note that in the diagram of the upper DNA molecule, the strand with its end at the right is on the top, while in the diagram of the lower DNA molecule, this strand is drawn on the bottom. See the text for discussion. See T. L. Orr-Weaver and J. W. Szostak, 1985, Microbiol. Rev. 49:33. Description The illustration shows two horizontal double-stranded D N A with arrows at the 3 prime ends. One of the double-stranded D N A's is with a double-strand break. The steps involved in the repair mechanism are as follows: Step 1: Ends digested by 5 prime exonucleases leaving 3 prime single-stranded ends. Step 2: RecA-mediated (prokaryotes) or Rad51-mediated (eukaryotes) strand invasion of homologous chromosome. Step 3: 3 prime end of invading strand is extended by DNA polymerase until the displaced single-strand (dark blue) base-pairs with the other 39 single strand generated initially (light green). Step 4: 3 prime end is extended by DNA polymerase. Step 5: Ends are ligated to form two Holliday structures. Step 6: Each Holliday structure shown here produces a homologous recombination event. Next the new ends are ligated (step 5 ) to the exonuclease-digested ends. This generates two Holliday structures in the paired molecules (step 5 ). Branch migration of these Holliday structures can occur in either direction (not diagrammed). Finally, cleavage of the strands at the positions shown by the arrows, and ligation of the alternative and
ends at each cleaved Holliday structure, generates two recombinant chromosomes that contain the DNA of one parent DNA molecule on one side of the initial break point (light and dark green strands), and the DNA of the other parent DNA molecule on the other side of the break point (light and dark blue) (step 6 ). The region in the immediate vicinity of the initial break point forms a heteroduplex, in which one strand from one parent is base-paired to the complementary strand of the other parent (light or dark green strand base-paired to light or dark blue strand). Basepair mismatches between the two parent strands are usually repaired by the repair mechanisms discussed above to generate a complementary base pair. In the process, sequence differences between the two parents are lost, a process referred to as gene conversion.
Figure 5-22 diagrams how cleavage of one or the other pair of strands at the four-way strand junction in the Holliday structure generates parent or recombinant molecules. This process, called resolution of the Holliday structure, separates DNA molecules initially joined by RecA/Rad51– catalyzed strand invasion. Each Holliday structure in the intermediate following Figure 5-21, step 5 , can be cleaved and religated in the two possible ways shown by the two sets of small black arrows in Figure 5-22 (step 1 or step 2 ). Consequently, there are four possible products of the recombination process shown in Figure 5-21. After ligation of the cleaved ends, two of these products regenerate the parent chromosomes, with the exception of the heteroduplex region at the break point, which is repaired into the sequence of one parent or the other by a process known as gene conversion. The other two possible products generate recombinant chromosomes as shown in Figure 5-21.
FIGURE 5-22 Alternative resolution of a Holliday structure. Diagonal and vertical lines represent a single phosphodiester bond. It is simplest to diagram the process by rotating the diagram of the bottom molecule so that the top and bottom molecules have the same strand orientations. Cutting the bonds as shown in step 1 and ligating the ends as indicated regenerates the original chromosomes. Cutting the strands as shown in step 2 and religating as shown at the bottom generates recombinant chromosomes. Description In the Holliday structure, a vertical and a horizontal cut cleave the two crossed over daughter DNA strands attached to two-parent strands. A horizontal cut on the crossed over daughter strand results in the regeneration of the original chromosome and a vertical cut on the parent strand results in the formation of the recombinant chromosome. KEY CONCEPTS OF SECTION 5.3 DNA Repair and Recombination Changes in the DNA sequence result from copying errors and the effects of various physical and chemical agents. Eukaryotic cells have three excision-repair systems for correcting mispaired bases and for removing chemical adducts from DNA. Base excision repair, mismatch repair, and nucleotide-excision repair operate with high accuracy and generally do not introduce errors. Damaged DNA bases in single-stranded template DNA at a replication fork will block the progress of a replicative DNA polymerase. These blocks can be overcome by a
translesion polymerase that has relaxed requirements for proper base pairing during replication. Translesion polymerases are error prone and tend to generate mutations of their own in the vicinity of the original lesion. Repair of double-strand breaks by the nonhomologous end joining pathway can link segments of DNA from different chromosomes, possibly forming an oncogenic chromosomal translocation. This repair mechanism also produces a small deletion, even when segments from the same chromosome are joined. Error-free repair of double-strand breaks in DNA is accomplished by homologous recombination using the undamaged sister chromatid as a template. Inherited defects in the nucleotide excision-repair pathway, as in individuals with xeroderma pigmentosum, predispose them to skin cancer. Inherited colon cancer is frequently associated with mutant forms of proteins essential for the mismatch repair pathway. Defects in repair by homologous recombination are associated with inheritance of one mutant allele of the BRCA1 or BRCA2 gene and result in predisposition to breast and uterine cancer.
5.4 Transcription of Protein-Coding Genes and Formation of mRNA
5.4 Transcription of ProteinCoding Genes and Formation of mRNA The simplest definition of a gene is a unit of DNA that contains the information to specify synthesis of a single polypeptide chain or functional RNA (such as a tRNA). The DNA molecules of small viruses contain only a few genes, whereas the single DNA molecule in each of the chromosomes of higher animals and plants may contain several thousand genes. The vast majority of genes carry information used to build protein molecules, and it is the RNA copies of such protein-coding genes that constitute the mRNA molecules of cells. In its most elemental form, the process of transcription is the formation of an RNA copy of the information carried in DNA for one gene. RNA synthesis, catalyzed by an RNA polymerase and based on the same basepairing mechanism as DNA replication copies, or transcribes, the fourbase language of DNA containing A, G, C, and T into the four-base language of RNA, which is identical except that U replaces T. Because of the inherent instability of RNA within the cell, the amount of transcribed RNA can readily be regulated by alterations in the rate of transcription initiation. This allows distinct sets of genes to be expressed in the multiple different types of cells that make up a multicellular organism. Regulation of transcription is addressed in Chapter 8.
A Template DNA Strand Is Transcribed into a Complementary RNA Strand by RNA Polymerase
For protein encoding genes, the product of transcription is an mRNA, which is subsequently translated into the 20–amino acid language of proteins. In this section, we focus on the formation of functional mRNAs from protein-coding genes (see Figure 5-1, step 1 ). A similar process yields precursors of rRNAs and tRNAs, encoded by rRNA and tRNA genes; these precursors are then further modified to yield functional rRNAs and tRNAs (see Chapters 8 and 9). A Template DNA Strand Is Transcribed into a Complementary RNA Strand by RNA Polymerase The process of transcription is closely related to that of DNA replication, in which one DNA strand acts as a template and, in the case of transcription, determines the order in which ribonucleoside triphosphate (rNTP) monomers are linked together to form a complementary RNA chain. Incoming rNTPs base-pair with complementary bases in the template DNA strand, and are then joined to a growing RNA strand in a polymerization reaction catalyzed by RNA polymerase. The polymerization reaction involves a nucleophilic attack by the oxygen in the growing RNA chain on the α phosphate of the next nucleotide precursor to be added, which results in the formation of a phosphodiester bond and the release of pyrophosphate . As with DNA replication, RNA molecules are always synthesized in the direction (Figure 523a).
FIGURE 5-23 RNA is synthesized . (a) Polymerization of ribonucleotides by RNA polymerase during transcription. The ribonucleotide to be added at the end of a
growing RNA strand is specified by base pairing between the next base in the template DNA strand and the complementary incoming ribonucleoside triphosphate (rNTP). A phosphodiester bond is formed when RNA polymerase catalyzes a reaction between the oxygen of the growing strand and the α phosphate of a correctly base-paired rNTP. RNA strands are always synthesized in the direction and are opposite in polarity to their template DNA strands. (b) The DNA nucleotide where RNA polymerase begins transcription is designated . The direction the polymerase travels on the DNA is termed downstream, and downstream bases are marked with positive numbers. The opposite direction is termed upstream, and upstream bases are marked with negative numbers. Some important gene features lie upstream of the transcription start site, including the promoter sequence that localizes RNA polymerase to the gene. (c) The DNA strand that is being transcribed is the template strand; its complement is the nontemplate strand. The RNA being synthesized is complementary to the template strand and is therefore identical with the nontemplate strand sequence, except with uracil in place of thymine. Description The illustration labeled A shows a vertical D N A template strand with R N A growing along with it from top to bottom. The illustration labeled B shows a horizontal doublestranded D N A. The positive 1 region in the D N A represents the site of transcription which travels toward the right of the D N A sequence. The region to the right of positive 1 and positive 30 are labeled as downstream and the region to the left of positive 1 and negative 30 are labeled as upstream. The promoters are labeled in the negative regions and the coding sequences are labeled in the positive regions. The illustration labeled C shows a template and nontemplate D N A strands. An arrow from the D N A points to the primary R N A transcript synthesized from the D N A template strand. By convention, the site on the DNA template at which RNA polymerase begins transcription is numbered (Figure 5-23b). Downstream denotes the direction in which a template DNA strand is transcribed; upstream denotes the opposite direction. Nucleotide positions in the DNA sequence
downstream from a start site are indicated by a positive (+) sign; those upstream, by a negative (−) sign. Because RNA is synthesized , RNA polymerase moves down the template DNA strand in a direction. The newly synthesized RNA is complementary to the template DNA strand; therefore, it is identical to the nontemplate DNA strand, with uracil in place of thymine (Figure 5-23c). Stages in Transcription To carry out transcription, RNA polymerase performs several distinct functions, as depicted in Figure 5-24. During transcription initiation, RNA polymerase, with the help of initiation factors (called s-factors in bacteria and general transcription factors in eukaryotes), recognizes and binds to a specific sequence of double-stranded DNA called a promoter (step 1 ). After binding, RNA polymerase and the initiation factors separate the DNA strands to make the bases in the template strand available for base pairing with the bases of the rNTPs that the RNA polymerase will polymerize (step 2 ). Approximately 12–14 base pairs of DNA around the transcription start site are separated, which allows the template strand to enter the active site of the enzyme. The active site is where catalysis of phosphodiester bond formation between rNTPs that are complementary to the template strand takes place. The 12–14 base-pair region of melted DNA in the active site of the polymerase is known as the transcription bubble. Transcription initiation is considered complete when the first two ribonucleotides of an RNA chain are linked by a phosphodiester bond (step 3 ).
FIGURE 5-24 Three stages in transcription. During initiation of transcription, RNA polymerase forms a transcription bubble and begins polymerization of ribonucleotides
(rNTPs) at the start site, which is located within the promoter region. Once a DNA region has been transcribed, the separated strands reassociate into a double helix. The nascent RNA is displaced from its template strand except at its end. The end of the RNA strand exits the RNA polymerase through a channel in the enzyme. Termination occurs when the polymerase encounters a specific termination sequence (stop site). See the text for details. For simplicity, the diagram depicts transcription of four turns of the DNA helix encoding some 40 nucleotides of RNA. Most RNAs are considerably longer, requiring transcription of a longer region of DNA. Description The illustration shows the three main stages of transcription in three panels. The first panel shows the initiation process in three steps. The steps involved in the initiation of transcription are as follows: Step 1: Polymerase binds to promoter sequence in duplex D N A. Closed complex. Step 2: Polymerase melts duplex D N A near transcription start site, forming a transcription bubble. Open complex. Step 3: Polymerase catalyzes phosphodiester linkage of two initial r N T P's. The second panel shows the elongation process in a single step. Step 4: Polymerase advances from 3 prime to 5 prime down the template strand, melting duplex D N A, and adding r N T P's to growing R N A. The third panel shows the termination process in a single step. Step 5: At transcription stop site, polymerase releases completed R N A and dissociates from D N A. After several ribonucleotides have been polymerized, RNA polymerase dissociates from both the promoter DNA and the initiation factors (called σ-factors in bacteria, and general transcription factors in archaea and eukaryotes). During the strand elongation stage, RNA polymerase moves
along the template DNA, opening the double-stranded DNA in front of its direction of movement and guiding the strands back together so that they reassociate at the upstream end of the transcription bubble (step 4 ). One ribonucleotide at a time is added by the polymerase to the end of the growing (nascent) RNA chain. During strand elongation, the enzyme maintains a melted region of approximately 14–20 base pairs in the transcription bubble. Approximately eight nucleotides at the end of the growing RNA strand remain base-paired to the template DNA strand in the transcription bubble. The elongation complex, comprising RNA polymerase, template DNA, and the nascent RNA strand, can be extraordinarily stable. For example, RNA polymerase transcribes the longest known mammalian gene, containing about 2 million base pairs, without dissociating from the DNA template or releasing the nascent RNA. RNA synthesis occurs at a rate of about 1000–2000 nucleotides per minute, so the elongation complex must remain intact for more than 24 hours to ensure continuous synthesis of pre-mRNA from this very long gene. Note that although biochemically similar to DNA replication, transcription of RNA occurs about 30 times more slowly in terms of the rate of nucleotides added. During transcription termination, the final stage in RNA synthesis, the completed RNA molecule is released from the RNA polymerase and the polymerase dissociates from the template DNA (step 5 ). Once it is released, an RNA polymerase is free to transcribe the same gene again or another gene. Structure of RNA Polymerases
The RNA polymerases of bacteria, archaea, and eukaryotic cells are fundamentally similar in structure and function. Bacterial RNA polymerases are composed of two related large subunits ( and β), two copies of a smaller subunit (α), and one copy of a fifth subunit (ω) that is not essential for transcription or cell viability, but that stabilizes the enzyme and assists in the assembly of its subunits. Archaeal and eukaryotic RNA polymerases have several additional small subunits associated with this core complex, which we describe in Chapter 8. Schematic diagrams of the transcription process generally show RNA polymerase bound to an unbent DNA molecule, as in Figure 5-24. However, x-ray crystallography and other studies of an elongating bacterial RNA polymerase indicate that the DNA bends at the transcription bubble (Figure 5-25).
FIGURE 5-25 Bacterial RNA polymerase. This structure corresponds to the polymerase molecule in the elongation stage of transcription. In this diagram, transcription is proceeding in the rightward direction. Arrows indicate where downstream DNA enters the polymerase and upstream DNA exits at an angle from the downstream DNA. The template strand is light violet; the nontemplate strand, dark violet; the nascent RNA, red. The RNA polymerase subunit is gold; the β subunit, light yellow; and the α subunits visible from this angle, brown. Nucleotides complementary to the template DNA are added to the end of the nascent RNA strand on the right side of the transcription bubble. The newly synthesized nascent RNA exits the polymerase at the upstream side through a channel formed by the β subunit. The ω subunit is also visible from this angle.
Eukaryotic Precursor mRNAs Are Processed to Form Functional mRNAs
[Data courtesy of Seth Darst; see N. Korzheva et al., 2000, Science 289:619–625, and N. Opalka et al., 2003, Cell 114:335–345.] Description The X-ray crystallography image shows the entry of D N A inside the R N A polymerase, synthesis of R N A in the transcription bubble, and the exit of R N A from the polymerase. The subunits of R N A polymerase alpha, alpha prime, beta, beta prime, and omega are highlighted in different colors. Eukaryotic Precursor mRNAs Are Processed to Form Functional mRNAs Early research on the structure of eukaryotic genes involved studies of viruses that infect animals. When researchers analyzed the regions of a viral DNA molecule that encode viral mRNAs, they were surprised to observe that the sequence of a single viral mRNA was encoded in several regions of the viral DNA separated by DNA sequences that are not present in the mRNA. Later, the development of gene cloning and DNA sequencing (see Chapter 6) allowed researchers to compare the genomic DNA sequences of multicellular organisms with the sequences of their mRNAs. This research revealed that most cellular mRNAs are also encoded in several separate regions of genomic DNA, called exons, separated by sequences of DNA called introns. Further studies showed that a gene is first transcribed into a long primary transcript that includes both the exon sequences and the intron sequences that separate them. Subsequently, the introns are removed and the exons are spliced together
(see Chapter 9). Although introns are common in multicellular eukaryotes, they are extremely rare in bacteria and archaea and uncommon in many unicellular eukaryotes, such as baker’s yeast. In eukaryotic cells, the site of RNA synthesis — the nucleus — is separated from the site of translation — the cytoplasm. In the nucleus, the primary transcripts of protein-coding genes — precursor mRNAs (premRNAs) — must undergo several modifications, collectively termed RNA processing, to yield a functional mRNA (see Figure 5-1, step 2). This mRNA must then be exported to the cytoplasm before it can be translated into protein. Thus transcription and translation cannot occur concurrently in eukaryotic cells. All eukaryotic pre-mRNAs are initially modified at the two ends, and these modifications are retained in mRNAs. As the end of a nascent RNA chain emerges from the surface of RNA polymerase, it is immediately acted on by several enzymes that together synthesize the cap, a 7-methylguanylate that is connected to the terminal nucleotide of the RNA by an unusual , triphosphate linkage (Figure 5-26). The cap protects an mRNA from enzymatic degradation and assists in its export to the cytoplasm, where it is bound by a protein factor required to begin translation.
FIGURE 5-26 Structure of the methylated cap. The distinguishing chemical features of the methylated cap on eukaryotic mRNA are (1) the linkage of 7methylguanylate to the initial nucleotide of the mRNA molecule and (2) the methyl group on the hydroxyl of the ribose of the first nucleotide (base 1). Both of these features occur in all animal cells and in cells of higher plants; yeasts lack the methyl group on nucleotide 1. The ribose of the second nucleotide (base 2) is also methylated in vertebrates. See A. J. Shatkin, 1976, Cell 9:645. Description The illustration shows the 5 prime to 5 prime linkages between 7-methyl guanylate and the first nucleotide of m R N A. The linkage is represented vertically with three parts. The first part shows 7-methyl guanylate with a methyl group in the N 7 position. The second part shows a linkage of 3 phosphate groups with 5 prime of 7-methyl guanylate at the top and with 5 prime of ribose at the bottom. The third part shows 2 bases labeled base 1 and 2 linked with a phosphodiester bond. The base 1 and 2 contain a methyl group on 2 prime of the ribose in m R N A. Processing at the end of a pre-mRNA involves cleavage by an endonuclease to yield a free -hydroxyl group, to which a string of adenylic acid residues is added one at a time by an enzyme called poly(A) polymerase. The resulting poly(A) tail contains 100–250 bases, and is shorter in yeasts and invertebrates than in vertebrates. Poly(A) polymerase is part of a complex of proteins that can locate and cleave a transcript at a specific site and then add the correct number of A bases, in a process that does not require a template. As discussed further in Section 5.6 and in
Chapter 10, the poly(A) tail has important functions both in translation of mRNA and in stabilizing pre-mRNAs in the nucleus and fully processed mRNAs in the nucleus and cytoplasm.
Another step in the processing of many different eukaryotic mRNA molecules is RNA splicing: the internal cleavage of a transcript to excise the introns and stitch together the coding exons. Figure 5-27 summarizes the basic steps in eukaryotic mRNA processing, using the β-globin gene as an example. We examine the cellular machinery for carrying out processing of mRNA, as well as tRNA and rRNA, in Chapter 9.
FIGURE 5-27 Overview of RNA processing. RNA processing produces functional mRNA in eukaryotes. The β-globin gene contains three protein-coding exons (constituting the coding region) and two intervening noncoding introns. The introns interrupt the proteincoding sequence between the codons for amino acids 31 and 32 and 105 and 106. Transcription of eukaryotic protein-coding genes starts before the sequence that encodes the first amino acid and extends beyond the sequence that encodes the last amino acid, resulting in noncoding regions at the ends of the primary transcript. These untranslated regions (UTRs) are retained during processing. The cap is added during formation of the primary RNA transcript, which extends beyond the poly(A) site. After cleavage at the poly(A) site and addition of multiple A residues to the end, splicing removes the introns and joins the exons. The small numbers refer to positions in the 147– amino acid sequence of β-globin. Description The illustration shows a double-stranded beta-globin genomic D N A with untranslated regions (U T R), exons, and introns represented in gray, red, and blue rectangular boxes, respectively. The D N A sequence contains exon-coding regions (red) 1 to 147, except 31, 32, 105, and 106, which codes for the introns (blue). An arrow pointing to the gray area before the exon is labeled as start site for R N A synthesis. An arrow pointing to the gray area at the end of the exon is labeled poly A site. The steps involved in the R N A processing are as follows: Step 1: Addition of m superscript 7 G P P P cap (represented in green circle) to U T R forms the primary t R N A transcript. Step 2: 3 prime cleavage and addition of poly-A tails. Step 3: Removal of introns. The R loops formed between the exons are removed. Step 4: Splicing of exons to form beta-globin m R N A. The beta-globin m R N A contains m superscript 7 G P P P cap, U T R, exons 1 to 147, U T R, and poly-A tails. The functional eukaryotic mRNAs produced by RNA processing retain noncoding regions, referred to as untranslated regions (UTRs), at each
Alternative RNA Splicing Increases the Number of Proteins That Can Be Expressed from a Single Eukaryotic Gene
end. In mammalian mRNAs, the UTR may be 100 or more nucleotides long, and the UTR may be several kilobases in length. Bacterial mRNAs also usually have and UTRs, but these regions are much shorter than those in eukaryotic mRNAs, generally containing fewer than 10 nucleotides. As discussed in Chapter 9, the UTR and UTR sequences participate in regulation of mRNA translation and stability, and UTRs also function in the localization of many mRNAs to specific regions of the cytoplasm. Alternative RNA Splicing Increases the Number of Proteins That Can Be Expressed from a Single Eukaryotic Gene The vast majority of genes in multicellular eukaryotes are made up of multiple exons separated by introns. As noted in Chapter 3, many proteins from higher eukaryotes have a multidomain tertiary structure (see Figure 3-10) and it is often the case that the boundaries between protein domains correspond to junctions between exons of the underlying gene. This tendency for exons to correspond to discrete folded domains in the encoded protein suggests that many eukaryotic proteins have evolved by genetic recombination events that bring exons together in new combinations, a process known as exon shuffling.
We will see many examples throughout this book of mammalian proteins that contain multiple repeated copies of a protein domain with identical or nearly identical amino acid sequences. Evidently these sequences arose by recombination events that led to a string of multiple copies of the same exon separated by introns. The presence of multiple intron genes permits expression of multiple variations of the protein from a single gene by means of alternative splicing. In higher eukaryotes, alternative splicing is an important mechanism for production of different forms of a protein, called isoforms, by different types of cells. Fibronectin, a multidomain protein found in mammals, provides a good example of alternative splicing (Figure 5-28). Fibronectin is a long, adhesive protein secreted into the extracellular space that can bind other proteins together. What and where it binds depends on which domains are spliced together. The fibronectin gene contains numerous exons, grouped into several regions corresponding to specific domains of the protein. Fibroblasts produce fibronectin mRNAs that contain exons EIIIA and EIIIB; these exons encode a protein domain that binds tightly to proteins in the fibroblast plasma membrane. Consequently, this fibronectin isoform adheres fibroblasts to the extracellular matrix. Alternative splicing of the fibronectin primary transcript in hepatocytes, the major type of cell in the liver, yields mRNAs that lack the EIIIA and EIIIB exons. As a result, the fibronectin secreted by hepatocytes into the blood does not adhere tightly to fibroblasts or to most other cell types, which allows it to circulate. During formation of blood clots, however, other fibrin-binding domains of hepatocyte fibronectin bind to fibrin, one of the principal constituents of blood clots. Yet another domain of the bound fibronectin then interacts
with integrins on the membranes of passing platelets, thereby expanding the clot by addition of platelets.
FIGURE 5-28 Alternative splicing. The ∼75-kb fibronectin gene (top) contains multiple exons; splicing of the fibronectin transcript varies by cell type. The EIIIB and EIIIA exons (green) encode binding domains for specific proteins on the surface of fibroblasts. The fibronectin mRNA produced in fibroblasts includes the EIIIA and EIIIB exons, whereas these exons are spliced out of fibronectin mRNA in hepatocytes. In this diagram, introns (black lines in the top diagram of the fibronectin gene) are not drawn to scale; most of them are much longer than any of the exons. Description The fibronectin gene contains multiple exons represented in rectangular boxes with different colors. The introns between the exons are represented in two horizontal closely arranged black parallel lines. The two exons in green at the middle of the fibronectin gene are labeled as E 3 A and E 3 B. Below the fibronectin gene is the fibroblast fibronectin m R N A from 5 prime to 3 prime. After splicing, the exons are retained without introns. Below the fibroblast fibronectin m R N A is the hepatocyte fibronectin m R N A from 5 prime to 3 prime. The exons E 3 A and E 3 B are spliced along with introns and other exons are retained. More than 20 different isoforms of fibronectin have been identified, each encoded by a different, alternatively spliced mRNA composed of a unique
combination of fibronectin gene exons. Sequencing of large numbers of mRNAs isolated from various tissues and comparison of their sequences with genomic DNA has revealed that nearly 90 percent of all human genes are expressed as alternatively spliced mRNAs. Clearly alternative RNA splicing greatly expands the number of protein variants encoded by the genomes of higher, multicellular organisms. KEY CONCEPTS OF SECTION 5.4 Transcription of Protein-Coding Genes and Formation of mRNA Transcription of DNA is carried out by RNA polymerase, which adds one ribonucleotide at a time to the end of a growing RNA chain (see Figure 5-23). The sequence of the template DNA strand determines the order in which ribonucleotides are polymerized to form an RNA chain. During transcription initiation, RNA polymerase binds to a specific site in DNA (the promoter), locally melts the double-stranded DNA to reveal the unpaired template strand, and polymerizes the first two nucleotides complementary to the template strand. The melted region of 12–14 base pairs is known as the transcription bubble. During strand elongation, RNA polymerase moves down the DNA, melting the DNA ahead of the polymerase so that the template strand can enter the active site of the enzyme, and allowing the complementary strands of the region just transcribed to reanneal behind it. The transcription bubble moves with the polymerase as the enzyme adds ribonucleotides complementary to the template strand to the end of the growing RNA chain. When RNA polymerase reaches a termination sequence in the DNA, the enzyme stops transcription, leading to release of the completed RNA and dissociation of the enzyme from the template DNA. In eukaryotic DNA, each protein-coding gene is transcribed from its own promoter. The initial primary transcript very often contains noncoding regions (introns) interspersed with coding regions (exons). Eukaryotic primary transcripts must undergo RNA processing to yield functional RNAs. During processing, the ends of nearly all primary transcripts from proteincoding genes are modified by addition of a cap and poly(A) tail. Transcripts from genes containing introns undergo splicing, the removal of the introns and joining of the exons (see Figure 5-27).
The individual domains of multidomain proteins found in higher eukaryotes are often encoded by individual exons or a small number of exons. Distinct isoforms of such proteins are often expressed in specific cell types as the result of alternative splicing of exons.
5.5 The Decoding of mRNA by tRNAs
5.5 The Decoding of mRNA by tRNAs As we saw in Chapter 3, the linear order of amino acids in each protein determines its three-dimensional structure and activity. Translation is the whole process by which the nucleotide sequence of an mRNA is used as a template to join the amino acids of a polypeptide chain in the correct order (see Figure 5-1, step 3 ). In eukaryotic cells, protein synthesis occurs in the cytoplasm, where three types of RNA molecules come together to perform different but cooperative functions (Figure 5-29): 1. Messenger RNA (mRNA) carries the genetic information transcribed from DNA in a linear form. The mRNA is read in sets of threenucleotide sequences, called codons, each of which specifies a particular amino acid. 2. Transfer RNA (tRNA) is the key to deciphering the codons in mRNA and serves as an adaptor to link three bases of RNA sequence to a particular amino acid. Each type of amino acid has its own subset of tRNAs, which are covalently bound to that amino acid and carry it to the growing end of a polypeptide chain when the next codon in the mRNA calls for it. The correct tRNA with its attached amino acid is selected at each step because each specific tRNA molecule contains a three-nucleotide sequence, an anticodon, that can base-pair with its complementary codon in the mRNA. 3. Ribosomal RNA (rRNA) associates with a set of proteins to form ribosomes. These complex structures form the scaffold for stepwise
alignment of the codons in mRNA with the anticodons in tRNA. The ribosome also catalyzes the sequential formation of peptide bonds between amino acids carried by tRNAs for the assembly of a polypeptide chain. Ribosomes are composed of a large and a small subunit, each of which contains its own rRNA molecule or molecules.
FIGURE 5-29 Three roles of RNA in protein synthesis. Messenger RNA (mRNA) is translated into protein by the joint action of transfer RNA (tRNA) and the ribosome, which
Messenger RNA Carries Information from DNA in a Three-Letter Genetic Code
is composed of numerous proteins and three (bacterial) or four (eukaryotic) ribosomal RNA (rRNA) molecules (not shown). Note the base pairing between tRNA anticodons and complementary codons in the mRNA. Formation of a peptide bond between the aminogroup N on the incoming aa-tRNA and the carboxy-terminal C on the growing protein chain (green) is catalyzed by one of the rRNAs. ; . Note that these are simplified diagrams of tRNAs and the ribosomal subunits. Their actual structures are shown in Figure 5-32b and Figure 5-35. Description The illustration shows the ribosomal R N A (r R N A) with 50 s and 30 s subunits. The codon regions of messenger R N A (m R N A) in the 30 s subunit of r R N A is attached to the anticodon regions of transfer R N A (t R N A). The growing polypeptide chain is catalyzed by 50 s subunit of r R N A. To the right of the r R N A is the arriving t R N A with side chains and to the left of the r R N A is the leaving t R N A. These three types of RNA participate in the synthesis of proteins in all organisms. In this section, we focus on the decoding of mRNA by tRNAs and how the structure of each of these RNAs relates to its specific task. How they work together with ribosomes and protein factors to synthesize proteins is detailed in the following section. Because translation is essential for protein synthesis, the two processes are commonly referred to interchangeably. However, the polypeptide chains resulting from translation must undergo additional steps after translation, including folding and often other changes (e.g., chemical modifications, association with other chains) that are required for the production of mature, functional proteins (see Chapter 3).
Messenger RNA Carries Information from DNA in a Three-Letter Genetic Code As noted previously, the genetic code used by cells is a triplet code, in which every three-nucleotide sequence, or codon, is read from a specified starting point in the mRNA. Of the 64 possible codons in the genetic code (one of four nucleotides at each of the three positions of a codon yields possible codons), 61 specify individual amino acids, and 3 are stop codons. Table 5-1 shows that most amino acids are encoded by more than one codon. Only two — methionine and tryptophan — have a single codon; at the other extreme, leucine, serine, and arginine are each specified by six different codons. The different codons that code for a given single amino acid are said to be synonymous. The code itself is termed degenerate, meaning that a particular amino acid can be specified by several codons.
TABLE 5-1 • The Genetic Code (Codons to Amino Acids)*
*AUG is the most common initiation codon; GUG usually codes for valine; and CUG for leucine, but rarely these codons can also code for methionine to initiate a protein chain. Description The table has 4 rows and 4 columns. The row headers read “U, C, A, and G” and represent the first position (5 prime end). The column headers read “U, C, A, and G” and represent the second position. Each row is further subdivided into four parts labeled U, C, A, and G that represent the third position (3 prime end). U U U - Phenylalanine (P h e) U U C - Phenylalanine (P h e) U U A - Leucine (L e u)
U U G - Leucine (L e u) Row 1 column 2 reads: U C U - Serine (S e r) U C C - Serine (S e r) U C A - Serine (S e r) U C G - Serine (S e r) Row 1 column 3 reads: U A U - Tyrosine (T y r) U A C - Tyrosine (T y r) U A A - Stop U A G - Stop Row 1 column 4 reads: U G U - Cysteine (C y s) U G C - Cysteine (C y s) U G A - Stop U G G - Tryptophan (T r p) Row 2 column 1 reads: C U U - Leucine (L e u) C U C - Leucine (L e u) C U A - Leucine (L e u) C U G - Leucine (L e u); Methionine (M e t) asterisk Row 2 column 2 reads:
C C U - Proline (P r o) C C C - Proline (P r o) C C A - Proline (P r o) C C G - Proline (P r o) Row 2 column 3 reads: C A U - Histidine (H I s) C A C - Histidine (H I s) C A A – Glutamine (G l n) C A G – Glutamine (G l n) Row 2 column 4 reads: C G U - Arginine (A r g) C G C - Arginine (A r g) C G A - Arginine (A r g) C G G - Arginine (A r g) Row 3 column 1 reads: A U U - Isoleucine (I l e) A U C - Isoleucine (I l e) A U A - Isoleucine (I l e) A U G - Start Methionine (M e t) Row 3 column 2 reads: A C U - Threonine (T h r) A C C - Threonine (T h r)
A C A - Threonine (T h r) A C G - Threonine (T h r) Row 3 column 3 reads: A A U - Asparagine (A s n) A A C - Asparagine (A s n) A A A - Lysine (L y s) A A G - Lysine (L y s) Row 3 column 4 reads: A G U - Serine (S e r) A G C - Serine (S e r) A G A - Arginine (A r g) A G G - Arginine (A r g) Row 4 column 1 reads: G U U - Valine (V a l) G U C - Valine (V a l) G U A - Valine (V a l) G U G - Valine (V a l); Methionine (M e t) asterisk Row 4 column 2 reads: G C U - Alanine (A l a) G C C - Alanine (A l a) G C A - Alanine (A l a) G C G - Alanine (A l a)
Row 4 column 3 reads: G A U - Aspartic acid (A s p) G A C - Aspartic acid (A s p) G A A - Glutamic acid (G l u) G A G - Glutamic acid (G l u) Row 4 column 4 reads: G G U - Glycine (G l y) G G C - Glycine (G l y) G G A - Glycine (G l y) G G G - Glycine (G l y) Synthesis of all polypeptide chains in prokaryotic and eukaryotic cells begins with the amino acid methionine. In bacteria, a specialized form of methionine with a formyl group linked to its amino group is used. In most mRNAs, the start (initiation) codon specifying this amino-terminal methionine is AUG. In a few bacterial mRNAs, GUG is used as the initiation codon, and CUG is occasionally used as an initiation codon for methionine in eukaryotes. The three codons UAA, UGA, and UAG do not specify amino acids, but rather constitute stop (termination) codons that mark the carboxyl terminus of polypeptide chains in almost all cells. The sequence of codons that runs from a specific start codon to a stop codon is called a reading frame. This precise linear array of ribonucleotides in groups of three in mRNA specifies the precise linear sequence of amino
acids in a polypeptide chain and also signals where synthesis of the chain starts and stops. The great utility of the genetic code table is that it allows the amino acid sequence of the encoded protein to be deduced from the sequence of an mRNA. Since the genetic code is read as a continuous sequence of triplet codons without divisions between them, a particular mRNA theoretically could be translated in three different reading frames (Figure 5-30). Once the sequence of an mRNA is known (usually determined from the DNA sequence of the transcribed gene), it is necessary to determine which of the three possible reading frames is translated by the ribosome into protein sequence. In principle, this could be done by identifying the AUG codon that is the start of translation. In practice, however, finding the correct initiation codon is error prone and the most often used method is to search for a long open reading frame. The idea behind this method is that in sequences that do not code for proteins, including the two incorrect reading frames of an mRNA, stop codons should appear randomly as 3 out of 64 codons, and thus noncoding reading frames should only have relatively short stretches (on average of about 20 amino acid codons) between stop codons. Thus a given mRNA will have two reading frames that are broken up by multiple stop codons and usually only have one long open reading frame not interrupted by stop codons representing the sequence of the encoded protein.
FIGURE 5-30 Three possible reading frames in an mRNA sequence. If translation of the mRNA sequence shown begins at three different upstream start sites (not shown), then three overlapping reading frames are possible. In this example, the codons are shifted one base to the right in the middle frame and two bases to the right in the third frame, which ends in a stop codon. As a result, the same mRNA nucleotide sequence can specify different amino acids. The correct reading frame for a given mRNA sequence can usually be readily identified as the longest open reading frame that is not interrupted by stop codons. Description In the illustration, three different frames with the codons of the same m R N A sequence from 5 prime direction; G C U U G U U U A C G A A U U A A. Frame 1 reads from the starting codon G C U; U G U; U U A; C G A; A U U, and A A and their respective amino acid chain in polypeptide 1 are Alanine; Cysteine; Leucine; Arginine, and Isoleucine. Frame 2 skips the first codon G and reads C U U; G U U; U A C; G A A; U U A, and A and their respective amino acid chain in polypeptide 2 are Leucine; Valine; Thyrosine; Glutamic acid, and Leucine.
Frame 3 skips the first and second codon G C and reads U U G; U U U; A C G; A A U, and U A A and their respective amino acid chain in polypeptide 3 are Leucine; Phenylalanine; Serine; Tyrosine, and Stop codon. A related use of the genetic code is to deduce the effect that a gene mutation will have on the encoded protein. To see the range of possibilities, imagine the effect of mutations in a serine codon UCG that occurs in the middle of a protein-coding sequence. A mutation changing would give a UCA codon, which also encodes serine and is thus a synonymous mutation, and would be predicted to have no effect on the function of the encoded protein. By contrast, a mutation changing would give a UUG codon, which codes for leucine: this is a missense mutation that may have an effect on the function of the protein depending on the importance of serine at that position for protein function. On the other hand, a mutation changing would give UAG, which is a stop codon and thus would cause a nonsense mutation that would terminate the protein sequence early and would likely cause a complete loss of function. Finally, an addition or subtraction of one or two bases will shift the reading frame. Known as a frameshift mutation, such a change in sequence would lead to a scrambled amino sequence with an early termination of the protein. Unless the frameshift mutation occurred near the end of the coding sequence, the likely consequence would be a complete loss of function. The genetic code shown in Table 5-1 is known as the universal code since meaning of each codon is the same in most known organisms — strong evidence that life on Earth evolved only once. The 20 amino acids are not
The Folded Structure of tRNA Promotes Its Decoding Functions
distributed among the 61 sense codons in haphazard fashion, and the underlying structure of the universal code provides clues to how an optimal code evolved. The most prominent pattern to emerge when the code is considered as a whole is that changing one pyrimidine for another ( or ) or changing one purine for another ( or ) in either the first or the third position typically either leads to a synonymous codon or changes to a codon for an amino acid with similar chemical properties. These types of sequence changes, as we saw in Section 5.3, are the most frequent kinds of mistakes to be made during DNA replication. Evidently the code evolved to minimize the deleterious effects on the amino acid sequence of proteins of the most common types of mutations in DNA. The Folded Structure of tRNA Promotes Its Decoding Functions The two information-carrying polymers, DNA and mRNA, perform their functions with the flexibility to accommodate a vast number of different sequences. On the other hand, tRNAs have specific fixed sequences that allow the tRNA molecules to fold into a three-dimensional structure that is crucial for their function. To participate in protein synthesis, a tRNA molecule must become chemically linked to a particular amino acid via a high-energy bond, forming an aminoacyl-tRNA (Figure 5-31). Enzymes called aminoacyl-tRNA synthetases are responsible for adding the correct amino acid to a given tRNA and, as will be described, they do this through the recognition of particular features of the tRNA structure. During the
process of translation, an aminoacyl-tRNA interacts with the ribosome and other protein factors so that the anticodon sequence is properly basepaired with a codon in mRNA. All of these interactions also depend on the folded structure of the tRNA (see Figure 5-29).
FIGURE 5-31 Translating nucleic acid sequence into amino acid sequence. (a) The process for translating nucleic acid sequences in mRNA into amino acid sequences in proteins involves two steps. Step 1 : An aminoacyl-tRNA synthetase first couples a specific amino acid, via a high-energy ester bond (yellow), to either the or hydroxyl of the terminal adenosine in the corresponding tRNA. Step 2 : A three-base sequence in the tRNA (the anticodon) then base-pairs with a codon in the mRNA specifying the attached amino acid. If an error occurs in either step, the wrong amino acid may be incorporated into a polypeptide chain. . (Note that this is a simplified diagram of ; its actual structure is shown in Figure 5-32b.) (b) Molecular model of the human mitochondrial aminoacyl-tRNA synthetase for Phe in complex with . [Data from L. Klipcan et al., 2012, J. Mol. Biol. 415:527, PDB ID 3tup.]
Description Step 1: Aminoacyl t R N A synthetase adds phenylalanine to the hydroxyl group of t R N A specific for phenylalanine (t R N A superscript phe) through high-energy ester bond and forms aminoacyl t R N A. The anticodon region of t R N A specific for phenylalanine is A A A. During the conversion process, A T P is converted into A M P and inorganic pyrophosphate. Step 2: The anticodon region A A A of aminoacyl t R N A binds to U U U codon of the m R N A. The illustration labeled B shows irregular shaped aminoacyl t R N A synthetase specific for phenylalanine attached to the distinct folded structure of t R N A specific for phenylalanine. Some 30–40 different tRNAs have been identified in bacterial cells and as many as 50–100 in animal and plant cells. Thus the number of tRNAs in most cells is more than the number of amino acids used in protein synthesis (20), but less than the number of amino acid codons in the genetic code (61). Consequently, many amino acids have more than one tRNA to which they can attach (explaining how there can be more tRNAs than amino acids); in addition, many tRNAs can pair with more than one codon (explaining how there can be more codons than tRNAs). tRNA molecules, which are 70–80 nucleotides long, are initially formed by transcription from a tRNA gene template. The maturation of the primary transcript to a functional tRNA molecule with precise threedimensional structure takes place in three stages. The first stage is the chemical modification of certain bases, creating nonstandard nucleotides, such as inosine, dihydrouridine, and pseudouridine. As we will see shortly,
some of these modified bases are known to play an important role in protein synthesis. The second stage involves complementary base pairing between antiparallel arrangements of segments of the same tRNA molecule to produce a form that resembles a cloverleaf when drawn in two dimensions (Figure 5-32a). The four short, double-helical segments that form are stabilized mostly by Watson-Crick base pairing, although other nonstandard base pairs, such as G·U, are accommodated by the helical structures that are much less uniform than that of DNA. Three of the four helical stems have loops containing seven or eight bases at their ends, while the remaining, unlooped stem contains the free and ends of the chain. The three nucleotides composing the anticodon are located at the center of the middle loop, in an accessible position that facilitates codonanticodon base pairing. In all tRNAs, the end of the unlooped acceptor stem, to which a specific amino acid is attached, has the sequence CCA, which in most cases is added after synthesis and processing of the tRNA are complete. Finally, the first and third loops join together by base pairing to form a compact, L-shaped structure when viewed in three dimensions. The anticodon loop and acceptor stem form the ends of the folded tRNA molecule (Figure 5-32b).
FIGURE 5-32 Structure of tRNAs. (a) Although the exact nucleotide sequence varies among tRNAs, they all fold into four base-paired stems and three loops. The CCA sequence at the end is also found in all tRNAs. Attachment of an amino acid to the A yields an aminoacyl-tRNA. Some of the A, C, G, and U residues are modified post-transcriptionally in most tRNAs (see key). Dihydrouridine (D) is nearly always present in the D loop; likewise, ribothymidine (T) and pseudouridine (ψ) are almost always present in the TψCG loop. Yeast alanine tRNA, represented here, also contains other modified bases. The triplet at the tip of the anticodon loop base-pairs with the corresponding codon in mRNA. See R. W. Holly et al., 1965, Science 147:1462. (b) Three-dimensional model of the generalized backbone of all tRNAs. Note the L shape of the molecule. [Part (b) Data from J. G. Arnez and D. Moras, 1997, Trends Biochem. Sci. 22:211, PDB ID 1vtq.] Description The illustration labeled A shows a cross-shaped t R N A with three loops. The loops at the right, left, and at the bottom are labeled as T pseudouridine C G loop, D loop, and anticodon loop, respectively. Below the t R N A is m R N A with codon C C G pairs with anticodon I G C in the anticodon loop. The acceptor arm at the top contains 5 prime and 3 prime ends with A C G and U nucleotides. The nucleotides at the 3 prime end start with A C C. The illustration labeled B shows the three-dimensional structure of t R N A in the shape of inverted upper case L. The labels on the L shaped t R N A is
Nonstandard Base Pairing Often Occurs Between Codons and Anticodons
acceptor stem, T pseudouridine C G loop, D loop, variable loop, anticodon loop, and anticodons. Nonstandard Base Pairing Often Occurs Between Codons and Anticodons If perfect Watson-Crick base pairing between codons and anticodons were required, cells would have to contain at least 61 different types of tRNAs, one for each codon that specifies an amino acid. As noted previously, however, many cells contain fewer than 61 tRNAs. The explanation for the smaller number lies in the capability of a single tRNA anticodon to recognize more than one, but not necessarily every, codon corresponding to a given amino acid. This broader recognition can occur because of nonstandard pairing between bases in the so-called wobble position: that is, the third base in an mRNA codon and the corresponding first base in its tRNA anticodon. The first and second bases of a codon almost always form standard Watson-Crick base pairs with the third and second bases, respectively, of the corresponding anticodon, but four nonstandard interactions can occur between bases in the wobble position. Particularly important is the G·U base pair, which fits into the short, 3-bp RNA-RNA double-stranded region formed between the codon and the anticodon almost as well as the standard G·C pair. Thus a tRNA anticodon with G in the first (wobble)
position can base-pair with the two corresponding codons that have either pyrimidine (C or U) in the third position (see Figure 5-26). For example, the phenylalanine codons UUU and UUC are both recognized by the tRNA that has GAA as its anticodon. In fact, any two codons of the type ; encode a single amino acid and are decoded by a single tRNA with G in the first (wobble) position of the anticodon. Although adenine is rarely found in the anticodon wobble position, many tRNAs in plants and animals contain inosine (I), a deaminated product of adenine, at this position. Inosine can form nonstandard base pairs with A, C, and U. A tRNA with inosine in the wobble position thus can recognize the corresponding mRNA codons with A, C, or U in the third (wobble) position (see Figure 5-33). For this reason, inosine-containing tRNAs are heavily employed in translation of the synonymous codons that specify a single amino acid. For example, four of the six codons for leucine (CUA, CUC, CUU, and UUA) are all recognized by the same tRNA with the anticodon ; the inosine in the wobble position forms nonstandard base pairs with the third base in each of these four codons.
FIGURE 5-33 Nonstandard base pairing at the wobble position. The base in the third (or wobble) position of an mRNA codon often forms a nonstandard base pair with the base in the first (or wobble) position of a tRNA anticodon. Wobble pairing allows a tRNA to recognize more than one mRNA codon, although each of those tRNAs will bear the same amino acid. Note that a tRNA with I (inosine) in the wobble position can read (become paired with) three different codons, and a tRNA with G or U in the wobble position can read two codons. Although A is theoretically possible in the wobble position of the anticodon, it is almost never found in nature. (Note that this is a simplified diagram of a tRNA. The actual structure of a tRNA is shown in Figure 5-32b.) Description An illustration shows the structure of t R N A with 5 prime and 3 prime ends. The anticodon regions in t R N A reads 1, 2, and 3 from right to left. Below the t R N A is m R N A with 5 prime and 3 prime ends. The codon regions in the m R N A reads 3, 2, and 1 from right to left. The corresponding text reads, if the bases are in first, or wobble, position of anticodon then the t R N A may recognize codons in m R N A having these bases in third position. A table next to the illustration has 5 columns with anticodons represented in purple and its complementary codons are represented in blue. The wobble pairings are as follows: Anticodon-C, codon-G; Anticodon-A, codon- U; Anticodon-G, codon- C U; Anticodon-U, codon- A G; Anticodon-I, codon- C A U.
Amino Acids Are Linked to Their Cognate tRNAs with Great Accuracy
Amino Acids Are Linked to Their Cognate tRNAs with Great Accuracy Recognition of the codon or codons specifying a given amino acid by a particular tRNA is actually the second step in decoding the genetic message. The first step, attachment of the appropriate amino acid to a tRNA, is catalyzed by a specific aminoacyl-tRNA synthetase. Each of the 20 different synthetases recognizes one amino acid and all its compatible, or cognate, tRNAs. These coupling enzymes link an amino acid to the free or hydroxyl of the adenosine at the terminus of the tRNA molecule by an ATP-requiring reaction. In this reaction, the amino acid is linked to the tRNA by a high-energy bond and is thus said to be activated. The energy of this bond subsequently drives the formation of the peptide bonds linking adjacent amino acids in a growing polypeptide chain. The equilibrium of the aminoacylation reaction is driven further toward activation of the amino acid by hydrolysis of the high-energy phosphoanhydride bond in the released pyrophosphate (see Figure 5-31a). Each aminoacyl-tRNA synthetase must specifically recognize two substrates; the amino acid and the corresponding set of cognate tRNAs. Any errors made in tRNA charging could lead to a deleterious error in translation and the aminoacyl-tRNA synthetases have evolved to have high fidelity, mischarging a tRNA only about one in times. As one would expect, most aminoacyl-tRNA synthetases recognize their cognate tRNAs by interacting with the anticodon loop. However, for amino acids that have multiple codons, other features common to all the cognate tRNAs
contribute to specific recognition. In an extreme case, in E. coli, the four tRNAs for alanine are not recognized by their anticodon loop at all. Instead, all four tRNAs contain a unique G·U base pair in the acceptor stem, which is used for specific recognition by the corresponding aminoacyl tRNA synthetase. In other instances, specific bases in incorrect tRNAs that are structurally similar to a cognate tRNA will inhibit charging of the incorrect tRNA. Thus recognition of the correct tRNA depends on both positive interactions and the absence of negative interactions. A second source of errors in charging of tRNAs by aminoacyl-tRNA synthetases is selection of the wrong amino acid. In cases in which mischarging by a structurally very similar amino acid is possible, the aminoacyl-tRNA synthetase has evolved a second active site with a proofreading activity. For example, a single active site in the aminoacyltRNA synthetase for isoleucine would not be able to effectively exclude valine, which is similar to isoleucine but lacks one methyl group. For this enzyme, there is a second proofreading site that can remove an incorrect amino acid that has been added to . The hydrophobic pocket for this site is too small to admit isoleucine, but it can admit valine that has been erroneously linked to and will remove it. This mechanism of proofreading by aminoacyl-tRNA synthetases is similar in outline to proofreading of incorrect nucleotide incorporation by the exonuclease activity of DNA polymerase. KEY CONCEPTS OF SECTION 5.5 The Decoding of mRNA by tRNAs
Genetic information from DNA is carried by mRNA in the form of a non-overlapping, degenerate triplet code. Each amino acid is encoded by one or more three-nucleotide sequences (codons) in mRNA. Each codon specifies one amino acid, but most amino acids are encoded by multiple codons (see Table 5-1). The AUG codon for methionine is the most common start codon, specifying the amino acid at the amino-terminus of a protein chain. Three codons (UAA, UAG, UGA) function as stop codons and specify no amino acids. There are three possible reading frames for the translation of a given mRNA sequence. The protein coding sequence can be identified as a long open reading frame that is not interrupted by stop codons. The genetic code can be used to determine the linear sequence of amino acids in a protein sequence and to deduce the effect of gene mutations on the encoded protein. All tRNAs have a similar three-dimensional structure that includes an acceptor stem for attachment of a specific amino acid and a stem-loop with a three-base anticodon sequence at its end (see Figure 5-32). The anticodon can base-pair with its corresponding codon in mRNA. Because of nonstandard interactions, a tRNA may base-pair with more than one mRNA codon; conversely, a particular codon may base-pair with multiple tRNAs. In each case, however, only the proper amino acid is inserted into a growing polypeptide chain. Each of the 20 aminoacyl-tRNA synthetases recognizes a single amino acid and covalently links it to a cognate tRNA, forming an aminoacyl-tRNA (see Figure 5-31). This reaction activates the amino acid so that it can participate in peptide bond formation.
Ribosomes Are Protein-Synthesizing Machines
5.6 Stepwise Synthesis of Proteins on Ribosomes The previous sections have introduced two of the major participants in protein synthesis: mRNAs and aminoacyl-tRNAs. Here we first describe the third key player in protein synthesis — the rRNA-containing ribosome — before taking a detailed look at how all three components are brought together to carry out the biochemical events leading to the formation of polypeptide chains by ribosomes. The complex process of translation can be divided into three stages — initiation, elongation, and termination — which we consider in order. We focus our description on translation in eukaryotic cells, but the mechanism of translation is fundamentally the same in all cells. Ribosomes Are Protein-Synthesizing Machines In simple terms, the ribosome serves both as a scaffold to facilitate the binding aminoacyl-tRNAs to sequential codons in the mRNA and as an enzyme to catalyze the peptidyl transfer reaction for the stepwise formation of peptide bonds in the growing polypeptide chain. The ribosome is responsible for all protein synthesis and is the most abundant RNA-protein complex in the cell. Elongation of polypeptides occurs at a rate of 3 to 5 amino acids added per second, so that although small
proteins may be made in a minute or less, the formation of very large proteins such as titin, a muscle protein of about 30,000 amino acid residues, takes 2–3 hours. The ribosome must therefore be precise, and it must be persistent. With the aid of the electron microscope, ribosomes were first discovered as small, discrete, RNA-rich particles found to be most abundant in those cells most active in protein synthesis. However, their direct role in protein synthesis was not recognized until reasonably pure ribosome preparations were obtained. In vitro radiolabeling experiments with such preparations showed that radioactive amino acids were first incorporated into growing polypeptide chains associated with ribosomes before appearing in finished proteins. Although there are differences between the ribosomes of bacteria, archaea, and eukaryotes, the great structural and functional similarities between ribosomes from all species reflect the common evolutionary origin of the most basic constituents of living cells. A ribosome is composed of three (in bacteria and archaea) or four (in eukaryotes) distinct rRNA molecules and as many as 80 proteins, organized into a large subunit and a small subunit (Figure 5-34 and Table 5-2). The ribosomal subunits and the rRNA molecules are commonly designated in Svedberg units (S), a measure of the sedimentation rate of macromolecules centrifuged under standard conditions — essentially, a logarithmic measure of size. The small ribosomal subunit contains a single rRNA molecule, referred to as small rRNA. The large subunit contains a molecule of large rRNA and one molecule of 5S rRNA, plus an additional molecule of 5.8S rRNA in
eukaryotes. The lengths of the rRNA molecules, the numbers of proteins in each subunit, and consequently, the sizes of the subunits differ between bacterial and eukaryotic cells (see Table 5-2). The assembled ribosome is 70S in bacteria and 80S in vertebrates.
FIGURE 5-34 Structure of the bacterial ribosome. Model of the Thermus thermophilus ribosome viewed along the interface between the large (50S) and small (30S) subunits. The 16S rRNA and proteins in the small subunit are dark gray. RNA is depicted as a tube model
and protein surfaces are shown. The 23S rRNA and proteins in the large subunit are light gray, and the 5S rRNA is an intermediate shade of gray. The surface of the ribosome is made partially transparent to display the positions of bound tRNAs. Note that the ribosomal proteins are located primarily on the surface of the ribosome. [Data from A. Korostelev et al., 2006, Cell 126:1065–1077, PDB ID 4v4i.] Description The illustration shows 50 s (top) and 30 s (bottom) ribosomal subunits with t R N A. The sites of t R N A are represented in different colors. E, green; P, yellow; A, blue. A polypeptide (light green) is attached to the P site of the t R N A. m R N A (red) from 5 prime to 3 prime direction represented in the form of a tube is attached to E P A.
TABLE 5-2 • Ribosome Components Description The table has four columns and ten rows. The column headers are common core, E.coli, S. cerevisiae, and human. The row entries are as follows. Row 1: Common core,
2.0 Mega Dalton; E.coli, 2.3 Mega Dalton; S. cerevisiae, 3.3 Mega Dalton; human, 4.3 Mega Dalton. Row 2: Common core, 34 proteins; E.coli, 54 proteins; S. cerevisiae, 79 proteins; human, 80 proteins. Row 3: Common core, 3 r R N A’s; E.coli, 3 r R N A’s; S.
cerevisiae, 4 r R N A’s; human, 4 r R N A’s. Row 4: Common core, large subunit; E.coli, 50 S; S. cerevisiae, 60 S; human, 60 S. Row 5: Common core, 19 proteins; E.coli, 33 proteins; S. cerevisiae, 46 proteins; human, 47 proteins. Row 6: Common core, 23 S r R N A: 2843 bases; E.coli, 23 S r R N A: 2904 bases; S. cerevisiae, 25 S r R N A: 3396 bases asterisk, 5.8 S r R N A: 158 bases asterisk; human, 28 S r R N A: 5034 bases asterisk, 5.8 S r R N A: 156 bases asterisk. Row 7: Common core, 5 S r R N A: 121 bases; E.coli, 5 S r R N A: 121 bases; S. cerevisiae, 5 S r R N A: 121 bases; human, 5 S r R N A: 121 bases. Row 8: Common core, small subunit; E.coli, 30 S; S. cerevisiae, 40 S; human, 40 S. Row 9: Common core, 15 proteins; E.coli, 21 proteins; S. cerevisiae, 33 proteins; human, 33 proteins. Row 10: Common core, 16 S r R N A: 1458 bases; E.coli, 16 S r R N A: 1542 bases; S. cerevisiae, 18 S r R N A: 1800 bases; human, 18 S r R N A: 1870 bases. Note: Asterisk, 5.8 S r R N A in eukaryotes is basepaired to 25 S or 28 S r R N A. Source: Data from G. Yusupov and M. Yusupov, Ann. Rev. Biochem., 2014, 83:467. The sequences of the small and large rRNAs from thousands of organisms are now known. Although the primary nucleotide sequences of these rRNAs vary considerably, the same parts of each type of rRNA contain complementary sequences that can form internally base-paired segments, indicating that they all share the same core structure. Since every organism has ribosomes, differences between their rRNA sequences have proven to be the most reliable means to determine the evolutionary distances between organisms. Even today, when whole genome sequences are often available, phylogenetic comparisons usually begin with the sequence of the respective rRNA genes. The three-dimensional structures of bacterial and yeast ribosomes (see
Figure 5-35) and of the large subunit of an archaeal ribosome have been determined by x-ray crystallography. The structures of human (see Figure 5-35d) and plant ribosomes have also been determined by cryoelectron
microscopy. The structure of the rRNAs in the common core, where mRNAs and tRNAs are bound and where peptide bond formation is catalyzed, is similar in all three domains of life. However, archaeal rRNAs and proteins are more similar to those of eukaryotic ribosomes than to those of bacterial ribosomes, reflecting their later divergence from a common ancestor (see Figure 1-1). For the most part, the multiple ribosomal proteins are much smaller than the rRNAs and associate with the surface of the ribosomes. Although the number of protein molecules in ribosomes greatly exceeds the number of RNA molecules, RNA constitutes about 60 percent of the mass of a bacterial ribosome and about 50 percent of the mass of a human ribosome. Eukaryotic ribosomes are generally similar to bacterial ribosomes but are larger because of eukaryote-specific insertions of RNA segments into regions of the common core rRNAs as well as the presence of a larger number of proteins (see Figure 5-35 and Table 5-2). Basic aspects of protein synthesis are thought to be similar among all three domains of life, although initiation of translation in eukaryotes, discussed later, is more complex and subject to additional mechanisms of regulation.
FIGURE 5-35 Comparison of the common core structure at the center of ribosomes from all domains of life and bacterial, yeast, and human ribosomes. (a) RNA in the
Methionyl-tRNAiMet Recognizes the AUG Start Codon
common core structure is shown in light blue and protein domains common to all ribosomes are shown in pink. Additions to the common core structure are shown in dark blue for RNA and red for proteins in ribosomes from T. thermophilus (b) and S. cerevisiae (c). (d) Human ribosome structure from cryoelectron microscopy. A tRNA visible in the E site is shown in green. [Parts (a, b, c) Data from G. Yusupova and M. Yusupov, 2014, Annu. Rev. Biochem. 83:467; data for (d) from H. Khatter et al., 2015, Nature 520:640; PDB ID 4ug0.] Description The illustration labeled A, B, and C shows the X-ray crystallography images of ribosomes and proteins in common core, bacteria (Thermus thermophilus), and lower eukaryotes (Saccharomyces cerevisiae) at 2, 2.3, and 3.3 Mega Dalton, respectively. The illustration labeled D shows the cryoelectron microscopy image of ribosomes in higher eukaryotes (Homo sapiens) at 4.3 Mega Dalton. The most therapeutically effective antibiotics target processes in bacterial cells that are essential for life and are fundamentally different between bacterial and mammalian cells. Because ribosomes satisfy both criteria, the bacterial ribosome has been a major target for the development of new antibiotics. The high-resolution structures of ribosomes are providing new insights into the mechanism by which many antibiotics inhibit bacterial protein synthesis without affecting the function of mammalian ribosomes. These insights are providing important clues for the design and synthesis of new antibiotics. Such research is desperately needed as the occurrence of bacteria resistant to currently available antibiotics becomes increasingly more common, especially in hospitals, where antibiotic-resistant bacteria are under positive selection.
Eukaryotic Translation Initiation Usually Occurs at the First AUG Downstream from the 5′ End of an mRNA
MethionylRecognizes the AUG Start Codon As noted earlier, the AUG codon for methionine functions as the start codon in the vast majority of mRNAs. A critical aspect of translation initiation is to begin protein synthesis at the start codon, thereby establishing the correct reading frame for the entire mRNA. Both bacteria and eukaryotes contain two different methionine can initiate protein synthesis, and can incorporate methionine only into a growing protein chain. The same aminoacyl-tRNA synthetase (MetRS) charges both tRNAs with methionine, but only (i.e., activated methionine attached to ) can bind at the appropriate site on the small ribosomal subunit, the P site, to begin synthesis of a polypeptide chain. The regular , and all other charged tRNAs, bind only to the A site, as described later. tRNAs enter the exit or E site after transferring their covalently bound amino acids to the growing polypeptide chain. Eukaryotic Translation Initiation Usually Occurs at the First AUG Downstream from the 5 End of an mRNA During the first stage of translation, the small and large ribosomal subunits assemble around an mRNA that has a correctly ′
positioned at the start codon in the ribosomal P site. In eukaryotes, the assembly of this complex is mediated by a special set of proteins known as eukaryotic translation initiation factors (eIFs). As each individual component joins the complex, it is guided by interactions with specific eIFs. Several of the initiation factors bind GTP, and the hydrolysis of GTP to GDP functions as a switch to ensure that the assembly pathway proceeds in an orderly and irreversible fashion. By coupling of assembly to the essentially irreversible step of GTP hydrolysis ensures that, once formed, the appropriate complexes are stable. The current model for initiation of translation in vertebrates is depicted in
Figure 5-36. Large and small ribosomal subunits released from a previous round of translation are kept apart by the binding of eIFs 1, 1A, and 3 to the small 40S subunit (Figure 5-36, top). The first step of translation initiation is formation of a 43S preinitiation complex. This preinitiation complex is formed when the 40S subunit with eIFs 1, 1A, and 3 associates with eIF5 and a ternary (three-part) complex consisting of the and eIF2 bound to GTP (Figure 5-36, steps 1 and 2 ). The initiation factor eIF2 alternates between association with GTP and GDP; it can bind only when it is associated with GTP. Cells can inhibit protein synthesis by phosphorylating a serine residue on the eIF2 bound to GDP; the phosphorylated complex is unable to exchange the bound GDP for GTP and cannot bind , so protein synthesis cannot occur.
FIGURE 5-36 Initiation of translation in eukaryotes. The current model of eukaryotic initiation involves eight steps. Step 1 : An eIF2 ternary complex forms when eIF2·GTP binds a . Step 2 : When a ribosome dissociates at the termination of translation, the 40S subunit is bound by eIF1, eIF1A, and eIF3. A 43S preinitiation complex forms when this subunit associates with an eIF2 ternary complex and eIF5. Step 3 : An mRNA is activated when a multisubunit eIF4 complex binds: subunit eIF4E binds to the cap, and subunit eIF4G binds multiple copies of the cytoplasmic poly(A)-binding protein (PABPC) bound to the mRNA poly(A) tail. For simplicity, binding of only one PABPC to eIF4G is shown. Then eIF4B, which stimulates eIF4A helicase activity, also joins this circular complex in which both the mRNA cap and poly(A) tail are associated with the eIF4 complex. Step 4 : The 43S preinitiation complex binds an eIF4-mRNA complex. Step 5 : The RNA helicase activity of subunit eIF4A unwinds any RNA secondary structure at the end of the mRNA as the 40S subunit scans in the direction until it recognizes the initiation codon. For simplicity, eIF4E is diagrammed as releasing from the remainder of the eIF4 complex, but in reality, it remains associated, forming a loop in the mRNA between the cap and the scanning eIF4 complex. Step 6 : Recognition of the initiation codon causes eIF5 to stimulate hydrolysis of eIF2-bound GTP. This switches the conformation of the scanning complex to a 48S initiation complex with the anticodon of basepaired to the initiator AUG in the 40S-subunit P site. Step 7 : The 60S subunit joins the 40S subunit, leading to the release of most of the earlier-acting eIFs as eIF5B-GTP binds to eIF1A in the ribosomal A site. The released eIF4 complex and eIF4B associate with the cap and PABPC as shown in step 3 to prepare for interaction with another 43S preinitiation complex. For simplicity, this association is not shown. Step 8 : Correct association of the 40S and 60S subunits results in hydrolysis of eIF5B-bound GTP, release of eIF5B-GDP and eIF1A, and formation of the 80S initiation complex with base-paired to the initiation codon in the ribosomal P site. See R. J. Jackson et al., 2010, Nat. Rev. Mol. Cell Biol. 11:113. Description The steps involved in the initiation of translation in eukaryotes are as follows: Step 1: e l F 2 ternary complex formation. Step 2: 43 S complex formation.
Step 3: m R N A activation. Step 4: Attachment to m R N A. Step 5: 5 prime to 3 prime scanning. Step 6: Initiation codon recognition hydrolysis of e l F 2 bound G T P and inorganic phosphate release. Step 7: Subunit joining and factor displacement. Step 8: Hydrolysis of e l F 5 B bound G T P and release of e l F 5 B and e l F 1 A. The mRNA to be translated is bound by the multisubunit eIF4 complex, which interacts with both the cap and the cytoplasmic poly(A)-binding protein (PABPC) bound in multiple copies to the mRNA poly(A) tail. Both interactions are required for translation of most mRNAs. This binding results in the formation of a circular complex (Figure 5-36, step 3 ). The eIF4 cap-binding complex consists of several subunits with different functions. The eIF4E subunit binds the cap on mRNAs (see Figure 513). The large eIF4G subunit binds cooperatively to several PABPC proteins bound to the mRNA poly(A) tail and also forms a scaffold to which the other eIF4 subunits bind. The mRNA-eIF4 complex then associates with the preinitiation complex through an interaction between eIF4G and eIF3 (step 4 ). The initiation complex slides along, or scans, the associated mRNA, with eIF4A, stimulated by eIF4B, unwinding the RNA secondary structure by using energy from ATP hydrolysis (step 5 ). Scanning stops when the anticodon recognizes the start codon, which is the first AUG
downstream from the end in most eukaryotic mRNAs. Recognition of the start codon leads to hydrolysis of the GTP associated with eIF2, an irreversible step that prevents further scanning, resulting in formation of the 48S initiation complex (step 6 ). This commitment to the correct initiation codon is facilitated by eIF5, an eIF2 GTPase–activating protein (GAP, see Figure 3-35). Selection of the initiating AUG is facilitated by specific surrounding nucleotides called the Kozak sequence, for Marilyn Kozak, who defined it:
. The A preceding the AUG (in bold) and the G immediately following it are the most important nucleotides affecting translation initiation efficiency. Association of the large (60S) subunit with the small subunit, which is mediated by eIF5B bound to GTP, results in displacement of many of the initiation factors (step 7 ). Correct association between the ribosomal subunits results in hydrolysis of the eIF5B-bound GTP to GDP and the release of eIF5B-GDP and eIF1A (step 8 ), completing the formation of an 80S initiation complex. Coupling of the ribosome subunit–joining reaction to GTP hydrolysis by eIF5B allows the initiation process to continue only when the subunit interaction has occurred correctly. It also makes this an irreversible step, so that the ribosomal subunits do not dissociate until the entire mRNA is translated and protein synthesis is terminated. The eukaryotic protein-synthesizing machinery begins translation of most cellular mRNAs within about 100 nucleotides of the -capped end, as just described. However, some cellular mRNAs contain an internal ribosome entry site (IRES) located far downstream from the end. It is thought that
During Chain Elongation Each Incoming Aminoacyl-tRNA Moves Through Three Ribosomal Sites
cellular IRESs form RNA structures that interact with a complex of eIF4A and eIF4G, which then associates with eIF3 bound to a 40S subunit with eIF1 and eIF1A. This assembly then binds an eIF2 ternary complex to assemble an initiation complex directly on a neighboring AUG codon. In addition, translation of some viral RNAs, which lack a cap, is initiated at viral IRES sequences. These RNAs fall into different classes depending on how many of the standard eIFs are required for initiation. In the case of cricket paralysis virus, the ∼200-nt-long IRES folds into a complex structure that interacts directly with the 40S ribosomal subunit and leads to initiation without any of the eIFs or even the initiator . During Chain Elongation Each Incoming Aminoacyl-tRNA Moves Through Three Ribosomal Sites The correctly positioned ribosome– complex is now ready to begin the task of stepwise addition of amino acids by in-frame translation of the mRNA. As is the case with initiation, a set of specialized proteins, termed translation elongation factors (EFs), is required to carry out this process of chain elongation. The key steps in elongation are the entry of each succeeding aminoacyl-tRNA with an anticodon complementary to the next codon, the formation of a peptide bond, and the movement, or translocation, of the ribosome one codon at a time along the mRNA. At the completion of translation initiation, as noted already, is bound to the P site on the assembled 80S ribosome (Figure 5-37, top).
This region of the ribosome is called the P site because the tRNA chemically linked to the growing polypeptide chain is located here. The second aminoacyl-tRNA is brought into the ribosome as a ternary complex in association with EF1α·GTP and becomes bound to the A site, so named because it is where aminoacyl-tRNAs bind (step 1 ). EF1α·GTP bound to various aminoacyl-tRNAs diffuse into the A site, but the next step in translation proceeds only when the tRNA anticodon base-pairs with the second codon in the coding region. When that occurs properly, the GTP in the associated EF1α·GTP is hydrolyzed. The hydrolysis of GTP promotes a conformational change in EF1α that leads to release of the resulting EF1α·GDP complex and tight binding of the aminoacyl-tRNA in the A site (step 2 ). This conformational change also positions the aminoacylated end of the tRNA in the A site close to the end of the in the P site. GTP hydrolysis, and hence tight binding, does not occur if the anticodon of the incoming aminoacyl-tRNA cannot base-pair with the codon at the A site. In this case, the ternary complex diffuses away, leaving an empty A site that can associate with other aminoacyl-tRNA– EF1α·GTP complexes until a correctly base-paired tRNA is bound. Thus GTP hydrolysis by EF1α is another proofreading step that allows protein synthesis to proceed only when the correct aminoacyl-tRNA is bound to the A site. This phenomenon contributes to the fidelity of protein synthesis.
FIGURE 5-37 Chain elongation in eukaryotes. Once the 80S ribosome with Metin the ribosome P site is assembled (top), a ternary complex bearing the second amino acid coded by the mRNA binds to the A site (step 1 ). Following a conformational change in the ribosome induced by hydrolysis of GTP in EF1α·GTP (step 2 ), the large rRNA catalyzes peptide bond formation between and (step 3 ). Hydrolysis of GTP in EF2·GTP causes another conformational change in the ribosome that results in its translocation one codon along the mRNA and shifts the unacylated to the E site and the tRNA with the bound peptide to the P site (step 4 ). The cycle can begin again with binding of a ternary complex bearing to the now open A site. In the second and subsequent elongation cycles, the tRNA at the E site is ejected during step 2 as a result of the conformational change induced by hydrolysis of GTP in EF1α·GTP. Description The steps involved in the elongation of translation in eukaryotes are as follows: Step 1: Entry of aminoacyl t R N A at A site. Step 2: G T P hydrolysis and ribosome conformational change. Step 3: Peptide bond formation. Step 4: Translocation. With the initiating at the P site and the second aminoacyltRNA tightly bound at the A site, the α-amino group of the second amino acid reacts with the activated (ester-linked) methionine on the initiator tRNA, forming a peptide bond (Figure 5-37, step 3 ; see Figure 5-29). Since the vast majority of cellular catalysts are protein enzymes, it was long assumed that the catalyst for peptide transfer would be one of the ribosomal proteins. It therefore came as quite a surprise when the highresolution crystal structure of the bacterial large ribosomal subunit
revealed that no proteins lie near the site of peptide bond synthesis and that the peptidyltransferase reaction was likely catalyzed by the large rRNA. The catalytic ability of the large rRNA in bacteria has been demonstrated by carefully removing the vast majority of the protein from large ribosomal subunits. The nearly pure bacterial 23S rRNA can catalyze a peptidyltransferase reaction between analogs of aminoacyl-tRNA and peptidyl-tRNA. The -hydroxyl of the terminal A of the peptidyl-tRNA in the P site also participates in catalysis. Following peptide bond synthesis, the ribosome translocates a distance equal to one codon along the mRNA. This translocation step is monitored by hydrolysis of the GTP in eukaryotic EF2·GTP. Once translocation has occurred correctly, the bound GTP is hydrolyzed, another irreversible process that prevents the ribosome from moving along the RNA in the wrong direction or from translocating an incorrect number of nucleotides. As a result of conformational changes in the ribosome that accompany proper translocation and the resulting GTP hydrolysis by EF2, , now without its activated methionine, is moved to the E (exit) site on the ribosome; concurrently, the second tRNA, now covalently bound to a dipeptide (a peptidyl-tRNA), is moved to the P site (Figure 5-37, step 4 ). Translocation thus returns the ribosome conformation to a state in which the A site is open and able to accept another aminoacyl-tRNA complexed with EF1α·GTP, beginning another cycle of chain elongation. Repetition of the elongation cycle depicted in Figure 5-37 adds amino acids one at a time to the carboxyl terminus of the growing polypeptide, as directed by the mRNA sequence, until a stop codon is encountered. In
Translation Is Terminated by Release Factors When a Stop Codon Is Reached
subsequent cycles, the conformational change that occurs in step 2 ejects the unacylated tRNA from the E site. As the nascent polypeptide chain becomes longer, it threads through a channel in the large ribosomal subunit, exiting at a position opposite the side that interacts with the small subunit (Figures 5-34). In the absence of the ribosome, the three-base-pair RNA-RNA hybrid between the tRNA anticodons and the mRNA codons in the A and P sites would not be stable; RNA-RNA duplexes between separate RNA molecules must be considerably longer to be stable under physiological conditions. However, multiple interactions between the large and small rRNAs and the general domains of tRNAs (e.g., the D and TψCG loops, see Figure 5-32) stabilize the tRNAs in the A and P sites, while other RNA-RNA interactions sense correct codon-anticodon base pairing, ensuring that the genetic code is read properly. Then, interactions between rRNAs and the general domains of all tRNAs result in the movement of the tRNAs between the A, P, and E sites as the ribosome translocates along the mRNA one three-nucleotide codon at a time. Translation Is Terminated by Release Factors When a Stop Codon Is Reached The final stages of translation, like initiation and elongation, require highly specific molecular signals that decide the fate of the mRNA– ribosome–peptidyl-tRNA complex. Two types of eukaryotic protein
release factors (RFs) have been discovered. The first, eRF1, whose shape is similar to that of tRNAs, acts by binding to the ribosomal A site and recognizing stop codons directly. Like some of the initiation and elongation factors discussed previously, the second release factor, eRF3, is a GTP-binding protein. The eRF3·GTP complex acts in concert with eRF1 to promote cleavage of the peptidyl-tRNA bond, thus releasing the completed protein chain and terminating translation (Figure 5-38). The peptidyl-tRNA bond of the tRNA in the P site is not cleaved until one of the three stop codons is correctly recognized by eRF1, another example of a proofreading step in protein synthesis.
FIGURE 5-38 Termination of translation in eukaryotes. When a ribosome bearing a nascent protein chain reaches a stop codon (UAA, UGA, UAG), release factor eRF1 enters the A site together with eRF3·GTP. Hydrolysis of the bound GTP is accompanied by cleavage of the peptide chain from the tRNA in the P site and ejection of the tRNA in the E site, forming a post-termination complex. The ribosomal subunits are separated by the action of the ABCE1 ATPase together with eIF1, eIF1A, and eIF3. The 40S subunit is released bound to these eIFs, ready to initiate another cycle of translation (see Figure 5-36). Description The steps involved in the termination of translation in eukaryotes are as follows: Step 1: Stop codon recognition. Step 2: G T P hydrolysis and peptide release. Step 3: Ribosome recycling. Release of the completed protein leaves a free tRNA in the P site and the mRNA still associated with the 80S ribosome, to which eRF1 and eRF3·GDP are still bound in the A site. In eukaryotes, ribosome recycling occurs when this post-termination complex is bound by a protein called ABCE1, which uses energy from ATP hydrolysis to separate the subunits and release the mRNA and tRNA in the P site. Initiation factors eIF1, eIF1A, and eIF3, which are also required for separation of the subunits, load onto the 40S subunit, making it ready for another round of initiation (see Figure 5-36, top). In reality, a free mRNA is never released, as diagrammed in Figure 5-25 for simplicity. Rather, the mRNA has other ribosomes associated with it in various stages of elongation, PABPC bound to the poly(A) tail, and the eIF4 complex associated with the cap, ready to associate with another 43S preinitiation complex (see Figure 5-36).
Polysomes and Rapid Ribosome Recycling Increase the Efficiency of Translation
In addition to these functions in protein synthesis, ribosomes also associate transiently with protein chaperones that assist in folding the polypeptide chain as it emerges from the ribosome surface (see Figure 319). As we will see in Chapter 13, ribosomes that synthesize proteins destined to be inserted into the endoplasmic reticulum (ER) associate with a ribonucleoprotein complex called SRP (signal recognition particle) that arrests protein synthesis until the nascent polypeptide encounters specialized channels for insertion into the ER. SRP also assists with the insertion and threading of these proteins through these ER channels when protein synthesis is permitted to resume. Polysomes and Rapid Ribosome Recycling Increase the Efficiency of Translation Translation of a single eukaryotic mRNA molecule to yield a typically sized protein takes 1 to 2 minutes. Two phenomena significantly increase the overall rate at which cells can synthesize a protein: the simultaneous translation of a single mRNA molecule by multiple ribosomes, and rapid recycling of ribosomal subunits after they disengage from a stop codon. Simultaneous translation of an mRNA by multiple ribosomes is readily observable in electron micrographs and by sedimentation analysis, revealing mRNA molecules attached to multiple ribosomes bearing nascent growing polypeptide chains. These structures, referred to as polyribosomes or polysomes, were seen to be circular in electron micrographs of some tissues.
Subsequent studies with purified initiation factors explained the circular shape of polyribosomes and suggested the mechanism by which ribosomes recycle efficiently. These studies revealed that multiple copies of the cytoplasmic poly(A)-binding protein (PABPC) interact with both an mRNA poly(A) tail and the eIF4G subunit of eIF4. Since the eIF4E subunit of eIF4 binds to the cap structure on the end of an mRNA, the two ends of an mRNA molecule are bridged by the intervening proteins, forming circular mRNA (Figure 5-39a). Because the two ends of a polysome are relatively close together, ribosomal subunits that disengage from a stop codon are positioned near the end, facilitating reinitiation by the interaction of the 40S subunit and its associated initiation factors with eIF4 bound to the cap. The circular pathway depicted in Figure 539b is thought to enhance ribosome recycling and thus increase the efficiency of protein synthesis. EXPERIMENTAL FIGURE 5-39 Circular structure of mRNA increases translation efficiency. Eukaryotic mRNA forms a circular structure owing to interactions of three proteins. (a) In the presence of purified yeast poly(A)-binding protein [PABP; there is only one PABP in S. cerevisiae, rather than a nuclear (PABPN) and cytoplasmic (PABPC) protein as in higher eukaryotes], eIF4E, and eIF4G, eukaryotic mRNAs form circular structures,
GTPase-Superfamily Proteins Function in Several Quality-Control Steps of Translation
visible in this force-field electron micrograph. In these structures, protein-protein and protein-mRNA interactions form a bridge between the and ends of the mRNA. (b) Model of protein synthesis on circular polysomes and recycling of ribosomal subunits. Multiple individual ribosomes can simultaneously translate a eukaryotic mRNA, shown here in a circular form stabilized by interactions between proteins bound at the and ends. When a ribosome completes translation and dissociates from the end, the separated subunits can rapidly find the nearby cap and PABPC-bound poly(A) tail and initiate another round of synthesis. GTPase-Superfamily Proteins Function in Several Quality-Control Steps of Translation We have now seen that one or more GTP-binding proteins participate in each stage of translation. These proteins belong to the GTPase superfamily of switch proteins that cycle between a GTP-bound form and a GDP-bound form (see Figure 3-35). Usually correct execution of some preceding assembly step is required to trigger hydrolysis of the bound GTP. The hydrolysis of bound GTP to bound GDP is essentially irreversible and causes a conformational change in the GTPase itself, thus ensuring that the next interdependent assembly step associated with translation proceeds rapidly, unidirectionally, and with high fidelity. As the details of translation have been worked out, two distinct but not mutually exclusive functions of GTP hydrolysis have been identified. The first is to make an assembly event unidirectional by locking associated proteins into place once components have come together in the correct manner. A good example of this is successful association of the large and small ribosomal
Nonsense Mutations Can Be Bypassed by Suppressing tRNA Mutations
subunits coupled to hydrolysis of GTP by eIF5B (Figure 5-36, step 8 ). Recall that if the correct complex does not form, eIF5B·GTP does not hydrolyze the GTP and the complex is unstable, free to diffuse apart and try again. When the precise alignment of the subunits required for elongation occurs, eIF5B hydrolyzes the GTP to GDP, locking the correct complex in place. Energy released from the high energy β-γ bond in GTP drives the reaction in one direction. A second function of GTP hydrolysis is to create an additional step by which the correct fit for a molecular association can be checked to increase the fidelity of translation. The best example of such a proofreading activity occurs during the crucial process of matching the correct aminoacyl-tRNA with the next available codon in an mRNA. An initial match is made when the anticodon of an aminoacyltRNA in complex with EF1α·GTP docks with the codon in the A site. Then as a distinct step, hydrolysis of EF1α·GTP to EF1α·GDP occurs only after correct base pairing between anticodon and codon is confirmed. GTP hydrolysis causes a conformational change in EF1α that results in the release of its bound tRNA, allowing the aminoacylated end of the charged tRNA to move into the position required for peptide bond formation (see Figure 5-37, step 2 ). In effect, the base pairing between anticodon and codon is checked twice; first in the initial selection of an amino acyl-tRNA and again in a step coupled to hydrolysis of EF1α·GTP. Molecular simulations estimate that this second proofreading step increases the fidelity of translation about 15-fold. Nonsense Mutations Can Be Bypassed by Suppressing tRNA Mutations
As we have seen, a mutation that converts a codon normally encoding an amino acid into a stop codon will lead to premature termination of translation. Nonsense mutations, as they are called, result in a truncated protein that is usually nonfunctional. In genetic studies with the bacterium E. coli, it was discovered that the effect of a nonsense mutation can be suppressed by a second mutation in a tRNA gene. This occurs when the sequence in a tRNA gene that encodes the anticodon is changed to a triplet that is complementary to a stop codon. If, for example, a nonsense mutation in a gene converted a UCG (serine) codon into a UAG (stop) codon, the stop may be read often enough to produce some full-length protein if the anticodon of a tRNA were changed to CUA, which can basepair with the UAG stop codon. One way to do this would be to mutate the gene for in a way that changes its anticodon from GUA to CUA. Importantly, the mutant can still be recognized by the tyrosine aminoacyl-tRNA synthetase, since the anticodon sequence is not a necessary feature for recognition by the corresponding aminoacyl-tRNA synthetase. Cells that have both the original nonsense mutation and the second mutation in the anticodon of the gene consequently can insert a tyrosine at the position of the mutant stop codon, allowing protein synthesis to continue past the original nonsense mutation. This mechanism is not highly efficient, so translation of normal mRNAs with a UAG stop codon terminates at the normal position in most instances. If enough of the protein encoded by the original gene with the nonsense mutation is produced to provide its essential functions, the effect of the first mutation is said to be suppressed by the second mutation in the anticodon of the tRNA gene.
This mechanism of nonsense suppression is a powerful tool in genetic studies of bacteria. For example, it allows us to isolate mutant bacterial viruses that cannot grow in normal cells but can grow in cells expressing a nonsense-suppressing tRNA because the mutant virus has a nonsense mutation in an essential gene. Such mutant viruses grown on nonsensesuppressing cells can then be used in experiments to analyze the function of the mutant gene by infecting normal cells that do not suppress the mutation and analyzing what step in the viral life cycle is defective in the absence of the mutant protein. KEY CONCEPTS OF SECTION 5.6 Stepwise Synthesis of Proteins on Ribosomes Bacterial, archaeal, and eukaryotic ribosomes — the large ribonucleoprotein complexes on which translation occurs — consist of a small and a large subunit. Each subunit contains numerous different proteins and one major rRNA molecule (small or large). The large subunit also contains one accessory 5S rRNA in bacteria and archaea and two accessory rRNAs in eukaryotes (5S and 5.8S). Analogous rRNAs from many different species fold into quite similar threedimensional structures containing numerous stems and loops, as well as binding sites for proteins, mRNAs, and tRNAs. Much smaller ribosomal proteins are associated with the periphery of the rRNAs. Of the two methionine tRNAs found in all cells, only one functions in initiation of translation. Each stage of translation — initiation, chain elongation, and termination — requires specific protein factors, including GTP-binding proteins that hydrolyze their bound GTP to GDP when a step has been completed successfully. During initiation, the ribosomal subunits assemble near the translation start site in an mRNA molecule on which a tRNA carrying the amino-terminal methionine has base-paired with the start codon (see Figure 5-36). Chain elongation entails a repetitive four-step cycle: loose binding of an incoming aminoacyl-tRNA to the A site on the ribosome; tight binding of the correct aminoacyl-tRNA to the A site accompanied by release of the previously used tRNA
from the E site; transfer of the growing polypeptide chain to the incoming amino acid catalyzed by large rRNA; and translocation of the ribosome to the next codon, thereby moving the peptidyl-tRNA in the A site to the P site and the now unacylated tRNA in the P site to the E site (see Figure 5-38). In each cycle of chain elongation, the ribosome undergoes two conformational changes monitored by two GTP-binding proteins. The first of these proteins (EF1α) permits tight binding of the incoming aminoacyl-tRNA to the A site and ejection of a tRNA from the E site, and the second (EF2) monitors translocation. Termination of translation is carried out by two types of termination factors: those that recognize stop codons and those that promote hydrolysis of peptidyl-tRNA (see
Figure 5-39). Once again, correct recognition of a stop codon is monitored by a GTPase (eRF3). The efficiency of protein synthesis is increased by the simultaneous translation of a single mRNA by multiple ribosomes, forming a polyribosome or polysome. In eukaryotic cells, protein-mediated interactions bring the two ends of a polyribosome close together, thereby promoting the rapid recycling of ribosomal subunits, which further increases the efficiency of protein synthesis (see Figure 5-39b).
5.7 Viruses: Parasites of the Cellular Genetic System
5.7 Viruses: Parasites of the Cellular Genetic System We wrap up this chapter with a discussion of viruses, which are radically different from the organisms we’ve so far discussed, but that nevertheless depend on fundamental genetic processes. Viruses carry instructions encoded in DNA or RNA that are copied by polymerization using standard Watson-Crick base pairing, and their genes are expressed according to the universal genetic code. The viral genome can be considered to be a selfreplicating genetic program. In addition, viruses can mutate and they can evolve through the process of natural selection. Thus viruses have an autonomous existence in terms of the biological informational that they carry, but just as the instructions of a computer program cannot fully come into being without a computer to run on, the genetic instructions of a virus need a cellular host to synthesize viral proteins and to replicate the viral genome. Viruses are categorized according to the type of nucleic acid that makes up their genome. RNA viruses, which usually replicate in the hostcell cytoplasm, have an RNA genome, and DNA viruses, which commonly replicate in the host-cell nucleus, have a DNA genome (see Figure 5-1). Viral genomes may be single or double stranded, depending on the specific type of virus. The entire infectious virus particle, called a virion, consists of nucleic acid encased in a shell of protein, known as the capsid, which both protects the viral nucleic acid and functions in the process of hostcell infection. The genomes of the simplest known viruses only encode
Most Viral Host Ranges Are Narrow
four proteins; the most complex can encode some two hundred proteins. In addition to their obvious importance as causes of disease, viruses are extremely useful as research tools in the study of basic biological processes, such as those discussed in this chapter. Most Viral Host Ranges Are Narrow The process of infection usually begins when a protein on the surface of the virion binds to a specific protein on the surface of the host cell that the virus hijacks for use as a receptor. This interaction is usually highly specific and thus limits the host range — the types of cells that a virus can infect — to a very narrow range of cell types in particular species. A virus that infects only bacteria is called a bacteriophage, or simply a phage. Viruses that infect animal or plant cells are referred to generally as animal viruses or plant viruses. Most animal viruses have host ranges restricted to certain phyla, and some infect only closely related species, such as primates. Some animal viruses, such as vesicular stomatitis virus, can infect insects as well as many different types of mammals. For viruses that can grow not only in plants or animals but also in the insects that feed on them, the highly mobile insects can serve as vectors for transferring the virus between susceptible hosts. The host-cell range of some animal viruses is further restricted to a limited number of cell types because only those cells have surface receptors to which the virions can attach. One example is poliovirus, which infects only cells in the intestine and, unfortunately for its host, motor neurons in the spinal cord, causing
Viral Capsids Are Regular Arrays of One or a Few Types of Protein
paralysis. Another is HIV-1, discussed further in this section, which infects cells called T lymphocytes that are essential for the immune response (see Chapter 24) as well as certain neurons and other cells of the central nervous system called glial cells. Viral Capsids Are Regular Arrays of One or a Few Types of Protein The viral capsid is usually composed of multiple copies of only one protein or a few different proteins, each of which is encoded by a single viral gene. Because of this structure, a virus is able to encode all the information for making a relatively large capsid in a small number of genes. This efficient use of genetic information is important because only a limited amount of DNA or RNA, and therefore a limited number of genes, can fit into a virion capsid. Nature has found two basic ways of packaging a viral genome within a regular structure composed of multiple copies of capsid proteins. In some viruses, multiple copies of a single capsid protein form a helical structure that encloses and protects the viral RNA or DNA, which runs in a helical groove within the protein tube. Viruses with such a helical structure, such as tobacco mosaic virus, have a rodlike shape (Figure 5-40a). The other major structural type is based on the icosahedron, a solid, approximately spherical object built of 20 identical faces (Figure 5-40b). Since each face is an equilateral triangle that must be composed of at least three protein subunits, an icosahedral capsid must be composed of at least 60 subunits.
During infection, some icosahedral viruses interact with host-cell receptors via clefts between the capsid subunits. Others interact via long fibrous proteins extending from the vertices of the icosahedron.
FIGURE 5-40 Virion structures. (a) Helical tobacco mosaic virus. (b, left) Diagram of the structure of poliovirus, a small icosahedral virus, made of 20 equilateral triangular faces, one of which is outlined in red. Each face is composed of three outlined structural elements called capsomeres. The numbers show how five capsomeres associate at the 12 vertices of the icosahedron. (b, right) Space-filling model of poliovirus based on x-ray crystallography.
The model is color-coded according to distance from the center of the virion, red farthest, blue closest. The virion binds to host cell receptors (not shown), which are long, narrow cell surface proteins that enter the blue “canyons” around each vertex. (c) Bacteriophage T4. (d) Influenza virus, an example of an enveloped virus. Part (b) data from D. J. Filman et al., 1989. EMBO J. 8:1567, PDB ID 2plv. Description The micrograph labeled A shows helical tobacco mosaic virus. The 3 D model and a space-filling model of x-ray crystallography labeled B shows the structure of poliovirus. The electron micrograph labeled C shows bacteriophage T 4 and the electron micrograph labeled D shows avian influenza virus. The scale bar reads 50 nanometers for labels a, c, and d and 10 nanometers for label b. In many DNA bacteriophages, the viral DNA is located within an icosahedral head that is attached to a rodlike tail. During infection, viral proteins at the tip of the tail bind to host-cell receptors, and the viral DNA then passes down the tail into the cytoplasm of the host cell (Figure 540c). In some viruses, a symmetrically arranged nucleocapsid composed of the viral genome associated with multiple copies of one or a few proteins is covered by an external membrane, or viral envelope, which consists mainly of a phospholipid bilayer but also contains one or more types of virus-encoded glycoproteins (Figure 5-40d). The phospholipids in the viral envelope are similar to those in the plasma membrane of an infected host cell. The viral envelope is, in fact, derived by budding from that membrane, but contains mainly viral glycoproteins, as we will discuss shortly.
Lytic Viral Growth Cycles Lead to Death of Host Cells
Lytic Viral Growth Cycles Lead to Death of Host Cells Although details vary among different types of viruses, those that exhibit a lytic cycle of growth proceed through the following stages: 1. Adsorption — Virion interacts with a host cell by binding of multiple copies of capsid protein to specific receptors on the cell surface. 2. Entry — The virus enters the cytoplasm of the host cell. Membrane enveloped viruses may enter through the endocytic pathway described in Chapter 14 while others fuse directly with the host plasma membrane. Viruses with a protein capsid must enter cells through the endocytic pathway so as not to disrupt the integrity of the plasma membrane. 3. Early gene expression — The first viral genes to be expressed are usually involved in the replication of the viral genome and the efficient expression of viral structural genes. For positive strand RNA viruses, early genes are expressed by direct translation of the genomic RNA in the cytoplasm. For DNA viruses, viral DNA enters the cell nucleus where viral mRNAs are produced with the aid of the host-cell transcription machinery. Viral mRNAs are translated by the host-cell translation machinery. 4. Genome replication — Production of multiple copies of the viral genome is carried out either by viral proteins alone or with the help of host-cell proteins. RNA viruses usually replicate in the cytoplasm whereas DNA viruses must replicate within the nucleus. 5. Structural gene expression — The virus uses host cell machinery for transcription and translation of viral structural proteins. These
include proteins of the nucleocapsid and abundant membrane glycoprotein for membrane enveloped viruses. 6. Assembly — Viral proteins and replicated genomes associate to form progeny virions in the cytoplasm. 7. Release — The host cell may rupture suddenly (lysis), releasing all the newly formed virions at once, or disintegrate gradually, releasing the virions slowly. For membrane enveloped viruses the last stage of assembly involves entry into membrane bounded organelles the secretory pathy and then release from the cell by exocytosis or by budding directly from the plasma membrane. We will use coronavirus, which is the cause of the COVID-19 pandemic, as an example to show how a lytic virus navigates the compartments of a eukaryotic cell. Note that the features of the coronavirus life cycle depicted in Figure 5-41 are governed by the basic properties of the coronavirus as a membrane enveloped virus with an RNA genome. Coronavirus begins entry into a target cell by binding of the spike protein of the viral envelope to a receptor protein on the surface of the host cell. Binding to the receptor allows the virus to enter endocytic vesicles which bud into the interior of the cell as described in Chapter 14. Within an endocytic compartment, the relatively low triggers a proteolytic cleavage of the viral spike protein, activating the spike protein for fusion of the viral membrane with the endosome membrane and releasing the nucleocapsid into the cytoplasm.
FIGURE 5-41 Lytic replication cycle of a membrane enveloped RNA coronavirus. Coronavirus has a genome of ~30 kb of single stranded RNA, encoding about 30 viral proteins. Within the virion, the RNA genome is in a complex with the viral nucleocapsid (N) protein forming a helical nucleocapsid. The nucleocapsid is packaged within the viral membrane whose major constituents are the membrane (M) and spike (S) proteins. Infection begins with adsorbtion of the virus to a target cell by binding of the S protein to a specific cell surface receptor (step 1 ), leading to engulfment into an endosome (step 2 ). The S protein can catalyze fusion of membranes after it has been activated by a proteolytic cleavage that takes place within the acidic interior of the endosome. S protein catalyzed fusion of the viral membrane with the membrane of the endosome enables the nucleocapsid to enter the cytoplasm (step 3 ). Host cell ribosomes in the cytoplasm recognize the viral genomic RNA as an mRNA and translate early sequences encoding the viral Replicase (step 4 ). The Replicase produces complementary copies of the genome that are acted upon by the Replicase to produce viral mRNAs for expression of the viral structural proteins as well as many copies of the full length viral RNA genome (step 5 ). Translated viral N protein (step 6 ) assembles with newly replicated viral genomic RNA to form nucleocapsid
complexes (step 7 ). At the same time, the viral membrane proteins M and S are translated and assembled in the membrane of the ER (step 8 ). In membranes derived from the ER, these viral membrane proteins assemble into a precursor of the viral membrane (step 9 ), which then envelopes the nucleocapsid as the final stage of viral assembly (step 10 ). Mature virions formed by budding into the interior of the ER are secreted from the cell by exocytosis (step 11 ), ready to carry out another round of cellular infection and growth. Description The steps involved in the lytic replication cycle of membrane enveloped R N A coronavirus are as follows: Step 1: Adsorption. Step 2: Endocytosis. Step 3: Entry. Step 4: Translation of replicase. Step 5: R N A genome replication. Step 6: Translation of N protein. Step 7: Nucleocapsid assembly. Step 8: Translation of S and M proteins. Step 9: Assembly of viral membrane. Step 10: Viral assembly. Step 11: Exocytosis. Coronavirus is a plus stranded RNA virus, meaning the single stranded RNA genome can immediately engage host-cell ribosomes in the cytoplasm for translation of early viral gene products. The early genes of
coronavirus encode a protein complex known as Replicase, which is responsible for replication of the viral RNA genome. Replicase, which contains an RNA template dependent RNA polymerase, first uses the RNA genome as a template to produce complementary RNA strands. These complementary strands, in turn, are used as templates for the production of more copies of the RNA genome as well as of viral mRNAs suitable for translation of the viral structural proteins. The translated viral nucleocapsid protein assembles with viral RNA to form mature helical nucleocapsid structures. Meanwhile, coronavirus membrane structural proteins are synthesized on the ER membrane (see Chapter 13) and cluster in regions of ER membrane that are precursors of the viral membrane. Complete virions are assembled by envelopment of nucleocapsid by viral membrane as the virus buds into the interior of the ER. Finally, the mature virions are released from the cell by exocytosis (see Chapter 14). Some membrane enveloped viruses form by a similar envelopment process but are released from the cell by budding directly from the plasma membrane, as shown in an electron micrographs of a cell infected with measles virus in Figure 5-42.
EXPERIMENTAL FIGURE 5-42 Release of progeny virions by budding. Membrane enveloped viruses are released from infected cells by budding from the plasma membrane. In this transmission electron micrograph of a cell infected with measles virus, virion buds are clearly visible protruding from the cell surface. Measles virus is an enveloped RNA virus with a helical nucleocapsid.
Viral DNA Is Integrated into the Host-Cell Genome in Some Nonlytic Viral Growth Cycles
We have seen that for RNA viruses, replication of the RNA genome and the production and translation of viral mRNAs can take place entirely within the cytoplasm. For DNA viruses, the DNA genome must enter the nucleus in order to be transcribed into viral mRNA and to be replicated by nuclear DNA polymerases. Soon after entry into a host cell, the viral DNA genome is transported into the cell nucleus through nuclear pores, as described in Chapter 13. Once inside the nucleus, the viral DNA is transcribed into RNA by the host’s transcription machinery. Processing of the viral RNA primary transcript by host-cell enzymes yields viral mRNA, which is transported to the cytoplasm and translated into viral proteins. The early viral proteins are then transported back into the nucleus, where they collaborate with the cellular replication machinery to replicate viral DNA. Viral proteins that comprise the capsid are usually expressed later in the infection process. Association of the capsid proteins with the newly replicated viral DNA occurs in the nucleus, yielding mature progeny virions. After the synthesis of hundreds to many thousands of new virions has been completed, depending on the type of virus and host cell, the host cells lyse, releasing all the virions at once, or the host cell releases the virions gradually as the cell progressively disintegrates. Viral DNA Is Integrated into the HostCell Genome in Some Nonlytic Viral Growth Cycles
The genomes of a number of animal viruses can integrate into the host-cell genome. Among the most important of these are the retroviruses, which are enveloped viruses with a genome consisting of two identical strands of RNA. These viruses are so named because their RNA genome acts as a template for the formation of a DNA molecule — a flow of genetic information that is opposite to the more common transcription of DNA into RNA. In the retroviral life cycle (Figure 5-43), a viral enzyme called reverse transcriptase initially copies the viral RNA genome into singlestranded DNA that is complementary to the viral RNA; the same enzyme then catalyzes the synthesis of a complementary DNA strand. (This complex reaction is detailed in Chapter 7, where we consider closely related intracellular parasites called retrotransposons.) The resulting double-stranded DNA is integrated into the chromosomal DNA of the infected cell by an integrase enzyme in the virion. Finally, the integrated DNA, called a provirus, is transcribed by the host cell’s own RNA polymerase into RNA, which is either translated into viral proteins or packaged within virion capsid proteins to form progeny virions that are released by budding from the host-cell membrane. Because most retroviruses do not kill their host cells, infected cells can replicate, producing daughter cells with integrated proviral DNA. These daughter cells continue to transcribe the proviral DNA and bud progeny virions.
FIGURE 5-43 Retroviral life cycle. Retroviruses have a genome of two identical copies of single-stranded RNA and an outer envelope. Step 1 : After viral glycoproteins in the retroviral envelope interact with a specific host-cell membrane protein, the envelope fuses directly with the plasma membrane, allowing entry of the nucleocapsid into the cytoplasm of the cell. Step 2 : Viral reverse transcriptase and other proteins copy the viral ssRNA genome into a double-stranded DNA. Step 3 : The viral dsDNA is transported into the nucleus and integrated into one of many possible sites in the host-cell chromosomal DNA. For simplicity, only one host-cell chromosome is depicted. Step 4 : The integrated viral DNA (provirus) is transcribed by the host-cell RNA polymerase, generating viral mRNAs (dark red) and viral genomic RNA molecules (bright red). The host-cell machinery translates the viral mRNAs into glycoproteins and nucleocapsid proteins. Step 5 : Progeny virions then assemble and are released by budding, as illustrated in Figure 5-42. Description The steps involved in the life cycle of the retrovirus are as follows:
Step 1: Fusion. The nucleocapsid with genomic s s R N A and reverse transcriptase enters the host cell cytoplasm. The host cell has chromosomal D N A inside the nucleus. Step 2: Reverse transcription. The enzyme reverse transcriptase synthesizes the viral D N A in the cytoplasm of the host cell. Step 3: Integration. The viral D N A (blue) is incorporated into the host cell chromosomal D N A (light blue) in the nucleus. Step 4: Transcription. Provirus synthesizes viral m R N A and viral genomic s s R N A molecules. By the process of translation, the retroviral proteins (blue spirals) are synthesized from viral m R N A in the cytoplasm of the host cell. Step 5: Budding. The newly formed immature viruses are released from the host cell. Some retroviruses contain cancer-causing genes (oncogenes), and cells infected by such retroviruses are oncogenically transformed into tumor cells. Studies of oncogenic retroviruses (mostly viruses of birds and mice) have revealed a great deal about the processes that lead to transformation of a normal cell into a cancer cell (see Chapter 25). Among the known human retroviruses are human T-cell lymphotrophic virus (HTLV), which causes a form of leukemia, and human immunodeficiency virus (HIV-1), which causes acquired immune deficiency syndrome (AIDS). Both of these viruses can infect only specific cell types, primarily certain cells of the immune system and, in the case of HIV-1, some central nervous system neurons and glial cells. Only those cells have cell-surface receptors that interact with viral envelope proteins. Unlike most other retroviruses, HIV-1 eventually kills
its host cells. The eventual death of large numbers of immune-system cells results in the defective immune response characteristic of AIDS. Retroviruses, such as HIV-1, make a DNA replica of the information in viral RNA. The enzyme responsible for this copying reaction is reverse transcriptase, which functions similarly to DNA polymerase but uses the viral RNA as a template. Reverse transcriptase, encoded as a viral gene, lacks a proofreading activity and is much more error prone than cellular DNA polymerases. In this case, a low fidelity of replication works to the advantage of the virus because frequent mutations in the viral capsid genes allow the virus to continually alter the antigens on the surface of the virion and thus evade the human immune system. Some DNA viruses can also integrate into a host-cell chromosome. One example is the human papillomaviruses (HPVs), which most commonly cause warts and other benign skin lesions. These viruses replicate without integrating into a host-cell chromosome. However, the genomes of certain HPV serotypes occasionally integrate into the chromosomal DNA of infected cervical epithelial cells. Unlike retroviruses and temperate bacteriophages, this integration is not carried out by viral proteins, but rather is the consequence of host-cell DNA repair processes. Integration is a dead-end for HPV. The integrated viral DNA cannot replicate and generate progeny virions, but oncogenic (cancer-causing) viral proteins can be expressed from the integrated viral genome, initiating development of cervical cancer. A vaccine for the types of HPV associated with cervical cancer has been developed and can protect against the initial infection by these viruses, and consequently, against development of cervical cancer.
KEY CONCEPTS OF SECTION 5.7 Viruses: Parasites of the Cellular Genetic System Viruses are small packages of genetic information that can move from one cell to another and that depend on the host cell for their gene expression and replication. Viral genomes may be either DNA (DNA viruses) or RNA (RNA viruses) and may be either single or double stranded. The capsid, which surrounds the viral genome, is composed of multiple copies of one or a small number of virus-encoded proteins. Some viruses also have an outer envelope, which is similar to the plasma membrane of the host cell but contains viral transmembrane proteins. Most animal and plant DNA viruses require host-cell nuclear enzymes to carry out transcription of the viral genome into mRNA and production of progeny genomes. In contrast, most RNA viruses encode enzymes that can transcribe the RNA genome into viral mRNA and produce new copies of the RNA genome. Host-cell ribosomes, tRNAs, and translation factors are used in the synthesis of all viral proteins in infected cells. Lytic viral infection entails adsorption, penetration, synthesis of viral proteins and progeny genomes (replication), assembly of progeny virions, and release of hundreds to thousands of virions, leading to death of the host cell (see Figure 5-41). Release of enveloped viruses occurs by budding through the host-cell plasma membrane (see
Figure 5-42). Retroviruses are enveloped animal viruses containing a single-stranded RNA genome. After a host cell is penetrated, reverse transcriptase, a viral enzyme carried in the virion, converts the viral RNA genome into double-stranded DNA, which is integrated into chromosomal DNA by an integrase enzyme that enters the cell inside the virion (see Figure 5-43). Unlike infection by other retroviruses, HIV-1 infection eventually kills host cells, causing the defects in the immune response characteristic of AIDS. Tumor viruses, which contain oncogenes, may have an RNA genome (e.g., human Tcell lymphotrophic virus) or a DNA genome (e.g., human papillomaviruses). Integration of the genomes of these viruses into a host-cell chromosome can cause transformation of the host cell into a tumor cell.
Key Terms
End of Chapter Visit Achieve to access study tools and learn more about the content of this chapter: Perspectives for the Future Analyze the Data Chapter References Additional study tools including videos, animations, and quizzes Key Terms alternative splicing aminoacyl-tRNA anticodon base pairs capsid codon complementary denaturation (melting) deoxyribonucleic acid (DNA) DNA polymerase double helix elongation factors (EFs) eukaryotic translation initiation factors (eIFs) excision-repair systems gene conversion
genetic code GTPase superfamily helicase Holliday structure homologous recombination hybridization isoform lagging strand leading strand messenger RNA (mRNA) mutation nonhomologous end joining nucleocapsid Okazaki fragments open reading frame phosphodiester bond point mutation polymer polyribosome precursor mRNA (pre-mRNA) primase primer promoter purine pyrimidine reading frame recombination release factors (RFs) replication fork retrovirus
Review the Concepts
reverse transcriptase ribosomal RNA (rRNA) ribosome RNA polymerase RNA splicing transcription transfer RNA (tRNA) translation viral envelope virion virus xeroderma pigmentosum Review the Concepts 1. What are Watson-Crick base pairs? Why are they important? 2. Preparing plasmid DNA (double stranded, circular) for sequencing involves annealing a complementary, short, single-stranded oligonucleotide DNA primer to one strand of the plasmid template. This is routinely accomplished by heating the plasmid DNA and primer to and then slowly bringing the temperature down to . Why does this protocol work? 3. What difference between RNA and DNA helps to explain the greater stability of DNA? What implications does this have for the function of DNA? 4. What are the major differences in the synthesis and structure of prokaryotic and eukaryotic mRNAs?
5. While investigating the function of a specific growth factor receptor gene from humans, researchers found that two types of proteins are synthesized from this gene. A larger protein containing a membrane-spanning domain recognizes growth factors at the cell surface, stimulating a specific downstream signaling pathway. In contrast, a related, smaller protein is secreted from the cell and binds available growth factor circulating in the blood, thus inhibiting the downstream signaling pathway. Speculate on how the cell synthesizes these disparate proteins. 6. How would a mutation in the poly(A)-binding protein gene affect translation? How would an electron micrograph of polyribosomes from such a mutant differ from the normal pattern? 7. What characteristic of DNA results in the requirement that some DNA synthesis be discontinuous? How are Okazaki fragments and DNA ligase used by the cell? 8. Eukaryotes have repair systems that prevent mutations due to copying errors and exposure to mutagens. What are the three excision-repair systems found in eukaryotes, and which one is responsible for correcting thymine-thymine dimers that form as a result of UV light damage to DNA? 9. DNA-repair systems are responsible for maintaining genomic fidelity in normal cells despite the high frequency with which mutational events occur. What type of DNA mutation is generated by (a) UV radiation and (b) ionizing radiation? Describe the system responsible for repairing each of these types of mutations in mammalian cells. Postulate why a loss of function in one or more DNA repair systems is present in many cancers. 10. What is the name given to the process that can repair DNA damage and generate genetic diversity? Briefly describe the similarities and differences of the two processes.
11. a. Which of the following DNA strands, the top or bottom, would serve as a template for RNA transcription if the DNA molecule were to unwind in the indicated direction? ACGGACTGTACCGCTGAAGTCATGGACGCTCGA TGCCTGACATGGCGACTTCAGTACCTGCGAGCT Direction of DNA unwinding b. What would be the resulting RNA sequence (written )? 12. Contrast prokaryotic and eukaryotic gene characteristics. 13. You have learned about the events surrounding DNA replication and the central dogma. Identify the steps associated with these processes that would be adversely affected in the following scenarios. a. Helicases unwind the DNA, but stabilizing proteins are mutated and cannot bind to the DNA. b. The mRNA molecule forms a hairpin loop on itself via complementary base pairing in an area spanning the AUG start site. c. The cell is unable to produce functional . 14. Use the accompanying key to determine the amino acid sequence of the polypeptide produced from the following DNA sequence. Intron sequences are highlighted. Note: Not all amino acids in the key will be used. TTCTAAACGCATGAAGCACCGTCTCAGAGCCAGTGA AAGATTTGCGTACTTCGTGGCAGAGTCTCGGTCACT Direction of DNA unwinding
15. Description The illustration shows D N A replication fork with an arrow pointing toward the left labeled direction of the replication fork. A continuous arrow complementary to the 3 prime to 5 prime of parental D N A strand (light blue) is the leading strand (Green) and three short discontinuous arrows complementary to the 5 prime to 3 prime parental D N A strand (Blue) is the lagging strand (light green). a. Look at the figure above. Explain why it is necessary for Okazaki fragments to be formed as the lagging strand is produced (instead of a continuous strand). b. If the DNA polymerase in the figure above could bind only to the lower template strand, under what condition(s) would it be able to produce a leading strand? 16. The DNA repair systems preferentially target the newly synthesized strand. Why is this important? 17. Identify the specific types of point mutations below (you are viewing the direct DNA version of the RNA sequence). Original sequence: AUG TCA GGA CGT CAC TCA GCT Mutation A: AUG TCA GGA CGT CAC TGA GCT Mutation B: AUA TCA GGA CGT CAC TCA GCT 18. The genome of a retrovirus can integrate into the host-cell genome. A number of retroviruses can infect certain types of human cells.
a. What gene is unique to retroviruses, and why is the protein encoded by this gene absolutely necessary for maintaining the retroviral life cycle? b. List two retroviruses that infect specific cell types, briefly describe the medical implications resulting from these infections, and describe why only certain cells are infected. 19. a. Detail the key differences between lytic and nonlytic viral infection and provide an example of each. b. Which of the following processes occurs in both lytic and nonlytic viral infections? i. Infected cell ruptures to release viral particles. ii. Viral mRNAs are transcribed by the host-cell translation machinery. iii. Viral proteins and nucleic acids are packaged to produce virions.