Introduction
Chapter 13 Moving Proteins into Membranes and Organelles

A three-dimensional reconstruction of the internal membranes of a yeast cell using scanning electron microscopy. The cell wall has been removed and the organelles highlighted with false color to reveal the endoplasmic reticulum (yellow), mitochondria (red), and nucleus (blue). Cell diameter is . [From D. Wei et al., 2012, “High-Resolution Three-Dimensional Reconstruction of a Whole Yeast Cell Using Focused-Ion Beam Scanning Electron Microscopy,” Biotechniques 53(1):41–48.]
13.1 Targeting Proteins to and Across the ER Membrane
13.3 Protein Modifications, Folding, and Quality Control in the ER
13.4 Targeting of Proteins to Mitochondria and Chloroplasts
13.6 Transport into and out of the Nucleus A typical mammalian cell contains up to 10,000 different kinds of proteins; a yeast cell, about 5000. The vast majority of these proteins are synthesized by cytosolic ribosomes, and although many newly synthesized proteins remain within the cytosol (see Chapter 5), as many as half of the different kinds of proteins produced in a typical cell are delivered to one or another of the various membrane-bounded compartments of the cell. For example, many receptor proteins and transport proteins must be delivered to the plasma membrane, digestive enzymes and polypeptide signaling molecules must be directed to the cell surface for secretion from the cell, and enzymes such as RNA and DNA polymerases must be targeted to the nucleus. These and all the other proteins produced by a cell must reach their correct locations for the cell to function properly. The delivery of newly synthesized proteins to their proper cellular destinations, usually referred to as protein targeting or protein sorting, encompasses two very different kinds of processes: signal-based targeting to a variety of organelles and vesicle-based trafficking in the secretory pathway. The first kind of process involves the targeting of a newly synthesized protein
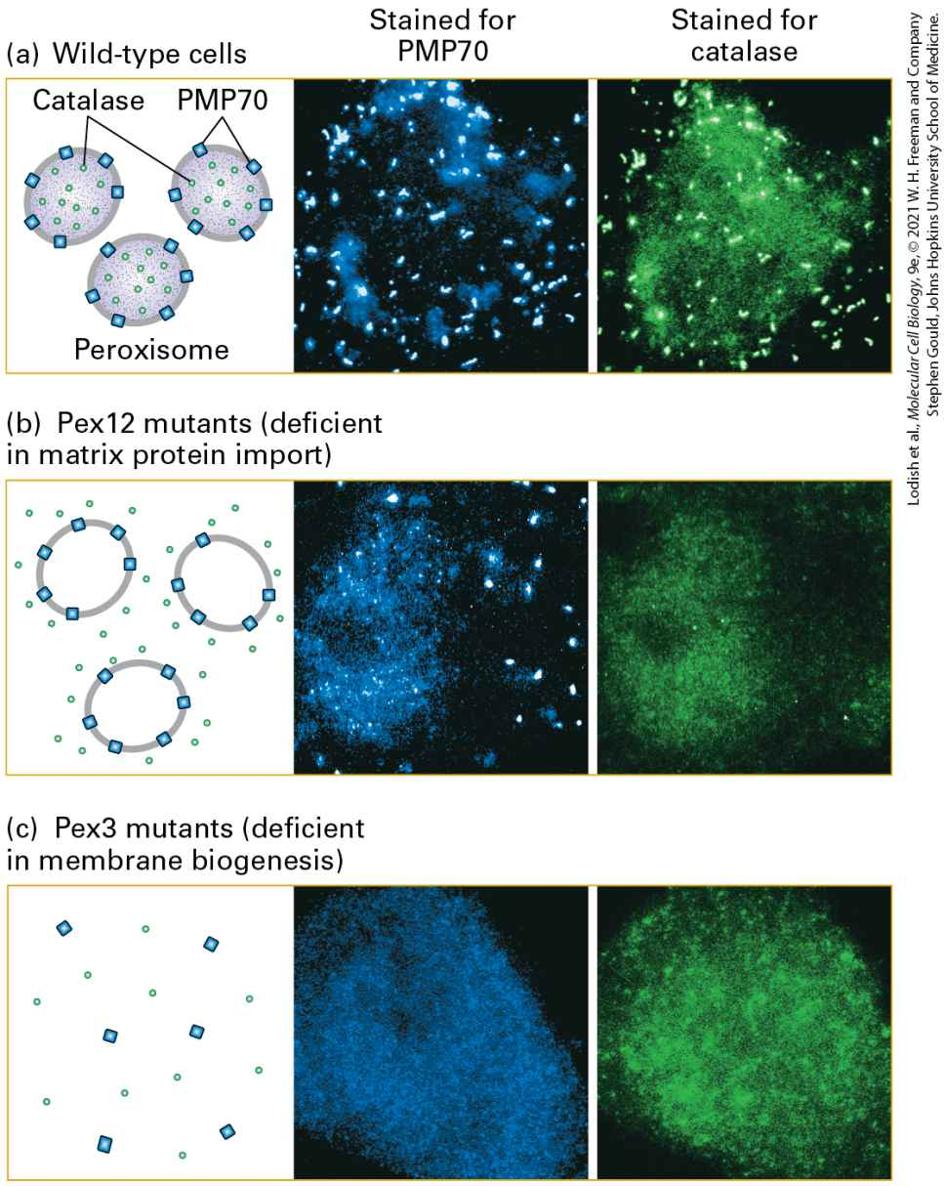
from the cytoplasm to an intracellular organelle. Targeting can occur during translation or soon after synthesis of the protein is complete. For membrane proteins, targeting leads to insertion of the protein into the lipid bilayer of the membrane, whereas for water-soluble proteins, targeting leads to translocation of the entire protein across the membrane into the aqueous interior of the organelle. Proteins are sorted to the endoplasmic reticulum (ER), mitochondria, chloroplasts, peroxisomes, and nucleus by this general process (Figure 13-1).
FIGURE 13-1 Overview of major protein-sorting pathways in eukaryotes. All nuclear DNA– encoded mRNAs are translated on cytosolic ribosomes (step 1 ), but proteins can be directed to different intracellular locations according to targeting signals within the polypeptide sequence. Left (secretory pathway): Ribosomes synthesizing nascent secretory proteins are directed to the
rough endoplasmic reticulum (ER) by an ER signal sequence (pink; step 2 ). After translation is completed on the ER, these proteins can move by vesicle-based transport processes to the Golgi complex and on to the plasma membrane or to lysosomes. The vesicle-based processes underlying the secretory pathway (shaded box) are discussed in Chapter 14. Right (signal-based targeting): Synthesis of proteins that contain no targeting sequence are released into the cytosol and remain there (step 3 ). Proteins with an organelle-specific targeting sequence (pink) are first released into the cytosol but are then imported into mitochondria, chloroplasts, peroxisomes, or the nucleus (steps 4 – 7 ). Mitochondrial and chloroplast proteins typically pass through the outer and inner membranes to enter the matrix or stromal space, respectively. Other proteins are sorted to other subcompartments of these organelles by additional sorting steps. Nuclear proteins enter and exit through visible pores in the nuclear envelope. Description The steps are as follows: Step 1. Signal-based targeting. Four ribosomes bound m R N A produces peptide chains. An arrow toward left lead to step 2 while toward right lead to steps 3 through 7. Ste 2. A ribosome bound m R N A produces a peptide chain with E R signal sequence in start. This reaches rough endoplasmic reticulum where the ribosome bound m R N A attaches to its surface. The signal sequence along with peptide chain are separated in the rough endoplasmic reticulum. Next, they reach Golgi complex and are transported to plasma membrane and lysosome by vesicle-based trafficking. Ste 3. A ribosome bound m R N A produces a cytosolic protein and a peptide chain with targeting sequence. Steps 4, 5, 6, and 7 show that this peptide chain with targeting sequence can be transported to mitochondrion, chloroplast, peroxisome, and nucleus, respectively. The second general sorting process, known as the secretory pathway, involves transport of proteins from the ER to their final destination within membraneenclosed vesicles. For many proteins, including those that make up the extracellular matrix, the final destination is the outside of the cell (hence the name); integral membrane proteins are also transported to the Golgi complex, lysosomes, and plasma membrane by this process. The secretory pathway begins in the ER; thus all proteins slated to enter the secretory pathway are initially targeted to this organelle.
Targeting to the ER usually involves nascent proteins still in the process of being synthesized on a ribosome. Newly made proteins are thus extruded from the ribosome directly into the ER membrane. Once translocated across the ER membrane, proteins are assembled into their native conformation by proteinfolding catalysts present in the lumen of the ER. Indeed, the ER is the location where about one-third of the proteins in a typical cell fold into their native conformations, and most of the resident ER proteins either directly or indirectly contribute to the folding process. As part of the folding process, proteins also undergo specific post-translational modifications in the ER. These processes are monitored carefully, and only after their folding and assembly is complete are proteins permitted to be transported out of the ER to other destinations along the secretory pathway. Proteins whose final destination is the lysosome, plasma membrane, or cell exterior are transported along the secretory pathway by the action of small vesicles that bud from the membrane of one organelle and then fuse with the membrane of another (see
Figure 13-1 [shaded box] and Figure 14-1). We discuss vesicle-based protein trafficking in Chapter 14 because mechanistically it differs significantly from non-vesicle-based protein targeting to intracellular organelles. In this chapter, we examine how proteins are targeted to five intracellular organelles: the ER, mitochondria, chloroplasts, peroxisomes, and nucleus. Two features of this protein-targeting process were initially quite baffling: How could a given protein be directed to only one specific membrane, and how could relatively large hydrophilic protein molecules be translocated across a hydrophobic membrane without disrupting the function of the bilayer as a barrier to ions and small molecules? Using a combination of biochemical purification methods and genetic screens for identifying mutants unable to execute particular translocation steps, cell biologists have identified many of
the cellular components required for translocation across each of the different intracellular membranes. In addition, many of the major translocation processes in the cell have been reconstituted by incorporating their purified protein components into artificial lipid bilayers, using in vitro systems that can be freely manipulated experimentally. These studies have shown that, despite some variations, the same basic mechanisms govern protein sorting to all the various intracellular organelles. As shown in Table 13-1, the mechanism of targeting to five organelles considered in this chapter can be described by four basic elements. (1) The information to target a protein to a particular organelle destination is encoded within the amino acid sequence of the protein itself, usually within a sequence of about 20 amino acids, known generically as a targeting sequence; these sequences are also called signal sequences or signal peptides. Such targeting sequences are usually located at the N-terminus of a protein and are thus the first part of the protein to be synthesized. More rarely, targeting sequences are located at either the C-terminus or within the interior of a protein sequence. (2) Each organelle carries a set of receptor proteins that bind (directly or indirectly) only to specific kinds of targeting sequences, thus ensuring the specificity of targeting. (3) Once a protein containing a targeting sequence has interacted with the corresponding receptor, the polypeptide chain is transferred to some kind of translocation channel that allows the protein to pass into or through the membrane bilayer. (4) Finally, the unidirectional transfer of a protein into an organelle, without its sliding back out into the cytoplasm, is usually achieved by coupling translocation to an energetically favorable process such as hydrolysis of GTP or ATP. In some cases, proteins are sorted further to reach a subcompartment within the target organelle; such sorting depends on yet other signal sequences and other receptor proteins.
TABLE 13-1 • Targeting Sequences Direct Proteins from the Cytosol to Organelles Target Organelle Targeting Sequence Receptor Translocation Channel Energy Source Endoplasmic reticulum (lumen) At N-terminus, 6– hydrophobic amino acids, often preceded by one or more basic amino acids (Arg, Lys) SRP (ribonucleoprotein complex associated with ribosome) and SRP receptor in the ER membrane; SRP and SRP receptor are GTPases Sec61 complex; proteins translocate as unfolded chain, as channel remains sealed to small molecules Translation elongation powered by GTP hydrolysis Mitochondrion (matrix) At N-terminus an amphipathic helix, 20–50 residues in length, with Arg and Lys residues on one side and hydrophobic residues on the other Tom20/22 import receptor in the outer mitochondrial membrane Channels composed of Tom40 in the outer mitochondrial membrane and Tim23/17 in the inner membrane ATP hydrolysis by Hsp70 in the matrix Chloroplast (stroma) At N-terminus, but no common motifs; generally rich in Ser, Toc159 and Toc34, GTPases in the outer membrane Channels composed Toc75 in the outer membrane and Tic20 in ATP hydrolysis by Hsp70 in the stroma i i
Thr, and small hydrophobic residues and poor in Glu and Asp the inner membrane Peroxisome (matrix) PTS1 signal (Ser-LysLeu) at extreme C-terminus; PTS2 signal at N-terminus Pex5, which cycles between the cytoplasm and peroxisomal membrane A complex of Pex5 and the peroxisomal membrane protein Pex14; cargo proteins can be transported in a folded state ATP hydrolysis coupled to ubiquitination and deubiquitination of Pex5 Nucleus (nucleoplasm) NLS sequences can function at any location within a protein sequence; a common motif includes a short segment rich in Lys and Arg residues Nuclear transport receptors, which cycle between the cytoplasm and the nuclear interior Central channel of the nuclear pore complex which is filled with a fluidlike phase of FGrepeat proteins; proteins and RNA can traverse in folded state when bound to a nuclear transport receptor GTP hydrolysis coupled to cycling of Ran GTPase into and out of the nucleus
Targeting to subcompartments of the mitochondria (such as the inner membrane) and the chloroplast (such as the thylakoid) require additional targeting sequences, receptors, and translocation channels as discussed in this chapter. In the first part of the chapter, we cover targeting of proteins to the ER, including the post-translational modifications that proteins undergo as they enter the secretory pathway. Targeting of proteins to the ER is the best understood example of protein targeting and will serve as a model of the process in general. We then describe targeting of proteins to mitochondria, chloroplasts, and peroxisomes. Finally, we cover the transport of proteins into and out of the nucleus through nuclear pores. An important consequence of knowing the nature of the different kinds of targeting sequences is that from just the amino acid sequence encoded by a gene it is possible to reliably deduce the final cellular location of the gene product. Indeed, the cellular location of most proteins encoded by the human genome has been accurately predicted using the information on the nature of different kinds of targeting sequences that we will discuss in this chapter. i
13.1 Targeting Proteins to and Across the ER Membrane
13.1 Targeting Proteins to and Across the ER Membrane All eukaryotic cells have an endoplasmic reticulum (ER). The ER is a convoluted organelle, made up of tubules and flattened sacs, whose membrane is continuous with the membrane of the nucleus. The ER usually has a very large surface area, and its membrane is where cellular lipids are synthesized (see Chapter 10). The ER is also where most membrane proteins are assembled, including those of the plasma membrane and the membranes of the lysosomes, ER, and Golgi complex. In addition, all soluble proteins that will eventually be secreted from the cell — as well as those destined for the lumen of the ER, Golgi complex, or lysosomes — are initially delivered to the ER lumen (see Figure 13-1). Since the ER plays such an important role in protein secretion, we refer to the pathway of protein trafficking that flows through the ER as the secretory pathway. For simplicity, we will refer to all proteins initially targeted to the ER as secretory proteins, but keep in mind that not all proteins that are targeted to the ER are actually secreted from the cell. In this first section of the chapter, we discuss how proteins are initially identified as secretory proteins and how such proteins are translocated across the ER membrane. We deal first with soluble proteins — those that pass all the way through the ER membrane, into the lumen. In the next
Pulse-Chase Experiments with Purified ER Membranes Demonstrated That Secreted Proteins Cross the ER Membrane
section, we discuss integral membrane proteins, which are inserted into the ER membrane. Pulse-Chase Experiments with Purified ER Membranes Demonstrated That Secreted Proteins Cross the ER Membrane Although all cells secrete a variety of proteins (e.g., extracellular matrix proteins), certain types of cells are specialized for secretion of large amounts of specific proteins. Pancreatic acinar cells, for instance, synthesize large quantities of several digestive enzymes, which are secreted into ductules that lead to the intestine. Because such secretory cells contain the organelles of the secretory pathway (e.g., ER and Golgi complex) in great abundance, they have been widely used in studying this pathway, including the initial steps that occur at the ER membrane. The sequence of events that occur immediately after the synthesis of a secretory protein was first elucidated by pulse-chase experiments with pancreatic acinar cells. In these experiments, radioactively labeled amino acids were incorporated into secretory proteins as they were synthesized on ribosomes bound to the surface of the ER. The portion of the ER that receives proteins entering the secretory pathway is known as the rough ER because it is so densely studded with ribosomes that its surface appears morphologically distinct from other ER membranes (Figure 13-2).
From these experiments, it became clear that during or immediately after their synthesis on the ribosome, secretory proteins translocate across the ER membrane into the lumen of the ER.
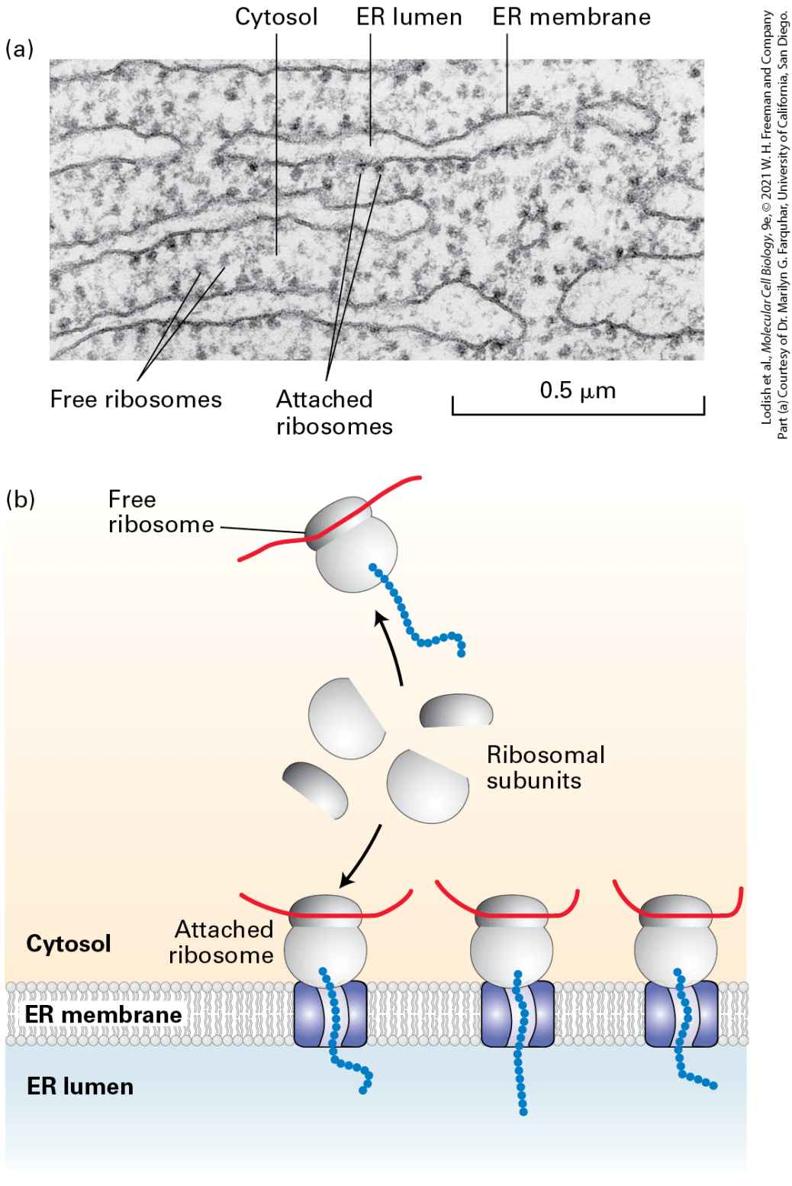
FIGURE 13-2 Structure of the rough ER. (a) Electron micrograph of ribosomes attached to the rough ER in a pancreatic acinar cell. Most of the proteins synthesized by this type of cell are secretory proteins and are formed on membrane-attached ribosomes. A few unattached (free) ribosomes are evident; presumably, these ribosomes are synthesizing cytosolic or other nonsecretory proteins. (b) Schematic representation of protein synthesis on the ER. Note that membrane-bound and free cytosolic ribosomes are identical. Membrane-bound ribosomes are recruited to the ER during synthesis of a polypeptide containing an ER signal sequence. Description The electron micrograph (a) shows several linear wavy-type endoplasmic reticula in the cytosol. The E R membrane enclose the E R lumen and have several small spheres of ribosomes attached on the outer surface. Many small spheres of ribosomes are also present freely in the cytosol. The illustration (b) shows three channels in the E R membrane separating cytosol from E R lumen. Each channel is attached to a ribosome bound m R N A on cytosol side that are formed from ribosomal subunits. A free ribosome bound m R N A also formed from ribosomal subunits is also present in the cytosol. To delineate the steps in the translocation process, it was necessary to isolate the ER from the rest of the cell. Isolation of intact ER, with its delicate lacelike structure and its interconnectedness with other organelles, is not feasible. However, scientists discovered that when cells are homogenized, the rough ER breaks up into small closed vesicles with ribosomes on the outside, termed microsomes, which retain most of the biochemical properties of the intact ER, including the capability of protein translocation. The experiments depicted in Figure 13-3, in which microsomes isolated from pulse-labeled cells were treated with a protease, demonstrate that although secretory proteins are synthesized on ribosomes
bound to the cytosolic face of the ER membrane, the polypeptides produced by these ribosomes end up within the lumen of a microsome. Experiments such as these raised the question of how polypeptides are recognized as secretory proteins shortly after their synthesis begins and how a nascent secretory protein is threaded across the ER membrane.
A Hydrophobic N-Terminal Signal Sequence Targets Nascent Secretory Proteins to the ER
FIGURE 13-3 Secretory proteins enter the ER lumen. Labeling experiments demonstrated that secretory proteins are localized to the ER lumen shortly after synthesis. Cells are incubated for a brief time with radiolabeled amino acids so that only newly synthesized proteins become labeled. The cells are then homogenized, fracturing the plasma membrane and shearing the rough ER into small vesicles called microsomes. Because they have bound ribosomes, microsomes have a much greater buoyant density than other membranous organelles and can be separated from them by a combination of differential and sucrose density-gradient centrifugation (see Chapter 4). The purified microsomes are treated with a protease in the presence or absence of a detergent. The labeled secretory proteins associated with the microsomes are digested by the protease only if the microsomal membrane is first destroyed by treatment with detergent. This finding indicates that the newly made proteins are inside the microsomes, equivalent to the lumen of the rough ER. Description The illustration starts with a rough E R having several ribosomes bound to m R N A attached to the ER membrane producing peptide chains into the rough E R lumen. Several labeled secretory proteins are also present in the lumen. Next, homogenization occurs leading to the formation of spherical microsomes with attached ribosomes. These microsomes first treated with detergent and then protease is added, resulting in digestion of secretory protein. The protease may directly be added to microsomes without detergent treatment, thus secretory protein digestion does not occur. A Hydrophobic N-Terminal Signal Sequence Targets Nascent Secretory Proteins to the ER After synthesis of a secretory protein begins on free ribosomes in the cytosol, a 16–30-residue ER targeting sequence in the nascent protein directs the ribosome to the ER membrane and initiates translocation of the
growing polypeptide across the ER membrane (see Figure 13-1, left). An ER targeting sequence, located at the N-terminus of the protein, is usually known as a signal sequence. The signal sequences of different secretory proteins all contain one or more positively charged amino acids adjacent to a continuous stretch of 6–12 hydrophobic residues (known as the hydrophobic core) but otherwise have little in common. The signal sequence is cleaved from most secretory proteins while they are still elongating on the ribosome; thus signal sequences are usually not present in the mature proteins found in cells. The hydrophobic core of an ER signal sequence is essential for its function. For instance, the specific deletion of several of the hydrophobic amino acids from a signal sequence or the introduction of charged amino acids into the hydrophobic core by mutation can abolish the ability of the N-terminus of a protein to function as a signal sequence. As a consequence, the modified protein remains in the cytosol, unable to cross the ER membrane into the lumen. Conversely, signal sequences can be added to normally cytosolic proteins using recombinant DNA techniques. Provided the added sequence is sufficiently long and hydrophobic, such a modified cytosolic protein can acquire the ability to be translocated into the ER lumen. The hydrophobic residues in the core of an ER signal sequence form a binding site that is critical for the interaction of the signal sequence with the machinery responsible for targeting the protein to the ER membrane. Biochemical studies using a cell-free protein-synthesizing system, mRNA encoding a secretory protein, and microsomes stripped of their own bound
ribosomes have elucidated how ER signal sequences function during protein translocation. Initial experiments with this system demonstrated that a typical secretory protein is incorporated into microsomes and has its signal sequence removed only if the microsomes are present during protein synthesis. If instead of being present during translation, the addition of microsomes to the system is delayed until after protein synthesis is completed, no protein transport into the microsomes occurs (Figure 13-4). Subsequent experiments were designed to determine the precise stage of protein synthesis at which microsomes must be present in order for translocation to occur. In these experiments, microsomes were added to the reaction mixtures at different times after protein synthesis had begun. These experiments showed that microsomes must be added before the first 70 or so amino acids are translated in order for the completed secretory protein to be localized in the microsomal lumen. At this point, the first 40 or so amino acids protrude from the ribosome, including the signal sequence that will later be cleaved off, and the next 30 or so amino acids are still buried within a channel in the ribosome (see
Figure 5-34). Thus the transport of most secretory proteins into the ER lumen begins while the incompletely synthesized (nascent) protein is still bound to the ribosome, a process referred to as cotranslational translocation.
FIGURE 13-4 Translation and translocation occur simultaneously. Cell-free experiments demonstrate that translocation of secretory proteins into microsomes is coupled to translation. Treatment of microsomes with EDTA, which chelates ions, strips them of associated ribosomes, allowing isolation of ribosome-free microsomes, which are equivalent to ER membranes (see Figure 13-3). Protein synthesis is carried out in a cell-free
Cotranslational Translocation Is Initiated by Two GTP-Hydrolyzing Proteins
system containing functional ribosomes, aminoacyl-tRNAs, ATP, GTP, and cytosolic enzymes, to which mRNA encoding a secretory protein is added. (a) When microsomes are added after synthesis of secretory proteins is complete, the secretory proteins that have already been formed will not enter microsomes. (b) However, when microsomes are present during protein synthesis, secretory proteins will be translocated across the vesicle membrane and lose their signal sequences (resulting in a decrease in molecular weight) as they are synthesized. Description The first part (a) shows cell-free protein synthesis; no microsomes present. An m R N A bound to three ribosomes produces complete proteins with N-terminal signal sequences. Microsomal membranes are added that results in no incorporation into microsomes; no removal of signal sequence. The second part (b) shows cell-free protein synthesis; microsomes present. An illustration of a spherical microsome shows an m R N A bound to ribosomes attached on its outer surface. Polypeptide chain with N-terminal signal sequences are produced into the lumen where the sequences are cleaved. A text below reads, cotranslational transport of protein into microsome and removal of signal sequence. Next, it leads to a spherical microsome containing mature protein chains without signal sequences. Cotranslational Translocation Is Initiated by Two GTP-Hydrolyzing Proteins Given that secretory proteins are synthesized in association with the ER membrane but not with any other cellular membrane, a signal-sequence recognition mechanism must target them there. The two key components in this targeting are the signal recognition particle (SRP) and its receptor.
The SRP is a cytosolic ribonucleoprotein particle that transiently binds to both the ER signal sequence in a nascent protein and the large (60S) ribosomal subunit, forming a large complex. The SRP then targets the nascent protein–ribosome complex to the ER membrane by binding to the SRP receptor, which is located in the membrane. The SRP is made up of six proteins bound to a 300-nucleotide RNA, which acts as a scaffold for the hexamer. One of the SRP proteins (P54) can be chemically cross-linked to ER signal sequences, which shows that this subunit is the one that binds to the signal sequence in a nascent secretory protein. A region of P54 known as the M domain, containing many methionine and other amino acid residues with hydrophobic side chains, forms a cleft or groove whose inner surface is lined by hydrophobic side chains (Figure 13-5a). The hydrophobic core of the signal sequence binds to this cleft via hydrophobic interactions. Other polypeptides in the SRP interact with the ribosome or are required for protein translocation into the ER lumen.
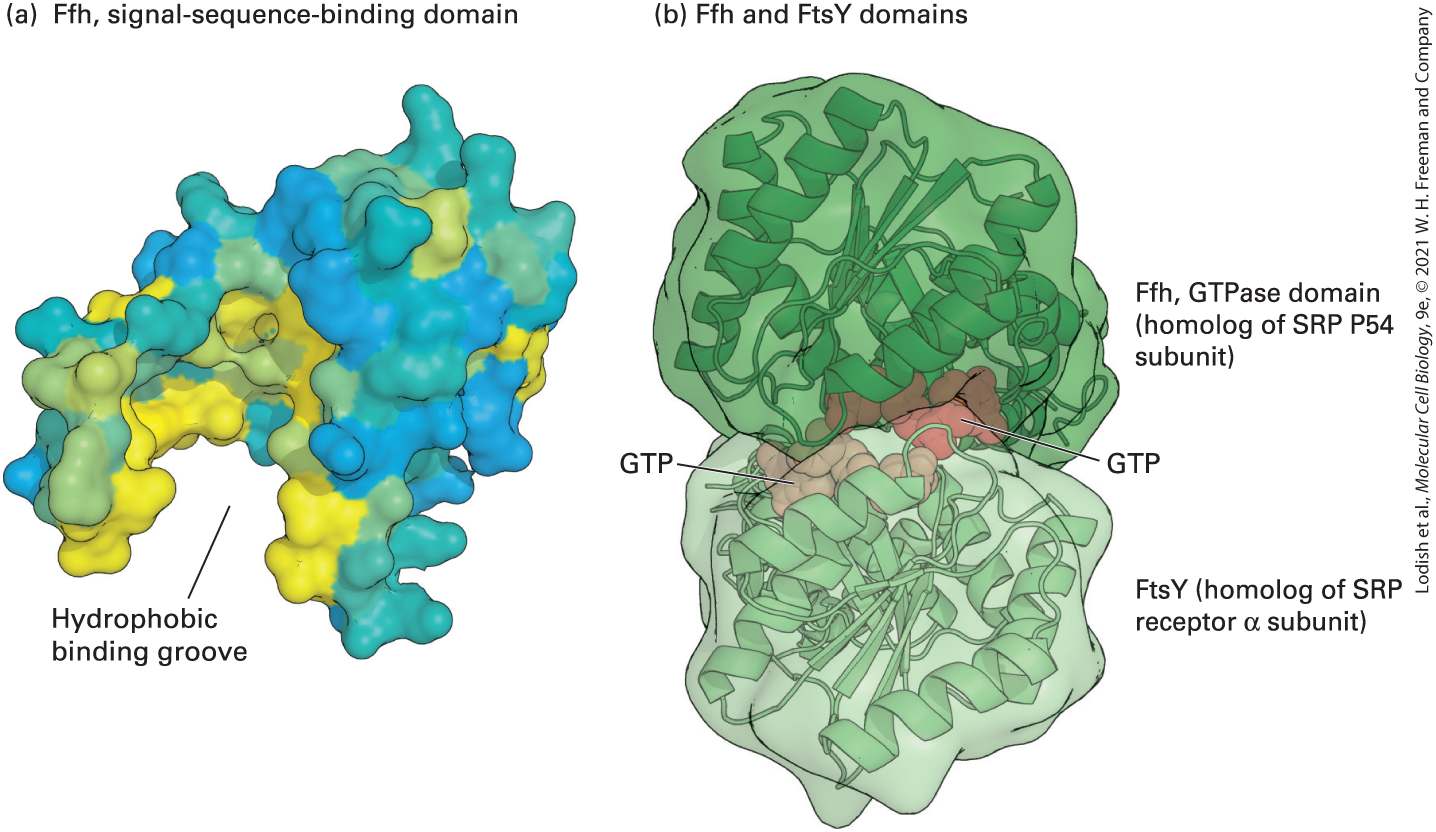
FIGURE 13-5 Structure of the signal recognition particle (SRP). (a) The signalsequence-binding domain: the bacterial Ffh protein is homologous to the portion of P54 that binds ER signal sequences in eukaryotes. This surface model shows the binding domain in Ffh, which contains a large cleft or groove lined with hydrophobic amino acids (yellow) whose side chains interact with signal sequences. (b) GTP- and receptor-binding domain: the structure of GTP bound to FtsY (the archaeal homolog of the α subunit of the SRP receptor) and Ffh subunits from Thermus aquaticus illustrate how the interaction between these proteins is controlled by GTP binding and hydrolysis. Ffh and FtsY each can bind to one molecule of GTP, and when they bind to each other, the two bound molecules of GTP fit in the interface between the protein subunits and stabilize the dimer. Assembly of the pseudosymmetric dimer allows formation of two active sites for the hydrolysis of both bound GTP molecules. [Part (a) Data from R. J. Keenan et al., 1998, Cell 94:181, PDB ID 2ffh. Part (b) data from P. J. Focia et al., 2004, Science 303:373, PDB ID 1okk.] Description The first illustration (a) shows F f h, signal-sequence-binding domain. The surface model is color coded oval-shaped structure with a deep hydrophobic binding groove at the bottom. The second illustration (b) shows F f h and F t s Y domains. The surface
model shows two spheres attached together and each having a G T P site at the point of attachment. The top sphere is labeled F f h, G T Pase domain (homolog of S R P P 54 subunit) and the bottom sphere is labeled F t s Y (homolog of S R P receptor alpha subunit). The SRP and the nascent polypeptide chain–ribosome complex bind to the ER membrane by docking with the SRP receptor, an integral protein of the ER membrane made up of two subunits: an α subunit and a smaller β subunit. Interaction of the SRP–nascent chain–ribosome complex with the SRP receptor is strengthened when both the P54 subunit of the SRP and the α subunit of the SRP receptor are bound to GTP. The structure of the SRP P54 subunit and the SRP receptor α subunit (FtsY) from the archaean Thermus aquaticus provides insight into how a cycle of GTP binding and hydrolysis can drive the binding and dissociation of these proteins. Figure 13-5b shows that P54 and FtsY, each bound to a single molecule of GTP, come together to form a pseudosymmetric heterodimer. Neither subunit alone contains a complete active site for the hydrolysis of GTP, but when the two proteins come together, they form two complete active sites that are capable of hydrolyzing both bound GTP molecules. Hydrolysis to GDP destabilizes the interface, causing disassembly of the dimer.
Figure 13-6 summarizes our current understanding of secretory protein synthesis and the role of the SRP and its receptor in this process. Hydrolysis of the bound GTP accompanies disassembly of the SRP and SRP receptor and initiates transfer of the nascent chain and ribosome to a site on the ER membrane, where translocation can take place. After dissociating from each other, the SRP and its receptor each release their
bound GDP, SRP is recycled back to the cytosol, and both are ready to initiate another round of interaction between ribosomes synthesizing nascent secretory proteins and the ER membrane.
FIGURE 13-6 Cotranslational translocation. Steps 1 – 2 : Once the ER signal sequence emerges from the ribosome, it is bound by a signal recognition particle (SRP). Step 3 : The SRP and the nascent polypeptide chain–ribosome complex bind to the SRP receptor in the ER membrane. This interaction is strengthened by the binding of GTP to both the SRP and its receptor. Step 4 : Transfer of the nascent polypeptide–ribosome to the translocon leads to opening of this translocation channel to admit the growing polypeptide adjacent to the signal sequence. The hydrophobic signal sequence itself is transferred to a hydrophobic binding site next to the central pore. Both the SRP and SRP receptor, once dissociated from the translocon, hydrolyze their bound GTP and then are ready to initiate the insertion of another polypeptide chain. Step 5 : As the polypeptide chain elongates, it passes through the translocon channel into the ER lumen. The signal peptidase, whose active site faces the ER lumen, cleaves the signal peptide as soon as the recognition site enters the lumen. Step 6 : The peptide chain continues to elongate as the mRNA is translated toward the end. Because the ribosome is attached to the translocon, the growing chain is extruded through the translocon into the ER lumen. Steps 7 – 8 : Once translation is complete, the ribosome
Passage of Growing Polypeptides Through the Translocon Is Driven by Translation
is released, the remainder of the protein is drawn into the ER lumen, the translocon closes, and the protein assumes its native folded conformation. Description The illustration shows an S R P receptor adjacent to the translocon embedded in the E R membrane separating cytosol from E R lumen. The steps are as follows: Step 1. A ribosome in the cytosol translates an m R N A producing a polypeptide chain with an N-terminal (N H 3 plus) signal sequence. Step 2. An S R P binds to the signal sequence and the ribosome. Step 3. The ribosome-bound S R P binds to the S R P receptor on E R membrane, after G T P binds to both S R P and the receptor. The translocon is closed. Step 4. The translocon opens. The ribosome binds to the open translocon, continues translating the peptide sequence, which is translocated into the ER lumen. G T P bound to the S R P and S R P receptor is hydrolyzed and the S R P detaches from the receptor. Step 5. A signal peptidase bound in the membrane cleaves the signal sequence from the translocating peptide sequence in the E R lumen. Step 6. Protein synthesis continues. Step 7. At the end of protein synthesis, the ribosome leaves the translocon. Step 8. The protein in the E R lumen folds to obtains its native conformation. What is the purpose of coupling GTP hydrolysis by SRP and SRP receptor to delivery of a signal sequence to the translocon — the channel across the membrane? It is clear that this GTP hydrolysis does not provide the motive energy for ongoing translocation since it occurs only at the initiation of the translocation process. The GTPase cycle depicted in
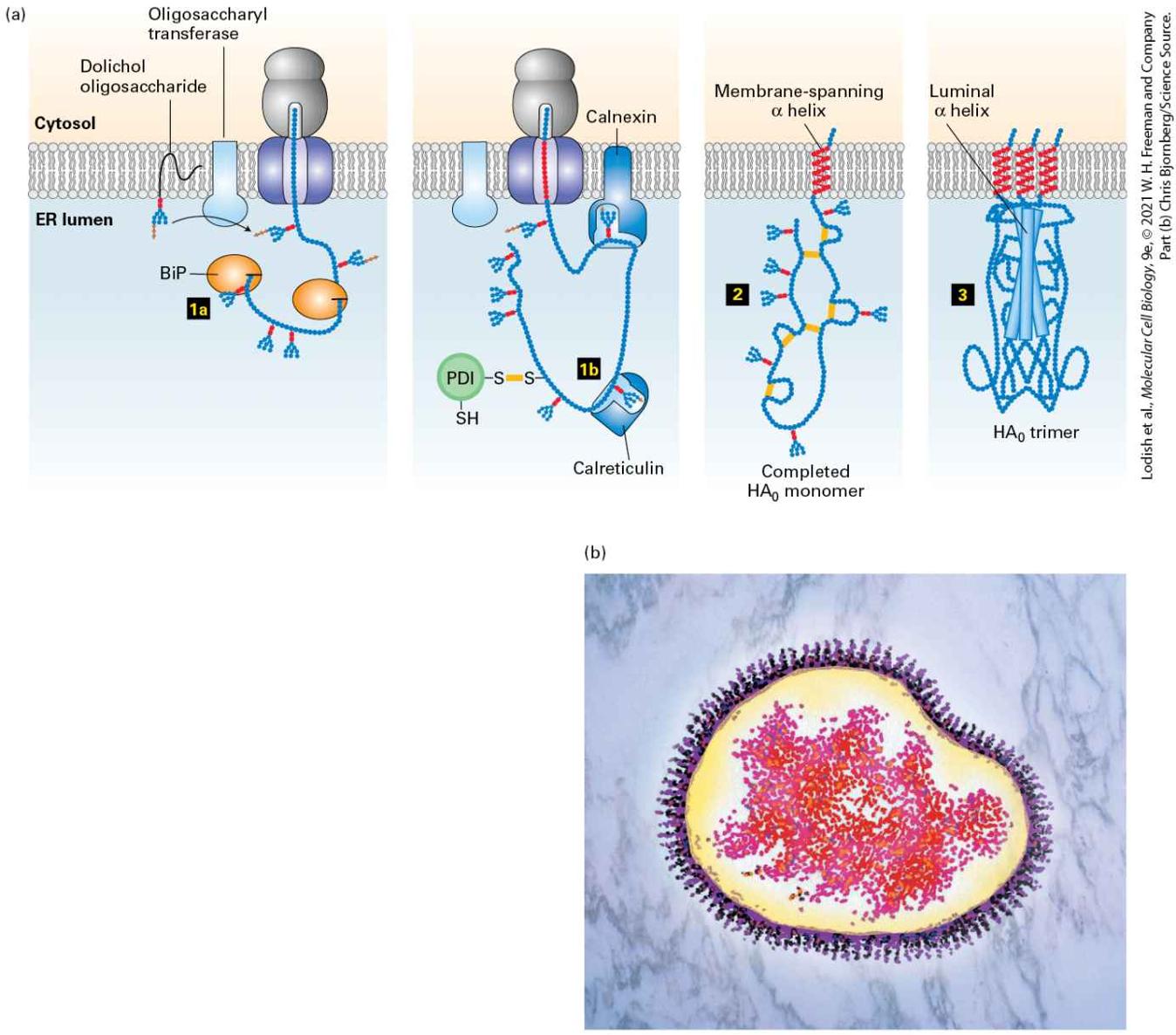
Figure 13-6 appears to be analogous to the GTP hydrolysis coupled to the initiation of translation on the ribosome as depicted in Figure 5-36. By coupling to GTP hydrolysis, a complex macromolecular assembly reaction can be made more efficient by adding a proofreading step that can increase the fidelity of the reaction.
Passage of Growing Polypeptides Through the Translocon Is Driven by Translation Once the SRP and its receptor have targeted a ribosome synthesizing a secretory protein to the ER membrane, the ribosome and nascent polypeptide chain are rapidly transferred to the translocon, a complex of proteins that forms a channel embedded within the ER membrane. As translation continues, the elongating chain passes directly from the large ribosomal subunit into the central pore of the translocon. The large ribosomal subunit is aligned with the pore of the translocon in such a way that the growing chain is never exposed to the cytoplasm and is prevented from folding until it reaches the ER lumen (see Figure 13-6). The translocon was first identified through mutations in the yeast gene encoding a protein called Sec61α, which caused a block in the translocation of secretory proteins into the lumen of the ER. Subsequently, three proteins, collectively called the Sec61 complex, were found to form the mammalian translocon: Sec61α, an integral membrane protein with 10 membrane-spanning α helices, and two smaller proteins, termed Sec61β and Sec61γ. Chemical cross-linking experiments in a cell-free translocation system — in which amino acid side chains from a nascent secretory protein became covalently attached to the Sec61α subunit — demonstrated that the translocating polypeptide chain comes into contact with the Sec61α protein, confirming its identity as the translocon pore (Figure 13-7).
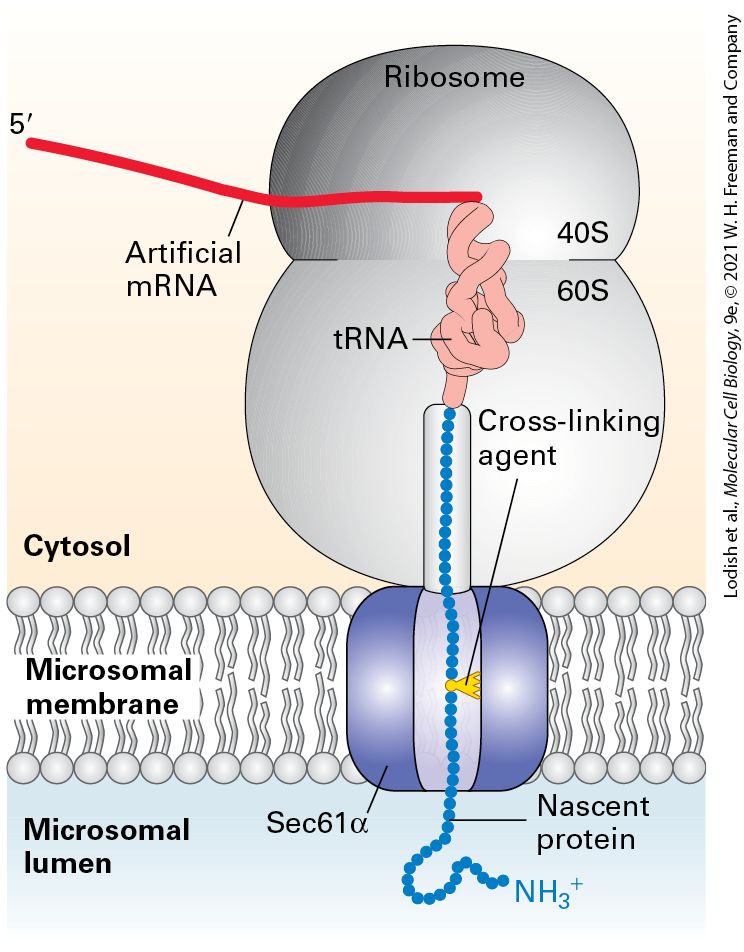
FIGURE 13-7 Sec61α is a translocon component. Experiments using added chemical cross-linkers show that nascent secretory proteins come into close proximity of the translocon component Sec61α as they pass into the ER lumen. An mRNA encoding the N-
terminal 70 amino acids of the secreted protein prolactin was translated in a cell-free system containing microsomes (see Figure 13-4b). The mRNA lacked a chain-termination codon and contained one lysine codon near the middle of the sequence. The reaction mixtures contained a chemically modified lysyl-tRNA in which a light-activated cross-linking reagent was attached to the lysine side chain. Although the entire mRNA was translated, the completed polypeptide could not be released from the ribosome without a chain-termination codon and thus became “stuck” crossing the ER membrane. The reaction mixtures were then exposed to intense light, which caused the nascent polypeptide chain to become covalently bound to whatever proteins were near it in the translocon. When the experiment was performed using microsomes from mammalian cells, the nascent chain became covalently linked to Sec61α. Different versions of the prolactin mRNA were created so that the modified lysine residue would be placed at different distances from the ribosome; crosslinking to Sec61α was observed only when the modified lysine was positioned within the translocation channel. See T. A. Rapoport, 1992, Science 258:931; and D. Görlich and T. A. Rapoport, 1993, Cell 75:615. Description The illustration shows a sec 61 alpha translocon present in the microsomal membrane separating cytosol from microsomal lumen. The ribosome is positioned on the cytosolic side, a protein is translated and translocated into the microsomal lumen. The nascent protein end in the lumen has an N H 3 plus terminal. Within the ribosome, a t R N A is bound to the end of a strand of artificial m R N A and to the C-terminal of the nascent protein. A cross-linking agent is present between the peptide in the translocon channel. When microsomes in the cell-free translocation system were replaced with reconstituted phospholipid vesicles containing only the SRP receptor and the Sec61 complex, nascent secretory proteins were translocated from their SRP-ribosome complex into the vesicles. This finding indicates that the SRP receptor and the Sec61 complex are the only ER-membrane proteins that are absolutely required for translocation. The energy derived
from chain elongation at the ribosome appears to provide the driving force to push the polypeptide chain across the membrane in one direction. The translocon must be able to allow passage of a polypeptide chain while remaining sealed to small molecules, such as ATP, in order to maintain the permeability barrier of the ER membrane. Furthermore, there must be some way to regulate the translocon so that it is closed in its default state, opening only when a nascent polypeptide chain–ribosome complex is bound. A high-resolution structural model of the archaeal Sec61 complex shows how the translocon preserves the integrity of the membrane (Figure 13-8). The 10 transmembrane helices of Sec61α form a central channel through which the translocating polypeptide chain passes. Two different gating steps are required for Sec61α to accept a translocating polypeptide. The 10 transmembrane helices are organized into two 5-helix bundles. In the first gating step, the bundles hinge apart like an opening clamshell to expose a hydrophobic binding pocket for the hydrophobic core of the signal sequence at the open edge. The signal sequence binds to Sec61α with its N-terminus facing the cytosol and the elongating polypeptide doubling back through the central channel. The structural model of the Sec61 complex, which was isolated without a translocating peptide and is therefore presumed to be in a closed conformation, reveals a short helical peptide plugging the central channel. Biochemical studies of the Sec61 complex have shown that, in the absence of a translocating polypeptide, the peptide that forms the plug effectively seals the translocon to prevent the passage of ions and small molecules. In the second gating step, after the signal sequence has bound to the opened channel, the translocating peptide enters the central pore of the channel, forcing away the plug
peptide and allowing translocation to proceed. The middle of the central pore is lined with hydrophobic isoleucine residues that in effect form a gasket, preventing leakage of small polar molecules around the translocating peptide even as translocation proceeds.
FIGURE 13-8 Structure of an archaeal Sec61 complex. The structure of the detergentsolubilized Sec61 complex from the archaeon Methanocaldococcus jannaschii (also known as the SecY complex) was determined by x-ray crystallography. (a) A side view shows the hourglass-shaped channel through the center of the pore. A ring of isoleucine residues at the constricted waist of the pore forms a gasket that keeps the channel sealed to small molecules even as a translocating polypeptide passes through the channel. When no translocating peptide is present, the channel is closed by a short helical plug (red). This plug moves out of the channel during translocation. In this view, the front half of protein has been removed to better show the pore. (b) A view looking through the center of the channel shows a region (on the left side) where helices may separate, allowing lateral passage of a hydrophobic transmembrane domain into the lipid bilayer. [Data from B. van den Berg et al., 2004, Nature 427:36–44, PDB ID 1rhz and 1rh5.] Description The side view (a) of sec 61 complex shows a small ribbon-shaped structure in the central pore labeled, plug in place. An arrow points to another ribbon-shaped coiled structure away from the pore labeled, plug removed. The top view (b) shows a pore
ATP Hydrolysis Powers Post-Translational Translocation of Some Secretory Proteins in Yeast
ring in the center of the sec 61 complex. The pore at the bottom left is labeled, lateral exit to lipid bilayer. A ribbon-shaped removed plug is present at a distance from the pore. As the growing polypeptide chain enters the lumen of the ER, the signal sequence is cleaved by signal peptidase, which is a transmembrane ER protein associated with the translocon (see Figure 13-6, step 5 ). Signal peptidase recognizes a sequence on the C-terminal end of the hydrophobic core of the signal peptide and cleaves the chain specifically at this sequence once it has emerged into the luminal space of the ER. After the signal sequence has been cleaved, the growing polypeptide moves through the translocon into the ER lumen. The translocon remains open until translation is complete and the entire polypeptide chain has moved into the ER lumen. After translocation is complete, the plug peptide reseals the translocon channel. ATP Hydrolysis Powers PostTranslational Translocation of Some Secretory Proteins in Yeast In most eukaryotes, secretory proteins enter the ER by cotranslational translocation. In yeast, however, some secretory proteins enter the ER lumen after translation has been completed. In such post-translational translocation, the translocating protein passes through the same Sec61 translocon that is used in cotranslational translocation. However, the SRP and SRP receptor are not involved in post-translational translocation, and
in such cases a direct interaction between the translocon and the signal sequence of the completed protein appears to be sufficient for targeting to the ER membrane. In addition, the driving force for unidirectional translocation across the ER membrane is provided by an additional protein complex known as the Sec63 complex and a member of the Hsp70 family of molecular chaperones known as BiP (see Chapter 3 for further discussion of molecular chaperones). The tetrameric Sec63 complex is embedded in the ER membrane in the vicinity of the translocon, whereas BiP is within the ER lumen. Like other members of the Hsp70 family, BiP has a peptide-binding domain and an ATPase domain. These chaperones bind and stabilize unfolded or partially folded proteins (see Figure 3-17). The current model for post-translational translocation of a protein into the ER is outlined in Figure 13-9. Once the N-terminal segment of the protein enters the ER lumen, signal peptidase cleaves the signal sequence just as in cotranslational translocation (step 1 ). Interaction of BiP⋅ATP with the luminal portion of the Sec63 complex causes hydrolysis of the bound ATP, producing a conformational change in BiP that promotes its binding to an exposed polypeptide chain (step 2 ). Since the Sec63 complex is located near the translocon, BiP is thus activated at sites where nascent polypeptides can enter the ER. Certain experiments suggest that, in the absence of binding to BiP, an unfolded polypeptide can freely slide back and forth within the translocon channel. Such random sliding motions rarely result in the entire polypeptide’s crossing the ER membrane. Binding of a molecule of BiP⋅ADP to the luminal portion of the polypeptide prevents backsliding of the polypeptide out of the ER. As further inward random sliding exposes more of the polypeptide on the
luminal side of the ER membrane, successive binding of BiP⋅ADP molecules to the polypeptide chain acts as a ratchet, ultimately drawing the entire polypeptide into the ER within a few seconds (steps 3 and 4 ). After a delay of some time, the BiP molecules spontaneously exchange their bound ADP for ATP, leading to release of the polypeptide, which can then fold into its native conformation (steps 5 and 6 ). The recycled BiP⋅ATP is then ready for another interaction with Sec63. BiP and the Sec63 complex are also required for cotranslational translocation. The details of their role in this process are not well understood, but they are thought to act at an early stage of the process, such as the threading of the signal peptide into the pore of the translocon.
FIGURE 13-9 Post-translational translocation. This mechanism is fairly common in yeast and probably occurs occasionally in higher eukaryotes. Step 1 : For yeast proteins that can be translocated post-translationally, the signal sequence of the fully translated protein can engage with the translocon, causing the signal sequence and N-terminal portion of the protein to enter the ER where the signal sequence is cleaved. Small arrows inside the translocon represent the ability of the translocating polypeptide to slide randomly inward and outward. Step 2 : Within the lumen of the ER, BiP⋅ATP is converted into BiP⋅ADP by the Sec63 complex. BiP⋅ADP has a high affinity for binding to exposed hydrophobic segments of the translocating polypeptide. Once BiP⋅ADP is bound, the polypeptide is no longer free to slide out toward the cytosol but can still slide inward as shown by the inward pointing arrow. Steps 3 – 5 : BiP⋅ADP binds to successive segments of the polypeptide as they enter the ER, progressively ratcheting the polypeptide inward until it has fully entered the ER lumen. Step 6 : Relatively slow exchange of ATP for ADP regenerates BiP⋅ATP, which releases the translocated peptide, allowing it to complete folding in the ER lumen. See K. E. Matlack et al., 1997, Science 277:938. Description The illustration shows a sec 63 complex adjacent to the translocon embedded in the E R membrane separating cytosol from E R lumen. Step 1. A translocating polypeptide chain moves through the translocon into the E R lumen. The signal sequence has been cleaved inside the E R lumen. Step 2. A BiP bound to A T P binds to the sec 63 complex, loses inorganic phosphate and binds to the emerging polypeptide chain as BiP-A D P complex. Steps 3 and 4. Successive BiP-A D P complexes binds to the polypeptide chain, preventing random slippage back into the cytosol. Step 5. The protein is fully transported into the lumen through the translocon. A D P is lost from the BiP-A D P complexes separating BiP from the polypeptide chain. The BiP reattaches to A T P and move back to step 2. Step 6. The translocated protein folds into its native conformation. The overall reaction carried out by BiP is an important example of how the chemical energy released by the hydrolysis of ATP can power the mechanical movement of a protein across a membrane. As we will see,
translocation of proteins into mitochondria and chloroplasts can be powered by ATP hydrolysis by ATPase chaperone proteins located inside these organelles and also typically occurs by post-translational translocation. This explains why ribosomes are typically not found bound to these other organelles, as they are to the rough ER. KEY CONCEPTS OF SECTION 13.1 Targeting Proteins to and Across the ER Membrane Synthesis of secreted proteins, integral plasma-membrane proteins, and proteins destined for the ER, Golgi complex, or lysosome begins on cytosolic ribosomes, which become attached to the membrane of the ER, forming the rough ER (see Figure 13-1, left). The ER signal sequence on a nascent secretory protein is located at the N-terminus and contains a sequence of hydrophobic amino acids. In cotranslational translocation, the signal recognition particle (SRP) first recognizes and binds the ER signal sequence on a nascent secretory protein, then is bound in turn by an SRP receptor on the ER membrane, thereby targeting the nascent polypeptide chain–ribosome complex to the ER. The SRP and SRP receptor then mediate insertion of the nascent secretory protein into the translocon (Sec61 complex). Hydrolysis of two molecules of GTP by the SRP and its receptor cause the dissociation of SRP (see Figures 13-5 and 13-6). Coupling GTP hydrolysis to the loading of signal-sequence bearing proteins to the translocon is thought to increase the fidelity of this assembly reaction. As the ribosome attached to the translocon continues translation, the unfolded protein chain is extruded into the ER lumen. The translocon contains a central channel lined with hydrophobic residues that allows transit of an unfolded protein chain while remaining sealed to ions and small hydrophilic molecules. In addition, the channel is gated so that it is open only when a polypeptide is being translocated. In post-translational translocation, a completed secretory protein is targeted to the ER membrane by interaction of the signal sequence with the translocon. The polypeptide chain is then pulled into the ER by a ratcheting mechanism that requires ATP hydrolysis by the chaperone BiP, which stabilizes the entering polypeptide (see Figure 13-9).
In both cotranslational and post-translational translocation, a signal peptidase in the ER membrane cleaves the ER signal sequence from a secretory protein soon after the N-terminus enters the lumen.
13.2 Insertion of Membrane Proteins into the ER
13.2 Insertion of Membrane Proteins into the ER In previous chapters, we have encountered many of the vast array of integral membrane (transmembrane) proteins that are present throughout the cell. Each such protein has a unique orientation with respect to the membrane’s phospholipid bilayer. Integral membrane proteins located in the ER, Golgi complex, and lysosomes, as well as in the plasma membrane, which are all synthesized on the rough ER, remain embedded in the membrane as they move to their final destinations along the same pathway that is followed by soluble secretory proteins (see Figure 13-1, left). During this transport, the orientation of a membrane protein is preserved; that is, the same segments of the protein always face the cytoplasmic side of the membrane, whereas other segments always face in the opposite direction, which could be considered the exoplasmic side of the membrane. Thus the final orientation of these membrane proteins is established during their biosynthesis on the ER membrane. As we will see in this section, insertion of membrane proteins into the ER membrane employs the same mechanism that has already been described for entry of soluble secretory proteins into the lumen of the ER. Membrane proteins first engage the translocon, made up of the Sec61 complex, by the interaction of a hydrophobic signal sequence with SRP and SRP receptor. Once engaged with the translocon, hydrophilic portions of the membrane protein can enter the ER lumen by translocation, whereas the hydrophobic
Several Topological Classes of Integral Membrane Proteins Are Synthesized on the ER
portions become embedded in the ER membrane by lateral opening of the translocon gate. The final topological orientation of membrane segments and hydrophilic regions of a membrane protein depends on the arrangement of hydrophobic sequences and neighboring positively charged amino acid residues. These sequences, known collectively as topogenic sequences, direct the membrane insertion and orientation of various classes of integral membrane proteins. Several Topological Classes of Integral Membrane Proteins Are Synthesized on the ER The topology of a membrane protein refers to the number of times its polypeptide chain spans the membrane and the orientation of those membrane-spanning segments within the membrane. The key elements of a protein that determine its topology are the membrane-spanning segments themselves, which are usually α helices containing 20–25 hydrophobic amino acids that contribute to energetically favorable interactions within the hydrophobic interior of the phospholipid bilayer. Most integral membrane proteins fall into one of the five topological classes illustrated in Figure 13-10. Topological classes I, II, III, and the tail-anchored proteins are single-pass membrane proteins, which have only one membrane-spanning α-helical segment. Type I proteins have a cleaved N-terminal ER signal sequence and are anchored in the membrane with their hydrophilic N-terminal region on the luminal face (also known
as the exoplasmic face) and their hydrophilic C-terminal region on the cytosolic face. Type II proteins do not contain a cleavable ER signal sequence and are oriented with their hydrophilic N-terminal region on the cytosolic face and their hydrophilic C-terminal region on the exoplasmic face (i.e., opposite to type I proteins). Type III proteins have a hydrophobic membrane-spanning segment at their N-terminus and thus have the same orientation as type I proteins, but do not contain a cleavable signal sequence. Finally, tail-anchored proteins have a hydrophobic segment at their C-terminus that spans the membrane. These different topologies reflect distinct mechanisms used by the cell to establish the orientation of transmembrane segments, as we will see shortly.
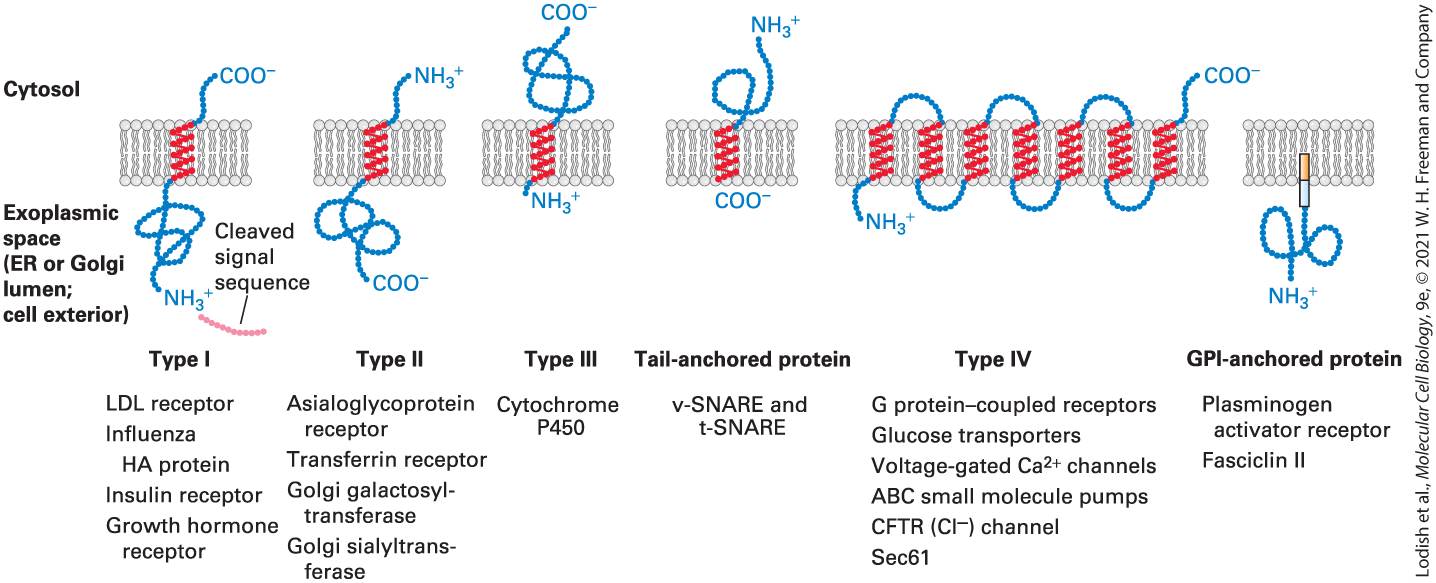
FIGURE 13-10 Classes of ER membrane proteins. Five topological classes of integral membrane proteins are synthesized on the rough ER, as is a sixth type tethered to the membrane by a phospholipid anchor. These membrane proteins are classified by their orientation in the membrane and the types of signals they contain to direct them there. In the integral membrane proteins, hydrophobic segments of the protein chain form α helices embedded in the membrane bilayer; the regions outside the membrane are hydrophilic and fold into various conformations. All type IV proteins have multiple transmembrane α helices. The type IV topology depicted here corresponds to that of G protein–coupled receptors: seven α helices, the N-terminus on the exoplasmic side of the membrane, and the
C-terminus on the cytosolic side. Other type IV proteins may have a different number of helices and various orientations of the N-terminus and C-terminus. See E. Hartmann et al., 1989, P. Natl. Acad. Sci. USA 86:5786; and C. A. Brown and S. D. Black, 1989, J. Biol. Chem. 264:4442. Description Each illustration shows a section of E R membrane separating cytosol from exoplasmic space (E R or Golgi lumen; cell exterior). The classes are as follows: Type 1 which includes L D L receptor, influenza H A protein, insulin receptor, and growth hormone receptor, consists of a helical portion in the membrane with the C-terminal in the cytosol and the N-terminal with folded hydrophilic portion in the exoplasmic space along with a cleaved signal sequence. Type 2, which includes asialoglycoprotein receptor, transferrin receptor, Golgi galactosyl-transferase, and Golgi sialyltransferase, consists of a helical portion embedded in the membrane with the N-terminal on the cytosolic side and the C-terminal and hydrophilic folded portion in the exoplasmic space. Type 3, which includes cytochrome p 450, consists of the N-terminal in the exoplasmic space and the C-terminal and folded hydrophilic portion in the cytosol. Tail-anchored protein, which includes v-SNARE and t-SNARE, consists of a helical portion embedded in the membrane with C-terminal at the exoplasmic face and folded hydrophilic portion and N-terminal in the cytosol. Type 4, which includes G-coupled receptors, glucose transporters, voltage-gated calcium channels, A B C small molecule pumps, C F T R chloride channels, and sec 61, consists of multiple alpha helices embedded in the membrane, connected by chains alternately on the cytosolic and exoplasmic faces, with the N-terminal on the exoplasmic face and the C-terminal in the cytosol. G P I-anchored protein, which includes plasminogen activator receptors and fasciclin 2, consists of a hydrophilic portion in the exoplasmic space attached by the C-terminal to glycosylphosphatidylinositol in the membrane.
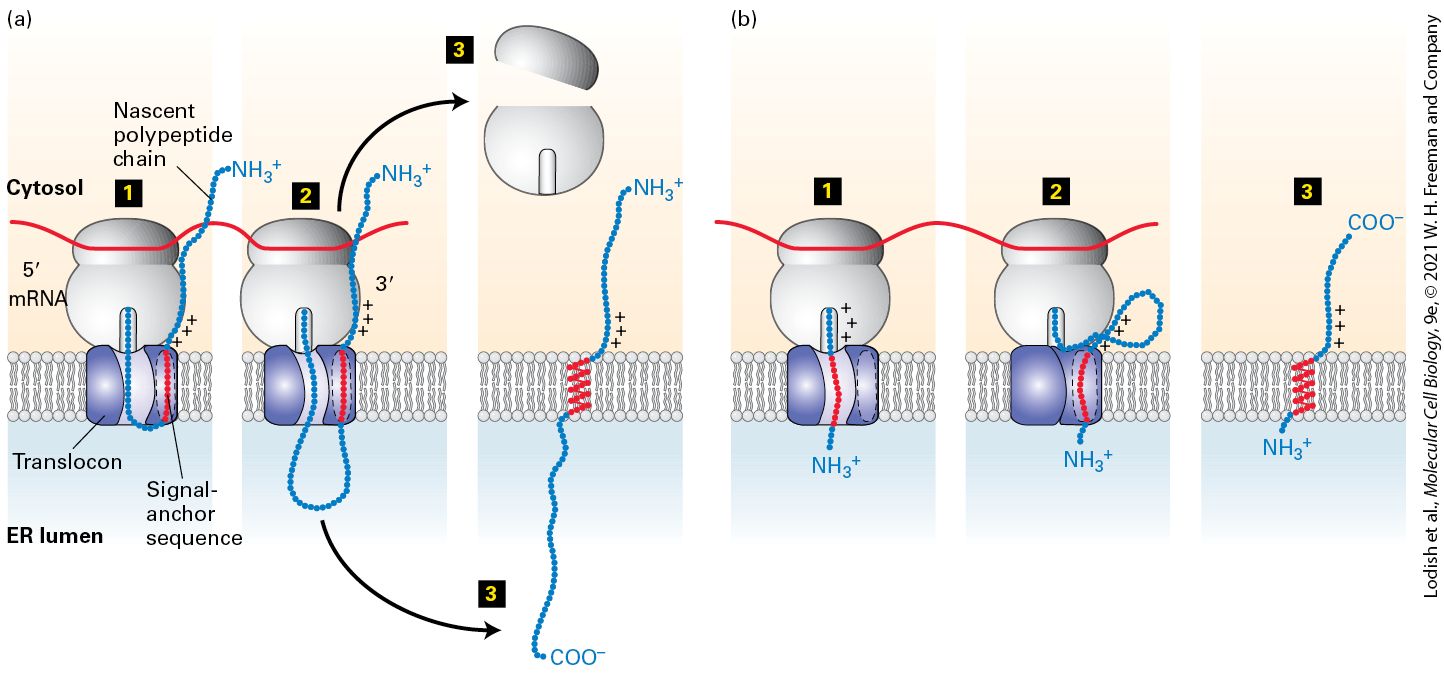
Internal Stop-Transfer Anchor and Signal-Anchor Sequences Determine Topology of Single-Pass Proteins
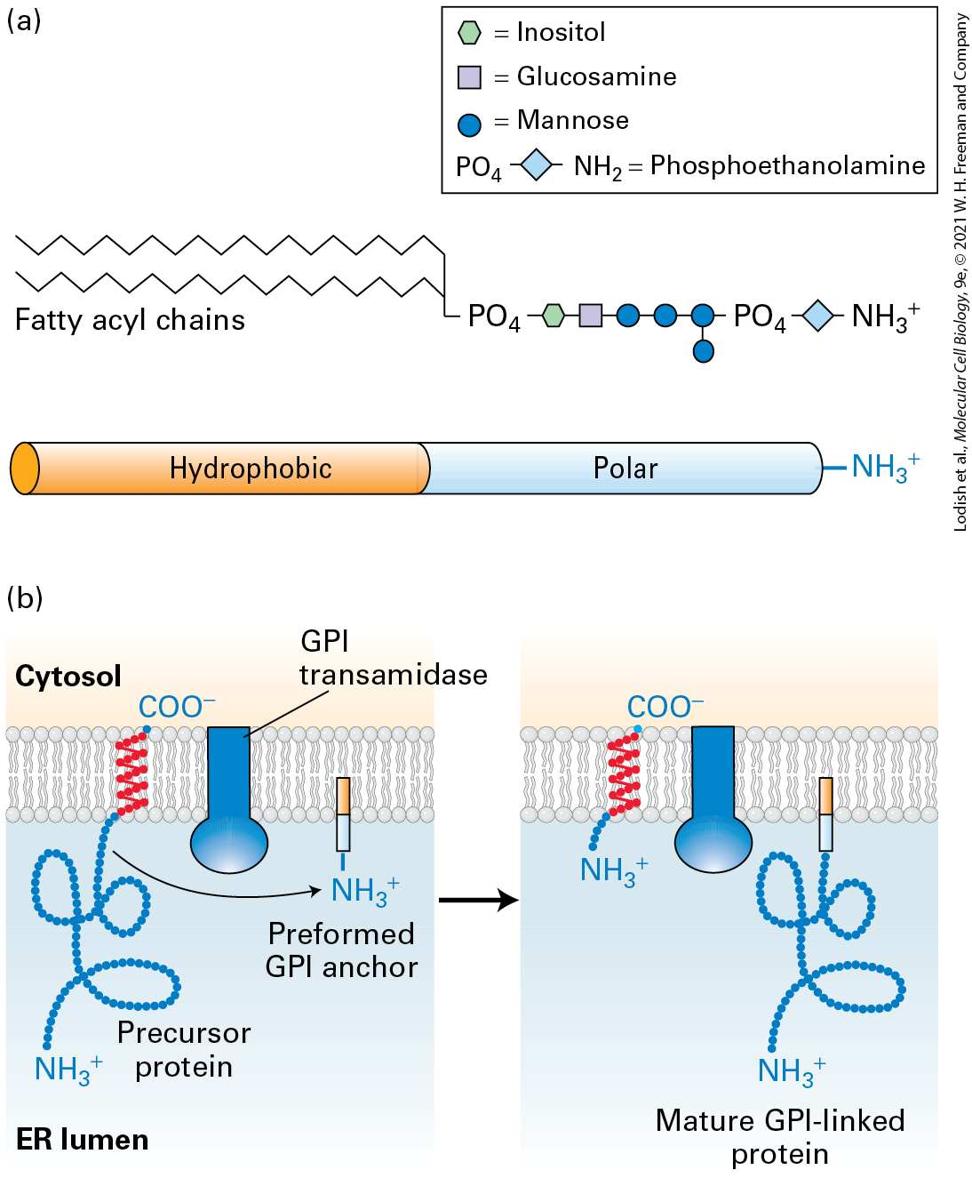
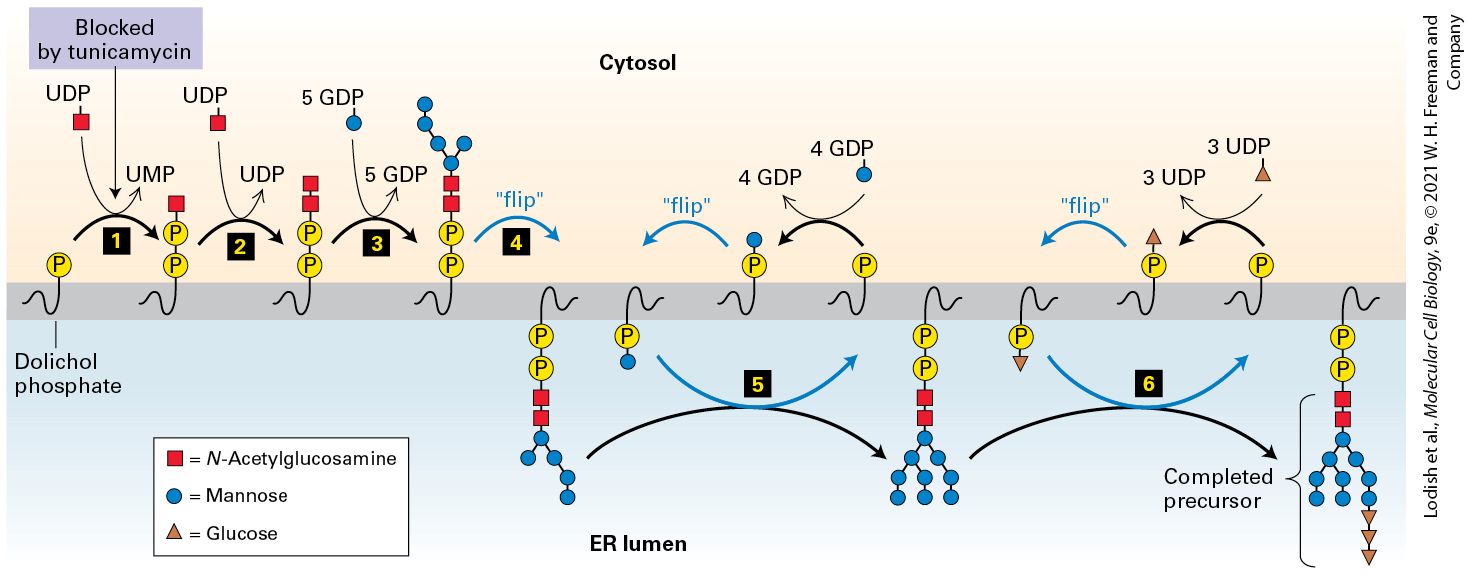
The proteins forming topological type IV contain two or more membranespanning segments and are sometimes called multipass membrane proteins. For example, many of the membrane transport proteins discussed in Chapter 11 and the numerous G protein–coupled receptors covered in Chapter 15 belong to this class. When considering the mechanism of assembly of integral membrane proteins it is often useful to consider the topology of an individual membrane spanning segment. An transmembrane segment is oriented with the N-terminal end facing the inside of the cytoplasm and the C-terminal end facing exoplasmic side of the membrane that would be the lumen of the ER or the topological equivalent that is the outside of the cell. The other possible orientation is . Thus the transmembrane span of a type I membrane protein is , whereas the transmembrane span of a type II protein is . Note that for a type IV protein with multiple spans, the orientation strictly alternates between and . Some lipid-anchored membrane proteins are also synthesized on the ER. These membrane proteins lack a hydrophobic membrane-spanning segment altogether; instead, they are linked to an amphipathic phospholipid anchor that is embedded in the membrane (Figure 13-10, right). Internal Stop-Transfer Anchor and Signal-Anchor Sequences Determine
Topology of Single-Pass Proteins We begin our discussion of how membrane protein topology is determined with the insertion of integral membrane proteins that contain a single hydrophobic membrane-spanning segment. As we will see, three main types of topogenic sequences are used to direct proteins to the ER membrane and to orient them within it. We have already introduced one, the N-terminal signal sequence. The other two types of sequences, which we will introduce here, are internal sequences known as stop-transfer anchor sequences and signal-anchor sequences. Unlike signal sequences, these two types of internal topogenic sequences end up in the mature protein as membrane-spanning segments. However, the two types differ in their final orientation in the membrane. Type I Proteins In addition to an N-terminal signal sequence that targets them to the ER, all type I transmembrane proteins possess an internal hydrophobic sequence of approximately 22 amino acids, which becomes the membranespanning α helix. The N-terminal signal sequence of a nascent type I protein, like that of a soluble secretory protein, initiates cotranslational translocation of the protein through the combined action of the SRP and SRP receptor. Once the N-terminus of the growing polypeptide enters the lumen of the ER, the signal sequence is cleaved, and the growing polypeptide chain continues to be extruded across the ER membrane. However, when the sequence that will become a transmembrane domain
enters the translocon, transfer of the protein through the channel stops and the transmembrane domain is allowed to move laterally from the channel into the membrane (Figure 13-11). The gating mechanism that allows lateral movement is the same as that for the opening of the translocon to accept the hydrophobic core of a signal sequence: two five-helix bundles of Sec61α hinge open to allow the hydrophobic transmembrane segment to move laterally past the hydrophobic signal-sequence-binding site through the opened edge of the translocon (see Figure 13-8). When the peptide exits the translocon in this manner, the hydrophobicity of the transmembrane segment anchors it in the hydrophobic interior of the membrane. Because such a sequence functions both to stop passage of the polypeptide chain through the translocon and to become a hydrophobic transmembrane segment in the membrane bilayer, it is called a stoptransfer anchor sequence (STA).
FIGURE 13-11 Membrane insertion and orientation of type I single-pass transmembrane proteins. Step 1 : After the nascent polypeptide chain–ribosome complex becomes associated with a translocon in the ER membrane, the N-terminal signal sequence is cleaved. This process occurs by the same mechanism as the one for soluble secretory
proteins (see Figure 13-6). Steps 2 – 3 : The chain is elongated until the hydrophobic stop-transfer anchor sequence is synthesized and enters the translocon, where it prevents the nascent chain from extruding farther into the ER lumen. Step 4 : The stop-transfer anchor sequence moves laterally through a hydrophobic cleft between translocon subunits and ultimately becomes anchored in the phospholipid bilayer. At this time, the translocon probably closes. Step 5 : As synthesis continues, the elongating chain may loop out into the cytosol through the small space between the ribosome and translocon. Step 6 : When synthesis is complete, the ribosomal subunits are released into the cytosol, leaving the protein free to diffuse laterally in the membrane. See H. Do et al., 1996, Cell 85:369; and W. Mothes et al., 1997, Cell 89:523. Description The illustrations show a translating ribosome attached to the cytosolic face of translocon embedded in the E R membrane separating cytosol from E R lumen. Step 1. A nascent N-terminal polypeptide chain enters the E R lumen. The signal peptidase is present adjacent to the translocon in the membrane and the signal sequence has been cleaved. Step 2. The polypeptide continues to grow. Step 3. A stop-transfer anchor sequence enters the translocon behind the growing polypeptide chain. Step 4. The stoptransfer anchor sequence stops the further entry of polypeptide chain. The growing chain on the cytosolic side begins to emerge into the cytosol. Step 5. The polypeptide chain in the cytosol continues to grow. Step 6. The ribosome disassociates from the membrane. The type 1 transmembrane protein is now embedded in the membrane with N-terminal chain in lumen, C-terminal in cytosol, and stop-transfer anchor sequence in the membrane. Once translocation is interrupted, translation continues at the ribosome, which is still anchored to the now unoccupied and closed translocon. As the C-terminus of the protein chain is synthesized, it loops out on the cytosolic side of the membrane. When translation is complete, the ribosome is released from the translocon, and the C-terminus of the newly synthesized type I protein remains in the cytosol. Ultimately, the stop-
transfer anchor sequence resides in the membrane in an orientation. Support for this mechanism has come from studies in which cDNAs encoding various mutant receptors for human growth hormone (HGH) were expressed in cultured mammalian cells. The wild-type HGH receptor, a typical type I protein, is normally transported to the plasma membrane. However, a mutant receptor that has charged residues inserted into the single membrane-spanning segment, or that is missing most of this segment altogether, is translocated entirely into the ER lumen and is eventually secreted from the cell as a soluble protein. These kinds of experiments have established that the hydrophobic membrane-spanning segment of the HGH receptor, and of other type I proteins, functions both as a stop-transfer sequence and as a membrane anchor that prevents the C-terminus of the protein from crossing the ER membrane. Type II and Type III Proteins Unlike type I proteins, type II and type III proteins lack a cleavable N-terminal ER signal sequence. Instead, both possess a single internal hydrophobic signal-anchor sequence (SA) that functions as both an ER signal sequence and a membrane anchor. Recall that type II and type III proteins have opposite orientations in the membrane (see Figure 13-10); this difference depends on the orientation that their respective signalanchor sequences assume within the translocon. The internal signal-anchor sequence in type II proteins directs insertion of the nascent polypeptide chain into the ER membrane so that the N-terminus of the chain faces the
cytosol, using the same SRP-dependent mechanism described for signal sequences (Figure 13-12a). However, the internal signal-anchor sequence does not have a recognition sequence for signal peptidase and therefore is not cleaved. Because of its hydrophobicity, the signal-anchor sequence can move laterally from the signal-sequence-binding site at the edge of the translocon directly into the phospholipid bilayer, where it functions as a membrane anchor. As elongation continues, the C-terminal region of the growing chain is extruded through the translocon into the ER lumen by cotranslational translocation.
FIGURE 13-12 Membrane insertion and orientation of type II and type III single-pass transmembrane proteins. (a) Type II proteins. Step 1 : After the internal signal-anchor sequence is synthesized on a cytosolic ribosome, it is bound by an SRP (not shown), which binds the SRP receptor on the ER membrane. This process is similar to the targeting of soluble secretory proteins except that the hydrophobic signal sequence is not located at the N-terminus and is not subsequently cleaved. The nascent polypeptide chain becomes oriented in the translocon with its N-terminal portion toward the cytosol. This orientation is dictated by the positively charged residues shown N-terminal to the signal-anchor sequence. Step 2 : As the chain is elongated and extruded into the lumen, the internal signal-anchor sequence moves laterally through a hydrophobic cleft between translocon subunits and
anchors the chain in the phospholipid bilayer. Step 3 : Once protein synthesis is complete, the C-terminus of the polypeptide is released into the lumen and the ribosomal subunits are released into the cytosol. (b) Type III proteins. Step 1 : Insertion is by a process similar to that of type II proteins, except that positively charged residues on the C-terminal side of the signal-anchor sequence cause the transmembrane segment to be oriented within the translocon with its C-terminal portion toward the cytosol and the N-terminal end in the ER lumen. Note that when the signal-anchor segment engages with the translocon, the preceding hydrophilic segment is short enough to spontaneously pass through the translocon channel. Steps 2 – 3 : Elongation of the C-terminal portion of the polypeptide chain is completed in the cytosol, and the ribosomal subunits are released. See M. Spiess and H. F. Lodish, 1986, Cell 44:177; and H. Do et al., 1996, Cell 85:369. Description Both (a) and (b) illustrations show a translating ribosome attached to the cytosolic face of translocon embedded in the E R membrane separating cytosol from E R lumen. The series (a) shows the process of type 2 proteins as follows: Step 1. A growing polypeptide sequence enters and exits the E R lumen through the translocon where the positively-charged signal-anchor sequence gets bound in the side pocket of translocon. Step 2. Polypeptide synthesis continues and the chain grows in the lumen. Step 3. The ribosome dissociates leaving behind the transmembrane protein with the N-terminal in the cytosol, C-terminal in the exoplasm, and signal-anchor sequence in the E R membrane. The series (b) shows the process of type 3 proteins as follows: Step 1. A growing polypeptide sequence enters the translocon followed by the positively-charged signalanchor sequence that gets bind into the translocon. Step 2. Polypeptide synthesis continues and the chain grows in the cytosol. Step 3. The ribosome exits leaving behind the transmembrane protein with the N-terminal in the exoplasm, C-terminal in the cytoplasm, and signal-anchor sequence in the E R membrane. In the case of type III proteins, the signal-anchor sequence, which is located near the N-terminus, directs insertion of the nascent chain into the ER membrane with its N-terminus facing the lumen, in an orientation
opposite to that of the signal-anchor in type II proteins. The signal-anchor sequence of type III proteins also functions like a stop-transfer sequence and prevents further extrusion of the elongating chain into the ER lumen (Figure 13-12b). Continued elongation of the chain C-terminal to the signal-anchor sequence proceeds as it does for type I proteins, with the hydrophobic sequence eventually moving laterally out of the translocon to anchor the polypeptide in the ER membrane (see Figure 13-11). The key difference between type II and type III proteins is the orientation of the hydrophobic transmembrane segment as it binds to the hydrophobic signal-sequence-binding site at the edge of Sec61α. The membrane span of a type II protein is oriented whereas the span of a type III protein is . The most important feature that determines whether a protein with a single transmembrane span will assume a type II or type III orientation is the length of the hydrophilic sequence that precedes the transmembrane span. If the hydrophilic segment is more than a few amino acids in length, the energetic cost of transferring this segment across the membrane is too great for the transmembrane segment to assume an orientation of a type III protein. The orientation of signalanchor sequences that are preceded by only a few hydrophilic amino acids is determined by the arrangement of positively charged amino acids adjacent to one end or the other of the hydrophobic segment. These positively charged residues tend to remain on the cytosolic side of the membrane, rather than traversing the membrane into the ER lumen. Thus the position of the charged residues dictates the orientation of the signalanchor sequence within the translocon as well as whether the rest of the polypeptide chain continues to pass into the ER lumen: type II proteins
tend to have positively charged residues on the N-terminal side of their signal-anchor sequence, orienting the N-terminus in the cytosol and allowing passage of the C-terminal side into the ER (see Figure 13-12a), whereas type III proteins tend to have positively charged residues on the C-terminal side of their signal-anchor sequence, which restrict the C-terminus to the cytosol (see Figure 13-12b). Note that the hydrophobic segment of a type II signal-anchor sequence assumes the same orientation as the signal sequence of a secreted protein and that in most respects these signal-anchor sequences behave exactly like signal sequences, although they are not cleaved. A striking experimental demonstration of the importance of the flanking charge in determining orientation in the membrane is provided by neuraminidase, a type II protein in the surface coat of the influenza virus. Three arginine residues are located just N-terminal to the internal signalanchor sequence in neuraminidase. Mutation of these three positively charged residues to negatively charged glutamate residues causes neuraminidase to acquire the reverse orientation. Similar experiments have shown that other proteins, with either type II or type III orientation, can be made to “flip” their orientation in the ER membrane by mutating charged residues that flank the internal signal-anchor segment. Tail-Anchored Proteins For all the topological classes of proteins we have considered so far, membrane insertion begins when the SRP recognizes a hydrophobic topogenic sequence as it emerges from the ribosome. Recognition of tail-
anchored proteins, which have a single hydrophobic topogenic sequence at the C-terminus, presents a unique challenge because the hydrophobic C-terminus becomes available for recognition only after translation has been completed and the protein has been released from the ribosome. Insertion of tail-anchored proteins into the ER membrane does not employ an SRP, SRP receptor, or the translocon, but instead depends on a pathway dedicated to this purpose, as depicted in Figure 13-13. This pathway involves a dimer of an ATPase known as Get3, which binds to the C-terminal hydrophobic segment of a tail-anchored protein. The Get3 dimer has two ATP-binding sites at the dimer interface in a structure that is similar to the two GTP-binding sites in the heterodimeric interface between SRP and SRP receptor shown in Figure 13-5b. The complex of a Get3 dimer bound to a tail-anchored protein is recruited to the ER by a dimeric integral membrane receptor known as . The tailanchored protein is released from Get3, and the transmembrane portion of participates in the insertion of the tail-anchor into the ER membrane. This process is mechanistically similar to the targeting of type II and type III signal-anchor sequences to the ER by the SRP and SRP receptor; the underlying structural similarity indicates that the mechanism of targeting is fundamentally related, despite the fact that Get3 couples targeting of tail-anchored proteins to ATP hydrolysis, whereas SRP couples protein targeting to GTP hydrolysis.
FIGURE 13-13 Insertion of tail-anchored proteins. For C-terminal tail-anchored proteins, the hydrophobic C-terminus is not available for membrane insertion until protein synthesis is complete and the protein has been released from the ribosome. Step 1 : Dimeric Get3 contains a hydrophobic binding pocket at the interface between dimer subunits and Get3 in an ATP-bound state binds to the hydrophobic C-terminal tail of the protein. This binding reaction is facilitated by a complex of three other proteins, Sgt2, Get4, and Get5, which sequester the hydrophobic C-terminal tail before transferring it to Get3⋅ATP (not shown). Step 2 : The ternary complex Get3⋅ATP bound to the protein docks onto the dimeric receptor, which is embedded in the ER membrane. Step 3 : In succession, ATP is hydrolyzed and ADP is released from Get3. At the same time, the hydrophobic C-terminal tail is released from Get3 and ultimately becomes embedded in the ER membrane in a process that is facilitated by . Step 4 : Get3 binds to ATP and Get3⋅ATP is released from in a soluble form, ready for another round of binding to a hydrophobic C-terminal tail.
Type IV (Multipass) Proteins
Description The illustrations show a dimeric Get 1 slash Get 2 receptor embedded in the E R membrane separating cytosol from E R lumen. Step 1. A hydrophobic C-terminal tail of polypeptide binds in the pocket of a dimeric Get 3. Each subunit of Get 3 has an A T P attached to it. Step 2. The polypeptide bound dimeric Get 3 binds on the cytosolic surface of the dimeric Get 1 slash Get 2 receptor. Step 3. The polypeptide detaches from dimeric Get 3 as A T Ps are converted to A D P and P i. The C-terminal tail of the detached polypeptide gets embedded in the E R membrane beside the dimeric Get 1 slash Get 2 receptor. Step 4. A D Ps leave the dimeric Get 3, detaches from dimeric Get 1 slash Get 2 receptor, and binds to two A T Ps to get back to its initial structure. Type IV (Multipass) Proteins As described in Chapter 11, many physiologically important proteins, such as channel proteins, membrane transporters and pumps, and some receptor proteins, can contain 12 or more membrane-spanning α helices. Although their three-dimensional structure can be quite complex, the membrane topology of multipass proteins can usually be deduced from the same principles used to predict the topology of single-pass proteins. Figure 1314 summarizes the arrangements of topogenic sequences in single-pass and multipass transmembrane proteins. Two key principles govern the assembly of the vast majority of multipass proteins: (1) the membranespanning segments pass from the translocon into the membrane cotranslationally in the sequential order by which they emerge from the ribosome and (2) the first topogenic segment engages the translocon in an SRP and SRP receptor-dependent manner as for single-pass proteins, whereas all subsequent transmembrane segments engage the translocon
independently of SRP. Based on these principles, the first transmembrane segment of a multipass protein acts as a topogenic sequence in the ways that we have already discussed for type I, type II, and type III proteins. Once the orientation of the first transmembrane segment is established ( as with type I and type III proteins or as for type II proteins), each subsequent transmembrane segment assumes the opposite orientation as the one before. Thus an transmembrane segment is always followed by an transmembrane segment and an transmembrane segment is always followed by an transmembrane segment. This strict alternation of orientation is a simple consequence of the fact that the hydrophilic segments cannot move across the membrane unless threaded through the channel of the translocon. If a multipass protein has an even number of transmembrane α helices, its N-terminus and C-terminus will be oriented toward the same side of the membrane (Figure 13-14d). Conversely, if a type IV protein has an odd number of α helices, its two ends will have opposite orientations (Figure 13-14e).
FIGURE 13-14 Topogenic sequences determine the orientation of ER membrane proteins. Topogenic sequences are shown in red; soluble, hydrophilic sequences in blue. The internal topogenic sequences form transmembrane α helices that anchor proteins or segments of proteins in the membrane. (a) Type I proteins contain a cleaved signal sequence and a single internal stop-transfer anchor (STA) sequence. (b, c) Type II and type III proteins contain a single internal signal-anchor (SA) sequence. The difference in the orientation of these protein types depends largely on whether there is a high density of positively charged amino acids on the N-terminal side (type II) or on the C-terminal side of the SA sequence (type III). (d, e) Nearly all multipass proteins lack a cleavable signal sequence, as depicted in the examples shown here. Type IV-A proteins, whose N-terminus faces the cytosol, begin with an SA sequence similar to a type II protein. Type IV-B proteins, whose N-terminus faces the lumen, begin with an SA sequence similar to a type III protein. After the first topogenic sequence all subsequent transmembrane segments insert into the membrane with the opposite polarity as the segment before. Description The sequences have S T A for internal stop-transfer anchor sequence, S A for internal signal anchor sequence, cytosol sequences, and lumen sequences. The topogenic sequences of different classes of transmembrane proteins starting from the N-terminal to C-terminal are as follows: (a) Type 1: N-terminal, signal sequence, lumen, S T A, cytosol, C-terminal.
(b) Type 2: N-terminal, cytosol, positively charged region, S A, lumen, C-terminal. (c) Type 3: N-terminal, very small lumen, S A, positively charged region, cytosol, C-terminal. (d) Type 4-A: N-terminal, cytosol, positively charged region, S A, lumen, anchor sequence, cytosol, anchor sequence, lumen, anchor sequence, cytosol, C-terminal. (e) Type 4-B: N-terminal, S A, positively charged region, cytosol, anchor sequence, lumen, anchor sequence, cytosol, anchor sequence, lumen, anchor sequence, cytosol, anchor sequence, lumen, anchor sequence, cytosol, C-terminal. Type IV Proteins with N-Terminus in the Cytosol Among the multipass proteins whose N-terminus extends into the cytosol are the various glucose transporters (GLUTs) and most ion-channel proteins, discussed in Chapter 11. In these proteins, the hydrophobic segment closest to the N-terminus functions like the internal signal-anchor sequence of a type II protein and is inserted into the ER membrane with an orientation (see Figure 13-12a). As the nascent chain following the transmembrane segment elongates, it moves through the translocon until the second hydrophobic segment is formed. This helix prevents further extrusion of the nascent chain through the translocon; thus its function is similar to that of the stop-transfer anchor sequence in a type I protein (see Figure 13-11). After synthesis of the first two transmembrane α helices, both ends of the nascent chain face the cytosol, and the loop between them extends into the ER lumen. As the C-terminus of the nascent chain then continues to grow into the cytosol, as it does in synthesis of type III proteins, the ribosome
A Phospholipid Anchor Tethers Some Cell-Surface Proteins to the Membrane
remains attached to the translocon, and hydrophobic sequences that subsequently emerge from the ribosome are threaded into the translocon without the need for the SRP and the SRP receptor. As new hydrophobic topogenic sequences engage the translocon, the previously engaged sequences move laterally out of the translocon using the same mechanism as for type I, type II, and type III membrane proteins. Type IV Proteins with N-Terminus in the Exoplasmic Space The large family of G protein–coupled receptors, all of which contain seven transmembrane α helices, constitute the most numerous type IV-B proteins, whose N-terminus extends into the exoplasmic space. In these proteins, the hydrophobic transmembrane segment closest to the N-terminus is often followed by a cluster of positively charged amino acids, like a type III signal-anchor sequence (see Figure 13-12b). As a result, the nascent polypeptide chain is inserted into the translocon with the N-terminus extending into the lumen (see Figure 13-14e). As the chain is elongated, it is inserted into the ER membrane in alternating orientation, as just described for type IV-A proteins. A Phospholipid Anchor Tethers Some Cell-Surface Proteins to the Membrane
Some cell-surface proteins are anchored to the phospholipid bilayer not by a sequence of hydrophobic amino acids, but by a covalently attached amphipathic molecule, glycosylphosphatidylinositol (GPI) (Figure 13-15a; see also Chapter 7). These proteins are synthesized and initially anchored to the ER membrane exactly like type I transmembrane proteins, with a cleaved N-terminal signal sequence and an internal stop-transfer anchor sequence directing the process (see Figure 13-11). However, a short sequence of amino acids in the luminal domain, adjacent to the membrane-spanning domain, is recognized by a transamidase located within the ER membrane. This enzyme simultaneously cleaves off the original stop-transfer anchor sequence and transfers the luminal portion of the protein to a preformed GPI anchor in the membrane (Figure 13-15b).
FIGURE 13-15 GPI-anchored proteins. (a) Structure of a glycosylphosphatidylinositol (GPI) molecule from yeast. The hydrophobic portion of the molecule is composed of fatty acyl chains, whereas the polar (hydrophilic) portion is composed of carbohydrate residues and phosphate groups. In other organisms, both the length of the acyl chains and the
carbohydrate moieties may vary somewhat from the structure shown. (b) Formation of GPIanchored proteins in the ER membrane. The protein is synthesized and initially inserted into the ER membrane like a type I transmembrane protein, as shown in Figure 13-11. A specific transamidase simultaneously cleaves the precursor protein within the exoplasmic-facing domain, near the stop-transfer anchor sequence (red), and transfers the carboxyl group of the new C-terminus to the terminal amino group of a preformed GPI anchor. See C. Abeijon and C. B. Hirschberg, 1992, Trends Biochem. Sci. 17:32; and K. Kodukula et al., 1992, P. Natl. Acad. Sci. USA 89:4982. Description A two-part illustration (a) shows the structure of G P I-anchored proteins. The first part two long wavy fatty acyl chains bound to a phosphate which is further attached to inositol, glucosamine, four mannoses, and phosphoethanolamine at the N H 3 plus end. The second part shows a horizontal rod with left half labeled hydrophobic and right half labeled polar with an N H 3 plus end. A two-part illustration (b) shows the formation of G P I-anchored proteins on E R membrane. The first part shows a G P I transamidase embedded in the E R membrane adjacent (left side) to a precursor protein having its C-terminal end embedded in E R membrane and N-terminal end with a long chain in E R lumen. A pre-formed G P I anchor is embedded in E R membrane, to the right side of G P I transamidase and have its polar N H 3 plus end protruding in the lumen. An arrow from precursor protein points to pre-formed G P I anchor. The second part shows the lumen precursor protein now bound to the polar N H 3 plus end of the pre-formed G P I anchor which is now labeled, mature G P I-linked protein. The C-terminal end of protein is still anchored in the E R membrane. Why change one type of membrane anchor for another? Attachment of the GPI anchor, which results in removal of the cytosol-facing hydrophilic domain from the protein, can have several consequences. Proteins with GPI anchors, for example, can diffuse relatively rapidly in the plane of the phospholipid bilayer. In contrast, many proteins anchored by membrane-
The Topology of a Membrane Protein Can Often Be Deduced from Its Sequence
spanning α helices are impeded from moving laterally in the membrane because their cytosol-facing segments interact with the cytoskeleton. In addition, the GPI anchor targets the attached protein to the apical domain of the plasma membrane (instead of the basolateral domain) in certain polarized epithelial cells, as we discuss in Chapter 14. The Topology of a Membrane Protein Can Often Be Deduced from Its Sequence As we have seen, various topogenic sequences in integral membrane proteins synthesized on the ER govern the interaction of the nascent polypeptide chain with the translocon. When scientists begin to study a protein of unknown function, the identification of potential topogenic sequences within the corresponding gene sequence can provide important clues about the protein’s topological class and function. Suppose, for example, that the gene for a protein known to be required for a cell-to-cell signaling pathway contains nucleotide sequences that encode an apparent N-terminal signal sequence and an internal hydrophobic sequence. These findings suggest that the protein is a type I integral membrane protein and therefore may be a cell-surface receptor for an extracellular ligand. Furthermore, the implied type I topology suggests that the N-terminal segment that lies between the signal sequence and the internal hydrophobic sequence probably constitutes the extracellular domain with a role in ligand binding, whereas the C-terminal segment that lies after the
internal hydrophobic sequence is probably cytosolic and may play a role in intracellular signaling. Identification of topogenic sequences requires a way to scan sequence databases for segments that are sufficiently hydrophobic to be either signal sequences or transmembrane anchor sequences. Topogenic sequences can often be identified with the aid of computer programs that generate a hydropathy profile for the protein of interest. The first step is to assign a value known as the hydropathic index to each amino acid in the protein. By convention, hydrophobic amino acids are assigned positive values and hydrophilic amino acids negative values. Although different scales for the hydropathic index exist, all assign the most positive values to amino acids with side chains made up of mostly hydrocarbon residues (e.g., phenylalanine and methionine) and the most negative values to charged amino acids (e.g., arginine and aspartate). The second step is to identify long segments of sufficient overall hydrophobicity to be N-terminal signal sequences or internal stop-transfer anchor sequences and signal-anchor sequences. To accomplish this, the total hydropathic index for each segment of 20 consecutive amino acids is calculated along the entire length of the protein. Plots of these calculated values against position in the amino acid sequence yield a hydropathy profile.
Figure 13-16 shows the hydropathy profiles for three different membrane proteins. The prominent peaks in such plots identify probable topogenic sequences as well as their positions and approximate lengths. For example, the hydropathy profile of the human growth hormone receptor reveals the presence of both a hydrophobic signal sequence at the extreme N-terminus
of the protein and an internal hydrophobic stop-transfer anchor sequence (Figure 13-16a). On the basis of this profile, we can deduce, correctly, that the HGH receptor is a type I integral membrane protein. The hydropathy profile of the asialoglycoprotein receptor, a cell-surface protein that mediates removal of abnormal extracellular glycoproteins, reveals a prominent internal hydrophobic signal-anchor sequence, but gives no indication of a hydrophobic N-terminal signal sequence (Figure 13-16b). Thus we can predict that the asialoglycoprotein receptor is a type II or type III membrane protein. Because the N-terminal segment of the asialoglycoprotein receptor contains 30 hydrophilic residues that could not move across the membrane without a signal sequence to initiate engagement with the translocon, we can deduce that this segment must remain in the cytosol and we can correctly predict that this is a type II protein.
FIGURE 13-16 Hydropathy profiles for three types of proteins. Hydropathy profiles can identify likely topogenic sequences in integral membrane proteins. They are generated by plotting the total hydrophobicity of each segment of 20 contiguous amino acids along the length of a protein. Positive values indicate relatively hydrophobic portions of the protein; negative values relatively hydrophilic portions. Probable hydrophobic topogenic sequences
are marked. (a) The human growth hormone receptor evidently has two topogenic sequences, a signal sequence at the N-terminus and an internal hydrophobic stop-transfer sequence. Note that the membrane-spanning stop-transfer sequence is significantly more hydrophobic than the signal sequence, which is typically the case. From this arrangement it would be possible to correctly deduce that this is a type I membrane protein with an N-terminal domain that binds the growth hormone. (b) The asialoglycoprotein receptor has a single hydrophobic topogenic sequence. This sequence can be correctly predicted to be a signal-anchor sequence for a type II protein because it is preceded by a hydrophilic segment of 30 amino acids. As a type II protein, the C-terminal domain is predicted to be the asialoglycoprotein binding domain. The complex profiles for multipass (type IV) proteins, such as GLUT1 in part (c), must often be supplemented with other analyses to determine the topology of these proteins. Description The vertical axis of all three graphs ranges from negative 3 to 4, in increments of 1. Graph (a) shows the human growth hormone receptor (type 1). The horizontal axis is labeled N-terminus on left end and C-terminus on right end and ranges from 100 to 500, in increments of 100. The curve starts at (0 or N-terminus, 1) and ends at (640 or C-terminus, 0). The curve shows several ups and downs with peaks not going beyond 2 and dips not going below negative 3. A shaded region representing signal sequence ranges from N-terminus to 20 with a peak at 2; and another shaded region representing stop-transfer sequence ranges from 260 to 290 with a peak at 3. Graph (b) shows the asialoglycoprotein receptor (type 2). The horizontal axis ranges from 0 to 300, in increments of 100. The curve starts at (0, 1) and ends at (290, negative 0.5). The curve shows several ups and downs with peaks not going beyond 1 and dips not going below negative 3. A shaded region representing signal-anchor sequence ranges from 40 to 60 with a peak at 3.5. Graph (c) shows the GLUT 1 (type 4). The horizontal axis ranges from 0 to 500, in increments of 100. The curve starts at (0, negative 1) and ends at (490, 0.3). The curve shows several ups and downs with 12 high peaks going beyond 2. Twenty sequences around the peak regions are shaded and represent transmembrane sequences.
The hydropathy profile of the GLUT1 glucose transporter, a multipass transmembrane protein, shows the presence of many segments that are sufficiently hydrophobic to be membrane-spanning helices (Figure 1316c). The complexity of this profile illustrates the difficulty both in predicting the orientation of the first topogenic sequence and in unambiguously identifying all the membrane-spanning segments. More sophisticated computer algorithms have been developed that take into account the presence of positively charged amino acids adjacent to hydrophobic segments as well as the length of and spacing between segments. Using all this information, the best algorithms can predict the complex topology of multipass proteins with an accuracy of greater than 80 percent. Finally, sequence homology to a known protein may permit accurate prediction of the topology of a newly discovered multipass protein. For example, the genomes of multicellular organisms encode a very large number of multipass proteins with seven hydrophobic transmembrane segments. The similarities between the sequences of these proteins strongly suggest that all have the same topology as the well-studied G protein–coupled receptors, which have the N-terminus oriented to the exoplasmic side and the C-terminus oriented to the cytosolic side of the membrane. KEY CONCEPTS OF SECTION 13.2 Insertion of Membrane Proteins into the ER
Proteins synthesized on the rough ER include five topological classes of integral membrane proteins as well as a lipid-anchored type protein (see Figure 13-10). Topogenic sequences — N-terminal signal sequences, internal stop-transfer anchor sequences, and internal signal-anchor sequences — direct the insertion of nascent proteins into the ER membrane and their orientation within it. This orientation is retained during transport of the completed membrane protein to its final destination — for example, the plasma membrane. Single-pass membrane proteins that are initiated by interaction with SRP and are synthesized on ribosomes bound to the translocon are of three topological varieties: type I, type II, and type III. The topological type depends on the presence or absence of a signal sequence, the length of the hydrophilic N-terminal segment, and the arrangement of positively charged residues adjacent to the hydrophobic transmembrane segment (see Figure 13-14). Tail-anchored proteins that have a single C-terminal transmembrane domain are inserted into the membrane post-translationally. Their insertion depends on a cytosolic receptor, Get3, which is an ATPase with similarity to the GTPase domains of SRP and SRP receptor. The topology of multipass (type IV) proteins is established by the orientation of the first transmembrane segment according to the same principles as for single-pass proteins. Each additional transmembrane segment engages the translocon in sequential order as they emerge from the ribosome and thus are inserted in the membrane in alternating orientation. Some cell-surface proteins are initially synthesized as type I proteins, but then cleaved, and their luminal domains transferred to a GPI anchor (see Figure 13-15). The topology of membrane proteins can often be correctly predicted by computer programs that identify hydrophobic topogenic segments within the amino acid sequence and generate hydropathy profiles (see Figure 13-16).
13.3 Protein Modifications, Folding, and Quality Control in the ER
13.3 Protein Modifications, Folding, and Quality Control in the ER Membrane and soluble secretory proteins synthesized on the rough ER undergo four principal modifications before they reach their final destinations: (1) covalent addition and processing of carbohydrates (glycosylation) in the ER and Golgi complex; (2) formation of disulfide bonds in the ER; (3) proper folding of polypeptide chains and assembly of multisubunit proteins in the ER; and (4) specific proteolytic cleavages in the ER, Golgi complex, and secretory vesicles. Generally speaking, these modifications promote the folding of secretory proteins into their native structures and add structural stability to proteins exposed to the extracellular environment. Modifications such as glycosylation also allow the cell to produce a vast array of chemically distinct molecules at the cell surface that are the basis of specific molecular interactions used in cell-tocell adhesion and communication. The majority of proteins that are synthesized on the rough ER and enter the lumen of the ER are modified by the addition of one or more carbohydrate chains. Proteins with attached carbohydrates are known as glycoproteins. Carbohydrate chains in glycoproteins attached to the hydroxyl group in serine and threonine residues are referred to as O-linked oligosaccharides, and carbohydrate chains attached to the amide
nitrogen of asparagine are referred to as N-linked oligosaccharides. The various types of O-linked oligosaccharides include the mucin-type O-linked chains (named after the abundant glycoproteins found in mucus) and the carbohydrate modifications on proteoglycans described in Chapter 20. O-linked chains typically consist of sugar residues in a linear chain, which are added to proteins by enzymes known as glycoslytransferases, located in the lumen of the Golgi complex. The more common N-linked oligosaccharides have a branched structure and are initially added to proteins in the ER. In this section, we focus on N-linked oligosaccharides, which are synthesized as a branched precursor in the ER. After the initial transfer of N-linked precursor to a protein in the ER, the oligosaccharide chain is modified in the ER and commonly in the Golgi complex as well. Disulfide bond formation, protein folding, and assembly of multimeric proteins, which take place exclusively in the rough ER, are also discussed in this section. Only properly folded and assembled proteins are transported from the rough ER to the Golgi complex and ultimately to the cell surface or other final destination through the secretory pathway. Unfolded, misfolded, or partly folded and assembled proteins are selectively retained in the rough ER and marked for degradation. We consider several features of such quality control in the latter part of this section. As discussed previously, N-terminal ER signal sequences are cleaved from soluble secretory proteins and type I membrane proteins in the ER. Some proteins also undergo other specific proteolytic cleavages in the Golgi complex or secretory vesicles. We cover these cleavages, as well as
A Preformed N-Linked Oligosaccharide Is Added to Many Proteins in the Rough ER
carbohydrate modifications that occur primarily or exclusively in the Golgi complex, in the next chapter. A Preformed N-Linked Oligosaccharide Is Added to Many Proteins in the Rough ER Biosynthesis of all N-linked oligosaccharides begins in the rough ER with a preformed oligosaccharide precursor containing 14 residues (Figure 1317). The structure of this precursor is the same in plants, animals, and single-celled eukaryotes: a branched oligosaccharide, containing three glucose (Glc), nine mannose (Man), and two N-acetylglucosamine (GlcNAc) molecules, which can be written as . Once added to a protein, this branched carbohydrate structure is modified by addition or removal of monosaccharides in the ER and Golgi complex. The modifications to N-linked chains differ from one glycoprotein to another and among different organisms, but a core of 5 of the 14 residues is conserved in the structures of all N-linked oligosaccharides on secretory and membrane proteins.
FIGURE 13-17 Biosynthesis of the oligosaccharide precursor. Dolichol phosphate is a strongly hydrophobic lipid, containing 75–95 carbon atoms, that is embedded in the ER membrane. Two N-acetylglucosamine (GlcNAc) and five mannose residues are added one at a time to a dolichol phosphate on the cytosolic face of the ER membrane (steps 1 – 3 ). The nucleotide-sugar donors in these and later reactions are synthesized in the cytosol. Note that the first sugar residue is attached to dolichol by a high-energy pyrophosphate linkage. Tunicamycin, which blocks the first enzyme in this pathway, inhibits the synthesis of all N-linked oligosaccharides in cells. After the seven-residue dolichol pyrophosphoryl intermediate is flipped to the luminal face (step 4 ), the remaining four mannose residues and all three glucose residues are added one at a time (steps 5 – 6 ). In the later reactions, the sugar to be added is first transferred from a nucleotide sugar to a carrier dolichol phosphate on the cytosolic face of the ER; the carrier is then flipped to the luminal face, where the sugar is transferred to the growing oligosaccharide, after which the “empty” carrier is flipped back to the cytosolic face. See C. Abeijon and C. B. Hirschberg, 1992, Trends Biochem. Sci. 17:32. Description The illustration shows an E R membrane separating cytosol from E R lumen. The process starts with a Dolichol phosphate embedded in the E R membrane with terminal phosphate group facing the cytosol. Step 1: Dolichol phosphate is enzymatically phosphorylated by U D P-N-acetylglucosamine, transferring a phosphate and N-acetylglucosamine to the dolichol phosphate and losing U M P. The step can be blocked by tunicamycin. Step 2: Another U D P-N-acetylglucosamine adds an additional n-acetylglucosamine to the growing chain. Step 3: 5 G D P-mannose transfer
the mannose units to the growing Dolichol chain toward cytosol. Step 4: The chain flips causing the attached parts to enter the E R lumen. Step 5: A Dolichol phosphate on the cytosol coupled to 4 mannoses also flips and transfers these mannose molecules to previous flipped chain that now has 9 mannoses. Step 6: Another Dolichol phosphate on the cytosol coupled to three glucose also flips and transfers these glucose molecules to 9 mannoses chain. The completed precursor has two phosphates, two N-acetylglucosamines, nine mannoses, and three glucose in the E R lumen. Prior to transfer to a nascent chain in the lumen of the ER, the oligosaccharide precursor is assembled on a membrane-attached anchor called dolichol phosphate, a long-chain polyisoprenoid lipid (see Chapter 10). After the first sugar, GlcNAc, is attached to the dolichol phosphate by a pyrophosphate bond, the other sugars are added by glycosidic bonds in a complex set of reactions catalyzed by enzymes attached to the cytosolic or luminal faces of the rough ER membrane (see Figure 13-17). The final dolichol pyrophosphoryl oligosaccharide is oriented so that the oligosaccharide portion faces the ER lumen. The three glucose residues, which are the last residues added during synthesis of the precursor on the dolichol carrier, appear to act as a signal that the oligosaccharide is complete and ready to be transferred to a protein. The entire 14-residue precursor is transferred from the dolichol carrier to an asparagine residue on a nascent polypeptide as it emerges into the ER lumen (Figure 13-18, step 1 ). Only asparagine residues in the tripeptide sequences Asn-X-Ser and Asn-X-Thr (where X is any amino acid except proline) are substrates for oligosaccharyl transferase, the enzyme that catalyzes this reaction. Two of the three subunits of this enzyme are ER membrane proteins whose cytosol-facing domains bind to the ribosome,
localizing a third subunit of the transferase, the catalytic subunit, near the growing polypeptide chain in the ER lumen. Not all sequences become glycosylated, and it is not possible to predict from the amino acid sequence alone which potential N-linked glycosylation sites will be modified; for instance, rapid folding of a segment of a protein containing an sequence may prevent transfer of the oligosaccharide precursor to it. Immediately after the entire precursor, , is transferred to a nascent polypeptide, three different enzymes, called glycosidases, remove all three glucose residues and one particular mannose residue (see Figure 13-18, steps 2 – 4 ).
FIGURE 13-18 Addition and initial processing of N-linked oligosaccharides. In the rough ER of vertebrate cells, the precursor is transferred from the dolichol carrier to a susceptible asparagine residue on a nascent protein as soon as the asparagine crosses to the luminal side of the ER (step 1 ). In three separate reactions, first one glucose residue (step 2 ), then two glucose residues (steps 3a and 3b ), and finally one mannose residue (step 4 ) are removed. Re-addition of one glucose residue (step 3c ) plays a role in the correct folding of many proteins in the ER, as discussed later. The process of N-linked glycosylation of a soluble secretory protein is shown here, but the luminal portions of an integral membrane protein can be modified on asparagine residues
Oligosaccharide Side Chains May Promote Folding and Stability of Glycoproteins
by the same mechanism. See R. Kornfeld and S. Kornfeld, 1985, Annu. Rev. Biochem. 45:631; and M. Sousa and A. J. Parodi, 1995, EMBO J. 14:4196. Description The illustration shows an E R vesicle in the cytosol with the E R lumen enclosed by E R membrane. The process starts with a N-linked oligosaccharide in E R lumen, embedded in the membrane by its Dol-phosphate end. Step 1: A translating ribosome attaches to the outer surface of E R membrane and polypeptide chain enters the lumen. The oligosaccharide chain with three glucose, nine mannoses, and two N-acetylglucosamines binds to the polypeptide chain. Step 2: The ribosome detaches and one glucose unit degrades. Step 3 a: The second glucose unit degrades. Step 3 b: The third glucose unit degrades. Step 3 c: reversible of step 3 b wherein one glucose adds. Step 4: One mannose degrades. This results in a polypeptide chain attached to eight mannoses and two N-acetylglucosamines; and leaves the E R to travel to cis-Golgi. Oligosaccharide Side Chains May Promote Folding and Stability of Glycoproteins The oligosaccharides attached to glycoproteins serve various functions. For example, some proteins require N-linked oligosaccharides in order to fold properly in the ER. This function has been demonstrated in studies with the antibiotic tunicamycin, which blocks the first step in the formation of the dolichol-linked oligosaccharide precursor and therefore inhibits synthesis of all N-linked oligosaccharides in cells (see Figure 1317, top left). For example, in the presence of tunicamycin, the influenza virus hemagglutinin precursor polypeptide is synthesized, but it
cannot fold properly and form a normal trimer; in this case, the misfolded protein remains in the rough ER. Moreover, mutation of a particular asparagine in the hemagglutinin sequence to a glutamine residue prevents addition of an N-linked oligosaccharide to that site and causes the protein to accumulate in the ER in an unfolded state. In addition to promoting proper folding, N-linked oligosaccharides confer stability on many secreted glycoproteins. Many secretory proteins fold properly and are transported to their final destination even if the addition of all N-linked oligosaccharides is blocked, for example, by treatment with tunicamycin. However, such nonglycosylated proteins have been shown to be less stable than their glycosylated forms. For instance, glycosylated fibronectin, a normal component of the extracellular matrix, is degraded much more slowly by tissue proteases than is nonglycosylated fibronectin. Oligosaccharides on certain cell-surface glycoproteins also play a role in cell-cell adhesion. For example, the plasma membrane of white blood cells (leukocytes) contains cell-adhesion molecules (CAMs) that are extensively glycosylated. The oligosaccharides in these molecules interact with a sugar-binding domain in certain other CAMs found on endothelial cells lining blood vessels. This interaction tethers the leukocytes to the endothelium and assists in their movement into tissues during an inflammatory response to infection (see Figure 20-40). Other cell-surface glycoproteins possess oligosaccharide side chains that can induce an immune response. A common example is the ABO blood group antigens, which are O-linked oligosaccharides attached to glycoproteins and glycolipids on the surfaces of erythrocytes and other cell types (see Figure
Disulfide Bonds Are Formed and Rearranged by Proteins in the ER Lumen
10-20). In both cases, oligosaccharides are added to the luminal face of these membrane proteins, in a manner similar to what is shown in Figure 13-18 for soluble proteins. The luminal face of these membrane proteins is topologically equivalent to the exterior face of the plasma membrane, where these proteins eventually end up. Disulfide Bonds Are Formed and Rearranged by Proteins in the ER Lumen In Chapter 3, we learned that both intramolecular and intermolecular disulfide bonds help stabilize the tertiary and quaternary structure of many proteins. These covalent bonds form by the oxidative linkage of sulfhydryl groups (–SH), also known as thiol groups, on two cysteine residues in the same or different polypeptide chains. This reaction can proceed only when a suitable oxidant is present. In eukaryotic cells, disulfide bonds are formed in the lumen of the rough ER and are typically found in soluble secretory proteins and in the exoplasmic domains of membrane proteins. The cytosol does not contain enzymes for disulfide bond formation and therefore cytosolic proteins and most organelle proteins (i.e., those destined for mitochondria, chloroplasts, peroxisomes, etc.) usually lack disulfide bonds. The efficient formation of disulfide bonds in the lumen of the ER depends on the enzyme protein disulfide isomerase (PDI), which is present in all eukaryotic cells. This enzyme is especially abundant in the ER of
secretory cells in organs such as the liver and pancreas, where large quantities of proteins that contain disulfide bonds are produced. As shown in Figure 13-19a, the disulfide bond in the active site of PDI can be readily transferred to a protein by two sequential thiol-disulfide transfer reactions. The reduced PDI generated by this reaction is returned to an oxidized form by the action of an ER-resident protein, called Ero1, which carries a disulfide bond that can be transferred to PDI. Ero1 itself becomes oxidized by reaction with molecular oxygen that has diffused into the ER.
FIGURE 13-19 Action of protein disulfide isomerase (PDI). PDI forms and rearranges disulfide bonds via an active site with two closely spaced cysteine residues that are easily interconverted between the reduced dithiol form and the oxidized disulfide form. Numbered red arrows indicate the sequence of electron transfers. Yellow bars represent disulfide
bonds. (a) In the formation of disulfide bonds, the ionized form of a cysteine thiol in the substrate protein reacts with the disulfide (S–S) bond in oxidized PDI to form a disulfide-bonded PDI–substrate protein intermediate. A second ionized thiol in the substrate protein then reacts with this intermediate, forming a disulfide bond within the substrate protein and releasing reduced PDI. PDI, in turn, transfers electrons to a disulfide bond in the luminal protein Ero1, thereby regenerating the oxidized form of PDI. (b) Reduced PDI can catalyze rearrangement of improperly formed disulfide bonds by similar thiol-disulfide transfer reactions. In this case, reduced PDI both initiates and is regenerated in the reaction pathway. These reactions are repeated until the most stable conformation of the protein is achieved. See M. M. Lyles and H. F. Gilbert, 1991, Biochemistry 30:619. Description The part (a) shows formation of disulfide bonds. An oxidized form of P D I contains a disulfide bond. This oxidized P D I interacts with a reduced substrate protein containing two cysteine residues. Next, the P D I disulfide bond is reduced, and a disulfide bond between P D I and the protein substrate is formed. Arrows indicate the direction of electron flow that occurs from the disulfide bond to the sulfur of P D I, and electrons flow from the unbounded sulfide of the substrate protein, forming a disulfide bond between the cysteine residues in the substrate, and resulting in reduced P D I. Next, the reduced P D I can be oxidized by Ero 1. The part (b) shows rearrangement of disulfide bonds. The illustration shows a protein substrate with two incorrect disulfide bonds. Electrons are transferred from reduced P D I to one of the sulfur atoms, breaking one of the disulfide bonds and forming a disulfide bond between P D I and the substrate protein. Subsequently, electrons are transferred from the reduced sulfide to the second disulfide bond in the substrate protein, forming a bond between these two sulfurs. Consequently, electrons flow from the second disulfide bond to the sulfur atom of protein substrate, breaking the disulfide bond between reduced P D I and the substrate. Thus, the two disulfide bonds are rearranged and reduced PDI is regenerated. In proteins that contain more than one disulfide bond, the proper pairing of cysteine residues is essential for normal structure and activity. Disulfide
Chaperones and Other ER Proteins Facilitate Folding and Assembly of Proteins
bonds are commonly formed between cysteines that occur sequentially in the amino acid sequence while a polypeptide is still growing on the ribosome. Such sequential formation, however, sometimes yields disulfide bonds between the wrong cysteines. For example, proinsulin, a precursor to the peptide hormone insulin, has three disulfide bonds that link cysteines 1 and 4, 2 and 6, and 3 and 5. In this case, a disulfide bond that initially formed sequentially (e.g., between cysteines 1 and 2) would have to be rearranged for the protein to achieve its proper folded conformation. In cells, the rearrangement of disulfide bonds is also accelerated by PDI, which acts on a broad range of protein substrates, allowing them to reach their most thermodynamically stable conformations (Figure 13-19b). Disulfide bonds generally form in a specific order, first stabilizing small domains of a polypeptide, then stabilizing the interactions of more distant segments; this phenomenon is illustrated by the folding of the influenza hemagglutinin (HA) protein, discussed in the next section. Chaperones and Other ER Proteins Facilitate Folding and Assembly of Proteins Although many denatured proteins can spontaneously refold into their native state in vitro, such refolding usually requires hours to reach completion. Yet proteins produced in the ER generally fold into their proper conformation within minutes after their synthesis. The rapid folding of these newly synthesized proteins in cells depends on the sequential action of several proteins present within the ER lumen. We have
already seen how the molecular chaperone BiP can drive post-translational translocation in yeast by binding fully synthesized polypeptides as they enter the ER (see Figure 13-9). BiP can also bind transiently to nascent polypeptide chains as they enter the ER during cotranslational translocation. Bound BiP is thought to prevent segments of a nascent chain from misfolding or forming aggregates, thereby promoting folding of the entire polypeptide into the proper conformation. PDI also contributes to proper folding because the correct three-dimensional conformation is stabilized by disulfide bonds in many proteins. As illustrated in Figure 13-20, two other ER proteins, the homologous lectins (carbohydrate-binding proteins) calnexin and calreticulin, bind selectively to certain N-linked oligosaccharides on growing polypeptide chains. The ligand for these two proteins, which resembles the N-linked oligosaccharide precursor but has only a single glucose residue , is generated by a specific glucosyltransferase in the ER lumen (see Figure 13-18, step 3a ). This enzyme acts only on polypeptide chains that are unfolded or misfolded, and in this respect, the glucosyltransferase acts as one of the primary surveillance mechanisms to ensure quality control of protein folding in the ER. Unfolded proteins often expose hydrophobic segments that in a properly folded state are buried in the hydrophobic core of the protein. The glucosyltransferase is thought to specifically recognize unfolded proteins by binding to these exposed hydrophobic segments. Binding of calnexin and calreticulin to unfolded nascent chains marked with glucosylated N-linked oligosaccharides prevents aggregation of adjacent segments of a protein as
it is being made on the ER. Thus calnexin and calreticulin, like BiP, help prevent premature, incorrect folding of segments of a newly made protein.
FIGURE 13-20 Hemagglutinin folding and assembly. (a) Mechanism of trimer assembly. Transient binding of the chaperone BiP (step 1a ) to the nascent polypeptide chain and of two lectins, calnexin and calreticulin, to certain oligosaccharide chains (step 1b ) promotes proper folding of adjacent segments of . A total of seven N-linked oligosaccharide chains are added to the luminal portion of the nascent chain during cotranslational translocation, and PDI catalyzes the formation of six disulfide bonds per monomer. Completed monomers are anchored in the membrane by a single membrane-spanning α helix with the N-terminus in the lumen (step 2 ). Interaction of three chains with one another, initially via their transmembrane α helices, apparently
triggers formation of a long stem containing one α helix from the luminal part of each polypeptide. Finally, interactions occur among the three globular heads, generating a stable trimer (step 3 ). (b) Electron micrograph (false color) of a complete influenza virion showing trimers of HA protein protruding as spikes from the surface of the viral membrane. See U. Tatu et al., 1995, EMBO J. 14:1340; and D. Hebert et al., 1997, J. Cell Biol. 139:613. Description The series of illustrations (a) highlights a 3-step process of hemagglutinin (H A) trimer assembly. The illustrations show a translating ribosome attached toward cytosolic face of a translocon embedded in the E R membrane. An oligosaccharyl transferase is embedded on the left side of translocon with a Dolichol oligosaccharide next to it. Step 1 a: Polypeptide chain emerge from the ribosome into the E R lumen and the oligosaccharyl transferase transfers oligosaccharide from Dolichol to the growing polypeptide chain attached to two BiP molecules. Step 1 b: A calnexin embedded on the right side of the translocon transfers calreticulin around the oligosaccharide and a P D I forms disulfide bonds in the polypeptide chain. Step 2: Ribosome detaches leaving behind membrane-spanning ¬helix of the completed H A 0 monomer having seven N-linked oligosaccharides and six disulfide bonds. Step 3: A luminal alpha helix is present between three H A 0 monomers resulting in a H A 0 trimer. The micrograph (b) shows an oval-shaped influenza virion surrounded by spikes on the outer surface of the membrane and consisting of large genome inside. Other important protein-folding catalysts in the ER lumen are peptidylprolyl isomerases, a family of enzymes that accelerate the rotation about peptidyl-prolyl bonds at proline residues in unfolded segments of a polypeptide:
Such isomerizations are sometimes the rate-limiting step in the folding of protein domains. Many peptidyl-prolyl isomerases can catalyze the rotation of exposed peptidyl-prolyl bonds indiscriminately in numerous proteins, but some have very specific protein substrates. Many important soluble secretory and membrane proteins synthesized on the ER are built of two or more polypeptide subunits. In all cases, the assembly of the subunits constituting these multisubunit (multimeric) proteins occurs in the ER. The immunoglobulins, which contain two heavy (H) and two light (L) chains, all linked by intrachain disulfide bonds, are assembled in this way. Hemagglutinin (HA) is another multimeric protein that provides a good illustration of folding and subunit assembly (see
Figure 13-20). This trimeric protein forms the spikes that protrude from the surface of an influenza virus particle. The HA trimer is formed within the ER of an infected host cell from three copies of a precursor protein termed , which has a single membrane-spanning α helix. In the Golgi complex, each of the three proteins is cleaved to form two
Improperly Folded Proteins in the ER Induce Expression of Protein-Folding Catalysts
polypeptides, and ; thus each HA molecule that eventually resides on the viral surface contains three copies of and three of (see Figure 3-10). The trimer is stabilized by interactions between the large exoplasmic domains of the constituent polypeptides, which extend into the ER lumen; after HA is transported to the cell surface, these domains extend into the extracellular space. Interactions between the smaller cytosolic and membrane-spanning portions of the HA subunits also help stabilize the trimeric protein. Studies have shown that it takes just 10 minutes for the polypeptides to fold and assemble into their proper trimeric conformation. Improperly Folded Proteins in the ER Induce Expression of Protein-Folding Catalysts Wild-type proteins that are synthesized on the rough ER cannot exit this compartment until they achieve their completely folded conformation. Likewise, almost any mutation that prevents proper folding of a protein in the ER also blocks the movement of that protein from the ER lumen or membrane to the Golgi complex. The mechanisms that retain unfolded or incompletely folded proteins within the ER probably increase the overall efficiency of folding by keeping intermediate forms in proximity to folding catalysts, which are most abundant in the ER. Improperly folded proteins retained within the ER are generally seen bound to the ER chaperones BiP and calnexin. Thus these luminal folding catalysts perform two related functions: assisting in the folding of normal proteins by
preventing their aggregation and binding to misfolded proteins to retain them in the ER. Both mammalian cells and yeasts respond to the presence of unfolded proteins in the rough ER by increasing transcription of several genes that encode ER chaperones and other folding catalysts. A key participant in this unfolded-protein response is Ire1, an ER membrane protein that exists both as a monomer and as a dimer. The dimeric form, but not the monomeric form, promotes formation of Hac1, a transcription factor in yeast that activates expression of the genes induced in the unfoldedprotein response. As depicted in Figure 13-21, binding of BiP to the luminal domain of monomeric Ire1 prevents formation of the Ire1 dimer. Thus the quantity of free BiP in the ER lumen determines the relative proportions of monomeric and dimeric Ire1. Accumulation of unfolded proteins within the ER lumen sequesters BiP molecules, making them unavailable for binding to Ire1. As a result, the level of dimeric Ire1 increases, leading to an increase in the level of Hac1 and production of proteins that assist in protein folding.
FIGURE 13-21 The unfolded-protein response. Ire1, a transmembrane protein in the ER membrane, has a binding site for BiP on its luminal domain; the cytosolic domain contains a specific RNA endonuclease. Step 1 : Accumulating unfolded proteins in the ER lumen bind BiP molecules, releasing them from monomeric Ire1. Dimerization of Ire1 then activates its endonuclease activity. Steps 2 – 3 : The unspliced mRNA precursor encoding the transcription factor Hac1 is cleaved by dimeric Ire1, and the two exons are joined to form functional Hac1 mRNA. Current evidence indicates that this processing occurs in the cytosol, although pre-mRNA processing generally occurs in the nucleus. Step 4 : Hac1 is
translated into Hac1 protein, which then moves back into the nucleus and activates transcription of genes encoding several protein-folding catalysts. See U. Ruegsegger et al., 2001, Cell 107:103; A. Bertolotti et al., 2000, Nat. Cell Biol. 2:326; and C. Sidrauski and P. Walter, 1997, Cell 90:1031. Description The illustration shows two Ire 1 monomers embedded in the E R membrane with a BiP molecules attached to at their base in the E R lumen. Two unfolded proteins are present in the lumen. Step 1: BiP detaches from Ire 1 monomers and binds to the unfolded proteins. Step 2: An unspliced Hac 1 m R N A binds to the Ire 1 dimer at the endonuclease site toward cytosol. Step 3: Endonuclease-cut Hac 1 m R N A. Step 4: Translation of spliced Hac 1 m R N A results in Hac 1 transcription factor. Mammalian cells contain an additional regulatory pathway that operates in response to unfolded proteins in the ER. In this pathway, accumulation of unfolded proteins in the ER triggers proteolysis of ATF6, a transmembrane protein in the ER membrane, at a site within the membrane-spanning segment. The cytosolic domain of ATF6 released by proteolysis then moves to the nucleus, where it stimulates transcription of the genes encoding ER chaperones. Activation of a transcription factor by such regulated intramembrane proteolysis also occurs in the Notch signaling pathway and during activation of the cholesterol-responsive transcription factor SREBP (see Figures 16-25 and 21-6). A hereditary form of emphysema illustrates the detrimental effects that can result from misfolding of proteins in the ER. This disease is caused by a point mutation in -antitrypsin, which is normally secreted
Unassembled or Misfolded Proteins in the ER Are Often Transported to the Cytosol for Degradation
by hepatocytes and macrophages. The wild-type protein binds to and inhibits trypsin as well as the blood protease elastase. In the absence of normal -antitrypsin, elastase degrades the fine tissue in the lung that participates in the absorption of oxygen, eventually producing the symptoms of emphysema. Although the mutant -antitrypsin is synthesized on the rough ER, it does not fold properly, forming an almost crystalline aggregate that is not exported from the ER. In hepatocytes, the secretion of other proteins also becomes impaired as the rough ER is filled with aggregated -antitrypsin. Unassembled or Misfolded Proteins in the ER Are Often Transported to the Cytosol for Degradation Misfolded soluble secretory and membrane proteins, as well as the unassembled subunits of multimeric proteins, are often degraded within an hour or two after their synthesis in the rough ER. Initially, it was thought that proteolytic enzymes within the ER lumen catalyzed degradation of misfolded or unassembled polypeptides, but such proteases were never found. More recent studies have shown that misfolded secretory proteins are recognized by specific ER membrane proteins and are targeted for transport from the ER lumen into the cytosol by a process known as dislocation. The dislocation and degradation of misfolded proteins depends on a set of proteins located in the ER membrane and in the cytosol that perform three
basic functions. The first function is recognition of misfolded proteins. One mechanism for recognition involves the trimming of N-linked carbohydrate chains by mannosidases located in the ER (Figure 13-22). Trimmed glycans with the structure are recognized by a protein known as OS-9, which targets the trimmed glycoprotein for dislocation. It is not known precisely how the ER α-mannosidases distinguish the partially folded states of defective proteins that can never completely fold properly and are thus legitimate substrates for the dislocation process, from normal proteins that have not yet acquired their fully folded conformation. One possibility is that the ER α-mannosidases act slowly, so that only those glycoproteins that remain misfolded in the ER lumen for a sufficiently long time are trimmed and therefore targeted for degradation. Luminal proteins that lack oligosaccharides altogether can also be targeted for degradation, indicating that other processes for the recognition of unfolded proteins must also exist that do not involve trimming of N-linked oligosaccharides.
FIGURE 13-22 Modifications of N-linked oligosaccharides are used to monitor folding and for quality control. After a N-linked oligosaccharide is added to a newly synthesized and incompletely folded protein in the ER, three glucose residues are removed by Glucosidases I and II to form (see
Figure 13-18, steps 2 and 3 ). This modified N-linked oligosaccharide binds the lectins calnexin (CNX) and calreticulin (CRT) for retention in the ER and engagement of folding chaperones. When the N-linked oligosaccharide is released from CRT, removal of glucose by Glucosidase II yields , which is further processed to before entering a COPII vesicle bound for the cis-Golgi (see Figure 13-18, steps 3 and 4 ). If the protein has not folded completely, a glucosyltransferase known as UGGT can add a glucose residue to reform , which will bind again to CNX and CRT for another attempt at folding. Proteins that cannot fold properly, and are therefore retained in the ER for longer times, eventually undergo mannose trimming by ER α-mannosidases to form , which is recognized by OS-9. Recognition by OS-9 leads to dislocation of the misfolded protein out of the ER, ubiquitinylation, and degradation by proteasomes. Description The illustration shows a translating ribosome attached on the cytosolic face of E R membrane and producing a polypeptide chain into the E R lumen. The polypeptide chain has an N-linked oligosaccharide attached to it that is made of 2 N-Acetylglucosamine, 9 mannose, and 3 glucose. Next, glucosidase 1 and 2 act on the structure, ribosome leaves, and the folded polypeptide chain is now attached to 2 N-Acetylglucosamine, 9 mannose, and 1 glucose with a C N X slash C R T molecule at the base. This step is labeled folding or retention. Next, glucosidase 2 acts and removes the remaining glucose unit. This process can be reversed by U G G T. This structure enters into COP 2 vesicle (if properly folded). Next, E R alpha-mannosidases act and remove 3 or 4 mannose units. This results in folded polypeptide chain now attached to 2 N-Acetylglucosamine and 5 or 6 mannose with a O S 9 molecule at the base. This causes degradation. Misfolded ER proteins that are marked for degradation are transported across the ER membrane by a process known as dislocation, since it occurs in the reverse direction from translocation, which occurs during synthesis. A complex of at least four integral membrane proteins, known
as the ERAD (ER-associated degradation) complex, enables dislocation of misfolded proteins across the ER membrane, but there is no evidence that the ERAD complex forms a protein channel for dislocation, suggesting other possible mechanisms. One possibility is that the Sec61 translocation channel can be repurposed for the dislocation of misfolded proteins from the ER lumen to the cytosol. Dislocation involves powerful pulling and unfolding protein machines in the cytosol and it is possible that misfolded proteins are dragged directly through the lipid bilayer without a channel dedicated for this purpose. As segments of a dislocated polypeptide emerge on the cytosolic face of the membrane, they encounter p97, a member of a protein family known as the AAA ATPase family that couples the energy of ATP hydrolysis to disassembly of protein complexes. In dislocation of polypeptides from the ER, hydrolysis of ATP by p97 may provide the driving force to pull misfolded proteins from the ER membrane into the cytosol. As the misfolded proteins enter the cytosol, specific ubiquitin ligase enzymes that are components of the ERAD complex add ubiquitin residues to these dislocated peptides. Like the action of p97, ubiquitinylation is coupled to ATP hydrolysis and this reaction may also contribute to the energetically favorable trapping of the proteins in the cytosol. The resulting polyubiquitinylated polypeptides, now fully in the cytosol, are targeted for degradation in proteasomes. The role of polyubiquitinylation in targeting proteins to proteasomes is discussed more fully in Chapter 3 (see Figure 332 and Figure 3-39). KEY CONCEPTS OF SECTION 13.3
Protein Modifications, Folding, and Quality Control in the ER All N-linked oligosaccharides, which are bound to asparagine residues, contain a core of two N-acetylglucosamine and at least three mannose residues and usually have several branches. O-linked oligosaccharides, which are bound to serine or threonine residues, are generally short, often containing only one to four sugar residues. Formation of N-linked oligosaccharides begins with assembly of a conserved 14residue high-mannose oligosaccharide precursor on dolichol, a lipid in the membrane of the rough ER (see Figure 13-17). After this preformed oligosaccharide is transferred to specific asparagine residues of nascent polypeptide chains in the ER lumen, three glucose residues and one mannose residue are removed (see Figure 1318). Oligosaccharide side chains may assist in the proper folding of glycoproteins, help protect the mature proteins from proteolysis, participate in cell-cell adhesion, and function as antigens. Disulfide bonds are added to many soluble secretory proteins and to the exoplasmic domain of membrane proteins in the ER. Protein disulfide isomerase (PDI), present in the ER lumen, catalyzes both the formation and the rearrangement of disulfide bonds (see Figure 13-19). The chaperone BiP, the lectins calnexin and calreticulin, and peptidyl-prolyl isomerases work together to ensure proper folding of newly made secretory and membrane proteins in the ER. The subunits of multimeric proteins also assemble in the ER (see Figure 13-20). Only properly folded proteins and assembled subunits are transported from the rough ER to the Golgi complex in vesicles. The accumulation of abnormally folded proteins and unassembled subunits in the ER can induce increased expression of ER protein-folding catalysts via the unfoldedprotein response (see Figure 13-21). Unassembled or misfolded proteins in the ER are often transported back to the cytosol, where they are degraded in the ubiquitin-proteasome pathway (see Figure 1322).
13.4 Targeting of Proteins to Mitochondria and Chloroplasts
13.4 Targeting of Proteins to Mitochondria and Chloroplasts In the remainder of this chapter, we examine how proteins synthesized on cytosolic ribosomes are sorted to discrete organelles: mitochondria, chloroplasts, peroxisomes, and the nucleus (see Figure 13-1). Mitochondria and chloroplasts are closely related organelles that contain an internal lumen — called the matrix in mitochondria and the stroma in chloroplasts — that is surrounded by a double membrane. Within the stroma, chloroplasts contain a third membrane known as the thylakoid, which is where the light-harvesting reactions of photosynthesis take place. In contrast to mitochondria and chloroplasts, peroxisomes are bounded by a single membrane and have a single luminal matrix compartment. The mechanism of protein transport into and out of the nucleus, which differs in many respects from sorting to other organelles, is discussed in the last section of the chapter. In addition to being bounded by two membranes, mitochondria and chloroplasts share similar types of electron-transporting proteins and use an F-class ATPase to synthesize ATP (see Figure 12-26). Remarkably, these characteristics are shared by gram-negative bacteria. Also like bacterial cells, mitochondria and chloroplasts contain their own DNA, which encodes organelle rRNAs, tRNAs, and some proteins (see Chapter 8). Moreover, growth and division of mitochondria and chloroplasts are
not coupled to nuclear division. Rather, these organelles grow by the incorporation of cellular proteins and lipids, and new organelles form by division of preexisting organelles. These numerous similarities to freeliving bacterial cells have led to the understanding that mitochondria and chloroplasts arose when bacteria were incorporated into ancestral eukaryotic cells, forming endosymbiotic organelles (see Figure 12-7). The sequence similarity of the many membrane translocation proteins shared by mitochondria, chloroplasts, and bacteria provides the most striking evidence for this ancient evolutionary relationship. In this section, we examine these membrane translocation proteins in detail. Proteins encoded by mitochondrial DNA or chloroplast DNA are synthesized on ribosomes within the organelles and directed to the correct subcompartment within the parent organelle immediately after synthesis. However, the majority of proteins located in mitochondria and chloroplasts are not encoded by organelle DNA — they are encoded by genes in the nucleus and are imported into the organelles after their synthesis in the cytosol. Apparently, as eukaryotic cells evolved over a billion years, much of the genetic information from the ancestral bacterial DNA in these endosymbiotic organelles moved, by an unknown mechanism, to the nucleus. Precursor proteins synthesized in the cytosol that are destined for the matrix of mitochondria or the equivalent stroma in chloroplasts usually contain specific N-terminal targeting sequences that specify binding to receptor proteins on the organelle surface. Generally these sequences are cleaved once the protein reaches the matrix or stroma. Clearly these targeting sequences are similar in their location and general function to the signal sequences that direct nascent proteins to
Amphipathic N-Terminal Targeting Sequences Direct Proteins to the Mitochondrial Matrix
the ER lumen. Although the three types of signals share some sequence features, their specific sequences differ considerably, as summarized in
Table 13-1. In both mitochondria and chloroplasts, protein import requires energy and occurs at points where the outer and inner organelle membranes are in direct contact. Because mitochondria and chloroplasts contain multiple membranes and membrane-limited spaces, the sorting of many proteins to their correct locations requires the sequential action of two targeting sequences and two membrane-bound translocation systems: one to direct the protein into the organelle and the other to direct it into the correct organelle subcompartment or membrane. As we will see, the mechanisms for sorting various proteins to mitochondria and chloroplasts are related to SRP-dependent recognition of a signal sequence as discussed previously. Amphipathic N-Terminal Targeting Sequences Direct Proteins to the Mitochondrial Matrix All proteins that travel from the cytosol to the same mitochondrial destination have targeting signals that share common motifs, although the signal sequences are not identical. Thus the receptors that recognize such signals are able to bind to a number of different but related sequences. The most extensively studied sequences for localizing proteins to mitochondria are the matrix-targeting sequences. These sequences, located at the N-terminus, are usually 20–50 amino acids in length. They are rich in
hydrophobic amino acids, positively charged basic amino acids (arginine and lysine), and hydroxylated ones (serine and threonine) but tend to lack negatively charged acidic residues (aspartate and glutamate). Mitochondrial matrix–targeting sequences are thought to assume an α-helical conformation in which positively charged amino acids predominate on one side of the helix and hydrophobic amino acids on the other. Sequences such as these that contain both hydrophobic and hydrophilic regions are said to be amphipathic. Mutations that disrupt the amphipathic character of these sequences usually disrupt targeting to the matrix, although many other amino acid substitutions do not. These findings indicate that the amphipathicity of matrix-targeting sequences is critical to their function. The cell-free assay outlined in Figure 13-23 has been widely used to define the biochemical steps in the import of mitochondrial precursor proteins. Respiring (energized) mitochondria extracted from cells can incorporate mitochondrial precursor proteins that have been synthesized in the absence of mitochondria if they are carrying appropriate targeting sequences. Successful incorporation of the precursor into the organelle can be assayed by resistance to digestion by an added protease such as trypsin. In other assays, successful import of a precursor protein can be shown by the proper cleavage of the N-terminal targeting sequences by specific mitochondrial proteases. The uptake of completely presynthesized mitochondrial precursor proteins by the organelle in this system contrasts with the cell-free cotranslational translocation of secretory proteins into
the ER, which generally occurs only when microsomal (ER-derived) membranes are present during synthesis (see Figure 13-4).
FIGURE 13-23 Import of mitochondrial precursor proteins is assayed in a cell-free system. Mitochondrial precursor proteins with attached uptake-targeting signals can be synthesized on ribosomes in a cell-free reaction. When respiring mitochondria are added to these synthesized mitochondrial precursor proteins (top), the proteins are taken up by mitochondria. Inside mitochondria, the proteins are protected from the action of proteases such as trypsin. When no mitochondria are present (bottom), the mitochondrial proteins are degraded by added protease. Only energized (respiring) mitochondria, which have a proton electrochemical gradient (proton-motive force) across the inner membrane, take up these proteins. Furthermore, the imported proteins must contain an appropriate uptake-targeting sequence. Uptake also requires ATP and a cytosolic extract containing chaperone proteins that maintain the precursor proteins in an unfolded conformation. This assay has been used to study targeting sequences and other features of the translocation process.
Mitochondrial Protein Import Requires Outer-Membrane Receptors and Translocons in Both Membranes
Description The first illustration shows a test tube having Yeast mitochondrial proteins made by cytoplasmic ribosomes in a cell-free system. The mitochondrial proteins are tagged by Uptake-targeting sequence. Next, energized yeast mitochondria is added resulting the mitochondria to take up proteins; uptake-targeting sequence are removed and degraded. Next, trypsin is added and shows the mitochondria unaffected by trypsin activity. Proteins sequestered within mitochondria are resistant to trypsin. However, when trypsin is added to the initial test tube, uptake-targeting sequence and mitochondrial proteins are degraded. Mitochondrial Protein Import Requires Outer-Membrane Receptors and Translocons in Both Membranes
Figure 13-24 presents an overview of protein import from the cytosol into the mitochondrial matrix, the route into the mitochondrion followed by most imported proteins. Here we discuss in detail each step of protein transport into the matrix, then consider how some proteins are subsequently targeted to other compartments of the mitochondrion.
FIGURE 13-24 Protein import into the mitochondrial matrix. Precursor proteins synthesized on cytosolic ribosomes are maintained in an unfolded or partially folded state by bound chaperones, such as cytosolic Hsp70 (step 1 ). After a precursor protein binds to
an import receptor near a site of contact with the inner membrane (step 2 ), it is transferred into the general import pore (step 3 ). The translocating protein then moves through this channel and an adjacent channel in the inner membrane (steps 4 – 5 ). Note that translocation occurs at rare “contact sites” at which the inner and outer membranes appear to touch. Binding of the translocating protein by matrix Hsp70 and subsequent ATP hydrolysis by Hsp70 helps drive import into the matrix. Once the targeting sequence is removed by a matrix protease and Hsp70 is released from the newly imported protein (step 6 ), the protein folds into its mature, active conformation within the matrix (step 7 ). Folding of some proteins depends on matrix chaperonins. See G. Schatz, 1996, J. Biol. Chem. 271:31763; and N. Pfanner et al., 1997, Annu. Rev. Cell Dev. Biol. 13:25. Description The steps are as follows: Step 1. A precursor protein in the cytosol is bound by cytosolic H s p 70 and has a matrix targeting sequences at the N-terminal. The A T P of cytosolic H s p 70 hydrolysis into A D P and P i. Step 2. The cytosolic H s p 70 detaches and the matrix targeting sequence binds to an import receptor, TOM 20 slash 22, embedded in the outer membrane of the mitochondrion. Step 3. The matrix targeting sequence is transferred to a general import pore TOM 40 lying adjacent to import receptor. Step 4. The protein is translocated through TOM 40 into intermembrane space. Step 5. The protein is transferred through a second pore (Tim 23 slash 17) embedded in the inner membrane at a site where the outer and inner membranes are in close contact. A protein complex, Tim 44, attached to Tim 23 slash 17 attaches a matrix H s p 70 to the entering protein chain as A T P is hydrolyzed to A D P and P i. A matrix processing protease cleaves the targeting sequence. Steps 6 and 7. The protein folds resulting into an active protein. After synthesis in the cytosol, the soluble precursors of mitochondrial proteins (including hydrophobic integral membrane proteins) can interact directly with the mitochondrial membrane. Import of an unfolded mitochondrial precursor protein is initiated by the binding of a mitochondrial targeting sequence to an import receptor in the outer
mitochondrial membrane. These import receptors were first identified by experiments in which antibodies to specific proteins of the outer mitochondrial membrane were shown to inhibit protein import into isolated mitochondria. Subsequent genetic experiments in which the genes for specific mitochondrial outer-membrane proteins were mutated showed that specific receptor proteins were responsible for the import of different classes of mitochondrial proteins. For example, N-terminal matrixtargeting sequences are recognized by a complex of Tom20 and Tom22 (proteins in the outer mitochondrial membrane involved in targeting and import, designated Tom proteins for translocon of the outer membrane). Many proteins can be imported into the mitochondrion only in an unfolded state. Chaperone proteins such as cytosolic Hsp70 and Hsp90 use energy derived from ATP hydrolysis to keep nascent and newly made proteins in a disaggregated state so that they are available to be taken up by mitochondria. For some mitochondrial precursor proteins, the mitochondrial outer-membrane protein Tom70 serves as an import receptor by binding to both Hsp90 and portions of the unfolded precursor protein. The import receptors subsequently transfer the precursor protein to an import channel in the outer membrane. This channel, composed mainly of the Tom40 protein, is known as the general import pore because all known mitochondrial precursor proteins gain access to the interior compartments of the mitochondrion through it. When Tom40 is purified and incorporated into liposomes, it forms a transmembrane channel with a pore wide enough to accommodate an unfolded polypeptide chain. The
Studies with Chimeric Proteins Demonstrate Important Features of Mitochondrial Protein Import
general import pore forms a largely passive channel through the outer mitochondrial membrane; the driving force for unidirectional transport comes from within the mitochondrion, as we will see shortly. In the case of precursors destined for the mitochondrial matrix, transfer through the outer membrane occurs simultaneously with transfer through an innermembrane channel composed of the Tim23 and Tim17 proteins. (Tim stands for translocon of the inner membrane.) Translocation into the matrix thus occurs at contact sites where the outer and inner membranes are in close proximity. Soon after the N-terminal matrix-targeting sequence of a protein enters the mitochondrial matrix, it is removed by a protease that resides within the matrix. The emerging protein is also bound by matrix Hsp70, a chaperone that is localized to the translocation channels in the inner mitochondrial membrane by interaction with transmembrane protein Tim44. Together, Tim44 and Hsp70 are thought to power translocation of proteins into the matrix. Some imported proteins can fold into their final, active conformation without further assistance. Final folding of many matrix proteins, however, requires chaperonins. As discussed in Chapter 3, chaperonin proteins actively facilitate protein folding by a process that depends on ATP. Yeast mutants defective in Hsc60, a chaperonin in the mitochondrial matrix, can import matrix proteins and cleave their targeting sequences normally, but the imported polypeptides fail to fold and assemble into their native tertiary and quaternary structures.
Studies with Chimeric Proteins Demonstrate Important Features of Mitochondrial Protein Import Dramatic evidence for the ability of mitochondrial matrix–targeting sequences to direct the import of any protein to the matrix was shown by adding the matrix-targeting sequence of alcohol dehydrogenase to the enzyme dihydrofolate reductase (DHFR), which normally resides in the cytosol. Cell-free translocation assays show that in the presence of chaperones, which prevent the DHFR segment from folding in the cytosol, the chimeric protein is transported into the matrix (Figure 13-25a). However, the DHFR inhibitor methotrexate, which binds tightly to the active site of DHFR and greatly stabilizes its folded conformation, renders the chimeric protein resistant to unfolding by cytosolic chaperones and prevents the chimeric protein from completely entering the translocon of the matrix. This finding demonstrates that a precursor must be unfolded in order to traverse the import pores in both mitochondrial membranes.
EXPERIMENTAL FIGURE 13-25 Experiments with chimeric proteins elucidate mitochondrial protein import processes. These experiments show that a matrix-targeting sequence alone directs proteins to the mitochondrial matrix and that only unfolded proteins are translocated across both mitochondrial membranes. The chimeric protein in these experiments contained a matrix-targeting signal at its N-terminus (red), followed by a spacer sequence of no particular function (black), and then by dihydrofolate reductase (DHFR), an enzyme normally present only in the cytosol. (a) When the DHFR segment is unfolded, the chimeric protein moves across both membranes to the matrix of an energized mitochondrion, and the matrix-targeting signal is then removed. (b) When the C-terminus of the chimeric protein is locked in the folded state by binding of methotrexate, translocation is blocked. If the spacer sequence is long enough to extend across both transport channels, a stable translocation intermediate with the targeting sequence cleaved off is generated in the presence of methotrexate, as shown here. (c) The C-terminus of the translocation intermediate in (b) can be detected by incubating the mitochondria with antibodies that bind to the DHFR segment, followed by gold particles coated with bacterial protein A, which binds nonspecifically to antibody molecules (see Figure 4-33). An electron micrograph of a sectioned sample reveals gold particles (red arrowhead) bound to the translocation intermediate at a contact site between the inner and outer membranes. Other contact sites (black arrows) are also evident. See J. Rassow et al., 1990, FEBS Lett. 275:190. [Part (c) M. Schwaiger, V. Herzog, and W. Neupert, 1987, J. Cell Biol. 105:235–246; https://doi.org/10.1083/jcb.105.1.235.]
Three Energy Inputs Are Needed to Import Proteins into Mitochondria
Description The illustration (a) shows an unfolded D H F R protein entering mitochondrial matrix from cytosol through pores in outer and inner membrane at the contact site. The C-terminal lies in cytosol, the N-terminal having target sequence lies in matrix, while the spacer sequence is present in the pores. Next, targeting sequence is cleaved and the D H F R having spacer sequence is folded. The illustration (b) shows a folded D H F R protein with a methotrexate inhibitor bound to it entering mitochondrial matrix from cytosol through pores in outer and inner membrane at the contact site. The C-terminal lies in cytosol and the N-terminal having spacer sequence lies in matrix. A cleaved targeting sequence is present in the mitochondrial matrix. The black-and-white micrograph (c) shows an outer and inner membrane of mitochondria with mitochondrial matrix inside and cytosol outside. A pink arrow points to a contact site where protein translocation occurs and two black arrows point to two contact sites. Additional studies revealed that if a sufficiently long spacer sequence separates the N-terminal matrix-targeting sequence and the DHFR portion of the chimeric protein, then, in the presence of methotrexate, a translocation intermediate that spans both membranes can be trapped (Figure 13-25b). Microscopy studies of these stable trapped intermediates show that they accumulate at sites where the inner and outer mitochondrial membranes are close together (Figure 13-25c). Since roughly a thousand stuck chimeric proteins can be observed at these contact sites in a typical yeast mitochondrion, it is thought that mitochondria have approximately a thousand general import pores for the uptake of mitochondrial proteins.
Three Energy Inputs Are Needed to Import Proteins into Mitochondria As noted previously and as indicated in Figure 13-24, ATP hydrolysis by Hsp70 chaperone proteins in both the cytosol and the mitochondrial matrix is required for the import of mitochondrial proteins. Cytosolic Hsp70 expends energy to maintain bound precursor proteins in an unfolded state so that they can be translocated into the matrix. The importance of ATP to this function was demonstrated in studies in which a mitochondrial precursor protein was purified and then denatured (unfolded) by urea. When tested in the cell-free mitochondrial translocation system, the denatured protein was incorporated into the matrix in the absence of ATP. In contrast, the same precursor protein in its native, undenatured state was not imported in the absence of ATP, even in the presence of cytosolic chaperones. The sequential binding to and ATP-driven release of multiple matrix Hsp70 molecules from a translocating protein may simply trap the unfolded protein in the matrix. In this case, the functions of matrix Hsp70 and Tim44 would be analogous to those of the chaperone BiP and Sec63 complex, respectively, in post-translational translocation into the ER lumen (see Figure 13-9). The third energy input required for mitochondrial protein import is an electrochemical gradient, or proton-motive force, across the inner membrane. Recall from Chapter 12 that protons are pumped from the
Multiple Signals and Pathways Target Proteins to Submitochondrial Compartments
matrix into the intermembrane space during electron transport, creating an electric potential across the inner membrane. In general, only mitochondria that are actively undergoing respiration, and therefore have generated a proton-motive force across the inner membrane, are able to translocate precursor proteins from the cytosol into the mitochondrial matrix. Treatment of mitochondria with inhibitors or uncouplers of oxidative phosphorylation, such as cyanide or dinitrophenol, dissipates this proton-motive force. Although precursor proteins can still bind tightly to receptors on such poisoned mitochondria, the proteins cannot be imported, either in intact cells or in cell-free systems, even in the presence of ATP and chaperone proteins. Once a protein is partially inserted into the inner membrane, it is subjected to a membrane potential of 200 mV (matrix negative). This seemingly small potential is established across the very narrow hydrophobic core of the lipid bilayer, which gives an enormous electrochemical gradient, equivalent to about 400,000 The positive charges in the amphipathic matrix-targeting sequence may simply be pulled into the matrix by the inside-negative membrane potential. Multiple Signals and Pathways Target Proteins to Submitochondrial Compartments Unlike targeting to the matrix, targeting of proteins to the intermembrane space, inner membrane, or outer membrane of mitochondria generally requires more than one targeting sequence and occurs via one of several
pathways. Figure 13-26 summarizes the organization of targeting sequences in proteins sorted to different mitochondrial locations.
FIGURE 13-26 Targeting sequences in imported mitochondrial proteins. Most mitochondrial proteins have an N-terminal matrix-targeting sequence (pink) that is similar, though not identical, in different proteins. Proteins destined for the inner membrane, the intermembrane space, or the outer membrane have one or more additional targeting sequences that function to direct the proteins to these locations by several different pathways. The lettered pathways correspond to those illustrated in Figures 13-27 and 13-28. See W. Neupert, 1997, Annu. Rev. Biochem. 66:863. Description Each part highlights proteins, location of imported protein, and locations of targeting sequences in preprotein. The first part for location of imported protein in matrix has protein, alcohol dehydrogenase 3, and the location of targeting sequences shows cleavage by matrix protease between matrix-targeting sequence and mature protein sequence. The second part shows three paths for location of imported protein in inner membrane. Path A: The protein is Cytochrome oxidase subunit Cox V a and the location of targeting sequences shows a cleavage by matrix protease between matrix-targeting sequence and hydrophobic stop-transfer anchor sequence which is attached to a mature protein sequence. Path C: The protein is A T P synthase subunit 9 and the location of targeting sequences shows a cleavage by matrix protease between matrix-targeting sequence and mature protein sequence. The mature protein sequence has two internal sequences recognized by Oxa 1. Path C: The protein is A D P slash A T P antiporter and the location of targeting sequences shows a mature protein having six internal sequences recognized by Tom 70 receptor and Tim 22 complex. The third part shows two paths for location of imported protein in intermembrane space. Path A: The protein is Cytochrome b 2 and the location of targeting sequences shows a first cleavage by matrix protease between matrix-targeting sequence and intermembrane-space-targeting sequence followed by a second cleavage by protease in intermembrane space between intermembrane-space-targeting sequence and mature protein sequence. Path B: The protein is Tim 9, Tim 10 and the location of targeting sequences shows a targeting sequence for the general import pore between two mature protein sequences.
The fourth part for location of imported protein in outer membrane has protein, Porin P 70, and the location of targeting sequences shows a stop-transfer and outer-membrane localization sequence between matrix-targeting sequence and mature protein sequence. Inner-Membrane Proteins Three separate pathways are known to target proteins to the inner mitochondrial membrane. One pathway makes use of the same machinery that is used for the targeting of matrix proteins (Figure 13-27, path A). A cytochrome oxidase subunit called CoxVa is one protein transported by this pathway. The precursor form of CoxVa, which contains an N-terminal matrix-targeting sequence recognized by the import receptor, is transferred through the Tom40 general import pore of the outer membrane and the inner-membrane translocation complex. In addition to the matrix-targeting sequence, which is cleaved during import, CoxVa contains a hydrophobic stop-transfer anchor sequence. As the protein passes through the channel, the stop-transfer anchor sequence blocks translocation of the C-terminus across the inner membrane. The membrane-anchored intermediate is then transferred laterally into the bilayer of the inner membrane, much like type I integral membrane proteins are incorporated into the ER membrane (see Figure 13-11).
FIGURE 13-27 Three pathways to the inner mitochondrial membrane from the cytosol. Proteins with different targeting sequences are directed to the inner membrane via different pathways. In all three pathways, proteins cross the outer membrane via the Tom40 general import pore. Proteins delivered by pathways A and B contain an N-terminal matrix-targeting sequence that is recognized by the import receptor in the outer membrane. Although both these pathways use the inner-membrane channel, they differ in that the entire precursor protein enters the matrix and is then redirected to the inner membrane in pathway B. Matrix Hsp70 plays a role similar to its role in the import of soluble matrix proteins (see Figure 13-23). Proteins delivered by pathway C contain internal sequences that are recognized by the import receptor; a different innermembrane translocation channel is used in this pathway. Two intermembrane proteins (Tim9 and Tim10) facilitate transfer between the outer and inner channels. See the text for discussion. See R. E. Dalbey and A. Kuhn, 2000, Annu. Rev. Cell Dev. Biol. 16:51; and N. Pfanner and A. Geissler, 2001, Nat. Rev. Mol. Cell Biol. 2:339.
Description The illustration shows from top to bottom: cytosol, outer mitochondrial membrane, intermembrane space, inner mitochondrial membrane, and mitochondrial matrix. Path A shows a precursor protein chain with a matrix-targeting sequence at N-terminal followed by a stop-transfer anchor sequence. The outer membrane has a Tom 40 channel next to a Tom 20, Tom 22 receptor; and the inner membrane has Tim 23 slash 17 channel attached to a Tim 44 receptor. Step 1: The precursor protein enters mitochondrial matrix via Tom 40, intermembrane space, and Tim 23 slash 17 where stop-transfer anchor sequence attaches to internal of Tim 23 slash 17 channel. H s p 70 in matrix cleaves the matrix-targeting sequence. Step 2 shows the protein embedded in the inner membrane with C-terminal in intermembrane space, stop-transfer anchor sequence in inner membrane, and N-terminal in matrix. Path B shows a precursor protein chain with a matrix-targeting sequence at N-terminal followed by two Oxa 1-targeting sequences. Step 1: The precursor protein enters mitochondrial matrix where the second Oxa 1-targeting sequence attaches to internal of Tim 23 slash 17 channel. H s p 70 in matrix cleaves after first Oxa 1-targeting sequence. Step 2 shows a protein chain in matrix with two Oxa 1-targeting sequences and a cleaved matrix-targeting sequence. Step 3: The protein gets embedded in the inner membrane next to an Oxa 1 embedded in the inner membrane. The protein embedded has both C-terminal and N-terminal in matrix, both Oxa 1-targeting sequence in inner membrane, and middle protein sequence in intermembrane space. Path C shows a protein chain with several internal targeting sequences. The outer membrane has a Tom 40 channel next to a Tom 22, Tom 70 receptor; and inner membrane has Tim 22 attached to a Tim 54 embedded in it. Step 1: The precursor protein enters intermembrane space via Tom 40, binds to Tim 9 slash 10 and enters Tim, Tim 54 of inner membrane. Step 2 shows the protein embedded in the inner membrane with both C-terminal and N-terminal in intermembrane space, internal targeting sequences in inner membrane, and middle protein sequence in matrix. A second pathway to the inner membrane is used by proteins such as ATP synthase subunit 9, whose precursors contain both a matrix-targeting sequence and internal hydrophobic domains recognized by an inner-
membrane protein termed Oxa1. This pathway is thought to involve translocation of at least a portion of the precursor into the matrix via the Tom40 and channels. After cleavage of the matrix-targeting sequence, the protein is inserted into the inner membrane by a process that requires interaction with Oxa1 and perhaps other inner-membrane proteins (see Figure 13-27, path B). Oxa1 is related to a protein involved in inserting some plasma-membrane proteins in bacteria. This relatedness suggests that Oxa1 may have descended from the translocation machinery in the endosymbiotic bacterium that eventually became the mitochondrion. However, the proteins that form the inner-membrane channels in mitochondria are not related to the proteins in bacterial translocons. Oxa1 also participates in the inner-membrane insertion of certain proteins, such as subunit II of cytochrome oxidase, that are encoded by mitochondrial DNA and synthesized in the matrix by mitochondrial ribosomes. The final pathway for insertion in the inner mitochondrial membrane is followed by multipass proteins that contain six membrane-spanning domains, such as the antiporter. These proteins, which lack the usual N-terminal matrix-targeting sequence, contain multiple internal mitochondrial targeting sequences. After the internal sequences are recognized by a second import receptor composed of outer-membrane proteins Tom70 and Tom22, the imported protein passes through the outer membrane via the general import pore (see Figure 13-27, path C). The protein then is transferred to a second translocation complex in the inner membrane composed of the Tim22, Tim18, and Tim54 proteins. Transfer to the complex depends on a multimeric complex of two
small proteins, Tim9 and Tim10, which reside in the intermembrane space. These small Tim proteins are thought to act as chaperones, guiding imported protein precursors from the general import pore to the complex in the inner membrane by binding to their hydrophobic regions, preventing them from forming insoluble aggregates in the aqueous environment of the intermembrane space. Ultimately the complex is responsible for incorporating the multiple hydrophobic segments of the imported protein into the inner membrane. Intermembrane-Space Proteins Two pathways deliver cytosolic proteins to the space between the inner and outer mitochondrial membranes. The major pathway is followed by proteins such as cytochrome , whose precursors carry two different N-terminal targeting sequences, both of which are ultimately cleaved. The most N-terminal of the two sequences is a matrix-targeting sequence, which is removed by the matrix protease. The second targeting sequence is a hydrophobic segment that blocks complete translocation of the protein across the inner membrane (Figure 13-28, path A). After the resulting membrane-embedded intermediate diffuses laterally away from the translocation channel, a protease in the membrane cleaves the protein near the hydrophobic transmembrane segment, releasing the mature protein in a soluble form into the intermembrane space. Except for the second proteolytic cleavage, this pathway is similar to that of innermembrane proteins such as CoxVa (see Figure 13-27, path A).
FIGURE 13-28 Two pathways to the mitochondrial intermembrane space. Pathway A, the major one for delivery of proteins from the cytosol to the intermembrane space, is similar to pathway A for delivery to the inner membrane (see Figure 13-26). The major difference is that the internal targeting sequence in these proteins, such as cytochrome , is recognized by an inner-membrane protease, which cleaves the protein on the intermembrane-space side of the membrane. The released protein then folds and binds to its heme cofactor within the intermembrane space. Pathway B is a specialized pathway for delivery of the proteins Tim9 and Tim10 to the intermembrane space. These proteins readily pass through the Tom40 general import pore, and once they are in the intermembrane space, they fold and form disulfide bonds that prevent reverse translocation through Tom40. The disulfide bonds are generated by Erv1 and are transferred to Tim9 and Tim10 by Mia40. See R. E. Dalbey and A. Kuhn, 2000, Annu. Rev. Cell Dev. Biol. 16:51; N. Pfanner and A. Geissler, 2001, Nat. Rev. Mol. Cell Biol. 2:339; and K. Tokatlidis, 2005, Cell 121:965–967. Description The illustration shows from top to bottom: cytosol, outer mitochondrial membrane, intermembrane space, inner mitochondrial membrane, and mitochondrial matrix. Path
A shows a preprotein chain with an intermembrane-space-targeting sequence and matrix-targeting sequence at N-terminal in the cytosol. The outer membrane has a Tom 40 channel next to a Tom 20, Tom 22 receptor; and the inner membrane has Tim 23 slash 17 channel attached to a Tim 44 receptor. Step 1: The preprotein enters mitochondrial matrix via Tom 40, intermembrane space, and Tim 23 slash 17 where intermembrane-space-targeting sequence attaches to internal of Tim 23 slash 17 channel. Step 2: A protease embedded in inner mitochondrial membrane cleaves matrix-targeting sequence leaving behind an embedded preprotein. Step 3: The preprotein chain present in the intermembrane space cleaves and folds by enclosing a heme within. Path B shows a Tim 9 or Tim 10 protein chain having an intermembrane-spacetargeting sequence toward N-terminal in the cytosol. The chain enters intermembrane space via Tom 40 channel where Mia 40 E r v 1 acts on it resulting in a U-shaped protein with two disulfide bonds. The small Tim9 and Tim10 proteins, which reside in the intermembrane space, illustrate a second pathway for targeting to the intermembrane space. Tim9 and Tim10 do not contain an N-terminal matrix-targeting sequence and are delivered directly to the intermembrane space via the general import pore without involvement of any inner-membrane translocation factors (see Figure 13-28, path B). Translocation through the Tom40 general import pore does not seem to be coupled to any energetically favorable process; however, once located in the intermembrane space, Tim9 and Tim10 proteins acquire two disulfide bonds each and fold into compact, stable structures. Apparently, the mechanism that drives their unidirectional translocation through the outer membrane involves passive diffusion through the outer membrane followed by folding and disulfide bond formation, which irreversibly traps the proteins in the intermembrane space. The process of disulfide bond
Import of Chloroplast Stromal Proteins Is Similar to Import of Mitochondrial Matrix Proteins
formation in the intermembrane space resembles that in the ER lumen and involves a disulfide bond–generating protein, Erv1, and a disulfide transfer protein, Mia40. Outer-Membrane Proteins Many of the proteins that reside in the mitochondrial outer membrane, including the Tom40 pore itself and mitochondrial porin, have a β-barrel structure in which antiparallel β strands form hydrophobic transmembrane segments surrounding a central channel. Such proteins are incorporated into the outer membrane by first interacting with the general import pore, Tom40; they are then transferred to the SAM (sorting and assembly machinery) complex, which is composed of at least three outer-membrane proteins. Presumably it is the very stable hydrophobic nature of β-barrel proteins that ultimately causes them to be stably incorporated into the outer membrane, but precisely how the SAM complex facilitates this process is not known. Import of Chloroplast Stromal Proteins Is Similar to Import of Mitochondrial Matrix Proteins The most abundant proteins found in the chloroplast stroma are the enzymes of the Calvin cycle, which function in fixing carbon dioxide during photosynthesis (see Chapter 12). The large (L) subunit of ribulose 1,5-bisphosphate carboxylase (rubisco) is encoded by chloroplast DNA
and synthesized on chloroplast ribosomes in the stromal space. The small (S) subunit of rubisco and all the other Calvin cycle enzymes are encoded by nuclear genes and transported to chloroplasts after their synthesis in the cytosol. The precursor forms of these stromal proteins contain an N-terminal stromal-import sequence (see Table 13-1). Experiments with isolated chloroplasts, similar to those with mitochondria, have shown that the general process of stromal import depicted in the top half of Figure 13-29 is similar to that of protein import into the mitochondrial matrix. At least three chloroplast outer-membrane proteins are known to be essential for directing proteins to the stroma. These include a receptor that binds the stromal-import sequence made up of proteins, known as Toc159 and Toc34, each of which has a GTPase domain on the cytosolic side of the outer membrane reminiscent of SRP and SRP receptor. The receptor transfers the stromal-import sequence to a translocation channel made up of Toc75. A complex of at least five chloroplast inner-membrane proteins known as the Tic complex forms the translocation channel for passage of proteins into the stromal space. Although these proteins are functionally analogous to the receptor and channel proteins of the mitochondria, they are not structurally homologous. The lack of sequence similarity between these chloroplast and mitochondrial proteins suggests that they may have arisen independently during evolution.
FIGURE 13-29 Transporting proteins to chloroplast thylakoids. Two of the four pathways for transporting proteins from the cytosol to the thylakoid lumen are shown here.
In these pathways, unfolded precursors are delivered to the stroma via the same outermembrane proteins that import stromal-localized proteins. Cleavage of the N-terminal stromal-import sequence by a stromal protease then reveals the thylakoid-targeting sequence (step 1 ). At this point the two pathways diverge. In the SRP-dependent pathway (left), plastocyanin and similar proteins are kept unfolded in the stromal space by a set of chaperones (not shown), and the thylakoid-targeting sequence binds to proteins that are closely related to the bacterial SRP, SRP receptor, and SecY translocon, which mediate movement into the thylakoid lumen (step 2a ). After the thylakoid-targeting sequence is removed in the thylakoid lumen by a separate endoprotease, the protein folds into its mature conformation (step 3a ). In the pH-dependent pathway (right), metal-binding proteins fold in the stroma, and complex redox cofactors are added (step 2b ). Two arginine residues (RR) at the N-terminus of the thylakoid-targeting sequence and a pH gradient across the inner membrane are required for transport of the folded protein into the thylakoid lumen (step 3b ). The translocon in the thylakoid membrane is composed of at least four proteins related to proteins in the bacterial plasma membrane. The thylakoid-targeting sequence containing the two arginine residues is cleaved in the thylakoid lumen (step 4b ). See R. Dalbey and C. Robinson, 1999, Trends Biochem. Sci. 24:17; R. E. Dalbey and A. Kuhn, 2000, Annu. Rev. Cell Dev. Biol. 16:51; and C. Robinson and A. Bolhuis, 2001, Nat. Rev. Mol. Cell Biol. 2:350. Description The illustration from top to bottom shows cytosol, outer chloroplast membrane, intermembrane space, inner chloroplast membrane, stroma, thylakoid membrane, and thylakoid lumen. The outer chloroplast membrane has Toc 75 channel attached to Toc 159-G T P and Toc 34-G T P; and the inner chloroplast membrane has Tic complex channel. The first part shows a plastocyanin precursor chain with stromal-import sequence at N-terminal followed by a thylakoid-targeting sequence in cytoplasm. Step 1: The precursor chain enters stroma through Toc 75, intermembrane space, and Tic complex. H s p 70 acts on the chain and cleaves stromal-import sequence leaving behind plastocyanin with thylakoid-targeting sequence. Step 2 a, S R P-dependent pathway: Chloroplast S R P binds to chloroplast S R P receptor embedded in thylakoid membrane next to a channel. This binding cause plastocyanin chain to enter thylakoid lumen. Step 3 a: The thylakoid-targeting sequence is cleaved from now matured plastocyanin.
Proteins Are Targeted to Thylakoids by Mechanisms Related to Bacterial Protein Translocation
The second part shows a metal-binding precursor chain with stromal-import sequence at N-terminal followed by a thylakoid-targeting sequence in cytoplasm. Step 1 is similar to first part except the thylakoid-targeting sequence of metal-binding protein has R R terminal. Step 2 b, p H-dependent pathway: This protein chain binds to two metal ions. Step 3 b: This enters thylakoid lumen via pore. Step 4 b: The thylakoid-targeting sequence with R R terminal is cleaved from now matured metal-binding protein bound to metal ions. The available evidence suggests that chloroplast stromal proteins, like mitochondrial matrix proteins, are imported in the unfolded state. Import into the stroma depends on ATP hydrolysis catalyzed by a stromal Hsp70 chaperone whose function is similar to that of Hsp70 in the mitochondrial matrix and BiP in the ER lumen. Unlike mitochondria, chloroplasts do not generate an electrochemical gradient (proton-motive force) across their inner membrane. Thus protein import into the chloroplast stroma appears to be powered solely by ATP hydrolysis. In addition to Hsp70, the stroma contains two other ATPases that may participate in protein import. These are a chloroplast version of the chaperone Hsp90, and a ring forming ATPase known as Hsp93. Homologs of both proteins are known to catalyze energy-dependent protein-folding reactions in other contexts and it may be that chloroplast Hsp90 and Hsp93 participate in protein folding in the stroma, protein import into the stroma, or both. Proteins Are Targeted to Thylakoids by Mechanisms Related to Bacterial
Protein Translocation In addition to the double membrane that surrounds them, chloroplasts contain a series of internal interconnected membranous sacs, the thylakoids (see Figure 12-33). All of the chemical reactions of photosynthesis take place in the thylakoid membrane or lumen and are catalyzed by the proteins that are localized to this specialized subcompartment. Many of these proteins are synthesized in the cytosol as precursors containing multiple targeting sequences. For example, plastocyanin and other proteins destined for the thylakoid lumen require the successive action of two targeting sequences. The first is an N-terminal stromal-import sequence that directs the protein to the stroma by the same pathway that imports the rubisco S subunit. The second sequence targets the protein from the stroma to the thylakoid lumen. The role of these targeting sequences has been shown in experiments measuring the uptake of mutant proteins generated by recombinant DNA techniques into isolated chloroplasts. For instance, mutant plastocyanin that lacks the thylakoid-targeting sequence but contains an intact stromal-import sequence accumulates in the stroma and is not transported into the thylakoid lumen. Four separate pathways for transporting proteins from the stroma into the thylakoid have been identified. All four pathways have been found to be closely related to analogous transport mechanisms in bacteria, illustrating the close evolutionary relationship between the thylakoid membrane and the bacterial plasma membrane. Transport of plastocyanin and related
proteins into the thylakoid lumen from the stroma occurs by an SRPdependent pathway that uses a translocon similar to SecY, the bacterial version of the Sec61 complex (Figure 13-29, left). A second pathway for transporting proteins into the thylakoid lumen involves a protein related to bacterial protein SecA, which uses the energy from ATP hydrolysis to drive protein translocation through the SecY translocon. A third pathway, which targets proteins to the thylakoid membrane, depends on a protein related to the mitochondrial Oxa1 protein and the homologous bacterial protein (see Figure 13-27, path B). Some proteins encoded by chloroplast DNA and synthesized in the stroma or transported into the stroma from the cytosol are inserted into the thylakoid membrane via this pathway. Finally, thylakoid proteins that bind metal-containing cofactors follow another pathway into the thylakoid lumen (see Figure 13-29, right). The unfolded precursors of these proteins are first targeted to the stroma, where the N-terminal stromal-import sequence is cleaved off, and the protein then folds and binds its cofactor. A set of thylakoid-membrane proteins assists in translocating the folded protein and bound cofactor into the thylakoid lumen. This process is powered by the electrochemical gradient normally maintained across the thylakoid membrane. The thylakoid-targeting sequence that directs a protein to this pathway includes two closely spaced arginine residues that are crucial for recognition. Bacterial cells also have a mechanism for translocating folded proteins with a similar arginine-containing sequence across the plasma membrane, known as the Tat (twin-arginine translocation) pathway. The molecular mechanism whereby these large folded globular proteins are translocated across the thylakoid membrane is not fully understood, but
the presence of a folded protein with an appropriate twin arginine signal appears to induce the oligomerization of Tat proteins in the membrane to form pore-like structures. In this respect, the Tat pathway resembles the pathway for the import of folded proteins into peroxisomes, described in the next section. KEY CONCEPTS OF SECTION 13.4 Targeting of Proteins to Mitochondria and Chloroplasts Most mitochondrial and chloroplast proteins are encoded by nuclear genes, synthesized on cytosolic ribosomes, and imported post-translationally into the organelles. All the information required to target a precursor protein from the cytosol to the mitochondrial matrix or chloroplast stroma is contained within its N-terminal targeting sequence. After protein import, the targeting sequence is removed by proteases within the matrix or stroma. Cytosolic chaperones maintain the precursors of mitochondrial and chloroplast proteins in an unfolded state. Only unfolded proteins can be imported into the organelles. Proteins destined for the mitochondrial matrix bind to receptors on the outer mitochondrial membrane and are then transferred to the general import pore (Tom40) in the outer membrane. Translocation takes place at sites where the outer and inner membranes of the organelles are close together and occurs through the outer and inner membranes concurrently. Translocation is driven by ATP hydrolysis by Hsp70 in the matrix (see Figure 13-24) and by the proton-motive force across the inner membrane. Proteins sorted to mitochondrial destinations other than the matrix usually contain two or more targeting sequences, one of which may be an N-terminal matrix-targeting sequence (see Figure 13-26). Some mitochondrial proteins destined for the intermembrane space or inner membrane are first imported into the matrix and then redirected; others never enter the matrix, but go directly to their final location. Protein import into the chloroplast stroma occurs through outer-membrane and innermembrane translocation channels that are analogous in function to mitochondrial
channels, but composed of proteins unrelated in sequence to the corresponding mitochondrial proteins. Proteins destined for the thylakoid have secondary targeting sequences. After entry of these proteins into the stroma, cleavage of the stromal-targeting sequences reveals the thylakoid-targeting sequences. The four known pathways for moving proteins from the chloroplast stroma to the thylakoid closely resemble translocation across the bacterial plasma membrane (see
Figure 13-29). One of these systems can translocate folded proteins.
13.5 Targeting of Peroxisomal Proteins
13.5 Targeting of Peroxisomal Proteins Peroxisomes are small organelles bounded by a single membrane which surrounds a luminal space known as the peroxisomal matrix. Although peroxisomes have a simple structure, the biogenesis of peroxisomes is similar in fundamental outline to the biogenesis of the more complicated mitochondria and chloroplasts. Peroxisomal proteins, encoded by nuclear genes and synthesized on free ribosomes in the cytosol, are recognized by specific peroxisomal-targeting sequences. These sequences bind to a peroxisome-specific receptor protein and are then delivered either to the peroxisomal matrix or to the peroxisomal membrane by an energydependent targeting process. The size and enzyme content of peroxisomes vary considerably among different kinds of cells. However, all peroxisomes contain enzymes that use molecular oxygen to oxidize various substrates such as fatty acids, breaking them down into smaller components for use in biosynthetic pathways. The hydrogen peroxide generated by these oxidation reactions is extremely reactive and potentially harmful to cellular components; however, the peroxisome contains other enzymes, such as catalase, that efficiently convert into . In mammals, peroxisomes are most abundant in liver cells, where they constitute about 1–2 percent of the cell volume.
A Cytosolic Receptor Targets Proteins with an SKL Sequence at the C-Terminus to the Peroxisomal Matrix
A Cytosolic Receptor Targets Proteins with an SKL Sequence at the C-Terminus to the Peroxisomal Matrix Peroxisomal-targeting sequences were first identified by testing of peroxisomal proteins with deletions for a specific defect in peroxisomal targeting. In one early study, the gene for firefly luciferase was expressed in cultured insect cells, and the resulting protein was shown to be properly targeted to the peroxisome. However, expression of a gene missing a small portion of the sequence encoding the C-terminus of the protein led to luciferase that failed to be targeted to the peroxisome and remained in the cytoplasm. By testing various mutant luciferase proteins in this system, researchers discovered that the sequence Ser-Lys-Leu (SKL in one-letter code) or a related sequence at the C-terminus is necessary for peroxisomal targeting. Furthermore, addition of the SKL sequence to the C-terminus of a normally cytosolic protein leads to uptake of the altered protein by peroxisomes in cultured cells. All but a few of the many different peroxisomal proteins bear a sequence of this type, known as peroxisomaltargeting sequence 1, or simply PTS1. The pathway for the import of catalase and other PTS1-bearing proteins into the peroxisomal matrix is depicted in Figure 13-30. In the cytosol, PTS1 binds to a receptor called Pex5. Pex5 has the remarkable property of being able to switch from a monomeric soluble form to an oligomeric form embedded in the peroxisomal membrane in a complex with the membrane protein Pex14. In a manner that is not well understood, the
PTS1-bearing protein is released from the oligomeric form of Pex5 into the interior of the peroxisome. This peroxisome import machinery, unlike most systems that mediate protein import into the ER, mitochondria, and chloroplasts, can translocate folded proteins across the membrane. For example, catalase assumes a folded conformation and binds to heme in the cytoplasm before traversing the peroxisomal membrane. Cell-free studies have shown that the peroxisome import machinery can transport large macromolecular objects, including gold particles about 9 nm in diameter, as long as they have a PTS1 tag attached to them. There is evidence that the size of oligomers of Pex5 bound to PTS1-bearing cargo molecules and Pex14 adjusts according to the size of the PTS1-bearing cargo molecules. The dynamic formation of oligomers is apparently the key mechanism by which PTS1-bearing cargo molecules can be accommodated without the formation of large stable pores that would disrupt the integrity of the peroxisomal membrane.
FIGURE 13-30 PTS1-directed import of peroxisomal matrix proteins. Step 1 : Most peroxisomal matrix proteins contain a C-terminal PTS1 targeting sequence (red), which binds to the cytosolic receptor Pex5. Step 2 : Pex5 with the bound matrix protein forms a multimeric complex with the Pex14 receptor located on the peroxisomal membrane. Step 3 : After assembly of the matrix protein-Pex5-Pex14 complex, the matrix protein dissociates from Pex5 and is released into the peroxisomal matrix. Steps 4 and 5 : Pex5 is then returned to the cytosol by a process that involves ubiquitinylation by the membrane proteins Pex2, Pex10, and Pex12, followed by ATP-dependent removal from the membrane by the AAA-ATPase proteins Pex1 and Pex6. Note that folded proteins can be imported into
peroxisomes and that the targeting sequence is not removed in the matrix. See P. E. Purdue and P. B. Lazarow, 2001, Annu. Rev. Cell Dev. Biol. 17:701; S. Subramani et al., 2000, Annu. Rev. Biochem. 69:399; and V. Dammai and S. Subramani, 2001, Cell 105:187. Description The illustration shows a peroxisomal membrane separating cytosol from peroxisomal matrix. Step 1: The P T S 1 peroxisomal-targeting sequence at C-terminal of a protein chain binds in a pocket of a Pex 5 receptor. Step 2: Two such complexes get embedded between two Pex 14 in the peroxisomal membrane with protein chain inside the matrix. Step 3: Peroxisomal matrix protein detaches from the Pex 5 receptor in to the matrix. Step 4: A Pex 2, Pex 10, and Pex 12 complex embedded in the peroxisomal membrane transfers Ubiquitin to two embedded Pex 5 receptors. Step 5: A Pex 1-Pex 6 complex hydrolysis A T P to A D P and P i causing the detachment of Pex 5 receptors into cytosol. Steps repeat. Once the PTS1-bearing cargo molecule is released into the interior of the peroxisome, the oligomeric complex of Pex5 and Pex14 is actively disassembled, thus releasing Pex5 back into the cytoplasm in a soluble state. Pex5 recycling involves modification of membrane-bound Pex5 by ubiquitinylation. A complex of the peroxisomal membrane proteins Pex10, Pex12, and Pex2 transfers a ubiquitin moiety to Pex5. The AAA ATPases Pex1 and Pex6, anchored to the peroxisomal membrane by Pex15, recognize ubiquitinylated Pex5 and use the energy from ATP hydrolysis to remove it from the oligomeric complex with Pex14 and release it into the cytosol. After removal of the ubiquitin modification, cytosolic Pex5 is ready to carry out another cycle of binding to a PTS1-bearing protein. Peroxisomal import studies with purified components have shown that the binding of Pex5 to a PTS1-bearing protein, the assembly of an oligomeric
Peroxisomal Membrane and Matrix Proteins Are Incorporated by Different Pathways
complex of Pex5 and Pex14, and release of the PTS1-bearing protein into the interior of the peroxisome can all occur spontaneously without a source of chemical energy such as ATP. In contrast, both ubiquitin modification of Pex5 and the recycling of Pex5 by the AAA-ATPase require ATP hydrolysis. Evidently, in the process by which Pex5 delivers PTS1-bearing proteins to the peroxisome matrix by cycling between the cytosol and the peroxisomal membrane, it is the recycling step that uses energy to power unidirectional translocation of cargo molecules into the peroxisomal matrix. A few peroxisomal matrix proteins, such as thiolase, are synthesized as precursors with an N-terminal targeting sequence known as PTS2. These proteins bind to a different cytosolic receptor protein, but otherwise their import is thought to occur by the same mechanism as for PTS1-containing proteins. Peroxisomal Membrane and Matrix Proteins Are Incorporated by Different Pathways Autosomal recessive mutations that cause defective peroxisome assembly occur naturally in the human population. Such mutations can lead to severe developmental defects often associated with craniofacial abnormalities. In Zellweger syndrome and related disorders, for example, the transport of many or all proteins into the peroxisomal matrix is
impaired: newly synthesized peroxisomal enzymes remain in the cytosol and are eventually degraded. Genetic analyses of cultured cells from patients with Zellweger syndrome and of yeast cells carrying similar mutations have identified more than 20 genes that are required for peroxisome biogenesis. Studies with peroxisome-assembly mutants have shown that different pathways are used for importing peroxisomal matrix proteins and for inserting proteins into the peroxisomal membrane (Figure 13-31). For example, analysis of cells from some patients with Zellweger syndrome led to the identification of the gene encoding Pex5 as well as many of the Pex genes needed for recycling of Pex5. Mutant cells that are defective in any one of these proteins cannot incorporate matrix proteins into peroxisomes; nonetheless, the cells contain empty peroxisomes that have a normal complement of peroxisomal membrane proteins (Figure 13-31b). Mutations in any one of three other genes were found to block insertion of peroxisomal membrane proteins as well as import of matrix proteins (Figure 13-31c). These findings demonstrate that one set of proteins translocates soluble proteins into the peroxisomal matrix, but a different set is required for insertion of proteins into the peroxisomal membrane. This situation differs markedly from that of the ER, mitochondrion, and chloroplast, whose membrane proteins and soluble proteins share many of the same components for their import into these organelles.
EXPERIMENTAL FIGURE 13-31 Studies reveal different pathways for incorporation of peroxisomal membrane and matrix proteins. Cells were stained with fluorescent antibodies to PMP70, a peroxisomal membrane protein, or with fluorescent antibodies to catalase, a peroxisomal matrix protein, then viewed in a fluorescence microscope. (a) In
wild-type cells, both peroxisomal membrane and matrix proteins are visible as bright foci in numerous peroxisomal bodies. (b) In cells from a Pex12-deficient patient, PMP70 is localized normally to peroxisomal bodies, but catalase is not delivered to peroxisomes and is distributed uniformly throughout the cytosol with no bright foci, whereas (c) in cells from a Pex3-deficient patient, peroxisomal membranes cannot assemble, and as a consequence, peroxisomal bodies do not form. Thus both catalase and PMP70 are mislocalized to the cytosol. Description The part (a) shows wild-type cells. The diagram shows three spherical peroxisomes with catalase inside and P M P 70 on the surface. First micrograph stained for P M P 70 shows blue fluorescence with numerous white speckles. Second micrograph stained for catalase shows green fluorescence with numerous white speckles. The part (b) shows Pex 12 mutants (deficient in matrix protein import). The diagram shows three ring-shaped peroxisomes with P M P 70 on the surface and catalase in cytoplasm. First micrograph stained for P M P 70 shows blue fluorescence with few white speckles. Second micrograph stained for catalase shows green fluorescence with no white speckle. The part (c) shows Pex 3 mutants (deficient in membrane biogenesis). The diagram shows P M P 70 and catalase in cytoplasm and no peroxisome. First micrograph stained for P M P 70 shows blue fluorescence and second micrograph stained for catalase shows green fluorescence without any white speckle. KEY CONCEPTS OF SECTION 13.5 Targeting of Peroxisomal Proteins All luminal peroxisomal proteins are synthesized on free cytosolic ribosomes and incorporated into the organelle post-translationally. Most peroxisomal matrix proteins contain a C-terminal targeting sequence known as PTS1; a few have an N-terminal PTS2 targeting sequence. Neither targeting sequence is cleaved after import.
All proteins destined for the peroxisomal matrix bind to a cytosolic receptor protein, which is Pex5 for PTS1-bearing proteins, and then are directed to common translocation machinery on the peroxisomal membrane (see Figure 13-30). Translocation of matrix proteins across the peroxisomal membrane depends on ATP hydrolysis, which is coupled to recycling of Pex5 from the peroxisomal membrane back to the cytosol. Unlike proteins imported to the ER, mitochondrion, or chloroplast, many peroxisomal matrix proteins fold in the cytosol and traverse the membrane in a folded conformation. Proteins destined for the peroxisomal membrane contain different targeting sequences than peroxisomal matrix proteins and are imported by a different pathway.
Large and Small Molecules Enter and Leave the Nucleus via Nuclear Pore Complexes
13.6 Transport into and out of the Nucleus The nucleus is separated from the cytoplasm by two membranes, which form the nuclear envelope (see Figure 1-12a). The nuclear envelope is continuous with the ER and forms a part of it. Transport of proteins from the cytoplasm into the nucleus and movement of macromolecules, including mRNAs, tRNAs, and ribosomal subunits, out of the nucleus occurs through nuclear pores, which span both membranes of the nuclear envelope. Import of proteins into the nucleus shares some fundamental features with protein import into other organelles. For example, imported nuclear proteins carry specific targeting sequences known as nuclearlocalization signals (NLSs). However, proteins are imported into the nucleus in a folded state, and thus nuclear import differs fundamentally from protein translocation across the membranes of the ER, mitochondrion, and chloroplast, during which proteins are unfolded. In this section, we discuss the main mechanism by which proteins enter and exit the nucleus. We also discuss the process by which mRNAs and other ribonuclear protein complexes are exported from the nucleus, which differs mechanistically from nuclear protein import. Large and Small Molecules Enter and Leave the Nucleus via Nuclear Pore
Complexes Numerous nuclear pores perforate the nuclear envelope in all eukaryotic cells. Each nuclear pore is formed from an elaborate structure termed the nuclear pore complex (NPC), which is one of the largest discrete protein assemblages in the cell. The total mass of the pore structure is 60,000– 80,000 kDa in vertebrates, which is about 16 times larger than a ribosome. An NPC is made up of many copies of some 30 different proteins called nucleoporins. Electron micrographs of nuclear pore complexes reveal a membrane-embedded ring structure that surrounds a largely aqueous pore (Figure 13-32). Eight approximately 100-nm long filaments extend into the nucleoplasm with the distal ends of these filaments joined by a terminal ring, forming a structure called the nuclear basket. Cytoplasmic filaments extend from the cytoplasmic side of the NPC into the cytosol.
FIGURE 13-32 Nuclear pore complex at different levels of resolution. (a) Nuclear envelopes from the large nuclei of Xenopus oocytes, visualized by scanning electron microscopy. Top: View of the cytoplasmic face reveals the octagonal shape of the membrane-embedded portion of nuclear pore complexes. Bottom: View of the nucleoplasmic face shows the nuclear basket that extends from the membrane-embedded portion. (b) Cutaway model of the nuclear pore complex, showing the major structural features formed by membrane nucleoporins, structural nucleoporins, and FG-nucleoporins. (c) Sixteen copies of the Y-complex form a major part of the structural scaffold of the nuclear pore complex. The three-dimensional structure of the Y-complex is modeled into the pore structure. Note the twofold symmetry across the double membrane of the nucleus
(left) and the eightfold rotational symmetry around the axis of the pore (right). (d) The FGnucleoporins have extended disordered structures that are composed of repeats of the sequence Phe–Gly interspersed with hydrophilic regions (left). The FG-nucleoporins are most abundant in the central part of the pore, and the FG-repeat sequences are thought to fill the central channel with a gel-like matrix (right). See K. Ribbeck and D. Görlich, 2001, EMBO J. 20:1320–1330; and M. P. Rout and J. D. Atchison, 2001, J. Biol. Chem. 276:16593. [Part (a) Republished with permission from Elsevier, from V. Doye and E. Hurt, 1997, Nucleoporins to Nuclear Pore Complexes, Curr. Opin. Cell Biol. 9(3):401–411; permission conveyed through Copyright Clearance Center, Inc.] Description The part (a) shows two micrographs of nuclear pore complexes. The top micrograph shows octagonal shaped structures and the bottom micrograph shows basket shaped structures. The illustration (b) of a nuclear pore complex shows a nuclear envelope with outer membrane, lumen, and inner membrane separating cytoplasm from nucleoplasm. A pore between the nuclear envelope has membrane nucleoporins attached to nuclear envelope ends. Projecting into the cytoplasm from the border of the pore are several cytoplasmic filaments. At the base of these filaments and surrounded the pore like a crown on the cytosolic side are the structural nucleoporins (Y-complex). A mesh of F G-nucleoporins covers the pore aperture. Projecting into the nucleoplasm is a cage shaped nuclear basket, capped with the terminal ring. Nuclear lamina is present on the nucleoplasmic side of the inner nuclear membrane. The illustration (c) shows a ringlike structure from side and top embedded in the outer and inner membrane of nucleus. The ring is made of eight pairs of Y-complexes. A close-up illustration of the center of the ring shows a matrix of F G-repeats in central channel of pore. The illustration (d) of F G-nucleoporin shows a wave-like structure with three beads on it. The wave is labeled, hydrophilic region, and the bead is labeled, F G-repeat (hydrophobic). Ions, small metabolites, and globular proteins up to about 40 kDa can diffuse passively through the central aqueous region of the nuclear pore complex. However, large proteins and ribonucleoprotein complexes cannot
diffuse in and out of the nucleus. Rather, these macromolecules are actively transported through the NPC with the assistance of soluble transport proteins that bind macromolecules and also interact with nucleoporins. The capacity and efficiency of the NPC for such active transport is remarkable. In one minute, each NPC is estimated to import 60,000 protein molecules into the nucleus, while exporting 50–250 mRNA molecules, 10–20 ribosomal subunits, and 1000 tRNAs out of the nucleus. In general terms, the nucleoporins are of three types: structural nucleoporins, membrane nucleoporins, and FG-nucleoporins. The structural nucleoporins form the scaffold of the nuclear pore, which is a ring of eightfold rotational symmetry that traverses both membranes of the nuclear envelope, creating an opening in the middle for movement of macromolecules into and out of the nucleus. The inner and outer membranes of the nuclear envelope are connected at the NPC by a highly curved region of membrane that contains the embedded membrane nucleoporins (Figure 13-32b). A set of seven structural nucleoporins forms a Y-shaped structure about the size of the ribosome, known as the Ycomplex. Sixteen copies of the Y-complex form the basic structural scaffold of the pore, which has bilateral symmetry across the nuclear envelope and eightfold rotational symmetry in the plane of the envelope (Figure 13-32c). A structural motif repeated several times within the Ycomplex is closely related to a structure found in the COPII proteins that drive the formation of coated vesicles within cells (see Chapter 14). This primordial relationship between structural nucleoproteins and vesicle coat proteins suggests that the two types of membrane coat complexes share a
Nuclear Transport Receptors Escort Proteins Containing Nuclear-Localization Signals into the Nucleus
common origin. The basic function of this element may be to form a protein lattice that deforms the membrane into a highly curved structure. The FG-nucleoporins, which line the channel of the nuclear pore complex and are also found associated with the nuclear basket and the cytoplasmic filaments, contain multiple repeats of short hydrophobic sequences that are rich in phenylalanine (F) and glycine (G) residues (FG-repeats). The hydrophobic FG-repeats are thought to occur in regions of extended, otherwise hydrophilic polypeptide chains that fill the central transporter channel. The FG-nucleoporins are essential for the function of the NPC, but their function appears to depend on a general property of these proteins rather than on a specific structure, since the NPC remains functional even if up to half of the FG-repeats have been deleted. Biophysical studies and dynamic computer modeling show that FG-repeats can have a disordered structure similar to that of an unfolded protein and are highly dynamic. Because of these properties, the central channel of the NPC can be thought of as being filled with a fluidlike phase that allows the diffusion of small molecules while excluding unchaperoned hydrophilic proteins larger than 40 kDa (Figure 13-32d). Nuclear Transport Receptors Escort Proteins Containing NuclearLocalization Signals into the Nucleus All proteins found in the nucleus — such as histones, transcription factors, and DNA and RNA polymerases — are synthesized in the cytoplasm and
imported into the nucleus through nuclear pore complexes. Such proteins contain a nuclear-localization signal (NLS) that directs their selective transport into the nucleus. NLSs were first discovered through the analysis of mutations of the gene for large T-antigen encoded by simian virus 40 (SV40). The wild-type form of large T-antigen is localized to the nucleus in virus-infected cells, whereas some mutated forms of large T-antigen accumulate in the cytoplasm. The mutations responsible for this altered cellular localization all occur within a specific seven-residue sequence rich in basic amino acids near the C-terminus of the protein: Pro-Lys-Lys- Lys-Arg-Lys-Val. Experiments with chimeric proteins in which this sequence was fused to a cytosolic protein demonstrated that it directs transport into the nucleus and consequently functions as an NLS (Figure 13-33). NLS sequences were subsequently identified in numerous other proteins imported into the nucleus. Many of these sequences are similar to the basic NLS in SV40 large T-antigen, whereas others are chemically quite different. For instance, an NLS in the RNA-binding protein hnRNP A1 is hydrophobic. Accordingly, there are multiple mechanisms for the recognition of these very different sequences.
EXPERIMENTAL FIGURE 13-33 Nuclear-localization signals (NLSs) direct proteins to the cell nucleus. Cytoplasmic proteins can be transported to the nucleus if they are fused to a nuclear-localization signal. (a) Normal pyruvate kinase, here visualized by immunofluorescence after cultured cells were treated with a specific antibody (yellow), is localized to the cytoplasm. This very large cytosolic protein functions in carbohydrate metabolism. (b) When a chimeric pyruvate kinase containing the SV40 NLS at its N-terminus was expressed in cells, it was localized to the nucleus. The chimeric protein was expressed from a transfected engineered gene produced by fusing a viral gene fragment encoding the SV40 NLS to the pyruvate kinase gene. [Republished with permission from Elsevier, from D. Kalderon et al., 1984, “A Short Amino Acid Sequence Able to Specify Nuclear Location,” Cell 39(3 Pt 2):499–509; permission conveyed through Copyright Clearance Center, Inc.] Early work on the mechanism of nuclear import showed that proteins containing a positively charged NLS, similar to the one in SV40 large T-
antigen, were efficiently transported into isolated nuclei only if they were provided with a cytosolic extract (Figure 13-34). Using this assay system, researchers purified two required cytosolic components, a nuclear transport receptor and Ran GTPase. Nuclear transport receptors bind to the NLS on a cargo protein to be transported into the nucleus; they also have affinity for FG-repeats on nucleoporins. Because of their affinity for FG-repeats, nuclear transport receptors and NLS-containing cargo proteins to which they are bound readily partition into the fluid phase of FGrepeats in the central channel of the NPC, whereas proteins of similar size that lack this property are excluded from the central channel. Nuclear transport receptors can be monomeric, consisting of a single polypeptide that can bind both to an NLS and to FG-repeats, or they can be dimeric, with one subunit binding to the NLS and the other binding to FG-repeats.
EXPERIMENTAL FIGURE 13-34 Cytosolic proteins are required for nuclear transport. The failure of nuclear transport to occur in permeabilized cultured cells in the absence of cell lysate demonstrates the involvement of soluble cytosolic components in the process. (a) Phase-contrast micrographs of untreated and digitonin-permeabilized HeLa cells. Treatment of a monolayer of cultured cells with the mild non-ionic detergent digitonin permeabilizes the plasma membrane so that cytosolic constituents leak out, but leaves the nuclear envelope and NPCs intact. (b) Fluorescence micrographs of digitonin-permeabilized HeLa cells incubated with a fluorescent protein chemically coupled to a synthetic SV40 T-
antigen NLS peptide in the presence and absence of cell lysate (cytosol). Accumulation of the transport substrate in the nucleus occurred only when cytosol was present (right). [S. A. Adam, R. S. Marr, and L. Gerace, 1990, J. Cell Biol. 111: 807–816; https://doi.org/10.1083/jcb.111.3.807.] Description The first pair (a) effect of digitonin shows one micrograph of HeLa cells not treated by digitonin and second micrograph of HeLa cells treated by digitonin. The first micrograph shows an intact HeLa cell and the second micrograph shows plasma degraded HeLa cell. The second pair (a) nuclear import by permeabilized cells shows digitonin-permeabilized HeLa cells. In absence of lysate, the left micrograph shows black surface. In presence of lysate, the right micrograph shows several brightly lit spheres against a dark background. Ran is a small monomeric G protein that exists in either a GTP-bound or a GDP-bound conformation (see Figure 3-35). It is the cycling of Ran between GTP-bound and GDP-bound conformations, leading to the net hydrolysis of GTP to GDP, that ultimately provides the energy to drive unidirectional transport of macromolecules through the nuclear pore. The mechanism for the import of cytoplasmic cargo proteins mediated by a nuclear transport receptor known as importin is shown in Figure 13-35. Free importin in the cytoplasm binds to its cognate NLS in a cargo protein, forming an importin-cargo complex. The cargo complex then efficiently partitions into the NPC channel as the importin interacts with FG-repeats. Once in the channel, the importin-cargo complex can rapidly reach the nucleoplasmic side of the channel by random diffusion, and there the importin interacts with , which causes a conformational change
in the importin that displaces the NLS, releasing the cargo protein into the nucleoplasm. The importincomplex then diffuses back through the NPC. Once the importincomplex reaches the cytoplasmic side of the NPC, Ran interacts with a specific GTPase-activating protein (Ran-GAP) that is a component of the NPC cytoplasmic filaments. This interaction stimulates Ran to hydrolyze its bound GTP to GDP, which causes it to convert to a conformation that has low affinity for the importin, so that the importin is released into the cytoplasm, where it can participate in another cycle of import. travels back through the pore to the nucleoplasm, where it encounters a specific guanine nucleotide exchange factor (Ran-GEF) that causes Ran to release its bound GDP in favor of GTP. The net result of this series of reactions is the coupling of the hydrolysis of GTP to the transfer of an NLS-bearing protein from the cytoplasm to the nuclear interior, thus providing the energy to drive nuclear import.
FIGURE 13-35 Mechanism for nuclear import of proteins. In the cytoplasm (top), free importin binds to the NLS of a cargo protein, and the importin-cargo complex thus formed diffuses through the NPC by transiently interacting with FG-nucleoporins. In the
nucleoplasm, Ran⋅GTP binds to the importin, causing a conformational change that decreases the importin’s affinity for the NLS and releasing the cargo. To support another cycle of import, the importin-Ran⋅GTP complex is transported back to the cytoplasm. A GTPase-activating protein (GAP) associated with the cytoplasmic filaments of the NPC stimulates Ran to hydrolyze the bound GTP. This generates a conformational change that causes Ran to dissociate from the nuclear transport receptor, which can then initiate another round of import. Ran⋅GDP is returned to the nucleoplasm, where a guanine nucleotide exchange factor (GEF) causes release of GDP and rebinding of GTP. Description The illustration shows F G-nucleoporins in two N P Cs in the nuclear membrane separating cytoplasm from nucleoplasm. In cytoplasm, importin binds to cargo having N E S to form a complex that enters nucleoplasm via N P C. In nucleoplasm, Ran-G D P is converted to Ran-G T P by G E F and this Ran-G T P binds to importin as cargo is released. The Ran-G T P and importin complex leaves nucleoplasm and enters cytoplasm via N P C. In cytoplasm, Ran-G T P hydrolyzes by GAP which converts H 2 o to P i and thus separating importin from now formed Ran-G D P. Although the importin-cargo complex travels through the pore by random diffusion, the overall process of transport of cargo into the nucleus is unidirectional. Because of the rapid dissociation of the complex when it reaches the nucleoplasm, there is a concentration gradient of importincargo complex across the NPC: high in the cytoplasm, where the complex assembles, and low in the nucleoplasm, where it dissociates. This concentration gradient is responsible for the unidirectional nature of nuclear import. A similar concentration gradient is responsible for driving importin from the nucleus back into the cytoplasm. The concentration of the importin-Ran·GTP complex is higher in the nucleoplasm, where it assembles, than on the cytoplasmic side of the NPC, where it dissociates. Ultimately, the direction of the transport processes depends on the
A Second Type of Nuclear Transport Receptor Escorts Proteins Containing Nuclear-Export Signals out of the Nucleus
localization of the Ran-GEF predominantly in the nucleoplasm and the Ran-GAP predominantly in the cytoplasm. Ran-GEF in the nucleoplasm maintains Ran in the Ran·GTP state, where it promotes dissociation of the cargo complex. Ran-GAP on the cytoplasmic side of the NPC converts Ran·GTP to Ran·GDP, dissociating the importin-Ran·GTP complex and releasing free importin into the cytosol. A Second Type of Nuclear Transport Receptor Escorts Proteins Containing Nuclear-Export Signals out of the Nucleus A mechanism very similar to the one we have just described is used to export proteins, tRNAs, and ribosomal subunits from the nucleus to the cytoplasm. This mechanism was initially elucidated by studies of certain ribonuclear protein complexes that shuttle between the nucleus and the cytoplasm. Such shuttling proteins contain a nuclear-export signal (NES) that stimulates their export from the nucleus to the cytoplasm through nuclear pores, in addition to an NLS that results in their uptake into the nucleus. Experiments with engineered hybrid genes encoding a nucleusrestricted protein fused to various segments of a shuttling protein have identified at least three different types of NESs: a leucine-rich sequence found in PKI (an inhibitor of protein kinase A) and in the Rev protein of human immunodeficiency virus (HIV), and two other sequences identified in two different heterogeneous ribonucleoprotein particles (hnRNPs). The
precise structural features that determine the recognition of each type of sequence for nuclear export remain poorly understood. The mechanism whereby shuttling proteins are exported from the nucleus is best understood for those containing a leucine-rich NES. According to the current model, shown in Figure 13-36a, a specific nuclear transport receptor, called exportin 1, first forms a complex with in the nucleus and then binds the NES in a cargo protein. Binding of exportin 1 to causes a conformational change in exportin 1 that increases its affinity for the NES, so that a trimolecular cargo complex is formed. Like other nuclear transport receptors, exportin 1 interacts transiently with FG-repeats in FG-nucleoporins and diffuses through the NPC. The cargo complex dissociates when it encounters the Ran-GAP associated with the NPC cytoplasmic filaments, which stimulates Ran to hydrolyze the bound GTP, shifting it into a conformation that has low affinity for exportin 1. After dissociates from the trimolecular cargo complex, exportin 1 changes its conformation to one that has low affinity for the NES, releasing the cargo into the cytosol. The direction of the export process is driven by this dissociation of the cargo from exportin 1 in the cytoplasm, which causes a concentration gradient of the cargo complex across the NPC that is high in the nucleoplasm and low in the cytoplasm. Exportin 1 and are then transported back into the nucleus through the NPC.
FIGURE 13-36 Ran-dependent and Ran-independent nuclear export. (a) Ran-dependent mechanism for nuclear export of cargo proteins containing a leucine-rich nuclear-export signal (NES). In the nucleoplasm (bottom), the protein exportin 1 binds cooperatively to the NES of the cargo protein to be transported and to . After the resulting cargo complex diffuses through an NPC via transient interactions with FG-repeats in FGnucleoporins, the GAP associated with the NPC cytoplasmic filaments stimulates GTP hydrolysis, converting to . The accompanying conformational change in Ran leads to dissociation of the complex. The NES-containing cargo protein is released into the cytosol, whereas exportin 1 and are transported back into the nucleus through an NPC. Ran-GEF in the nucleoplasm then stimulates conversion of to . (b) Ran-independent nuclear export of mRNAs. The heterodimeric complex binds to mRNA-protein complexes (mRNPs) in the nucleus. acts as a nuclear transport receptor and directs the associated mRNP to the central channel of the NPC by transiently interacting with FG-nucleoporins. An RNA helicase (Dbp5) located on the cytoplasmic side of the NPC removes NXF1 and NXT1 from the mRNA in a reaction that is powered by ATP hydrolysis. Free NXF1 and NXT1 proteins are recycled back into the nucleus by the Ran-dependent import process depicted in Figure 13-35. Description Both series show two N P Cs in the nuclear membrane separating cytoplasm from nucleoplasm. Part (a) Ran-dependent nuclear transport shows an exportin 1 and Ran-G D P present in cytoplasm enters nucleoplasm through one N P C and Ran-G D P gets converted to Ran-G T P by G E F. The exportin 1 and Ran-G T P binds to cargo having N E S to form cargo complex which is transported to cytoplasm through another N P C. In cytoplasm, the Ran-G T P in cargo complex hydrolysis, separating all three complexes. Part (b) Ran-independent nuclear transport shows N X F 1 and N X T 1 in the nucleoplasm where they assemble or bind on processed m R N A having 5-prime capped end and poly (A) tail. This structure moves from nucleoplasm to cytosol as D b p 5 bound to N P C hydrolysis A T P to A D P. In cytoplasm, N X F 1 and N X T 1 are separated from m R N A and m R N P remodeling occurs resulting in a cellular m R N A bound to two ribosomes.
By comparing this model for nuclear export with that in Figure 13-35 for nuclear import, we can see one obvious difference: is part of the cargo complex during export, but not during import. Apart from this difference, the two transport processes are remarkably similar. In both processes, association of a nuclear transport receptor with in the nucleoplasm causes a conformational change that affects its affinity for the transport signal. During import, the interaction causes release of the cargo, whereas during export, the interaction promotes association with the cargo. In both export and import, stimulation of hydrolysis in the cytoplasm by Ran-GAP produces a conformational change in Ran that releases the nuclear transport receptor. During nuclear export, the cargo is also released. Localization of Ran-GAP and Ran-GEF to the cytoplasm and nucleus, respectively, is the basis for the unidirectional import and export of cargo proteins across the NPC. In keeping with their similarity in function, the two types of nuclear transport receptors — importins and exportins — are highly homologous in sequence and structure. The family of nuclear transport receptors has 14 members in yeast and more than 20 in mammalian cells. The NESs or NLSs to which they bind have been determined for only a fraction of them. Some individual nuclear transport receptors function in both import and export. A similar shuttling mechanism has been shown to export other cargoes from the nucleus. For example, exportin-t functions to export tRNAs. Exportin-t binds fully processed tRNAs in a complex with that diffuses through NPCs and dissociates when it interacts with Ran-GAP in
Most mRNAs Are Exported from the Nucleus by a Ran-Independent Mechanism
the NPC cytoplasmic filaments, releasing the tRNA into the cytosol. A Ran-dependent process is also required for the nuclear export of ribosomal subunits through NPCs once the protein and RNA components have been properly assembled in the nucleolus. Likewise, certain specific mRNAs that associate with particular hnRNP proteins can be exported by a Randependent mechanism. Most mRNAs Are Exported from the Nucleus by a Ran-Independent Mechanism Once the processing of an mRNA is completed in the nucleus, it remains associated with specific proteins in a messenger ribonuclear protein complex, or mRNP. The principal transporter of mRNPs out of the nucleus is the mRNP exporter, a heterodimeric protein composed of a large subunit called nuclear export factor 1 (NXF1) and a small subunit called nuclear export transporter 1 (NXT1). Multiple dimers bind to nuclear mRNPs through cooperative interactions with the RNA and other mRNP adapter proteins that associate with nascent pre-mRNAs during transcriptional elongation and pre-mRNA processing. This process adds a degree of quality control to mRNA export since dimers do not assemble onto the mRNP until it has all of the attributes of a properly and completely processed mRNA. Once incorporated into an mRNP, the subunits of in many respects act like a nuclear transport receptor that binds to an NLS or NES, since both NXF1 and
NXT1 interact with the FG-repeats of FG-nucleoporins, and this interaction allows them to diffuse through the central channel of the NPC. The process of mRNP export does not require Ran, and thus the unidirectional transport of mRNA out of the nucleus requires a source of energy other than GTP hydrolysis by Ran. Once the mRNPcomplex reaches the cytoplasmic side of the NPC, NXF1, and NXT1 dissociate from the mRNP with the help of the RNA helicase Dbp5, which is associated with cytoplasmic NPC filaments. Recall that RNA helicases use the energy derived from hydrolysis of ATP to move along RNA molecules, separating double-stranded RNA chains and dissociating RNAprotein complexes (see Chapter 5). This leads to the simple idea that Dpb5, which is associated with the cytoplasmic side of the nuclear pore complex, acts as an ATP-driven motor to remove from the mRNP complexes as they emerge on the cytoplasmic side of the NPC. The portions of an mRNP that have emerged into the cytosol and have been stripped of lack the ability to interact with FG-repeat nucleoporins. Thus an extended mRNP that transits the NPC cannot slide back into the nucleus, and the process of energy dependent removal of by Dbp5 helicase causes the mRNP to be progressively ratcheted into the cytoplasm. After being removed from the mRNP, the free NXF1 and NXT1 subunits are imported back into the nucleus by a process that depends on Ran and a nuclear transport receptor (Figure 1336b). KEY CONCEPTS OF SECTION 13.6
Transport into and out of the Nucleus The nuclear envelope contains numerous nuclear pore complexes (NPCs), which are large, complex structures composed of multiple copies of 30 different proteins called nucleoporins (see Figure 13-32). FG-nucleoporins, which contain multiple repeats of a short hydrophobic sequence (FG-repeats), fill the central transporter channel with a gel-like matrix that allows small molecules to pass but excludes macromolecules larger than 40 kDa. Active transport plays a role in the transport of all large proteins and RNAs through nuclear pores. This process requires the assistance of nuclear transport receptors that interact with both the transported molecule and FG-repeats of FG-nucleoporins. Proteins imported into or exported from the nucleus contain a specific amino acid sequence that functions as a nuclear-localization signal (NLS) or a nuclear-export signal (NES). Nucleus-restricted proteins contain an NLS but not an NES, whereas proteins that shuttle between the nucleus and cytoplasm contain both signals. Several different types of NESs and NLSs have been identified. Each type of nucleartransport signal is thought to interact with a specific nuclear transport receptor belonging to a family of homologous proteins. A cargo protein bearing an NES or NLS translocates through nuclear pores bound to its cognate nuclear transport receptor. The transient interactions between nuclear transport receptors and FG-repeats allow very rapid diffusion of nuclear transport receptor–cargo complexes through the central channel of the NPC, which is filled with a hydrophobic matrix of FG-repeats. The unidirectional nature of protein export and import through nuclear pores results from the participation of Ran, a monomeric G protein that exists in different conformations when bound to GTP or GDP. Localization of the Ran guanine nucleotide exchange factor (Ran-GEF) in the nucleus and of the Ran GTPaseactivating protein (Ran-GAP) in the cytoplasm creates a gradient with high concentrations of in the nucleoplasm and of in the cytoplasm. The interaction of a cargo complex with in the nucleoplasm causes dissociation of the complex, releasing the cargo into the nucleoplasm (see
Figure 13-35), whereas the assembly of an export cargo complex is stimulated by interaction with in the nucleoplasm (see Figure 13-36). Most mRNPs are exported from the nucleus by binding to a heterodimeric mRNP exporter in the nucleoplasm that interacts with FG-repeats. The direction of transport (nucleus to cytoplasm) results from the action of an RNA helicase associated with the cytoplasmic filaments of the NPC that removes the heterodimeric mRNP exporter once the transport complex has reached the cytoplasm.
Key Terms
End of Chapter Visit Achieve to access study tools and to learn more about the concepts in this chapter: Perspectives for the Future Analyze the Data Chapter References Additional tools including videos, animations, and quizzes Key Terms AAA ATPase family cotranslational translocation dislocation disulfide bonds (–S–S–) dolichol phosphate ERAD (ER-associated protein degradation) FG-nucleoporin general import pore glycoprotein hydropathy profile microsome mRNP exporter multipass membrane protein N-linked oligosaccharide
Review the Concepts
nuclear pore complex (NPC) nuclear transport receptor nucleoporins O-linked oligosaccharide post-translational translocation protein disulfide isomerase (PDI) Ran rough ER secretory pathway signal-anchor sequence (SA) signal-based targeting signal recognition particle (SRP) single-pass membrane protein SRP receptor stop-transfer anchor sequence (STA) targeting sequence thylakoids topogenic sequence topology translocon unfolded-protein response vesicle-based trafficking Review the Concepts 1. The following results were obtained in early studies on the translation of secretory proteins. Based on what we now know of
this process, explain the reason why each result was observed. a. An in vitro translation system consisting only of mRNA and ribosomes resulted in secretory proteins that were larger than the identical protein when translated in a cell. b. A similar system that also included microsomes produced secretory proteins that were identical in size to those found in a cell. c. When the microsomes were added after in vitro translation, the synthesized proteins were again larger than those made in a cell. 2. Describe the source or sources of energy needed for unidirectional translocation across the membrane in (a) cotranslational translocation into the endoplasmic reticulum (ER); (b) post-translational translocation into the ER; and (c) translocation into the mitochondrial matrix. 3. Translocation into most organelles usually requires the activity of one or more cytosolic proteins. Describe the basic functions of three different cytosolic factors required for translocation into the ER, mitochondria, and peroxisomes, respectively. 4. Describe the typical principles used to identify topogenic sequences within proteins and how these principles can be used to develop computer algorithms. How does the identification of topogenic sequences lead to prediction of the membrane arrangement of a multipass protein? What is the importance of the arrangement of positive charges relative to the membrane orientation of a signal-anchor sequence? 5. An abundance of misfolded proteins in the ER can result in the activation of the unfolded-protein response (UPR) and ER-
associated degradation (ERAD) pathways. UPR decreases the abundance of unfolded proteins by altering gene expression of what type of genes? What is one manner in which ERAD may identify misfolded proteins? Why is dislocation of these misfolded proteins to the cytoplasm necessary? 6. Temperature-sensitive yeast mutants have been isolated that block each of the enzymatic steps in the synthesis of the dolichol-linked oligosaccharide precursor for N-linked glycosylation. Propose an explanation for why mutations that block synthesis of the intermediate with the structure dolicholPPcompletely prevent addition of N-linked oligosaccharide chains to secretory proteins, whereas mutations that block conversion of this intermediate into the completed precursor — dolichol-PP- — allow the addition of N-linked oligosaccharide chains to secretory glycoproteins. 7. Name four different proteins that facilitate the modification or folding of secretory proteins within the lumen of the ER. Indicate which of these proteins covalently modifies substrate proteins and which brings about only conformational changes in substrate proteins. 8. Describe what would happen to the precursor of a mitochondrial matrix protein in the following types of mitochondrial mutants: (a) a mutation in the Tom22 signal receptor; (b) a mutation in the Tom70 signal receptor; (c) a mutation in the matrix Hsp70; and (d) a mutation in the matrix signal peptidase. 9. Describe the similarities and differences between the mechanism of import into the mitochondrial matrix and the chloroplast
stroma. 10. Design a set of experiments using chimeric proteins, composed of a mitochondrial precursor protein fused to dihydrofolate reductase (DHFR), that could be used to determine how much of the precursor protein must protrude into the mitochondrial matrix in order for the matrix-targeting sequence to be cleaved by the matrix-processing protease. 11. Peroxisomes contain enzymes that use molecular oxygen to oxidize various substrates, but in the process, hydrogen peroxide — a compound that can damage DNA and proteins — is formed. What is the name of the enzyme responsible for the breakdown of hydrogen peroxide to water? What is the mechanism of the import of this protein into the peroxisome, and what other proteins are involved? 12. Suppose that you have identified a new mutant cell line that lacks functional peroxisomes. Describe how you could determine experimentally whether the mutant is primarily defective for of peroxisomal membrane proteins or matrix proteins. 13. The nuclear import of proteins larger than 40 kDa requires the presence of what amino acid sequence? Describe the mechanism of nuclear import. How are nuclear transport receptors able to get through the nuclear pore complex? 14. Why is localization of Ran-GAP in the nucleus and Ran-GEF in the cytoplasm necessary for unidirectional transport of cargo proteins containing an NES?