Introduction
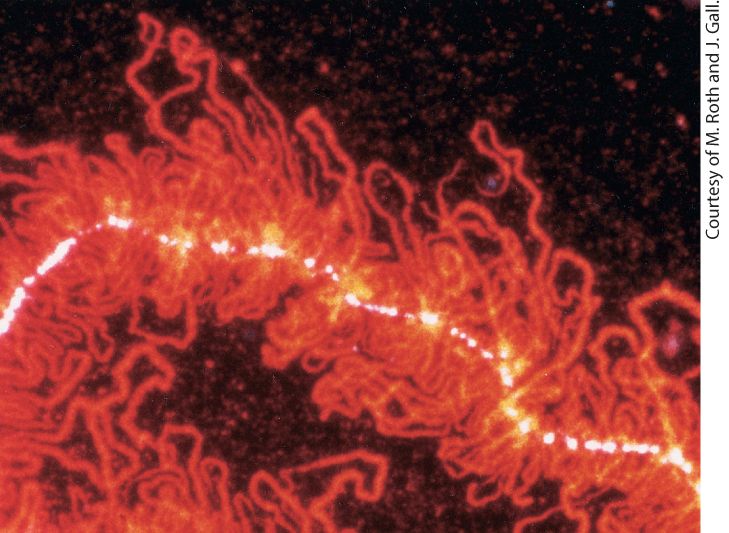
Chapter 9 Post-Transcriptional Gene Control Portion of a “lampbrush chromosome” from an oocyte of the newt Nophthalmus viridescens. The hnRNP protein associated with nascent RNA transcripts fluoresces red after staining with a monoclonal antibody.
9.4 Cytoplasmic Mechanisms of Post-Transcriptional Control
9.6 Nuclear Bodies Are Functionally Specialized Nuclear Domains In the previous chapter, we saw that most genes are regulated at the first step in gene expression — transcription — by regulating the assembly of the transcription preinitiation complex on a promoter-DNA sequence and regulating transcription elongation in the promoter-proximal region. Once transcription has been initiated, synthesis of the encoded RNA requires that RNA polymerase transcribe the entire gene and not terminate prematurely. Moreover, the initial primary transcripts produced from eukaryotic genes must undergo various processing reactions to yield the corresponding functional RNAs. For mRNAs, the cap structure necessary for translation must be added (see Figure 5-26), introns must be spliced out of pre-mRNAs (Table 9-1), and the end must be polyadenylated (see Figure 5-27). Once formed in the nucleus, mature, functional RNAs are exported to the cytoplasm as components of ribonucleoproteins. Processing of mRNAs in the nucleus, their export from the nucleus, and their transport to specific loci in the cytoplasm offer opportunities for further regulating gene expression after the initiation of transcription.
TABLE 9-1 • RNAs Discussed in Chapter 9 mRNA Fully processed messenger RNA with cap, introns removed by RNA splicing, and a poly(A) tail. premRNA An mRNA precursor containing introns and not cleaved at the poly(A) site. hnRNA Heterogeneous nuclear RNAs. These RNAs include pre-mRNAs and RNAprocessing intermediates containing one or more introns. snRNA Five small nuclear RNAs that function in the removal of introns from premRNAs by RNA splicing, plus two small nuclear RNAs that substitute for the first two at rare introns. pretRNA A tRNA precursor containing additional transcribed bases at the and ends compared with the mature tRNA. Some pre-tRNAs also contain an intron in the anticodon loop. prerRNA The precursor to mature 18S, 5.8S, and 28S ribosomal RNAs. The mature rRNAs are processed from this long precursor RNA molecule by cleavage, removal of bases from the ends of the cleaved products, and modification of specific bases. snoRNA Small nucleolar RNAs. These RNAs base-pair with complementary regions of the pre-rRNA molecule, directing cleavage of the RNA chain and modification of bases during maturation of the rRNAs. siRNA Short interfering RNAs, bases long, that are each perfectly complementary to a sequence in an mRNA. Together with associated proteins, siRNAs cause cleavage of the target RNA, leading to its rapid degradation. miRNA MicroRNAs, bases long, that base-pair extensively, but not completely, with mRNAs, especially over bases 2 to 7 at the end of the miRNA (the “seed” sequence). This pairing inhibits translation of the target mRNA and targets it for degradation.
Recently, the vast amount of sequence data of human mRNAs expressed in different tissues and at various times during embryogenesis and cellular differentiation has revealed that percent of human genes give rise to alternatively spliced mRNAs. These alternatively spliced mRNAs encode related proteins with differences in sequences limited to specific functional domains. In many cases, alternative RNA splicing is regulated to meet the need for a specific protein isoform in a specific cell type. Given the complexity of pre-mRNA splicing, it is not surprising that mistakes are occasionally made, giving rise to mRNA precursors with improperly spliced exons. However, eukaryotic cells have evolved RNAsurveillance mechanisms that prevent the transport of incorrectly processed RNAs to the cytoplasm or lead to their degradation if they are transported. Additional control of gene expression can occur in the cytoplasm. In the case of protein-coding genes, for instance, the amount of protein produced depends on the stability of the corresponding mRNAs in the cytoplasm and the rate of their translation. For example, during an immune response, lymphocytes communicate by secreting polypeptide hormones called cytokines that signal neighboring lymphocytes through cytokine receptors that span their plasma membranes (Chapter 24). It is important for lymphocytes to synthesize and secrete cytokines in short bursts. This is possible because cytokine mRNAs are extremely unstable. Consequently, the concentration of the mRNA in the cytoplasm falls rapidly once its synthesis is stopped. In contrast, mRNAs encoding proteins required in large amounts that function over long periods, such as ribosomal proteins,
are extremely stable so that multiple polypeptides can be transcribed from each mRNA. In addition to regulation of pre-mRNA processing, nuclear export, and translation, the cellular locations of many, if not most, mRNAs are regulated so that newly synthesized protein is concentrated where it is needed. Particularly striking examples of this occur in the nervous systems of multicellular animals. Some neurons in the human brain generate more than 1000 separate synapses with other neurons. During the process of learning, synapses that fire more frequently than others increase in size many times, while other less active synapses made by the same neuron do not. This can occur because mRNAs encoding critical synapse proteins are stored at all synapses, but translation of these localized, stored mRNAs is regulated at each synapse independently by the frequency at which that synapse fires. In this way, synthesis of synapse-associated proteins can be regulated independently at each of the many synapses made by the same neuron (see Chapter 23). Another type of gene regulation involves microRNAs (miRNAs), which regulate the stability and translation of specific target mRNAs to which they base pair in multicellular animals and plants. Analyses of these short miRNAs in various human tissues indicate that miRNAs are expressed in the multiple types of human cells. Although some have recently been discovered to function through inhibition of target gene expression in the appropriate tissue and at the appropriate time in development, the functions of the vast majority of human miRNAs are unknown and are the subject of a growing area of research. A closely
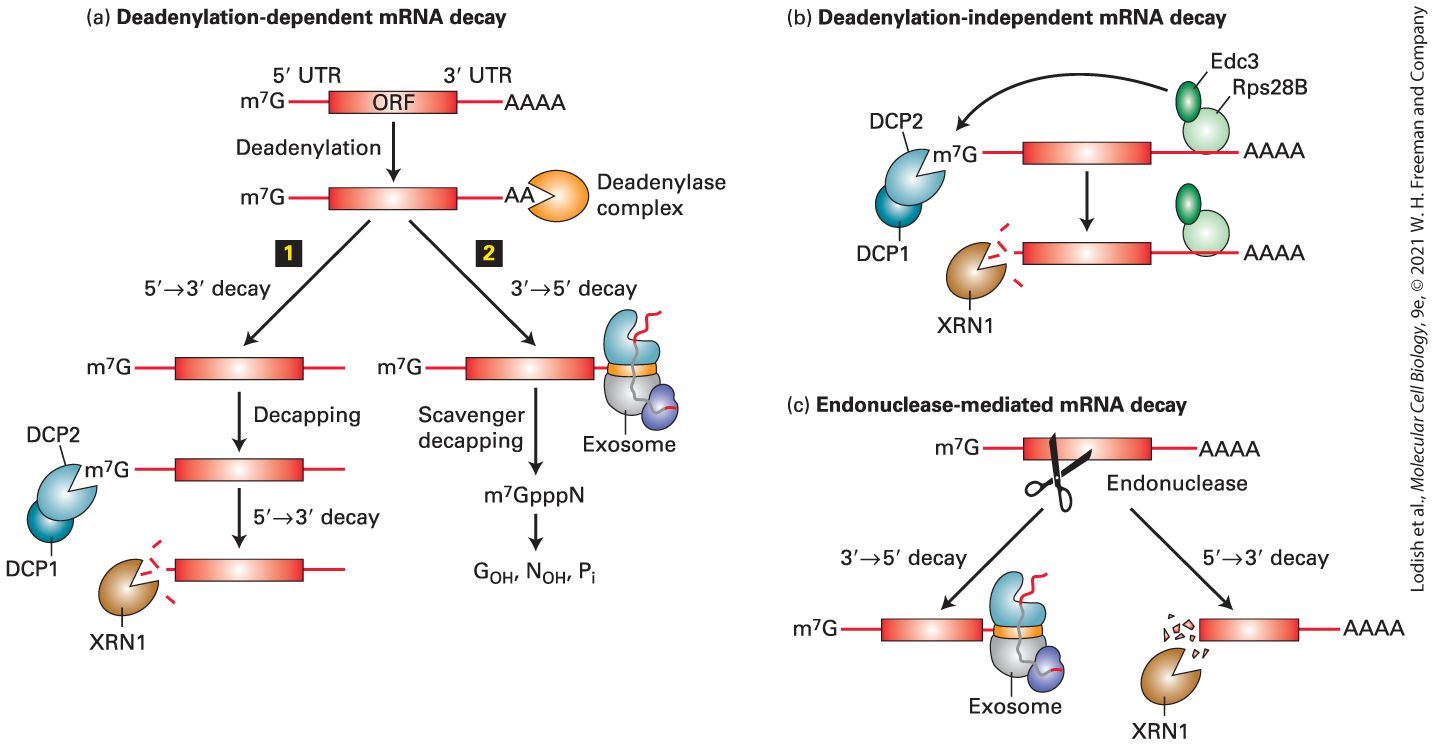
related process called RNA interference (RNAi) is used by some organisms to degrade viral RNAs following infection and in the degradation of transposon-encoded RNAs. RNAi is extensively utilized in biomedical research: designed short interfering RNAs (siRNA) can be used to inhibit the translation of specific mRNAs experimentally by a process called RNA knockdown. This makes it possible to inhibit the function of any desired gene, even in organisms that are not amenable to classic genetic methods for isolating mutants. We refer to all the mechanisms that regulate gene expression following transcription as post-transcriptional gene control (Figure 9-1). Because the stability and translation rate of an mRNA contribute to the amount of protein expressed from a gene, these post-transcriptional processes are important components of gene control. Indeed, the protein output of a gene is regulated at every step in the life of an mRNA from the initiation of its synthesis to its degradation. Thus genetic regulatory processes act on RNA as well as DNA. In this chapter, we consider the events that occur in the processing of mRNA following transcription initiation and promoterproximal elongation as well as mechanisms discovered that regulate these steps in mRNA processing. In the last section, we briefly discuss the processing of primary transcripts produced from genes encoding rRNAs and tRNAs.
FIGURE 9-1 Overview of RNA processing and post-transcriptional gene control. Nearly all cytoplasmic RNAs are processed from primary transcripts in the nucleus before they are exported to the cytoplasm. Major processing steps are diagrammed for RNAs transcribed by RNA polymerases I, II, and III. For protein-coding genes transcribed by RNA polymerase II, post-transcriptional gene control can be exerted through (step 1 ) the choice of alternative exons during pre-mRNA splicing and (step 2 ) the choice of alternative poly(A) sites. Improperly processed mRNAs are blocked from export to the cytoplasm and degraded (step 3 ) by a large complex called the exosome that contains internal ribonucleases. Once exported to the cytoplasm (step 4 ), translation initiation factors bind to the mRNA -cap cooperatively with poly(A)-binding protein C bound to the poly(A) tail and initiate translation (see Figure 5-36) (step 5 ). (Step 6 ) mRNA is degraded in the cytoplasm by deadenylation and decapping followed by degradation by cytoplasmic exosomes. Much of this mRNA degradation takes place in regions of the cytoplasm with high RNA concentration called “P bodies.” The degradation rate of each mRNA is controlled, thereby regulating the mRNA concentration and, consequently, the amount of protein translated. Some mRNAs are synthesized without long poly(A) tails. Their translation is regulated by (step 7 ) controlling the synthesis of a long poly(A) tail by a cytoplasmic poly(A) polymerase. Translation is also regulated by other mechanisms including miRNAs (step 8 ). When expressed, these -nucleotide RNAs inhibit translation of mRNAs to which they hybridize, usually in the untranslated region. tRNAs and rRNAs are also synthesized as precursor RNAs that must be (step 9a , b ) processed before they are functional. Regions of precursors cleaved from the mature RNAs are degraded by nuclear exosomes (step 10 ). See J. Houseley et al., 2006, Nat. Rev. Mol. Cell Biol. 7:529. Description The post-transcriptional gene control is divided into processes occurring in the nucleus and those occurring the cytoplasm. The series starts with three double-helical D N A being processed by polymerase 2, polymerase 3, and polymerase 1 in the nucleus. The polymerase 2 causes pre-m R N A transcription resulting into a pre-m R N A strand with 2 hair-pin loops and cap on one end. The polymerase 3 causes pre-t R N A transcription resulting into a pre-t R N A or acts in the formation of 5 S r R N A that enters the nucleolus. The polymerase 1 in nucleolus causes pre-r R N A transcription; base modification, cleavage, and ribosomal subunit synthesis in nucleolus; and excised pre-r R N A. The pre-m R N A undergoes several steps before entering cytoplasm; pre-t
R N A undergoes processing before entering cytoplasm; and ribosomal subunits are directly transported to the cytoplasm.
9.1 Processing of Eukaryotic Pre-mRNA
9.1 Processing of Eukaryotic PremRNA In this section, we take a closer look at how eukaryotic cells convert the initial primary transcript synthesized by RNA polymerase II into a functional mRNA. Three major events occur during the process: capping, cleavage/polyadenylation, and RNA splicing (Figure 9-2). Adding these specific modifications to the and ends of the pre-mRNA is important to protect it from enzymes that quickly digest uncapped RNAs generated by RNA processing, such as spliced-out introns and RNA transcribed downstream from a polyadenylation site. The cap and poly(A) tail distinguish pre-mRNA molecules from the many other kinds of RNAs in the nucleus. Pre-mRNA molecules (see Table 9-1) are bound by nuclear proteins that function in mRNA export to the cytoplasm. After mRNAs are exported to the cytoplasm, they are bound by a set of cytoplasmic proteins that stimulate translation and are critical for mRNA stability in the cytoplasm. Prior to nuclear export, introns must be removed to generate the correct coding region of the mRNA. In vertebrates, including humans, alternative splicing is intricately regulated in order to include alternative functional domains in closely related proteins, producing a considerable expansion of the proteome of these organisms.
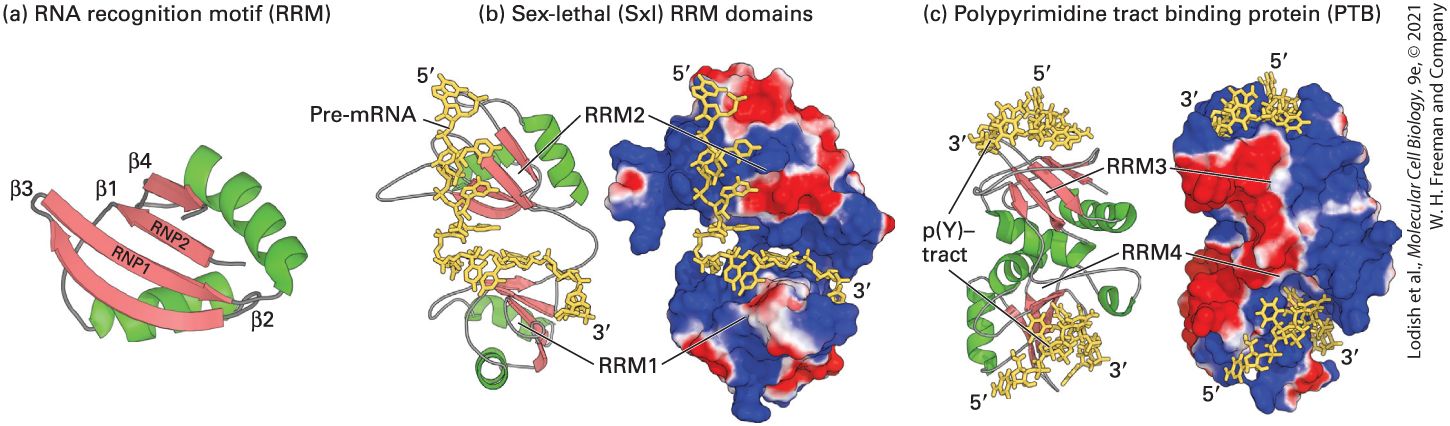
FIGURE 9-2 Overview of mRNA processing in eukaryotes. Shortly after RNA polymerase II initiates transcription at the first nucleotide of the first exon of a gene, the end of the nascent RNA is capped with 7-methylguanylate (step 1 ). Transcription by RNA polymerase II terminates at any one of multiple termination sites downstream from the poly(A) site, which is located at the end of the final exon. After the primary transcript is cleaved at the poly(A) site (step 2 ), a string of adenosine (A) residues is added (step 3 ). The poly(A) tail contains A residues in mammals, in insects, and in yeasts. For short primary transcripts with few introns, splicing (step 4 ) usually follows cleavage and polyadenylation, as shown. For large genes with multiple introns, introns often are spliced out of the nascent RNA during its transcription, i.e., before transcription of the gene is complete. Note that the cap and sequence adjacent to the poly(A) tail are retained in mature mRNAs. The diagram shown represents processing of human β-globin RNA. Description The process starts with a bar-shaped D N A with the following sequence: exon, intron, exon, intron, exon, poly (A) site, D N A segment, termination site, D N A segment, and termination site. A four-step pre-m R N A processing occurs resulting in an m R N A strand with 5-prime capped end and 3-prime end with approximately 250 adenosine residues.
The 5′ Cap Is Added to Nascent RNAs Shortly After Transcription Initiation
The pre-mRNA processing events of capping, splicing, and polyadenylation occur in the nucleus as the nascent mRNA precursor is being transcribed. Thus pre-mRNA processing is co-transcriptional: it occurs while the RNA is being transcribed. As the RNA emerges from the surface of RNA polymerase II, its end is immediately modified by the addition of the cap structure found on all mRNAs (see Figure 5-26). As the nascent pre-mRNA continues to emerge from the surface of the polymerase, it is immediately bound by members of a complex group of abundant RNA-binding proteins called hnRNP proteins that assist in RNA splicing and export of the fully processed mRNA through nuclear pore complexes into the cytoplasm. Some of these proteins remain associated with the mRNA in the cytoplasm, but most either remain in the nucleus or shuttle back into the nucleus shortly after the mRNA is exported to the cytoplasm. Cytoplasmic RNA-binding proteins are exchanged for the nuclear ones. Consequently, eukaryotic mRNAs never occur as free RNA molecules in the cell but are always associated with protein as ribonucleoprotein (RNP) complexes. Nuclear nascent pre-mRNAs associated with hnRNP proteins are referred to as pre-mRNPs. They are capped and spliced as they are transcribed. Then, following cleavage and polyadenylation at the end, they are referred to as nuclear mRNPs. Following the exchange of proteins that accompany export to the cytoplasm, they are called cytoplasmic mRNPs. Although we frequently refer to pre-mRNAs and mRNAs, it is important to remember that in eukaryotes they are always associated with proteins as RNP complexes.
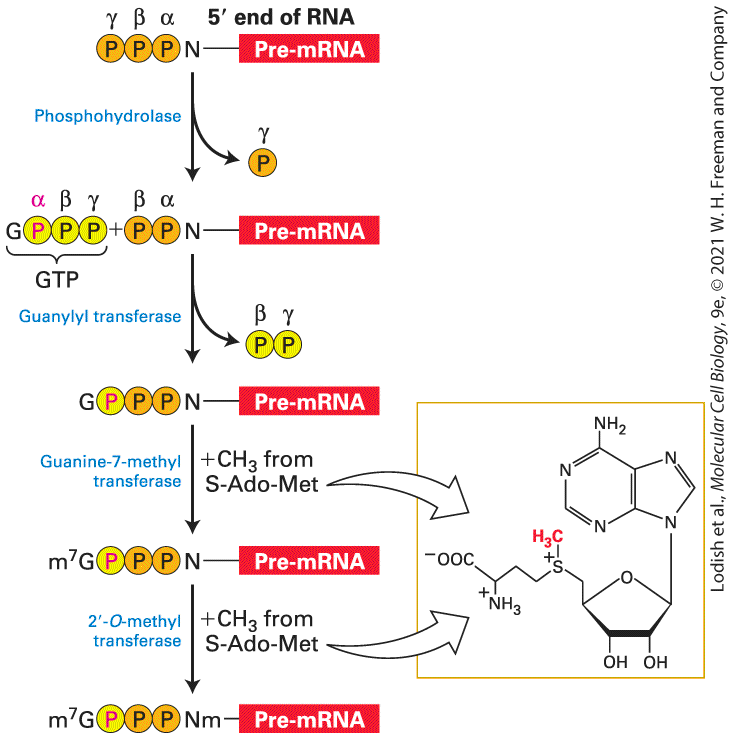
The Cap Is Added to Nascent RNAs Shortly After Transcription Initiation As the nascent RNA transcript emerges from the RNA exit channel of RNA polymerase II and reaches a length of nucleotides, a protective cap composed of 7-methylguanosine and methylated riboses is added to the end of eukaryotic mRNAs (see Figure 5-26). The cap marks RNA molecules as mRNA precursors and protects them from RNA-digesting enzymes ( -exoribonucleases) in the nucleus and cytoplasm. This initial step in RNA processing is catalyzed by a dimeric capping enzyme, which associates with the phosphorylated carboxyl-terminal domain (CTD) of RNA polymerase II. Recall that the CTD becomes phosphorylated by the TFIIH general transcription factor at multiple serines at the 5 position in the CTD heptapeptide (seven residue) repeat during transcription initiation (see Figure 8-7(b) red arrow, Figure 8-8). Binding to the phosphorylated CTD stimulates the activity of the capping enzymes so that they target RNAs containing a -triphosphate that emerge from RNA polymerase II and not RNAs transcribed by RNA polymerases I or III, which do not have a CTD. This is important because pre-mRNA synthesis accounts for only percent of the total RNA synthesized in replicating cells. The other 20 percent is pre-ribosomal RNA (pre-rRNA), which is transcribed by RNA polymerase I, and 5S rRNA, tRNAs, and other stable small RNAs transcribed by RNA polymerase III. One subunit of the capping enzyme removes the γ-phosphate from the end of the nascent RNA (Figure 9-3). Another domain of this subunit
transfers the GMP moiety from GTP to the -diphosphate of the nascent transcript, creating the unusual guanosine , -triphosphate ester structure. In the final steps, separate enzymes transfer methyl groups from S-adenosylmethionine to the position of the guanine and one or two of the oxygens of the ribose at the end of the nascent RNA.
FIGURE 9-3 Synthesis of -cap on eukaryotic mRNAs. The end of a nascent RNA contains a -triphosphate from the initiating NTP. The γ-phosphate is removed in the first
step of capping, while the remaining α- and β-phosphates (orange) remain associated with the cap. The third phosphate of the -triphosphate ester is derived from the α-phosphate of the GTP that donates the guanine. The methyl donor for methylation of the cap guanine and the first one or two riboses of the mRNA is S-adenosylmethionine (S-Ado-Met). See S. Venkatesan and B. Moss, 1982, Proc. Nat’l. Acad. Sci. USA 79:304. Description The process starts with a pre-m R N A having triphosphate group (alpha, beta, and gamma) attached to the N T P at its 5-prime end. Phosphohydrolase cleaves the gamma phosphate. Next step shows a G T P being added to the beta phosphate of the pre-m R N A. The G T P has a guanine attached to triphosphate group (alpha, beta, and gamma) with alpha highlighted. Guanylyl transferase cleaves the beta and gamma phosphates of the G T P. Next step shows alpha phosphate of guanine attached to the beta phosphate of pre-m R N A. Guanine-7-methyl transferase transfers a methyl group (C H 3) from S-adenosylmethionine to the guanine. Next step shows m 7 attached to the guanine. 2prime-O-methyl transferase also transfers a methyl group (C H 3) from Sadenosylmethionine to the 5-prime end of pre-m R N A. This finally results into m 7 guanine with alpha phosphate attached to the beta and gamma phosphates of methylated N of pre-m R N A. Considerable evidence indicates that capping of the nascent transcript is coupled to elongation by RNA polymerase II so that all transcripts generated by this polymerase are capped during the earliest phase of elongation. As discussed in Chapter 8, during the initial phase of transcription in metazoans, the polymerase elongates the nascent transcript very slowly due to association of NELF (negative elongation factor) with a complex of RNA polymerase II and DSIF in the promoterproximal region (see Figure 8-3 bottom, Figure 8-15a). Once the end of the nascent RNA is capped, phosphorylation of the RNA polymerase CTD at position 2 in the heptapeptide repeat and of NELF and DSIF by the
Chain Elongation by RNA Polymerase II Is Coupled to the Presence of RNA Processing Factors
cyclin T-CDK9 protein kinase causes the release of NELF (Figure 8-16). This allows the PAF elongation complex to associate with RNA polymerase II (Figure 8-15b), which then enters into a faster mode of elongation that rapidly transcribes away from the promoter. The net effect of this mechanism is that the polymerase waits for the nascent RNA to be capped before elongating at a rapid rate. Chain Elongation by RNA Polymerase II Is Coupled to the Presence of RNA Processing Factors How is RNA processing coupled with the transcription of a pre-mRNA so that only Pol II transcripts are subjected to capping, RNA splicing, and polyadenylation? The key lies in the long carboxyl-terminal domain (CTD) of RNA polymerase II, which, as discussed in Chapter 8, is composed of multiple repeats of a heptapeptide sequence. When fully extended, the CTD domain in the yeast enzyme is about 65 nm long (Figure 9-4); the CTD in human RNA polymerase II is about twice as long. The remarkable length of the CTD apparently allows multiple proteins to associate simultaneously with a single RNA polymerase II molecule. As mentioned earlier, the enzymes that add the cap to nascent transcripts associate with the CTD phosphorylated on multiple serines at the fifth position in the heptapeptide repeat (Ser-5) during or shortly after transcription initiation by a subunit of TFIIH. In addition, RNA splicing and polyadenylation factors associate with the phosphorylated CTD. As a consequence, these processing factors are present at high local
concentrations when splice sites and poly(A) signals are transcribed by the polymerase, enhancing the rate and specificity of RNA processing. In a reciprocal fashion, the association of hnRNP proteins, discussed below, with the nascent RNA enhances the interaction of RNA polymerase II with elongation factors such as DSIF and cyclin T-CDK9 (also called “PTEFb”) (Figure 8-16), increasing the rate of transcription. As a consequence, the rate of transcription is coordinated with the rate of nascent RNA association with hnRNP proteins and RNA-processing factors. This mechanism ensures that a pre-mRNA is not synthesized unless the machinery for processing it is properly positioned.
A Diverse Set of Proteins with Conserved RNA-Binding Domains Associate with Pre-mRNAs
FIGURE 9-4 Schematic diagram of the length of the RNA polymerase II CTD relative to the globular domain of the polymerase. The length of the yeast RNA polymerase II carboxyl-terminal domain (CTD) and the linker region that connects it to the polymerase is shown relative to the globular domain of the polymerase. The CTD of mammalian RNA polymerase II is twice as long. In its extended form, the CTD can associate with multiple RNA-processing factors simultaneously. See P. Cramer, D. A. Bushnell, and R. D. Kornberg, 2001, Science 292:1863. A Diverse Set of Proteins with Conserved RNA-Binding Domains Associate with Pre-mRNAs As noted earlier, neither nascent RNA transcripts from protein-coding genes nor the intermediates of mRNA processing, collectively referred to as pre-mRNA, exist as free RNA molecules in the nuclei of eukaryotic cells. From the time nascent transcripts first emerge from RNA polymerase II until mature mRNAs are transported into the cytoplasm, the RNA molecules are associated with nuclear proteins. The most abundant of these are the major protein components of heterogeneous ribonucleoprotein particles (hnRNPs), which contain heterogeneous nuclear RNA (hnRNA), a collective term referring to pre-mRNA- and mRNA-processing intermediates of various sizes. These hnRNP proteins contribute to further steps in RNA processing, including splicing, polyadenylation, and export through nuclear pore complexes to the cytoplasm.
Researchers identified hnRNP proteins by first exposing cultured cells to high-dose UV irradiation, which causes covalent cross-links to form between RNA bases and closely associated proteins. Chromatography of nuclear extracts from treated cells on an oligo-dT cellulose column, which binds RNAs with a poly(A) tail, was used to recover the proteins that had become cross-linked to nuclear polyadenylated RNA. These included a complex set of abundant hnRNP proteins ranging in size from to kDa, collectively referred to as hnRNP proteins. Like transcription factors, most hnRNP proteins have a modular structure. They contain one or more RNA-binding domains and at least one other domain, often an intrinsically disordered protein domain (see Figure 3-13 and associated text), that interacts with other proteins. Several different classes of RNA-binding domains are associated with hnRNP proteins, identified by testing deletion mutants for their ability to bind RNA. Functions of hnRNP Proteins These RNA-associated proteins have three key functions. First, the association of pre-mRNAs with the RNA-binding domains of hnRNP proteins prevents the pre-mRNAs from forming short secondary structures dependent on base pairing of closely spaced, short complementary regions. Pre-mRNAs associated with hnRNP proteins present a more uniform substrate for subsequent processing steps than would free, unbound premRNAs, in which each mRNA forms a unique secondary structure due to its specific sequence. The RNA-binding domains of hnRNPs usually interact preferentially with short RNA sequences of three to four bases.
The second key function is in regulating pre-mRNA splicing. In mammals, alternatively spliced mRNAs are expressed from almost all genes ( percent). Regulation of the use of one or the other of the alternative splice sites present in a pre-mRNA transcribed from these genes depends in part on binding of specific hnRNP proteins. The binding of hnRNP proteins to a pre-mRNA near an RNA splice site can inhibit binding of factors required for pre-mRNA splicing and hence direct splicing to an alternative splice site (see below). The third function is in mRNA transport. Cell-fusion experiments have shown that some hnRNP proteins remain localized in the nucleus, whereas others cycle in and out of the cytoplasm. This was the initial evidence suggesting that the hnRNPs that cycle in and out of the nucleus function in the transport of mRNA into the cytoplasm (Figure 9-5).
FIGURE 9-5 Human hnRNP A1 protein can cycle in and out of the nucleus, but human hnRNP C protein cannot. Cultured HeLa cells and Xenopus cells were fused by treatment with polyethylene glycol, producing heterokaryons containing nuclei from each cell type. Cyclohexamide was added immediately after fusion to prevent protein synthesis. After 2 hours, the cells were fixed and stained with fluorescent-labeled antibodies specific for human hnRNP C (green) and A1 proteins (red). These antibodies do not bind to the homologous Xenopus proteins. (a) A fixed preparation viewed by phase-contrast microscopy includes one unfused HeLa cell (arrowhead) and one unfused Xenopus cell (dotted arrow). Note that the nucleus in the Xenopus cell contains one prominent nucleolus
and is more oval in shape than the round HeLa cell nucleus containing three nuclei. A fused heterokaryon (solid arrow) in this micrograph contains a round HeLa cell nucleus to the right of the oval-shaped Xenopus nucleus with one nucleolus. (b, c) When the same preparation was viewed by fluorescence microscopy, human hnRNP C protein appeared green and human hnRNP A1 protein appeared red. Note that the unfused Xenopus cell on the left is unstained, confirming that the antibodies are specific for the human proteins. In the heterokaryon, human hnRNP C protein (green) appears only in the HeLa-cell nucleus (b), whereas the human A1 protein (red) appears in both the HeLa-cell nucleus and the Xenopus nucleus in the heterokaryon (c). Since protein synthesis was blocked after cell fusion, some of the human hnRNP A1 protein must have left the HeLa-cell nucleus, moved through the cytoplasm, and entered the Xenopus nucleus in the heterokaryon. [Reprinted by permission from Nature Publishing Group, from R. S. Piñol and G. Dreyfuss, 1992, “Shuttling of Pre-mRNA Binding Proteins Between Nucleus and Cytoplasm,” Nature 355(6362):730–732; permission conveyed through the Copyright Clearance Center, Inc.] RNA-Binding Domains The RNA recognition motif (RRM) is the most common RNA-binding domain in hnRNP proteins. This -residue domain, which occurs in many other RNA-binding proteins, contains two highly conserved sequences (RNP1 and RNP2) that are found across organisms ranging from yeast to human — indicating that like many DNA-binding domains, they arose early in eukaryotic evolution. Structural analyses have shown that the RRM domain consists of a fourstranded β sheet flanked on one side by two α helices (Figure 9-6a). To interact with the negatively charged RNA phosphates, the β sheet forms a positively charged surface. The conserved RNP1 and RNP2 sequences lie side by side on the two central β strands, and their side chains make
multiple contacts with a single-stranded region of RNA that lies across the surface of the β sheet. This is seen in the x-ray crystal structure of the two RRM domains of Drosophila Sex-lethal protein (Sxl) bound to its specific site in RNA (Figure 9-6b), and the two RRM domains of human polypyrimidine tract-binding protein (PTB) bound to its specific RNA (Figure 9-6c). Sxl and PTB are examples of the strikingly different orientations of RRM domains in different hnRNPs.
FIGURE 9-6 Structure of the RRM domain and its interaction with RNA. (a) Diagram of the RRM domain showing the two α helices (green) and four β strands (red) that characterize this motif. The conserved RNP1 and RNP2 regions are located in the two central β strands. (b) Surface representation of the two RRM domains in Drosophila Sexlethal (Sxl) protein, which together bind a continuous nine-base sequence in transformer pre-mRNA (yellow). In both (b) and (c), positively charged regions are shown in shades of blue; negatively charged regions, in shades of red; RNA is yellow. The two RRMs in Sxl are oriented like the two parts of an open pair of castanets, with the β sheets of the RRMs facing toward each other. The pre-mRNA is bound to the surfaces of the positively charged β sheets, making most of its contacts with the RNP1 and RNP2 regions of each RRM. (c) Strikingly different orientation of RRM domains in the polypyrimidine tract-binding protein (PTB), illustrating that RRMs are oriented in different relative positions in different hnRNPs. p(Y)-tract is a polypyrimidine tract. In PTB, the two RRMs associate through their α helices, so that the positively charged β sheets face away from each other, upward for RRM3 and downward for RRM4. The structure of CUCUCU single-stranded RNA bound to each of the two RRMs was determined, explaining how PTB can bind to two tracts of six pyrimidines in a single RNA if they are separated by a loop of or more nucleotides. This ability of
PTB to form a small loop in a pre-mRNA probably contributes to its ability to function as a splicing repressor at exons where the upstream splice site or the downstream splice site is flanked by two polypyrimidine tracts. Part (a) See K. Nagai et al., 1995, Trends Biochem. Sci. 20:235. [Part (b) Data from N. Harada et al., 1999, Nature 398:579. Part (c) Data from F. C. Oberstrass et al., 2006, Science 309:2054.] Description (a) R N A recognition motif, R R M shows four beta-sheets, labeled beta-one to betafour, and two alpha-helices. (b) sex-lethal or S x l R R M domains show 5-prime to 3prime pre-m R N A interacting with the proteins. A color-coded surface model of the protein shows pre-m R N A binding at regions colored blue, indicating negative surface charge. (c) polypyrimidine tract binding protein (P T B) shows two R R M domains, labeled R R M 3 and R R M 4. The pre-m R N A interacts at the top and bottom of the protein. The 45-residue KH domain is found in the hnRNP K protein and several other RNA-binding proteins. The three-dimensional structure of representative KH domains is similar to that of the RRM domain but smaller, consisting of a three-stranded β sheet supported from one side by two α helices. Nonetheless, the KH domain interacts with RNA much differently than does the RRM domain. RNA binds to the KH domain by interacting with a hydrophobic surface formed by the α helices and one β strand. The RGG box, another RNA-binding motif found in hnRNP proteins, contains five Arg-Gly-Gly (RGG) repeats with several interspersed aromatic amino acids. Its arginine-rich nature is similar to the RNA-binding domains of the HIV Tat protein. KH domains and RGG repeats are often interspersed in two or more sets in a single RNA-binding protein.
Splicing Occurs at Short, Conserved Sequences in Pre-mRNAs via Two Transesterification Reactions
Splicing Occurs at Short, Conserved Sequences in Pre-mRNAs via Two Transesterification Reactions During formation of a mature, functional mRNA, the introns are removed and exons are spliced together. For short transcription units, RNA splicing usually follows cleavage and polyadenylation of the end of the primary transcript, as depicted for β-globin in Figure 9-2. However, for long transcription units containing multiple exons, splicing of exons in the nascent RNA begins before transcription of the gene is complete. Early pioneering research on the nuclear processing of mRNAs revealed that mRNAs are initially transcribed as much longer RNA molecules than the mature mRNAs in the cytoplasm. It was also shown that RNA sequences near the cap added shortly after transcription initiation are retained in the mature mRNA and that RNA sequences near the polyadenylated end of mRNA processing intermediates in hnRNA also are retained in the mature mRNA in the cytoplasm. How could an RNA be shorter than the primary transcript, but have the same and ends? The solution to this apparent conundrum came from the discovery of introns by electron microscopy of RNA-DNA hybrids of adenovirus DNA and the mRNA-encoding hexon, a major virion capsid protein (Figure 9-7). Other studies revealed nuclear viral RNAs that were colinear with the viral DNA (primary transcripts) and RNAs with one or two of the introns removed (processing intermediates). These results, together with the earlier findings that the cap and poly(A) tail at each end of long mRNA
precursors are retained in shorter mature cytoplasmic mRNAs, led to the realization that introns are removed from primary transcripts as exons are spliced together. EXPERIMENTAL FIGURE 9-7 Electron microscopy of mRNA-template DNA hybrids shows that introns are spliced out during pre-mRNA processing. (a) Diagram of the EcoRI A fragment of adenovirus DNA, which extends from the left end of the genome to just before the end of the final exon of the hexon gene. The gene consists of three short exons and one long exon separated by three introns of , 2.5, and 9 kb. (b) Electron micrograph (left) and schematic drawing (right) of a hybrid between an EcoRI A DNA fragment and a hexon mRNA. The loops marked A, B, and C correspond to the introns indicated in (a). Since these intron sequences in the viral genomic DNA are not present in the mature hexon mRNA, they loop out between the exon sequences that hybridize to their complementary sequences in the mRNA. (c) Consensus sequences around splice sites in vertebrate pre-mRNAs. The only nearly invariant bases are the GU and the
AG of the intron (blue), although the flanking bases indicated are found at frequencies higher than expected based on a random distribution. A pyrimidine-rich region (hatch marked) near the end of the intron is found in most cases. The branchpoint A, also invariant, usually is 20–50 bases from the splice site. The central region of the intron, which may range from 40 bases to 50 kilobases in length, generally is unnecessary for splicing to occur. See R. A. Padgett et al., 1986, Ann. Rev. Biochem. 55:1119, and E. B. Keller and W. A. Noon, 1984, Proc. Nat’l. Acad. Sci. USA 81:7417. [Micrograph in (b) Data from S. M. Berget et al., 1977, Proc. Nat’l. Acad. Sci. USA 74:3171;] Description The illustration (a) shows a bar-shaped D N A with following sequence in the adenovirus hexon gene from 5-prime to 3-prime end: exon, intron A, exon, intron B, exon, intron C, and exon. The length of exon and intron increases from 5-prime to 3prime end. The region from 5-prime end to more than half of the last exon is labeled Eco R 1 a and the remaining region of exon is of 1 kilobases. The illustration (b) shows and electron micrograph and similar schematic of hybrid D N A-m R N A structure. Both show A, B, C introns forming a loop as an m R N A is transcribed from 3-prime to 5-prime. The illustration (c) shows bar-shaped pre-m R N A with following sequence from 5-prime to 3-prime end: 5-prime exon, 5-prime splicing site, intron, scale, break, branch point, polypyrimidine tract (10-12 bases), 3-prime splicing site, and 3 prime exon. There are 20-50 bases between branch point and 3-prime splice site. The frequency of occurrence (in percent) is as follows: In the 5-prime exon: A or C, 70 percent; A, 60 percent; and G, 80 percent. In the intron: G, 100 percent; U, 100 percent; A or G, 95 percent; A, 70 percent; G, 80 percent; U, 45 percent; scale break; C, 80 percent; U, 90 percent; A or G, 80 percent; branch point, A 100 percent; C or U, 80 percent; polypyrimidine tract; any nucleotide; C, 80 percent; A, 100 percent; and G, 100 percent. In the 3-prime exon: G, 60 percent. The location of splice sites — that is, exon-intron junctions — in a premRNA can be determined by comparing the sequence of genomic DNA with that of the cDNA prepared from the corresponding mRNA (see Figure
6-17). Sequences that are present in the genomic DNA but absent from the cDNA represent introns and indicate the positions of splice sites. Such analysis of a large number of different mRNAs revealed moderately conserved, short consensus sequences at the splice sites flanking introns in vertebrate pre-mRNAs; a pyrimidine-rich region just upstream of the splice site is also common (Figure 9-7c). Studies of mutant genes with deletions introduced into introns showed that much of the central portion of introns can be removed without affecting splicing; generally, only 30– 40 nucleotides at each end of an intron are necessary for splicing to occur at normal rates. Analysis of the intermediates formed during splicing of pre-mRNAs in vitro led to the discovery that splicing of exons proceeds via two sequential transesterification reactions (Figure 9-8). Introns are removed as a lariat structure in which the G of the intron is joined in an unusual -phosphodiester bond to an adenosine near the end of the intron. This adenosine (A) residue is called the branch-point A because it forms an RNA branch in the lariat structure. In each transesterification reaction, one phosphoester bond is exchanged for another. Since the number of phosphoester bonds in the molecule is not changed in either transesterification reaction, no energy is consumed. The net result of these two reactions is that two exons are ligated and the intervening intron is released as a branched lariat structure.
During Splicing, snRNAs Base-Pair with Pre-mRNA to Select Splice Sites and Guide the Transesterification Reactions
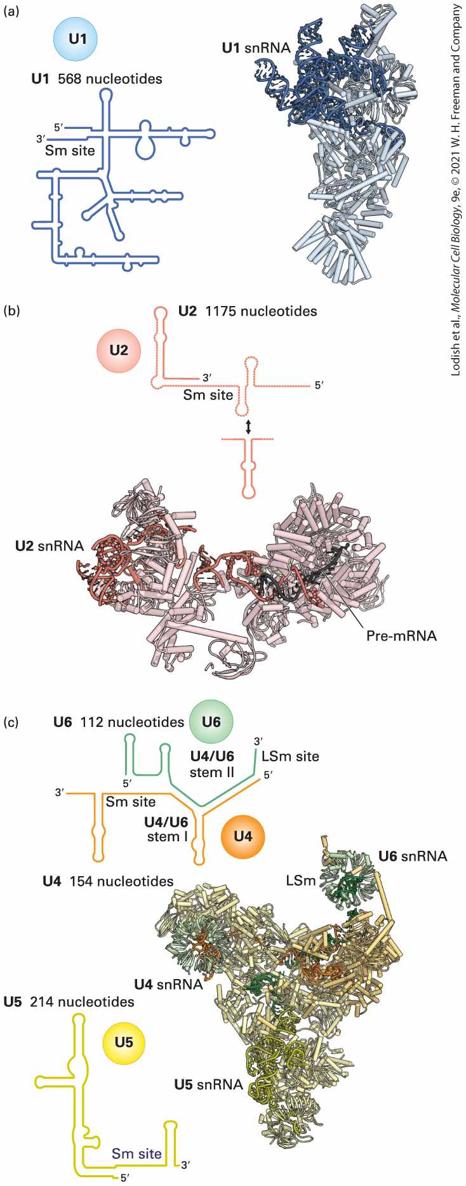
FIGURE 9-8 Two transesterification reactions that result in splicing of exons in premRNA. In the first reaction, the ester bond between the phosphorus of the intron and the oxygen (dark red) of exon 1 is exchanged for an ester bond with the oxygen (blue) of the branch-site A residue. In the second reaction, the ester bond between the phosphorus of exon 2 and the oxygen (orange) of the intron is exchanged for an ester bond with the oxygen of exon 1, releasing the intron as a lariat structure and joining the two exons. Arrows show where activated hydroxyl oxygens react with phosphorus atoms. Description The process starts with two exons with an intervening intron. It shows 3-prime oxygen of exon 1 bound to 5-prime triphosphate of intron having branch point A and the 3prime oxygen of intron is bound to the 5-prime triphosphate of exon 2. An arrow from 2-prime O H of branch-point A points to phosphate of 5-prime triphosphate of intron. First transesterification occurs resulting in separation of exon 1 bound to 3-prime O H group. An arrow from 3-prime oxygen of exon 1 points to phosphate of 5-prime triphosphate of exon 2. Second transesterification occurs resulting in an excised lariat intron and spliced exons. During Splicing, snRNAs Base-Pair with Pre-mRNA to Select Splice Sites and Guide the Transesterification Reactions Splicing requires highly abundant small nuclear RNAs (snRNAs), important for base pairing and other interactions with the pre-mRNA, as well as splicing factor proteins. Five U-rich snRNAs restricted to the nucleus, designated U1, U2, U4, U5, and U6, participate in pre-mRNA
splicing. Ranging in length from 107–210 nucleotides in vertebrates, these snRNAs are associated with 5–12 proteins each in the abundant small nuclear ribonucleoprotein particles (snRNPs) in the nuclei of eukaryotic cells (see Figure 9-10). Definitive evidence for the function of U1 snRNA in splicing came from experiments indicating that base pairing between the splice site of a premRNA and the region of U1 snRNA (Figure 9-9a) is required for RNA splicing (Figure 9-9b). In vitro experiments showed that a synthetic oligonucleotide that hybridizes with the -end region of U1 snRNA blocks RNA splicing. In vivo experiments showed that base pairing–disrupting mutations in the pre-mRNA splice site (red A in Figure 9-9b, left) also block RNA splicing; in this case, however, splicing can be restored by expression of a mutant U1 snRNA with a compensating mutation (red U in
Figure 9-9b) that restores base pairing to the mutant pre-mRNA splice site. These results and similar experiments with other mutations in premRNA bases near a splice site and compensating mutations in U1 snRNA that restore base pairing to the mutant pre-mRNA, showed that, except for the invariant GU at the end of nearly all introns, base pairing with the U1 region, rather than a specific RNA sequence, is required for RNA splicing.
FIGURE 9-9 Base pairing between pre-mRNA, U1 snRNA, and U2 snRNA early in the splicing process. (a) In this diagram, secondary structures in the snRNAs that are not altered during splicing are depicted schematically. The rectangles labeled Sm are binding sites for a ring of seven Sm proteins that encircle the snRNA at the positions shown in U1 and U2 snRNAs. The same Sm proteins also form a ring around U4 and U5 snRNAs (not shown). The invariant yeast branch-point sequence UACUAAC is shown here, with the bulged branch-point A indicated. Note that U2 snRNA base-pairs with a sequence that includes the branch-point A, although the branch-point A is not base-paired. (b) Only the ends of U1 snRNAs and splice sites in pre-mRNAs are shown. Left: A mutation (red A) in a premRNA splice site that interferes with base pairing to the end of U1 snRNA blocks splicing. Right: Expression of a U1 snRNA with a compensating mutation (red U) that restores base pairing also restores splicing of the mutant pre-mRNA. (c, d) Structure of a bulged A in an RNA-RNA helix, as in the pre-mRNA branch-point A base paired to U2 snRNA in (a). The structure of an RNA duplex with the sequence shown (c), containing bulged A residues (red) at position 5 in the RNA helix, was determined by x-ray crystallography (d). The bulged A residues extend from the side of an A-form RNA-RNA helix. The phosphate backbone of one strand is shown in green; the other strand in blue.
The structure on the right is turned 90° for a view down the axis of the helix. See M. J. Moore et al., 1993, in R. Gesteland and J. Atkins, eds., The RNA World, Cold Spring Harbor Press, pp. 303–357. See also Y. Zhuang and A. M. Weiner, 1986, Cell 46:827. [Parts (c) and (d) Data from J. A. Berglund et al., 2001, RNA 7:682.] Description (a) Schematic shows complementary base pairing between U 1 and U 2 small nuclear R N As (s n R N A) and pre-m R N A during splicing. The sequence on pre-m R N A from 5-prime to 3-prime end is as follows: exon 1, nucleotide sequence C A G G U A A G U (with C A G part of exon), pre-m R N A fragment, nucleotide sequence U A C U A (branch point) C, polypyrimidine tract, C A G G, exon 2. The U 1, formed of two stem-loops, base pairs with its nucleotides toward 5-prime cap end with the nucleotides of the pre-m R N A toward exon 1. The U 2, formed of three stem-loops, base pairs with its nucleotides between second and third loop with the nucleotides of the pre-m R N A around the branch point. (b) The effect of mutations on base pairing during splicing. A mutation in the pre-m R N A 5-prime splice site blocks splicing. The illustration of a mutant pre-m R N A shows the nucleotide sequence C A G G U A A A (highlighted) U toward exon 1. This blocks base pairing with the wild type U 1 s n R N A. A compensatory mutation in the U 1 restores splicing. The illustration shows a mutant pre-m R N A base paired with a mutant U 1 s n R N A having a complementary nucleotide for the mutated A of pre-m R N A. (c) Self-complementary sequences with bulging A. A 5-prime to 3-prime sequence of the top strand is: U A C U (bulging A) A C G U (space) A G U A. The complementary sequence of the bottom strand is A U G A (space) U G C A (bulging A) U C A U. Spaces correspond to the location of the bulging A in the complementary strand. (d) An X-ray crystallography structure shows side view and top view of a double-helix of R N A with bulging A nucleotides. In the top view, the diameter of the helix is indicated as 18.5 angstroms and the bulging A's of the strands are labeled, top and bottom.
Spliceosomes Catalyze Pre-mRNA Splicing
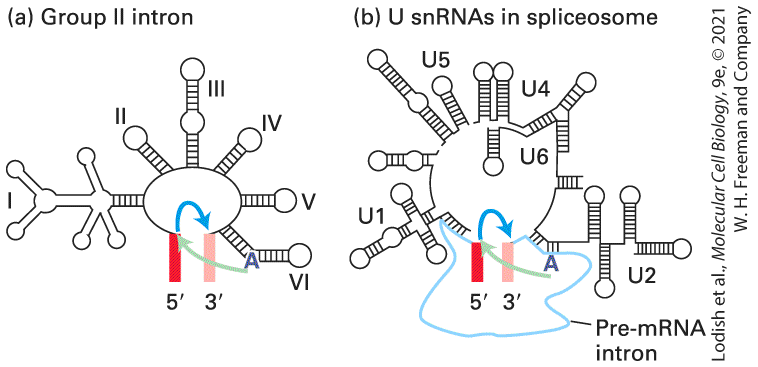
Involvement of U2 snRNA in splicing was initially suspected when it similarly was found to have an internal sequence that is largely complementary to the consensus sequence flanking the branch-point A in pre-mRNAs (see Figure 9-7c). Compensating mutation experiments, similar to those conducted with U1 snRNA and splice sites, demonstrated that base pairing between U2 snRNA and the branch-point sequence in pre-mRNA (see Figure 9-7c) is also critical to splicing. Significantly, the branch-point A itself, which is not base-paired to U2 snRNA, bulges out (Figure 9-9a), allowing its hydroxyl to participate in the first transesterification reaction of RNA splicing (see Figure 9-8). The structure of a model RNA-RNA duplex with two bulged As (Figure 9-9c) was determined by x-ray crystallography, showing the structure of a bulged-out A similar to the bulged-out A generated when U2 snRNA base pairs with the branch-point region of pre-mRNA (Figure 9-9d). Spliceosomes Catalyze Pre-mRNA Splicing Much of our understanding of the mechanisms regulating pre-mRNA splicing has come from biochemical studies with extracts of cultured mammalian cells and yeast and from genetic studies in yeast. The two transesterification reactions of RNA splicing are catalyzed by a 1.3 megadalton RNA-protein complex called the spliceosome, similar in size to the small ribosomal subunit. The spliceosome is assembled using five smaller ribonucleoprotein complexes called snRNPs (small ribonuclear
particles) U1, U2, U4, U5, and U6 assembled on the snRNAs described above with the same names (Figure 9-10). (U3 RNA, discussed later, is not involved in pre-mRNA splicing; rather, it functions in pre-ribosomal RNA processing; see Figure 9-46.)
FIGURE 9-10 Structures of Saccharomyces cerevisiae snRNPs involved in pre-mRNA splicing. The secondary structures of the snRNAs are diagrammed at the left, and the threedimensional structures determined by cryo-EM are shown at the right, with the locations of associated proteins indicated by ribbon diagrams. Each of the five small nuclear ribonucleoprotein particles (snRNPs) is shown with shades of distinct colors (U1, blue; U2, pink; U4, orange; U5, yellow; U6, green). “Sm site” in the RNA secondary structure diagrams of the U1, U2, U4, and U5 snRNAs are sites where a ring of seven Sm proteins encircle the snRNA. An alternative seven-protein ring composed of proteins LSM2-8 forms around the end of the U6 snRNA. [Data from C. Plaschka, A. J. Newman, and K. Nagai, 2019, Cold Spring Harb. Perspect. Biol. 11:a032391.] Description The illustration (a) shows U 1 made of 568 nucleotides with 3-prime end lying close to 5-prime end. The illustration (b) shows U 2 made of 1175 nucleotides and the ribbon model shows pre-m R N A bound to the U 2 s n R N A. The illustration (c) shows one ribbon diagram made of U 4, U 5, and U 6 together. The U 6 is labeled at the top right, the U 5 is labeled at the bottom of the structure, and the U 4 is labeled at the left with L S m lying near U 6. The schematics shows a common U 4 and U 6 structure with U 4 made of 154 nucleotides and U 6 made of 112 nucleotides. The structure has S m site on U 4, L S m site on U 6, U 4 slash U 6 stem 1 in U 4, and U 4 slash U 6 stem 2 on U 6. A schematic of U 5 is made of 214 nucleotides. The U1, U2, U4, U5, and U6 snRNPs associate transiently with each other and with splice sites in pre-mRNAs to assemble an active spliceosome that includes only the U2, U5, and U6 snRNPs (Figure 9-11a). The active spliceosome also includes two large multiprotein complexes called NTR and NTC (Figure 9-11a). As can be seen from Figure 9-11b highlighting the snRNAs, the active spliceosome is composed primarily of protein. The fully assembled, active spliceosome performs the two-step splicing
reaction and then dissociates. Studies of yeast temperature-sensitive mutants identified essential protein-coding genes required for RNA splicing, most of which have clear orthologs in humans. Most of these encode polypeptides that are transiently bound and then dissociate from spliceosome assembly intermediates, as well as factors released in association with the excised lariat intron. The released snRNPs and splicing-protein factors then reassemble onto another pre-mRNA splice site to catalyze another two-transesterification splicing reaction.
FIGURE 9-11 Structure of the catalytically active yeast spliceosome. (a) U5 snRNP (yellow) forms a scaffold onto which the other three major subcomplexes of the catalytically active spliceosome (U2 snRNP (pink) and protein complexes NTR (light purple) and NTC (dark purple) dock around the RNA molecules and the catalytic center. The four RNA molecules visible in this space-filling model are colored orange for U2, yellow for U5, green for U6, and red for the intron lariot RNA. (b) Positions of U6, U5, two portions of U2 snRNAs, and the lariat intron RNA (red) in the catalytically active spliceosome, determined at high resolution by cryo-EM.
[Republished with permission from the American Association for the Advancement of Science, from C. Yan et al., “Structure of a Yeast Spliceosome at 3.6-Angstrom Resolution.” Science 349 (6253):1182–1191; permission conveyed through Copyright Clearance Center, Inc.] Description The illustration (a) of a yeast spliceosome has the following labels: top left, U 2; middle left, Lariot R N A; bottom left, U 5; bottom right, U 6 s n R N A; and top right, N T C or N T R. The illustration (b) shows a U 2 s n R N A another structure with U 6 s n R N A at the top right, U 5 s n R N A at the bottom, Lariot R N A at the right, and U 2 s n R N A at the top left. Unlike the ribosome during protein synthesis, the spliceosome is not a stable complex. During assembly of the active spliceosome, some snRNPs and additional splicing factor proteins initially associate with assembly intermediates and then are released. As discussed above, the RNA-splicing reaction results in no change in free energy since there is no change in the number of phosphodiester bonds before and after the reaction. However, during RNA splicing in the cell, the process is driven toward ligation of the and exons by RNA-helicase enzymes (proteins), utilizing energy from ATP hydrolysis, as discussed below. In yeast, the splice site and branch-point sequences are stringently conserved for almost all introns (Figure 9-12). In contrast, splice sites and branch-point sequences are highly degenerate in mammals, except for the GU and AG at the and ends of most introns and the defining A at the branch point (Figure 9-12). This is because compared to yeast, mammals utilize multiple additional sequence-specific RNA-binding proteins that
assist and regulate snRNP U1 and U2 binding to splice sites and the branch point, respectively. This allows alternative splicing of primary transcripts in mammals by regulating the activities of these RNA-binding proteins. Comparison of the more simplified splicing system in yeast with the more complex system in mammals facilitated studies in both systems and an understanding of how additional nuclear proteins in mammals participate in and regulate alternative RNA splicing.
FIGURE 9-12 RNA sequences at the splice sites and branch-point A in yeast and humans. Yeast exhibit more conserved splice site and branch-point sequences than mammalian cells [note the high number of N’s (any base) in the mammalian sequence]. Description The regions in yeast R N A from left to right are as follows: exon 1; 5-prime splice site; nucleotide sequence G U A U G U; scale break; branch-point sequence U A C U A A C; 3-prime end of intron with sequence Y A G; 3-prime splice site; and exon 2. The regions in human R N A from left to right are as follows: exon 1; N N or A G; 5-prime splice site; nucleotide sequence G U R A G N or G U N N N N; scale break; branchpoint sequence Y N Y U R A Y; 3-prime end of intron with sequence Y subscript N, A G; 3-prime splice site; and exon 2. Region between 5-prime splice site and 3-prime splice site is intron. Pre-mRNA Splicing Cycle
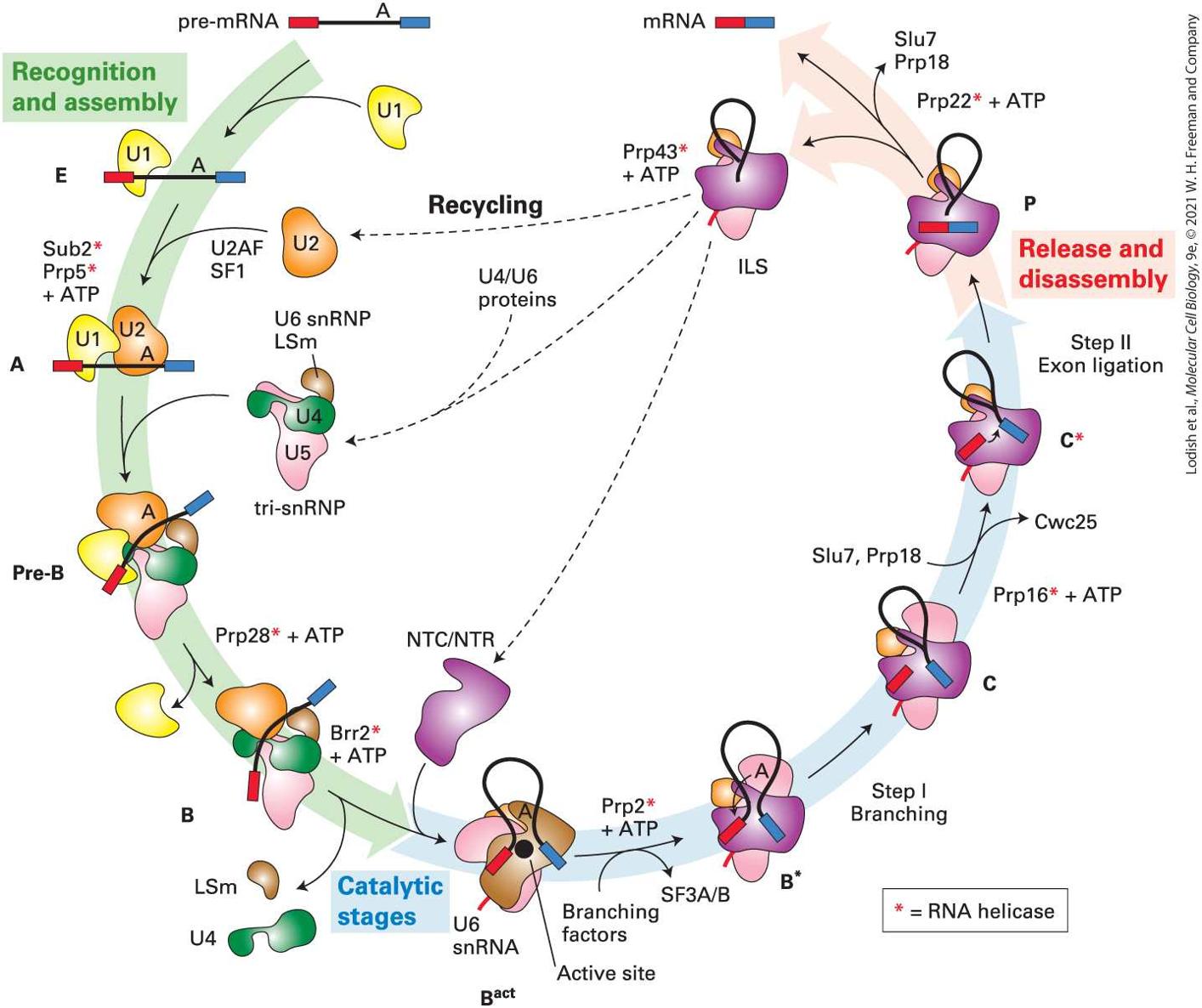
In yeast, 10 complexes of snRNPs and protein splicing factors have been distinguished during a splicing cycle (Figure 9-13). As discussed above (Figure 9-9a), spliceosome assembly initiates through base pairing interactions between the U1 snRNA in a U1 snRNP and a splice site in a pre-mRNA (Figure 9-9a, left). Next, in mammalian cells, a heterodimeric splicing factor protein called U2 associated factor (U2AF), together with another protein factor, splicing factor 1 (SF1), promote association of a U2 snRNP with a branch-point sequence near the end of the intron (Figure 9-9a, right). The small U2AF subunit binds the splice site AG, and the large U2AF subunit binds the polypyrimidine tract in metazoans. SF1 interacts transiently with the branch-point A, and then is released as U2 snRNA base pairs to the branch-point region (Figure 9-9a). Together, these interactions specify the location of a splice site used in a splicing reaction. Other protein splicing factors not shown associate with and stabilize these U1 and U2 snRNA interactions with the pre-mRNA.
FIGURE 9-13 Ten complexes of pre-mRNAs with snRNPs and protein splicing factors have been distinguished during the splicing cycle in yeast. The names of each complex are indicated around the circumference of the diagram. See text for explanation. [Data from C. Plaschka, A. J. Newman, and K. Nagai, 2019, Cold Spring Harb. Perspect. Biol. 11:a032391.] Description The cycle starts with a pre-m R N A made of red exon, intron with branch point A, and blue exon. During recognition and assembly stage: Structure E shows U 1 bound to 5prime splicing site. Next, U 2 enters the cycle along with U 2 A F, S F 1, Sub 2 (red asterisk), P r p 5 (red asterisk) plus A T P. Structure A shows U 2 bound to branch point A. Next, tri-s n R N P enters (U 6 L s m, U 4, and U 5 complex). Structure pre-B shows tri-s n R N P bound to the structure A. P r p 28 (red asterisk) plus A T P causes the
removal of U 1 resulting in the formation of structure B. Next: B r r 2 (red asterisk) plus A T P causes the removal of L s m and U 4. The cycle then enters catalytic stage: N T C slash N T R enters the cycle. Structure B superscript act shows pre-m R N A forming an inverted U with its end near the active site. Next, branching factors enter causing the release of S F 3 A slash B and P r p 2 (red asterisk) plus A T P enters the cycle. Structure B asterisk shows an arrow pointing from branching point to 5-prime splice site. Next, step 1 branching occurs resulting in a structure C showing excised red exon. S l u 7, P r p 18 causes the removal of C w C 25 and P r p 16 (red asterisk) plus A T P enters the cycle. Structure C (red asterisk) shows an arrow pointing from red exon to blue exon. Next, step 2 exon ligation occurs. The cycle then enters release and disassembly stage: The structure P shows red and blue exons bound together with a separated intron. Next, P r p 22 (red asterisk) plus A T P causes the removal of S l u 7, P r p 18. The step results in an I L S complex and an m R N A made of blue and red exons. Next, P r p 43 (red asterisk) plus A T P acts on I L S causing the separation of of N T C slash N T R, recycled U 2, and formation of tri-s n R N P by addition of U 4 slash U 6 proteins. A textbox at the bottom states, red asterisk equals R N A helicase. U2 snRNP association is followed by large conformational rearrangements of the snRNPs, including association of a pre-assembled tri-snRNP complex of U4, U5, and U6 snRNPs (Figure 9-10) during the transition from the A to pre-B complex (Figure 9-13). This is followed by dissociation of the U1 snRNP to form the B-complex. The U4 snRNP then dissociates as the large multisubunit protein complexes NTC (composed of seven polypeptides) and NTR (composed of six polypeptides) associate, generating the complex. The snRNP rearrangements that occur during the transition from the B complex to the complex generate new interactions between U6 and U2 and between U6 and the splice site
(Figure 9-14a) that are required for catalysis of the two splicingtransesterification reactions.
FIGURE 9-14 Rearrangements of snRNPs generate an active catalytic site. (a) The network of RNA interactions in the precatalytic B-complex (top) and the catalytically active complex (bottom). During activation, base pairing interactions in the B complex indicated by dashed red ovals including regions in U4, U6 and U2 undergo major rearrangements. U1 and U4 snRNPs dissociate from the spliceosome, allowing the end of the U6 snRNA to base pair through its highly conserved ACAGAG motif to the splice site (dashed red oval). Other critical base-pairing interactions in the Bact complex are indicated (dashed red ovals). (b) Structures of the RNAs in the precatalytic B-complex visualized at high resolution by cryo-EM. (c) RNA elements at the splicing active site of the Bact complex. ions are held by U6 snRNA in the correct geometry relative to the and splice sites and the branchpoint A and elements of U2 and U5 snRNAs to catalyze the two splicing transesterification reactions. [Part (a) Data from M. C. Wahl, C. L. Will, and R. Luhrmann, 2009, Cell 136:701–718. Part (b) Data from C. Plaschka, A. J. Newman, and K. Nagai, 2019, Cold Spring Harb. Perspect. Biol. 11:a032391. Part (c) Data from C. Yan, R. Wan, and Y. Shi, 2019, Cold Spring Harb. Perspect. Biol. 11:a032409.]
Description Two illustration in (a) show B-complex, precatalytic spliceosome and B activatedcomplex, catalytically active spliceosome. The B-complex shows an arrangement of U 1, U 2, U 4, U 5, U 6, and Lariot R N A with branch point A. The B activated-complex shows an arrangement of U 2, U 5, U 6, and Lariot R N A with branch point A. An arrow from branch point A points to 5-prime splice site of Lariot R N A. The illustration (b) shows a ribbon model of B-complex, precatalytic spliceosome. The complex shows U 1 bound to 5-prime end of Lariot R N A, U 2 bound to branch point A, U 2 slash U 4 slash U 6 bound to 3-prime end of R N A. An unbound U 5 is near the 5-prime end. The illustration (c) shows a ribbon model of B activated-complex, conserved splicing active site. The complex shows a bound structure made of U 2 and U 6 with M 1 and M 2 sites in the center and an unbound U 5 near the complex. These rearrangements of the snRNPs and protein splicing factors generate a catalytic active site (Figure 9-14b) where the first transesterification is catalyzed, followed by the second transesterification joining the and exons. The two reactions take place without significant rearrangement of the active site other than the positions of the phosphoester bonds at the splice sites (distinguishing complexes B, C, and C; Figure 9-13). Unlike the ribosome, in which RNA accounts for percent of the molecular mass, the three snRNAs (U2, U5, and U6) of the catalytically active human spliceosome represent no more than 5 percent of its total molecular mass (Figure 9-11). This seems consistent with the important functions of proteins at many steps in the splicing process. Spliceosomal protein components organize the splicing active site, deliver the reactive chemical groups into the active site, drive the reaction toward exon joining, and regulate the splicing process.
The splicing process is completed by disassembly of the spliceosome, which requires the NTC and NTR protein complexes (Figure 9-13, complex P → ILS). This results in release of the product spliced RNA with the exon ligated to the exon by a standard to phosphodiester bond, and release of the intron lariat RNA associated with U2, U5, and U6 snRNPs, and the NTC and NTR protein complexes (labeled ILS in Figure 9-13 for intron lariat spliceosome). Degradation of the intron lariat by a debranching enzyme that cleaves the phosphodiester bond with the branch-site A and by and exonucleases releases the U2, U5, and U6 snRNPs and splicing factors for use in another cycle of splicing (Figure 913, Recycling). Several RNA helicases are required in the splicing process (indicated by * in Figure 9-13). For example, the RNA helicase Brr2 is required for the critical transition from the catalytically inactive B complex that contains all the snRNPs required for splicing (but in inactive conformations), to the catalytically active complex. Many RNA helicases are proposed to break RNA-RNA base pairs by binding single-stranded RNA together with ATP next to an RNA-RNA duplex. ATP hydrolysis is then coupled to a change in the helicase conformation that pulls the single-stranded RNA through the active site of the helicase in a direction. This dislodges a complementary single strand or a protein bound to the RNA. Brr2 is postulated to bind the single-stranded U4 snRNA in the U4/U6/U5 trisnRNP next to the U4/U6 duplex (Figure 9-10, U4/U6 stem 1) and then pull on the RNA in the direction, causing unwinding of the U4/U6 duplex and dissociation of the associated proteins. This allows U4 to be released from the complex and U6 to change conformation in order to
make interactions with the splice site and to bind two ions in the proper geometry relative to the splice sites and branch-point A to catalyze the exchange of phosphodiester bonds (Figure 9-14c). RNA is central to the catalytic mechanism. The transesterification reactions are catalyzed by the two ions bound by snRNA U6. The critical branch-site A -OH is exposed through RNA-RNA interactions. However, proteins stabilize and guide these RNA-RNA interactions, and RNA helicases use energy from ATP hydrolysis to push the reaction cycle in favor of exon joining. Consequently, the spliceosome is considered to be a protein-directed metalloribozyme. Exon Junction Complexes in Vertebrates Following RNA splicing in vertebrates, a specific set of hnRNP proteins remain bound to the spliced RNA approximately 20 nucleotides to each exon-exon junction, thus forming an exon-junction complex. One of the hnRNP proteins associated with the exon-junction complex is the RNA export factor (REF), which functions in the export of fully processed mRNPs from the nucleus to the cytoplasm, as will be discussed in Section 9.3. Other proteins associated with the exon-junction complex function in a quality-control mechanism that leads to the degradation of improperly spliced mRNAs, known as nonsense-mediated decay (Section 9.4). Rare -AU … AC- Introns A small fraction of pre-mRNAs ( percent in humans) contain introns whose splice sites do not conform to the standard consensus sequence.
3′ Cleavage and Polyadenylation of Pre-mRNAs Are Tightly Coupled
This class of introns begins with AU and ends with AC rather than following the usual “GU-AG rule” (see Figure 9-7c). Splicing of this special class of introns occurs via a splicing cycle analogous to that shown in Figure 9-13, except that four novel, low-abundance snRNPs, together with the standard U5 snRNP, are involved. Trans-Splicing Nearly all functional mRNAs in vertebrate, insect, and plant cells are derived from a single molecule of the corresponding pre-mRNA by removal of internal introns and splicing of exons. However, in two types of protozoans — trypanosomes and euglenoids — mRNAs are constructed by splicing together separate RNA molecules. This process, referred to as trans-splicing, is also used in the synthesis of 10–15 percent of the mRNAs in the nematode (roundworm) Caenorhabditis elegans, an important model organism for studying embryonic development. Transsplicing is carried out by snRNPs and splicing factors by a process similar to the splicing of exons in a single pre-mRNA (Figure 9-8), except that there is a break in the loop representing the intron. Cleavage and Polyadenylation of Pre-mRNAs Are Tightly Coupled In eukaryotic cells, all mRNAs, except histone mRNAs, have a poly(A) tail. Early studies of pulse-labeled adenovirus and SV40 RNA demonstrated that the viral primary transcripts extend beyond the
sequence that is polyadenylated. These results suggested that A residues are added to a hydroxyl generated by endonucleolytic cleavage of a longer transcript. But the predicted downstream RNA fragments were never detected in vivo, presumably because of their rapid degradation. However, detection of both predicted cleavage products was observed in the in vitro processing reactions performed with nuclear extracts of cultured human cells. The cleavage/polyadenylation process and degradation of the RNA downstream of the cleavage site occurs much more slowly in these in vitro reactions, simplifying detection of the downstream cleavage product. Early sequencing of cDNA clones from animal cells showed that nearly all mRNAs contain the sequence AAUAAA 10–35 nucleotides upstream from the poly(A) tail (Figure 9-15). Polyadenylation of RNA transcripts is virtually eliminated when the corresponding sequence in the template DNA is mutated to any other sequence, except the closely related sequence (AUUAAA). The unprocessed RNA transcripts produced from such mutant templates do not accumulate in nuclei but are rapidly degraded. Further mutagenesis studies revealed that a second signal downstream from the cleavage site is required for efficient cleavage and polyadenylation of most pre-mRNAs in animal cells. This downstream signal is not a specific sequence but rather a GU-rich or simply a U-rich region within nucleotides of the cleavage site.
FIGURE 9-15 Model for cleavage and polyadenylation of pre-mRNAs in mammalian cells. Cleavage and polyadenylation specificity factor (CPSF) binds to the upstream AAUAAA poly(A) signal. CStF interacts with a downstream GU- or U-rich sequence and with bound CPSF, forming a loop in the RNA; binding of CFI and CFII helps stabilize the complex. Binding of poly(A) polymerase (PAP) then stimulates cleavage at a poly(A) site, which usually is 10–35 nucleotides of the upstream poly(A) signal. The cleavage factors are released, as is the downstream RNA cleavage product, which is rapidly degraded. Bound PAP then adds residues at a slow rate to the -hydroxyl group generated by the cleavage reaction. Binding of poly(A)-binding protein (PABPNI) to the initial short poly(A) tail accelerates the rate of addition by PAP. After 200–250 A residues have been added, PABPN1 signals PAP to stop polymerization. Description The series starts with a pre-m R N A showing 2 poly (A) signal areas with poly (A) site in between. The poly (A) signal toward 5-prime capped end shows sequence A A U A A A and the poly (A) signal toward 3-prime end shows sequence G slash U. Next, C P S F, C S t F, C F 1, and C F 2 enters the process resulting in bending of pre-m R N A. C P S F binds to 5-prime end poly (A) signal, C S t F binds to 3-prime end poly (A) signal, and C F 1 and C F 2 binds to poly (A) site. Next, P A P enters the process and binds between C P S F and C F 1 and 2. Next, cleavage occurs at the P A P bound site with O H end at 5-prime capped m R N A. Next, slow polyadenylation occurs wherein A T P enters causing the release of P P I, pre-m R N A strand with G slash U poly (A) signal plus C S t F, C F 1, and C F 2. This resulting 5-capped pre-m R N A strand show a poly A tail at the end of poly (A) signal with the P A P bound to the O H end. Next, P A B P N 1 enters and binds between poly (A) signal and P A P with approximately 12 adenosines. Next, rapid, processive polyadenylation occurs wherein P A B P N 1 and A T P enters the process and P P I is released. The final resulting m R N A shows a 5prime capped end, C P S F bound to poly (A) signal, three P A B P N 1 with each consisting approximately 12 adenosines, approximately 200 adenosines at the 3-prime O H end. The P A P dissociates when tail reaches approximately 250 adenosines.
Purification of the proteins required for cleavage and polyadenylation of pre-mRNA precursors in vitro led to the initial identification of proteins involved in the process. This was followed by isolation and sequencing of the genes encoding them, purification of the recombinant proteins expressed from these genes, and then further identification of associated proteins by binding to the overexpressed proteins and by co-purification of epitope-tagged versions of the proteins expressed in vivo. Studies with these purified proteins led to the model of cleavage and polyadenylation in
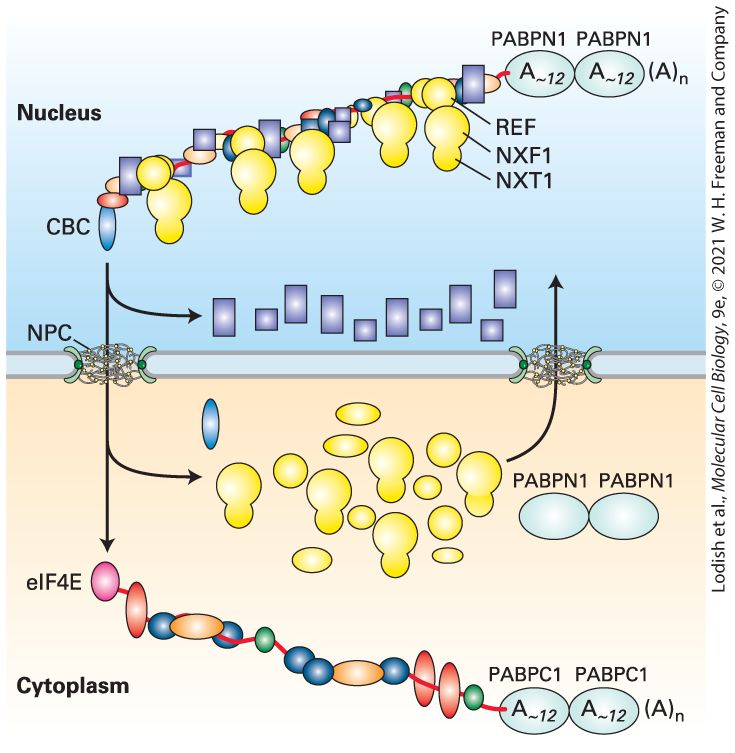
Figure 9-15. In S. cerevisiae, 14 or 15 polypeptides assemble into an million Dalton complex with three modules that perform cleavage and polyadenylation at the poly(A) site. One module binds the AAUAAA RNA sequence in the pre-mRNA and cleaves the RNA 10–35 bases downstream. The variation in distance may be due to different secondary structures in the RNA between the AAUAAA and the cleavage site for each pre-mRNA. The second module of the cleavage polyadenylation complex includes two phosphatases that appear to regulate elongation by RNA polymerase II and polyadenylation. The third module includes the poly(A) polymerase enzyme that adds A’s to the end of the RNA generated when the first module cleaves downstream of AAUAAA sequence. Following cleavage at the poly(A) site, polyadenylation proceeds in two phases. Addition of the first 12 or so A residues occurs slowly, followed by rapid addition of up to 200–250 more A residues. The rapid phase requires the binding of multiple copies of a poly(A)-binding protein containing an RRM motif. This protein is designated PABPNI to distinguish it from the
poly(A)-binding proteins present in the cytoplasm (PABPC1,2,3). PABPN1 binds to the short A tail initially added by PAP, stimulating the rate of polymerization of additional A residues by PAP, resulting in a much faster phase of polyadenylation. PABPN1 is also responsible for signaling poly(A) polymerase to terminate polymerization when the poly(A) tail reaches a length of 200–250 residues, although the mechanism for controlling the length of the tail is not yet understood. Binding of PABPN1 to the poly(A) tail is essential for mRNA export into the cytoplasm where it is replaced by a PABC1, 2, or 3 and shuttled back into the nucleus. Alternative Polyadenylation Sites In addition to alternative splicing, mRNA regulation is also mediated by alternative polyadenylation, which occurs for percent of human mRNAs. Alternative polyadenylation results from the use of two or more alternative cleavage/polyadenylation signals in alternative cell types. In some cases, this appears to be due to different concentrations of the polyadenylation factors in alternative cell types coupled with alternative poly(A) sites that have higher or lower affinity for the cleavage polyadenylation complex that binds the downstream G/U-rich portion of the cleavage/polyadenylation signal (Figure 9-15). In these cases, when the concentration of the factor is low, only the highest affinity cleavage/polyadenylation sites are used. But in alternative cell types where the factor concentration is higher, an upstream low-affinity site is used preferentially because it is bound by the cleavage/polyadenylation factor at the higher concentration, and once the pre-mRNA is cleaved, the downstream site cannot be used. In other cases, sequence specific RNA-
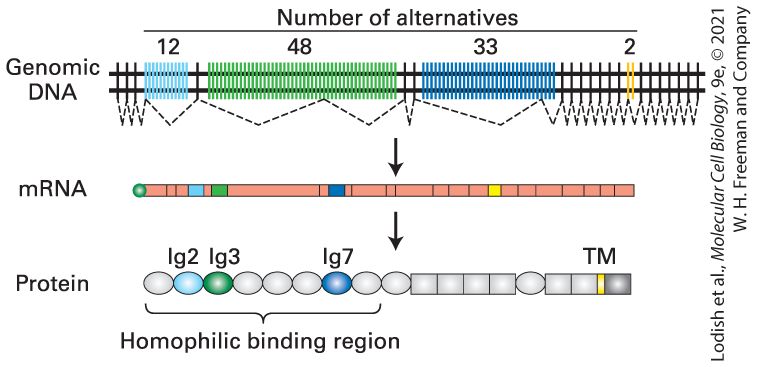
binding proteins may block or enhance binding of the cleavage/polyadenylation factors, similarly to the way they regulate the binding of splicing repressors and activators. Alternative sites of polyadenylation can also be coupled to alternative splicing of the final exon in an mRNA. As a consequence, protein isoforms can be expressed that have alternative C-terminal amino acid sequences. This is observed in the expression of alternative immunoglobulin molecules during B-lymphocyte development (see Figure 24-19). Initially, an antibody is produced with a transmembrane domain that anchors the antibody in the plasma membrane, and a cytoplasmic domain that signals when the antigen-binding extracellular domain encounters antigen — the molecule bound by an antibody. When that occurs, processing of the premRNA is modified so that an alternative exon is included in the mRNA. The resulting antibody molecules translated from this alternatively processed mRNA lack the membrane-spanning domain and, as a consequence, are secreted into the extracellular space where they can neutralize pathogens (see Chapters 14 and 24). RNA Polymerase II Transcription Termination Pol II generally terminates transcription at any one of several alternative sites within of the poly(A) site. Experiments with SV40 and adenovirus, both DNA viruses, showed that when the polyadenylation signal is mutated, RNA polymerase does not terminate transcription but continues transcription until the next poly(A) site in the viral genome is encountered. Similar results were observed when a wild-type copy of the
β-globin gene was inserted downstream of a viral promoter in the genome of a DNA virus. These experiments showed that transcription termination by RNA polymerase II is coupled to cleavage and polyadenylation of the transcript, and that the RNA fragment downstream of the cleavage site is rapidly degraded. This is thought to be due to the de-protection of the end of the nascent transcript. Because no cap is present on the end of the cleaved RNA, it is susceptible to digestion by the major exoribonuclease in the nucleus, XRN1. It is thought that when XRN1 reaches the still transcribing polymerase, it triggers termination, either by pulling the end of the nascent RNA out of the polymerase active site, or by introducing a conformational change in the polymerase that causes transcription termination. Once the nascent RNA is removed from the elongating polymerase, the contacts between the RNA polymerase II clamp and the RNA-DNA hybrid within the polymerase (see Figure 8-9b) are lost, allowing the clamp to swing open and release the polymerase from the DNA template. KEY CONCEPTS OF SECTION 9.1 Processing of Eukaryotic Pre-mRNA In the nucleus of eukaryotic cells, pre-mRNAs are associated with hnRNP proteins and processed by capping, cleavage/polyadenylation, and RNA splicing to remove internal introns before being transported to the cytoplasm (see Figure 9-2). In multicellular organisms, splicing of exons in long genes with multiple exons usually begins as the pre-mRNA is still being transcribed. Cleavage and polyadenylation to form the end of the mRNA occur after the poly(A) site is transcribed. Transcription and RNA processing are coupled via the carboxy-terminal domain (CTD) of RNA polymerase II. -capping enzymes interact with the CTD
phosphorylated at Ser-5 by TFIIH during transcription initiation. The cap protects the nascent pre-mRNA from exonucleases. Factors involved in RNA splicing and cleavage and polyadenylation associate with the Pol II CTD phosphorylated on Ser-2 by cyclin T-CDK9 during release of the paused polymerase early in gene transcription (Figures 8-13a, 8-16). These interactions with the phosphorylated CTD allow the processing factors to interact with the nascent pre-mRNA shortly after it emerges from the surface of the polymerase. Splicing occurs at short, conserved splice sites in pre-mRNAs (Figure 9-7c), via two transesterification reactions (Figure 9-8). The first of these generates a lariat intron with an unusual phosphoester bond at the branch-point A. Five different abundant small nuclear RNAs (snRNAs) are found in snRNA-protein complexes (snRNPs). These interact via base pairing of their snRNAs with one another and with pre-mRNA to form the 1.3 MDa spliceosome (see Figure 9-11). Multiple specific proteins are also associated with the catalytically active spliceosome, comprising 95 percent of its mass (Figure 9-11b). The spliceosome is not a stable complex. During assembly of the active spliceosome, some snRNPs and additional splicing factor proteins initially associate with assembly intermediates, and then are released. Ten complexes of snRNPs and protein splicing factors have been distinguished during a splicing cycle (Figure 9-13). Several RNA helicases are required in the splicing process. They utilize energy from ATP hydrolysis to drive the transesterification reactions in the direction of exon ligation. Since the basic catalytic mechanism of the spliceosome is performed by two ions bound by U6 (Figure 9-14c), and the substrate splice sites are delivered to the RNA-based active site by interactions with both snRNAs and proteins, the spliceosome is considered to be a protein-directed metalloribozyme. Following RNA splicing in vertebrates, a specific set of hnRNP proteins remain bound to the spliced RNA approximately 20 nucleotides to each exon-exon junction forming an exon-junction complex (EJC). As discussed in later sections, EJCs participate in mRNA export to the cytoplasm and in quality-control surveillance mechanisms that eliminate improperly processed mRNAs. The end of an mRNA is generated by cleavage of the pre-mRNA at a poly(A) site, an AAUAAA 10-35 bases upstream of the cleavage site and a GU- or U-rich sequence nucleotides downstream from the cleavage site. A poly(A) polymerase in a multisubunit cleavage-polyadenylation complex that performs the cleavage reactions adds a poly(A) tail of nucleotides in vertebrates, nucleotides in yeast, to the end. The poly(A) tail is then immediately bound by
nuclear poly(A) binding protein (PABPN1), protecting the end from digestion by an exosome.
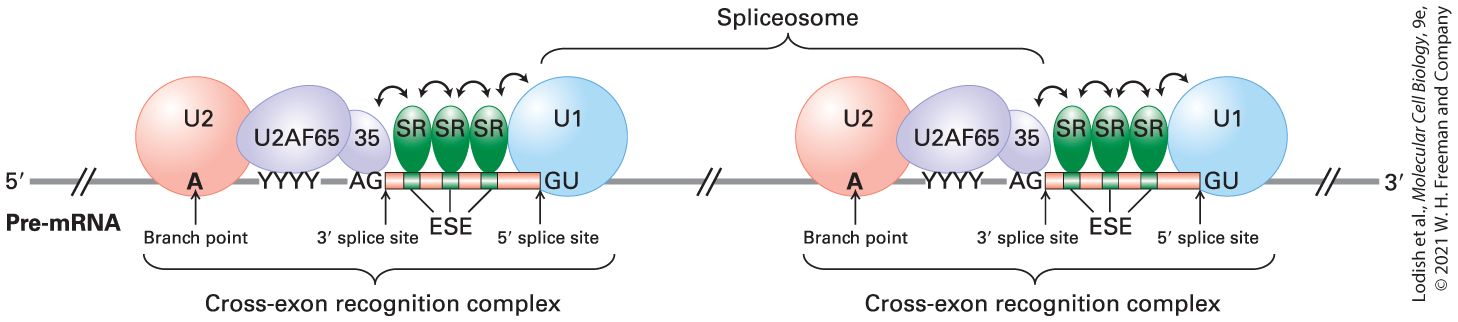
Additional Nuclear Proteins Contribute to Splice-Site Selection in the Long Pre-mRNAs of Humans and Other Vertebrates
9.2 Regulation of Pre-mRNA Processing Now that we’ve seen how pre-mRNAs are processed into mature, functional mRNAs, we consider how regulation of this process can contribute to control of gene expression. Recall from Figure 7-3 that multicellular eukaryotes contain both simple and complex transcription units encoded in their DNA. The primary transcripts produced from the former contain one poly(A) site and exhibit only one pattern of RNA splicing, even if multiple introns are present; thus simple transcription units encode a single mRNA. In contrast, the primary transcripts produced from complex transcription units ( percent of all human transcription units) can be processed in alternative ways to yield different mRNAs that encode distinct proteins (see Figure 7-3). This process of alternative RNA splicing greatly expands the number of functionally distinct proteins encoded by the human genes (where one gene equals one transcription unit for a primary transcript). The increase in complexity of mRNAs resulting from alternative splicing in metazoans reaches its zenith in the central nervous system, where regulation of complex patterns of alternative RNA splicing is required for generating synaptic connections between neurons, and consequently, appropriate behavior (see Chapter 23). Additional Nuclear Proteins Contribute to Splice-Site Selection in
the Long Pre-mRNAs of Humans and Other Vertebrates How are splice sites recognized during processing of eukaryotic premRNAs so that exons can be joined precisely to generate long open reading frames? As mentioned above, almost all splice sites in yeast conform to the sequence/GUAUGU, which is perfectly complementary to the sequence at the end of U1 snRNA (Figure 9-9). As first shown in the compensatory base pairing experiment described in Figure 9-9b, WatsonCrick base pairing between U1 snRNA and the first six bases of the intron are required for pre-mRNA splicing. In yeast, perfect complementarity between a six-base sequence near the middle of U2 snRNA and the branchpoint A sequence near the end of the intron (Figures 9-9a and 9-12) is also essential for splicing. Only percent of S. cerevisiae genes contain an intron, and most of these contain a single intron. In this organism, premRNA splice sites are selected primarily by base-pairing to U1 snRNA for the splice site at the beginning of an intron, and to U2 snRNA at the branch-point sequence just upstream of the splice site (Figure 9-9a). However, in humans, RNA sequences at and splice sites and the branch site are far more degenerate (Figure 9-12). Most introns begin with GU and end with AG preceded by a polypyrimidine tract of 10–12 bases, and a very short, degenerate six-base sequence encompassing the branch-point A. These consensus splice-site sequences are so degenerate that they cannot be used to predict splice sites in the human genome sequence; the vast majority of sequences that match these degenerate
sequences are not used as splice sites. Consequently, other aspects of human pre-mRNA sequences must contribute to splice site selection. Extensive mutational analysis of sequences within of splice sites of a number of specific mammalian genes identified sequences in some pre-mRNAs required for splicing in addition to the short, degenerate splice sites, branch point, and splice sites (Figure 9-12, bottom). These were different specific sequences in different genes within bases of the and splice sites. Characterization of nuclear RNA-binding proteins that bind to these regulatory sequences in RNA, and cDNA cloning and sequencing of related genes, have led to the discovery of at least 1500 human RNA-binding proteins that bind to short 3–4 base RNA sequences. These are hnRNP proteins discussed earlier, and, consequently, consist of one or more RNA-binding domains (RRMs, Figure 9-6) fused to other functional domains. These additional functional domains often either stimulate or inhibit splicing at a splice site within bases of the binding site for the regulatory hnRNP. This allows relatively short exons (average length bases in humans) to be recognized in long primary transcripts that can have introns of tens of kb. This is because the splice sites are determined by the interactions of these RNA-binding proteins with specific sequences in the pre-mRNA in addition to the interactions of the snRNAs with splice sites and branch points, and of the U2AF protein splicing factor with splice sites. The RNA-sequences bound by RNAbinding proteins that stimulate RNA splicing are referred to as splicing enhancers, and the RNA-sequences bound by RNA-binding proteins that inhibit RNA splicing are referred to as splicing silencers.
In humans and other metazoans, the information for defining some splice sites is encoded by splicing enhancers within the sequence of an exon, consequently called exonic splicing enhancers. A subset of hnRNP proteins called SR proteins, bind exonic splicing enhancers. SR proteins contain one or more RRM RNA-binding domain. They also contain intrinsically disordered regions of polypeptide rich in arginine (R) and serine (S) residues, called RS domains, which can interact with RS domains in other RNA-binding proteins. When bound to exonic splicing enhancers, SR proteins mediate the cooperative binding of U1 snRNP to a splice site and U2 snRNP to a branch point, through a network of protein-protein interactions that span the exon (Figure 9-16). The complex of SR proteins, snRNPs, and other splicing factors (e.g., U2AF) that assemble across an exon, called a cross-exon recognition complex, permits precise specification of some exons in long pre-mRNAs. Many other exons are properly spliced without the assistance of SR proteins because they are demarcated by constitutive splice sites with good complementarity to U1 and U2 snRNAs and polypyrimidine tracts and splice site RNA sequences with high affinity for U2AF.
FIGURE 9-16 Exon recognition through cooperative binding of SR proteins and splicing factors to pre-mRNA. The correct -GU and -AG splice sites are recognized by splicing factors on the basis of their proximity to exons. The exons contain exonic splicing enhancers (ESEs) that are binding sites for SR proteins. When bound to ESEs, the SR
proteins interact with one another and promote the cooperative binding of the U1 snRNP to the splice site of the downstream intron, SF1 and then the U2 snRNP to the branch point of the upstream intron, the 65- and 35-kD subunits of U2AF to the pyrimidine-rich region and AG splice site, respectively, of the upstream intron, and other splicing factors (not shown). The resulting RNA-protein cross-exon recognition complex spans an exon and activates the correct splice sites for RNA splicing. Note that the U1 and U2 snRNPs in this unit do not become part of the same spliceosome. The U2 snRNP on the right forms a spliceosome with the U1 snRNP bound to the end of the same intron. The U1 snRNP shown on the right forms a spliceosome with the U2 snRNP bound to the branch point of the downstream intron (not shown), and the U2 snRNP on the left forms a spliceosome with a U1 snRNP bound to the splice site of the upstream intron (not shown). Double-headed arrows indicate protein-protein interactions. See T. Maniatis, 2002, Nature 418:236; see also S. M. Berget, 1995, J. Biol. Chem. 270:2411. Description The pre-m R N A structure from 5-prime to 3-prime end shows: a scale break, U 2 at branch point A, U 2 A F 65 at Y Y Y Y sequence, U 2 A F 35 at the A G sequence, 3prime splice site, three S Rs at three E S E sequences, 5-prime splice site, U 1 at G U sequence, scale break, similar complex, and a scale break. A double headed arrow points to U 2 A F 35 and S R; second double headed arrow points to S R and S R; third double headed arrow points to S R and S R; and fourth double headed arrow points to S R and U 1. The complex from U 2 to U 1 is labeled, cross-exon recognition complex. The structure from U 1 of first complex to U 2 A F 35 of second complex is labeled, spliceosome. This participation of regulatory proteins in the selection of and splice sites allows metazoans to regulate the use of alternative splice sites. The sequences of the first cDNAs cloned, including mRNAs for alpha and beta globins, antibody light chains, and chicken ovalbumin, revealed only a single type of spliced mRNA expressed from the genes encoding them. However, this is a very unusual situation in vertebrates that applies to
Regulation of RNA Splicing Through Splicing Enhancers and Silencers Controls Drosophila Sexual Differentiation
their position along the cochlea. This remarkable arrangement suggests that splicing of the -activated -channel pre-mRNA is regulated in response to extracellular signals that inform the cell of its position along the cochlea. Other studies demonstrated that splicing at one of the alternative splice sites in the -activated -channel pre-mRNA in the rat is suppressed when a specific protein kinase is activated by neuron depolarization in response to synaptic activity from interacting neurons. This observation raises the possibility that a splicing repressor specific for this site may be activated when it is phosphorylated by this protein kinase, whose activity in turn is regulated by synaptic activity. Since hnRNP and SR proteins are extensively modified by phosphorylation and other post-translational modifications, it seems likely that complex regulation of alternative RNA splicing through post-translational modifications of splicing factors has a significant function in modulating neuron function. Regulation of RNA Splicing Through Splicing Enhancers and Silencers Controls Drosophila Sexual Differentiation One of the earliest and best understood examples of regulated alternative splicing of pre-mRNA came from studies of sexual differentiation in Drosophila. Genes required for normal Drosophila sexual differentiation
were first characterized by isolating Drosophila mutants defective in the process. When the proteins encoded by the wild-type genes were characterized biochemically, two of them were found to regulate a cascade of alternative RNA splicing in Drosophila embryos. Subsequent research provided insight into how these proteins regulate RNA processing and ultimately lead to the expression of one or the other of the two sexspecific transcriptional repressors that suppress the development of characteristics of the opposite sex. The Sxl protein, encoded by the sex-lethal gene, is the first protein to act in the cascade (Figure 9-18). The Sxl protein is present only in female embryos. Early in development, the Sxl gene is transcribed from a promoter that functions only in early female embryos. Later in development, this female-specific promoter is shut off and another promoter for sex-lethal becomes active in both male and female embryos. However, in the absence of early Sxl protein, the sex-lethal pre-mRNA in male embryos is spliced to produce an mRNA that contains a stop codon early in the sequence (Figure 9-18a, blue shading). The net result is that male embryos produce no functional Sxl protein either early or later in development.
FIGURE 9-18 Cascade of regulated splicing that controls sex determination in Drosophila embryos. For clarity, only the exons (boxes) and introns (black lines) where regulated splicing occurs are shown. Splicing is indicated by red dashed lines above (female) and blue dashed lines below (male) the pre-mRNAs. Vertical red lines in exons indicate in-frame stop codons, which prevent synthesis of functional protein. Only female embryos produce functional Sxl protein, which represses splicing between exons 2 and 3 in sxl pre-mRNA (a) and between exons 1 and 2 in tra pre-mRNA (b). (c) In contrast, the cooperative binding of Tra protein and two SR proteins, Rbp1 and Tra2, activates splicing between exons 3 and 4 and cleavage/polyadenylation at the end of exon 4 in dsx pre-mRNA in female embryos. In male embryos, which lack functional Tra, the SR proteins do not bind to exon 4, and consequently exon 3 is spliced to exon 5. The distinct Dsx proteins produced in female and male embryos as the result of this cascade of regulated splicing repress transcription of genes required for sexual differentiation of the opposite sex. See M. J. Moore et al., 1993, in R. Gesteland and J. Atkins, eds., The RNA World, Cold Spring Harbor Press, pp. 303–357. Description
The illustration (a) shows s x l. The pre-m R N A show regions from 5-prime to 3-prime end: intron, exon 2, intron, exon 3 with in-frame stop codon, intron, exon 4, and intron. Exons 2 and 4 are linked by a pink dashed line, indicating that these comprise the final mature m R N A expressed in females. All three exons are connected by a blue dashed line, indicating that these comprise the final m R N A expressed in males. The female m R N A undergoes translation to produce s x l protein. The female s x l protein represses splicing between exons 2 and 3. The illustration (b) shows t r a. The pre-m R N A show regions from 5-prime to 3-prime end: exon 1, intron, exon 2 with in-frame stop codon, exon 3, and intron. The female m R N A comprises exons 1 and 3 that results into female T r a protein. The female s x l protein represses splicing between exons 1 and 2. The male m R N A comprises exons 1, exon 2, and exon 3. The illustration (c) shows d s x. The pre-m R N A show regions from 5-prime to 3prime end: intron, exon 3, intron, exon 4 with poly (A) tail, intron, exon 5, and intron. The female t r a protein along with R b p 1 plus T r a 2 encourages splicing of exons 3 and 4. The female m R N A comprises exons 3 and 4 with poly (A) tail that results into female D s x protein. The male m R N A comprises exons 3 and 5 that results into male D s x protein. In contrast, the Sxl protein expressed in early female embryos directs splicing of the sex-lethal pre-mRNA so that a functional sex-lethal mRNA is produced (Figure 9-18a). Sxl accomplishes this by binding to a sequence in the pre-mRNA near the end of the intron between exon 2 and exon 3, thereby blocking the proper association of U2AF and U2 snRNP with the branch-point A and the AG (Figure 9-16). As a consequence, the U1 snRNP bound to the end of exon 2 assembles into a spliceosome with U2 snRNP bound to the branch point at the end of the intron between exons 3 and 4, leading to splicing of exon 2 to exon 4 and skipping of exon 3 (Figure 9-18a). The binding site for Sxl in the Sxl pre-mRNA is called an intronic splicing silencer because of its location in an intron and its
function in blocking, or “silencing,” use of a splice site. The resulting female-specific sex-lethal mRNA is translated into functional Sxl protein, which reinforces its own expression in female embryos by continuing to cause skipping of exon 3. The absence of Sxl protein in male embryos allows the inclusion of exon 3 and, consequently, of the stop codon that prevents translation of functional Sxl protein. Sxl protein also regulates alternative RNA splicing of the transformer gene pre-mRNA, the second gene in the regulatory cascade (Figure 9-18b). In male embryos, where no Sxl is expressed, tra exon 1 is spliced to tra exon 2, which contains a stop codon that prevents synthesis of a functional transformer protein, very similar to the inhibition of Sxl protein in male cells (Figure 9-18a). In female embryos, however, binding of Sxl protein to an intronic splicing silencer at the end of the intron between exons 1 and 2 blocks binding of U2AF at this site. The interaction of Sxl with transformer pre-mRNA is mediated by two RRM domains in the Sxl protein (see Figure 9-6b). When Sxl is bound, U2AF binds to a lower affinity site farther in the pre-mRNA. As a result, exon 1 is spliced to this alternative splice site, eliminating exon 2 with its stop codon. The resulting female-specific transformer mRNA, which contains additional constitutively spliced exons, is translated into functional Transformer (Tra) protein. In the last step in the regulatory cascade, Tra protein regulates the alternative processing of pre-mRNA transcribed from the double-sex gene (Figure 9-18c). In female embryos, a complex of Tra and two constitutively expressed proteins, Rbp1 and Tra2, directs splicing of exon
3 to exon 4 and also promotes cleavage/polyadenylation at the alternative poly(A) site at the end of exon 4 — leading to a short, female-specific version of the Dsx protein. In male embryos, which produce no Tra protein, exon 4 is skipped, so that exon 3 is spliced to exon 5. Exon 5 is constitutively spliced to exon 6, which is polyadenylated at its end — leading to a longer, male-specific version of the Dsx protein. The RNA sequence to which Tra binds in exon 4 is called an exonic splicing enhancer since it enhances splicing at a nearby splice site. As a result of this cascade of regulated RNA processing depicted in Figure 9-18, different Dsx proteins are expressed in male and female embryos. The male Dsx protein is a transcriptional repressor that inhibits the expression of genes required for female development. Conversely, the female Dsx protein represses transcription of genes required for male development. In wild-type Drosophila, no Sxl protein is expressed in cells of male embryos, whereas it is expressed in female embryos. This is an example of “on-off switch” regulation. The initial expression of Sxl in early female embryos positively regulates its own expression. Other examples of nuclear RNA-binding proteins that generate switch-like control of alternative RNA splicing in neurons have been identified and termed master splicing factors.
Figure 9-19 illustrates how the Tra/Tra2/Rbp1 complex is thought to interact with double-sex (dsx) pre-mRNA. Rbp1 and Tra2 are SR proteins, but they do not interact with exon 4 in the absence of the Tra protein. Tra protein interacts with Rbp1 and Tra2, resulting in the cooperative binding of all three proteins to six exonic splicing enhancers in exon 4. The bound
Tra2 and Rbp1 proteins then promote the binding of U2AF and U2 snRNP to the end of the intron between exons 3 and 4 (see Figure 9-17). The Tra/Tra2/Rbp1 complexes may also enhance binding of the cleavage/polyadenylation complex (Figure 9-15) to the end of exon 4.
FIGURE 9-19 Model of splicing activation by Tra protein and the SR proteins Rbp1 and Tra2. In female Drosophila embryos, splicing of exons 3 and 4 in dsx pre-mRNA is activated by binding of Tra/Tra2/Rbp1 complexes to six sites in exon 4. Because Rbp1 and Tra2 cannot bind to the pre-mRNA in the absence of Tra, exon 4 is skipped in male embryos. See the text for discussion. represents polyadenylation. See T. Maniatis and B. Tasic, 2002, Nature 418:236. Description The pre-m R N A from 5-prime to 3-prime end shows: exon 1, intron, exon 2, intron, exon 3, intron, exon 4 with poly (A) tail, intron, exon 5, intron, and exon 6 with poly (A) tail. In females, the m R N A comprise exon 3 and 4 and in males, the m R N A comprise exon 3 and 5. The exon 4 shows six T r a slash T r a 2 slash R b p 1 complexes. Each complex shows a T r a globular head bound to an R b p 1 and T r a 2 tail.
Expression of Dscam Isoforms in Drosophila Retinal Neurons
Splicing Repressors and Activators Control Splicing at Alternative Sites As is evident from Figure 9-18, the Drosophila Sxl protein and Tra protein have opposite effects: Sxl prevents splicing, causing exons to be skipped, whereas Tra promotes splicing. The action of similar proteins may explain the cell-type-specific expression of fibronectin isoforms in humans. For instance, an Sxl-like splicing repressor expressed in hepatocytes might bind to splice sites for the EIIIA and EIIIB exons in the fibronectin premRNA, causing them to be skipped during RNA splicing (Figure 5-28). Alternatively, a Tra-like splicing activator expressed in fibroblasts might activate the splice sites associated with the fibronectin EIIIA and EIIIB exons, leading to inclusion of these exons in the mature mRNA. Experimental examination in some systems has revealed that inclusion of an exon in some cell types versus skipping of the same exon in other cell types results from the combined influence of several splicing repressors and enhancers. RNA binding sites for repressors can also occur in exons, where they are called exonic splicing silencers. And binding sites for splicing activators, usually SR proteins, can also occur in introns, where they are called intronic splicing enhancers. Expression of Dscam Isoforms in Drosophila Retinal Neurons
The most extreme example of regulated alternative RNA processing yet uncovered occurs in expression of the Dscam gene in Drosophila. Mutations in this gene interfere with the normal synaptic connections made between axons and dendrites during fly development. Analysis of the Dscam gene showed that it contains 95 alternatively spliced exons that could be spliced to generate 38,016 possible isoforms (Figure 9-20)! One of 12 possible exons encoding domain Ig2 of the protein is included in each Dscam mRNA. One of 48 possible exons encoding domain Ig3 is included in each Dscam mRNA, and one of 33 possible exons encoding domain Ig7 and one of 2 possible exons encoding the Dscam transmembrane domain are included in each Dscam RNA. Exons diagrammed at the top as vertical black lines are constitutively spliced into all Dscam mRNAs. As a result, the Dscam pre-mRNA is processed into one of possible isoforms. Drosophila mutants with a version of the gene that can be spliced in only about 22,000 different ways have specific defects in connectivity between neurons. These results indicate that expression of most of the possible Dscam isoforms through regulated RNA splicing helps to specify the tens of millions of different specific synaptic connections between neurons in the Drosophila brain. In other words, the correct wiring of neurons in the brain requires regulated RNA splicing.
FIGURE 9-20 The Drosophila Dscam gene is processed into a vast number of alternative isoforms. Dscam encodes a cell surface protein on neurons. The protein (bottom) is composed of 10 different immunoglobulin domains (ovals), six different fibronectin type III domains (rectangles), one transmembrane domain (yellow), and a C-terminal cytoplasmic domain. One of 12 possible Ig2 domains, one of 48 possible Ig3 domains, one of 33 possible Ig7 domains, and one of two possible transmembrane domains, each encoded by one exon, are included in the protein because of the alternative RNA splicing diagrammed at the top. The fully processed mRNA contains one of the alternative exons encoding an Ig2 domain diagrammed in light blue in genomic DNA, one of the alternative exons encoding Ig3 shown in green in genomic DNA, and one of the exons shown in yellow. The exons shown in black are constitutively spliced into each of the messages, generating possible isoforms. See M. R. Sawaya et al., 2008, Cell 134:1007. Description The genomic D N A shows a double stranded D N A with the following vertical lines on them: three black, 12 light blue, 1 black, 48 green, two black, 33 dark blue, seven black, two yellow, and seven black. A dashed line at the bottom connects the three black, first light blue, 1 black, mid green, two black, almost last dark blue, seven black, first yellow, and seven black lines. A horizontal line at the top from starting of light blue to end of yellow indicates number of alternatives.
Abnormal RNA Splicing and Disease
This leads to a capped m R N A made of different sized rectangular blocks constituting one of light blue, green, dark blue, and yellow. This further leads to a protein made of nine oval structures connected to four squares, then one oval, and four more squares. The second oval structure is light blue I g 2, third is green I g 3, seventh is dark blue I g 7, and the second last square is yellow T M. The starting oval chain is labeled, homophilic binding region. Abnormal RNA Splicing and Disease Multiple examples of human diseases result from abnormalities in RNA splicing. Here we look at examples of mutations that affect exon definition and defects in microexon splicing. Mutations Affecting Exon Definition Cause Disease Mutations that affect exon definition are responsible for approximately 15 percent of the single-base-pair mutations that cause human genetic diseases. Some single-base-pair mutations occur in or splice sites, often resulting in the use of nearby alternative cryptic splice sites present in the normal gene sequence. In the absence of the normal splice site, the cross-exon recognition complex (Figure 9-16) recognizes these alternative sites, which generally are a poorer match to consensus splice-site sequences than the splice site in the wild-type gene that was mutated. Other mutations that cause abnormal splicing result in a new - or - consensus sequence in a pre-mRNA that becomes recognized in place of the normal splice site. This generally results in processing of an abnormal
mRNA that does not encode a functional protein. Finally, some mutations can interfere with binding of a pre-mRNA to a specific SR protein. These mutations interfere with the assembly of a cross-exon recognition complex on the mutated exon, and thus lead to exon skipping. Understanding the mechanisms of exon definition has led to strategies to treat Duschenne Muscular Dystrophy (DMD). Mutations in the sex-linked DMD gene encoded on the X-chromosome are responsible for muscular dystrophy in approximately one in 5000 males, making it one of the most common human genetic diseases. The gene encodes dystrophin, a protein that participates in linking the actin-cytoskeleton to the muscle cell plasma membrane (see Figure 20-39). When dystrophin is defective, the connection is weakened and muscle cell contraction by movement of actin fibers damages the plasma membrane leading to apoptosis in the mutant muscle cells. DMD mutations in many patients result from a frame shift or stop codon in a DMD exon that evolved from exon duplication (e.g., the tan-colored exons in the fibronectin gene in
Figure 5-28). Synthetic oligonucleotides that hybridize to the splice site of the exon with the mutation prevent U1 snRNP from binding to the splice site and cause skipping of the mutant exon and in-frame splicing of the preceding exon to the exon following the mutant exon. The resulting mRNA lacks any chain terminating mutations. It encodes a protein lacking one copy of the repeated protein domain, but that protein functions almost as well as the wild-type protein. Current work to develop more efficient membrane-permeant synthetic oligonucleotide derivatives and delivery
methods are an active area of research for treatment of DMD due to mutations in repeated exons arising from exon duplication. Spinal Muscular Atrophy is one of the most common genetic causes of childhood mortality. This disease results from mutations in a region of the genome containing two closely related genes, SMN1 and SMN2, that arose by gene duplication during human evolution. SMN2 encodes a protein identical with SMN1; however, correctly spliced SMN2 mRNA is expressed at a much lower level than SMN1 mRNA because of a mutation in an exonic splicing enhancer in the duplicated SMN2 gene. This mutation resulted in a synonymous codon that does not change the encoded amino acid sequence but does inhibit binding of an SR protein to this region of the SMN2 pre-mRNA. This leads to skipping of the exon during processing of most SMN2 primary transcripts because a crossexon-recognition complex (Figure 9-16) cannot assemble across the affected exon, and consequently cannot stimulate association of U1 snRNP and U2 snRNP with and splice sites for this exon in the SMN2 premRNA. This has little consequence in unaffected individuals because most Smn protein is expressed from the SMN1 gene. Genetic studies in the mouse indicate that expression of some Smn protein is required for normal mammalian embryonic development. In the mouse, there is only one Smn gene. Mouse embryos with inactivating mutations in both the maternal and paternal copies of the Smn gene die early during embryogenesis. However, heterozygous mutants that retain one functional copy of the Smn gene develop normally because the lower level of Smn
protein expressed from the single copy of the functional gene is sufficient for viability. In other words, inactivating mutations in the mouse Smn gene are recessive, as is the case for the majority of protein-coding genes. The human disease spinal muscular atrophy results when a patient inherits inactivating mutations in both the maternal and paternal alleles of SMN1, the gene that expresses most Smn protein. In these patients, the low level of Smn protein translated from the small fraction of SMN2 mRNA that is correctly spliced is sufficient to maintain cell viability during embryogenesis and fetal development. However, it is not sufficient to maintain viability of spinal cord motor neurons in childhood, resulting in their death and the associated disease. Microexons and Their Association with Autism Spectrum Disorder Several types of alternative pre-mRNA splicing have been observed in vertebrate cells (Figure 9-21a). The most common and extensively conserved type of alternatively spliced exons in the central nervous system (CNS) are called microexons because they are unusually short: 3 to 27 bases long in multiples of three so that they maintain the reading frames of up- and downstream exons of more typical longer lengths (average bases in humans). The discovery of these microexons was surprising because their very short length was expected to prevent the previously understood standard cross-exon recognition complex interactions needed for exon definition (Figure 9-16). The short stretches of amino acids encoded by microexons (1–9 amino acids) generally reside on protein
surfaces that participate in protein-protein interactions, and are significantly enriched in genes with critical functions in synaptic biology (Figure 9-21b). Of critical importance here, of patients with autism spectrum disorder (ASD) exhibit abnormally low levels of microexon splicing in the CNS.
FIGURE 9-21 Splicing changes in autism spectrum disorder (ASD). (a) Types of alternative splicing. Thin black lines indicate splicing between the indicated splice sites. RNA sequencing analyses (RNA-seq) revealed that cassette exons and microexons (3–27 nucleotides) are much more common forms of alternative RNA splicing in the normal human central nervous system than alternative and splice sites, retained introns, or complex events. In one-third of patients with autism spectrum disorder (ASD), more skipping of cassette exons and microexons are observed than in normals. Abnormalities in the frequency of splicing of alternative and splice sites are observed only rarely in ASD. Complex splicing events, while present, occur at too low an incidence to determine if their incidence is altered significantly in ASD patients. (b) Genes containing microexons
with abnormal inclusion in ASD patients are listed in the context of molecular hubs of ASD pathogenesis (colored boxes) or their functions in neuronal biology (grey box). [Data from M. Quesnel-Vallières et al., 2019, Nat. Rev. Genet. 20:51.] Description The part (a) shows types of splicing namely alternative cassette exon, microexons, alternative 5-prime and 3-prime sites, retained intron, and complex events. Alternative cassette exon shows exon 1, intron, exon 2, intron, and exon 3 which results into controls with exons 1, 2, and 3; and A S D with exons 1 and 3. Microexons shows exon 1, intron, exon mu, intron, and exon 3 which results into controls with exons 1, mu, and 3; and A S D with exons 1 and 3. Alternative 5-prime and 3-prime sites shows splicing of either the introns or the entire segment between exon 1 and 3. Retained intron shows a thick intron between exon 1 and 2 and splicing between exon 2 and 3. Complex events shows a thick intron between exon 1 and 2 and splicing of either the introns or the entire segment between exon 1 and 3 or some part of exon 3. Four tables in (b) list synaptic function, W N T signaling, translation, and function of genes. The first table lists synaptic function as follows: A G R N, A N K 2, C A D M 2, C A D P S, C A S K, D T N A, E N A H, G O P C, M I N K 1, P R K D 1, P T P R D, P T P R S, R E L N, S H A N K 2, T R I M 9, and U N C 13 B. The second table lists W N T signaling as follows: A R V C F, M A C F 1, M I N K 1, P T K 2, S L I T 2, S P O C K 1, and V A V 2. The third table lists translation as follows: C P E B 4, E I F 4 G 3, and E P R S. The fourth table lists gene and their function as follows: T A F 1, axon growth; N A V 2, axon growth; N R C A M, axon growth; R O B O 1, axon growth; N C K A P 1, neuronal migration; S L C 38 A 10, neurotransmitter transport; and T R A P P C 9, neronal differentiation. Further research on the mechanism underlying this defect in microexon splicing focused on a particular, neuron-specific SR-protein, SRRM4. Human SRRM4 mRNA was expressed at low level compared to normal in the same of ASD patients with abnormally low levels of included microexons. Further, the number of microexons that were skipped in these
patients correlated with the severity of the decrease in SRRM4 expression. The importance of SRRM4 in some forms of ASD was further supported by genetic experiments in mice. Homozygous knock-out of the Srrm4 gene in mice is lethal in most pups due to postnatal defects in respiration linked to abnormal innervation of the diaphragm and other neurological defects. Heterozygous Srrm4 +/- mice are viable, but they express approximately one-half the level of Srrm4 mRNA than Srrm4 +/+ littermates. This is associated with a significant decrease in microexon inclusion in the CNS and with multiple autistic-like features, including altered social behaviors, altered synaptic density, and signaling. These results provide evidence that misregulation of human SRRM4 is causally linked to a substantial fraction of autism cases. Mouse Srrm4 protein has been found to stimulate the inclusion of microexons by binding to RNA sequences found upstream from microexons. From this position, Srrm4 is able to stimulate snRNP U1 association with the microexon splice site in much the same way that SR-proteins bound to exonic splicing enhancers at the end of an exon stimulate snRNP U1 association with splice sites in exons of typical length (see Figure 9-16). Once U1 snRNP is associated with the microexon splice site, it can interact with U2 snRNP associated with the branch point preceding the next splice site in the pre-mRNA and participate in a splicing reaction that joins the microexon to the downstream exon. Myelodysplastic syndromes (MDS) are a group of blood cell diseases characterized by deregulated production of dysplastic (abnormal)
myelocytes (phagocytic white blood cells). A high percentage of MDS patients progress to acute myeloid leukemia (AML), which is terminal if not controlled. Remarkably, sequencing of all exons (the exome) from MDS patients revealed that mutations in genes encoding a few specific RNA splicing factors occur in percent of all MDS patients. The vast majority of these mutations occur in genes for the splicing factors U2AF35, SRSF2, ZRSR2, or SF3B1 (Figure 9-22). U2AF35 is the small subunit of the heterodimeric U2AF splicing factor required for association of the U2 snRNP with the branch-point A near the end of an intron (Figure 9-13). SRSF2 and ZRSR2 are SR proteins that associate with exonic splicing enhancers in many genes. SF3B1 is a protein component of human U2 snRNP. Specific mutations in other splicing factors (U2AF65, SF1, SF3A1 and PRPF40B) also occur in MDS patients, but less frequently. U2AF65 is the large subunit of U2AF that associates with the pyrimidine tract just upstream of the splice site. SF1 is required along with U2AF for association of U2 snRNP with the branchpoint A. SF3A1 is another protein component of human U2 snRNP. PRP40B is an essential splicing factor, but its precise function is not yet understood.
FIGURE 9-22 Specific amino acid changes in some proteins required for U2 snRNP binding to the branch-point A are associated with myelodysplastic syndrome and some leukemias. Spliceosome components mutated in myelodysplasic syndromes, related diseases causing reduced cell replication and abnormal development of myelocytes (phagocytic white blood cells). RNA splicing is initiated by the recruitment of U1 snRNP to a splice site. SF1 binds the branch-point sequence, and the larger subunit of the U2 auxiliary factor (U2AF65) binds the downstream polypyrimidine tract. The smaller subunit of U2AF (U2AF35) binds to the AG dinucleotide of the splice site. U2 snRNP association with the branch-point A also requires two protein splicing factors, SF3A1 and SF3B1. U2AF interactions with an SR-protein bound near the end of the downstream exon are also required (see Figure 9-15). SRSF2 and ZRSR2 are SR-proteins that fulfill this function at a large number of splice sites. Specific amino acid changes in one of these splicing factors indicated by a lightning bolt is found in the abnormal myelocytes in 60–70 percent of patients with myelodysplastic syndrome. These mutations lead to abnormal inclusion of alternatively spliced exons and introns in a large number of pre-mRNAs. The mutations are lethal when homozygous, but somehow interfere with normal replication and development when half of the splicing factors have these specific amino acid changes in heterozygous cells. See text. [Data from K. Yoshida et al., 2011, Nature 478:64.]
Description A bent pre-m R N A shows following regions from 5-prime to 3-prime end: E S E sequence; U 1 s n R N P bound to 5-prime splice site; dashed line; S F 1 bound to Y N C U R A Y sequence; three U 2 A F 65 bound to intron; U 2 A F 35 bound to 3-prime splice site; S R S F 2 bound to E S E sequence; and U 1 s n R N P bound to the 3-prime end. An arrow from the structure of Z R S R 2 points to U 2 A F 35 and an arrow from the structure of U 2 s n R N P bound to S F 3 A 1 and S F 3 B 1 points to third U 2 A F 65. Several wiggly red arrows point to S F 1, second U 2 A F 65, U 2 A F 35, S R S F 2, Z R S R 2, S F 3 A 1, and S F 3 B 1. Only one of these mutations in a splicing factor gene is observed in the abnormal myeloctyes from any one MDS patient, and the MDS cells always retain a wild-type allele of the mutant gene. That is, the myelodysplastic cells are always heterozygous for the splicing factor mutation. Most remarkable of all: the same specific mutations recur in different patients. For example, all MDS-associated mutations in SRSF2 change proline 95 into another amino acid, either histidine, leucine, or arginine. MDS-associated mutations in the small subunit of U2AF, which interacts with the AG at the end of most introns, either change serine 34 to a phenylalanine or a tyrosine, or change glutamine 157 to an arginine or a proline. Genetic techniques described in Chapter 6 permitted researchers to substitute MDS-associated mutations for the wild-type splicing factor genes in cultured cells. Cells with homozygous MDS-associated mutations were not viable, indicating that co-expression of the wild-type splicing factor is essential. This is presumably to generate a sufficient number of normally spliced mRNAs for the heterozygous cells to survive. Analysis
Self-Splicing Group II Introns Provide Clues to the Evolution of snRNAs
of RNA splicing in the heterozygous cells showed global abnormalities, as in the abnormal myeloid cells from MDS-patients. The very specific mutations recovered from MDS patients appear to generate a change of splicing factor function rather than loss of function. For example, loss of SRSF2 results in failure of cassette exon splicing (Figure 9-22), while expression of MDS-associated SRSF2 mutants result in a change in cassette exon splicing in a sequence-specific manner based on the sequence of exonic splicing enhancer (ESE) motifs present in the abnormally spliced exon. In contrast, mutations in U2AF1, which interacts with the pyrimidine tract upstream from the splice site, alter splicing by promoting the recognition of abnormal splice sites. Mutations in SF3B1 promote usage of cryptic splice sites. How these abnormalities in pre-mRNA splicing influence the phenotype of myelodysplastic cells is not known. In particular, it is not known if the abnormal phenotype is due to (1) expression of one or a few abnormally spliced mRNAs, influencing the function of one or a small number of proteins that regulate myeloid blood cell replication and differentiation, or (2) whether the phenotype results from the pleiotropic effects of abnormal expression of a large number of protein isoforms. Self-Splicing Group II Introns Provide Clues to the Evolution of snRNAs Under certain nonphysiological in vitro conditions, pure preparations of some RNA transcripts slowly splice out introns in the absence of any
protein. This observation led to the recognition that some introns are selfsplicing. Two types of self-splicing introns have been discovered: group I introns, present in nuclear rRNA genes of protozoans, and group II introns, present in protein-coding genes and some rRNA and tRNA genes in mitochondria and chloroplasts of plants and fungi. Discovery of the catalytic activity of self-splicing introns revolutionized concepts about the functions of RNA. As discussed in Chapter 4, RNA is now known to catalyze peptide-bond formation during protein synthesis in ribosomes. Here we discuss the probable role of group II introns, now found only in mitochondrial and chloroplast DNA, in the evolution of snRNAs; the functioning of group I introns is considered in the later section on rRNA processing. Even though their precise sequences are not highly conserved, all group II introns fold into a conserved, complex secondary structure containing numerous stem loops (Figure 9-23a). Self-splicing by a group II intron occurs via two transesterification reactions, involving intermediates and products analogous to those found in nuclear pre-mRNA splicing. The mechanistic similarities between group II intron self-splicing and spliceosomal splicing led to the hypothesis that snRNAs function analogously to the stem loops in the secondary structure of group II introns. According to this hypothesis, snRNAs interact with and splice sites of pre-mRNAs and with each other to produce a threedimensional RNA structure functionally analogous to that of group II selfsplicing introns (Figure 9-23b).
FIGURE 9-23 Comparison of group II self-splicing introns and the spliceosome. The schematic diagrams compare the secondary structures of (a) group II self-splicing introns and (b) U snRNAs present in the spliceosome. The first transesterification reaction is indicated by light green arrows; the second reaction, by blue arrows. The branch-point A is boldfaced. The similarity in these structures suggests that the spliceosomal snRNAs evolved from group II introns, with the trans-acting snRNAs being functionally analogous to the corresponding domains in group II introns. The colored bars flanking the introns in (a) and (b) represent exons. See P. A. Sharp, 1991, Science 254:663. Description The structure of (a) group 2 intron shows a rounded structure with six stem-loop branches, labeled 1 through 6, from 5-prime to 3-prime end. Both 5-prime to 3-prime ends show exons and stem-loop 6 shows branch point A. An arrow points from branch point A to 5-prime splice site and another from 5-prime splice site to 3-prime splice site. The structure of (b) U s n R N As in spliceosome shows a rounded structure with stemloop branches made of U 1, U 5, U 4, U 6, and U 2 from 5-prime to 3-prime end. Both 5-prime to 3-prime ends show exons and stem-loop U 2 shows branch point A. An arrow points from branch point A to 5-prime splice site and another from 5-prime splice site to 3-prime splice site. A thread-like pre-m R N A intron in opposite side of
the rounded structure starts from 5-prime splice site, connecting U 1 to branch point A of U 2, and ending at 3-prime splice site. An extension of this hypothesis is that introns in ancient pre-mRNAs evolved from group II self-splicing introns through the progressive loss of internal RNA structures, which concurrently evolved into trans-acting snRNAs that perform the same functions. Support for this type of evolutionary model comes from experiments with group II intron mutants in which domain V and part of domain I are deleted (Figure 9-23a). RNA transcripts containing such mutant introns are defective in self-splicing, but when RNA molecules equivalent to the deleted regions are added to the in vitro reaction, self-splicing occurs. This finding demonstrates that these domains in group II introns can be trans-acting, like snRNAs. Further evidence that spliceosomal splicing evolved from group II introns comes from recent cryo-EM structures of group II introns showing that the RNA interactions that occur during group II intron splicing are very similar to the interactions at the spliceosome active site (see Figure 9-14b, c). Although group II introns can self-splice in vitro at elevated temperatures and concentrations, under in vivo conditions proteins called maturases, which bind to group II intron RNA, are required for rapid splicing. Maturases are thought to stabilize the precise threedimensional interactions of the intron RNA required to catalyze the two splicing transesterification reactions. By analogy, we can now see from the cryo-EM structures of catalytically active spliceosomes (see Figure 9-11) that proteins associated with spliceosomes also stabilize the precise
Nuclear Exonucleases and the Exosome Degrade RNA That Is Processed out of Pre-mRNAs
geometry of snRNAs and intron nucleotides required to catalyze premRNA splicing. The evolution of snRNAs may have been an important step in the rapid evolution of multicellular eukaryotes. As internal intron sequences were lost and their functions in RNA splicing supplanted by trans-acting snRNAs, the remaining intron sequences would be free to diverge. This in turn likely facilitated the evolution of new genes through exon shuffling since there are few constraints on the sequence of new introns generated in the process (see Figures 7-18 and 7-19). It also permitted the increase in protein diversity that results from alternative RNA splicing and an additional level of gene control resulting from regulation of alternative RNA splicing. Nuclear Exonucleases and the Exosome Degrade RNA That Is Processed out of Pre-mRNAs Because the human genome contains long introns, only percent of the nucleotides that are polymerized by RNA polymerase II during transcription are retained in mature, processed mRNAs. Although this process appears inefficient, it probably evolved in multicellular organisms because the process of exon shuffling facilitated the evolution of new genes in organisms with long introns (Chapter 7). The introns that are spliced out and the region downstream from the cleavage and
polyadenylation site are degraded by nuclear exoribonucleases that hydrolyze one base at a time from either the or end of an RNA strand. As mentioned earlier, the -phosphodiester bond in excised introns (see
Figure 9-13, ILS) is hydrolyzed by a debranching enzyme, yielding a linear molecule with unprotected ends that can be attacked by exonucleases. The predominant nuclear decay pathway for RNAs removed from premRNAs by RNA splicing, and the transcribed RNA downstream from a cleavage/polyadenylation site, is hydrolysis by a large protein complex called the exosome (Figure 9-24). The exosome consists of a nine-subunit barrel referred to as Exo-9, comprised of nine similar polypeptides (gray in Figure 9-24). Although these have sequences homologous to bacterial RNA phosphorylases, the Exo-9 barrel has no ribonuclease activity. Rather, it forms a channel through which the RNA substrate is threaded end first to finally reach the tenth subunit in the exosome, RRP44, which has the only ribonuclease activity of any subunit of the complex. The major site of RNA digestion is in the exonuclease active site at the end of the channel (Figure 9-24b, marked “Exo.”) Since this active site is within the exosome, RNAs outside of the exosome are protected from digestion by the highly active RRP44 exonuclease. The long channel through the large exosome is thought to hold the RNA in optimal conformation for removal of one nucleotide at a time by the rapid exonuclease activity of the complex, without releasing the substrate RNA. This results in what is known as processive degradation; the RNA
substrate is digested completely (to a tetranucleotide) without being released from the enzyme.
FIGURE 9-24 Structure of the exosome. (a) Structure of the 420 kDa exosome core complex, termed Exo 10. It consists of the catalytically inactive Exo 9 complex composed of nine closely related subunits (gray) and the S1 and KH ring (orange). The Exo 9 complex associates with RRP44 (violet), which contains sites of endonuclease (endo) and exonuclease (exo) activity. (b) Schematic representation showing the multisubunit exosome lid, which includes RNA helicases that push RNA substrates through a narrow channel in the lid (blue) that continues into the Exo 10 complex until the end of the substrate RNA reaches the processive catalytic exonuclease site in RRP44. [Data from D. Makino, F. Halbach, and E. Conti, 2013, Nat. Rev. Mol. Cell Biol. 14:655.]
Description The three-dimensional model (a) shows a structure with A T Pase base at the top, Exo 9 in the middle, and R R P 44 at the bottom. A thread-like R N A starts from top A T Pase base, passes through Exo 9, and ends in the mid of R R P 44. The illustration (b) shows substrate unwinding by R N A helicases. The structure shows a regulatory lid at the top followed by A T Pase base, Exo 9, and R R P 44 at the bottom. A funnel-shaped R N A helicase with its opening in the regulatory lid ends at the bottom of A T Pase base and is continued as an irregular shaped structure to end in the mid of R R P 44 and is labeled Exo. A small opening in the R R P 44 is labeled Endo. A thread-like R N A substrate starts from the top outside and passes through the R N A helicase to end at Exo. In addition to an exonuclease site, RRP44 has an endonuclease site on its surface. This is thought to be utilized when RNA degradation at the RRP44 exonuclease site is inhibited because of a highly stable RNA structure or chemical damage such as oxidation at the end of the RNA. In this case, the RRP44 endonuclease site can cleave the substrate RNA to the block to exonuclease digestion when the blocked substrate RNA diffuses out of the exosome channel. This generates a new end susceptible to rapid digestion that can be threaded into the exosome, resulting in complete digestion of the RNA. Since the exonuclease active site is located at the end of a long tunnel in the exosome with dimensions just adequate to accommodate an extended RNA chain, the highly active enzymatic activity does not have access to other nuclear or cytoplasmic RNAs. Rather, substrates must be recognized and loaded into the pore at the top of the exosome before RNA helicases in the lid push the end of the RNA down the channel until it reaches the exonuclease active site in RRP44 (Figure 9-24b). This regulatory lid on
nuclear exosomes is called the TRAMP complex and contains the RNA helicase, Mtr4. Exosomes in the cytoplasm are associated with a regulatory lid called the SKI complex containing the Ski2 RNA helicase. Note that basic design principles of the exosome are similar to those of the major protein degrading multiprotein complex in eukaryotic cells, the proteasome (see Figure 3-32). In both degradative complexes, degradation takes place inside a protein barrel that excludes other potential cellular substrates. The selection of substrates to be rapidly degraded is made by proteins in a regulatory cap structure that binds one end of the substrate polymer and then, using energy from ATP hydrolysis, pushes it into the digestive cavity as an extended polymer highly susceptible to the degradative enzymes. In addition to degrading introns, nuclear exosomes appear to degrade premRNAs that have not been properly spliced or polyadenylated, thus providing a quality-control mechanism. It is not yet clear how the exosome recognizes improperly processed pre-mRNAs. But in yeast cells with temperature-sensitive mutant poly(A) polymerase (see Figure 9-15), pre-mRNAs are retained at their sites of transcription in the nucleus at the nonpermissive temperature. These abnormally processed pre-mRNAs are released in cells with a second mutation in a subunit of the exosome found only in nuclear and not in cytoplasmic exosomes (EXOSC10; 100 kD in humans). Also, exosomes are found concentrated at sites of transcription in Drosophila polytene chromosomes, where they are associated with RNA polymerase II elongation factors. These results suggest that the exosome participates in an as yet poorly understood quality-control
RNA Processing Solves the Problem of Pervasive Transcription of the Genome in Mammalian Cells
mechanism that recognizes aberrantly processed pre-mRNAs, preventing their export to the cytoplasm and ultimately leading to their degradation. To avoid being degraded by nuclear exonucleases, nascent transcripts, premRNA processing intermediates, and mature mRNAs in the nucleus must have their ends protected. As discussed above, the end of a nascent transcript is protected by addition of the cap structure as soon as the end emerges from the polymerase. The cap is protected because it is bound by a nuclear cap-binding complex, which protects it from exonucleases and also functions in export of mRNA to the cytoplasm. The end of a nascent transcript lies within the RNA polymerase and thus is inaccessible to exonucleases (see Figure 8-9b). As discussed previously, the free end generated by cleavage of a pre-mRNA downstream from the poly(A) signal is rapidly polyadenylated by the poly(A) polymerase in a 900 kDa cleavage and polyadenylation complex, and the resulting poly(A) tail is bound by PABPN1 (see Figure 9-15). This tight coupling of cleavage and polyadenylation protects the end from exonuclease attack. RNA Processing Solves the Problem of Pervasive Transcription of the Genome in Mammalian Cells As discussed in Chapter 8, analysis of the location of transcribing RNA polymerase II in metazoan cells revealed the surprising result that the polymerases transcribe in both the downstream direction into coding regions and in the upstream direction away from coding regions at nearly
equal frequency from most promoters. This finding was also confirmed by deep sequencing of small RNAs isolated from mammalian cells that revealed low levels of short, capped RNAs transcribed from both the sense and antisense strands at CpG-island promoters ( percent of mammalian promoters). Indeed, deep sequencing of all cellular RNAs showed that both strands of nearly the entire genome are transcribed, although much of this RNA is present at extremely low concentrations of less than one molecule per cell. This raised the question of how the cell deals with such pervasive transcription of the genome. Sequence analysis of these low-abundance, short RNAs indicates that they are likely prevented from reaching high concentrations by RNA processing and nuclear surveillance of abnormally processed RNAs. Sequencing of RNAs from several cell types revealed that the antisense RNAs have a higher frequency of AAUAAA polyadenylation site sequences than transcripts transcribed in the sense direction into gene coding regions. This is probably because the high AT composition of mammalian DNA ( percent in humans) results in occasional transcription of AAUAAA in antisense transcripts ( percent of all six base sequences with an average of 60 percent AT). However, AAUAAA sequences in proteincoding genes occur at much lower frequency because the genes have evolved to have one or a small number of poly(A) sites. Consequently, the enrichment of poly(A) sites in antisense transcripts leads to cleavage of most antisense transcripts within a few kb of their transcription start sites by cleavage/polyadenylation factors, followed by termination of transcription. Most cleaved antisense transcripts are degraded by nuclear exosomes; evidence for this comes from experiments in which
RNA Editing Alters the Sequences of Some Pre-mRNAs
temperature-sensitive mutations in exosome subunits cause a rapid increase in short antisense promoter-proximal RNAs when the mutant cells are shifted to the nonpermissive temperature. As a result, even though a large number of RNA polymerase II molecules transcribe in the “wrong” direction, most of the transcripts generated are degraded before they reach a length of a few kb. RNA Editing Alters the Sequences of Some Pre-mRNAs In the mid-1980s, sequencing of numerous cDNA clones and corresponding genomic DNAs from multiple organisms led to the unexpected discovery of another type of pre-mRNA processing. In this type of processing, called RNA editing, the sequence of a pre-mRNA is altered; as a result, the sequence of the corresponding mature mRNA differs from the exons encoding it in genomic DNA. RNA editing is widespread in the mitochondria of protozoans and plants and also in chloroplasts. In the mitochondria of certain pathogenic trypanosomes, more than half the sequence of some mRNAs is altered from the sequence of the corresponding primary transcripts. Additions and deletions of specific numbers of U’s follow templates provided by basepaired short “guide” RNAs. These RNAs are encoded by thousands of mini-mitochondrial DNA circles catenated to many fewer large mitochondrial DNA molecules. The reason for this baroque mechanism for encoding mitochondrial proteins in such protozoans is not clear. But this
system does represent a potential target for drugs to inhibit the complex processing enzymes essential to the microbe that do not exist in the cells of their human or other vertebrate hosts. In multicellular eukaryotes, RNA editing is much rarer, and, thus far, only single-base changes have been observed in mammals. Such minor editing, however, turns out to have significant functional consequences in some cases. An important example of RNA editing in mammals involves the apoB gene. This gene encodes two alternative forms of a serum protein central to the uptake and transport of cholesterol. Consequently, it is important in the pathogenic processes that lead to atherosclerosis, the arterial disease that is the major cause of death in the developed world. The apoB gene expresses both the serum protein apolipoprotein B-100 (apoB-100) in hepatocytes, the major cell type in the liver, and apoB-48, expressed in intestinal epithelial cells. The apoB-48 corresponds to the N-terminal region of the apoB-100. As we detail in Chapter 14, both apoB proteins are components of large lipoprotein complexes that transport lipids in the serum. However, only low-density lipoprotein (LDL) complexes, which contain apoB-100 on their surface, deliver cholesterol to body tissues by binding to the LDL receptor present on all cells. The cell-type-specific expression of the two forms of apoB results from editing of apoB pre-mRNA so as to change the nucleotide at position 6666 in the sequence from a C to a U. This alteration, which occurs only in intestinal cells, converts a CAA codon for glutamine to a UAA stop codon, leading to synthesis of the shorter apoB-48 (Figure 9-25). Studies with the
partially purified enzyme that performs the post-transcriptional deamination of to U shows that it can recognize and edit an RNA as short as 26 nucleotides with the sequence surrounding in the apoB primary transcript, but it does not deaminate C’s in RNAs with other sequences.
FIGURE 9-25 RNA editing of apoB pre-mRNA. The apoB mRNA produced in the liver has the same sequence as the exons in the primary transcript. This mRNA is translated into apoB-100, which has two functional domains: an N-terminal domain (green) that associates with lipids and a C-terminal domain (orange) that binds to LDL receptors on cell membranes. In the apo-B mRNA produced in the intestine, the CAA codon in exon 26 is edited to a UAA stop codon. As a result, intestinal cells produce apoB-48, which corresponds to the N-terminal domain of apoB-100. See P. Hodges and J. Scott, 1992, Trends Biochem. Sci. 17:77. Description The series starts with a A P O B gene constituting 29 exons with a large exon 26 having C A A codon and a small exon 29 with T A A codon toward the 3-prime end. The gene transcribes into different types of A P O B m R N A in liver and intestine. The A P O B m R N A in liver is made of 5-prime end, C A A codon in the mid, U A A codon toward 3-prime end, and a poly (A) tail. The A P O B m R N A in intestine is made of 5-prime end, C A A to U A A codon in the mid, U A A codon toward 3-prime end, and a poly (A) tail. The A P O B m R N A in liver translates into a p o B-100 protein made of 4536 residues with one end labeled N H 2 and other end labeled C O O H. The half
side toward N H 2 is green and toward C O O H is orange. The A P O B m R N A in intestine translates into green a p o B-48 protein made of 2152 residues with one end labeled N H 2 and other end labeled C O O H. KEY CONCEPTS OF SECTION 9.2 Regulation of Pre-mRNA Processing Because of alternative splicing of primary transcripts, the use of alternative promoters, and cleavage at different poly(A) sites, different mRNAs may be expressed from the same gene in different cell types or at different developmental stages (see Figure 918). This vastly expands the number of distinct proteins encoded in the genomes of multicellular animals and plants compared to eukaryotes that exhibit little alternative RNA splicing such as Saccharomyces cerevisiae. SR proteins that bind to exonic splicing enhancer sequences present in some exons are critical for defining the splice sites of these exons in the large pre-mRNAs of vertebrates. A network of interactions between SR proteins, snRNPs, and splicing factors forms a cross-exon recognition complex over the short vertebrate exon that specifies correct localization of splice sites (see Figure 9-16). Alternative splicing can be regulated by RNA-binding proteins that bind to specific sequences near regulated splice sites. Splicing repressors may sterically block the binding of splicing factors to specific sites in pre-mRNAs or inhibit their function. Splicing activators enhance splicing by interacting with splicing factors, thus promoting their association with a regulated splice site. The RNA sequences bound by splicing repressors are called intronic or exonic splicing silencers, depending on their location in an intron or exon. RNA sequences bound by splicing activators are called intronic or exonic splicing enhancers. Excised introns and RNA downstream from the cleavage/polyadenylation site are degraded primarily by exosomes, multiprotein complexes that contain an internal exonuclease. Exosomes also degrade improperly processed pre-mRNAs. Multiple human genetic diseases result from abnormal splicing. A base pair change in a pre-mRNA can create a consensus or splice site sequence, leading to splicing of an abnormal mRNA that does not encode a functional protein. A base pair change in an exonic splicing enhancer can cause skipping of the exon. Mutations causing specific amino acid changes in a subset of splicing factors cause myelodysplastic syndromes that are precursors to acute myeloid leukemia (AML).
Group II self-splicing introns in mitochondria and chloroplasts of protozoa, plants, and fungi may represent evolutionary precursors of snRNAs and their ability to act in trans at the short splice site sequences of nuclear pre-mRNAs. In RNA editing, the nucleotide sequence of a pre-mRNA is altered in the nucleus. In vertebrates, this process is relatively rare, and only single-base C to U changes have been observed, but those changes can have important consequences by altering the amino acid encoded by an edited codon (see Figure 9-25). In pathogenic trypanosome mitochondria (called kinetoplasts) RNA editing using separately transcribed guide RNAs leads to insertions and base changes comprising more than half of the length of some kinetoplast mRNAs.
9.3 Transport of mRNA Across the Nuclear Envelope
9.3 Transport of mRNA Across the Nuclear Envelope Before an mRNA can be translated into its encoded protein, it must be exported out of the nucleus into the cytoplasm. The nucleus is separated from the cytoplasm by the nuclear envelope, a double membrane that is continuous with the endoplasmic reticulum (ER) (see Figure 1-16). Like the plasma membrane surrounding cells, the inner and outer nuclear membranes consist of water-impermeable phospholipid bilayers and multiple associated proteins. Fully processed mRNAs in the nucleus remain bound by hnRNP proteins in complexes referred to as nuclear mRNPs. mRNPs and other macromolecules including tRNAs and ribosomal subunits traverse the nuclear envelope through nuclear pores. This section will focus on the export of mRNPs through the nuclear pore and the mechanisms that allow some level of regulation of this step. Transport of other cargoes across the nuclear pore is discussed in Chapter 13. Embedded in the nuclear envelope, nuclear pore complexes (NPCs) are cylindrical in shape with a diameter of (see Figure 13-32). Proteins and RNPs larger than must be selectively transported across NPCs with the assistance of soluble transporter proteins. These nuclear transporters bind cargo to be transported across an NPC and also interact reversibly with short hydrophobic FG-repeat regions in random
coil polypeptides extending from the NPC walls into the central channel. As a consequence of these reversible interactions with FG-domains, the transporter and its stably bound cargo can be passed from FG-domain to FG-domain, allowing the complex of transporter and cargo to diffuse down a concentration gradient: the concentration of the transporter-cargo protein complex is high in the nucleus where the complexes form, and low in the cytoplasm where the transporter-cargo complexes dissociate. Most types of mRNPs are transported through the NPC by the mRNP exporter, a heterodimer consisting of a large subunit, called nuclear export factor 1 (NXF1), and a small subunit, nuclear export transporter 1 (NXT1). NXF1 binds nuclear mRNPs through associations with both RNA and other proteins in the mRNP complex. One of the most important of these is REF (RNA export factor), a component of the exon-junction complexes discussed earlier, which is bound approximately 20 nucleotides to each exon-exon junction. The NXF1/NXT1 mRNP exporter also associates with SR proteins bound to exonic splicing enhancers. Thus SR proteins associated with exons function to direct both the splicing of premRNAs and the export of fully processed mRNAs through NPCs to the cytoplasm. mRNPs are probably bound along their length by multiple NXF1/NXT1 mRNP exporters, which interact with the FG-domains of FGnucleoporins to facilitate export of mRNPs through the NPC central channel. Protein filaments extend from the NPC core scaffold into the nucleoplasm, forming a nuclear basket (see Figure 13-32b). Protein filaments also extend into the cytoplasm. These filaments assist in mRNP export. Gle2,
an adapter protein that reversibly binds both NXF1 and a protein in the nuclear basket, brings nuclear mRNPs to the pore in preparation for export. A protein in the cytoplasmic filaments of the NPC binds an RNA helicase (Dbp5) that functions in the dissociation of NXF1/NXT1 and other hnRNP proteins from the mRNP as it reaches the cytoplasm. In a process called mRNP remodeling, the proteins associated with an mRNA in the nuclear mRNP complex are exchanged for a different set of proteins as the mRNP is transported through the NPC (Figure 9-26). Some nuclear mRNP proteins, like hnRNPC (see Figure 9-5) dissociate early in transport, remaining in the nucleus to bind to newly synthesized nascent pre-mRNA. Other nuclear mRNP proteins remain with the mRNP complex as it traverses the pore and do not dissociate from the mRNP until the complex reaches the cytoplasm, like hnRNPA1 (see Figure 9-5). Proteins in this category include the NXF1/NXT1 mRNP exporter, cap-binding complex (CBC) bound to the cap, and PABPN1 bound to the poly(A) tail. They dissociate from the mRNP on the cytoplasmic side of the NPC through the action of the Dbp5 RNA helicase that associates with cytoplasmic NPC filaments, as discussed above. These proteins are then imported back into the nucleus where they can function in the export of another mRNP. In the cytoplasm, the cap-binding translation initiation factor eIF4E replaces CBC bound to the cap of nuclear mRNPs. In humans, the nuclear poly(A)-binding protein PABPN1 is replaced with the cytoplasmic poly(A)-binding proteins PABPC1, PABPC2, or PABPC3. Only a single PABP is found in budding yeast, in both the nucleus and the cytoplasm.
FIGURE 9-26 Remodeling of mRNPs during nuclear export. Some mRNP proteins (purple) dissociate from nuclear mRNP complexes before export through an NPC. Some (yellow) are exported through the NPC associated with the mRNP but dissociate in the cytoplasm and are shuttled back into the nucleus through an NPC. In the cytoplasm, translation initiation factor eIF4E replaces CBC bound to the cap and PABPCI, 2, or 3 replaces PABPNI. Description
SR Proteins Mediate Nuclear Export of mRNA
A series starts from a nuclear messenger ribonucleoprotein (m R N P) complex made of chain of different proteins including C B C at 5-prime end, N X F 1, N X T 1, R E F, and two terminal P A B P N 1 of 12 adenosines ending into poly (A) tail. Some proteins disassociate from the m R N P and remain in the nucleus while the remaining enter cytoplasm and dissociate. C B C, N X F 1, N X T 1, R E F, and both P A B P N 1 enter back to nucleus. The remaining complex chain in cytoplasm is now made of e l F 4 E at 5-prime end and two 3-prime end P A B P C 1 proteins of 12 adenosines ending into poly (A) tail. SR Proteins Mediate Nuclear Export of mRNA Studies of S. cerevisiae indicate that the direction of mRNP export from the nucleus into the cytoplasm is controlled by phosphorylation and dephosphorylation of mRNP adapter proteins such as REF that assist in the binding of the NXF1/NXT1 exporter to mRNPs. For example, a yeast SR protein (Npl3) functions as an adapter protein that promotes the binding of the yeast mRNP exporter (Figure 9-27). The SR protein initially binds to nascent pre-mRNAs in its phosphorylated form. When cleavage and polyadenylation are completed, the adapter protein is dephosphorylated by a specific nuclear protein phosphatase essential for mRNP export. Only the dephosphorylated adapter protein can bind the mRNP exporter, thereby coupling mRNP export to correct polyadenylation. This is one form of mRNA quality control. If the nascent mRNP is not correctly processed, it is not recognized by the phosphatase that dephosphorylates Npl3. Consequently, it is not bound by the mRNA exporter and not exported from the nucleus. Instead, it is degraded by exosomes, the multiprotein
complexes that degrade unprotected RNAs in the nucleus and cytoplasm (see Figure 9-24).
FIGURE 9-27 Reversible phosphorylation of mRNP proteins controls the direction of mRNP nuclear export. Step 1 : The yeast SR protein Npl3 binds nascent pre-mRNAs in its phosphorylated form. Step 2 : When polyadenylation has occurred successfully, the Glc7 nuclear phosphatase essential for mRNP export dephosphorylates Npl3, promoting the binding of the yeast mRNP exporter, NXF1/NXT1. Step 3 : The mRNP exporter allows diffusion of the mRNP complex through the central channel of the nuclear pore complex (NPC). Steps 4 and 5 : The cytoplasmic protein kinase Sky1 phosphorylates Npl3 in the cytoplasm, causing dissociation of the mRNP exporter and phosphorylated Npl3, probably through the action of an RNA helicase associated with NPC cytoplasmic filaments. Step 6 : The mRNA transporter and phosphorylated Npl3 are transported back into the nucleus through NPCs. Step 7 : Transported mRNA is available for translation in the cytoplasm.
See E. Izaurralde, 2004, Nat. Struct. Mol. Biol. 11:210–212. Data from W. Gilbert and C. Guthrie, 2004, Mol. Cell 13:201–212. Description The process starts with R N A polymerase 2 transcribing an m R N A which gets attached with a phosphorylated N p l 3. Series of steps show export of m R N P to cytoplasm where it is translated and the m R N P exporter, N X F 1 slash N X T 1 enters back the nucleus along with a phosphorylated N pl 3. Following export to the cytoplasm, the Npl3 SR protein is phosphorylated by a specific protein kinase localized in the cytoplasm. This causes it to dissociate from the mRNP, along with the mRNP exporter. In this way, dephosphorylation of adapter mRNP proteins in the nucleus once RNA processing is complete, and their phosphorylation and resulting dissociation in the cytoplasm drives mRNP nuclear export. This results in a higher concentration of mRNP exporter–mRNP complexes in the nucleus, where they form, and a lower concentration of these complexes in the cytoplasm, where they dissociate. Consequently, the direction of mRNP export may be driven by simple diffusion down a concentration gradient of the transport-competent mRNP exporter–mRNP complex across the NPC, from high concentration in the nucleus to low concentration in the cytoplasm. Nuclear Export of Balbiani Ring mRNPs The larval salivary glands of the insect Chironomous tentans provide a good model system where assembly of an mRNP and its transport across
an NPC can be visualized by electron microscopy. In these larvae, genes in large chromosomal puffs called Balbiani rings are abundantly transcribed into nascent pre-mRNAs that associate with hnRNP proteins and are processed into coiled mRNPs with a final mRNA length of (Figure 9-28a, b). These giant mRNAs encode large glue proteins that adhere the developing larvae to a leaf. After processing of the pre-mRNA in Balbiani ring hnRNPs, the resulting mRNPs move through nuclear pores to the cytoplasm. Electron micrographs of sections of these cells show mRNPs that appear to uncoil during their passage through nuclear pores and then bind to ribosomes as they enter the cytoplasm. This uncoiling is probably a consequence of the remodeling of mRNPs as the result of phosphorylation of mRNP proteins by cytoplasmic kinases and the action of the RNA helicase associated with NPC cytoplasmic filaments, as discussed in the previous section. The observation that mRNPs become associated with ribosomes during transport indicates that the end leads the way through the nuclear pore complex. Detailed electron microscopic studies of the transport of Balbiani ring mRNPs through nuclear pore complexes led to the model depicted in Figure 9-28c.
FIGURE 9-28 Formation of heterogeneous ribonucleoprotein particles (hnRNPs) and export of mRNPs through NPCs in Chironomus tentans salivary gland cells during metamorphosis. (a) Model of a single chromatin transcription loop and assembly of Balbiani ring (BR) mRNP in Chironomus tentans. Nascent RNA transcripts produced from the template DNA rapidly associate with proteins, forming hnRNPs. The gradual increase in size of the hnRNPs reflects the increasing length of RNA transcripts at greater distances from the transcription start site. The model was reconstructed from electron micrographs of serial thin sections of salivary gland cells. (b) Model of the morphology of the BR hnRNP as it grows in length during its transcription. Following processing and cleavage/polyadenylation of the pre-mRNA, the resulting ribonucleoprotein particle, referred to as an mRNP, appeared like a microscopic croissant by EM. (c) Model for the transport of BR mRNPs through the nuclear pore complex (NPC) based on electron microscopic studies. Note that the curved mRNPs appear to uncoil as they pass through nuclear pores. As the mRNA enters the cytoplasm, it rapidly associates with ribosomes, indicating that the end passes through the NPC first. Parts (b) and (c) see B. Daneholt, 1997, Cell 88:585. See also B. Daneholt, 2001, Proc. Nat’l. Acad. Sci. USA 98:7012.
Pre-mRNAs Associated with Spliceosomes Are Not Exported from the Nucleus
[Part (a) Republished with permission from Elsevier, from C. Erricson et al., 1989, “The Ultrastructure of Upstream and Downstream Regions of an Active Balbiani Ring Gene,” Cell 56(4):631–639; permission conveyed through Copyright Clearance Center, Inc.] Description (a) The structure of single chromatin transcription loop shows an inverted U-shaped structure with multiple thread-like h n R N Ps arising on both sides and increasing in size from one end to another. (b) The structure of growing B R h n R N P shows a vertical template D N A strand with h n R N P structure arising from it and growing in size from top to bottom. The h n R N P curves as it grows and the m R N P dissociates from the template. (c) Transport of B R m R N Ps through N P C shows a series of illustration starting from a comma-shaped m R N P coming in contact with an N P C. First the tail enters the pore and m R N A is transcribed as the tail point reaches the cytoplasm. The m R N P remains in the nuclear pore while m R N A grows and is translated. Pre-mRNAs Associated with Spliceosomes Are Not Exported from the Nucleus It is critical that only fully processed, mature mRNAs be exported from the nucleus because translation of incompletely processed pre-mRNAs containing introns would produce defective proteins that might interfere with normal cellular functions. To prevent this, pre-mRNAs associated with snRNPs in spliceosomes are usually prevented from being transported to the cytoplasm.
In one type of experiment demonstrating this restriction, levels of cytoplasmic and nuclear RNAs were determined for RNAs expressed from a derivative of the rabbit β–globin gene containing only the second intron (see Figure 9-2). This modified β–globin gene expressed high levels of fully processed β–globin mRNA in the cytoplasm and only low levels of the unspliced hnRNA in the nucleus, presumably the initial transcript before being subjected to RNA splicing and export to the cytoplasm. However, when the invariant GT at the end of the globin intron, or the invariant AG at the end of the intron were mutated, splicing of this premRNA was blocked, and it was retained in the nucleus. Remarkably, when both the and splice sites in the pre-mRNA were mutated, splicing did not occur, as expected, but the unspliced mutant β–globin RNA was exported to the cytoplasm. Further studies revealed that transcripts with only one intact splice site were bound by snRNPs to form intermediates in spliceosome assembly that could not complete the splicing process because of the absence of the second splice site. However, RNA expressed from the mutant gene in which both the - and -splice site–invariant bases were mutated was not assembled into spliceosome assembly intermediates with U1 or U2 snRNPs, but as mentioned above, these RNAs were exported. Further studies in yeast have shown that a nuclear protein that associates with a protein in the NPC nuclear basket is required to retain pre-mRNAs in the nucleus that are associated with spliceosome assembly intermediates. If the gene encoding this protein or the nuclear pore basket protein to which it binds is deleted, unspliced pre-mRNAs are exported. Thus intermediates in spliceosome assembly are actively prevented from being exported to the cytoplasm. Another important observation made in these experiments was that the mutant hnRNAs
HIV Rev Protein Regulates the Transport of Unspliced Viral mRNAs
retaining only one or splice site that are retained in the nucleus do not build up to high concentration. Consequently, some mechanism exists that causes degradation of the hnRNAs that cannot be spliced. Understanding how such RNAs are selected for degradation is a goal of current research. Many cases of thalassemia, an inherited disease that results in abnormally low levels of adult globin proteins, result from mutations in adult globin-gene splice sites that decrease the efficiency of splicing and cause the mutant pre-mRNA to be retained in the nuclei of developing red blood cells in the bone marrow (erythrocyte precursors). The resulting unspliced nuclear globin pre-mRNAs are recognized by mechanisms still being elucidated, and degraded by nuclear exosomes (see Figure 9-24). HIV Rev Protein Regulates the Transport of Unspliced Viral mRNAs As discussed earlier, transport of fully processed nuclear mRNPs containing mature, functional mRNAs from the nucleus to the cytoplasm entails a complex mechanism that is crucial to gene expression (see Figures 9-26 and 9-27). Regulation of this transport could theoretically provide another means of gene control, although it appears to be relatively rare. The only known examples of regulated mRNA export occur during the cellular response to conditions that cause protein denaturation (e.g., heat shock) or during viral infection when virus-induced alterations in nuclear transport maximize viral replication.
One example of the regulation of mRNP export comes from a protein encoded by human immunodeficiency virus (HIV). A retrovirus, HIV integrates a DNA copy of its RNA genome into the host-cell DNA (see
Figure 5-43). The integrated viral DNA, or provirus, contains a single transcription unit, which is transcribed into a single primary transcript by cellular RNA polymerase II. The HIV transcript can be spliced in alternative ways to yield three classes of mRNAs: a 9-kb unspliced mRNA, mRNAs formed by removal of one intron, and mRNAs formed by removal of two or more introns (Figure 9-29). After their synthesis in the host-cell nucleus, all three classes of HIV mRNAs are transported to the cytoplasm and translated into viral proteins; some of the 9-kb unspliced RNA is used as the viral genome in progeny virions that bud from the cell surface.
FIGURE 9-29 Transport of HIV mRNAs from the nucleus to the cytoplasm. The HIV genome, which contains several coding regions, is transcribed into a single 9-kb primary transcript. Several mRNAs result from alternative splicing out of any one of several introns (dashed lines) and several mRNAs from splicing out of two or more alternative introns. After transport to the cytoplasm, the various RNA species are translated
into different viral proteins. Rev protein, encoded by a 2-kb mRNA, interacts with the Revresponse element (RRE) in the unspliced and singly spliced mRNAs, stimulating their transport to the cytoplasm. See B. R. Cullen and M. H. Malim, 1991, Trends Biochem. Sci. 16:346. Description In the nucleus, an H I V provirus gene with R R E site is transcribed into 9 kilobase unspliced m R N A which can be spliced into 4 kilobase singly spliced m R N A or 2 kilobase multiple spliced m R N A. The 9 kilobase and 4 kilobase m R N As are transported to cytoplasm in presence of Rev protein and their transport is inhibited in absence of Rev protein. The 2 kilobase m R N A is transported to cytoplasm in absence of Rev where it is translated into Rev protein. This Rev protein enters back to the nucleus and attaches to the R R E site of 9 kilobase and 4 kilobase m R N A to help their transport. Since the 9-kb and 4-kb HIV mRNAs contain splice sites, they can be viewed as incompletely spliced pre-RNAs. Association of such incompletely spliced mRNAs with snRNPs in spliceosome assembly intermediates normally blocks their export from the nucleus. Thus HIV, as well as other retroviruses, must have some mechanism for overcoming this block, permitting export of the longer viral mRNAs. Studies with HIV mutants showed that transport of unspliced 9-kb and singly spliced 4-kb viral mRNAs from the nucleus to the cytoplasm requires the virus-encoded Rev protein. Biochemical experiments show that Rev binds to a specific Rev-response element (RRE) present in HIV RNA. Early in an infection, before any Rev protein is synthesized, only the multiply spliced 2-kb mRNAs can be exported. One of these 2-kb mRNAs encodes Rev, which contains a leucine-rich nuclear export signal that
interacts with the transporter Exportin 1. After translation and nuclear import of Rev, Rev binds to the RRE sequence in unspliced and singly spliced HIV RNAs. Rev then associates with Exportin 1, resulting in export of the larger unspliced and singly spliced HIV mRNAs through the nuclear pore complex without the assistance of NXF1/NXT1, the major mRNP exporter for cellular mRNPs. In cells infected with HIV mutants lacking the RRE, unspliced and singly spliced viral mRNAs remain in the nucleus, confirming that the RRE is required for Rev-mediated stimulation of nuclear export. KEY CONCEPTS OF SECTION 9.3 Transport of mRNA Across the Nuclear Envelope Most mRNPs are exported from the nucleus by a heterodimeric mRNP exporter that interacts with the central channel of the nuclear pore complex (NPC). The direction of transport (nucleus to cytoplasm) is controlled by phosphorylation of mRNP adaptor proteins. For example, the yeast Npl3 adapter protein enters nuclei only when it is phorphorylated and associates with nascent pre-mRNAs as they are transcribed (see Figure 9-27). When polyadenylation of the pre-mRNA is complete, a nuclear protein phosphatase (Glc7) removes the phosphates from Npl3, allowing the pre-mRNA to interact with the NXF1/NXT1 mRNP exporter, causing export of the pre-mRNP to the cytoplasm. When the mRNP reaches the cytoplasm, the Sky1 protein kinase restricted to the cytoplasm phosphorylates Npl3, causing dissociation of the mRNP transporter, completing delivery of the mRNP to the cytoplasm. The mRNP exporter binds to most mRNAs cooperatively with SR proteins bound to exonic splicing enhancers and with REF associated with exon-junction complexes as well as with additional mRNP proteins. Pre-mRNAs bound by a spliceosome normally are not exported from the nucleus, ensuring that only fully processed, functional mRNAs reach the cytoplasm for translation.
The Concentration of an mRNA in the Cytoplasm Is Determined by Its Rate of Synthesis and Its Rate of Degradation
9.4 Cytoplasmic Mechanisms of Post-Transcriptional Control Before proceeding, let’s quickly review the steps in gene expression at which control is exerted. We saw in the previous chapter that regulation of transcription initiation and transcription elongation in the promoterproximal region are the initial mechanisms for controlling the expression of genes in the gene expression pathway of DNA → RNA → protein. In preceding sections of this chapter, we learned that the expression of protein isoforms is controlled by regulating alternative RNA splicing and cleavage and polyadenylation at alternative poly(A) sites. Although nuclear export of fully and correctly processed mRNPs to the cytoplasm is rarely regulated, the export of improperly processed or aberrantly remodeled pre-mRNPs is prevented, and such abnormal transcripts are degraded by the exosome. However, retroviruses, including HIV, have evolved mechanisms that permit pre-mRNAs that retain splice sites to be exported and translated. In this section, we consider other mechanisms of post-transcriptional control that contribute to regulating the expression of many genes. Most of these mechanisms operate in the cytoplasm, controlling the stability or localization of mRNA or its translation into protein.
The Concentration of an mRNA in the Cytoplasm Is Determined by Its Rate of Synthesis and Its Rate of Degradation The most stable mRNAs encode proteins required in large numbers, such as the ribosomal proteins; these can accumulate to very high copy number per cell. In contrast, highly unstable mRNAs encoding proteins expressed in short bursts — such as cytokines, secreted proteins that regulate the immune response — rarely achieve such high concentrations even when transcribed, processed, and exported from the nucleus at high rates. We begin by discussing the major pathways that degrade mRNAs. Next we discuss two related mechanisms of gene control that provide powerful new techniques for manipulating the expression of specific genes for experimental and therapeutic purposes. These two mechanisms are controlled by short, -nucleotide, singlestranded RNAs called microRNAs (miRNAs) and short interfering RNAs (siRNAs). Both base-pair with specific target mRNAs, causing their rapid degradation (siRNAs) or inhibiting their translation and inducing a slower form of degradation (miRNAs). Approximately 1900 human miRNAs have been characterized. Most of these are expressed in specific cell types at particular times during embryogenesis and after birth. Many miRNAs can target more than one mRNA. Consequently, these mechanisms contribute significantly to the regulation of gene expression.
Degradation of mRNAs in the Cytoplasm Occurs by Several Mechanisms
siRNAs, involved in the process called RNA interference, are an important cellular defense against viral infection and excessive transposition by retrotransposons. We also discuss mechanisms that control the overall rate of protein synthesis, and highly specific regulation of the translation and stability of particular mRNAs by proteins that bind them with high specificity. Finally, we discuss mechanisms that control the localization of mRNAs in the cytoplasm of asymmetric cells so that the encoded protein is translated at sites in the cell where it is needed. Degradation of mRNAs in the Cytoplasm Occurs by Several Mechanisms The concentration of an mRNA is a function of both its rate of synthesis and its rate of degradation. For this reason, if two genes are transcribed at the same rate, the steady-state concentration of the corresponding mRNA that is more stable will be higher than the concentration of the other. The stability of an mRNA also determines how rapidly the synthesis of the encoded protein can be shut down. For a stable mRNA, synthesis of the encoded protein persists long after transcription of the gene is repressed. Most bacterial mRNAs are unstable, decaying exponentially with a typical half-life of a few minutes. For this reason, a bacterial cell can rapidly adjust the synthesis of proteins to accommodate changes to the cellular environment. Most cells in multicellular organisms, on the other hand, exist in a fairly constant environment and carry out a specific set of functions over periods of days to months or even the lifetime of the
organism (nerve cells, for example). Accordingly, most mRNAs of multicellular eukaryotes have half-lives of many hours. Some proteins in eukaryotic cells are required only for short periods and must be expressed in bursts. For example, certain signaling molecules called cytokines, which are involved in regulating the immune response of mammals, are synthesized and secreted in short bursts (see Chapter 24). Similarly, many of the transcription factors that regulate the onset of the S phase of the cell cycle, such as Fos and Jun, are synthesized for only brief periods (see Chapter 19). Expression of such proteins occurs in short bursts because transcription of their genes can be rapidly turned on and off, and their mRNAs have unusually short half-lives, on the order of 30 minutes or less, so that concentration of the mRNA falls rapidly when its transcription is repressed. Cytoplasmic mRNAs are degraded by one of the three pathways shown in
Figure 9-30. For most mammalian mRNAs, the deadenylation-dependent pathway is followed (Figure 9-30a). The length of the poly(A) tail gradually decreases with time through the action of a deadenylating nuclease complex in the cytoplasm. When it is shortened sufficiently, PABPC molecules can no longer bind and stabilize the interaction of the cap and translation initiation factors (see Figure 5-36). The exposed cap then is removed by a heterodymeric decapping enzyme (Dcp1/Dcp2), leaving the unprotected mRNA susceptible to degradation by a exonuclease (Xrn1) (Figure 9-30 step 1 ). Removal of the poly(A) tail also makes mRNAs susceptible to degradation by cytoplasmic exosomes containing exonuclease activity (Figure 9-30 step 2 ). The
exonuclease pathway step 1 predominates in yeast, and the exosome pathway step 2 predominates in mammalian cells.
FIGURE 9-30 Pathways for degradation of eukaryotic mRNAs. (a) In the most common pathway of mRNA degradation, the deadenylation-dependent pathway, the poly(A) tail is progressively shortened by a deadenylase complex in the cytoplasm (orange) until it reaches a length of 20 or fewer A residues, at which point the interaction with PABPC1 and the remaining poly(A) is destabilized. This leads to weakened interactions between the cap and translation initiation factors (see Figures 5-36, step 3 , and Figure 5-39). The deadenylated mRNA then may either 1 be decapped by the DCP1/DCP2 deadenylation complex and degraded by XRN1, a exonuclease, or 2 be degraded by exonucleases in cytoplasmic exosomes. (b) Other mRNAs are decapped before they are deadenylated and then degraded by the XRN1 exonuclease. (c) Some mRNAs are cleaved internally by an endonuclease and the fragments degraded by a cytoplasmic exosome and the XRN1 exonuclease. See N. L. Garneau, J. Wilusz, and C. J. Wilusz, 2007, Nat. Rev. Mol. Cell Biol. 8:113. Description The deadenylation-dependent m R N A decay (a) shows an O R F segment with 5prime U T R at m 7 G end and 3-prime U T R at poly (A) end. During deadenylation,
the deadenylase complex digests the poly (A) tail. The deadenylated m R N A undergoes either 5-prime to 3-prime decay or 3-prime to 5-prime decay. The deadenylation-independent m R N A decay (b) shows E d c 3 and R p s 28 B complex bound to poly (A) tail and an arrow from this complex points to D C P 2 and D C P 1 complex near m 7 G. Next, X R N 1 digests the m 7 G, 5-prime U T R region of the O R F segment. The endonuclease-mediated m R N A decay (c) shows endonuclease cleaving the O R F segment at the center. This cleaved m R N A may undergo either 5-prime to 3-prime decay where an exonuclease digests it or 3-prime to 5-prime decay where X R N 1 digests it. The decapping enzymes and exonuclease are concentrated in cytoplasmic P bodies (processing bodies), regions of the cytoplasm with unusually high concentrations of RNPs. These regions of highly concentrated proteins are not surrounded by a lipid bilayer membrane. Their formation is dependent on multivalent, low-affinity interactions between simple short repeats of amino acid sequence within intrinsically disordered regions (IDRs) of cytoplasmic RNA-binding proteins associated with some RNAs. These are similar to the transcriptional condensates discussed in Chapter 8 (see Figure 8-38). The process that generates such protein concentrates in both the nucleus and cytoplasm has been called a “liquid-liquid phase separation.” This process was first studied for RNA-binding proteins in the cytoplasm that generate P bodies and large cytoplasmic mRNP complexes that are transported to specific regions of the cell by myosin motor proteins that “walk” on actin filaments (see Figure 9-40, and Chapter 17) or by kinesin and dynein motor proteins that walk on microtubules (Chapter 18).
The rate of mRNA deadenylation varies inversely with the frequency of translation initiation for an mRNA: the higher the frequency of initiation, the slower the rate of deadenylation. This relation probably is due to the reciprocal interactions between translation initiation factors bound at the cap and PABPC bound to the poly(A) tail. For an mRNA that is translated at a high rate, initiation factors are bound to the cap much of the time, stabilizing the binding of PABPC and thereby protecting the poly(A) tail from deadenylation exonuclease complexes. AU-Rich Elements Many short-lived mRNAs in mammalian cells encoding proteins — such as cytokines and transcription factors whose concentrations are changed rapidly — contain multiple, sometimes overlapping copies of the sequence AUUUA in their untranslated region. These are known as AU-rich elements. Specific RNA-binding proteins have been found that both bind to these -AU-rich sequences and also interact with a deadenylating enzyme and with the exosome. This causes rapid deadenylation and subsequent degradation of these mRNAs (Figure 9-30a). In this mechanism, the rate of mRNA degradation is uncoupled from the frequency of translation. Thus mRNAs containing AU-rich elements can be translated at high frequency yet also be degraded rapidly, allowing the encoded proteins to be expressed in short bursts. Some mRNAs are degraded primarily by a deadenylation-independent decapping pathway (see Figure 9-30b). This is because certain sequences
MicroRNAs Repress Translation and Induce Degradation of Specific mRNAs
at the end of an mRNA make the cap sensitive to the decapping enzyme. For these mRNAs, the rate at which they are decapped controls the rate at which they are degraded because once the cap is removed, the RNA is rapidly hydrolyzed by the exonuclease Xrn1. Finally, as shown in Figure 9-30c, some mRNAs are degraded by an endonucleolytic pathway that does not involve decapping or significant deadenylation. One example of this type of pathway is the RNAi pathway discussed below. Each siRNA-RISC complex can degrade multiple targeted RNA molecules. The fragments generated by internal cleavage then are degraded by exonucleases. MicroRNAs Repress Translation and Induce Degradation of Specific mRNAs MicroRNAs (miRNAs) were first discovered during analysis of mutations in the lin-4 and let-7 genes of the nematode C. elegans, which influence development of the organism. Cloning and analysis of wild-type lin-4 and let-7 revealed that they do not encode protein but rather RNAs only 21 and 22 nucleotides long, respectively. The RNAs hybridize to the untranslated regions of specific target mRNAs. For example, the lin-4 miRNA, which is expressed early in larval development, hybridizes to the untranslated regions of both the lin-14 and lin-28 mRNAs in the cytoplasm, thereby destabilizing these mRNAs and repressing their translation. Expression of let-7 miRNA occurs at comparable times during
embryogenesis of all bilaterally symmetric animals, including insects and mammals. miRNA-mediated regulation appears to be widespread in all multicellular plants and animals. In the past few years, small RNAs of 20–26 nucleotides have been isolated, cloned, and sequenced from various tissues of multiple-model organisms. Recent estimates suggest the expression of ∼60% of all human genes is regulated by one or more of the 556 wellvalidated human miRNAs that have been isolated from various tissues. The potential for regulation of multiple mRNAs by one miRNA is high because base pairing between the miRNA and the sequence in the ends of mRNAs that they regulate need not be perfect (Figure 9-31a). In fact, considerable experimentation with synthetic miRNAs has shown that complementarity between bases two to seven at the end of an miRNA (called the seed sequence) and its target mRNA untranslated region are most critical for target mRNA selection.
FIGURE 9-31 The extent of base pairing with the miRNA determines the fate of the target RNA. (a) When miRNAs hybridize imperfectly with their target mRNAs, deadenylation is induced and the mRNA is degraded by de-adenylation-dependent decay (Figure 9-30a). Nucleotides 2 to 7 of an miRNA (highlighted blue) are the most critical for targeting it to a specific mRNA. The CXCR4 miRNA shown at the bottom is a synthetic oligonucleotide introduced into cells by transfection. (b) miRNAs can also extensively pair to their target mRNAs, causing cleavage of the mRNA at the position indicated by the red arrow, triggering its rapid degradation. See P. D. Zamore and B. Haley, 2005, Science 309:1519. Description The part (a) shows a segment of m i R N A paired with the target R N A with loop-out regions of unpaired sites. Examples shows lin-4 m i R N A and lin-14 m R N A of C. elegans and C X C R 4 m i R N A and target m R N A of H. sapiens. The part (b) shows a large segment of s i R N A paired completely with the target R N A with no loop formation. Examples show m i R-196 a and HOX B 8 m R N A of H. sapiens and m i R-166 and P H A VOLUTA m R N A of A. thaliana. Most miRNAs are processed from RNA polymerase II transcripts of several hundred to thousands of nucleotides in length called pri (for primary transcript)-miRNAs (Figure 9-32). Pri-miRNAs can contain the sequence of one or more miRNAs. miRNAs are also processed out of some excised introns. Within these long transcripts are sequences that fold into hairpin structures of nucleotides in length with imperfect base pairing in the stem. A nuclear RNase specific for double-stranded RNA called Drosha acts in concert with a nuclear double-stranded RNA-binding protein called DGCR8 in humans and cleaves most of the hairpin region out of the long precursor RNA, generating a 60 nucleotide pre-miRNA. Pre-miRNAs are recognized and bound by a specific nuclear export factor,
Exportin5, which allows the complex to diffuse through the inner channel of the nuclear pore complex. Once in the cytoplasm, a cytoplasmic doublestranded RNA-specific RNase called Dicer acts with a cytoplasmic double-stranded RNA-binding protein called TRBP in humans (for Tar binding protein) to further process the pre-miRNA into a double-stranded miRNA. The double-stranded miRNA is approximately two turns of an Aform RNA helix in length, with strands 21–23 nucleotides long and with two unpaired nucleotides at each end.
FIGURE 9-32 miRNA processing. This diagram shows transcription and processing of the miR-1-1 miRNA. The primary miRNA transcript (pri-miRNA) is transcribed by RNA polymerase II. The nuclear double-stranded RNA-specific endoribonuclease Drosha, with its partner double-stranded RNA-binding protein DGCR8, make the initial cleavages in the pri-miRNA, generating a -nucleotide pre-miRNA (step 1 ) that is exported to the cytoplasm by nuclear transporter Exportin5 (step 2 ). The pre-miRNA is further processed in the cytoplasm to a double-stranded miRNA with two-base single-stranded ends by Dicer in conjunction with the double-stranded RNA-binding protein TRBP (step 3 ). Finally, one of the two strands is incorporated into an RISC complex, where it is bound by an Argonaute protein (step 4 ). See P. D. Zamore and B. Haley, 2005, Science 309:1519. Description The process starts in nucleus with an m i R-1-1 gene being transcribed into pre-m i R-1- 1 which is transported to cytoplasm and undergoes stepwise processing to result into an R I S C complex wherein the mature m i R-1-1 is bound to an argonaute protein. Finally, one of the two strands is selected for assembly into a mature RNA-induced silencing complex (RISC) containing a single-stranded mature miRNA bound by a multidomain Argonaute protein, a member of a protein family with a recognizable conserved sequence. Several Argonaute proteins are expressed in some organisms, especially plants, and are found in distinct RISC complexes with different functions. Humans express four Argonaute proteins that can each associate with a miRNA to produce a RISC with a miRNA with a seed sequence that can base-pair to a target mRNA, stimulating its deadenyation and degradation (Figure 9-30a). Ago2 is also able to catalyze mRNA cleavage. The four human Argonaute proteins have partially overlapping functions, demonstrated by experiments that show that knock-out of all four human
Argonaute proteins is lethal to human embryonic stem cells, but any one of the four is sufficient for viability. The miRNA-RISC complexes associate with target mRNPs by base pairing between the Argonaute-bound mature miRNA and complementary regions in the untranslated regions ( UTRs) of target mRNAs (see Figure 931). Generally, the more RISC complexes are bound to the UTR of an mRNA, the greater the repression. This allows combinatorial regulation of by separately regulating the transcription of two or more different primiRNAs, which are processed to miRNAs required in combination to repress a specific target mRNA. Approximately 600 different human miRNAs have been observed, most of them expressed only in specific cell types. This has raised the question about the function of miRNAs in differentiation. In one example, a specific miRNA called miR-133 is induced when myoblasts differentiate into muscle cells. miR-133 suppresses the translation of PTB, a regulatory splicing factor that functions similarly to Sxl in Drosophila (see Figure 918). PTB binds to the splice-site region in the pre-mRNAs of many genes, leading to exon skipping or use of alternative splice sites. When miR-133 is expressed in differentiating myoblasts, the PTB concentration falls. As a result, alternative isoforms of multiple proteins important for muscle-cell function are expressed in the differentiated cells. Other examples of miRNA regulation in differentiation in various organisms are being discovered at a rapid pace. Knocking out the Dicer gene eliminates the generation of miRNA in mammals. This causes
embryonic death early in mouse development. However, when Dicer is knocked out only in limb primordia, the influence of miRNA on the development of limbs in the mouse can be observed because limbs are not essential for viability in a laboratory mouse (Figure 9-33). Although all major cell types differentiate and the fundamental aspects of limb patterning are maintained, development is abnormal — demonstrating the importance of miRNAs in regulating the proper level of translation of multiple mRNAs. In effect, miRNAs tune gene expression to the appropriate level for gene function in various cell types. Of the human miRNAs, 53 appear to be unique to primates. It seems likely that new miRNAs arose readily during evolution by the duplication of a primiRNA gene followed by mutation of bases encoding the mature miRNA. EXPERIMENTAL FIGURE 9-33 miRNA function in limb development. Micrographs comparing normal (left) and Dicer knockout (right) limbs of embryonic development day13 mouse embryos immunostained for the Gd5 protein, a marker of joint formation. Dicer is knocked out in developing mouse embryo limb buds by conditional expression of Cre to induce deletion of the Dicer gene only in these cells (see Figure 6-40). [From B. D. Harfe et al., 2005, “The RNaseIII Enzyme Dicer Is Required for Morphogenesis but Not Patterning of the Vertebrate Limb,” Proc. Natl. Acad. Sci. USA
RNA Interference Induces Degradation of Precisely Complementary mRNAs
102(31):10898–10903.] Alternative Polyadenylation and mRNA Regulation by miRNAs The use of alternative polyadenylation sites in mRNA processing, discussed earlier, adds another avenue of regulation of gene expression by miRNAs. For mRNAs expressed from the same gene that use alternative polyadenylation sites, additional miRNA-binding sites may be located in the mRNA with the longer exon. As a consequence, mRNAs with the same protein-coding sequence may be regulated differently in alternative cell types depending on the miRNAs expressed in the cells. Consequently, alternative polyadenylation can indirectly regulate mRNAs encoding the same protein as a consequence of miRNA control of translation and mRNA stability. RNA Interference Induces Degradation of Precisely Complementary mRNAs RNA interference (RNAi) was discovered unexpectedly during attempts to experimentally manipulate the expression of specific genes. Researchers tried to inhibit the expression of a gene in C. elegans by microinjecting a single-stranded, complementary RNA that would hybridize to the encoded mRNA and prevent its translation, a method
called antisense inhibition. But in control experiments, perfectly basepaired double-stranded RNA a few hundred base pairs long was much more effective at inhibiting expression of the gene than the antisense strand alone (see Figure 6-41). Similar inhibition of gene expression by an introduced double-stranded RNA soon was observed in plants. In each case, the double-stranded RNA induced degradation of all cellular RNAs containing a sequence that was exactly the same as one strand of the double-stranded RNA. Because of the specificity of RNA interference in targeting mRNAs for destruction, it has become a powerful experimental tool for studying gene function. Subsequent biochemical studies with extracts of Drosophila embryos showed that a long double-stranded RNA that mediates interference is initially processed into a double-stranded short interfering RNA (siRNA). The strands in siRNA contain 21–23 nucleotides hybridized to each other so that the two bases at the end of each strand are single stranded. Further studies revealed that the cytoplasmic double-stranded RNAspecific ribonuclease that cleaves long double-stranded RNA into siRNAs is the same Dicer enzyme involved in processing pre-miRNAs after their nuclear export to the cytoplasm (see Figure 9-32). This discovery led to the realization that RNA interference and miRNA-mediated translational repression and target mRNA degradation are related processes. In both cases, the mature, short single-stranded RNA, either mature siRNA or mature miRNA, is assembled into a RISC complex in which the short RNAs are bound by an Argonaute protein. When bound to a short RNA that base-pairs extensively with its target RNA, the RISC complex induces target mRNA cleavage. By contrast, a RISC complex associated with a
short RNA, which base pairs less extensively (see Figure 9-31a), results in inhibition of translation and a slower form of target mRNA degradation. In humans, the Argonaute 2 protein is responsible for cleavage of target RNA in an RNAi complex; one domain of the Argonaute protein is homologous to RNase H enzymes that degrade the RNA of an RNA-DNA hybrid. When the end of the short RNA of a RISC complex base-pairs precisely with a target mRNA, this domain cleaves the phosphodiester bond of the target RNA across from nucleotides 10 and 11 of the siRNA (see Figure 9-31b). The cleaved RNAs are released and subsequently degraded by cytoplasmic exosomes and the Xrn1 exoribonuclease. If base pairing is less extensive, the Argonaute domain does not cleave or release the target mRNA. Instead, translation is inhibited and the mRNA it is degraded by a different and slower mechanism than the degradation pathway initiated by RISC cleavage of a perfectly complementary target RNA. When double-stranded RNA is introduced into the cytoplasm of eukaryotic cells, it enters the pathway for assembly of siRNAs into a RISC complex because it is recognized by the cytoplasmic Dicer enzyme and TRBP double-stranded RNA-binding protein that process pre-miRNAs (see
Figure 9-32). This process of RNA interference is believed to be an ancient cellular defense against viruses and mobile genetic elements in both plants and animals. Plants with mutations in the genes encoding Dicer and RISC proteins exhibit increased sensitivity to infection by RNA viruses and increased movement of transposons within their genomes. The double-stranded RNA intermediates generated during replication of RNA
viruses are thought to be recognized by the Dicer ribonuclease, inducing an RNAi response that ultimately degrades viral mRNAs. During transposition, transposons are inserted into cellular genes in a random orientation, and their transcription from different promoters produces complementary RNAs that can hybridize with each other, initiating the RNAi system that then interferes with the expression of transposon proteins required for additional transpositions. In plants and C. elegans the RNAi response can be induced in all cells of the organism by introduction of double-stranded RNA into just a few cells. Such organism-wide induction requires production of a protein that is homologous to the RNA replicases of RNA viruses. It has been revealed that double-stranded siRNAs are replicated and then transferred to other cells in these organisms. In plants, transfer of siRNAs might occur through plasmodesmata, the cytoplasmic connections between plant cells that traverse the cell walls between them (see Figure 20-38). Organism-wide induction of RNA interference does not occur in Drosophila or mammals, presumably because their genomes do not encode RNA-replicase homologs. In mammalian cells, the introduction of long RNA-RNA duplex molecules into the cytoplasm results in the generalized inhibition of protein synthesis via the PKR pathway, discussed further below. This greatly limits the use of long double-stranded RNAs to experimentally induce an RNAi response against a specific targeted mRNA in mammalian cells as opposed to Drosophila cells. Fortunately, researchers discovered that double-stranded siRNAs 21–23 nucleotides in length with two-base -single-stranded
Cytoplasmic Polyadenylation Promotes Translation of Some mRNAs
regions lead to the generation of single-stranded RNAs that are incorporated into functional siRNA RISC complexes without inducing the generalized inhibition of protein synthesis. This has allowed researchers to use synthetic double-stranded siRNAs to “knock down” the expression of specific genes in human cells as well as in those of other mammals. Also, expression vectors for designed pri-miRNAs can be introduced into cells, leading to the processing of designed siRNAs that hybridize to specific, experimentally controlled target mRNAs, causing their degradation. These methods of siRNA knockdown are now widely used in studies of diverse processes, including the RNAi pathway itself! Cytoplasmic Polyadenylation Promotes Translation of Some mRNAs In addition to repression by miRNAs, other protein-mediated controls help regulate expression of some genes. Regulatory sequences, or elements, in mRNAs that interact with specific proteins to control translation are generally present in the untranslated region (UTR) at the or end of an mRNA. Here we discuss a type of protein-mediated translational control involving regulatory elements. A different mechanism involving RNAbinding proteins that interact with regulatory elements is discussed later. Translation of many eukaryotic mRNAs is regulated by sequence-specific RNA-binding proteins that bind cooperatively to neighboring sites in
UTRs. This allows them to function in a combinatorial manner, similar to the cooperative binding of transcription factors to regulatory sites in an enhancer or promoter region. In most cases studied, translation is repressed by protein binding to regulatory elements and regulation results from derepression at the appropriate time or place in a cell or developing embryo. The mechanism of such repression is best understood for mRNAs that must undergo cytoplasmic polyadenylation before they can be translated. Cytoplasmic polyadenylation is a critical aspect of gene expression in the early embryo of animals. The egg cells (oocytes) of multicellular animals contain many mRNAs, encoding numerous different proteins that are not translated until after the egg is fertilized by a sperm cell. Some of these “stored” mRNAs have a short poly(A) tail, consisting of only A residues, to which just a few molecules of cytoplasmic poly(A)-binding protein (PABPCI) can bind. Multiple PABPCI molecules bound to the long poly(A) tail of an mRNA interact with the eIF4G initiation factor, thereby stabilizing the interaction of the mRNA cap with eIF4E, which is required for translation initiation (see Figure 5-36). Because this stabilization cannot occur with mRNAs that have short poly(A) tails, such mRNAs stored in oocytes are not translated efficiently. At the appropriate time during oocyte maturation or after fertilization of an egg cell, usually in response to an external signal, approximately 150 A residues are added to the short poly(A) tails on these mRNAs in the cytoplasm, stimulating their translation.
Studies with mRNAs stored in Xenopus oocytes have helped elucidate the mechanism of this type of translational control. Experiments in which short-tailed mRNAs are injected into oocytes have shown that two sequences in their UTR are required for their polyadenylation in the cytoplasm: the AAUAAA poly(A) signal that is also required for the nuclear polyadenylation of pre-mRNAs, and one or more copies of an upstream U-rich cytoplasmic polyadenylation element (CPE) (Figure 934). This regulatory element is bound by a highly conserved CPE-binding protein (CPEB) that contains an RRM domain and a zinc-finger domain.
FIGURE 9-34 Model for control of cytoplasmic polyadenylation and translation initiation. Left: In immature oocytes, mRNAs containing the U-rich cytoplasmic polyadenylation element (CPE) have short poly(A) tails and are translationally dormant. CPE-binding protein (CPEB) mediates repression of translation through the interactions depicted, which prevent assembly of an initiation complex at the end of the mRNA. Right: Hormone stimulation of oocytes activates a protein kinase that phosphorylates CPEB, causing it to release Maskin. The cleavage and polyadenylation specificity factor (CPSF) then bind to the poly(A) site, interacting with both bound CPEB and the cytoplasmic form of poly(A) polymerase (PAP). After the poly(A) tail is lengthened, multiple copies of the cytoplasmic poly(A)-binding protein I (PABPC1) can bind to it and interact with eIF4G, which functions with other initiation factors to bind the 40S ribosome subunit and initiate translation (translationally active). See R. Mendez and J. D. Richter, 2001, Nat. Rev. Mol. Cell Biol. 2:521.
Description The translationally dormant m R N A shows a C-shaped m R N A structure with the top end having e l F 4 E bound to the capped region and Maskin bound to the e l F 4 E. The Maskin is further bound at the bottom to the C P E B which is in turn bound to the C P E region lying next to the poly (A) signal at the bottom end of m R N A. The coding region lies at the curve side of the m R N A. The translationally active m R N A shows a similar m R N A structure but with a long poly (A) tail. At the top end, e l F 4 E binds to the capped region and is further bound to e l F 4 G. The e l F 4 G is bound to e l F 3 with is in turn bound to the 40 S subunit attached to the capped region. The e l F 4 G is also bound to two P A B P C 1 at the bottom which are attached to the end of the poly (A) tail. A phosphorylated C P E B is bound to the C P E region and is attached adjacently to C P S F and P A P that lie above the poly (A) tail. In the absence of a stimulatory signal, CPEB bound to the U-rich CPE interacts with the protein Maskin, which in turn binds to eIF4E associated with the mRNA cap (Figure 9-34, left). As a result, eIF4E cannot interact with other initiation factors and the 40S ribosomal subunit, so translation initiation is blocked. During oocyte maturation, a specific CPEB serine is phosphorylated, causing Maskin to dissociate from the complex. This allows cytoplasmic forms of the cleavage and polyadenylation specificity factor (CPSF) and poly(A) polymerase to bind to the mRNA cooperatively with CPEB. Once the poly(A) polymerase catalyzes the addition of A residues, PABPCI can bind to the lengthened poly(A) tail, leading to the stabilized interaction of all the factors needed to initiate translation (Figure 9-34, right; see also Figure 5-36). In the case of Xenopus oocyte maturation, the protein kinase that phosphorylates CPEB is activated in response to the hormone progesterone. Thus timing
Protein Synthesis Can Be Globally Regulated
of the translation of stored mRNAs encoding proteins needed for oocyte maturation is regulated by this external signal. Considerable evidence indicates that a similar mechanism of translational control plays a role in learning and memory. In the central nervous system, the axons from a thousand or so neurons can make connections (synapses) with the dendrites of a single postsynaptic neuron (see Figure 23-3). When one of these axons is stimulated, the postsynaptic neuron “remembers” which one of these thousands of synapses was stimulated. The next time that synapse is stimulated, the strength of the response triggered in the postsynaptic cell differs from the first time. This change in response has been shown to result largely from the translational activation of mRNAs stored in the region of the synapse in the postsynaptic neuron, leading to the local synthesis of new proteins that increase the size and alter the neurophysiological characteristics of the synapse. The finding that CPEB is present in neuronal dendrites has led to the proposal that cytoplasmic polyadenylation stimulates translation of specific mRNAs in dendrites, much as it does in oocytes. In this case, presumably, synaptic activity (rather than a hormone) is the signal that induces local phosphorylation of CPEB and subsequent activation of translation in one specific synapse of the neuron. Protein Synthesis Can Be Globally Regulated
Like proteins involved in other processes, translation initiation factors and ribosomal proteins can be regulated by post-translational modifications such as phosphorylation. Such mechanisms affect the translation rate of most mRNAs and hence the overall rate of cellular protein synthesis. The TOR pathway, which represses translation when the supply of amino acids is low as sensed through communication between lysosomes and the cytosol, is discussed in detail in Chapter 21. Other mechanisms that control the global rate of cellular translation include mechanisms that control the activity of translation initiation factor eIF2. Here we will focus on the mechanisms that can regulate the initiation of translation. Global regulation of protein synthesis by regulating translation initiation is mediated by eIF2 kinases. Figure 5-36 summarizes the steps in translation initiation. Translation initiation factor eIF2 brings the charged initiator tRNA to the small ribosome subunit P site. eIF2 is a trimeric G protein and exists in either a GTP-bound or a GDP-bound conformation. Only the GTP-bound form of eIF2 is able to bind the charged initiator tRNA and associate with the small ribosomal subunit. The small subunit with bound initiation factors and charged initiator tRNA then interacts with the eIF4 complex bound to the cap of an mRNA via its eIF4E subunit. The small ribosomal subunit then scans down the mRNA in the direction until it reaches an AUG initiation codon that can base-pair with the initiator tRNA in its P site. When this occurs, the GTP bound by eIF2 is hydrolyzed to GDP and the resulting eIF2·GDP complex is released. GTP hydrolysis results in an irreversible “proofreading” step that prepares the small ribosomal subunit to associate with the large subunit only when an initiator tRNA is properly bound in the P site and is properly base-
paired with the AUG start codon. Before eIF2 can participate in another round of initiation, its bound GDP must be replaced with a GTP. This process is catalyzed by the translation initiation factor eIF2B, a guanine nucleotide exchange factor (GEF) specific for eIF2. A mechanism for inhibiting general protein synthesis in stressed cells involves phosphorylation of the eIF2α subunit at a specific serine. Phosphorylation at this site does not interfere with eIF2 function in protein synthesis directly. Rather, phosphorylated eIF2 has very high affinity for the eIF2 guanine nucleotide exchange factor, eIF2B, which cannot release the phosphorylated eIF2 and consequently is blocked from catalyzing GTP exchange by additional eIF2 factors. Since there is an excess of eIF2 over eIF2B, phosphorylation of a fraction of eIF2 results in inhibition of all the cellular eIF2B. The remaining eIF2 accumulates in its GDP-bound form, which cannot participate in protein synthesis, thereby inhibiting nearly all protein synthesis in the cell. However, some mRNAs have regions that allow translation initiation at the low eIF2-GTP concentration that results from eIF2 phosphorylation. These mRNAs include those for chaperone proteins that function to refold cellular proteins denatured as the result of cellular stress, additional proteins that help the cell to cope with stress, and transcription factors that activate transcription of the genes encoding these stress-induced proteins. Human cells contain four eIF2 kinases that phosphorylate the same inhibitory eIF2 α serine. Each of these is regulated by a different type of cellular stress, inhibiting protein synthesis and allowing cells to divert the
large fraction of cellular resources usually devoted to protein synthesis in growing cells for use in responding to the stress: The GCN2 (general control non-derepressible 2) eIF2-kinase is activated by binding uncharged tRNAs. The concentration of uncharged tRNAs increases when cells are starved for amino acids, activating GCN2 eIF2-kinase activity and greatly inhibiting protein synthesis. PEK (pancreatic eIF2 kinase) is activated when proteins translocated into the endoplasmic reticulum (ER) do not fold properly because of abnormalities in the ER lumen environment. Inducers include abnormal carbohydrate concentrations, because this inhibits the glycosylation of many ER proteins. Inactivating mutations in an ER chaperone required for proper folding of many ER proteins (Chapters 13 and 14) also results in PEK activation. Heme-regulated inhibitor (HRI) is another eIF2 kinase activated in developing red blood cells when the supply of the heme prosthetic group is too low to accommodate the rate of globin protein synthesis. This negative feedback loop lowers the rate of globin protein synthesis until it matches the rate of heme synthesis. HRI is also activated in other types of cells in response to oxidative stress or heat shock. Protein kinase RNA activated (PKR) is activated by double-stranded RNAs longer than base pairs. Under normal circumstances in mammalian cells, such double-stranded RNAs are produced only during a viral infection. Long regions of double-stranded RNA are generated in replication intermediates of RNA viruses or from hybridization of complementary regions of RNA transcribed from both strands of DNA virus genomes. Inhibition of protein synthesis
Sequence-Specific RNA-Binding Proteins Control Specific mRNA Translation
prevents the production of progeny virions, protecting neighboring cells from infection. Interestingly, adenoviruses evolved a defense against PKR: they express prodigious amounts of an -nucleotide virus-associated (VA) RNA with long double-stranded hairpin regions. VA RNA is transcribed by RNA polymerase III and exported from the nucleus by Exportin5, the exportin for pre-miRNAs (see
Figure 9-32). VA RNA binds to PKR with high affinity, inhibiting its protein kinase activity and preventing the inhibition of protein synthesis observed in cells infected with a mutant adenovirus from which the VA gene was deleted. Sequence-Specific RNA-Binding Proteins Control Specific mRNA Translation In contrast to global mRNA regulation, mechanisms have also evolved for controlling the translation of certain specific mRNAs. This is usually done by sequence-specific RNA-binding proteins that bind to a particular sequence or RNA structure in the mRNA. When binding is in the untranslated region ( UTR) of an mRNA, the 40S ribosomal subunit’s ability to scan to the first initiation codon is blocked, inhibiting translation initiation. Binding in other regions can either promote or inhibit mRNA degradation. Control of intracellular iron concentration is an elegant example of the protein-mediated regulation of the translation of one mRNA and the
degradation of another. This is accomplished by Iron Response Element Binding Proteins (IRE-BPs), including the bifunctional protein aconitase 1 (encoded by the ACO1 gene, alias IRE-BP1) and its paralog IRE-BP2 (encoded by the IREB2 gene). Precise regulation of cellular iron ion concentration is critical to the cell. Multiple enzymes and proteins contain as a cofactor, such as enzymes of the TCA cycle (see Figure 12-16) and electron-carrying proteins involved in the generation of ATP by mitochondria and chloroplasts (Chapter 12). On the other hand, excess generates free radicals that react with and damage cellular macromolecules. When intracellular iron stores are low, a dual-control system operates to increase the level of cellular iron; when iron is in excess, the system operates to store the iron in an inert form that prevents accumulation of toxic levels of free ions. IRE-BPs are 4Fe-4S cluster-containing proteins that act as cellular iron sensors. When the cytosolic concentration is too low, the iron in the Fe-S clusters dissociates from the protein and the IRE-BP becomes an active RNA-binding protein that binds to a specific nucleotide sequence within stem-loop secondary structures in the untranslated regions of the ferritin and transferrin receptor mRNAs. These Iron Response Elements (IREs) in these mRNAs are located in the untranslated region of the ferritin mRNAs (Figure 9-35a) and in the untranslated sequence in the transferrin receptor (TfR) mRNA. Ferritin is composed of 24 subunits of a ferritin heavy chain and an equal number of ferritin light chains. They form a large intracellular protein complex that binds and stores excess cellular iron. When an IRE-BP is in its RNA-binding conformation at low
cellular concentration, it recognizes and binds five specific bases in the IRE loop and the duplex nature of the stem in the mRNA for both heavy and light ferritin chains (Figure 9-35a). The bound IRE-BP blocks the small ribosomal subunit from scanning for the AUG start codon (see
Figure 5-36), thereby inhibiting translation initiation. This causes a decrease in ferritin protein production and results in less iron being stored within the ferritin complex, making more iron available to iron-requiring enzymes. At high iron concentrations, the IRE-BP is in a conformation where it cannot bind the IREs in the untranslated region of the ferritin mRNAs. Consequently, ferritin translation is not inhibited, and newly synthesized ferritin can store free , thus prevent its accumulation to harmful levels within the cell.
FIGURE 9-35 Iron-dependent regulation of mRNA translation and degradation. The iron response element–binding protein (IRE-BP) controls (a) translation of ferritin mRNA and (b) degradation of transferrin-receptor (TfR) mRNA. At low intracellular iron concentrations IRE-BP binds to iron-response elements (IREs) in the or untranslated region of these mRNAs. At high iron concentrations, IRE-BP undergoes a conformational change and cannot bind either mRNA. The dual control by IRE-BP precisely regulates the level of free iron ions within cells. See the text for discussion. Description
Part (a) shows ferritin m R N A with two step-loop shaped I R Es toward 5-prime capped end and high iron coding region toward poly (A) end. This produces a translated ferratin. However, when aconitase in inactive R N A-binding conformation is converted to aconitase in conformation active for R N A-binding, it binds to the I R Es and high iron coding region changes to low iron coding region. As a result, no translation initiation occurs. Part (b) shows T f R m R N A with three step-loop shaped I R Es with A U-rich elements in the step toward poly (A) end and high iron coding region toward 5-prime capped end. This results in degraded mononucleotides. However, when inactive I R EB P is converted to active I R E-B P, it binds to the I R Es and high iron coding region changes to low iron coding region. As a result, little degradation occurs. The second mRNA regulated by IRE-BPs is regulated differently: it encodes the transferrin receptor (TfR). In vertebrates, iron is carried through the circulatory system bound to a protein called transferrin. After binding to the transferrin receptor (TfR) in the plasma membrane of most cell types, the transferrin-iron complex is brought into cells by receptormediated endocytosis (see Chapter 14; Figure 14-31). The -untranslated region of TfR mRNA contains multiple IREs with AU-rich element destabilizing sequences interspersed between the stem-loop structures (see above). At high concentrations, when the IRE-BP is unable to bind to the IRE, these AU-rich elements promote degradation of TfR mRNA by the same mechanism that leads to rapid degradation of other short-lived mRNAs with AU-rich elements, as described previously. The resulting decrease in production of the transferrin receptor quickly reduces iron import, thus protecting the cell from excess iron. At low , concentrations, however, the IRE-BP binds to the IREs in TfR mRNA and blocks access of proteins to the destabilizing AU-rich elements. As a
Surveillance Mechanisms Prevent Translation of Improperly Processed mRNAs
result, TfR mRNA levels increase, leading to increased production of the transferrin receptor protein and more iron transport into the cell. As a consequence of these two mechanisms of post-translational control, regulation of translation of the ferritin mRNAs and regulation of stability of the transferrin mRNA, the intracellular concentration is held at the concentration required for binding of to iron-requiring enzymes and is prevented from reaching higher concentrations that would be toxic. Other regulated RNA-binding proteins may also function to control mRNA translation or degradation. For example, a heme-sensitive RNA-binding protein controls translation of the mRNA encoding aminolevulinate (ALA) synthase, a key enzyme in the synthesis of heme. Similarly, in vitro studies have shown that the mRNA encoding the milk protein casein is stabilized by the hormone prolactin and rapidly degraded in its absence. Surveillance Mechanisms Prevent Translation of Improperly Processed mRNAs Translation of an improperly processed mRNA could lead to production of a nonfunctional or abnormally functioning protein. This effect is equivalent to that resulting from dominant-negative mutations, discussed in Chapter 6 (see Figure 6-2). Several mechanisms collectively termed mRNA surveillance help cells avoid the translation of improperly processed mRNA molecules. We have previously mentioned two such surveillance mechanisms: the recognition of improperly processed pre-
mRNAs in the nucleus and their degradation by the nuclear exosome, and the general restriction against nuclear export of incompletely spliced premRNAs that remain associated with a spliceosome assembly intermediate. Nonsense-Mediated Decay (NMD) This mechanism of mRNA surveillance causes degradation of mRNAs in which one or more exons have been incorrectly spliced. Such incorrect splicing will often alter the open reading frame of the mRNA to the improper exon junction, resulting in introduction of an out-of-frame missense mutation and an incorrect stop codon. For nearly all properly spliced mRNAs, the stop codon is in the last exon. The process of nonsense-mediated decay results in the rapid degradation of mRNAs with stop codons that occur before the last splice junction in the mRNA, since in most cases such mRNAs arise from errors in RNA splicing. However, NMD can also result from a mutation creating a stop codon within a gene or a frame-shifting deletion or insertion. NMD was initially discovered during the study of patients with -thalasemia, who produce a low level of β-globin protein associated with a low level of β-globin mRNA (Figure 9-36).
FIGURE 9-36 Discovery of nonsense-mediated mRNA decay (NMD). (a) Patients with -thalassemia express very low levels of β-globin mRNA. A common cause of this syndrome is a single-base-pair deletion in exon 1 or exon 2 of the β-globin gene. Ribosomes translating the mutant mRNA read out of frame following the deletion and encounter a stop codon in the wrong reading frame before they translate across the last exon junction in the mRNA. Consequently, they do not displace an exon-junction complex (EJC) from the mRNA. Cytoplasmic proteins associate with the EJC and induce degradation of the mRNA. (b) Bone marrow was obtained from a patient with a wild-type β-globin gene and from a patient with -thalassemia. RNA was isolated from the bone marrow cells shortly after collection, or 30 minutes after incubation in media with Actinomycin D, a drug that inhibits transcription. The amount of -globin RNA was measured using the S1-nuclease protection method (arrow). The patient with -thalassemia had much less β-globin mRNA than the patient with a wild-type β-globin gene (−Act D). The mutant β-globin mRNA decayed rapidly when transcription was inhibited (+Act D), whereas the wild-type β-globin mRNA remained stable.
[Republished with permission from Elsevier, from L. E. Maquat et al., 1981, “Unstable β-Globin mRNA in mRNA-Deficient Thalassemia,” Cell 27(3):543–553; permission conveyed through Copyright Clearance Center, Inc.] Description (a) The structure if beta-globin genomic D N A starts with an A U G codon and terminates with a poly (A) site. Exons are present from 1 to 31, 32 to 105, and 106 to 147 with a C G deletion site in exon 2, between 32 and 105 base pairs. The illustration (b) shows two test spots comparing beta-globin m R N A. The patient with wild-type beta-globin gene shows large spots representing beta-globin m R N A in presence and absence of Actinomycin D. The patient with beta superscript 0-thalassemia shows a small spot in absence of Actinomycin D and a very faint spot in presence of Actinomycin D. A search for possible molecular signals that might indicate the positions of splice junctions in a processed mRNA led to the discovery of exonjunction complexes. As noted already, these complexes of several proteins (including a core complex of eIF4AIII, MLN51, MAGOH, and Y14 proteins and peripheral factors including REF), bind nucleotides to an exon-exon junction following RNA splicing, and stimulate export of mRNPs from the nucleus by interacting with the mRNA exporter, NXT1/NXF1 (see Figure 9-26). Analysis of yeast mutants indicated that one of the peripheral proteins associated with exon-junction complexes in the nucleus, UPF3, functions in nonsense-mediated decay (see Figure 937, step 1 ). UPF3 remains associated with the EJC as it is exported through a nuclear pore complex, and associates with the cytoplasmic NMD factor UPF2 (see Figure 9-37, step 2 ). During translation termination, eukaryotic release factors 1 and 3 (eRF1 and eRF3) associate with ribosomes with a stop codon in the A site (see Figure 5-38). NMD factors
UPF1 and SMG1, a protein kinase, associate with eRF1 and eRF3 forming a complex called the SURF complex (step 3 ) associated with the terminating ribosome. The SURF complex associates, via an interaction between UPF1 and UPF2, with any UPF2 that remains associated with EJC complexes on the mRNA that were not displaced from the mRNA by a translating ribosome (step 4 ). This leads to the phosphorylation of UPF1 by SMG1, which promotes the release of eRF1 and eRF3 and the binding of NMG factor SMG7. Finally, SMG7 causes the mRNP to associate with P bodies, inhibiting translation. UPF2 then binds a P body–associated deadenylase complex that rapidly removes the poly(A) tail from an associated mRNA, leading to its decapping and degradation by the P body–associated exonuclease Xrn1 (see Figure 9-30a).
FIGURE 9-37 (a) Nonsence Mediated Decay (NMD). Step 1 : An abnormal mRNA with a premature termination codon (PTC) is produced in the nucleus as the result of abnormal RNA splicing. An exon junction complex (EJC) is left at a splice junction following RNAsplicing and associates with NMD factor UPF3. Step 2 : After export to the cytoplasm, NMD factor UPF2 associates with UPF3 bound to the EJC. A ribosome also initiates
translation and reaches the PTC. Step 3 : Eukaryotic release factors 1 (eRF1) and 3 (eRF3) (see Figure 5-38) associate with the ribosome stopped at the termination codon and associate with UPF1 and protein kinase SMG1, forming a “SURF complex” associated with the terminating ribosome. Step 4 : Any EJCs associated with mRNA that were not displaced by a translating ribosome, interact with the terminating ribosome through an interaction between UPF2 associated with the EJC and UPF1 associated with the SURF complex, leading to SMG1 phorphorylation of UPF1. Step 5 : NMD factor SMG7 binds phosphorylated UPF1 as eRF1 and eRF3 are released. Step 6 : Bound SMG7 causes the mRNP complex to associate with P-bodies where translation is inhibited and the abnormal mRNA is degraded. (b) Non-stop decay. (c) No-go decay. Bottom, diagram of ribosomes “stacking up” to a stalled ribosome. See NL Garneau, J Wilusz, and CJ Wilusz Nature Reviews Mol Cell Biol 8:113 (2007); Part (c) Simms CL et al. (2017) Mol. Cell 68, 361– 373. Description The flowchart (a) titled nonsense-mediated decay shows a 6-step process starting from transport of an abnormal m R N A from nucleoplasm to cytoplasm where it is translated and finally decayed. The flowchart (b) titled non-stop decay shows an O R F segment with 5-prime U T R at m 7 G end and 3-prime U T R at poly (A) end. Several P A B P C 1 bound to the poly (A) tail. Next, ribosome translates through the poly (A) tail and stalls. The P A B P C 1 are removed and S k I 7 gets attached to the poly (A) tail. Next, either (1) S k I 7 recruits the exosome leading to 5-prime to 3-prime decay or (2) in the absence of S k I 7, loss of P A B P C 1 allows decapping and thus leading to 3-prime to 5-prime decay by X R N 1. The flowchart (c) no-go decay shows a strong stem-looped R N A structure in the middle of an O R F fragment. This leads to stalling of translation by Dom 34-H b s 1. Next, this leads to endonucleolytic cleavage and decay of fragments by exosome or X R N 1. Another illustration shows a ribosome being stalled near the 3-prime end that leads to stacking of trailing ribosomes behind it.
In the case of a properly spliced mRNA, the current hypothesis is that during translation of the mRNA by the first “pioneer” ribosome to translate the mRNA, EJCs are displaced from the mRNP. However, for mRNAs with a stop codon before the final exon junction, one or more EJCs remain associated with the mRNA, resulting in nonsense-mediated decay (Figure 9-37a). Non-Stop Decay This alternative mechanism of mRNA surveillance leads to the inhibition of translation and degradation of mRNAs that have been prematurely polyadenylated so that a translating ribosome does not encounter an inframe stop codon and translation continues to the end of the poly(A) where the ribosome arrests, still tightly bound to the mRNA (Figure 9-37b). Such ribosomes associated with the end of an mRNA without terminating translation by the normal mechanism involving peptide release factors (Figure 5-38) are recognized by the protein Ski7. The C-terminus of Ski7 is similar in structure to the GTPase domains of elongation factor-1A (EF1A) (see Figure 5-37) and peptide release factor eRF3 (see Figure 538) that bind to the empty A site of the ribosome. Ski7 is proposed to bind to the A site similarly, causing release of the ribosome from the mRNA. Ski7 then recruits the exosome to deadenylate and then rapidly decay the transcript in the direction. No-Go Decay
This is the final mechanism of mRNA quality control in the cytoplasm we will discuss in this chapter. This releases ribosomes that are stalled during translation due to damage to the mRNA template or the presence of an unusually large and stable secondary structure in the mRNA of the stop codon (see Figure 9-37c). A recent study indicates that ribosome collision is a critical trigger for mRNA cleavages that occur in the initial step of nogo decay. Ribosome collision occurs when ribosomes stack-up along the mRNA to the block to ribosome translocation (see Figure 9-37c, bottom). This was detected experimentally because mRNAs with engineered long, stable stem loops that block ribosome translocation (“nogo mRNAs”) exhibit regions of protection in 30 base intervals to the block when RNase is added to mildly cross-linked, permeabilized cells treated as in the procedure for chromatin immunoprecipitation (Figure 812a, Step 1 ). This protection of the no-go mRNA in 30-base intervals presumably results from protection of the mRNA in the channel of the 30S ribosomal subunits (Figure 5-34) stacked up along the mRNA. In vivo, this stacking up of ribosomes to the block to ribosome translocation causes endonucleolytic cleavage of the no-go mRNA. Although the endonuclease that does this has not been identified, it seems likely to be associated with the ribosome since deep sequencing revealed that cleavage sites in no-go mRNAs occur in regions of the mRNA within the mRNA channels of 30S subunits (see Figure 5-34) phased behind the block to translocation. In humans, the E3 ubiquitin ligase ZNF598 polyubiquitinylates several ribosomal proteins leading to their proteosomal degradation as well as degradation of nascent polypeptide and release of the ribosomal subunits from the cleaved ends of the no-go
Localization of mRNAs Permits Production of Proteins at Specific Regions Within the Cytoplasm
mRNA. This is followed by degradation of the fragment of the cleaved no-go mRNA by cytoplasmic exosomes and degradation of the fragment by cytoplasmic XRN1 (Figure 9-37c). Localization of mRNAs Permits Production of Proteins at Specific Regions Within the Cytoplasm Many cellular processes depend on localization of particular proteins to specific structures or regions of the cell. In later chapters, we examine how some proteins are transported after their synthesis to their proper cellular location. Alternatively, protein localization can be achieved by localization of mRNAs to specific regions of the cytoplasm in which their encoded proteins function. In most cases examined thus far, such mRNA localization is specified by sequences in the untranslated region of the mRNA. A recent genomic-level study of mRNA localization in Drosophila embryos revealed that percent of 3000 mRNAs analyzed were localized to specific subcellular regions, raising the possibility that this is a much more general phenomenon in all metazoa than previously appreciated. Localization of mRNAs to the Bud in S. Cerevisiae The most thoroughly understood example of mRNA localization occurs in the budding yeast S. cerevisiae. As discussed in Chapter 8, whether a haploid yeast cell exhibits the a or α mating type is determined by whether
a or α genes are present at the expressed MAT locus on chromosome III (see Figure 8-31). The process that transfers a or α genes from a silent mating-type locus near the telomeres of chromosome III to the expressed MAT locus is initiated by a sequence-specific endonuclease called HO. Transcription of the HO gene is dependent on the SWI/SNF chromatinremodeling complex (see Section 8.4). Daughter yeast cells that arise by budding from mother cells contain a transcriptional repressor called Ash1 (for Asymmetric synthesis of HO) that prevents recruitment of the SWI/SNF complex to the HO gene, thereby preventing its transcription. The absence of Ash1 from mother cells allows them to transcribe the HO gene. As a consequence, mother cells switch their mating type, while daughter cells generated by budding do not (Figure 9-38).
FIGURE 9-38 Mating-type switching in haploid budding yeast cells. Following nuclear DNA synthesis in S. cerevisiae, a haploid yeast cell undergoes mitosis and generates a small daughter cell from a daughter cell bud (Figure 1-24) and a larger mother cell [Cell division 1]. The resulting daughter and mother cells have the same mating type as the parent cell (α in this example, represented by purple color). Before the next cell division, the smaller daughter cell grows in size to the size of a mother cell and then divides [Cell division 2] into a smaller daughter cell and a larger mother cell that have the same mating type as the parental cell (α in this example). In contrast, the larger mother cell generated in Cell division 1 switches its mating type before DNA synthesis and divides into a mother and daughter cell with the opposite mating type (a in this example, represented by orange). This process continues in further divisions so that daughter cells have progeny of the same mating type, while the mother cells have progeny that have switched their mating type compared to their mother. Description The flowchart starts with a yeast cell with mating type alpha undergoing budding. Division results into a large mother cell and a small daughter cell, both with mating type alpha. This daughter cell undergoes division in the presence of A S H 1 wherein no H O transcription and no switching takes place, resulting into a large mother cell and a small daughter cell, both with mating type alpha. While the mother cell undergoes division in the absence of A S H 1 wherein H O transcription and switching takes place resulting into large mother cell and a small daughter cell, both with mating type a. This mother cell divides to produce a large mother cell and a small daughter cell, both with mating type alpha; while the daughter cell divides to produce a large mother cell and a small daughter cell, both with mating type a. Ash1 protein accumulates only in daughter cells because the mRNA encoding it is localized to daughter cells. The localization process requires three proteins: She2 (for SWI-dependent HO expression), an RNA-binding protein that binds specifically to a localization signal with a specific RNA structure in the ASH1 mRNA; Myo4, a myosin motor protein that moves
cargos on actin filaments (see Chapter 17); and She3, which links She2 and therefore ASH1 mRNA to Myo4 (Figure 9-39). ASH1 mRNA is transcribed in the nucleus of the mother cell before mitosis. Movement of Myo4 with its bound ASH1 mRNA along actin filaments that extend from the mother cell into the bud carries the ASH1 mRNA into the growing bud before cell division.
FIGURE 9-39 Model for restriction of mating-type switching to mother cells in S. cerevisiae. Ash1 protein prevents a cell from transcribing the HO gene whose encoded protein initiates the DNA rearrangement that results in mating-type switching from α to a or a to α. Switching occurs only in the mother cell, after it separates from a newly budded daughter cell, because the Ash1 protein is present only in the daughter cell. The molecular basis for this differential localization of Ash1 is the one-way transport of ASH1 mRNA into the bud. A linking protein, She2, binds to specific untranslated sequences in the ASH1 mRNA and also binds to She3 protein. This protein in turn binds to a myosin motor, Myo4, which moves along actin filaments into the bud. See S. Koon and B. J. Schnapp, 2001, Curr. Biol. 11:R166.
At least 23 other mRNAs were found to be transported by the She2, She3, Myo4 system. All have an RNA localization signal to which She2 binds, usually in their UTR. The process can be visualized in live cells by the experiment shown in Figure 9-40. RNAs can be fluorescently labeled by including in their sequence high-affinity binding sites for RNA-binding proteins, such as bacteriophage proteins MS2 coat protein and bacteriophage λN protein, that bind to different stem loops of specific sequence (Figure 9-40a). When such engineered mRNAs are expressed in budding yeast cells along with the bacteriophage proteins fused to proteins that fluoresce different colors, the fusion proteins bind to these specific RNA sequences, thereby labeling the RNAs that contain them with different colors. In the experiment shown in Figure 9-40b, ASH1 mRNA was labeled by the binding of green fluorescent protein fused to λN. Another mRNA localized to the bud by this system, the IST2 mRNA, encoding a component of the growing bud membrane, was labeled by the binding of red fluorescent protein fused to MS2 coat protein. Video of a budding cell showed that the differently labeled ASH1 and IST2 mRNAs accumulated in the same large cytoplasmic RNP particle containing multiple mRNAs in the mother cell cytoplasm, as can be seen from the merge of the green and red fluorescent signals. The RNP particle was then transported into the bud within about one minute.
EXPERIMENTAL FIGURE 9-40 Transport of mRNP particles from a yeast mother cell into the bud. (a) Yeast cells were engineered to express an Ash1 mRNA with binding sites for the bacteriophage λN protein in its untranslated region and an IST2 mRNA with binding sites for bacteriophage MS2 coat protein in its untranslated region. A fusion of green fluorescent protein to λN protein (GFP-λN) and a fusion of red fluorescent protein to MS2 coat protein (RFP-MS2) also were expressed in the same cells. In other experiments,
these fluorescently tagged sequence-specific RNA-binding proteins were shown to bind to their own specific binding sites engineered into the ASH1 and IST2 mRNAs, and not to each others’ binding sites. Both fluorescently tagged proteins also contained a nuclear localization signal so that the fluorescent proteins that were not bound to their high-affinity binding sites in these mRNAs were transported into nuclei through nuclear pore complexes (see Chapter 13). This was necessary to prevent high fluorescence from excess GFP-λN and RFP-MS2 in the cytoplasm. At the right, GFP-λN and RFP-MS2 were independently visualized by using millisecond alternating laser excitation of GFP and RFP. (b) Frames from a video of fluorescing cells are shown. The nucleus next to the large vacuole in the mother cell near the center of the micrographs, as well as nuclei in neighboring cells, was observed by green and red fluorescence as shown in the top and middle rows. A merge of the two images is shown in the bottom row, which also indicates the time elapsed between images. An RNP particle containing both the ASH1 mRNA with λN-binding sites and the IST2 mRNA with MS2-binding sites was observed in the mother cell cytoplasm in the left column of images (arrow). The particle increased in intensity between 0.00 and 46.80 seconds, indicating that more of these mRNAs joined the RNP particle. The RNP particle was transported into the bud between 46.80 and 85.17 seconds and then became localized to the bud tip. [Republished with permission of John Wiley & Sons, Inc., from Lange, S. et al., ”Simultaneous transport of different localized mRNA species revealed by live-cell imaging,” 2008, Traffic, 9:(8)1256–67; permission conveyed through Copyright Clearance Center, Inc.] Description The two illustration in part (a) show an A S H 1 m R N A and an I S T 2 m R N A structure with poly (A) tail highlighting binding sites for G F P-lambda N in A S H 1 and R F P-M S 2 in I S T 2 m R N As. The part (b) shows three rows with six video frames of fluorescing cells. Row 1 shows green colored cells, row 2 shows red colored cells, and row 3 shows yellow colored cells. An arrow in each frame point to a corresponding colored dot of R N P particle with color intensity increasing from left to right.
As discussed above in the section on P bodies, cytoplasmic sites of mRNA decay, formation of the large cytoplasmic granules of RNPs observed here (Figure 9-40b) and in other examples of transported RNA granules in cells of multicellular eukaryotes requires sequences in the RNA-binding protein composed of a low complexity amino acid sequence such as repeats of [G/S]Y[G/S]. Peptides containing these low complexity sequences spontaneously associate in vitro by a process referred to as “liquid-liquid phase separation” discussed in Chapter 8 in regard to formation of transcriptional condensates in the nucleus (Figure 8-38). The condensates formed in vitro from these peptides with simple sequence repeats can be dissociated by phosphorylation of serines within them. Such liquid-liquid phase separated droplets are probably involved in the formation of the large RNP complexes transported on actin cables in yeast and on microtubules (see Chapters 17 and 18) in large asymmetric cells in higher eukaryotes, such as in the axons and dendrites of nerve cells (see Chapter 23) described in the next section. Regulated phosphorylation of these low complexity sequences in RNA-binding proteins associated with RNP granules may well account for the regulated formation and dissociation of RNP granules such as those observed in Figure 9-40b. Localization of mRNAs to Synapses in the Mammalian Nervous System As mentioned earlier, in neurons, localization of specific mRNAs at synapses far from the nucleus in the cell body plays an essential function in learning and memory (Figure 9-41). Like the localized mRNAs in yeast,
these mRNAs contain RNA localization signals in their untranslated region. Some of these mRNAs are initially synthesized with short poly(A) tails that do not allow translation initiation. Once again, large RNP particles containing multiple mRNAs bearing localization signals form in the cytoplasm near the cell nucleus. In this case, the RNP particles are transported down the axon to synapses by kinesin motor proteins that travel down microtubules extending the length of the axon (see Chapter 18). Membrane depolarization at a given synapse by the regulated opening of ion channels in the plasma membrane (see Chapter 23) may then stimulate the mRNAs’ polyadenylation in the region of the synapse, activating the translation of encoded proteins that increase the size and alter the neurophysiological properties of the one synapse while leaving unaffected the hundreds to thousands of other synapses made by the same neuron. EXPERIMENTAL FIGURE 9-41 A specific neuronal mRNA localizes to synapses. Sensory neurons from the sea slug Aplysia californica were cultured with target motor neurons so that axons from the sensory neurons formed synapses with dendrites from the motor neurons in culture. The micrograph at the left shows motor neuron axons visualized
with a blue fluorescent dye. GFP-VAMP (green) was expressed in sensory neurons and marks the location of synapses formed between sensory neuron axons and motor neuron dendrites (arrows). The micrograph on the right shows red fluorescence from in situ hybridization of an anti-sensorin mRNA probe. Sensorin is a neurotransmitter expressed only by the sensory neuron; sensory neuron axons are not otherwise visualized in this preparation, but they lie adjacent to the motor neuron dendrites. The in situ hybridization results indicate that sensorin mRNA in the sensory neuron is localized to synapses. [Republished with permission of Elsevier, from Lyles, V., et al., “Synapse formation and mRNA localization in cultured Aplysia neurons,” Neuron, 2006, 49(3):349–356; permission conveyed through Copyright Clearance Center, Inc.] KEY CONCEPTS OF SECTION 9.4 Cytoplasmic Mechanisms of Post-Transcriptional Control Most mRNAs are degraded in the cytoplasm as the result of the gradual shortening of the poly(A) tail (deadenylation) followed by exosome-mediated digestion, or removal of the cap and digestion by a exoribonuclease, XRN1 (see
Figure 9-30). Eukaryotic mRNAs encoding proteins that are expressed in short bursts generally have repeated copies of an AU-rich sequence (AU-rich element) in their UTR. Specific proteins that bind to these elements also interact with a deadenylating enzyme complex and cytoplasmic exosomes, promoting rapid mRNA degradation. Translation can be repressed by microRNAs (miRNAs), which form imperfect hybrids with sequences in the untranslated region (UTR) of specific target mRNAs. mRNAs bound by several miRNAs are concentrated in P bodies in the cytoplasm, where they are degraded by decapping followed by digestion by the cytoplasmic exosome. The related phenomenon of RNA interference (RNAi), which probably evolved as an early defense system against viruses and transposons, leads to rapid degradation of mRNAs that form perfect hybrids with short interfering RNAs (siRNAs). Both miRNAs and siRNAs are 21–23 nucleotides in length, are generated from longer precursor molecules and are bound by an Argonaute protein and assembled into a multiprotein RNA-induced silencing complex (RISC). RISC complexes either repress translation of target mRNAs and induce their localization to P bodies, where they are degraded (miRNAs), or cleave them (siRNAs), generating unprotected ends that are
rapidly degraded by cytoplasmic exosomes and the exonuclease XRN1 (see Figures 9-31 and 9-32). Cytoplasmic polyadenylation is required for the translation of mRNAs with a short poly(A) tail. This step is regulated in cells: binding of a specific protein to regulatory elements in the UTRs represses translation of these mRNAs, and phosphorylation of this RNA-binding protein leads to lengthening of the poly(A) tail and thus translation (see Figure 9-34). The global rate of cellular protein synthesis is regulated in part by controlling the activity of eukaryotic initiation factor 2 (eIF2) required at an early step in selecting mRNAs for translation (see Figure 5-24). eIF2 kinases respond to various types of cellular stress by phosphorylating a regulatory site in eIF2, inhibiting eIF2 exchange of GTP for GDP and consequently inhibiting an early step in protein synthesis. In addition to regulation of the global rate of protein synthesis by most mRNAs, the translation, stability, and cytoplasmic localization of specific mRNAs can be controlled. This is generally accomplished by sequence-specific RNA-binding proteins that bind to regulatory elements in the or less commonly the UTRs of some mRNAs. For example, translation of ferritin mRNA and degradation of transferrin receptor (TfR) mRNA are both regulated by the same iron-sensitive RNAbinding proteins, IREBP1 and 2. At low iron concentrations, these proteins have an active conformation that binds to specific sequences that form stem-loops in the mRNAs, inhibiting ferritin mRNA translation and degradation of TfR mRNA (see
Figure 9-35). This dual control precisely regulates the intracellular concentration. Nonsense-mediated decay and other mRNA surveillance mechanisms prevent the translation of improperly processed mRNAs encoding abnormal proteins that might interfere with the functioning of the corresponding normal proteins. Many mRNAs are transported to specific subcellular locations by sequence-specific RNA-binding proteins that bind localization sequences usually found in the UTR. These RNA-binding proteins then associate, directly or via intermediary proteins, with motor proteins that carry large RNP particles, containing many mRNAs bearing the localization signals, on actin or microtubule fibers to specific locations in the cytoplasm.
9.5 Processing of rRNA and tRNA
9.5 Processing of rRNA and tRNA Approximately 80 percent of the total RNA in rapidly growing mammalian cells (e.g., cultured HeLa cells) is rRNA, and 15 percent is tRNA; protein-coding mRNA thus constitutes only a small portion of the total RNA. The primary transcripts produced from most rRNA genes and from tRNA genes, like pre-mRNAs, are extensively processed to yield the mature, functional forms of these RNAs. The ribosome is a highly evolved, complex structure (see Figure 5-34), optimized for its function in protein synthesis. Ribosome synthesis requires the function and coordination of all three nuclear RNA polymerases, RNA pol I, II, and III. The 28S and 5.8S rRNAs associated with the large ribosomal subunit and the single 18S rRNA of the small subunit are transcribed by RNA polymerase I. The 5S rRNA of the large subunit is transcribed by RNA polymerase III, and the mRNAs encoding the ribosomal proteins are transcribed by RNA polymerase II. In addition to the four rRNAs and ribosomal proteins, at least 150 other RNAs and proteins interact transiently with the two ribosomal subunits during their assembly through a series of coordinated steps. Furthermore, multiple specific bases and riboses of the mature rRNAs are modified to optimize their function in protein synthesis. Although most of the steps in ribosomal subunit synthesis and assembly occur in the nucleolus, some occur in the nucleoplasm during passage from the nucleolus to nuclear pore complexes. A quality-control step occurs before nuclear export so
Pre-rRNA Genes Are Similar in All Eukaryotes and Function as Nucleolar Organizers
that only fully functional subunits are exported to the cytoplasm, where the final steps of ribosome subunit maturation occur. tRNAs also are processed from precursor primary transcripts in the nucleus and modified extensively before they are exported to the cytoplasm and used in protein synthesis. First, we’ll discuss the processing and modification of rRNA and the assembly and nuclear export of ribosomes. Then we’ll consider the processing and modification of tRNAs. Pre-rRNA Genes Are Similar in All Eukaryotes and Function as Nucleolar Organizers The 28S and 5.8S rRNAs associated with the large (60S) ribosomal subunit and the 18S rRNA associated with the small (40S) ribosomal subunit in multicellular eukaryotes (and the functionally equivalent rRNAs in all other eukaryotes) are encoded by a single type of pre-rRNA transcription unit. In human cells, transcription by RNA polymerase I yields a 45S primary transcript (pre-rRNA), which is processed into the mature 28S, 18S, and 5.8S rRNAs found in cytoplasmic ribosomes. The fourth rRNA, 5S, is encoded separately and transcribed outside the nucleolus by RNA polymerase III. Sequencing of the DNA encoding the 45S pre-rRNA from many species showed that this DNA shares several properties in all eukaryotes. First, the pre-rRNA genes are arranged in long tandem arrays separated by nontranscribed spacer regions ranging in length from in frogs to in humans (Figure 9-42). Second, the genomic regions corresponding to the three mature rRNAs are
always arranged in the same order: 18S, 5.8S, and 28S. Third, in all eukaryotic cells (and even in bacteria), the pre-rRNA gene codes for regions that are removed during processing and rapidly degraded. These regions probably contribute to proper folding of the rRNAs but are not required once the folding has occurred. The general structure of pre-rRNA genes in several organisms is diagrammed in Figure 9-43.
EXPERIMENTAL FIGURE 9-42 Electron micrograph of pre-rRNA transcription units from the nucleolus of a frog oocyte. Each “feather” represents multiple pre-rRNA molecules associated with protein in a pre-ribonucleoprotein complex (pre-rRNP) emerging from a transcription unit. Note the dense “knob” at the end of each nascent pre-RNP thought to be a processome. Pre-rRNA transcription units are arranged in tandem, separated by nontranscribed spacer regions of nucleolar chromatin.
Description The micrograph shows a slightly curved vertical strand, labeled nuclear chromatin, with feather-like structure, labeled transcription unit, at the top and at the bottom. The feather branches end into bulb-like structures and are labeled nascent pre-r R N P. The central region of the strand is labeled nontranscribed spacer. An upward arrow indicates direction of transcription.
FIGURE 9-43 General structure of eukaryotic pre-rRNA transcription units. The three coding regions (red) encode the 18S, 5.8S, and 28S rRNAs found in ribosomes of multicellular eukaryotes or their equivalents in other species. The order of these coding regions in the genome is always . Variations in the lengths of the transcribed spacer regions (blue) account for the major difference in the lengths of pre-rRNA transcription units in different organisms. Description The illustration shows bar models of four transcription units along with their sizes as follows: Human, about 13.7 kilobases; X. laevis (frog), about 7.9 kilobases; D. melanogaster (fruit fly), about 7.7 kilobases; and S. cerevisisae (yeast), about 6.6
Small Nucleolar RNAs Assist in Processing Pre-rRNAs
kilobases. All pre-r R N A transcription units contain alternative transcribed spacer and regions preserved in r R N A. Three coding regions highlighted in the human unit are 18 S, 5.8 S, and 28 S, respectively. The synthesis and most of the processing of pre-rRNA occurs in the nucleolus. When pre-rRNA genes initially were identified in the nucleolus by in situ hybridization, it was not known whether any other DNA was required to form the nucleolus. Subsequent experiments with transgenic Drosophila strains demonstrated that a single complete pre-rRNA transcription unit induces formation of a small nucleolus. Thus a single pre-rRNA gene is sufficient to be a “nucleolar organizer,” and all the other components of the ribosome diffuse to the newly formed pre-rRNA. The structure of the nucleolus observed by light and electron microscopy results primarily from the processing of pre-RNA and the assembly of ribosomal subunits. Small Nucleolar RNAs Assist in Processing Pre-rRNAs Ribosomal subunit assembly, maturation, and export to the cytoplasm are best understood in the yeast S. cerevisiae. However, nearly all the proteins and RNAs involved are highly conserved in multicellular eukaryotes, where the fundamental aspects of ribosome biosynthesis are likely to be the same. As we have seen with pre-mRNAs, nascent pre-rRNA transcripts are immediately bound by proteins, forming pre-ribosomal ribonucleoprotein particles (pre-rRNPs). Cleavage of the pre-rRNA does
not begin until transcription of the pre-rRNA is nearly complete. In yeast, it takes approximately 6 minutes for a pre-rRNA to be transcribed. Once transcription is complete, the rRNA is cleaved, and bases and riboses are modified in about 10 seconds. In a rapidly growing yeast cell, pairs of ribosomal subunits are synthesized, processed, and transported to the cytoplasm every second. This extremely high rate of ribosome synthesis despite the seemingly long period required to transcribe a pre-rRNA is possible because pre-rRNA genes are packed with RNA polymerase I molecules transcribing the same gene simultaneously (see Figure 9-42) and because there are 100–200 such genes on chromosome XII, the yeast nucleolar organizer. The primary transcript of is cut in a series of cleavage and exonucleolytic steps that ultimately yield the mature rRNAs found in ribosomes (Figure 9-44). During processing, pre-rRNA is also extensively modified, mostly by methylation of the -hydroxyl group of specific riboses and conversion of specific uridine residues to pseudouridine. Virtually all of these modifications occur in the most conserved core structure of the ribosome, which is directly involved in protein synthesis. The positions of the specific sites of -O-methylation and pseudouridine formation are determined by approximately 150 different small nucleolusrestricted RNA species, called small nucleolar RNAs (snoRNAs), which hybridize transiently to pre-rRNA molecules. Like the snRNAs that function in pre-mRNA processing, snoRNAs associate with proteins, forming ribonucleoprotein particles called snoRNPs. One class of more than 40 snoRNPs (box C/D containing snoRNAs, see Figure 9-45a) positions a methyltransferase enzyme near methylation sites in the pre-
mRNA. The multiple different box C/D snoRNAs direct methylation at multiple sites through a similar mechanism. They share common sequence and structural features, including the C and D box sequences (Figure 945a) and are bound by a common set of proteins. One or two regions of each of these snoRNAs are precisely complementary to sites on the prerRNA and direct the methyltransferase to specific riboses in the hybrid region (see Figure 9-45a). A second major class of snoRNPs (box H/ACA containing snoRNAs, Figure 9-45b) positions the enzyme that converts uridine to pseudouridine by rotation of the pyrimidine ring (Figure 9-45c). Bases on either side of the modified uridine in the pre-rRNA base-pair with bases in the bulge of a stem in the H/ACA snoRNAs, leaving the uridine to be modified bulged out of the helical double-stranded region, like the branch-point A bulges out in pre-mRNA spliceosomal splicing (see Figure 9-9d). Other modifications of pre-rRNA nucleotides, such as adenine dimethylation, are carried out by specific proteins without the assistance of guiding snoRNAs.
FIGURE 9-44 rRNA processing. Endoribonucleases that make internal cleavages are represented as scissors. Exoribonucleases that digest from one end, either or , are shown as Pac-Men. Most -O-ribose methylation and generation of pseudouridines in the rRNAs occurs following the initial cleavage at the end, before the initial cleavage at the end. Proteins and snoRNPs known to participate in these steps are indicated. See J. Venema and D. Tollervey, 1999, Ann. Rev. Genetics 33:261.
Description The process starts with a primary transcript made of alternative axons and introns. An endoribonuclease cleaves the 3-prime end intron which is digested by exoribonuclease Rat 1. Co-transcriptional endonucleolytic cleavage results in a 35 S r R N A. Box C plus D s n o R N Ps and Box H plus A C A s n o R N Ps enter the process, and methylation and pseudouridylation takes place. An endoribonuclease cleaves the 5prime end intron which is digested by an exosome. Next, cleavage occurs resulting in a 33 S r R N A which is further cleaved by an endoribonuclease at the 5-prime end and is digested by exoribonucleases X m 1 and Rat 1. This results in a 32 S r R N A which is further cleaved by an endoribonuclease M R P from the middle resulting in a 20 S r R N A and a 27 S A 2 r R N A. The 20 S r R N A is transported from the nucleus into the cytoplasm where it is further cleaved by exoribonuclease X r n 1 resulting in an 18 S r R N A. The 27 S A 2 which either gets 85 percent cleaved by endoribonuclease and digested by X R N 1 and Rat 1 exoribonuclease forming the 27 S A 3, or can be 15 percent cleaved by an exonuclease resulting in 27 S B l. The 27 S A 3 undergoes exonuclease processing and is digested by X R N 1 and Rat 1 exoribonuclease to result into 27 S B s. The 27 S B s undergoes processing cleavage by endoribonuclease to result into 25 S r R N A and 7 S s which is further digested by an exosome and exonuclease to finally result into 5.8 S s. Similarly, the The 27 S B l undergoes processing cleavage by endoribonuclease to result into 25 S r R N A and 7 S l which is further digested by an exosome and exonuclease to finally result into 5.8 S l.
FIGURE 9-45 snoRNP-directed modification of pre-rRNA. (a) SnoRNAs called box C/D snoRNAs are involved in ribose -O-methylation. Sequences in these snoRNAs hybridize to multiple different regions in the pre-rRNA, directing methylation at the indicated sites. (b) Box H/ACA snoRNAs fold into two stem loops with internal single-stranded bulges in the stems. Pre-rRNA hybridizes to the single-stranded bulges, demarcating a site of pseudouridylation. (c) Conversion from uridine to pseudouridine directed by the box H/ACA snoRNAs of part (b). See T. Kiss, 2001, EMBO J. 20:3617. Part (b) from U. T. Meier, 2005, Chromosoma 114:1. Description The illustration (a) shows box C slash D s n o R N As attached to the box C prime slash D prime s n o R N As with methylated pre-R N A strands present at both sides of attachment. The illustration (b) shows box H s n o R N A attached to another s n o R N A having box A C A. A pre-R N A strand passes through both s n o R N As with a uracil paired in each s n o R N A. The illustration (c) shows chemical structures of uridine and pseuouridine. The U3 snoRNA is assembled into a large snoRNP containing proteins called the small subunit (SSU) processome, which specifies cleavage at site , the initial cut near the end of the pre-rRNA (see
Figure 9-44). The processome helps to fold the 6.6-kb pre-rRNA into a structure required for the multiple cleavage events that follow transcription of the primary pre-rRNA transcript. U3 snoRNA base-pairs with an upstream region of the pre-rRNA to specify the location of the cleavage. The processome is thought to form the “ knob” visible in electron micrographs of pre-rRNPs (see Figure 9-42). Base pairing of other snoRNPs specify additional cleavage reactions that remove transcribed spacer regions. The first cleavage to initiate processing of the 5.8S and 25S rRNAs of the large subunit is performed by RNase MRP, a complex of nine proteins with an RNA. Once cleaved from pre-rRNAs, these sequences are degraded by the same exosome-associated nuclear exonuclease that degrades introns spliced from pre-mRNAs. Nuclear exoribonucleases (Rat1; Xrn1) also remove some regions of spacer. Some snoRNAs are expressed from their own promoters by RNA polymerase II or III. Remarkably, however, the large majority of snoRNAs are processed from spliced-out introns of genes encoding functional mRNAs for proteins involved in ribosome synthesis or translation. Some snoRNAs are processed from introns spliced from apparently nonfunctional mRNAs. The genes encoding these mRNAs seem to exist only to express snoRNAs from excised introns. Unlike 18S, 5.8S, and 28S genes, 5S rRNA genes are transcribed by RNA polymerase III in the nucleoplasm outside the nucleolus. With only minor additional processing to remove nucleotides at the end, 5S rRNA diffuses to the nucleolus, where it assembles with the pre-rRNA precursor
and remains associated with the region that is cleaved into the precursor of the large ribosomal subunit. Most of the ribosomal proteins of the small 40S ribosomal subunit associate with the nascent pre-rRNA during transcription (Figure 9-46). Cleavage of the full-length pre-rRNA in the 90S RNP precursor releases a pre-40S particle that requires only a few more remodeling steps, that is gain or loss of associated proteins, base modifications, and changes in conformation, before it is transported to the cytoplasm, as diagrammed in
Figure 9-46. Once the pre-40S particle leaves the nucleolus (purple in
Figure 9-46), it traverses the nucleoplasm (blue) quickly and is exported through nuclear pore complexes (NPCs) to the cytoplasm (tan), as discussed below. Final maturation of the small ribosomal subunit occurs in the cytoplasm: exonucleolytic processing of the 20S rRNA into mature small subunit 18S rRNA by the cytoplasmic exoribonuclease Xrn1 and the dimethylation of two adjacent adenines near the end of 18S rRNA by the cytoplasmic enzyme Dim1.
FIGURE 9-46 Ribosomal subunit assembly. Ribosomal proteins and RNAs in the maturing small and large ribosomal subunits are depicted in blue, with a shape similar to the icons for the mature subunits in the cytoplasm. Other factors that associate transiently with the maturing subunits are depicted in different colors, as shown in the key. See H. Tschochner and E. Hurt, 2003, Trends Cell Biol.13:255. Description In the nucleolus an r R N A is transcribed from an r D N A by R N A polymerase 1. This is translated into an S S U processome constituting U 3 s n o R N P and U 3associated factors which forms a pre-90 S large subunit. During the early stage, the pre90 S large subunit is cleaved into pre-60 S subunit and pre-40 S subunit along with removal of several U 3-associated factors and U 3 s n o R N P. Several r R N A processing or modification factors, G T Pases, helicases, and intranuclear transport (N o c proteins) attach to the pre-60 S subunit. This complex then enters the intermediate stage in the nucleoplasm by shedding all the r R N A processing or modification factors while attaching more intranuclear transport (N o c proteins).
Next, the pre-60 S subunit sheds all the intranuclear transport proteins and some helicases while attaching A A A A T Pase. Next, the complex enters the late stage in the nucleoplasm by shedding the remaining helicases while attaching export factors (N m d 3, N x t 1, Ran G T P) and some G T Pases. Next, the complex enters the cytoplasm by shedding the A A A A T Pase and some of the export factors and G T Pases. In the cytoplasm, the remaining export factors and G T Pases are removed resulting into a matured 60 S ribosomal subunit. Similarly, the pre-40 S subunit attached with few U 3-associated factors and export factors (N m d 3, N x t 1, Ran G T P) enters nucleoplasm where the remaining U 3associated factors are shed during the late stage and the pre-40 S subunit enters the cytoplasm. In the cytoplasm, the remaining export factors are shed by the pre-40 S subunit resulting into a matured 40 S ribosomal subunit. In contrast to the pre-40S particle, the precursor of the large subunit requires considerably more remodeling through many more transient interactions with nonribosomal proteins before it is sufficiently mature for export to the cytoplasm. Consequently, it takes a considerably longer period for the maturing 60S subunit to exit the nucleus (30 minutes compared to 5 minutes for export of the 40S subunit in cultured human cells). Multiple RNA helicases and small G proteins are associated with the maturing pre-60S subunits. Some RNA helicases are necessary to dislodge the snoRNPs that base-pair perfectly with pre-rRNA over up to 30 base pairs. Other RNA helicases may function in the disruption of protein-RNA interactions. The requirement for so many GTPases suggests that there are many quality-control checkpoints in the assembly and remodeling of the large subunit RNP, where one step must be completed before a GTPase is activated to allow the next step to proceed. Members of the AAA ATPase family also bind transiently. This class of proteins is often involved in large molecular movements and may be required to fold
the complex, large rRNA into the proper conformation. Some steps in 60S subunit maturation occur in the nucleoplasm (Figure 9-46, blue), during passage from the nucleolus to nuclear pore complexes. Much remains to be learned about the complex, fascinating, and essential remodeling processes that occur during formation of the ribosomal subunits. The large ribosomal subunit is one of the largest structures to pass through the nuclear pore complexes. Maturation of the large subunit in the nucleoplasm leads to the generation of binding sites for a nuclear export adapter called Nmd3. Nmd3 is bound by the nuclear transporter Exportin 1 (also called Crm1). This is another quality-control step, because only correctly assembled subunits can bind Nmd3 and be exported. The small subunit of the mRNP exporter (NXT1) also becomes associated with the nearly mature large ribosomal subunit. These nuclear transporters permit diffusion of the large subunit through the central channel of the NPC, which is filled with a cloud of unstructured protein domains that extend from the structured parts of proteins that line the wall of the channel (see
Chapter 13). Several additional subunits that form the walls of the NPC central channel are also required for ribosomal subunit export and may have additional functions specific for this task. The dimensions of ribosomal subunits ( in diameter) and the central channel of the NPC are comparable, so passage may not require distortion of either the ribosomal subunit or the channel. Final maturation of the large subunit in the cytoplasm includes removal of these export factors. As for the export of most macromolecules from the nucleus, including tRNAs and premiRNAs (but not most mRNPs), ribosome subunit export requires the function of a small G protein called Ran, as discussed in Chapter 13.
Self-Splicing Group I Introns Were the First Examples of Catalytic RNA
Self-Splicing Group I Introns Were the First Examples of Catalytic RNA During the 1970s, all the pre-rRNA genes of the protozoan Tetrahymena thermophila were discovered to contain an intron, indicating that splicing is required to produce mature rRNA in these organisms. In 1982, in vitro studies showing that the pre-rRNA was spliced at the correct sites in the absence of any protein provided the first indication that RNA can function as a catalyst, like protein enzymes. A whole raft of self-splicing sequences were subsequently found in prerRNAs from other single-celled organisms, in mitochondrial and chloroplast pre-rRNAs, in several pre-mRNAs from certain Escherichia coli bacteriophages, and in some bacterial tRNA primary transcripts. The self-splicing sequences in all these precursors, referred to as group I introns, use guanosine as a cofactor and can fold by internal base pairing to juxtapose closely the two exons that must be joined. As discussed earlier, certain mitochondrial and chloroplast pre-mRNAs and tRNAs contain a second type of self-splicing intron, designated group II. The splicing mechanisms used by group I introns, group II introns, and spliceosomes are generally similar, involving two transesterification reactions, which require no input of energy (Figure 9-47). Structural studies of the group I intron from Tetrahymena pre-rRNA combined with mutational and biochemical experiments revealed that the RNA folds into a precise three-dimensional structure that, like protein enzymes, contains
deep grooves for binding substrates and solvent-inaccessible regions that function in catalysis. The group I intron functions like a metalloenzyme to precisely orient the atoms that participate in the two transesterification reactions adjacent to catalytic ions. Considerable evidence now indicates that splicing by group II introns and by snRNAs in the spliceosome also involves bound catalytic ions. In both the group I and II self-splicing introns, RNA functions as a ribozyme, an RNA sequence with catalytic ability. As discussed above, spliceosomal splicing is also catalyzed by ions held in the proper positions by U6 snRNA and its interactions with U1 and U5 snRNAs (see Figure 9-14c), but only with considerable help from multiple proteins that also shape the catalytic site and help bind and release substrates.
FIGURE 9-47 Splicing mechanisms in group I and group II self-splicing introns and spliceosome-catalyzed splicing of pre-mRNA. The intron is shown in gray, the exons to be joined in red. In group I introns, a guanosine cofactor (G) that is not part of the RNA chain associates with the active site. The -hydroxyl group of this guanosine participates in a transesterification reaction with the phosphate at the end of the intron; this reaction is analogous to that involving the -hydroxyl groups of branch-site A’s in group II introns and pre-mRNA introns spliced in spliceosomes (see Figure 8-8). The subsequent transesterification that links the and exons is similar in all three splicing mechanisms. Note that spliced-out group I introns are linear structures, unlike the branched intron products in the other two cases. See P. A. Sharp, 1987, Science 235:769. Description The flowchart for self-splicing group 1 introns starts with a pre-m R N A having the following sequence from 5-prime to 3-prime end: dark pink exon with its phosphate linked to a looped intron whose other end is linked to the phosphate of a light pink exon. An arrow from 3-prime O H group of branch point G in the intron points to phosphate of dark pink exon. Next, an arrow from the 3-prime O H group on the spliced dark exon points to the phosphate of the light pink exon. The G is now bound to the open end of intron. Next, dark and light pink exons are linked together with a phosphate in between along with a spliced intron with P G on 5-prime end and O H on 3-prime end. The flowchart for self-splicing group 2 introns starts with a pre-m R N A having the following sequence from 5-prime to 3-prime end: dark pink exon with its phosphate linked to a looped intron whose other end is linked to the phosphate of a light pink exon. An arrow from 2-prime O H group of branch point A in the intron points to phosphate of dark pink exon. Next, an arrow from the 3-prime O H group on the spliced dark exon points to the phosphate of the light pink exon. The A is now paired to the open end of intron forming a loop. Next, dark and light pink exons are linked together with a phosphate in between along with a spliced looped intron with 5-prime phosphate paired with branch point A. The flowchart for spliceosome-catalyzed splicing of pre-m R N A is similar to selfsplicing group 2 introns except the first two steps occur inside the spliceosome.
Pre-tRNAs Undergo Extensive Modification in the Nucleus
Pre-tRNAs Undergo Extensive Modification in the Nucleus Mature cytosolic tRNAs, which average 75–80 nucleotides in length, are produced from larger precursors (pre-tRNAs) synthesized by RNA polymerase III in the nucleoplasm. Mature tRNAs also contain numerous modified bases that are not present in tRNA primary transcripts. Cleavage and base modification occur during processing of all pre-tRNAs; some pre-tRNAs also are spliced during processing. All of these processing and modification events occur in the nucleus. A sequence of variable length that is absent from mature tRNAs is present in all pre-tRNAs (Figure 9-48). This occurs because the end of mature tRNAs is generated by an endonucleolytic cleavage specified by the tRNA three-dimensional structure rather than the start site of transcription. These extra nucleotides are removed by ribonuclease P (RNase P), a ribonucleoprotein endonuclease. Studies with E. coli RNase P indicate that at high concentrations the RNA component alone can recognize and cleave E. coli pre-tRNAs. The RNase P polypeptide increases the rate of cleavage by the RNA, allowing it to proceed at physiological concentrations. A comparable RNase P functions in eukaryotes.
FIGURE 9-48 Changes that occur during the processing of tyrosine pre-tRNA. A 14nucleotide intron (blue) in the anticodon loop is removed by splicing. A 16-nucleotide sequence (green) at the end is cleaved by RNase P. U residues at the end are replaced by the CCA sequence (red) found in all mature tRNAs. Numerous bases in the stem-loops are converted to characteristic modified bases (yellow). Not all pre-tRNAs contain introns that are spliced out during processing, but they all undergo the other types of changes shown here. D = dihydrouridine; Ψ = pseudouridine. Description The first illustration shows a tyrosine pre-t R N A with two uracil bases at the 3-prime O H end, 14-nucleotide intron sequence in the anticodon loop, and 16-nucleotide sequence at the 5-prime end. After processing, the these sequences are removed in the matured tyrosine t R N A that now shows modified dihydrouridine bases in the D loop, modified pseudouridine and adenosine bases in the T Psi C G loop, modified pseudouridine bases in the anticodon loop, and A C G codon at the 3-prime O H end.
About 10 percent of the bases in pre-tRNAs are modified enzymatically during processing. Three classes of base modifications have been described (Figure 9-48): (1) U residues at the end of pre-tRNA are replaced with a CCA sequence. The CCA sequence is found at the end of all tRNAs and is required for their charging by amino-acyl-tRNA synthetases during protein synthesis. This step in tRNA synthesis likely functions as a quality-control point, since only properly folded tRNAs are recognized by the CCA addition enzyme. (2) Methyl and isopentenyl groups are added to the heterocyclic ring of purine bases, and the -OH groups in the ribose of specific residues are methylated. (3) Specific uridines are converted to dihydrouridine, pseudouridine, or ribothymidine residues. The functions of these base and ribose modifications are not well understood, but since they are highly conserved, they probably function in protein synthesis. As shown in Figure 9-48, the pre-tRNA expressed from the yeast tyrosine tRNA (tRNATyr) gene contains a 14-base intron that is not present in mature tRNATyr. Some other eukaryotic tRNA genes and some archaeal tRNA genes also contain introns. The introns in nuclear pre-tRNAs are shorter than those in pre-mRNAs and lack the consensus splice-site sequences found in pre-mRNAs (see Figure 9-7c). Pre-tRNA introns are also clearly distinct from the much longer self-splicing group I and group II introns found in chloroplast and mitochondrial pre-rRNAs. The mechanism of pre-tRNA splicing differs greatly from spliceosomal and self-splicing group I and group II intron splicing (see Figure 9-47). First, splicing of pre-tRNAs is catalyzed by proteins, not by RNAs. Second, a pre-tRNA intron is excised in one step that entails simultaneous cleavage
at both ends of the intron. Finally, hydrolysis of GTP and ATP is required to join the two tRNA halves generated by cleavage on either side of the intron. After pre-tRNAs are processed in the nucleoplasm, the mature tRNAs are transported to the cytoplasm through nuclear pore complexes by Exportint, a transporter (see Chapter 13) dedicated to the nuclear export of tRNAs. In the cytoplasm, tRNAs are passed between aminoacyl-tRNA synthetases, elongation factors, and ribosomes during protein synthesis (Chapter 5). Thus tRNAs generally are associated with proteins and spend little time free in the cell, as is also the case for mRNAs and rRNAs. KEY CONCEPTS OF SECTION 9.5 Processing of rRNA and tRNA A large precursor pre-rRNA (13.7 kb in humans) transcribed by RNA polymerase I undergoes cleavage, exonucleolytic digestion, and base modifications to yield mature 28S, 18S, and 5.8S rRNAs, which associate with ribosomal proteins into ribosomal subunits. Transcription and processing of pre-rRNA occur in the nucleolus. The 5S rRNA component of the large ribosomal subunit is synthesized in the nucleoplasm by RNA polymerase III. Approximately 150 snoRNAs, associated with proteins in snoRNPs, base-pair with specific sites in pre-rRNA, where they direct ribose methylation, modification of uridine to pseudouridine, and cleavage at specific sites during rRNA processing in the nucleolus. Group I and group II self-splicing introns, and probably snRNAs in spliceosomes, all function as ribozymes, or catalytically active RNA sequences, that carry out splicing by analogous transesterification reactions requiring bound ions (see Figure 947). Pre-tRNAs synthesized by RNA polymerase III in the nucleoplasm are processed by removal of the -end sequence, addition of CCA to the end, and modification of
multiple internal bases (see Figure 9-48). Some pre-tRNAs contain a short intron that is removed by a protein-catalyzed mechanism distinct from the splicing mechanisms used by pre-mRNAs and selfsplicing introns. All species of RNA molecules are associated with proteins in various types of ribonucleoprotein particles, both in the nucleus and after export to the cytoplasm.
9.6 Nuclear Bodies Are Functionally Specialized Nuclear Domains
9.6 Nuclear Bodies Are Functionally Specialized Nuclear Domains High-resolution visualization of plant- and animal-cell nuclei by electron microscopy and by staining with fluorescently labeled antibodies has revealed specialized nuclear domains, called nuclear bodies. These domains are not surrounded by membranes. Nonetheless, they are regions of high concentrations of specific proteins and RNAs that form distinct, often roughly spherical structures within the nucleus (Figure 9-49). The most prominent nuclear bodies are nucleoli, the sites of ribosomal subunit synthesis and assembly discussed earlier. Several other types of nuclear bodies also have been described in structural studies. Experiments with fluorescently labeled nuclear proteins have shown that the nucleus is a highly dynamic environment, with rapid diffusion of proteins through the nucleoplasm. Proteins associated with nuclear bodies are often also observed at lower concentrations in the nucleoplasm outside the nuclear bodies, and fluorescence studies indicate that they diffuse in and out of the nuclear bodies. Based on these measurements of molecular mobility in living cells, nuclear bodies can be mathematically modeled as the expected steady state for diffusing proteins that interact with sufficient affinity to form self-organized regions of high concentrations of specific proteins, but with low enough affinity for each other to be able to diffuse
Cajal Bodies
in and out of the structure. This is similar to the situation for transcriptional condensates discussed in Chapter 8 (Figure 8-38) and cytoplasmic ribonuclear protein particles involved in localization of mRNAs associated with motor proteins that “walk” on actin filaments and microtubules (see Figures 9-40b and 9-41). In electron micrographs, these structures appear to be a heterogeneous, spongelike network of interacting components. We discuss a few examples of nuclear bodies here. Cajal Bodies Cajal bodies are ∼0.2–1 μm spherical structures that have been observed in large nuclei for more than a century (Figure 9-49a). Current research indicates that like nucleoli, Cajal bodies are centers of RNP-complex assembly, in this case for spliceosomal snRNPs and other nuclear RNPs. Like rRNAs, snRNAs undergo specific modifications, such as the conversion of specific uridine residues to pseudouridine and addition of methyl groups to the -hydroxyl groups of specific riboses. These posttranscriptional modifications are important for the proper assembly and function of snRNPs in pre-mRNA splicing. These modifications occur in Cajal bodies, where they are directed by a class of snoRNA-like guide RNA molecules called scaRNAs (small Cajal body–associated RNAs). There is also evidence that the Cajal body is the site of reassembly of the U4/U6/U5 tri-snRNP complexes required for pre-mRNA splicing from the free U4, U5, and U6 snRNPs released during the removal of each intron (see Figure 9-13). Since Cajal bodies also contain a high concentration of the U7 snRNP involved in the specialized -end processing of the major
Nuclear Speckles
histone mRNAs, it is likely that this process also occurs in Cajal bodies, as may the assembly of the telomerase RNP. Nuclear Speckles Nuclear speckles were observed using fluorescently labeled antibodies to snRNP proteins and other proteins involved in pre-mRNA splicing, as approximately 25–50 irregular, amorphous structures 0.5–2 μm in diameter that are distributed through the nucleoplasm of vertebrate cells (Figure 9-49c, d). Since speckles are not located at sites of cotranscriptional pre-mRNA splicing, which are associated closely with chromatin, they are thought to be storage regions for snRNPs and proteins involved in pre-mRNA splicing that are released into the nucleoplasm when required.
FIGURE 9-49 Examples of nuclear bodies. (a, b) Cajal bodies and nucleoli in a HeLa cell nucleus. (a, left) DIC image showing four nucleoli and three Cajal bodies (arrowheads). Scale bar = 10 μm. (a, right) The same nucleus immunostained with antibodies against coilin (green) and fibrillarin (red). The three Cajal bodies appear yellow because they stain with both antibodies. The nucleoli stain only for fibrillarin. Fibrillarin is the methyltransferase for -O-methylation of rRNA in the nucleoli and snRNAs in the Cajal Bodies. (b) Transmission electron microscopy of nuclear bodies in a Xeopus oocyte nucleus. These extraordinarily large nuclei are larger than in most vertebrate cells. A nucleolus and a histone body with an associated speckle were relatively close to each other in this oocyte nucleus, and consequently were observed in the same electron micrograph. Scale bar = 1 μm. (c) HeLa cell stained with DAPI (blue), antibody to SC35 (red), a splicing factor stored in nuclear bodies called “speckles,” and antibody to PSPC1, a protein found in nuclear bodies called “paraspeckles” (white arrows) because they are most often observed close to speckles. Scale bar = 10 μm. (d) PML-nuclear bodies in an H1299 cell (a lung canrcinoma cell line) nucleus. DNA was stained with DAPI (blue) and PML-nuclear bodies were immunostained with antibody to the major protein in these bodies, PML. Scale bar = 1 μm. [Part (a) Reprinted with permission of Nature Publishing Group, from Gall, J.G., “The centennial of the Cajal body,” Nat Rev Mol Cell Biol., 2003, 4(12):975–980; permission conveyed through Copyright Clearance Center, Inc. Part (b) Republished with permission of Elsevier, from Handwerger, K. E. and Gall, J. G., “Subnuclear organelles: new insights into form and function,” Trends Cell Biol. 2006, 16(1):19–26; permission conveyed through Copyright Clearance Center, Inc. Part (c) Republished with permission from Cold Spring Harbor Laboratory Press, from A. H. Fox and A. I. Lamond, 2010, “Paraspeckles,” Cold Spring Harb. Perspect. Biol. 2(7):a000687. Part (d) Republished with permission from American Society for Microbiology, from M. A. Pennella et al., 2010, “Adenovirus E1B 55Kilodalton Protein Is a p53-SUMO1 E3 Ligase That Represses p53 and Stimulates Its Nuclear Export Through Interactions with Promyelocytic Leukemia Nuclear Bodies,” J. Virol. 84(23):12210–12225; permission conveyed through Copyright Clearance Center, Inc.] Description
Promyelocytic Leukemia (PML) Nuclear Bodies
The first pair (a) shows an oval-shaped HeLa nucleus with large nucleoli and arrows pointing to three small spheres of Cajal Bodies. The first micrograph shows a black and white image while the second shows red nucleoli and yellow Cajal Bodies against a black background. The second pair (b) shows a black and white image of a rounded nucleolus with dark speckled center and a large rounded histone locus body with a small rounded dark speckle atop. The micrograph (c) shows a blue colored oval-shaped HeLa cell with several patches of red colored speckles containing small spheres of yellow paraspeckles. The micrograph (d) shows several small red colored dots in a blue colored nucleus of H 1299 carcinoma cell. Nuclear Paraspeckles Paraspeckles are composed of RNPs formed by the interaction between a long nonprotein-coding RNA species (lncRNA), NEAT1, and members of the DBHS (Drosophila Behavior Human Splicing) family of proteins, P54NRB/NONO, PSPC1, and PSF/SFPQ. Paraspeckles are critical to the control of gene expression through the nuclear retention of RNA containing double-stranded RNA regions that have been subjected to adenosine-to-inosine editing. In this way, they may function in the incompletely understood mRNA quality-control mechanisms that operate in the nucleus. Promyelocytic Leukemia (PML) Nuclear Bodies The PML gene was originally discovered when chromosomal translocations within the gene were observed in the leukemic cells of
Nucleolar Functions in Addition to Ribosomal Subunit Synthesis
patients with the rare disease promyelocytic leukemia (PML). When antibodies specific for the PML protein were used in immunofluorescence microscopy studies, the protein was found to localize to roughly spherical regions 0.3–1 μm in diameter in the nuclei of mammalian cells. Multiple functions have been proposed for these PML nuclear bodies, but a consensus is emerging that they function as sites for the assembly and modification of protein complexes involved in DNA repair and the induction of apoptosis. For example, the important p53 tumor suppressor protein appears to be post-translationally modified by phosphorylation and acetylation in PML nuclear bodies in response to DNA damage, increasing its ability to activate the expression of DNA-damage response genes. PML nuclear bodies are also required for cellular defenses against DNA viruses that are induced by interferons, proteins secreted by virus-infected cells and T-lymphocytes involved in the immune response (see Chapter 24). PML nuclear bodies are also sites of protein post-translational modification through the addition of a small, ubiquitin-like protein called SUMO1 (small ubiquitin-like moiety-1), which can control the activity and subcellular localization of the modified protein. Many transcriptional activators are inhibited when they are sumoylated, and mutation of their site of sumoylation increases their activity in stimulating transcription. These observations indicate that PML nuclear bodies are involved in a mechanism of transcriptional repression that remains to be thoroughly understood.
Nucleolar Functions in Addition to Ribosomal Subunit Synthesis The first nuclear bodies to be observed, the nucleoli, have specialized regions of substructure (Figure 9-49b) that are dedicated to functions other than ribosome biogenesis. There is evidence that immature SRP ribonucleoprotein complexes involved in protein secretion and ER membrane insertion (Chapter 13) are assembled in nucleoli and then exported to the cytoplasm, where their final maturation takes place. The Cdc14 protein phosphatase that regulates processes in the final stages of mitosis is sequestered in nucleoli until chromosomes have been properly segregated into daughter cells (Chapter 19). Also, a tumor-suppressor protein called ARF, which is involved in the regulation of the protein encoded by the most frequently mutated gene in human cancers, p53, is sequestered in nucleoli and released in response to DNA damage (Chapter 25). In addition, heterochromatin often forms on the surface of nucleoli (see Figure 7-29a), suggesting that proteins associated with nucleoli also participate in the formation of this repressing chromatin structure. KEY CONCEPT OF SECTION 9.6 Nuclear Bodies Are Functionally Specialized Nuclear Domains Nuclear bodies are functionally specialized regions in the nucleus where interacting proteins form self-organized structures. Many of these bodies, including the nucleolus, are regions of assembly of RNP complexes.
Key Terms
End of Chapter Visit Achieve to access study tools and to learn more about the content in this chapter: Perspectives for the Future Analyze the Data Chapter References Additional study tools, including videos, animations, and quizzes Key Terms capping cleavage/polyadenylation alternative splice sites alternative splicing (alternative RNA splicing) Argonaute protein cross-exon recognition complex Dicer Drosha exons exon shuffling exosome group I introns group II introns introns
Iron Response Element Binding Proteins (IRE-BPs) microexons microRNAs (miRNAs) mRNA surveillance mRNP exporter nuclear bodies nuclear envelope nuclear pore complexes (NPCs) P bodies pervasive transcription poly(A) tail pre-mRNA pre-rRNA ribonucleoprotein (RNP) complexes ribozyme RNA editing RNA-induced silencing complex (RISC) RNA interference (RNAi) RNA splicing short interfering RNAs (siRNAs) siRNA knockdown small nuclear RNAs (snRNAs) small nucleolar RNAs (snoRNAs) snRNPs (small ribonuclear particles) U1, U2, U4, U5, and U6 spliceosome splicing enhancers splicing silencers SR proteins
Review the Concepts
Review the Concepts 1. Describe three types of post-transcriptional regulation of protein-coding genes. 2. True or false? The CTD is responsible for mRNA- processing steps that are specific for mRNA and not for other forms of RNA. Explain why you chose true or false. 3. There are a number of conserved sequences found in an mRNA that dictate where splicing occurs. Where are these sequences found relative to the exon-intron junctions? What is the significance of these sequences in the splicing process? One of these important regions is the branch-point A found in the intron. What is the role of the branch-point A in the splicing process, and can this be accomplished with the OH group on either the or the carbon? 4. What are the differences between hnRNAs, snRNAs, miRNAs, siRNAs, and snoRNAs? 5. What are the mechanistic similarities between group II intron self-splicing and spliceosomal splicing? What is the evidence that there may be an evolutionary relationship between the two? 6. You obtain the sequence of a gene containing 10 exons, 9 introns, and a UTR containing a polyadenylation consensus sequence. The fifth intron also contains a polyadenylation site. To test whether both polyadenylation sites are used, you isolate mRNA and find a longer transcript from muscle tissue and a shorter transcript from all other tissues. Speculate about the mechanism involved in the production of these different transcripts.
7. RNA editing is a common process in the mitochondria of trypanosomes and plants as well as in chloroplasts, and in rare cases it occurs in higher eukaryotes. What is RNA editing, and what benefit does it demonstrate in the documented example of ApoB in humans? 8. Because DNA is found in the nucleus, transcription is a nuclearlocalized process. Ribosomes responsible for protein synthesis are found in the cytoplasm. Why is hnRNP trafficking to the cytoplasm restricted to the nuclear pore complexes? 9. A protein complex in the nucleus is responsible for transporting mRNA molecules into the cytoplasm. Describe the proteins that form this exporter. What two protein groups are probably behind the mechanism involved in the directional movement of the mRNP and exporter into the cytosol? 10. RNA knockdown has become a powerful tool in the arsenal of methods used to repress gene expression. Briefly describe how gene expression can be knocked down. 11. Speculate about why plants deficient in Dicer activity show increased sensitivity to infection by RNA viruses. 12. mRNA stability is a key regulator of protein levels in a cell. Briefly describe the three mRNA degradation pathways. Suppose that a yeast cell has a mutation in the DCP1 gene, resulting in decreased uncapping activity. Would you expect to see a change in the P bodies found in this mutant cell? 13. mRNA localization now appears to be a common phenomenon. What benefit does mRNA localization have for a cell? What is the evidence that some mRNAs are directed to accumulate in specific subcellular locations?