Introduction
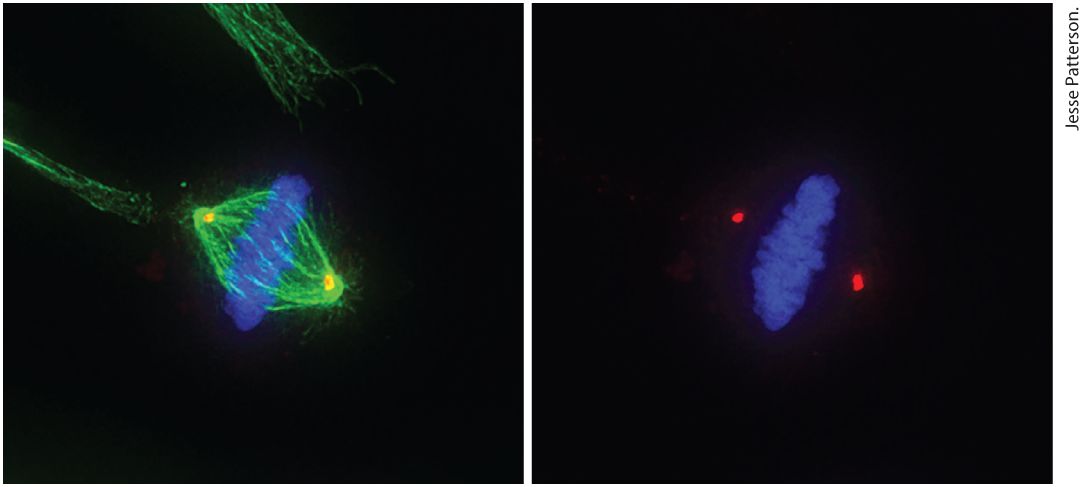
Chapter 19 The Eukaryotic Cell Cycle Micrograph on the left shows a human epithelial cell in metaphase. Following DNA replication, cells undergo mitosis to segregate their replicated chromosomes. The cell has aligned its chromosomes (blue) in the center of the mitotic spindle apparatus (green), prior to pulling them to opposite poles of the spindle, which will occur during anaphase. Thereafter, the cytoplasm of the cell is divided to produce two identical daughter cells. The micrograph on the right is the same cell without imaging the spindle apparatus in order to show the location of the centrosomes or spindle poles (red).
19.2 Model Organisms and Methods of Studying the Cell Cycle
19.3 Cell Cycle Progression and Control: Feedback Loops and Post-Translational Modification
19.4 The Transition from into S Phase and DNA Replication
19.5 The Transition and the Irreversible Engine of Mitosis
19.6 The Mitotic Spindle, Chromosome Segregation, and Exit from Mitosis
19.7 Surveillance Mechanisms in Cell Cycle Regulation
19.8 Meiosis: A Special Type of Cell Division The central tenet of life and the definition of living matter is inextricably linked to the concept of replication. The pioneering discoveries of Matthias Schleiden and Theodor Schwann, and those of Rudolf Virchow, that living matter was made of cells, and that cells arise only from the division of other cells, dictates that cells proliferate by going through repeated cycles of growth and division of a parent cell into two daughter cells. The daughter cells, in turn, repeat this process, giving rise to more cells. This behavior leads to the general concept of a cell cycle that incorporates each of the stages required for cells to replicate themselves: growth in size and nutrient content; duplication of subcellular organelles; DNA replication; chromosome segregation; and finally, separation of the cytoplasmic organelles, contents, and cell membrane between the two progeny. These steps in the cell cycle occur in distinct phases, and the transition from one phase to the next is abrupt and precise. Errors that
occur within one phase must be detected and corrected before the cell transits into the next phase. Surveillance mechanisms prevent the transition from one phase of the cell cycle into the next phase until all of the steps of a phase have been completed properly. It is through this process of repeated cell cycles that a fertilized egg — one cell — can eventually give rise to a fully formed animal containing hundreds of millions of cells or more. Proper control of cell division is vital to all organisms. First, cell division must be balanced with cell growth so that cell size is maintained. If several divisions occur before parent cells have reached the proper size, daughter cells eventually become too small to be viable. If cells grow too large before cell division, the cells function improperly and the number of cells increases too slowly for proper tissue, organ, and animal development during embryogenesis. Second, in developing multicellular organisms, the replication of each cell must be precisely controlled and timed with respect to the replication of other cells in order to faithfully and reproducibly complete the developmental program in every individual in order for complex organs (e.g., the brain, heart, intestine, and kidney) that are made of various cell types to develop properly. Finally, there is a balance between cell replication and cell differentiation, so that cells with high replicative potential are generally much less differentiated into specific cell types, while cells that have highly differentiated into specific lineages usually have quite limited replicative potential. Cells that have the potential to continuously replicate and have not yet differentiated (or have only partially differentiated) but have the ability to differentiate into one or more specific cell types at a later time are called stem cells. During
embryogenesis in animals, for example, there is a period of rapid cell division after fertilization of the egg, resulting in a small number of cells that have nearly unlimited replicative potential but have not yet undergone any type of differentiation (see Chapter 22). Their progeny, however, will later differentiate into every type of cell in the body. In this chapter, we first present an overview of the cell cycle and then describe the various experimental systems that have contributed to our current understanding of it. We discuss the molecular mechanisms responsible for regulating cell cycle progression, particularly the roles of cyclin-dependent kinases (CDKs) as master controllers, along with other protein kinases, phosphatases, phospho-binding modules, and ubiquitin ligases that modulate their activity and function. Next we examine each cell cycle phase in greater detail, with an emphasis on how the events that take place in each phase are governed by protein post-translational modifications, CDK activity, and regulation of downstream targets. We discuss the checkpoint pathways that establish the order of the cell cycle and ensure that each cell cycle phase has been properly and accurately completed prior to transition into the next phase. The chapter concludes with a discussion of meiosis, a special type of cell division that generates haploid germ cells (eggs and sperm), and the molecular mechanisms that distinguish it from mitosis. Throughout the chapter, we emphasize the general principles governing cell cycle progression and use a speciesspanning nomenclature when discussing examples of the factors controlling each cell cycle phase.
19.1 Overview of the Cell Cycle
19.1 Overview of the Cell Cycle The cell cycle in eukaryotes is a series of processes that occur in a specific sequence every time a cell divides. In cells that are continuously replicating, each process occurs on a schedule, as though the cells were following some type of molecular clock. This clock idea was part of the motivation that eventually led to the discovery of cyclin-CDK complexes as master regulators of the cell division cycle. The cell cycle is divided into four major phases (Figure 19-1). Cycling (replicating) mammalian somatic cells grow in size and synthesize the RNAs and proteins required for DNA synthesis during the (first gap) phase. When cells have reached the appropriate size and have synthesized the required proteins, they enter the cell cycle by traversing a point in known as START in yeast and the restriction point in mammals. Once this point has been crossed, cells are committed to cell division. The first step toward successful cell division is entry into the S (synthesis) phase, the period in which cells replicate their chromosomes. After progressing through a second gap phase, the phase, cells begin the complicated process of mitosis, also called the M (mitotic) phase.
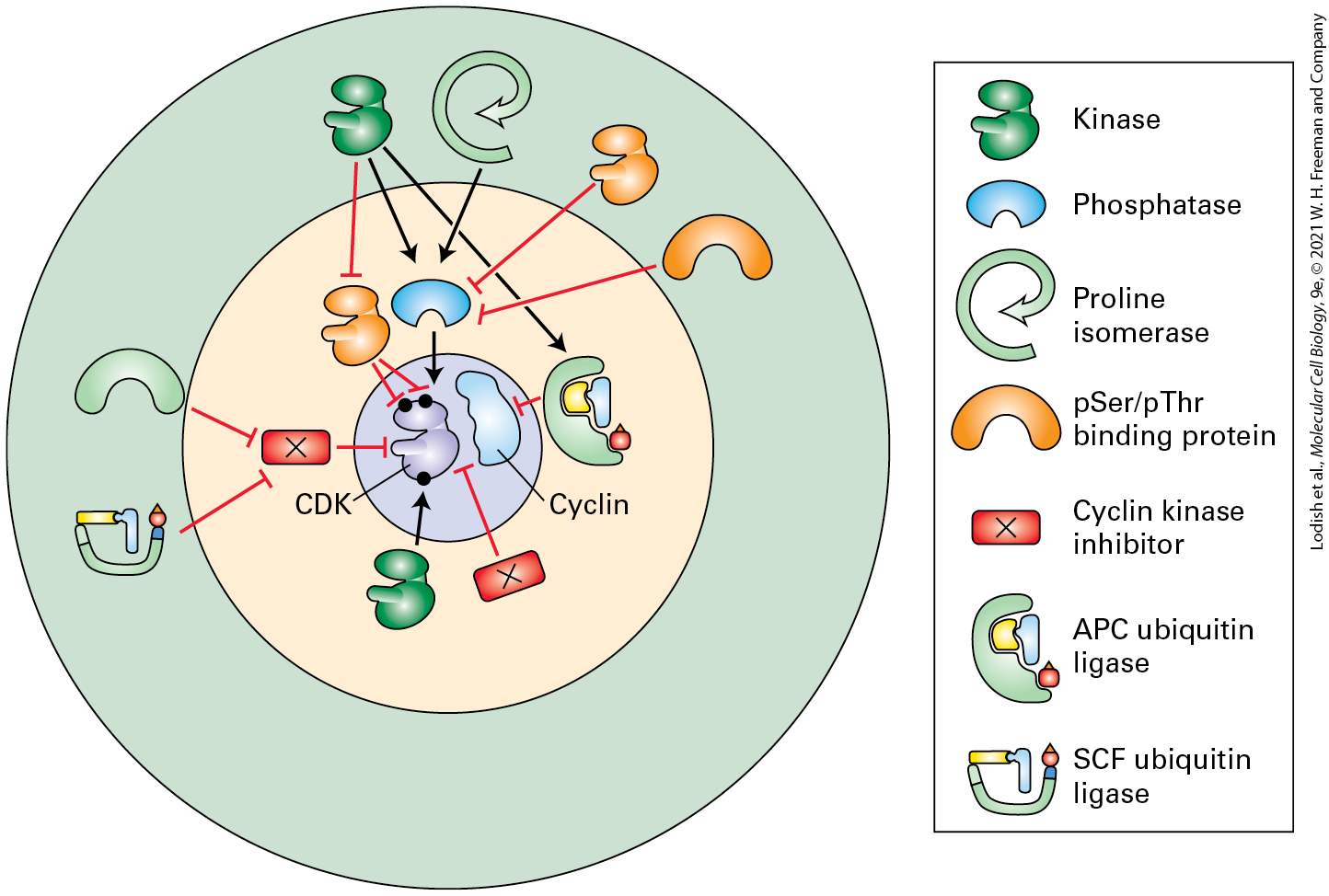
FIGURE 19-1 The eukaryotic cell cycle. The fate of a single chromosome during different stages of the cell cycle is depicted. Following mitosis (M), daughter cells contain 2n chromosomes in diploid organisms and 1n chromosomes in haploid organisms. In proliferating cells, is the period between the “birth” of a cell following mitosis and the initiation of DNA synthesis, which marks the beginning of the S phase. The point in late when cells become committed to enter S phase is called START or the restriction point. At the end of the S phase, cells enter containing twice the number of chromosomes they had as cells (4n in diploid organisms, 2n in haploid organisms). The end of is
marked by the onset of mitosis, during which numerous events leading to cell division occur. The , S, and phases are collectively referred to as interphase: the period between one mitosis and the next. Most nonproliferating cells in vertebrates leave the cell cycle in , entering the state. Although chromosomes condense only during mitosis, here they are shown in condensed form throughout the cell cycle to emphasize the number of chromosomes at each stage. The nuclear envelope surrounds the chromosomes during interphase in all cells but breaks down during mitosis in metazoan cells, but not in yeast. Description The illustration of the cell cycle starts at START or restriction point. A curved arrow labeled S D N A synthesis points at a cell having a chromosome with two chromatids. A curved arrow labeled G subscript 2 from this cell points to a cell undergoing mitosis. The cell is labeled M. The chromosome inside the cell is split to move to the end of the poles with the help of microtubules. Two arrows from this cell points towards two newly formed daughter cells with a homologous chromosome, respectively. A curved arrow labeled G subscript 1 points at a cell with a chromatid from which the cycle repeats. It has a small looped arrow along it labeled G subscript 0. A dot on this arrow is labeled start slash restriction point. Based on studying cell cycles in budding yeast, Lee Hartwell proposed that cells use specific surveillance mechanisms to ensure that each stage of the cell cycle is properly completed before cells enter the next stage. These surveillance programs establish checkpoints at the border between the different cell cycle phases. Cells must pass through the checkpoint, for example before they can enter S phase and begin performing DNA synthesis, and they must pass through the checkpoint before they can enter mitosis. The progression of cell cycle stages from to S to to M is the same for all eukaryotes, though the time it takes to complete one turn of the cycle varies considerably among organisms. Rapidly replicating human cells progress through the full cell cycle in about 24
G1 Controls Entry into S Phase
hours: takes 9 hours; the S phase, 10 hours; , 4.5 hours; and mitosis, 30 minutes. In contrast, the full cycle takes only 90 minutes in rapidly growing yeast cells. The cell divisions that take place during early embryonic development of the fruit fly Drosophila melanogaster are completed in as little as 8 minutes! Controls Entry into S Phase If the purpose of S phase is to duplicate the chromosomal DNA, and the purpose of M phase is to segregate the chromosomes to the daughter cells, what are and for? During , a cell must evaluate its status and decide whether it is appropriate to commit to doubling and cell division. This assessment includes an evaluation of the cell’s size, nutrient status, substrate attachment, density of neighboring cells, and the presence of extracellular growth factors and other chemicals that stimulate cell division. Growth factors and chemicals that promote cells to transit through the cell cycle and divide are generally referred to as mitogens. Specific signaling pathways monitor the state of the cell and its environment, and the outputs of these pathways are integrated to control the decision whether to replicate and divide. The TOR and Hippo signaling pathways, discussed in Chapter 21, monitor a cell’s nutrient status, size, attachment status, and the density of surrounding cells. Growth factor signal transduction pathways, the phosphoinositol-3 kinase pathway, and the mitogen-activated protein kinase (MAPK) pathway discussed in
Chapter 16, all report on whether mitogens are in the immediate vicinity of the cell.
Once cells commit to replicating their DNA, the decision is irreversible. That is, once cells enter S phase, they must complete the process of duplicating all of their chromosomes regardless of whether growth factors or nutrients are removed from the extracellular environment. The START or restriction point where cells commit to entering S phase occurs late in ; in the case of mammalian cells this point is 2–3 hours prior to S phase onset. The observation that growth factors could be removed after mammalian cells had passed the restriction point (or that nutrients could be removed or mating pheromones that normally arrest yeast cells in could be added after yeast cells had passed START) and the cells would still progress into S phase led scientists to posit that some mysterious factor was accumulating during , and that once enough of this labile “Rfactor” had accumulated, the cells were destined to begin DNA replication. When S phase was nearly completed, this factor would be destroyed, and would have to be re-synthesized during of the next cell cycle. This cyclic accumulation and degradation of R-factor could explain why cells only synthesized DNA during S phase, and how cells controlled the timing of S phase entry by regulating the levels of R-factor during , based on nutrients, mitogens, cell size, density, and substrate attachment. In mammalian cells, we now know that this cycling R-factor is a protein known as cyclin D (likely working in a feedback loop with another protein, cyclin E), and in yeast this factor turned out to be Cln3 working through a feedback loop involving Cln1, Cln2, and a transcriptional repressor called Whi5. The term cyclin or its abbreviated form, Cln, captures the central concept that this labile regulatory factor cycles in abundance as the cells go through the different stages of cell duplication. As we shall see, all of the cyclin proteins play critical roles in controlling the parts of the cell
G2 Phase Readies the Cell for Mitosis and Cell Division
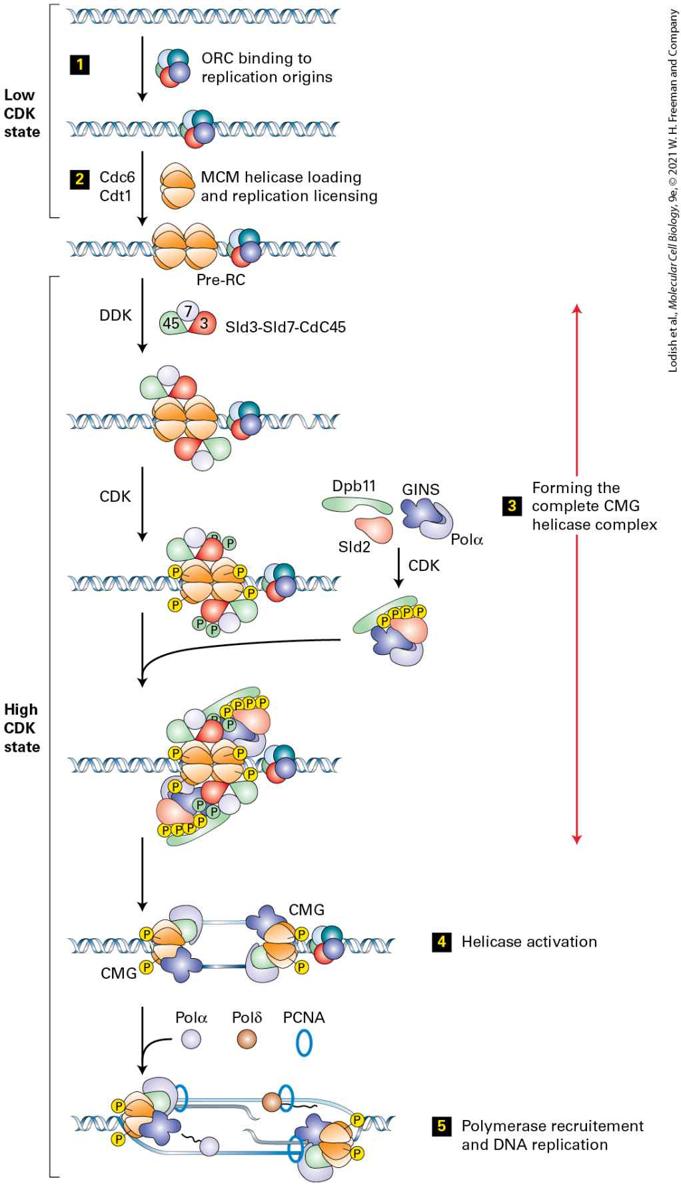
cycle. Cyclin proteins function at the molecular level by binding to one or more specific protein kinases — the cyclin-dependent kinases (CDKs) mentioned in the introduction — in order to increase the kinase’s activity, change the kinase substrate specificity, and localize the kinase to specific parts of the cell. Once in S phase, complex replication machinery is assembled at specific sites on the chromosomal DNA, at the origins of replication and precise duplication of each chromosome ensues. The centrosome, the main microtubule organizing center in animal cells (see Chapter 18) also duplicates during S phase. Once DNA replication has been completed, cells enter , the second gap phase. Phase Readies the Cell for Mitosis and Cell Division During the phase, the cell verifies that all of the DNA has been correctly duplicated and is sufficient for producing two cells, errors that were made during DNA copying are corrected, and any breaks in the DNA strands are repaired. In addition, the first signs of chromosome condensation occur, and there is some early reorganization of the cytoskeleton and microtubules as the cell prepares to enter mitosis. How does the cell know during when everything is ready for mitosis? As with the transition from into S phase, another labile cyclin protein accumulates during , which when present in sufficient amounts is able
to drive the cells from into mitosis. In a classic experiment, Tim Hunt added radioactive methionine to fertilized sea urchin eggs and noticed the gradual accumulation of a radiolabeled protein that was precipitously destroyed in mitosis. Work done in a variety of different organisms — budding yeast, frog eggs, sea urchin eggs, sea star, and surf clam embryos — expanded on this observation, and eventually led scientists to clone and characterize this protein, which we now know as cyclin B. The binding of cyclin B to another cyclin-dependent kinase, called CDK1, forms an active protein kinase complex that drives the entry of cells into M phase. Surveillance mechanisms at the checkpoint ensure that cells do not progress from into M phase in the presence of cell stress and damage, such as that caused by radiation, DNA-damaging agents, or drugs that interfere with microtubule polymerization. Intriguingly, certain organisms, including budding yeast (but not fission yeast), lack a discernable phase altogether. In other model organisms that have been used to study the cell cycle, such as the frog Xenopus laevis and the fruit fly Drosophila melanogaster, both the and phases appear to be absent during several of the rapid early embryonic cell cycles that occur when the egg first begins to divide, prior to the onset of development. In these cases, the cells actually divide without growth, making the cells progressively smaller in size. Nonetheless, later cell cycles in these organisms, and most cell cycles in adult organisms in general, require growth during and the establishment of a phase between S phase and M phase. Additional accumulation of cell mass including lipid and protein synthesis and mitochondrial division occurs in , while Golgi complex duplication occurs in late , S phase, and .
Mitosis and Cytokinesis Occur During M-Phase
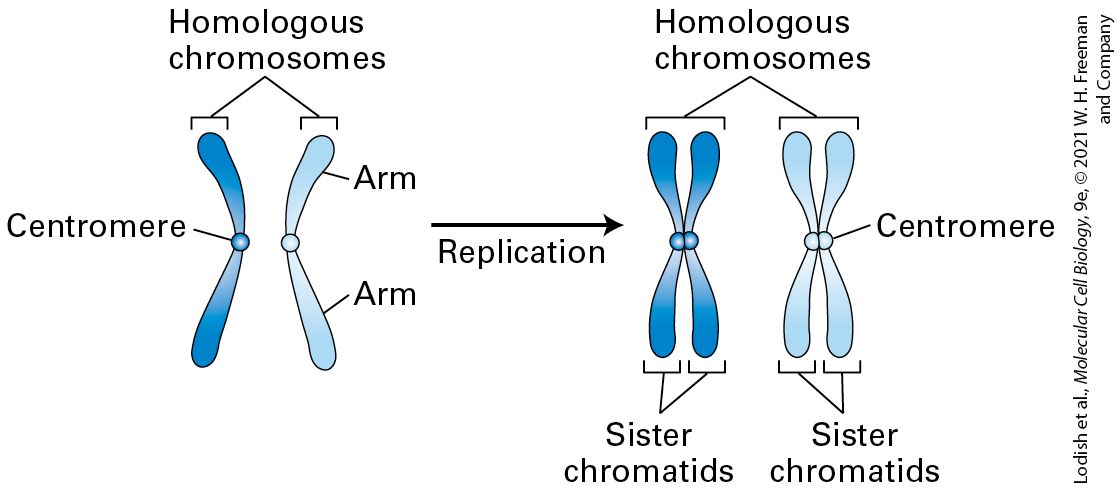
Mitosis and Cytokinesis Occur During M-Phase At the completion of S and , the DNA of each chromosome has been duplicated through the process of DNA replication, generating two identical DNA molecules, each of which is also coated with histones and other chromosome-associated proteins (see Figure 8-35). Each of the identical chromosomes is now referred to as a chromatid (Figure 19-2). The two sister chromatids are initially held tightly together all along their length by a protein complex called cohesin. The chromatid attachment points on mitotic chromosomes are densest at a knobby constricted central region of each chromosome called the centromere, which contains many repeated copies of certain DNA sequences. The parts of the chromatid on either side of the centromere are referred to as arms. During mitosis, the cohesin links between the two sister chromatids are dissolved, and the sister chromatids are separated from each other along a proteinaceous spindle made primarily from microtubules; the chromatids ultimately are distributed into each of the two daughter cells.
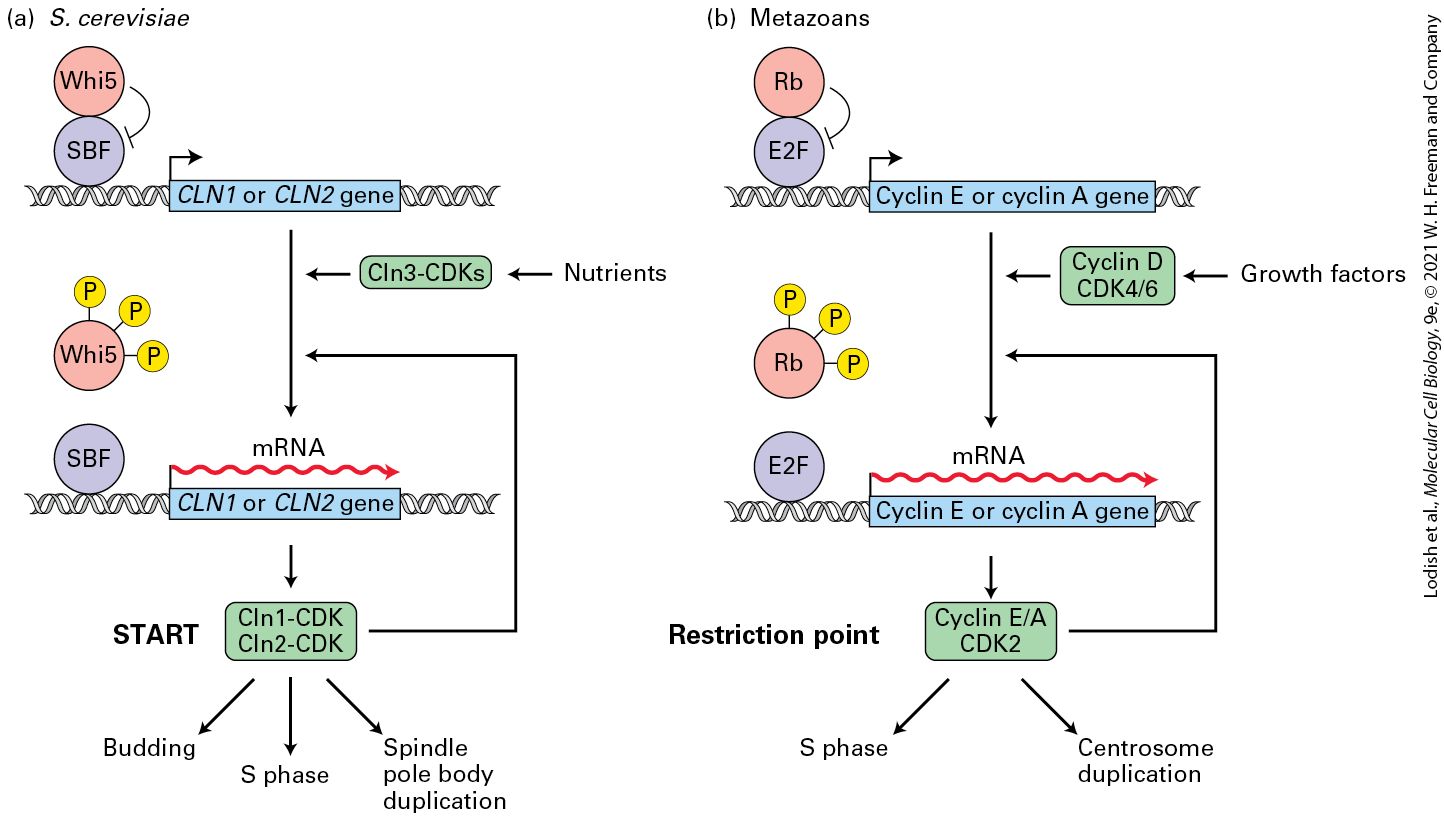
FIGURE 19-2 Duplication of chromosomes during S phase. Diploid cells contain two of each chromosome, referred to as chromosome homologs. At the end of S phase each homologous chromosome has two copies of itself, each copy of which is called a sister chromatid. The centromere and chromosome arms are indicated. For simplicity, the cohesin protein that holds the two sister chromatids together is not shown. Description The illustration shows two homologous chromosomes each with centromere and arms labeled. An arrow labeled replication points at two homologous chromosomes having two centromeres and the label sister chromatids respectively. The process of separating the two copies of the chromosome is rather elaborate and occurs in a series of six stages that can be recognized when dividing cells are viewed under the microscope, as described in Chapter 18. These stages are summarized in Figure 19-3. To reiterate the main events that occur at each stage: during prophase the chromatin fibers in the nucleus condense into the discrete chromosomes that can be visualized under the light microscope. The cohesin protein that connects the arms of the duplicated chromosomes to each other from S phase is degraded,
leaving the two sister chromatids connected to each other only through cohesin bridges near the centromere. Nucleoli, the sites in the nucleus where ribosome synthesis occurs, disappear. The interphase array of microtubules breaks down, and the centrosomes (which are sometimes now called mitotic asters or spindle poles) move away from each other as a mitotic spindle begins to form between them. A complex of proteins called the kinetochore, which will anchor each chromosome to some of the spindle microtubules, assembles at the centromere of each chromosome.
FIGURE 19-3 The stages of mitosis. The top panels show a diagrammatic representation of key events that occur in each stage of mitosis, the relatively short period of the cell cycle that follows the S and phases. The bottom panels are micrographs of dividing cells with the DNA stained blue and the microtubules stained green. Below are listed the major events that occur in each stage. Description
Prophase: The illustration shows a cell with an oval structure inside. The oval structure has several worm shaped structures inside. There are two tiny spheres from which thin microtubules extend to cover the cell. The corresponding micrograph shows a blue colored oval structure in the center with a web of green lines extending from the poles around the oval structure. A text below reads: Chromosomes condense and become visible, centrosomes move toward opposite poles and spindle begins to form, cohesin is shed from chromosome arms, and kinetochore assembles on centromere. Prometaphase: The illustration shows a cell with two tiny spheres at the opposite end of its poles with microtubules extending in all directions. The microtubules attached to chromosomes move it to the center of the cell. The corresponding micrograph shows a broken blue structure with large green spots on it from which thin green lines extend outwards. A text below reads: Chromosomes continue to condense, Mitotic spindle microtubules attach to kinetochores, and nuclear envelope breaks down. Metaphase: The illustration shows a cell with two tiny spheres at the opposite end of its poles with microtubules extending in all directions. The microtubules align the chromosomes at the center of the cell. The corresponding micrograph shows a blue colored structure in the center with green lines splaying at the poles of the cell. A text below reads: Chromosomes are lined up at the metaphase plate and each sister chromatid is attached to a spindle fiber originating from opposite poles. Anaphase: The illustration shows a cell with spindles as green lines extending from tiny spheres at the poles holding chromatids to move to their respective poles. The corresponding micrograph shows a blue center area with green spindles on each side. A text below reads: sister chromatids are pulled apart toward opposite poles and certain spindle fibers begin to elongate the spindle. Telophase: The illustration shows a cell with a red band around the center of the cell with the two areas separated. The cell is similar to the anaphase cell. The corresponding micrograph shows separate groups of blue structures and green lines extending over it. A text below reads: Chromosomes arrive at opposite poles and begin to decondense, nuclear envelope material surrounds each set of chromosomes, mitotic spindle breaks down, contractile ring assembles under plasma membrane, and nucleoli reappear. Cytokinesis: The illustration shows the red band almost pinching of the cell. Two circular structures form one on each side covered by microtubules. The corresponding
micrograph shows an area of green spindles in two groups at the center and blue structure forming on each side. A text below reads: In animal cells, a cleavage furrow separates the daughter cells, and in plant cells, a cell plate (precursor to a new cell wall) separates the daughter cells. Next, during prometaphase in higher eukaryotes, the nuclear membrane, the outer part of which was previously contiguous with the endoplasmic reticulum, begins to break down into vesicles and retract back into the endoplasmic reticulum, and the Golgi apparatus breaks down into vesicles. This allows the microtubules emanating from each centrosome to penetrate into the nuclear area and become captured by the kinetochores, becoming kinetochore microtubules. The chromosomes become even more condensed and now appear as the classic thick X-shaped mitotic chromosomes composed of paired sister chromatids (see Figure 1-17). Non-kinetochore microtubules reach across the cell to overlap with those arising from the other centrosome, while a second set of microtubules that face toward the cell membrane and away from the mitotic chromosomes becomes apparent. Following capture of the chromosomes by attachment of their kinetochores to microtubules, the chromosomes begin to move to and fro, with a net movement toward the center of the mitotic spindle. When the centrosomes have moved to the opposite ends of the cell and the chromosomes have congressed to the middle of the spindle, the cell has entered metaphase. At this point the centromeres at the center of each chromosome have aligned along an imaginary plane midway between the spindle poles called the metaphase plate. The kinetochores of the sister chromatids in each chromosome should now be attached to microtubules
coming from opposite poles. Once this correct series of attachments has been confirmed, the cell enters anaphase. During anaphase A, the cohesin glue that held the sister chromatids together at the kinetochores is dissolved; the individual sister chromatids separate from one another and move toward opposite poles of the spindle as the kinetochore microtubules shorten at a rate of about 1 μm/min. During anaphase B, the nonkinetochore microtubules lengthen, causing the cell to elongate as the spindle poles separate further. Once the chromosomes have fully separated to opposite ends of the cell, the cell enters telophase. The chromosomes decondense, the nuclear envelope reforms, nucleoli reappear, any remaining spindle microtubules disassemble, and in animal cells, an actin/myosin–based contractile ring forms just under the plasma membrane in the region of the former spindle midzone. The next step in cell division is cytokinesis, when the cytoplasm becomes divided between the two daughter cells. Historically, cytokinesis was considered a separate process from mitosis, but in this book we follow the current convention to include cytokinesis as the terminal stage of mitosis. Cytokinesis begins in late anaphase or telophase. In animal cells, it involves the formation of a cleavage furrow by narrowing of the contractile ring and severing of any remaining microtubules in the region between where the two daughter cells will separate. The ring slowly closes until the plasma membrane pinches off, forming two discrete cells. The process is somewhat different in yeast, since they must also synthesize a new carbohydrate-based cell wall called the septum at the same time that the actomyosin ring is contracting. In either case — animals or yeast, the end result of mitosis is the same — two daughter cells each containing a nucleus and cytoplasm
with a share of the organelles, membranes, and other structures that were present in the mother cell. All of the stages of mitosis are choreographed by the actions of mitotic protein kinases. As we shall see, cyclin B/CDK1 plays one of the most important roles in controlling the entry into mitosis and in orchestrating many of the molecular events that occur up until the onset of anaphase, when cyclin B is rapidly destroyed. A variety of other protein kinases, including Polo-like kinase, Aurora A and B kinases, and the Nek family of kinases, also play very important roles during the mitotic process. Once again, surveillance mechanisms ensure that each stage of mitosis has been completed properly before moving into the next stage. This is particularly evident at the transition from metaphase to anaphase, where the spindle assembly checkpoint monitors whether each of the chromosomes is properly positioned at the metaphase plate and whether the kinetochores of each sister chromatid are properly attached to microtubules arising from opposite spindle poles. At the completion of mitosis, the daughter cells can continue cycling or can exit the cell cycle and proceed down a pathway of differentiation. In multicellular organisms, most differentiating cells exit the cell cycle and survive for days, weeks, or in some cases (e.g., nerve cells and cells of the eye lens) even the lifetime of the organism without dividing again. Such postmitotic cells generally exit the cell cycle in and enter a phase called (see Figure 19-1). Some cells can return to the cell cycle and resume replicating; this re-entry is regulated, thereby providing control of cell proliferation.
KEY CONCEPTS OF SECTION 19.1 Overview of the Cell Cycle The eukaryotic cell cycle is divided into four phases: (the period between mitosis and the initiation of nuclear DNA replication), S (the period of nuclear DNA replication), (the period between the completion of nuclear DNA replication and mitosis), and M (mitosis and cytokinesis). Cells commit to a new cell division at a specific point in known as START or the restriction point. This commitment involves the integration of multiple signaling pathways that monitor cell size, nutrient status, and the conditions of the extracellular environment. Mitogens are growth factors and other molecules that induce cells to divide. Labile factors called cyclins drive the progression of cells through different stages of the cell cycle by binding to cyclin-dependent protein kinases (CDKs). The resulting cyclin-CDK complexes are present and active only in the cell cycle stage that they promote. Surveillance mechanisms, called checkpoint pathways, guarantee that each cell cycle step is completed correctly before the next one is initiated.
Budding and Fission Yeasts Are Powerful Systems for Genetic Analysis of the Cell Cycle
19.2 Model Organisms and Methods of Studying the Cell Cycle Unraveling the molecular mechanisms governing cell cycle progression in eukaryotes was remarkably rapid and was fueled by a powerful combination of genetic and biochemical approaches. In this section, we discuss the contributions of several model systems to the discovery of the molecular mechanisms of cell division. Three of the most important systems employed to study the cell cycle are the single-celled yeasts Saccharomyces cerevisiae (budding yeast) and Schizosaccharomyces pombe (fission yeast) and the oocytes and early embryos of the frog Xenopus laevis. Studies using mammalian tissue culture cells led to the characterization of cell cycle control in mammals. Studies of the cell cycle in many different experimental systems also led to two remarkable discoveries about general control of the cell cycle. First, complex molecular processes such as initiation of DNA replication and entry into mitosis are all regulated and coordinated by a small number of master cell cycle regulatory proteins. Second, these master regulators and the proteins that control them are highly conserved, so that cell cycle studies in fungi, sea urchins, insects, frogs, and other species are directly applicable to all eukaryotic cells, including human cells.
Budding and Fission Yeasts Are Powerful Systems for Genetic Analysis of the Cell Cycle Budding and fission yeasts have proved to be extraordinarily valuable systems for the study of the cell cycle because of the ease with which they can be genetically manipulated. Both budding and fission yeast belong to the kingdom Fungi, but they diverged from each other over 350 million years ago and are therefore only distantly related. In fact, the evolutionary distance between these yeast is similar to the evolutionary distance between each of them and mammals. As a result, studies in both types of yeast reveal valuable insights that directly apply to metazoan animals. Both yeast organisms can exist in the haploid state, carrying only one copy of each chromosome and therefore single copies of each gene. Haploid yeast are an ideal genetic system in which to screen or select for mutants with defects in cell proliferation, because doing the same experiment in diploid organisms would require mutating both copies of a gene. In addition, it is relatively easy to manipulate the expression of individual yeast genes, and yeasts are easy to cultivate and manipulate so that cultures of cells progress through the cell cycle in a synchronous manner. S. cerevisiae cells are ovoid in shape and divide by budding (Figure 194a). The bud, which is the future daughter cell, begins to form concomitant with the initiation of DNA replication and continues to grow throughout the cell cycle (Figure 19-4b). Cell cycle stage can therefore be inferred from the size of the bud, which makes S. cerevisiae a useful system for
identifying mutants that are blocked at specific steps in the cell cycle. As with mammalian cells, the budding yeast cell cycle has a long phase, and the study of the budding yeast cell cycle shaped our understanding of how the phase transition is controlled.
FIGURE 19-4 The cell cycle of budding yeast S. cerevisiae and fission yeast S. pombe. (a) Scanning electron micrograph of S. cerevisiae cells at various stages of the cell cycle.
The larger the bud that emerges at the end of the phase, the farther along in the cycle the cell is. Bud scars are also evident at sites where previous daughter cells budded off. (b) Main events in the S. cerevisiae cell cycle. The daughter cells must grow to a certain size before they enter START. is not well defined in budding yeast and is therefore indicated by a dashed arrow. In contrast to higher eukaryotes, the nuclear envelope does not disassemble during mitosis in S. cerevisiae and other yeasts, and they undergo a “closed mitosis.” Complicating matters further, the small S. cerevisiae chromosomes do not condense sufficiently to be visible by light microscopy. (c) Scanning electron micrograph of S. pombe cells at various stages of the cell cycle. Long cells are about to enter mitosis; short cells have just passed through cytokinesis. (d) Main events in the S. pombe cell cycle. This yeast has relatively short and S phases of the cell cycle and a prominent stage that is similar to that in metazoan cells. As in S. cerevisiae, the nuclear envelope does not break down during mitosis. Description The electron micrograph labeled (a) shows several spindle shaped structures each having tiny circular buds on one side and scars on the other side. The illustration labeled (b) shows the budding cycle. A cycle with four arrows is labeled G subscript 1, S, G subscript 2 (broken arrow), M, and G subscript 1. The budding cycle starts at G subscript 1, where an arrow labeled spindle body duplication points at a budding cell labeled bud emergence. An arrow labeled D N A replication points at a cell budding. An arrow from continues to points at a cell with a bigger bud. Another arrow labeled spindle formation; nuclear migration points at the cell undergoing mitosis with the spindle fibers highlighted. An arrow labeled chromosome segregation; nuclear division points at a cell almost dividing. Two arrows from the dividing cell each point at a parent cell and a daughter cell respectively. An arrow from the daughter cell labeled growth points at a cell. The cycle repeats at start. The electron micrograph labeled (b) shows long and short yeast cells at various cell stages. The illustration labeled (b) shows cell fission cycle. A cycle with four arrows is labeled G subscript 1, S, G subscript 2, M, and G subscript 1 again. The cell fission cycle starts at S phase. An arrow labeled D N A replication points at a cell. An arrow labeled cell growth points at a bigger cell labeled spindle pole body duplication. Another arrow
labeled chromosome condensation and spindle formation points at a cell in the metaphase stage. An arrow from this cell labeled chromosome segregation and nuclear division points at a dividing cell. Two arrows from the dividing cell points at two new cells, respectively. One of the new cells is labeled start. Fission yeast cells are rod-shaped, divide in the middle, and grow entirely by elongation at their ends (Figure 19-4c). Consequently, the cell cycle stage of this organism can be inferred from a simple measurement of its length. Cytokinesis occurs by the formation of a septum in the center of the cell, splitting it in two, hence the name fission yeast (Figure 19-4d). In contrast to budding yeast and mammalian cells, fission yeast have very short and S phases of the cell cycle, and instead spend most of the cell cycle in . The molecular mechanisms governing and entry into mitosis in fission yeast and in metazoan cells are very similar; studies with this organism by Paul Nurse and colleagues revealed the molecular events surrounding the phase transition. Studying yeast mutants that are blocked at specific steps in the cell cycle or that exhibit altered regulation of the cycle has been very useful. Because cell cycle progression is essential for viability, scientists isolated temperature-sensitive mutants whose genes encode proteins that are functional at one temperature but become inactive at a different, often higher, temperature (e.g., due to protein misfolding at the nonpermissive temperature; see Figure 6-6). These mutants, when arrested at a particular cell cycle stage, are easily distinguished from normally dividing cells by microscopic examination. Such cells are called cdc (cell division cycle) mutants. Identification of the genes mutated in these temperature-sensitive
Frog Oocytes and Early Embryos Facilitate Biochemical Characterization of the Cell Cycle Machinery
yeast strains provided a comprehensive list of genes critical for virtually all aspects of cell division. Frog Oocytes and Early Embryos Facilitate Biochemical Characterization of the Cell Cycle Machinery Although yeast are ideal for large-scale genetic screens, their small size and thick cell wall make them much less suitable for detailed biochemical analysis. To prepare cell extracts for biochemical studies of the cell cycle, the eggs and early embryos of amphibians and marine invertebrates have proven to be particularly suitable. These organisms typically have large eggs, and fertilization is followed by multiple synchronous cell cycles. By isolating large numbers of eggs from females and fertilizing them simultaneously by addition of sperm (or by treating them in ways that mimic fertilization), researchers can obtain extracts from cells at specific points in the cell cycle for analysis of proteins and enzymatic activities. To understand how X. laevis oocytes and eggs can be used for the analysis of cell cycle progression, let’s first look at oocyte maturation, which can be recapitulated in vitro. Oocytes undergo a meiotic division (see Figure 19-37 for an overview of meiosis). As oocytes develop in the frog ovary, they replicate their DNA and become arrested in for 8 months, during which time they grow in size to a diameter of 1 mm, stockpiling all the
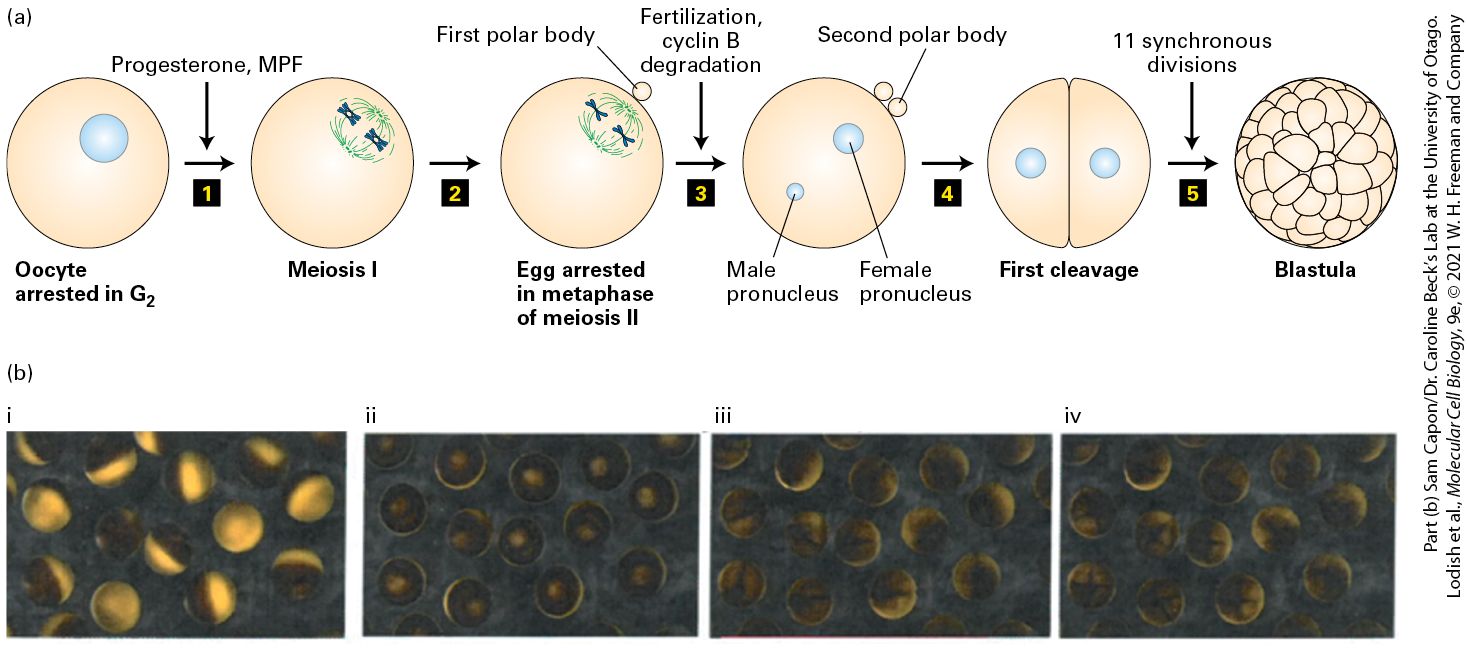
materials needed for the multiple cell divisions of the early embryo. When stimulated by a male, an adult female’s ovarian cells secrete the steroid hormone progesterone, which induces the -arrested oocytes to mature and enter meiosis. As we will see in Section 19.8, meiosis consists of two consecutive chromosome segregation phases known as meiosis I and meiosis II. Progesterone triggers oocytes to undergo meiosis I and progress to the second meiotic metaphase, where they arrest and await fertilization (Figure 19-5a). At this stage the cells are called eggs. When fertilized by sperm, the egg nucleus is released from its metaphase II arrest and completes meiosis. The resulting haploid egg nucleus then fuses with the haploid sperm nucleus, producing a diploid zygote nucleus. DNA replication follows, and the first mitotic division of embryogenesis begins (Figure 19-5b). The resulting embryonic cells proceed through 11 more rapid, synchronous cell cycles, generating a hollow sphere of cells called the blastula. Cell division then slows, and subsequent divisions are asynchronous, with cells at different positions in the blastula dividing at different times.
FIGURE 19-5 Progesterone stimulates maturation of Xenopus oocytes. (a) Step 1 : Progesterone treatment of -arrested Xenopus oocytes causes the oocytes to enter meiosis I. The same activity is seen if the oocytes are injected with MPF. Two pairs of synapsed homologous chromosomes (blue) connected to meiotic spindle microtubules (green) are shown schematically to represent cells in metaphase of meiosis I. Step 2 : Segregation of homologous chromosomes and a highly asymmetric cell division expels half the chromosomes into a small cell called the first polar body. The oocyte immediately commences meiosis II and arrests in metaphase II to yield an egg. Two chromosomes connected to spindle microtubules are shown schematically to represent egg cells arrested in metaphase of meiosis II. Step 3 : Fertilization by sperm releases eggs from their metaphase arrest, allowing them to proceed through anaphase of meiosis II and undergo a second highly asymmetric cell division that expels one chromatid of each chromosome into a second polar body. The resulting haploid female pronucleus fuses with the haploid sperm pronucleus to produce a diploid zygote. Step 4 : The zygote undergoes DNA replication and the first mitosis. Step 5 : The first mitosis is followed by 11 more synchronous divisions to form a blastula. (b) Micrographs of Xenopus eggs transitioning through the cell cycle. (i) -arrested oocytes. (ii) Following progesterone or MPF treatment the oocytes transit from into meiosis. (iii) Upon fertilization, there is a rise in intracellular that triggers cyclin B destruction, leading to completion of meiosis and the first cell division. (iv) The first of the next 11 rapid synchronous cycles of cell division is shown. Description The illustration labeled (a) shows the following sequence 1. An oocyte arrested in G 2 is represented by a round cell with a nucleus. An arrow labeled progesterone, M P F is points to a cell undergoing meiosis 1. 2. The cycle is arrested in metaphase of meiosis 2 and the first polar body has been expelled which is represented by a tiny sphere stuck to the cell membrane. 3. An arrow labeled fertilization cyclin B degradation points to a cell with the male pronucleus and female pronucleus represented by two spherical structures respectively. A second polar body is attached with the first one on the cell membrane. 4. First cleavage of the cell occurs, resulting in two cells. 5. Eleven synchronous divisions is the label above the arrow and results in a blastula. The four micrographs show cells in the G subscript 2 arrested stage to blastula formation.
The Study of Tissue Culture Cells Uncovers Cell Cycle Regulation in Mammals
The advantage of using X. laevis to study factors involved in mitosis is that large numbers of oocytes and eggs can be obtained, and following progesterone treatment and fertilization in the laboratory they will all proceed synchronously through the cell cycle in a manner that can be studied experimentally. Furthermore, the harvested eggs can be crushed by centrifugation in order to prepare reasonably large amounts of an extract that can be induced to undergo mitosis by the addition of sperm nuclei. This makes it possible to carry out biochemical experiments from cells that were all at the same point in the cell cycle. It was in this system that the cyclin-CDK complexes that trigger mitosis and the oscillatory nature of their activity were first discovered. This activity was called maturationpromoting factor (MPF) because of its ability to induce entry into meiosis and oocyte maturation when injected into -arrested oocytes and later called mitosis-promoting factor when it was shown to induce mitosis in all eukaryotic cells. The Study of Tissue Culture Cells Uncovers Cell Cycle Regulation in Mammals Cell cycle regulation in human cells is more complex than in nonmammalian systems. To understand this increased level of complexity, and to understand the cell cycle alterations that are the cause of cancer, it is important to study the cell cycle not only in model organisms but also in human cells. To study the properties of the human cell cycle, researchers use normal or tumor cells grown in plastic dishes, a method called tissue
culture or cell culture (see Chapter 4). However, note that many of the cell types used to study the human cell cycle have altered cell cycle properties due to genetic alterations that occurred during their culturing or because they were isolated from human tumors. Furthermore, in vitro culture conditions do not resemble those found in the organism and could lead to altered behavior of cells. Although some aspects of mammalian cell division are not recapitulated in cell culture conditions — such as tissue organization and developmental signals governing cell cycle control — cell culture systems nevertheless provide critical insights into the mammalian cell’s intrinsic mechanisms governing cell division. Researchers are also working toward establishing culture systems that more closely resemble the cell architecture in tissues. For example, mixtures of different cell types can be grown together to form tissue-like architectures, while embryonic or adult stem cells, or patient-derived tumor cells can be grown in the absence of serum on purified extracellular matrix from tissues, resulting in the formation of self-organizing structures called organoids that more closely resemble real organs (see
Figure 22-17). More chemically defined polymer lattices are currently being developed that also allow scientists to grow cells in threedimensional culture mimicking cellular organization within a tissue. Primary human cells and other mammalian cells have a finite life span when cultured in vitro. Normal human cells, for example, divide 25–50 times, but thereafter proliferation slows and eventually stops. This process is called replicative senescence. Some cells can escape this process and become immortalized, allowing researchers to establish cell lines. Although these cell lines harbor genetic alterations that affect some
Researchers Use Multiple Tools to Study the Cell Cycle
aspects of their proliferation, they are nevertheless a useful tool for studying cell cycle progression in human cells. These cell lines provide an inexhaustible supply of cells that, as we will see next, can be manipulated to progress through the cell cycle in a synchronous manner, allowing for the analysis of protein levels and enzymatic activity at different stages of the cell cycle. Researchers Use Multiple Tools to Study the Cell Cycle The experimental analysis of cell cycle properties requires that we be able to determine the cell cycle stage of individual cells. Light microscopy provides some estimate of cell cycle progression. For example, light microscopy allows a researcher to determine whether cultured mammalian cells are in interphase ( , S phase, and ) or in mitosis. Mammalian tissue culture cells are flat and adhere to their growth surface during interphase, but round up and form spherical structures as they undergo mitosis (Figure 19-6). Fluorescence microscopy of cellular structures or analysis of specific cell cycle markers — that is, proteins that are present only in certain cell cycle stages — allows for a more accurate determination of cell cycle stage.
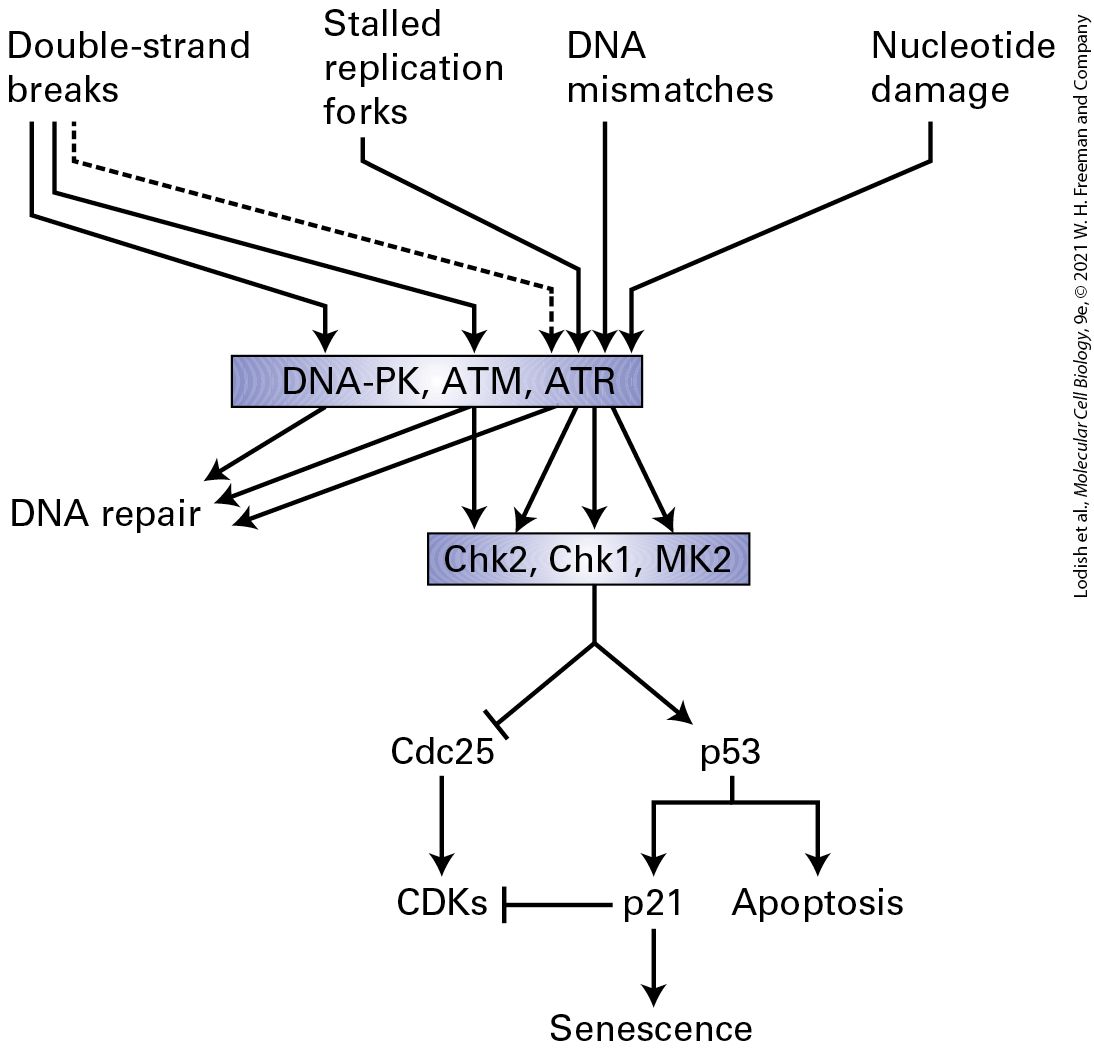
FIGURE 19-6 Human cells undergoing mitosis. HeLa Kyoto cells seen under phase contrast microscopy were filmed as they underwent mitosis. The images shown were taken every 20 minutes. Cells are flat during interphase. During mitosis, the cells round up and divide, and acquire a bright and refractile appearance. Subsequently, they flatten out again. In addition to microscopic tools, cell cycle researchers use flow cytometry to determine the DNA content of a cell population (Figure 19-7; see also
Figure 4-1). Cells are treated with a DNA-binding fluorescent dye, and the amount of dye that is incorporated into the DNA of cells is quantitatively assessed using a flow cytometer. Cells are recorded by their DNA content, and the percentages of cells in , S phase, and or mitosis can be assessed in this manner. Cells in have half as much DNA as cells in or mitosis. Cells undergoing DNA synthesis in S phase have an intermediate amount of DNA.
EXPERIMENTAL FIGURE 19-7 Analysis of DNA content by flow cytometry. Haploid yeast cells were grown in culture and stained with propidium iodide, a fluorescent dye that is incorporated into DNA. The x axis shows DNA content, the y axis the number of cells. The DNA content analysis shows two predominant populations of cells: cells with unreplicated DNA (1C) and with replicated DNA (2C). The cells between the two peaks represent cells that are in the process of undergoing DNA replication. Description The vertical axis of the graph represents number of cells ranging from 0 to 1200 in the increments of 300. The horizontal axis plots D N A content in units of chromosome
number; that is, 1 C represents normal cells and 2 C represents cells whose D N A content has doubled in preparation for replication. The curve shows two peaks, one at 1 C labeled unreplicated, where the maximum is around 1200, and the other at 2 C labeled replicated, where the maximum is around 400. Some cells in between 1 C and 2 C are indicated as cells undergoing replication. To characterize different cell cycle events, it is essential to examine cell populations that progress through the cell cycle in unison. Researchers can generate such populations by reversibly arresting cells in a particular cell cycle stage. This cell cycle arrest is usually accomplished by restricting nutrients or growth factors, causing cells to arrest in . In budding yeast, for example, cells treated with a mating pheromone arrest in . When the pheromone is removed from the cells (usually by washing them extensively), the cells exit and progress through the cell cycle in a synchronous manner. In mammalian cells, removal of growth factors by removing serum from the culture medium (serum starvation) arrests cells in . Re-addition of serum allows cells to re-enter the cell cycle. Other methods of arresting cells involve blocking a certain cell cycle step with chemicals. Hydroxyurea inhibits DNA replication, leading to arrest in S phase. After removing the drug, cells resume DNA synthesis in unison. Nocodazole disrupts the mitotic spindle and halts the cell cycle in mitosis. Once the drug is washed away, cells resume progression through mitosis in a synchronous manner. In budding and fission yeasts, when temperaturesensitive cdc mutants are incubated at the nonpermissive temperature, they arrest in a particular cell cycle stage because they are defective in a certain key cell cycle protein. Returning cells to the permissive
temperature allows them to continue with the cell division cycle in a synchronous fashion. KEY CONCEPTS OF SECTION 19.2 Model Organisms and Methods of Studying the Cell Cycle The ability to isolate mutants of budding and fission yeasts and the use of powerful genetic tools led to the isolation of key genetic factors important for cell cycle regulation. Frog eggs and early embryos from synchronously fertilized eggs provide sources of extracts for biochemical studies of cell cycle events, which led to the identification of the oscillatory nature of cyclin-CDK complexes. Human tissue culture cells are used to study the properties of the mammalian cell cycle. The generation of synchronized cell populations by reversibly arresting cells in a particular cell cycle stage allows researchers to examine the behavior of proteins and cellular processes during the cell cycle.
19.3 Cell Cycle Progression and Control: Feedback Loops and Post-Translational Modification
19.3 Cell Cycle Progression and Control: Feedback Loops and PostTranslational Modification Progression through the cell cycle is driven, in large part, by the action of cyclin-dependent kinases (CDKs). These protein kinases act as master controllers of the cell cycle by phosphorylating substrates that when properly phosphorylated directly cause the events in each cell cycle stage to take place. In certain phases of the cell cycle, other kinases also play important roles, but the importance of CDKs in each phase of the cell cycle is paramount. We know from many experimental observations that cells move irreversibly through the cell cycle, and they do so in only one direction (see Figure 19-1): from to S phase to to M phase. Cells have never been observed to go backward from into S phase, nor has anyone ever seen an S phase cell somehow unreplicate its DNA and revert back to a -phase cell. This commitment once cells initiate each phase of the cell cycle means that the activity of these master controllers, the CDKs, must be regulated with extraordinary precision — at just the right time and place and in a manner that is essentially irreversible. Although details of how this is accomplished vary for different cell cycle stages, we will see in subsequent sections that the general molecular basis for CDK regulation is the same no matter which cell cycle stage we are talking about. It
always involves the use of cyclins — the accessory proteins that bind to the CDKs to control their activity — in combination with protein phosphorylation and ubiquitination (see Section 3.4). The control of CDKs can be thought of as occurring in several layers, represented by the concentric circles in Figure 19-8. At the center are the CDKs themselves, along with their activating accessory proteins, the cyclins. In the second layer are the direct regulators of the cyclin-CDK complexes, which includes protein kinases and phosphatases that activate or inhibit the CDKs, together with CDK inhibitor proteins that bind directly to the CDKs to block their activity, transcription factors that increase the rate of cyclin synthesis to create active cyclin-CDK complexes at specific points in the cell cycle and protein kinases and ubiquitin ligases that work together to promote the destruction of the cyclins once cells have completed a specific part of the cell cycle. In the outer layer are the indirect regulators of the cyclin-CDK complexes that function by regulating the direct regulators (Figure 19-8). Like the second layer of control, this third layer of indirect regulators is also composed of protein kinases and ubiquitin ligases, which work together with phosphoserine/threonine-binding domains and proline isomerases to control the levels, activity, and subcellular location of the direct regulators of cyclin-CDK activity. This multilayered network of control allows a cell to evaluate the massive amount of information about the state a cell finds itself in. A cell then integrates the information into all-or-none step-function-like decisions that control the onset of DNA replication or the initiation of mitosis, for example. Note that this information is reflected by gradual linear changes in the activity of various signaling pathways. The combination of direct and indirect control afforded by this multilayered regulatory network
allows a very small set of basic biochemical reactions — protein phosphorylation and de-phosphorylation, molecular recognition, and ubiquitin-mediated protein destruction — to build feed-forward and feedback loops that provide fine control of whether a cell cycle transition takes place. In addition, the outermost circle of indirect regulation provides the molecular basis for cell cycle checkpoint control, as explored in Section 19.7.
FIGURE 19-8 A multilayered network controls the activity of cyclin-dependent kinases. Concentric circles indicate the CDKs and their activating cyclins in the center, surrounded by their direct regulators, and then their indirect regulators. As shown in the key, the shapes represent kinases, phosphatases, proline isomerases, phosphoserine/threonine-binding proteins and domains, cyclin kinase inhibitors, and ubiquitin ligases. Black lines generally denote activation of the indicated protein’s target, while red lines denote inhibition. Feedback loops involving phosphorylation of the regulators by the cyclin-CDKs are not
shown to maintain clarity. Note that there is substantial cross talk among the molecules and that the number of molecules and interactions are more complex than is shown here. Description The illustration shows a concentric circle. A key lists seven different structures of various molecules. In the innermost circle there are two molecules labeled C D K and cyclin. Certain molecules surrounding these two molecules in the center act as their direct regulators. Some help in activation and the rest in inhibition. Some more molecules surrounding these regulators act as indirect regulators. In the following sections, we describe the current model of eukaryotic cell cycle regulation, which is summarized in Figure 19-9. A key discovery in cell cycle studies was that cyclin-dependent kinases govern progression through the cell cycle. Three key features of these kinases are important to keep in mind throughout this chapter. Cyclin-dependent kinases (CDKs) are active only when bound to a regulatory cyclin subunit. Different types of cyclin-CDK complexes initiate different events. cyclin-CDKs and phase cyclin-CDKs promote entry into the cell cycle, S phase cyclin-CDKs trigger S phase, and mitotic cyclinCDKs initiate the events of mitosis (Figure 19-9). Multiple biochemical mechanisms ensure that the CDKs are active only in the stages of the cell cycle they trigger. These molecular mechanisms are organized into feedback loops that amplify the process of CDK activation and inhibition.
FIGURE 19-9 An overview of how CDKs regulate cell cycle progression. Cells harbor different types of CDKs that initiate different events of the cell cycle. Importantly, the CDKs are active only in the stages of the cell cycle that they trigger. phase CDKs are active at the phase transition to trigger entry into the cell cycle. S phase CDKs are active during S phase and trigger DNA replication. Mitotic CDKs are active during mitosis and trigger mitosis. The anaphase-promoting complex or cyclosome (APC/C) ubiquitin-protein ligase catalyzes two key cell cycle transitions by ubiquitinylating proteins, hence targeting them for degradation. APC/C initiates both anaphase and exit from mitosis. The names of the different CDKs and their associated cyclin subunits for yeast and humans are listed.
Cyclin-Dependent Kinases Are Small Protein Kinases That Require a Regulatory Cyclin Subunit for Their Activity
Description The illustration of the cell cycle is represented as a circle of arrows below. In G subscript 1, moving counterclockwise from the top right, G subscript 1 C D K kinases and G subscript 1slash S phase C D Ks prepare cells for S phase. In addition, S C F ubiquitin-protein ligase induces S phase. In the S phase, which is at the bottom of the cycle, C D Ks activate D N A replication. In G subscript 2, mitotic C D Ks induce mitosis. An expansion of the M phase shows cells in prophase, metaphase, anaphase, and telophase and cytokinesis. Between metaphase and anaphase, A P C-C ubiquitinprotein ligase induces anaphase. In telophase and cytokinesis, A P C-C and phosphatases induce late steps in mitosis. Below is a chart comparing budding yeast cells to human cells and the C D Ks involved in each phase. The first column is labeled G subscript 1 cyclin-C D Ks and shows C I n 3-C d c 28 for budding yeast and cyclin D-C D K 4 and cyclin D-C D K 6 for humans. The second column is labeled G subscript 1 slash S cyclin-C D Ks and shows C I n1 slash 2-C d c 28 for yeast and cyclin E-C D K 2 for humans. The third column is labeled S phase cyclin-C D Ks and shows C l b 5 slash 6-C d c 28 for yeast and cyclin A-C D K 2 for humans. The last column is labeled M phase cyclin-C D Ks and shows C I b 1 slash 2 slash 3 slash 4-C d c 28 for yeast and cyclin A-C D K 1 and cyclin B-C D K 1 for humans. In this section, we discuss the properties of CDKs and investigate the structural basis of their activation and regulation. We then describe how cyclins activate CDKs and investigate the general regulatory mechanisms that restrict different cyclins to the appropriate cell cycle stage. We will see that protein degradation plays an essential part in this process, as do post-translational modifications to CDKs and inhibitory proteins that directly bind to cyclin-CDK complexes. Cyclin-Dependent Kinases Are Small Protein Kinases That Require a
Regulatory Cyclin Subunit for Their Activity Cyclin-dependent kinases constitute a family of small (30–40 kDa) serine/threonine kinases that contain little more than the kinase domain. By themselves, monomeric CDKs are not active, and in general, their protein levels in the cell remain relatively constant throughout the cell cycle. The activity of these CDKs and the amounts of the substrates that they phosphorylate, however, change dramatically in the different cell cycle stages. These changes occur because the CDKs require the binding of an activating subunit — a cyclin — to become active protein kinases. In budding and fission yeasts, a single CDK controls progression through the cell cycle. Its activity, and the specific substrates that it phosphorylates, is specified by which of several cell-cycle-stage-specific cyclin subunits it is bound to. Mammalian cells, in contrast, contain as many as 20 different CDKs, with 4 of them, CDK1, CDK2, CDK4, and CDK6, having clear roles in regulating cell cycle progression. Each CDK binds to one or more cyclin subunits that control CDK activity and substrate preference. The different mammalian CDKs, together with their specific cyclins, promote different cell cycle transitions. Mammalian CDK4 and CDK6, for example are CDKs that promote entry into the cell cycle, CDK2 functions as a phase and S phase CDK, and CDK1 is the mitotic CDK. For historical reasons, the names of various cyclindependent kinases from yeasts and vertebrates differ. Whenever possible, we will use the general terms , phase, S phase, and mitotic CDKs
Cyclins Determine the Activity of CDKs
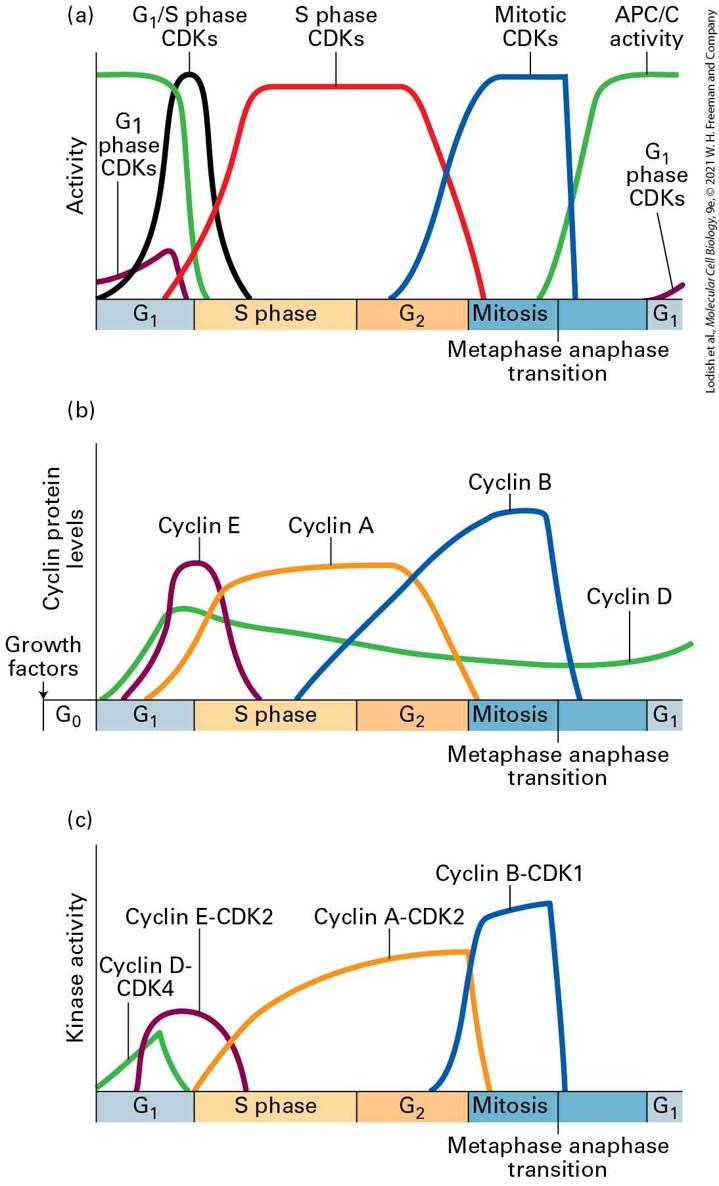
to describe CDKs instead of the species-specific terminology. The bottom of Figure 19-9 lists the names of the various mammalian and budding yeast cyclin-CDKs and indicates when in the cell cycle they are active. Cyclins Determine the Activity of CDKs Cyclins are so named because their concentrations change during the cell cycle. They form a family of proteins that is defined by three features. Cyclins bind to and activate CDKs. The activity and substrate specificity of any given CDK is primarily defined by the particular bound cyclin. Cyclins are present only during the cell cycle stage that they trigger and are absent from other cell cycle stages. In addition to triggering a particular cell cycle stage, cyclins also set in motion a series of events in preparation for the next cell cycle stage. In this way, they propel the cell cycle forward (Figure 19-10a).
FIGURE 19-10 CDK activity during the cell cycle is regulated by binding to different cyclins. (a) The activity of different CDKs varies as cells transit through the stages of the cell cycle, as indicated in Figure 19-9. (b) The protein levels of different cyclin subunits that form the cell-cycle-stage-specific CDK complexes rise and fall in a cell-cycle-stage-specific manner. (c) The activity of the different cyclin-CDK complexes that are formed by the different cyclin-CDK complexes varies in a manner that is very similar, but not exactly identical to the accumulation of the cyclins. The difference between cyclin abundance in panel (b) and kinase activity shown in panel (c) indicates that there must be additional levels of regulation of cyclin-CDK activity, such as phosphorylation and dephosphorylation in addition to cyclin binding. Description The vertical axis of the graph labeled (a) represents activity. It has no units. The horizontal axis represents the stages of a cell cycle which includes G subscript 1, S phase, G subscript 2, mitosis ( metaphase anaphase transition), and G subscript 1. There are differently colored curves that represent the activity of various C D Ks like the G subscript 1 phase C D Ks, G subscript 1 slash S phase C D Ks, S phase C D Ks, mitotic C D Ks, and A P C slash C activity. The vertical axis of the graph labeled (b) represents cyclin protein levels. It has no units. The horizontal axis represents the stages of a cell cycle which includes G subscript 0 (labeled growth factors), G subscript 1, S phase, G subscript 2, mitosis ( metaphase anaphase transition), and G subscript 1. There are differently colored curves that represent levels of cyclin E, A, B, and D respectively. The curves show a steady pattern except for the cyclin D curve which rises at G subscript 1 to steadily lower at mitosis stage to increase again at G subscript 1. The vertical axis of the graph labeled (c) represents kinase activity. The horizontal axis represents the stages of a cell cycle which includes G subscript 0 (labeled growth factors), G subscript 1, S phase, G subscript 2, mitosis (metaphase anaphase transition), and G subscript 1. There are differently colored curves that represent kinase activity of cyclin D-C D K 4, cyclin E-C D K 2, cyclin A-C D K 2, and cyclin B-C D K 1. The cyclin D- C D K 4 curve appears during G subscript 1 and does not appear after that. The other 3 curve patterns are similar to that of (a) and (b) graphs.
Similar to the nomenclature used for CDKs, cyclins are divided into four classes defined by their presence and activity during specific phases of the cell cycle: cyclins, phase cyclins, S phase cyclins, and mitotic cyclins (see Figure 19-9, bottom). Some cyclins can bind to and activate more than one CDK, while others can only bind to a single CDK. In mammalian cells, for example, cyclin A can form an activating complex with both CDK2 in S phase, and with CDK1 at the very beginning of mitosis. In contrast, cyclin B, the main mitotic cyclin, can only form a complex with CDK1. The cyclins are the linchpin in coordinating the cell cycle with extracellular events. Their activity is subject to regulation by signal transduction pathways that sense the presence of growth factors or cell proliferation inhibitory signals. In metazoans, cyclins are known as cyclin Ds, and they bind to CDK4 and CDK6. Unlike other cyclins, cyclins levels do not show strong, stage-dependent fluctuations. Instead, in response to macromolecule biosynthesis and extracellular signals, their levels gradually increase in , drop a bit during S and phase, and rise again during late M phase (Figure 19-10b). The cyclins accumulate during late , reach peak levels when cells enter S phase, and decline during S phase (Figure 19-10b). They are known as cyclin E in metazoans and bind to CDK2. The main function of cyclin E–CDK2 complexes, together with cyclin D–CDK4/6, is to trigger the phase transition. This transition, which we defined in Section 19.1 as START in yeast and the restriction point in mammalian cells, is defined as the point at which cells are irreversibly committed to cell division and can
no longer return to the state. In molecular terms, this means that cells initiate DNA replication as well as duplicating their centrosomes, which is the first step in the formation of the mitotic spindle that will be used during mitosis. S phase cyclins are synthesized concomitantly with cyclins, but their levels remain high throughout S phase and do not decline until early mitosis. Two types of S phase cyclins trigger S phase in metazoans: cyclin E, which can also promote entry into the cell cycle and is therefore also a cyclin, and cyclin A. Both cyclins bind CDK2 and are directly responsible for DNA synthesis. As we will see in Section 19.4, these protein kinases phosphorylate proteins that activate DNA helicases and load polymerases onto DNA. Mitotic cyclins bind to CDK1 to promote entry into and progression through mitosis. The metazoan mitotic cyclins are cyclins A and cyclins B (note that cyclin A can also trigger S phase when bound to CDK2). Mitotic cyclin-CDK complexes are synthesized during S phase and , but as we will see shortly, their activities are held in check until DNA synthesis is completed. In Section 19.5, we will see that once activated, mitotic CDKs promote entry into mitosis by phosphorylating and activating hundreds of proteins to promote chromosome condensation, nuclear envelope breakdown, mitotic spindle formation, and other aspects of mitosis. Their inactivation during anaphase prompts cells to exit mitosis, which involves the disassembly of the mitotic spindle, chromosome decondensation, the re-formation of the nuclear envelope, and eventually cytokinesis.
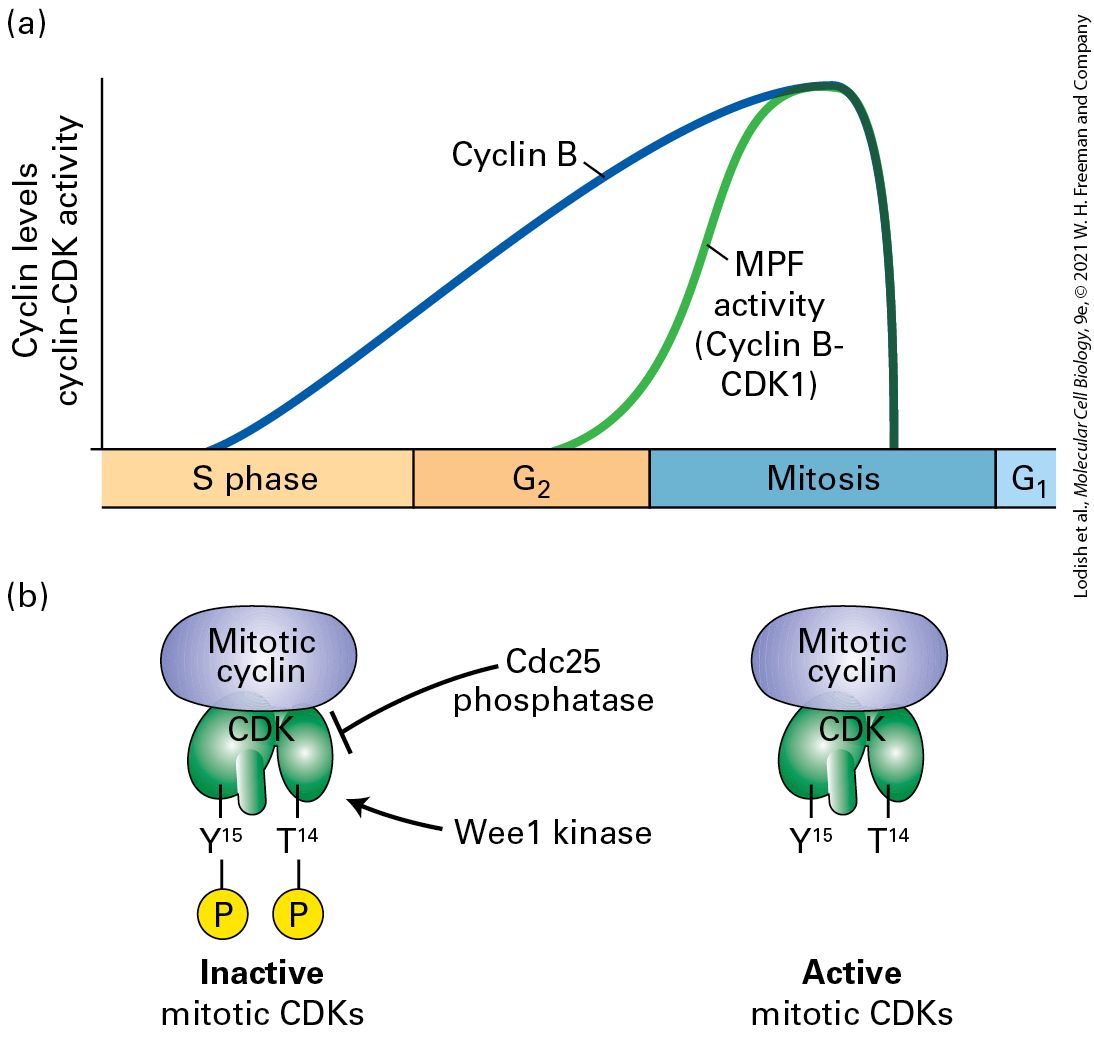
Mitotic cyclins were the first set of cyclins to be discovered, and it was their characterization that led to the discovery of the oscillatory nature of the activities that govern all of cell cycle progression. An impressive series of genetic and biochemical experiments in fission yeast by Paul Nurse and colleagues in the 1980s, building on related work from Lee Hartwell in budding yeast, had established that mitotic entry required the activity of a specific protein kinase, called Cdc2 in fission yeast and Cdc28 in budding yeast, and characterized some aspects of how this kinase was regulated. Independently, biochemical experiments in sea star embryos and eggs from frogs, surf clams, and sea urchins in the 1970s and 1980s had suggested that the maturation-promoting factor/mitosispromoting factor (MPF) in those organisms, which we described in Section 19.2, might involve this same protein kinase. In a remarkable experiment in 1982, while studying protein synthesis in sea urchin eggs after fertilization, Tim Hunt unexpectedly observed a protein that showed cycles of synthesis followed by rapid destruction. The timing of this protein’s destruction occurred just before the cells physically split into two, suggesting that its synthesis correlated with mitotic entry, and its destruction correlated with completion of mitosis (Figure 19-11). A similar pattern of cyclic protein synthesis and destruction was seen for two related proteins in clam eggs. These findings hinted that these proteins might be the mysterious labile factor(s) mentioned in Section 19.1 that control the switch from to mitosis. Cloning of their cDNAs further contributed to the identification of the proteins as sea urchin cyclin B and clam cyclin A. These cyclin proteins were not the critical protein kinases that drove budding yeast, fission yeast, or vertebrate eggs and embryos into mitosis themselves, but cyclin immunoprecipitates (see Section 3.5)
from the eggs had strong protein kinase activity. Furthermore, the addition of the sea urchin cyclin cDNA to RNase-treated frog egg extracts could drive the extracts through several rounds of cell cycles, indicating that these cyclin proteins were associating with and activating the protein kinases that were responsible for cell division. EXPERIMENTAL FIGURE 19-11 How cyclins were discovered. The discovery of cyclins was based on the observation that the levels of a newly translated protein, labeled A in the autoradiogram, peaked in abundance and then rapidly declined each time before a sea urchin cell divided. In this experiment, the eggs were fertilized and incubated with
radioactive methionine. Samples were then analyzed at 10-minute intervals using SDSPAGE and autoradiography, and the eggs were also examined microscopically for cleavage. Compare the levels of one protein on the gel, labeled B, which progressively accumulates, with the levels of protein A that seems to rise and fall just before the cells undergo cleavage. The band intensities and cleavage fraction, as a function of time, are quantified in the graph. Protein A is the mitotic cyclin. [Reprinted with permission from Elsevier, from Evans et al., 1983, “Cyclin: A Protein Specified by Maternal mRNA in Sea Urchin Eggs That Is Destroyed at Each Cleavage Division,” Cell 33:389–396; permission conveyed through Copyright Clearance Center, Inc.] Description The vertical-axis of the graph represents amount ranging from 0 to 75 in increments of 25. The horizontal-axis represents time ranging from 0 to 3 hours, in increments of 1. Three lines are plotted on the graph: A purple line labeled cleavage index is at zero until after one hour, when it rises and falls sharply between hours 1 and 2 and again after hour 2 starts. A green line is labeled intensity of band B, and starts at zero and rises continually throughout the graph. A third line, labeled intensity of band A, starts at 0 at about 15 minutes into hour 1, then rises sharply to 50, then lowers to almost 0 in one hour, rises again to 50 by beginning of hour 2, then lowers to about 10 at end of graph line. The autoradiograph shows two rows of bands, the top row labeled A and the bottom row labeled B. Both bands are dim at the left and increase to very dark bands at the right. Ultimately, the composition of MPF from frog eggs was resolved by biochemical purification of MPF followed by protein analysis using SDSPAGE. This revealed two bands, one at 32 kDa, similar in size to that of the yeast Cdc2 kinase, and a second band at 45 kDa that corresponded to cyclin B, indicating that frog egg MPF was, in fact, a complex between cyclin B and the mitotic CDK1 kinase. At around the same time, genetics experiments in fission yeast revealed a critical regulator for Cdc2, the
yeast CDK, to be another cell division cycle gene, Cdc13. Cloning and sequencing of Cdc13 revealed that it was the fission yeast homolog of cyclin B. The levels of Cdc13 were shown to vary during the yeast cell cycle, just as cyclin B did during the sea urchin cell cycle. In addition, just as cyclin B was required for CDK1 kinase activity, Cdc13 was required for Cdc2 protein kinase activity, and this complex, formed by a mitotic cyclin and a cyclin-dependent kinase (Cdc13 and Cdc2, respectively) was required for fission yeast to progress from into M phase. This combination of genetics and biochemical experiments performed in so many of the different organisms discussed in Section 19.2 ultimately revealed a universal mechanism that controls mitotic entry for all eukaryotic life forms: the formation of a complex between an unstable cyclin molecule and a small cyclin-dependent protein kinase whose activity is strictly dependent on the presence of the cyclin subunit. Subsequent studies showed that phase cyclins had similar properties. Their expression is sufficient to promote entry of resting cells from into S phase, and subsequent movement through S phase. How exactly does cyclin-binding convert CDKs from an inactive to an active state? The three-dimensional structure of CDKs and cyclin-CDK complexes, solved for cyclin B/CDK1 and cyclin A/CDK2 by x-ray crystallography, provides an answer (Figure 19-12). As discussed in
Chapter 3, cyclin-dependent kinases, like all protein kinases, are bilobed structures, with an active site cleft where both ATP and substrates bind. In the absence of cyclin binding, however, several of the catalytic residues in the CDK are not in the proper position to bind ATP and catalyze the transfer of a phosphate group from ATP to the substrate proteins. In
addition, the activation loop of the monomeric CDK blocks substrates from accessing the active site cleft. Cyclins are composed of two domains, each containing five α helices. The overall structure is called a cyclin fold, and the first domain corresponds to the cyclin box region that is required for CDK binding and activation. In the case of CDK1 and CDK2, binding of the cyclin box causes an α helix in the CDK to move, reorganizing the amino acids in the ATP-binding pocket of the CDK into a more productive conformation for catalysis. In addition, cyclin binding structures and repositions the activation loop, as we discuss shortly. Besides stimulating CDK activity, the cyclin subunit can also interact directly with substrate proteins of the kinase, thereby contributing to the substrate specificity of the cyclin-CDK complex.
FIGURE 19-12 Structural models of human CDK2. (a) Free, inactive CDK2 not bound to its cyclin subunit, cyclin A. In free CDK2, the A-loop blocks access of protein substrates to the γ phosphate of the bound ATP, shown as a stick model. The conformations of the Aloop and the region highlighted in yellow (α1 helix) are altered when CDK is bound to cyclin A. (b) Nonphosphorylated, low-activity cyclin A-CDK2 complex. Conformational changes induced by binding of a domain of cyclin A (blue) cause the A-loop to pull away from the active site of CDK2 so that substrate proteins can bind. The α1 helix in CDK2, which interacts extensively with cyclin A, moves several angstroms into the catalytic cleft,
CDKs Are Regulated by Activating and Inhibitory Phosphorylation
repositioning key catalytic side chains required for the phosphotransfer reaction. The black ball marks the position of the threonine (Thr-160) whose phosphorylation by CAK activates CDKs. The red balls mark the positions of phosphorylated Thr-14 and Tyr-15, which inhibit CDK activity. (c) Phosphorylated, high-activity cyclin A-CDK2 complex. The conformational changes induced by phosphorylation of the activating threonine (black ball) alter the shape of the substrate-binding surface, greatly increasing the affinity for protein substrates. Removal of the phosphate groups from Thr-14 and Tyr-15 enhances ATP binding and catalytic activity. See P. D. Jeffrey et al., 1995, Nature 376:313–320. [Data from A. A. Russo, P. D. Jeffrey, and N. P. Pavletich, 1996, Nat. Struct. Biol. 3:696, PDB ID 1jst.] Description The illustration labeled (a) shows a semi-transparent surface structure enclosing a ribbon. The ribbon is bound to a ball-and-stick model of A T P. A yellow ribbon in top right area is labeled alpha 1 helix. A red ribbon is attached to this and moves down and to the left where it curls and has 3 labels, from the top down: G-loop, T h r-160, T h r -14, and T h r-15. The ribbon continues down and to the right with the label A-loop. The illustration labeled (b) has a similar structure to that of the one in illustration (a) except that it is bound to another three-dimensional structure enclosed by a surface structure on its top right. T h r-160 is labeled and is represented by two spheres bound to the A-loop. The illustration labeled (c) has a similar structure to that of the one in illustration (b) except that the T h r-160 is bound to a phosphate group on the A-loop (P-T h r-160). CDKs Are Regulated by Activating and Inhibitory Phosphorylation As we will discuss in detail below, in addition to cyclin-binding, a variety of other mechanisms ensure that CDKs are only active at the right stage of
the cell cycle. Table 19-1 lists many of these additional key regulators of CDKs.
TABLE 19-1 • Regulators of Cyclin-CDK Activity Type of Regulator Function Kinases and Phosphatases CAK kinase Activates CDKs in all organisms Wee1 kinase Inhibits CDKs in all organisms Cdc25 phosphatase Activates CDKs in fission yeast Cdc14 phosphatase Activates Cdh1 to degrade mitotic cyclins in budding yeast Cdc25A phosphatase Activates vertebrate S phase CDKs Cdc25B and C phosphatases Activate vertebrate mitotic CDKs Inhibitory Proteins Sic1 Binds and inhibits S phase CDKs in budding yeast CKIs , , and Bind and inhibit CDKs in animal cells
Binds and inhibits CDKs in animal cells Rb Binds E2Fs, preventing transcription of multiple cell cycle genes in animal cells Ubiquitin-Protein Ligases
SCF Degradation of phosphorylated Sic1 or to activate S phase CDKs Degradation of securin, initiating anaphase. Induces degradation of B-type cyclins Degradation of B-type cyclins in and geminin in metazoans to allow loading of replicative helicases on DNA replication origins Phosphoserine/Threonine-Binding Proteins and Modular Domains WD40 domains, leucine-rich repeats Recognize phosphorylated substrates and target them for degradation by SCF ligases Cks subunits Target cyclin-CDKs for processive substrate phosphorylation 14-3-3 proteins Bind and inactivate regulators of cyclin-CDKs to establish cell cycle checkpoints. Also regulates cytokinesis. Pin1 Isomerizes phosphorylated substrates of CDKs to control mitotic entry and exit FHA and BRCT domains Form phospho-dependent molecular assemblies necessary for cell cycle surveillance mechanisms in all stages of the cell cycle, and regulates cytokinesis. Polo-box domains Target Polo kinases to sites of CDK phosphorylation to drive mitotic entry and progression If cyclin-binding was the only thing required for CDKs to become fully active, then one would expect the activity of each CDK to perfectly parallel the accumulation of its cyclin partner. However, that is not what is observed for most cyclin-CDK pairs. Figure 19-13a shows the
accumulation of cyclin B, and the activity of MPF (the cyclin B-CDK1 complex in Xenopus oocytes). The abrupt rise of MPF activity observed upon entering mitosis, despite the slow progressive rise in the amount of cyclin B present, indicates that additional types of regulation must be present besides cyclin binding. Indeed, at least two additional mechanisms are involved in controlling the activity of CDKs: activating and inhibitory phosphorylations on the CDK subunit, and the action of inhibitor proteins, called CKIs (cyclin kinase inhibitors) that bind to CDK to block its activity. These three regulatory events — cyclin binding, phosphorylation, and CKIs — together with cyclin synthesis and destruction, ensure that CDKs are active only at the appropriate time and cell cycle stage.
FIGURE 19-13 Phosphorylation of the CDK subunit restrains mitotic CDK activity during S phase and . (a) Mitotic cyclin B is synthesized during S phase and and binds to CDK1, but the complex does not become active until aound the time of mitotic entry. (b) The cyclin-CDK complex is not active because threonine 14 and tyrosine 15 of the CDK1 subunit are phosphorylated by the protein kinase Wee1, which inhibits the accumulation of the active form. Once DNA replication has been completed, the protein phosphatase Cdc25 is activated and de-phosphorylates CDK1, promoting its activity. A positive feedback loop, indicated by the black curved arrows and discussed in Section 19.5, controls the activation of Cdc25 and inhibits Wee1 in animal cells.
Description The vertical axis of the graph labeled (a) represents cyclin levels cyclin-C D K activity. It has no units. The horizontal-axis represents the stages of a cell cycle which includes S phase, G subscript 2, mitosis, and G subscript 1. A curve labeled cyclin B and starts low in S phase at left, and rises until a little more than halfway through mitosis, where it drops drastically to the bottom. Another curve labeled M P F activity (Cyclin B-C D K 1) and begins low in the middle of the G subscript 1 phase, rises up to the same height of cyclin B, then matches the cyclin B line as it drops down in mitosis. The illustration labeled (b) shows inactive and active mitotic C D Ks side by side. The inactive C D Ks has an oval structure labeled mitotic cyclin attached to another structure labeled C D K. An arrow labeled wee 1 kinase points to the bottom of the C D K structure. The left side of the C D K structure is labeled Y superscript 15 with a yellow phosphate circle attached, and the right side is labeled T superscript 14, also with a phosphate circle attached. A T-shaped line labeled C d c 25 phosphatase points towards C D K. The active C D Ks has a similar structure to the inactive C D K except the phosphorous molecules. Nonphosphorylated CDK bound to one of its cyclin partners has minimal but detectable protein kinase activity in vitro, although it may be essentially inactive in vivo. This is because the activation loop, though partially structured by cyclin binding, still largely blocks the catalytic cleft (see Figure 19-12b). Phosphorylation of a critical threonine residue in the activation loop, Thr-160, is needed to pull the activation loop out of the way by tethering it to several basic residues in the CDK, allowing substrates to access the active site of the CDK and increasing the activity of the phosphorylated cyclin-CDK complex by over a hundredfold compared to the nonphosphorylated complex (see Figure 19-12c). This threonine phosphorylation is mediated by CDK-activating kinase (CAK). Curiously, CAK activity is constant throughout the cell cycle and the
CDK Inhibitors Provide Additional Control of Cyclin-CDK Activity
activating phosphorylation of the CDK occurs either before or as soon as a cyclin-CDK complex is formed, so it is clear that CAK phosphorylation of the CDK is not a rate-limiting step in CDK activation. Why, then, does cyclin B/CDK1 (MPF) activity rise so rapidly when cells go from into M phase (Figure 19-13), if the threonine-phosphorylated cyclin B–bound kinase is already present? This behavior arises because the CDK also contains inhibitory phosphorylations. The two inhibitory phosphorylations on CDK play a critical role in controlling CDK activity. A highly conserved tyrosine (Y15 in human CDKs) and an adjacent threonine (T14 in humans) are subject to regulated phosphorylation (see Figures 19-12b and 19-13b). Importantly, both of these residues are situated in the G-loop of the CDK, containing the consensus sequence GXGXXGXV, that drapes over the phosphate groups in ATP (see Figures 19-13 and 3-38). In the case of CDKs, the G-loop sequence is . Phosphorylation of T14 and Y15 in this Gloop introduces significant negative charge that electrostatically interferes with the binding and positioning of ATP in the catalytic pocket. Changes in the phosphorylation of these sites are essential for the regulation of mitotic CDKs and have also been implicated in the control of and S phase CDKs. As we will see in Section 19.5, a highly conserved kinase called Wee1 brings about these inhibitory phosphorylations, and a family of highly conserved phosphatases called Cdc25, mediates their dephosphorylation (Figure 19-13b).
CDK Inhibitors Provide Additional Control of Cyclin-CDK Activity In addition to cyclin binding and reversible phosphorylations on the CDK itself, there is one more level of control of cyclin-CDK activity. A family of proteins known as CDK inhibitors or CKIs bind directly to CDKs and cyclin-CDK complexes to inhibit their activity. Different classes of CKIs act in , at the boundary, or in S and phases of the cell cycle to prevent premature activation of , S phase, and M phase CDKs. An important class of CKIs acting in are members of the INK4s (inhibitors of kinase 4) family. This family of proteins includes several small closely related proteins that bind to CDK4 and CDK6 monomers, blocking their interaction with cyclin D and thus rendering these CDKs inactive. As we will see in Section 19.4, these CDKs play an especially important role in the regulation of the phase transition, and its integration with extracellular signals. A second class of CKIs found in metazoan cells is the Cip/Kip family of CKIs. This family consists of three proteins — , , and . The Cip/Kip family members bind to cyclin-bound CDKs and block the activity of cyclin D-, E-, A-, and B-containing complexes. Cip/Kip CKIs must be sequestered away from their respective cyclin/CDK targets or degraded in order for DNA replication or mitosis to commence. Each of the three proteins appears to play a unique physiological role. The protein, encoded by the cdkn1a gene, is important in establishing
Cyclin Levels Are Regulated by Transcriptional Activation and Ubiquitin-Mediated Protein Degradation
and arrest in metazoan cells in response to DNA damage, and its transcription is controlled by the important tumor suppressor protein p53. The protein, encoded by the cdkn1b gene, is critical for blocking progression from into S phase in response to nutrient and growth factor limitation in order to limit organ and body size. Mice in whom the gene has been knocked out display increased body size and multiple organ hyperplasia (i.e., an excessive number of cells in their organs). The protein, encoded by the cdkn1c gene, plays an important role in regulating the cell cycle during embryonic development. Mouse embryos lacking cdkn1c display organ hyperplasia and impaired tissue differentiation, resulting in death soon after birth. Based on the crystal structure of bound to cyclin A-CDK2, the Cip/Kip proteins are thought to function both by blocking the site on the cyclin that interacts with cyclin-CDK substrates and occluding the active site cleft to block ATP binding and catalytic activity (see Figure 19-12). These proteins have other functions beyond cell cycle control, including effects on gene transcription, cell fate determination, cell migration, and apoptosis. Nonetheless, the Cip/Kip family members play critical roles in proper cell and tissue homeostasis. Cyclin Levels Are Regulated by Transcriptional Activation and Ubiquitin-Mediated Protein Degradation
The expression and degradation of cyclins are carefully regulated so that the appropriate cyclins are present in the cell cycle stage at which they are needed. In this section, we discuss how cyclin levels are regulated. Control of cyclin gene transcription is one mechanism that ensures proper temporal expression of the cyclins. In somatic cells and yeast, waves of transcription factor activity help establish waves of cyclin activity. A general principle is that an earlier wave of CDK phosphorylationdependent transcriptional activity helps produce the factors essential to generate a subsequent CDK phosphorylation-dependent transcriptional wave. As we will see in Section 19.4, transcription of phase cyclins is promoted by the E2F transcription factor complex that becomes activated at this point in the cell cycle. A very important mechanism that restricts cyclins to the appropriate cell cycle stage is ubiquitin-mediated, proteasome-dependent protein degradation. Because protein degradation is an irreversible process, in the sense that the protein can be replenished only through de novo protein synthesis, this regulatory mechanism is ideal to ensure that the cell cycle engine is driven forward and that cells cannot go backward in the cell cycle. Recall from Chapter 3 that ubiquitin-protein ligases, also called E3ligases, transfer ubiquitin from an E2 ubiquitin-conjugating enzyme to one or more lysine residues on the E3-ligase’s substrate proteins, making long polyubiquitin chains that mark these proteins for degradation by proteasomes (see Figure 3-32). Cyclins are degraded through the action of
two different E3-ligases, called SCF (named after the first letters of its constituent proteins, Skp1, Cullin, and F-box proteins) and the anaphasepromoting complex or cyclosome (abbreviated as APC/C in this chapter). The mechanism of regulation of these two E3 ligases that are so critical for cell cycle control is strikingly different, though both involve protein phosphorylation. In the case of the SCF complex, it is the substrates that are phosphorylated to target them for degradation. In the case of the APC/C, it is the APC/C itself that is phosphorylated, controlling its activity. SCF controls the transition by targeting the and phase cyclins for degradation. SCF controls the transition by targeting the CDK inhibitory kinase Wee1, along with the and CDK inhibitory proteins for degradation, as we shall see in more detail shortly. The APC/C, in contrast, becomes activated at the onset of mitosis and remains active through the next phase of the cell cycle. The APC/C targets both S phase and mitotic cyclins for degradation. Activation of the APC/C during mitosis controls the onset of chromosome segregation at the metaphase-anaphase transition by degrading an anaphase inhibitory protein (as discussed in Section 19.6) and promotes mitotic exit. APC/C activity during inhibits mitotic and S phase CDKs, which allows replication origins to be reset so that a new round of replication can take place during the subsequent S phase (as discussed in Section 19.4). SCF and APC/C are multisubunit ubiquitin-protein ligases that belong to the RING finger family of ubiquitin-protein ligases. Despite the fact that SCF and APC/C belong to the same ubiquitin-protein ligase family, their
structures and substrate targeting regulation are quite different. The structures of both complexes are shown in Figure 19-14. These molecular assemblies resemble U- or C-shaped clamps. Human SCF complexes contain three core components and a substrate-targeting F-box protein. As shown in Figure 19-14a and b, the Cul1 subunit forms a central structural scaffold that links RBX1 and Skp1. RBX1 is a RING finger–containing protein that binds the E2 ubiquitin-conjugating enzyme. Skp1 is a bridging protein that binds the F-box protein. A number of different interchangeable F-box proteins bind to Skp1, and each one recruits a distinct set of substrate proteins to the SCF, where they are ubiquitinated and targeted for destruction.
FIGURE 19-14 E3 ubiquitin ligases control cell cycle progression. (a) SCF ligases are built from a central Cullin scaffold (Cul1) with a RING finger–containing Rbx1 protein and an E2 ubiquitin-conjugating enzyme at one end, and a Skp1 protein bound to an F-box substrate-targeting protein at the other. In this example, the F-box protein is Skp2, which uses a series of leucine-rich repeats to bind to target proteins only when they are phosphorylated. (b) This general architecture is conserved in all SCF ligases, but different phosphoserine/threonine-binding F-box proteins determine the substrate specificity. (c) The APC/C E3 ligases are assembled from 13 or 14 subunits, which are organized like SCF complexes into scaffolding (green), substrate-binding (yellow), and catalytic (red) functions. (d) The APC/C is inactive until it binds to one of the substrate-targeting coactivators, either Cdc20 or Cdh1. This binding activates the catalytic components and positions the Cdc20- or Cdh1-bound target protein for conjugation to ubiquitin (orange triangles). Description The illustration labeled (a) shows a U-shaped ribbon structure. It has the following parts labeled from left to right: F-box, S k p 1, C u l 1, R b x 1, and E 2. All of the portions are colored differently. The illustration labeled (b) depicts the same structure in (a) but in a schematic form. The F-box protein is tubular in shape with a white rod like structure labeled Target protein attached at the right. At the left of the tube, a U- shaped structure starts with the label S k p 1, continues around the U with C u l 1, and ends with a portion labeled R b x 1. A sphere above R b x 1 is labeled E 2 and attached to the top of this is a cone labeled ubiquitin, which has an arrow pointing to the Target protein. The illustration labeled (c) shows a C-shaped ribbon structure. A large area of ribbon is labeled A P C slash C scaffold and is a letter C shape. Inside the C shape is a small area labeled substrate-targeting module. At the right end of the letter C shape is an area labeled catalytic module. The illustration labeled (d) depicts the same structure in (c) but in a schematic form. The C shaped structure is labeled A P C slash C scaffold. It is attached to a rectangular shaped structure labeled substrate-targeting module. Attached to this is a rod-like structure labeled target protein. At the bottom right of the C shape a sphere is labeled
catalytic module with a cone on top labeled ubiquitin. An arrow from the cone points to the target protein. The anaphase promoting complex or cyclosome (APC/C) consists of 14 core subunits in metazoans and 13 core subunits in yeast, together with one of two related substrate-targeting factors called Cdc20 and Cdh1 (Figure 19-14c and d). The activity of APC/C is entirely dependent on binding of the core complex to one of these two substrate-targeting subunits. At the metaphase to anaphase transition, APC/C bound to Cdc20 targets cyclins A and B for degradation, while during anaphase, APC/C bound to Cdc20 ubiquitinylates proteins that inhibit chromosome segregation. During late anaphase, telophase and , APC/C bound to Cdh1 targets different substrates for degradation. Like SCF complexes, the APC/C core is organized into three modules (Figure 19-14c): a scaffold module that structurally organizes the entire complex, a catalytic module that accepts activated ubiquitin from an E2 enzyme, and a substrate recognition module that determines which proteins the APC/C will ubiquitinate. In the absence of phosphorylation, a loop on one of the APC/C core subunits blocks the APC/C binding site for the substrate-targeting subunit Cdc20. Phosphorylation of this loop by mitotic CDKs shifts its position in a similar manner to how phosphorylation activates protein kinases (Section 3.4). This allows Cdc20 to bind and results in the formation of an active complex that can ubiquitinate and target cyclins A and B for degradation, thereby shutting off mitotic CDK activity. Mitotic CDKs also phosphorylate the Cdh1 substrate-targeting subunit, which
Phosphoserine/Threonine-Binding Domains Build Feedback Loops That Coordinate CDK Activation and Cell Cycle Progression
prevents it from competing with the Cdc20 subunit for binding during metaphase and early anaphase. After targets the mitotic cyclins for degradation, the APC/C and Cdh1 become dephosphorylated through the action of protein phosphatases, particularly PP2A in vertebrate cells or Cdc14 in yeast, preventing Cdc20 binding but instead allowing Cdh1 to bind and form an active E3-ligase complex in late anaphase that persists until late of the next cell cycle. Substrate-targeting subunits of the APC/C recognize specific sequence motifs on the proteins that the APC/C ubiquitinates. One motif, the destruction box or D-box, has the consensus sequence RXXLX[D/E]ΦΦΦXN[N/S], where X stands for any amino acid, Φ means any hydrophobic amino acid, and the brackets indicate that either of the two amino acids is found at that position in the sequence. Experimentally mutating or deleting the destruction box in mitotic cyclins prevented their ubiquitination and blocked cells from exiting mitosis, demonstrating that mitotic exit requires degradation of mitotic cyclins. Phosphoserine/Threonine-Binding Domains Build Feedback Loops That Coordinate CDK Activation and Cell Cycle Progression The final molecular components that we need to discuss in order to understand cyclin-CDK regulation are phosphoserine/threonine-binding
domains. We have seen that phosphorylation by protein kinases plays an inordinately important role in controlling cyclin abundance, cyclin degradation, and CDK activity. While some of this regulation is a direct consequence of allosteric changes in the shapes of the phosphorylated proteins themselves, another important way that protein phosphorylation regulates the cell cycle is through the creation of short phosphoserine- or phosphothreonine-containing sequence motifs that are targeted for recognition by phospho-binding proteins or protein domains. The functions of these phosphoserine/threonine-binding domains are summarized in Table 19-1. These phosphoserine/threonine-binding proteins and modules orchestrate nearly all aspects of cell cycle control. In the most general sense, binding of phosphoserine/threonine-binding domains to CDK substrates or to proteins that control CDK activation works by either targeting the bound proteins to specific subcellular locations, altering their three-dimensional conformation, changing their activity, or targeting them for specific modifications such as additional phosphorylation or ubiquitin-mediated destruction. One example of this is found in the F-box proteins that comprise the substrate-targeting subunits of SCF ligases mentioned previously. Many F-box proteins contain modular phosphoserine/threonine-binding domains that bind to a substrate only if the substrate contains a specific phosphoserine/threonine-containing sequence. Thus the substrate protein is targeted for ubiquitination and degradation only if it has been phosphorylated previously by a protein kinase. As we will see in the next section, CDK-mediated phosphorylation of a CDK inhibitor can target it for SCF-mediated degradation, leading to
Mass Spectrometry Studies and Genetically Engineered CDKs Led to the Discovery of New CDK Substrates and Functions
more CDK activity, followed by additional CKI degradation, creating a feed-forward loop that amplifies total CDK activity. Other phosphoserine/threonine-binding molecules, including 14-3-3 proteins that regulate the cell cycle, Cks proteins that promote CDK phosphorylation, a proline isomerase called Pin1 that changes the activity of phosphorylated cell cycle regulators, Polo-box domains that target Polo kinase 1 to its substrates in mitosis, and FHA and BRCT domains that provide surveillance for DNA damage and control cytokinesis. These phosphobinding modules are necessary to form the positive and negative feedback loops that control progression through the cell cycle (Section 19-6) and for surveillance mechanisms that stop the cell cycle in response to catastrophic events (Section 19-7). The importance of being able to specifically recognize short sequences created by protein kinases as a mechanism of cell cycle control is evident from the fact that many different types of protein structures are capable of this type of phosphospecific binding, indicating that the phospho-binding function likely arose in these different proteins by convergent evolution. Mass Spectrometry Studies and Genetically Engineered CDKs Led to the Discovery of New CDK Substrates and Functions Different CDKs initiate different cell cycle phases by phosphorylating specific proteins. It is now clear that CDKs phosphorylate a myriad of
substrates, thereby directly initiating all aspects of a given cell cycle phase. In recent years, there have been systematic efforts to identify all of the CDK substrates. The challenge in identifying the substrates of a particular kinase is to distinguish one kinase’s phosphorylation events from those carried out by other kinases. Examination of mapped phosphorylation sites on known substrates of cyclin-CDKs and studies using synthetic peptides have revealed that cyclin-CDK complexes preferentially phosphorylate serine and threonine residues when they are found within the consensus sequence [S/T]PX[R/K]K where [S/T] (labeled the P0 position) is the site of CDK phosphorylation, and the sequence contains either an arginine or lysine in the P3 position, a lysine in the P4 position, or both. A breakthrough in understanding which proteins are targets of CDKs was facilitated by the engineering of a CDK mutant that uses an ATP analog that is not bound by other kinases. The ATP analog has a bulky benzyl group attached to of the adenine, which makes the analog too large to fit into the ATP-binding pocket of wild-type protein kinases. However, the ATP-binding pocket of the mutant CDK accommodates this -benzyl ATP analog. Consequently, only the mutant CDK can use this ATP analog as a substrate for transferring its γ phosphate to a protein side chain. When the -benzyl ATP analog with a thiol-labeled γ phosphate was incubated with cell extracts containing a recombinant mitotic CDK with the altered ATP-binding pocket, multiple proteins were labeled and then purified and identified by mass spectrometry (Figure 19-15). In yeast, this procedure
identified most of the known CDK substrates plus more than 150 additional yeast proteins.
FIGURE 19-15 Identification of CDK substrates using genetically engineered CDK mutants. (a) The ATP-binding pocket of CDKs was engineered to accept a bulky - benzyl ATP analog also containing a sulfur atom in place of a γ phosphate oxygen atom. Substrates of the mutant kinase that have been labeled with the ATP analog will contain a sulfur atom. (b) After protease digestion of cell proteins, the sulfur-containing peptides are purified using iodoacetamide beads (which bind sulfur), and the peptides are identified using mass spectrometry. Description
The illustration labeled (a) shows a wild-type kinase represented by a comma-shaped structure which is bonded to the chemical structure of A T P. Below this is a mutant kinase represented by a comma-shaped structure with a hole in the center bonded to N 6-benzyl thio-A T P. The inset shows a purple crescent moon shaped structure being attached to a phosphate group with one of the oxygen atoms replaced with a negatively charged sulfur atom. Below there are two other crescent moon shaped structure each bonded to a phosphate group. The illustration labeled (b) starts with an arrow labeled digestion pointing at four squiggly lines. The first squiggly line remains as is. The second one is bonded to a phosphate group with one of the oxygen atoms replaced with a negatively charged sulfur atom, the third one is bonded to an S H group, and the fourth one is bonded to a phosphate group. A rightward arrow from these structures is labeled iodoacetamide beads. It shows a chemical structure of the same. The second squiggly line bonded to a phosphate group with one of the oxygen atoms replaced with a negatively charged sulfur atom is further bonded to iodoacetamide. An arrow from this structure points at a text that reads, “identify by mass spectrometry.” We have now discussed all of the core components necessary for the cell to circumnavigate around the cell cycle: cyclins, CDKs, the phosphoserine/threonine motifs that CDKs create on their substrates, transcription factors, other protein kinases, phosphatases, CKIs, E3 ligases and the sequence motifs that they recognize, and phospho-binding proteins and domains. In the following sections, we see how these components work together to enable the cell to transition from one stage of the cell cycle to the next. We also describe the critical events that occur during each stage of the cell cycle. KEY CONCEPTS OF SECTION 19.3 Cell Cycle Progression and Control: Feedback Loops and Post-Translational Modification
Cyclin-dependent kinases are activated by cyclin subunits. Their activity is controlled at multiple levels. Different cyclin subunits activate CDKs at different cell cycle stages. Cyclins are present only in the cell cycle stages that they promote. Activating and inhibitory phosphorylation of the CDK subunit contributes to the regulation of CDK activity. CDK inhibitors (CKIs) inhibit CDK activity by binding directly to the cyclin-CDK complex. Protein degradation is the key mechanism responsible for restricting cyclins to the appropriate cell cycle stage. This degradation is mediated by the ubiquitin-proteasome system and the ubiquitin-protein ligases APC/C and SCF. Phosphoserine/threonine-binding proteins and modular domains are used to construct positive feed-forward and negative feedback loops that allow rapid activation and inactivation of CDK activity by binding to CDK regulators and substrates. CDKs initiate every aspect of each cell cycle stage by phosphorylating many different target proteins. Systematic efforts using protein kinases engineered to bind only modified forms of ATP have led to the identification of many of these substrates.
The G1/S Transition in Budding Yeast Is Controlled by Cyclin-CDK Complexes
19.4 The Transition from into S Phase and DNA Replication The previous section described the multiple mechanisms that control the different cyclin-CDK complexes. In this and the following two sections, we examine each cell cycle stage carefully and discuss how it is induced and controlled. We examine how cells initiate DNA replication and mitosis and how chromosomes are segregated. We focus on how cyclinCDK complexes and other key cell cycle regulators affect each cell cycle phase, and we examine the mechanisms that coordinate their activities. This section investigates how cells decide whether or not to undergo cell division, the molecular basis for the transition from into S phase, and how DNA replication is initiated. We begin with yeast. The Transition in Budding Yeast Is Controlled by Cyclin-CDK Complexes The process of cell cycle entry is particularly well understood in budding yeast, and it was in this organism that the molecular mechanisms underlying this cell cycle transition were initially elucidated.
Recall from Section 19.1 that the decision to enter S phase occurs late in and requires the accumulation of a labile R-factor that drives the cells through the restriction point, called START in yeast. Elegant genetic experiments identified temperature-sensitive mutants in budding yeast that arrest in , failing to form a bud and to initiate DNA replication. One group of these mutants was in the gene encoding the yeast CDK. (As mentioned earlier, budding yeast has only one CDK that triggers all cell cycle transitions and is known as CDC28.) This observation proved that CDK activity is essential for entry into S phase. A second set of mutants involved the gene for the labile R-factor, which turned out to be the yeast cyclin known as CLN3. We now know that a CDK cascade triggers further progress from through S phase in yeast (Figure 19-16a). The cyclin-CDK complex (Cln3-CDK in yeast) stimulates the formation of phase cyclin-CDKs, which initiate bud formation, centrosome duplication, and DNA replication. The mRNA for the yeast cyclin gene CLN3 is produced at a nearly constant level throughout the cell cycle, but its translation is regulated in response to nutrient levels and, as we will see shortly, it is a linchpin in coupling cell cycle entry to nutrient signals. Once sufficient Cln3 is synthesized from its mRNA, Cln3-CDK complexes phosphorylate and inactivate the transcriptional repressor Whi5. Phosphorylation of Whi5 promotes its export out of the nucleus, allowing the transcription factor complex SBF to induce transcription of the phase cyclin genes CLN1 and CLN2 as well as other genes important for DNA replication. Once produced, Cln1/2-CDKs contribute to further Whi5 phosphorylation. This positive feedback loop ensures the rapid accumulation of phase cyclin-CDKs. The point in the cell cycle at which 50 percent of Whi5 has exited the nucleus marks the point when
cells are irreversibly committed to division and provides a molecular definition of START. Cln1/2-CDKs then trigger bud formation, entry into S phase, and the duplication of the centrosome (also known as the spindle pole body in yeast), which will organize the mitotic spindle.
FIGURE 19-16 Control of the phase transition. (a) In budding yeast, activity of the cyclin–CDK Cln3-CDK rises during and is controlled by nutrient availability. Once sufficiently active, the kinase phosphorylates the transcriptional repressor Whi5, promoting its export from the nucleus. This releases the transcription factor complex SBF, allowing it to induce the transcription of the phase cyclin genes CLN1 and CLN2 and of other genes whose products are needed for DNA replication. phase CDKs further phosphorylate Whi5, promoting further CLN1 and CLN2 transcription. Once sufficiently high levels of phase CDKs have been produced, START is traversed. Cells irreversibly enter the cell cycle: they initiate DNA replication, bud formation, and spindle pole body duplication. (b) In vertebrates, CDK activity rises during and is stimulated by the presence of growth factors. When signaling from growth factors is sustained, the resulting cyclin D–CDK4/6 complexes begin phosphorylating Rb, releasing some E2F, which stimulates transcription of the genes encoding cyclin E, CDK2, and E2F itself. The cyclin E–CDK2 complexes further phosphorylate Rb, resulting in a positive
The G1–S Phase Transition in Metazoans Involves Cyclin-CDK Control of the E2F Transcription Factor Through Its Regulator Rb
feedback loop that leads to a rapid rise in the expression and activity of both E2F and cyclin E–CDK2. Once phase CDKs are sufficiently high, cells pass through the restriction point; they irreversibly commence DNA replication and centrosome duplication. Description The illustration labeled (a) has the following sequence: 1. S B F, a transcription factor, is blocked by W h i-5 both represented by two circles attached to each other bound to the C L N 1 or C L N 2 gene. 2. Under the effects of nutrient availability, C l n 3-C D K kinases are activated. W h i-5 gets phosphorylated to unbind from S B F which results in the transcription of C L N 1 or C L N 2 gene, which are required for D N A replication. An m R N A is formed. 3. C L N 1 and 2 C D K s then induce budding, the S phase, and spindle pole body duplication. The illustration labeled (b) has the following sequence: 1. In metazoans, R b blocks transcription of cyclin E or A gene by the E 2 F transcription factor, both represented by two circles attached to each other bound to the gene. 2. Growth factors activate cyclin D-C D K 4 and 6 complexes resulting in phosphorylated R b unbinding from E 2 F which results in the transcription of cyclin E or A gene to form an m R N A. Once the cyclin E or A C D K 2 concentration is high, the restriction point is passed, and the cell enters the S-phase and centrosome duplication. The Phase Transition in Metazoans Involves Cyclin-CDK Control of the E2F Transcription Factor Through Its Regulator Rb The sequence of events we described in yeast is similar in mammalian — and in fact all metazoan — cells. Growth factor signaling through the mitogen-activated protein kinase (MAPK) pathway triggers increased
transcription of the cyclins, members of the cyclin D family. As long as levels of the Ink4 CKI family members are low, cyclin D binds to the CDKs, CDK4 and CDK6, to form active cyclin-CDK complexes. In turn, these CDKs activate members of a small family of related transcription factors, referred to collectively as E2F transcription factors (E2Fs). During , the E2Fs are maintained in an inactive state through tight association with the retinoblastoma protein (Rb; Figure 19-16b). When CDKs become activated, they phosphorylate Rb, causing it to dissociate from the E2Fs. Released E2Fs then activate genes encoding many of the proteins involved in DNA synthesis. Thus the E2Fs have a function in late that is similar to that of the S. cerevisiae transcription factor complex SBF [compare (a) and (b) in Figure 19-16]. The E2Fs also stimulate transcription of genes encoding the phase cyclin, cyclin E, and the S phase cyclin, cyclin A. Cyclin E forms a complex with the CDK, CDK2, which then further phosphorylates Rb in a positive feedback loop, much like how the Cln1/2-CDK complex further phosphorylates Whi5 in yeast (Figure 19-16), to reinforce S phase entry. As noted above, key to the regulation of E2F function is the Rb protein. When E2Fs are bound to Rb, not only are they transcriptionally inactive, but they also function to repress transcription. This is because Rb recruits chromatin-modifying enzymes that promote deacetylation and methylation of specific histone lysines, causing chromatin to assume a condensed, transcriptionally inactive form (see Section 8.5). RB was initially identified as the gene mutated in retinoblastoma, a childhood cancer of the retina. Subsequent studies found Rb to be inactivated in
Extracellular Signals Govern Cell Cycle Entry
many cancers, either by mutations in both alleles of RB or by abnormal regulation of Rb phosphorylation (see Chapter 25). Rb protein regulation by CDKs in mammalian cells is analogous to Whi5 regulation by Cln3-CDK in yeast. Phosphorylation on multiple sites by CDKs blocks Rb from associating with E2F and promotes its export out of the nucleus. This allows E2F to activate the transcription of genes required for entry into S phase. Once the expression of genes coding for the cyclins and CDK has been induced by phosphorylation of some of the Rb molecules, the resulting phase CDK complexes further phosphorylate Rb in late . This is one of the principal biochemical events responsible for passage through the restriction point (Figure 1916b). Since E2F stimulates its own expression as well as that of the cyclin-CDKs, cross-regulation of E2F and cyclin-CDKs through a positive feedback loop produces a rapid rise of both activities in late . As they accumulate, S phase CDKs as well as mitotic CDKs maintain Rb protein in the phosphorylated state throughout the S, , and early M phases. After cells complete anaphase and enter early or , a fall in all cyclin-CDK activities leads to the de-phosphorylation of Rb. As a consequence, poorly phosphorylated Rb is available to inhibit E2F activity during early of the next cycle and in -arrested cells. Thus phase CDK activity remains low until cells decide to enter a new cell cycle and CDKs break the inhibitory grip of Rb on E2F.
Extracellular Signals Govern Cell Cycle Entry Whether or not cells enter the cell cycle is influenced by extracellular as well as intracellular signals. Unicellular organisms such as yeasts, for example, enter the cell cycle only when they have reached an appropriate size, known as the critical cell size. This critical size, in turn, is controlled by nutrients available in the environment. Here we restrict our discussion to the fact that cyclin synthesis is responsive to the rate of protein synthesis, which is in turn controlled by pathways that are regulated by nutrients in the environment. This link between the macromolecule biosynthesis machinery and the cell cycle machinery is well understood in budding yeast. In this organism, the cyclin transcript CLN3 contains a short upstream open reading frame that inhibits translation initiation when nutrients are limited. This inhibition is diminished when nutrients are in abundance. In the presence of sufficient nutrients, the TOR signaling pathway, which senses nutrients and growth factor signals, is active and stimulates translational activity (see Figure 21-3b). Since Cln3 is an unstable protein, its concentration fluctuates with the translation rate of its mRNA. Consequently, the amount and activity of Cln3-CDK complexes, which depend on the concentration of Cln3 protein, are regulated by nutrient levels. In multicellular organisms, cells are surrounded by nutrients, and as such, nutrients do not usually limit the rate of cell proliferation. Rather, cell proliferation is controlled by the presence of growth-promoting factors
(mitogens) and growth-inhibiting factors (anti-mitogens) in the cell surroundings. As discussed in Chapter 16, addition of mitogens to - arrested mammalian cells induces receptor tyrosine kinase–linked signal transduction pathways that initiate signal transduction cascades. These signaling cascades ultimately influence transcription and cell cycle control. They do so in multiple ways. Mitogens activate the transcription of multiple genes. Most of these genes fall into one of two classes — early response or delayed response genes — depending on how soon their encoded mRNAs appear. Transcription of early response genes is induced within a few minutes after addition of growth factors by signal transduction cascades that activate preexisting transcription factors in the cytosol or nucleus (see Chapter 16). Many of the early response genes encode transcription factors, such as c-Fos and cJun, that stimulate transcription of the delayed response genes. The early response transcription factor Myc induces the transcription of cyclin and CDK genes. In addition to being controlled by transcription, CDKs are regulated by CKIs. The CKI is a potent CDK inhibitor. In some tissues, mitogens inhibit the production of this CKI by inhibiting its transcription. In many tissues, cell proliferation is also regulated by anti-mitogens, which prevent entry into the cell cycle. Similarly, during differentiation, cells cease to divide and enter . Some differentiated cells (e.g., fibroblasts and lymphocytes) can be stimulated to re-enter the cell cycle and replicate. Many postmitotic differentiated cells, however, never reenter the cell cycle to replicate again. Anti-mitogens and differentiation
Degradation of an S Phase CDK Inhibitor Triggers DNA Replication
pathways prevent the accumulation of CDKs. They antagonize the production of cyclins and induce the production of CKIs. Transforming growth factor β (TGF-β) is an important anti-mitogen. This hormone induces a signaling cascade that brings about arrest by inducing the expression of . As we will see in Chapter 25, the signaling pathways that regulate CDKs are often mutated in many human cancers. Degradation of an S Phase CDK Inhibitor Triggers DNA Replication Entry into S phase is defined by the unwinding of origins of DNA replication. phase CDKs play an essential role in this process by turning off the machinery that degrades S phase cyclins during exit from mitosis and . In addition, they induce the degradation of a CKI that inhibits S phase CDKs, initiating a rapid transition from low to high S phase CDK activity. One of the important substrates of the phase cyclin-CDK complexes is Cdh1. During late anaphase, Cdh1 directs APC/C to ubiquitinylate substrate proteins, including S phase and mitotic cyclins, marking them for proteolysis by proteasomes (see Figure 19-14d). This ensures that when cells exit mitosis, they arrive into a state of low cyclin-CDK activity, allowing them to assess their internal and external environments prior to committing to another round of cell division. The complex remains active throughout , preventing the premature
accumulation of S phase and mitotic cyclins. If conditions for another round of cell division are favorable, then a cyclin-CDK induced signaling pathway results in phosphorylation of Cdh1, causing it to dissociate from the APC/C complex, inhibiting further ubiquitinylation of S phase and mitotic cyclins during late and allowing them to progressively accumulate (Figure 19-17). This block in cyclin degradation, combined with the induced transcription of S phase cyclins during late , allows S phase cyclins to accumulate as cyclin-CDK levels rise. Later in the cell cycle, S phase and mitotic CDKs take over to maintain Cdh1 in the phosphorylated, and hence inactive, state. Only as mitotic CDKs decline and a protein phosphatase in yeast known as Cdc14 becomes activated are these inhibitory phosphates removed from Cdh1, leading to its reactivation, again targeting mitotic proteins including M and S phase cyclins for degradation. In mammalian cells, similar mechanisms are responsible for stabilizing S phase and mitotic cyclins, but the phosphatase(s) involved in de-phosphorylation of Cdh1 have not been identified.
FIGURE 19-17 Regulation of S phase and mitotic cyclin levels in budding yeast. In late anaphase, the anaphase-promoting complex (APC/C) ubiquitinylates S phase and mitotic cyclins. The activity of this ubiquitin-protein ligase is directed toward mitotic cyclins by a specificity factor called Cdh1. Cdh1 activity is regulated by phosphorylation. During exit from mitosis and , Cdh1 is de-phosphorylated and is active. During S phase and mitosis, Cdh1 is phosphorylated, Cdh1 dissociates from APC/C, and APC/C becomes inactive. The phase CDKs, which themselves are not substrates, phosphorylate Cdh1 at the phase transition. A specific yeast phosphatase called Cdc14 removes the regulatory phosphate from the specificity factor late in anaphase. Description The illustration on the left is labeled exit from mitosis and G subscript 1, and the right one is labeled S phase and mitosis. The structure on the left shows a C-shaped structure labeled A P C slash C with three other structures bound to it. Attached to this is a sphere labeled C d h 1. The title below this structure reads, A P C slash C superscript C d h 1 S phase and mitotic cyclins are unstable. An arrow labeled G subscript 1 slash S phase C D K s points at the same structure except that C d h 1 now bonded to three phosphorus groups exits the complex. The title below this structure reads: A P C slash C superscript C d h 1 inactive S phase and mitotic cyclins are stable. A left pointing
arrow goes from the separated C d h 1 to the left diagram and is labeled C d c 14 phosphates. In S. cerevisiae, as S phase cyclin-CDK accumulates in late , they are immediately inactivated by binding of a CKI called Sic1 that is expressed late in mitosis and early in (Figure 19-18a). Because Sic1 specifically inhibits S phase and M phase CDK complexes but has no effect on the CDK and phase CDK complexes, it functions as an S phase inhibitor. Initiation of DNA replication occurs when the Sic1 inhibitor is precipitously degraded following its ubiquitinylation by the SCF ubiquitin-protein ligase (see Figure 19-14a).
FIGURE 19-18 Switch-like control of S phase onset in S. cerevisiae by phosphoregulated proteolysis of the S phase inhibitor Sic1. (a) The S phase cyclin-CDK complexes begin to accumulate in but are inhibited by Sic1. This inhibition prevents initiation of DNA replication until the cells have completed all events. phase CDKs assembled in late phosphorylate Sic1 at multiple sites (step 1 ), marking it for phospho-dependent binding and ubiquitinylation by the SCF ubiquitin-protein ligase and subsequent proteasomal degradation (step 2 ). The active S phase CDKs then trigger initiation of DNA synthesis (step 3 ) by phosphorylating and recruiting MCM helicase activators to DNA replication origins (not shown). (b) A single optimal phase CDK phosphorylation site in Sic1 that was strongly recognized by the phospho-binding F-box in SCF ubiquitin-protein ligase would result in a sluggish phase transition. As phase CDKs accumulate during , Sic1 would be progressively degraded. As a result, S phase CDKs would slowly rise, and initiation of S phase would be a drawn-out event. (c)
Six suboptimal phosphorylation sites in Sic1 create a switch-like cell cycle entry. Only when all of the suboptimal sites in Sic1 are fully phosphorylated is the protein recognized by the phospho-binding F box in SCF. This can only occur when phase CDKs have reached high levels. This ensures that Sic1 degradation occurs rapidly, and only when phase CDKs have accomplished all their other tasks. See P. Nash et al., 2001, Nature 414:514–521; and D. O. Morgan, 2006. Description The first illustration labeled (a) shows the following sequence. 1. An oval structure labeled S phase cyclin is attached to two structures labeled S I c 1 and C D K. the entire structure is labeled G subscript 1. Step 1: An arrow from another structure made up of an oval structure labeled G subscript 1 slash S cyclin attached to a structure labeled C D K points at another arrow pointing from the first structure to another complex. This complex is similar to the first structure except that the S I c 1 IS bonded to six phosphate molecules. Step 2: an arrow labeled Polyubiquitinylation of phosphorylated S I c 1 by S C F Proteasomal degradation from the previous structure point at a structure similar to that in the previous step except that the S I c 1 gets disintegrated. Step 3: a D N A undergoes D N A replication. The vertical axis of the graph labeled (b) titled one optimal G subscript 1 slash S phase C D K site in S i c 1 represents protein levels or activity. It has no units. The horizontal axis represents the progression from G subscript 1 to the S phase. It units are not given. Three curves represent S i c 1 protein, G subscript 1 slash S phase C D K activity, and S-phase C D K activity respectively. The activity of S i c 1 protein decreases as the cell enters S phase. S phase C DK activity increases as the cell moves toward the S phase. G subscript 1 slash S phase C D K activity peaks at the intersection of the previous two curves. The vertical axis of the graph labeled (c) titled six suboptimal G subscript 1slash S phase C D K site in S i c 1 represents protein levels or activity. It has no units. The horizontal axis represents the progression from G subscript 1 to the S phase. It units are not given. Three curves represent S i c 1 protein, G subscript 1 slash S phase C D K activity, and S-phase C D K activity respectively. The activity of S i c 1 protein is constant until the level of G subscript 1 slash S phase C D K activity increases to its peak. At this point, S i c 1 protein activity falls and S-phase C D K activity rises.
Degradation of Sic1 is induced by its phosphorylation by phase CDKs (Figure 19-18a). It must be phosphorylated at no fewer than six sites, each of which is a relatively poor substrate for the phase CDKs, before it is bound sufficiently well by SCF to be ubiquitinylated. In contrast, if Sic1 were inactivated following the phosphorylation of a single site, Sic1 molecules would start to get phosphorylated and degraded as soon as the levels of phase CDK activity begin to rise, leading to a gradual decrease in Sic1 levels (Figure 19-18b) and poor control over S phase entry. Because six sites need to be phosphorylated, at low levels of phase CDK activity Sic1 is only partially phosphorylated and Sic1 is not destroyed. Only when phase CDK levels are high is Sic1 sufficiently phosphorylated to target it for degradation. Having multiple suboptimal phosphorylation sites therefore leads to an ultrasensitive, switch-like response in Sic1 degradation and hence precipitous activation of S phase CDKs (Figure 19-18c). Sic1 degradation occurs only when phase CDK activity has reached its peak and virtually all the other phase CDK substrates have been phosphorylated. An obvious advantage of proteolysis for controlling passage through this critical point in the cell cycle is that protein degradation is an irreversible process, ensuring that cells proceed in one direction through the cycle. Entry into S phase in metazoan cells is regulated by a switch-like mechanism similar to that in budding yeast. Like Sic1, the CKI p27 binds to and prevents the premature activation of S phase CDKs during . Unlike Sic1, however, this CKI inhibits both S phase CDKs and phase CDKs. Like Sic1 in yeast, p27 is removed from cyclin-CDK
Replication at Each Origin Is Initiated Once and Only Once During the Cell Cycle
complexes by ubiquitin-dependent protein degradation and a pathway, analogous to the one operating on Sic1, targets p27 for degradation at the phase transition. As phase CDKs and S phase CDKs reach high levels during late and early S phase, they begin to phosphorylate p27, targeting it for ubiquitinylation by . Degradation of p27 causes activation of phase and S phase CDKs. These kinases then initiate S phase by phosphorylating proteins important for the initiation of DNA replication. Replication at Each Origin Is Initiated Once and Only Once During the Cell Cycle As discussed in Chapter 5, eukaryotic chromosomes are replicated from multiple replication origins, specific sites on the chromosomes where the replication machinery is assembled, and where the copying of the two strands of DNA begins. Replication of all of the chromosomal DNA — starting from these origins — occurs during S phase. Each eukaryotic origin fires only once per S phase to initiate replication from that site, and S phase continues until replication from each of the multiple origins that lie along the length of each chromosome has resulted in complete replication of the entire chromosome. The DNA that is copied from a single origin is called a replicon. This process of creating and fusing the replicons ensures that each DNA strand is copied only once, maintaining the correct gene copy number each time a cell replicates.
S phase CDKs play an essential role regulating DNA replication. As we shall see in more detail shortly, these kinases allow DNA replication to be initiated only at the phase transition and prevent re-initiation from origins that have already fired. We first discuss how initiation of DNA replication is controlled, and the role of S phase CDKs in the process, before turning to the mechanisms these kinases use to prevent reinitiation. The powerful genetic methods available for experimental manipulation of budding yeast have led to the identification of specific mutants defective in DNA replication. Consequently, we have a fairly comprehensive understanding of the mechanisms by which yeast initiates and implements DNA replication. Although many details differ between yeast and mammalian cells, the basic processes are essentially the same, so we will focus our discussion first on how DNA replication is accomplished in budding yeast. It is important to remember that the five basic steps of genome replication are essentially the same in all eukaryotic species (Figure 19-19). The steps are: (1) recognizing the replication origins; (2) assembling replication initiation factors that then load a replicative helicase in order to form a so-called prereplication complexes, or pre-RCs; (3) recruiting critical helicase cofactors; (4) activation of the helicase to unwind the two DNA strands; and (5) recruitment of the replicative DNA polymerase machinery (i.e., the “replisome”) that then copies each strand.
FIGURE 19-19 The molecular mechanisms governing the initiation of DNA replication. Step 1 : During exit from mitosis and early , when CDK activity is low, ORC recognizes and binds to replication origins. In step 2 , the MCM loading factors Cdc6, and Cdt1 load the inactive replicative helicase, the MCM complex, onto DNA at replication origins, forming the pre-RC complex. Step 3 : Activation of S phase CDKs and DDK marks the onset of S phase. They phosphorylate the MCM helicase, Sld2, and Sld3 (depicted as yellow phosphorylation events), to facilitate the loading of MCM helicase activators — the Cdc45 and GINS complexes — onto sites of replication initiation. Step 4 : Loading of these activators leads MCM helicases to unwind DNA. S phase CDKs also prevent reloading of MCM helicases by phosphorylating Cdc6 and Cdt1 promoting their release from the replication origins and their nuclear export or degradation by ubiquitin ligases. S phase CDKs also phosphorylate MCM helicases (depicted as orange phosphorylation events), which leads to their export from the nucleus when the helicases disengage from the DNA when replication is complete. Step 5 : The additional DNA polymerases Pol α (the primase) and Pol δ, along with the processivity factor PCNA, are recruited to origins, which leads to the initiation of DNA replication on both the leading and lagging strands (see Figure 5-30). [Data from Y. Li and H. Araki, 2013, Genes Cells 18:266–277.] Description In the illustration, the first two steps are labeled together as low C D K state. Step 1: shows a D N A helix with a structure made of 5 spheres being attached to it. The label reads: O R C binding to replication origins. Step 2: shows two structures made of 5 oval shaped structures attach to the D N A helix to the left of the O R C. The label reads: M C M helicase loading and replication licensing. To the left of the arrow is another label: C d c 6 C d t1. The rest of the steps have a group label to the left reading high C D K state. Step 3 includes a group of steps where many structures are added to the M C M area. The step 3 label reads: forming the complete C M G helicase complex. A downward arrow from step 2 is labeled D D K and a structure labeled S l d 3-S l d 7-C d c 45 moves to get attached to the M C M. The next arrow is labeled C D K and shows the structures being added with phosphate circles (green). Into the next arrow is a line leading to a close up of structures, labeled D p b11, S l d 2 and the other labeled G I N 5 and Pol epsilon being attached to the C D K. Step 4: is labeled helicase activation,
and shows the C D K slash M C M structure stretching apart in the middle. Step 5: is labeled polymerase recruitment and D N A replication and shows the addition of 3 small structures labeled Pol alpha, Pol delta and P C N A. Even though DNA replication will not occur until S phase, the first 2 steps actually occur in phase, when the activity of and S phase CDKs is low. As shown in Figure 19-19, in step 1 , a complex of six proteins known as the origin-recognition complex (ORC) binds to all of the DNA replication origins. In budding yeast, an ORC binds to an 11-bp conserved core sequence found in every replication origin. The situation is more complicated in multicellular organisms because DNA replication origins lack any recognizable consensus sequence. Instead, it is thought that additional chromatin-associated factors target ORC to the DNA at replication origins. Once bound to DNA, ORC is thought to function as a landing pad for the assembly of additional factors necessary for DNA replication to commence. During step 2 , two replication initiation factors, Cdc6 and Cdt1, bind to ORC at the origins and then proceed to load another complex of six proteins, known as the MCM helicase (for mini chromosome maintenance), onto the DNA to form a pre-RC. The MCM complexes appear to be loaded as double hexamers facing in opposite directions, allowing DNA unwinding to progress in both directions from the origins. Once loaded onto the origin DNA in phase, the helicases remain inactive until the cell has entered S phase. This restriction on activity is conferred by CDKs, which regulate two opposing phosphorylation states. The MCM helicases can only be loaded onto the DNA in a state of low CDK activity that occurs when CDKs are inactivated during exit from mitosis and during early In other words,
MCM helicases are loaded onto DNA to form pre-RCs only when they are nonphosphorylated. In contrast to DNA loading, activation of the MCM helicases and the recruitment of DNA polymerases to the unwound origin DNA can only occur when the MCM helicase proteins are phosphorylated. This happens only in S phase, when they are phosphorylated by S phase CDKs and a second protein kinase called DDK (for Dbf4-dependent kinase). Just like CDKs, which require cyclin-binding for activity, the monomeric DDK kinase subunit (also known as Cdc7) is inactive in its monomeric form. Only when DDK binds to the Dbf4 subunit does it acquire kinase catalytic activity. During , the Dbf4 protein is continuously targeted for degradation by the APC/C. Only during S phase, when the APC/C is inactivated by CDK phosphorylation, does Dbf4 accumulate and activate DDK. (Recall that S phase CDKs become active only when phase CDK levels reach their peak, allowing the CKIs of S phase CDKs to be destroyed.) Putting all of this phosphorylation and ubiquitinationmediated control together, the key concept that emerges is that the increase in CDK and DDK activity during S phase enforces the strict temporal separation of pre-RC formation (i.e., loading of the MCM helicases), which only happens during , from firing of the origins to start DNA replication, which only happens during S phase. Formation of the pre-RCs is sometimes referred to as replication licensing, meaning that the MCM-loaded origins are now licensed to fire later in S phase. By only licensing origins during (Figure 19-19, steps 1 and 2 ), and only firing origins during S phase (Figure 19-19, steps 3 – 5 ), the cell ensures that each origin only triggers DNA replication once with each
passage through the cell cycle, thereby preventing inappropriate genomic re-replication. So how do S phase CDKs and DDK collaborate at the mechanistic level to initiate DNA replication and prevent re-replication? The process is rather complicated, and the exact order of events differs somewhat between yeast and metazoans, but the basic concept, which is the same in all cells, is to recruit helicase cofactors (step 3 ) that will activate the MCM helicases (step 4 ) at the same time that the replicative polymerase machinery (the replisome) is loaded onto the DNA (step 5 ). This coordination prevents excessive unwinding of DNA, which would result in the accumulation of large amounts of single-stranded DNA. The activation process involves binding of inactive MCM helicase to two other protein complexes, one containing Cdc45 and the other containing GINS, in order to form the active Cdc45-MCM-GINS (CMG) helicase complex. Formation of the CMG complex requires phosphorylation events catalyzed by both DDK and S phase CDKs, which are activated in late . In yeast it happens like this: DDK phosphorylates the N-termini of several subunits of the MCM helicase, altering their conformation, and leading to the recruitment of one of the two critical MCM helicase cofactors, a protein known as Cdc45. Cdc45 is required for MCM helicase activity, and binds to the MCM helicase as part of a complex with two other proteins Sld3 and Sld7 (Figure 19-19, step 3 ). Sld3 recruits another protein complex containing Pol ε (the replicative polymerase that synthesizes the leading strand), forming a fully active CMG helicase. The origins unwind (Figure 19-19, step 4 ), Pol α (the primase), Pol δ (the replicative polymerase that synthesizes the lagging strand), and PCNA (the DNA sliding clamp that
holds the polymerases anchored to the DNA to increase processivity) then bind, and replication ensues (Figure 19-19, step 5 ). S phase CDKs are not only essential for initiating DNA replication but also are responsible for ensuring that each origin fires only once during S phase. Refiring of origins during S phase is prevented by phosphorylation of several components of the MCM helicase-loading machinery and the MCM helicase complex itself. To distinguish these phosphorylation events from the ones required for the initiation of DNA replication, they are depicted in orange in Figure 19-19. Concomitant with activation of the MCM helicases, Cdc6 and Cdt1 dissociate from the sites of DNA replication initiation. Once they have done so, their phosphorylation leads to export of Cdt1 from the nucleus, and degradation of Cdc6 by the SCF ubiquitin-protein ligase in yeast. Phosphorylation of MCM helicases leads to the export of these proteins from the nucleus after they dissociate from the DNA on completion of DNA replication. Thus only after CDK activity is lowered by during exit from mitosis can MCM helicases be reloaded onto DNA. As a result, helicase loading is restricted to late stages of mitosis and early (see Figure 19-19, steps 1 and 2 ). The general mechanisms governing the initiation of DNA replication in metazoan cells parallel those in S. cerevisiae, although some differences are found in vertebrates. The helicases are loaded in , when CDK activity is low. Phosphorylation of MCM helicase activators by phase CDKs and S phase CDKs activates the helicases and promotes polymerase loading. As in yeast, phosphorylation of the MCM helicaseloading factors Cdc6 and Cdt1 prevents reloading of MCM helicases until
Duplicated DNA Strands Become Linked During Replication
the cell passes through mitosis, thereby ensuring that replication from each origin occurs only once during each cell cycle. In addition, vertebrates contain a small protein, geminin, that also contributes to the inhibition of re-initiation at origins until cells complete a full cell cycle. Geminin is expressed in late ; it binds to and inhibits Cdt1 (see Figure 19-19, step 2 ), thereby preventing reloading of the MCM helicase that is released from origins after DNA replication is initiated during S phase. Geminin contains a destruction box at its N-terminus that is recognized by , causing it to be ubiquitinylated in late anaphase and degraded by proteasomes. Its degradation frees the MCM helicase-loading factors, which are also de-phosphorylated as CDK activity declines, to bind to ORC on replication origins and load MCM helicases during the following phase. During S phase, the combined ubiquitin-mediated degradation of Cdt1 and the inactivation of any residual Cdt1 left over by geminin provides redundant regulation to ensure that there is no accidental chromosome re-replication. Duplicated DNA Strands Become Linked During Replication It is essential that the sister chromatids generated during S phase remain together until they separate during the metaphase-to-anaphase transition of mitosis. As chromosomes are duplicated to form sister chromatids, they become tethered to each other by protein links. The protein ring complexes that establish these linkages between sister chromatids are called cohesins. They are composed of four subunits: Smc1, Smc3, Scc1,
and Scc3 (Figure 19-20a). Smc1 and Smc3 are members of the structural maintenance of chromosomes (SMC) protein family, which is characterized by globular domains at the N- and C-termini connected by a long α helix. The helix is broken in the middle by a globular hinge domain, so that the two halves of the α helix in each protein fold back on themselves to form a coiled-coil structure. The globular head domains bind and hydrolyze ATP. The ATPase domains interact with the proteins Scc1 and Scc3, which together form a ring structure. These cohesin rings embrace one or both copies of the replicated DNA. When cohesins are experimentally inactivated, sister chromatids do not associate properly with each other.
FIGURE 19-20 Model for establishment of cohesin linkage of sister chromatids. Cohesin complexes form rings that link sister chromatids by embracing the two sister DNA molecules. (a) Schematic structure of the yeast cohesin complex. The structure of vertebrate cohesion is essentially the same, though some of the proteins have different names. (b) Mechanism whereby cohesins are loaded onto DNA and acquire their cohesive properties. Step 1 : Cohesins are loaded onto chromosomes during by the cohesin-loading complex Scc2-Scc4, but the cohesins do not possess cohesive (gluelike) properties. In this
state, cohesins are dynamic and can slide along the DNA or dissociate from the DNA with the help of the Pds5-Wapl complex, which associates with cohesins. Step 2 : Concomitant with DNA replication, closely behind the replication fork cohesins are converted into cohesive molecules, able to glue sister chromatids together through acetylation of Smc3 by cohesin acetyltransferases (CoATs). Acetylation, indicated by brown circles on SMC3, is accompanied in vertebrates by the binding of sororin to cohesin, which helps stabilize cohesins on chromosomes. In budding yeast, both sister chromatids are embraced within a single cohesin ring, as shown. (c) Cohesion-independent functions of cohesins during include partitioning of the genomic DNA into discrete compartments of the nucleus called topologically associated domains (TADs) and controlling gene expression by bringing enhancers close to promoters and transcription start sites through chromosomal DNA looping. In combination with CCCTC-binding factor (CTCF), cohesin-looping can activate some genes in a cluster, shown by arrows, while repressing others, indicated by the T symbol. [Part (c) Information from A. Losada, 2014, Nat. Rev. Cancer 14:389–393.] Description The illustration labeled (a) shows the structure of a cohesin ring. The left side of the ring is labeled S mc 1 and is blue, the right side is labeled S m c 3 and is red. They meet at the top where the top is labeled hinge region. The the bottom of this ring is labeled A T Pase heads. Below these are structures labeled, from top right around to top left: S c c 3, W a p l, Sororin, P d s 5, and S c c 1 represented by tiny spheres. The illustration labeled (b) shows a two-step mechanism where cohesin rings attach to the chromosome. The chromosome is represented by a rod shaped structure with a sphere attached in the center labeled centromere. Step 1: shows a backward arrow labeled P d s 5-W a p l to this structure and a forward arrow below this labeled S c c 2-S c c 4. There is a ring labeled cohesins in the vicinity. The forward and backward arrows point at a chromosome inside five cohesin rings. Step 2: A forward arrow labeled C o A T points at a structure. Below this is an oval labeled Soronin. The structure shows 3 cohesin rings at the top of the chromatids, of which there are now two labeled cohesed sister chromatids. The ring at the top has a label reading acetylated, and the bottom of the 3 rings has a structure labeled replication fork. A downward arrow indicates that the
replication will continue downward. Another ring is above the centromere and one is below it. The illustration labeled (c) shows three structures below G subscript 1. The first structure labeled genome compartmentalization shows genomic D N A, a thread-like structure with 6 tangles in it. The first tangle is labeled T A D. The second and third structures are labeled transcription regulation. The second structure shows two cohesin rings around a bend of the D N A and below are three ovals labeled, left to right: Enhancer, Transcription factor, Promoter. The third structure shows a closer view that shows 3 promotor and one enhancer structure above the ring, two pink circles below the ring labeled C T C F. Below these is another ring around a loop of D N A with three more enhancers attached. Cohesin-mediated cohesion between sister chromatids is established by a two-step process and is tightly tied to DNA replication. In the first step, which occurs during , cohesins associate with chromosomes, aided by cohesin loading factors Scc2 and Scc4 (Figure 19-20b, step 1 ). In , the chromosome-cohesin interaction is quite dynamic. In addition to Scc2Scc4–mediated binding, cohesins are unloaded continuously by a cohesinassociated complex composed of the Pds5 and Wapl proteins. This continual loading and unloading of cohesins is likely to be important in regulating interphase chromatin structure and gene expression (Figure 1920c). Cohesins acquire their cohesive properties during DNA replication. In the second step, the two duplicated DNA strands become entrapped within the cohesin rings as replication forks replicate the DNA (Figure 19-20b, step 2 ). Converting DNA-bound cohesins into cohesive complexes requires acetylation of the Smc3 subunit by cohesin acetyl transferases
(CoATs). This acetylation prevents unloading of cohesins by Pds5-Wapl, thereby stabilizing the cohesins on chromosomes. In vertebrates, stabilization of cohesins requires the cohesin-associated factor sororin (Figure 19-20a and b, step 2 ). As we will see in Section 19.6, cohesins are essential for accurate attachment of the replicated sister chromatids to the mitotic spindle and for their segregation during mitosis. Cohesins also regulate the topological localization of chromosomes within the interphase cell nucleus and help control gene expression (Figure 19-20c). In some instances, cohesins promote gene expression, whereas in others they restrain it. The mechanism cohesins use to accomplish gene expression control is, however, the same in both cases: cohesins promote chromatin loop formation, which brings enhancer or repressor elements close to the promoter and transcriptional start site. Defects of cohesin’s gene expression regulation function are the cause of a group of diseases collectively called cohesinopathies. In these diseases, mutations in cohesin subunits or cohesin-loading factors disrupt the expression of genes that are critical for development, causing limb and craniofacial abnormalities and intellectual disabilities. Cohesin’s sister chromatid cohesion function, however, is intact in these diseases. In contrast, as we will see in Section 19.8, defects in cohesin’s cohesion function during meiosis cause miscarriages and intellectual disabilities. KEY CONCEPTS OF SECTION 19.4 The Transition from into S Phase and DNA Replication
The molecular events promoting entry into the cell cycle are conserved across species. CDKs phosphorylate and inhibit a transcriptional repressor. This permits transcription of phase cyclin genes and other genes important for S phase (see
Figure 19-16). Extracellular signals such as nutritional state (in yeast) and the presence of mitogens and anti-mitogens (in vertebrates) regulate entry into the cell cycle. Various polypeptide growth factors called mitogens stimulate cultured mammalian cells to proliferate by inducing expression of early response genes. Many of these genes encode transcription factors that stimulate expression of genes encoding the phase cyclins and E2F transcription factors. The phase CDKs phosphorylate and inhibit Cdh1, the factor that directs the anaphase-promoting complex (APC/C) to ubiquitinylate S phase and M phase cyclins. This allows S phase cyclins to accumulate in late (see Figure 19-17). In yeast, S phase CDKs are initially inhibited by Sic1. A series of phosphorylation sites marks Sic1 for binding to a phosphoserine/threonine-binding domain on an SCF ubiquitin-protein ligase, leading to its ubiquitinylation and proteosomal degradation. This releases activated S phase CDKs that trigger an abrupt step-like onset of the S phase (see Figure 19-18). A similar phospho-dependent degradation of Cip/Kip CKI proteins functions in a similar manner to control S phase entry in animal cells. DNA replication is initiated from helicase loading sites known as replication origins. Loading and activation of MCM helicases occur in mutually exclusive cell cycle states: MCM helicase loading can occur only when CDK activity is low (during early ); MCM helicases are activated when CDK activity is high (during S phase). S phase CDKs and DDK trigger the initiation of DNA replication by recruiting MCM helicase activators to origins (see Figure 19-19). Initiation of DNA replication occurs at each origin only once during the cell cycle because S phase CDKs activate the helicases and at the same time prevent additional helicases from loading onto DNA. Cohesins establish linkages between the replicated DNA molecules (see Figure 1920), which are essential for their accurate segregation later in the cell cycle. This linking mechanism is coupled to DNA replication.
Precipitous Activation of Mitotic CDKs by Positive Feedback Loops Initiates Mitosis
19.5 The Transition and the Irreversible Engine of Mitosis Once S phase has been completed and the entire genome has been duplicated, the pairs of duplicated DNA chromosomes — the sister chromatids — are segregated to the future daughter cells. This process requires not only the formation of the apparatus that facilitates this segregation — the mitotic spindle — but essentially a complete remodeling of the cell. Chromosomes condense and attach to the mitotic spindle, the nuclear envelope is disassembled, and almost all organelles are rebuilt or modified. These events are triggered by mitotic CDKs and a handful of other key mitotic kinases, particularly the Aurora kinases and the Polo-like kinases. This section first discusses how the mitotic CDKs are precipitously activated during , following the completion of DNA replication, through a series of positive feedback loops involving the CDKs themselves, together with Aurora kinase A and Polo-like kinase 1 (Plk1). We then describe how these kinases bring about the dramatic changes in the cell necessary to facilitate sister chromatid segregation during anaphase, focusing on the events as they occur in metazoans. Precipitous Activation of Mitotic CDKs by Positive Feedback Loops Initiates Mitosis
Mitotic cyclin-CDKs initiate mitosis. Whereas levels of the catalytic CDK1 subunit (the mitotic CDK) are constant throughout the cell cycle, mitotic cyclins gradually accumulate during S phase. Most eukaryotes contain multiple mitotic cyclins, which for historical reasons are subdivided into the cyclin A and cyclin B families. As they assemble, mitotic cyclin-CDK complexes are maintained in an inactive state through inhibitory phosphorylation of the CDK1 subunit. Recall from Section 19.3 that two highly conserved tyrosine and threonine residues in mammalian CDKs are subject to regulated phosphorylation. In CDK1, phosphorylation of threonine 14 and tyrosine 15 maintains mitotic cyclin-CDK complexes in an inactivate state (see Figure 19-12). The phosphorylation state of T14 and Y15 is controlled by a dual-specificity protein kinase known as Wee1 and a pair of dual-specificity phosphatases, Cdc25B and Cdc25C). Dualspecificity kinases and phosphatases can phosphorylate and dephosphorylate serines, threonines, and tyrosines. Their regulation of mitotic CDK1 underlies the abrupt activation of CDK1 kinase activity at the phase transition and explains the observation that although mitotic cyclins gradually accumulate during S phase and , mitotic CDKs are not active until cells enter mitosis. The critical roles of Wee1 and Cdc25 in controlling the entry of cells into mitosis emerged first from studies in the fission yeast Schizosaccharomyces pombe. Recall from Section 19.2 that fission yeast coordinate cell size and cell division during (see Figure 19-4). This is in contrast to budding yeast and vertebrate cells, which coordinate cell size and cell division during and therefore spend most of the cell cycle in this phase. This means that the length of fission yeast at the time they
divide is a direct reflection of the duration of . In S. pombe the dualspecificity protein kinase Wee1 phosphorylates CDKs on the inhibitory tyrosine 15 (see Figure 19-13). (Threonine 14 is not phosphorylated in S. pombe CDK1.) Yeast cells with a defective gene activate mitotic CDKs prematurely and hence experience premature entry into mitosis. These wee1 mutants are therefore smaller than normal cells. In fact, the Scottish term for small is “wee,” hence the name for this gene that, when mutated, resulted in small cells. Consistent with the importance of Wee1 in phosphorylating Tyr-15 in CDK1, fission yeast cells carrying a CDK1 mutation in which the tyrosine-15 residue is replaced by phenylalanine (which is structurally similar to tyrosine but cannot be phosphorylated) show the same premature mitotic CDK activation and entry into mitosis. In contrast, fission yeast cells carrying mutations in the cdc25 gene arrest in , giving rise to very long cells. This indicates that this phosphatase, which opposes Wee1 activity, is essential for entry into mitosis. Vertebrates contain the same Wee1 protein kinase as S. pombe but have multiple Cdc25 phosphatases. Two family members, Cdc25B and Cdc25C, remove the inhibitory phosphorylation on CDK1 during . Cdc25B is thought to act as a starter phosphatase that initiates CDK1 activation, while Cdc25C is the workhorse phosphatase that activates the vast majority of cyclin-CDK1 complexes. Abrupt activation of mitotic CDKs at the transition is the consequence of rapid inactivation of Wee1 together with activation of Cdc25. Central to this rapid transition are two positive feedback loops built from CDKs, Polo-like kinase 1 (Plk1), and phosphoserine/threonine-
binding domains, in which mitotic CDKs and Plk1 simultaneously activate Cdc25 and inactivate Wee1 (Figure 19-21). The process begins with Cdc25B, the starter phosphatase, activating an initial small pool of cyclin B-CDK1 at the centrosome (Figure 19-21, step 1 ), which is thought to facilitate centrosome maturation. This initial pool of active cyclin BCDK1 that is generated then phosphorylates Cdc25C, the workhorse phosphatase, in the cytoplasm. This recruits Pin1 and Plk1, which recognize Cdc25C through their phospho-binding domains. These proteins further modify and phosphorylate Cdc25C to stimulate its phosphatase activity (Figure 19-21, step 2 ). This pool of active Cdc25C that is created, in turn activates the large pool of cytosolic cyclin B-CDK1, which activates more cytosolic Cdc25C as part of positive feedback loop 1. At the same time that Cdc25C is being activated, mitotic CDKs and Plk1 phosphorylate Wee1, which targets it for destruction through a phosphobinding SCF protein-ubiquitin ligase. This forms positive feedback loop 2 (Figure 19-21, step 3 ). This combination of two positive feedback loops — one that activates a CDK1 activator (Cdc25C), and one that inhibits a CDK1 inhibitor (Wee1) — results in the abrupt and irreversible transition from into mitosis.
FIGURE 19-21 Positive feedback loops involving CDKs, Plk1, and phospho-binding domains result in abrupt mitotic CDK activation and the transition from into M phase. Although mitotic cyclins are synthesized during late S phase and and bind to CDK1, the cyclin-CDK complex is not active because threonine 14 and tyrosine 15 of the CDK1 subunit are phosphorylated by the protein kinase Wee1 (shown as red kinases), while the activating protein phosphatase, Cdc25, is phosphorylated and bound and inhibited by the phosphoserine/threonine-binding protein 14-3-3. The entire process of mitotic cyclinCDK activation and entry into mitosis involves two positive feedback loops and coordination between events at the centrosome, in the cytoplasm, and in the nucleus. Step 1 : At the centrosome, an initial pool of cyclin B-CDK1 is activated by Cdc25B, which removes the inhibitory phosphorylations on threonine 14 and 15. This initial pool of active cyclin B-CDK1 then phosphorylates 14-3-3-bound Cdc25C on multiple serine-proline or threonine-proline sequences in the cytoplasm (shown as blue circles). Step 2 : The resulting phosphoserine/phosphothreonine-proline motifs created by cyclin B-CDK target
Cdc25 for recognition by specific phospho-binding domains within the proline isomerase Pin1 and the protein kinase Plk1, which then bind to and further activate Cdc25C. Pin1 changes the conformation of Cdc25C while Plk1 catalyzes additional activating phosphorylations (shown as black circles). This releases Cdc25C from 14-3-3, allowing it to further de-phosphorylate and activate the larger cytosolic pool of cyclin B-CDK1. The resulting cyclin B-CDK1 drives further release and activation of more 14-3-3-bound Cdc25C, forming positive feedback loop 1. Step 3 : At the same time, the active cyclin BCDK1 in the cytosol phosphorylates its inhibitory kinase Wee1 on serine-proline motifs, targeting it for recognition by the phospho-binding domain in Plk1. Plk1 then phosphorylates Wee1 on a motif that is recognized by the phospho-binding domain in the SCF-βTrCP ubiquitin protein ligase, targeting Wee1 for ubiquitination and destruction by the proteasome. This loss of Wee1 inhibition further enhances cyclin B-CDK1 activation, forming positive feedback loop 2. Step 4 : Active cytoplasmic cyclin B-CDK1 translocates into the nucleus, where it phosphorylates nuclear lamins to cause nuclear envelope breakdown and, together with Plk1 and the Aurora kinases, induces chromosome condensation and formation of the mitotic spindle. Description The four steps in this illustration are described in detail in the caption. The location of two positive feedback loops depicted. Positive feedback loop 1 is present in step 2 at the center of the illustration and Positive feedback loop 2 is present in step 3 at the bottom right of the illustration. Step four shows a nucleus with chromosome replication represented by blue shapes at the center, then an arrow pointing to a nuclear membrane that is a dotted line and two sets of chromatids in the center. Once the precipitous activation of mitotic CDKs is initiated, these protein kinases set in motion all the events necessary to ready the cell for chromosome segregation. The initial activation of mitotic CDKs at the centrosome, followed by activation in the cytoplasm, results in changes in the subcellular localization of these kinases. They enter the nucleus, where they bring about chromosome condensation and nuclear envelope
Mitotic CDKs Promote Nuclear Envelope Breakdown
breakdown (Figure 19-21, step 4 ). In what follows, we discuss how the mitotic CDKs accomplish the coordinated execution of mitosis. Just as S phase CDKs work together with DDK to promote MCM helicase activation during DNA replication initiation (see Section 19.4), mitotic CDKs collaborate in a similar manner with other protein kinases to bring about the mitotic events. Plk1 is critical for formation of the mitotic spindle as well as for chromosome segregation. The Aurora kinase family members Aurora A and Aurora B play key roles in mitotic spindle formation and in ensuring that chromosomes attach correctly to the mitotic spindle so that they are segregated accurately during mitosis. Their contributions to the various mitotic events will also be discussed. Mitotic CDKs Promote Nuclear Envelope Breakdown During interphase, chromosomes are surrounded by the nuclear envelope, while the centrosomes that nucleate the mitotic spindle are located in the cytoplasm. For nuclear chromosomes to interact with the cytoplasmic microtubules nucleated by the centrosomes, the nuclear envelope must be dismantled. The nuclear envelope is a double-membrane extension of the endoplasmic reticulum containing many nuclear pore complexes (Figure 19-22a; see also Figures 1-12, 1-15, 13-33, and 18-52). The lipid bilayer of the inner nuclear membrane is associated with the nuclear lamina, a meshwork of
lamin filaments adjacent to the inside face of the nuclear envelope (Figure 19-22b) that participate in maintenance of nuclear structure and chromosome positioning within the nucleus (Figure 19-22c; see also
Figure 1-15). The three nuclear lamins (A, B, and C) present in vertebrate cells belong to a class of cytoskeletal proteins, the intermediate filaments, that are critical in supporting cellular membranes. Once mitotic CDKs are activated at the end of , they phosphorylate specific serine residues in all three nuclear lamins. This phosphorylation, together with phosphorylation of lamin B by protein kinase C, causes depolymerization of the lamin intermediate filaments (Figure 19-22d). Depolymerization of the nuclear lamins leads to disintegration of the nuclear lamina and contributes to disassembly of the nuclear envelope (Figure 19-22e).
FIGURE 19-22 Dissolution of the nuclear envelope during mitosis. (a) The nuclear lamins form a meshwork between the inner nuclear membrane and the underlying chromatin (see
Figure 18-53). (b) Electron micrograph of the nuclear lamina from a Xenopus oocyte. (c) The nuclear lamin meshwork helps tether chromatin to nuclear pore complexes and to specific proteins in the nuclear membrane. (d) Phosphorylation of lamins adjacent to the head domains by cyclin B-CDK1 and protein kinase C, results in lamin disassembly and dissolution of the nuclear membrane into vesicles or retraction into the endoplasmic reticulum. Phosphorylation of lamins A and C results in their solubilization and dispersal in
the cytoplasm, while phosphorylation of lamin B dissociates it into membrane-bound subunits. (e) Human cells in which lamin A has been fused with GFP. The DNA has been stained blue with DAPI. The nuclear lamins are beginning to dissolve in early prometaphase. By metaphase, all of the lamin A fibers have been solubilized in the cytoplasm. (f) An overview of nuclear envelope breakdown during mitosis including disassembly of nuclear lamins and dispersal of the nuclear membrane. [Part (b) Republished with permission from Nature, from Electron micrograph of the nuclear lamina from a Xenopus oocyte U. Aebi et al., 323, 1986; permission conveyed through Copyright Clearance Center, Inc. Parts (c) and (d) Data from T. D. Pollard et al., 2016, Cell Biology, 3d ed., Elsevier, Fig. 44.6. Part (e) Republished with permission from Elsevier, from T. D. Pollard et al., 2016, Cell Biology, 3d ed.; permission conveyed through Copyright Clearance Center, Inc.] Description The illustration labeled (a) shows a nucleus with a thin inner nuclear membrane, and an outer nuclear membrane surrounded by endoplasmic reticulum with ribosomes. A nuclear pore complex is present in a space located in the membrane. Inside the nucleus is labeled chromatin and nuclear lamins line the nuclear membrane. The micrograph labeled (b) shows nuclear lamina that looks like a thin net. The illustration labeled (c) is a close up of the nuclear pore complex in the cytoplasm. Ribosomes are labeled to the left, with unlabeled red rod-like structures coming out of the pore complex and blue rod-like structures attaching to this within the nucleus. The lumen is present between the inner and outer membrane. Lamins are represented by yellow lines attached to thicker worm like chromatin inside the nucleus. The illustration labeled (d) shows a lamin structure represented by a thread like structure coiled around each other with tangled endings. Bothe the endings are bonded to a O H group. On addition of A T P in the presence of C D K P K C the hydrogen atom of the O H group gets replaced by a phosphate group. Two arrows labeled lamin A or C and lamin B from a net like structure points at short rod-like structures and, short connected rod-like structures enclosed by a dotted membrane line, respectively.
Centrosomes Duplicate During S phase and Separate During Mitosis
The micrograph labeled (e) shows a cell in Interphase (with D N A labeled as a blue cloud in nucleus), a cell in early prometaphase (D N A gathering together), and metaphase (D N A in rows). The illustration labeled (f) shows nuclear envelope breakdown in interphase and mitosis. In the interphase stage, the endoplasmic reticulum is represented by long ovals around the nucleus with lamins and membranes labeled. Inside the nucleus is blue blobs labeled decondensed chromosomes. The mitosis stage shows no membranes, the condensed chromosomes are labeled at the center. A label at top right reads: lamins A and C dispersed in cytoplasm. A label below this diagram reads: Lamin B on membrane vesicles from nuclear envelope and on endoplasmic reticulum. Mitotic CDKs also affect other nuclear envelope components. The CDKs phosphorylate specific nucleoporins, which causes nuclear pore complexes to dissociate during prophase. Phosphorylation of integral membrane proteins of the inner nuclear membrane is thought to decrease their affinity for chromatin and further contributes to the disassembly of the nuclear envelope. Weakening of the associations between the inner nuclear membrane proteins and the nuclear lamina and chromatin allows sheets of inner nuclear membrane to retract into the endoplasmic reticulum, which is continuous with the outer nuclear membrane (Figure 19-22f). Centrosomes Duplicate During S phase and Separate During Mitosis A key function of mitotic CDKs, together with other protein kinases and ubiquitin-protein ligases, is to induce the formation of the mitotic
spindle, also known as the mitotic apparatus. The function of the mitotic spindle is to segregate chromosomes so that the sister chromatids separate from each other and are moved to opposite poles of the cell (see Figure 18-39). In what follows, we describe how the mitotic spindle forms, how chromosomes attach to it, and how cells correct faulty attachments. As we saw in Chapter 18, the mitotic spindle is made of microtubules that attach to chromosomes via specialized protein structures known as kinetochores, which form at the central centromeric region of chromosomes. In most organisms, the mitotic spindle is built between two centrosomes (called spindle pole bodies in yeast), that lie at opposite ends (poles) of the spindle (Figure 19-23; also see Figure 18-39). Centrosomes contain a specialized type of tubulin, γ tubulin, which forms specific ringlike complexes (γ TuRCs) that, together with a variety of associated proteins, have the ability to nucleate microtubules and form a bipolar spindle. [Notable exceptions to these centrosome-based spindle assembly mechanisms are seen in higher plants and metazoan oocytes. In these cells, the (−) ends of microtubules are cross-linked, and the microtubules self-assemble into a spindle.] In either case, the ends of the mitotic spindle are always referred to as spindle poles, and in animal cells these typically arise from centrosomes.
FIGURE 19-23 The centrosome cycle is coordinated with the cell cycle to form a bipolar spindle that can correctly segregate chromosomes in mitosis. (a) Thin section electron micrograph of an interphase centrosome showing two centrioles at right angles to
each other surrounded by pericentriolar material. (b) Diagram of the centrosome showing each centriole composed of nine triplet outer microtubules embedded in pericentriolar material that contains γ-TuRC nucleating structures. The original mother centriole from the prior cell division is marked with additional appendages shown as blue spheres, and the centrioles are connected by a flexible protein linker. (c) Stages of the centrosome cycle. Step 1 : CDKs and Plk4 induce each centriole (green) to produce a daughter procentriole, which elongates during S phase. Step 2 : The kinase Nek2 drives cleavage of the protein linker connecting the two original centrioles, while CDK1, Plk1, and Aurora A induce accumulation of large amounts of PCM (blue). Step 3 : Each centriole-procentriole pair together with its associated PCM separates from the other, forming two individual, mature centrosomes located on opposite sides of the cell nucleus. The mitotic spindle that forms between them segregates the chromosomes during the next steps in mitosis. Step 4 : At the end of mitosis, Plk1, together with a protease called separase, promotes centriolar disengagement, separating the former centriole-procentriole pair from the orthogonal arrangement they had during mitosis. This Plk1-mediated separation so to speak licenses the centrosome, so that procentriolar formation and centrosome duplication can only happen once each cell cycle. This is conceptually similar to how CDK activity licenses replication origins (see Figure 19-19) so that they can only fire once each cell cycle. [Part (a) Reprinted with permission from Nature Publishing Group, from G. Sluder, 2005, “Two-Way Traffic: Centrosomes and the Cell Cycle,” Nat. Rev. Mol. Cell Biol. 6:743–748; permission conveyed through Copyright Clearance Center, Inc. Part (c) Data from P. T. Conduit, A. Wainman, and J. W. Raff, 2015, Nat. Rev. Mol. Cell Biol. 16:611–624.] Description The micrograph labeled (a) shows the top view and the side view of a centrosome represented by a circular and a cylindrical structure. Some arrows point at the area surrounding these structures. The illustration labeled (b) shows the structure of the centrosome. The mother centriole is composed of a ring of triplet microtubules (tubular structures), with subdistal and distal appendages attached to the outside of the microtubule ring. These appendages represented by spheres with hair like projections at the bottom. Gamma-T u R C is represented by tiny oval rings surrounding the mother centriole. A separate tubular structure is labeled microtubule. The daughter centriole lies at 90 degrees to the mother
centriole. It is also made of a ring of triplet microtubules. A thread-like structure extending from it is labeled protein linker. Both of these structures are suspended in pericentriolar material represented by tiny dots. The illustration labeled (c) shows the centrosome cycle in four steps: Step 1, starting at about 3 o-clock and moving counterclockwise, an arrow is labeled G 1slash S C D Ks P l k 4. It shows 2 centrioles as green cylinders surrounded by a blue round structure labeled P C M with a red linker line between. A daughter centriole is being formed. In step 2, labeled mitotic C D Ks N e k 2, P l k 1 and Aurora A, shows the P C M getting larger and a protein linker coming out, then the centriole is taking position at one end of the nucleus to receive chromosomes. Step 3 has a label: Aurora A, Aurora B, P l k 1 and shows both centrioles with chromosomes between them. Step 4 is labeled P l k 1 and goes back to the 2 centrioles with P C M around them. During , cells contain a single centrosome, which functions as the major microtubule nucleating center of the cell. In order for this single centrosome to create a mitotic spindle, it has to duplicate into two centrosomes and acquire additional proteins and modifications through a process called centrosome maturation. The two centrosomes then have to move to opposite sides of the cell and nucleate a mitotic spindle. Once mitosis is completed, the centrosome must re-form its -like structure. This sequence of events is sometimes called the centrosome cycle (Figure 19-23c), which is tightly coordinated with the cell cycle overall. Mitotic spindle formation actually begins at the phase transition with the duplication of the centrosome. How duplication occurs is not completely understood, but at the heart of this process is duplication of the pair of centrioles, short, microtubule-based structures arranged orthogonally to each other and surrounded by proteinaceous pericentriolar material (PCM). As discussed in Chapter 18, cells contain a single pair
Mitotic CDKs, Polo-like Kinases, and Aurora Kinases Drive Assembly of a Mitotic Spindle That Attaches to the Kinetochores of Condensed Chromosomes
of centrioles, which are connected together by a flexible protein linker (Figure 19-23a and b). As a cell enters S phase, each of the two centrioles begins to duplicate by growing a new daughter procentriole (Figure 1923c). This process is initiated by the cyclin-CDKs and is largely controlled by a member of the Polo-like kinase family, Plk4, which phosphorylates additional centriole-associated proteins. They recruit γ-tubulin complexes that help form the new procentrioles (Figure 19-23c, step 1 ). The procentrioles enlarge during S phase so that by each centriole has made an exact copy of itself. The two centriole-procentriole pairs remain in close proximity to each other. At the transition, the protein linker connecting the two original centrioles is dissolved (Figure 19-23c, step 2 ). This process, called centrosome disjunction, creates two separate centrosomes. Each centrosome expands its complement of pericentriolar material. The centrosomes recruit more γ TuRCs, allowing the centrosome to efficiently organize and nucleate the large number of microtubules that are required for mitotic spindle formation and function (Figure 19-23c, step 3 ). Once the cell enters prophase, the mature centrosomes separate from one another and move to opposite sides of the cell nucleus, where they create the mitotic spindle. This occurs in close proximity to the still-intact nuclear envelope through the actions of a kinesin motor protein (see Chapter 18), which pushes apart overlapping microtubules emanating from each centrosome. The specifics of microtubule formation, mitotic spindle assembly, and microtubule motors were discussed in Chapter 18. Here we briefly consider how chromosomes attach to the mitotic spindle and how mistakes in the process are corrected.
Mitotic CDKs, Polo-like Kinases, and Aurora Kinases Drive Assembly of a Mitotic Spindle That Attaches to the Kinetochores of Condensed Chromosomes For chromosomes to be accurately segregated during mitosis, they must attach to the mitotic spindle in such a way that one kinetochore of each sister chromatid pair attaches to microtubules emanating from opposite spindle poles. Once this happens, the sister chromatids are said to be bioriented (Figure 19-24a). This type of attachment, in which the kinetochores of each sister chromatid are attached to microtubules coming from opposing centrosomes, is called an amphitelic attachment. How is this accomplished? Once centrosomes have moved apart from each other during prophase, and the nuclear envelope has broken down during prometaphase (see Figure 19-3), microtubules, in a search-and-capture mechanism, begin to interact with the kinetochores of sister chromatid pairs. Initially, chromosomes glide along the length of microtubules, propelled by motor proteins. When a chromosome reaches the (+) end of a microtubule, the kinetochores attach to microtubules in an end-on attachment, the final configuration in which chromosomes are linked to the mitotic spindle (Figure 19-24b and c; also see Figure 18-42). Kinetochores of sister chromatids then bind microtubules emanating from the opposite spindle poles, allowing the chromosomes to separate by poleward pulling forces coming from motor proteins at the kinetochore.
FIGURE 19-24 Chromosome attachment to the mitotic spindle. (a) Chromosomes attach to the mitotic spindle and accumulate at the spindle center. They then attach, via their kinetochores, to the ends of microtubules (called end-on attachments), and these attachments are stabilized by additional microtubules. The final chromosome attachment, in which the chromosome is stably bi-oriented on the mitotic spindle, is shown. “(−)” indicates the minus end of the microtubule, “(+)” the plus end. The boxed region at the kinetochore is explained in parts b and c. (b) Electron micrograph of a microtubule bound end-on to a purified yeast kinetochore. (c) Schematic cartoon illustrating the key features shown in part (b) and the boxed area in part (a). The ring structure embracing the microtubule most likely represents the outer kinetochore Dam1 complex (called the Ska complex in metazoans) and part of the Ndc80 complex, also an outer kinetochore component. The globular structure at the end of the complex most likely reflects the inner kinetochore and protein complexes that link the inner kinetochore to the outer kinetochore. [Part (b) Reprinted with permission from Nature Publishing Group, from S. Gonen et al., 2012, Nat. Struct. Mol. Biol. 19:925–929, Fig. 2d; permission conveyed through Copyright Clearance Center, Inc. Part (c) Information from S. Gonen et al., 2012, Nat. Struc. Mol. Biol. 19:925–929, Fig. 1a.] Description The illustration labeled (a) shows the attachment of chromosomes to the mitotic spindle. A chromosome is shown with microtubules attached at their positive ends to the kinetochores at near the centromere. The poleward pilling forces are represented by arrows and are labeled. The electron micrograph labeled (b) shows a tube-like structure representing microtubule attached to another dark structure representing kinetochores. The illustration labeled (c) shows a microtubule with its negative and positive end labeled. The microtubule is made up of lines of tubulin units stacked together, which are represented by spheres of two colors arranged alternatively to form a tubular structure. The microtubule is attached to a set of two ring-like structures called the outer kinetochore. It is made of the N d c 80 complex. Five coiled coils from this complex labeled D a m 1 complex extend to attach to the inner kinetochore. It is made of five
oval structures attached to five other longer oval structures attached to the centromeric chromatin. Before the chromosomes can be separated by poleward pulling forces, each and every chromosome in the cell has to be properly attached to the mitotic spindle in a bi-oriented amphitelic attachment (Figure 19-25a). How does the cell know that this has occurred? Microscopic analysis of chromosome attachment has shown that, initially, many chromosomes attach to microtubules in faulty ways. A kinetochore can attach to microtubules emanating from both poles at the same time, a situation called merotelic attachment (Figure 19-25b). Alternatively, both kinetochores of a sister chromatid pair can attach to microtubules from the same pole (syntelic attachment; Figure 19-25c), or only one kinetochore can attach to microtubules (monotelic attachment; Figure 19-25d). Clearly none of these attachments would result in accurate chromosome segregation, since the sister chromatids would not be pulled apart to opposite spindle poles. Thus mechanisms must be in place that detect and correct such faulty kinetochore-microtubule attachments.
FIGURE 19-25 Stable and unstable chromosome attachments. When sister kinetochores attach to microtubules emanating from opposite spindle poles, they are stably attached. This configuration is called amphitelic attachment (a). Microtubules (green) pull kinetochores; cohesins resist this pulling force. The resulting tension leads to the outer kinetochore component Ndc80 (orange) being pulled away from the protein kinase Aurora B (colored red), which localizes to the inner kinetochore when chromosomes are near the spindle midzone. As a result, Aurora B can no longer phosphorylate Ndc80, and kinetochoremicrotubule attachments are stable. When one of the two sister chromatid kinetochores simultaneously attaches to microtubules emanating from two opposite spindle poles [merotelic attachment, (b)], or both sister kinetochores attach to microtubules emanating from the same spindle pole [syntelic attachment, (c)], or only one of the two sister kinetochores attaches to microtubules [monotelic attachment, (d)], Ndc80 is not pulled away from Aurora B. As a result, Aurora B phosphorylates Ndc80 (yellow circles labeled ‘P’), and Ndc80 can no longer form stable attachments to microtubules.
Description The illustration labeled (a) is titled amphitelic attachment of chromosomes. Sister chromatids are bound to the kinetochores, where N d c 80 on both sides is attached to three microtubules each. Four cohesin rings are bound to the chromosomes. In the chromosome center tiny structures called aurora B pull the chromatids apart. The illustration labeled (b) is titled merotelic attachment of chromosomes. The microtubules are attached asymmetrically, with one microtubule on left, and three on the right. The illustration labeled (c) is titled syntelic attachment; microtubule attachment to both kinetochores from one spindle pole occurs only at the right side, there are no microtubules on the left. The illustration labeled (c) is titled monotelic attachment. Only one set of kinetochores are attached to microtubules on the right. The kinetochores on the left have no microtubules. The sensing mechanism used by cells to detect incorrect chromosome attachments is based on tension. When sister chromatids are correctly attached to microtubules, their kinetochores are under tension (Figure 1925a). Microtubules attached to the kinetochores pull at them, while the cohesin molecules that hold the sister chromatids together withstand these forces, creating tension at the kinetochores. Merotelic, syntelic, or monotelic attachment leads to insufficient tension at kinetochores, allowing the cell to distinguish these faulty forms of attachment from the correct amphitelic one. How does the cell measure whether or not kinetochores are under tension? The Aurora protein kinase family members Aurora A and Aurora B and their associated regulatory factors sense kinetochores that are not under
tension and sever these microtubule attachments, giving cells a second chance to get the attachment right. The molecular basis for this sensing mechanism is reasonably well understood. Recall that outer kinetochore components, especially the Ndc80 complex, bind to microtubules (see
Figure 19-24c) in a manner that is stabilized by the Ska complex (or the Dam1 complex in yeast). Aurora kinases phosphorylate the N-terminal tail of Ndc80. When phosphorylated, Ndc80 loses its ability to form stable interactions with microtubules. Aurora B localizes to the inner kinetochore and is thought to be the dominant error-correcting kinase for chromosomes along most of the spindle. When kinetochores are not under tension, Ndc80 is in close proximity to Aurora B, and the protein kinase can phosphorylate the protein, destabilizing any kinetochore-microtubule attachments (Figure 19-25b–d). When microtubules are attached correctly to kinetochores, microtubule forces pull Ndc80 away from Aurora B, and the kinase can no longer phosphorylate Ndc80 (Figure 19-25a). Protein phosphatase 1 (PP1) localizes to the outer kinetochore and continuously de-phosphorylates Ndc80. Thus, when kinetochores are under tension and pulled away from Aurora B, Ndc80 is quickly de-phosphorylated by PP1 and microtubule–kinetochore attachments are stabilized. A similar mechanism is thought to operate on kinetochore-microtubule interactions close to the spindle poles, where Aurora A substitutes for Aurora B. Microtubules continuously pull on chromosomes. Once all the chromosomes have attached to microtubules in an amphitelic manner, only the cohesins that hold them back in the middle of the spindle (Figure 19-25a) prevent chromosomes from segregating to the poles. As we will see in Section 19.6, it is the severing of these cohesins that initiates
Chromosome Condensation Facilitates Chromosome Segregation
anaphase chromosome segregation and drives mitotic exit. Again, just as in S phase entry, mitotic entry, chromosome replication, and the centrosome cycle, protein phosphorylation and de-phosphorylation by protein kinases and phosphatases control all of the events leading to chromosome separation. Chromosome Condensation Facilitates Chromosome Segregation Chromosome segregation not only requires building the apparatus that segregates chromosomes, but it also requires that the DNA be compacted into travel-friendly structures. Any attempt to segregate the long and intertwined DNA-protein complexes present in interphase cells would lead to DNA breakage and loss of genetic material. To avoid this fate, cells compact their chromosomes during prophase into the dense structures we have become acquainted with through light and electron microscopy (Figure 19-26a and see Figure 19-2).
FIGURE 19-26 The Condensin complex packages chromosomes for efficient segregation on the mitotic spindle. (a) Electron micrograph of a metaphase chromosome. By metaphase, the chromosomes are fully condensed, and the two individual sister chromatids are visible. (b) The condensin complexes have a similar architecture to the cohesin complex discussed previously. (c) A model of cell cycle regulated chromosome compaction involving progressive chromosome looping. During prophase, prior to nuclear envelope breakdown, condensin II binds DNA and extrudes large loops. After nuclear envelope breakdown in prometaphase, condensin I forms additional loops nested within the loops formed by condensin II. [Part (a) Republished with permission from Nature Publishing Group, from E. J. DuPRAW and P. M. M. Rae, 1966, “Polytene Chromosome Structure in Relation to the ‘Folded Fibre’ Concept”. Nature 212:598–600; permission conveyed through Copyright Clearance Center, Inc.]
Description The electron micrograph labeled (a) shows two dark chromatids attached at the center. The illustration labeled (b) shows a cohesin ring with the S m c 1 on the left side of the ring, S m c 3 on the right side. Below it are four structures labeled, from top left around, are S c c 1, S c c 3, W a p l, and P d s 5. Next to it is a ring labeled condensin. The left side of the ring is labeled S m c 2, the right side is labeled S m c 4. At the bottom are 4 structures labeled, from top left around, C a p-H slash H 2, G slash G 2, W a p l, D 2 slash D 3. The illustration labeled (c) shows where these rings are located on chromatids during phases. In the first part, labeled G subscript 2 (sister chromatid tethering), the two chromosomes are present with four cohesin rings attached in groups of two. An arrow from this points at the second part, labeled prophase, there are loops in the chromatids coming to the left and right from the chromosomes and each has a ring on it at the neck of the loop labeled condensin 2. An arrow labeled N E B from this points towards the last part. The chromatids labeled prometaphase, the loops are elongating and now have a second ring attached around them. This ring is labeled condensin 1. Chromosome condensation results in a dramatic reduction in chromosome length, up to 10,000-fold in vertebrates. During chromosome compaction, the intertwined sister chromatids are also untangled. This process, called sister chromatid resolution, is mediated in part by topoisomerase II, which cuts one chromatid in a pair of sister chromatid molecules, passes the other one through, and closes the cut molecule. As part of this process, during prophase most of the cohesin proteins that tether sister chromatids together along the chromosome arms are shed through the actions of Plk1 and Aurora B. Central to the process of chromosome condensation is another ring-shaped protein complex known as condensin. Most eukaryotes have two closely
related condensin complexes, called condensin I and II. These protein complexes are closely related to the cohesins that link sister chromatids together (Figure 19-26b). Condensins were first identified based on their ability to promote chromosome condensation in frog egg extracts. Like cohesins, condensins are composed of two large coiled-coil SMC protein subunits that associate through their ATPase domains with non-SMC subunits. When condensin function is lost in cells, chromosomes do not condense and sister chromatid tangles are not resolved. Recent studies have shown that chromosome compaction occurs via the generation of consecutive loops that lead to the formation of a fiber at the bases of the loops (Figure 19-26c). These fibers are compressed, leading to further chromosome compaction. Condensins likely package chromosomes by creating these loops by forming intrachromosomal linkages, through the process explained in Figure 19-26c. Condensin association with chromosomes is facilitated by mitotic CDKs and Aurora B, which phosphorylate histone H2A (see Figure 7-26b), allowing condensins to bind chromatin. KEY CONCEPTS OF SECTION 19.5 The Transition and the Irreversible Engine of Mitosis Mitotic CDKs induce entry into mitosis in all eukaryotes. Mitotic CDKs are kept inactive by inhibitory phosphorylation of the CDK subunit until the completion of DNA replication (see Figure 19-21). Mitotic CDKs promote their own activation through two positive feedback loops that inactivate Wee1 kinase and activate Cdc25 phosphatase (see Figure 19-21). Mitotic CDKs induce nuclear envelope breakdown in most eukaryotes by phosphorylating lamins (see Figure 19-22).
Centrosome duplication occurs during S phase. Mitotic CDKs induce the separation of the duplicated centrosomes, which initiates mitotic spindle formation (see Figure 19-23). Sister chromatids attach to the mitotic spindle via their kinetochores in a bi-oriented manner, with one sister kinetochore attaching to microtubules emanating from one spindle pole and the other one to microtubules nucleated by the other spindle pole (see Figure 19-24). Cells sense bi-orientation of sister chromatids through a tension-based mechanism. When kinetochores are not under tension, the protein kinases Aurora A and Aurora B phosphorylate the microtubule-binding subunits of the kinetochore, which limits their ability to form stable long-lasting interactions (see Figure 19-24). Chromosomes must be compacted for segregation to occur. Condensins, protein complexes that are related to cohesins, facilitate chromosome condensation and are activated by mitotic CDKs (see Figure 19-26).
Separase-Mediated Cleavage of Cohesins Initiates Chromosome Segregation
19.6 The Mitotic Spindle, Chromosome Segregation, and Exit from Mitosis Once all chromosomes have condensed and have correctly attached to the mitotic spindle, chromosome segregation commences. In this section, we discuss how cleavage of cohesins by a protease known as separase triggers anaphase chromosome movement and how this cleavage is regulated by phosphorylation and protein ubiquitination. We then see how the same machinery that initiates cohesin cleavage at the metaphase-anaphase transition also initiates mitotic CDK inactivation. Next we describe how phosphatases activated at the end of mitosis also participate in mitotic CDK inactivation, bringing about the disassembly of mitotic structures and the resetting of the cell to the state. We end this section with a discussion of cytokinesis, the process that produces two daughter cells. Separase-Mediated Cleavage of Cohesins Initiates Chromosome Segregation As mentioned in the previous section, chromosome condensation and dissociation of cohesins along the chromosome arms leads to further compaction of chromosomes. Most of the cohesins along the chromosome
arms are removed from chromosomes during prophase (Figure 19-27a, steps 1 and 2 ). This process is mediated by phosphorylation of cohesins by Plk1 and Aurora B kinase. In most organisms, by metaphase, cohesins are maintained only around centromeres. These centromereassociated cohesins are specifically protected from phosphorylationdependent removal by protein phosphatase 2A (PP2A). This phosphatase is recruited to centromeric regions in human cells from through metaphase by a member of a family of PP2A targeting factors known as the Shugoshin family of proteins (Figure 19-27b). The protected pool of cohesins provides the resistance to the pulling force exerted by microtubules that are necessary to establish tension at bi-oriented kinetochores. As we will see in Section 19.8, this protection mechanism also plays an essential role in establishing the meiotic chromosome segregation pattern.
FIGURE 19-27 Cleavage of cohesin at centromeres by separase triggers anaphase onset. (a) A multistep process removes the cohesin that links sister chromatids. Step 1 : During , when sister chromatids are linked along their entire length by cohesins, the Shugoshin proteins begin recruiting the protein phosphatase 2A (PP2A) to centromeric regions. This recruitment process continues through the beginning of prophase when CDK1 triggers chromosome condensation. Step 2 : Later in prophase and early metaphase, cohesin along the chromosome arms is shed by the action of a protein complex in combination with phosphorylation of cohesins by Polo kinase and Aurora B kinase. However, cohesin remains tightly associated with centromeric regions of the chromosome because PP2A localized there removes any phosphates added by the Plk1 kinase. Finally, in step 3 separase cleaves the residual centromeric cohesin, triggering the onset of anaphase, releasing the sister chromatids, which move rapidly toward opposite spindle poles. (b) Diagrammatic representation of Shugoshin-PP2A recruitment and retention at centromeric chromatin. Description The illustration labeled (a) shows 3 steps in the separation of chromatids. The chromatids are have oval shaped structures labeled cohesin lining the center. The centromere region is labeled. A text below reads: end of S phase forward arrow G subscript 2 (uncondensed sister chromatids, arms are connected by cohesin, shugosin-P P 2 A recruited to centromere. Step 1: An arrow labeled C D K 1 points from the first structure to a chromatid where its centromere region is attached to an oval shaped structure on both sides. A text below reads: beginning of prophase (condensed sister chromatids, arms remain connected). Step 2: An arrow labeled P l k 1, aurora B points at a chromosome with splayed ends releasing the cohesins on both sides. The cohesin remains at the centromere. A text below reads: middle of prophase (arm cohesins removed). Step 3: An arrow labeled separase points at two separate chromatids with centromeres releasing cohesin. The text below reads: Anaphase (centromere cohesins cleaved, chromatids separate). Below, the illustration labeled (b) shows the location of cohesin rings. In the S phase, cohesed sister chromatids are surrounded by three cohesin rings. The replication fork is labeled below it. Two cohesin rings are present sandwiching the centromere. An arrow joins a line pointing from an oval structure labeled shugoshin-P P 2 A to point at the
chromatids in G subscript 2 phase. In G subscript 2, the chromatids have many cohesin rings around them. Two shugoshin-P P 2 A are bound to the centromere region which is now has a loop on both the side. Another arrow from this labeled polo kinase aurora B kinase points at chromatids in prophase stage. There are only four cohesin rings around the centromere loops which are attached to two shugoshin-P P 2 A on both sides. Each sister chromatid of a metaphase chromosome is attached to microtubules via its kinetochore (see Figure 19-24). Once all of the chromosomes have made stable amphitelic attachments to the spindle pole microtubules and congressed to the spindle midzone at metaphase, the chromosomes will be in a state of tension, with forces pulling the two kinetochores toward opposite spindle poles. Sister chromatids do not separate from each other yet, however, because they are held together at their centromeres by what remains of the cohesion attachments. In all organisms analyzed to date, the loss of this residual cohesin from the centromeric regions of chromosomes is what triggers anaphase chromosome movement (Figure 19-27a, step 3 ). The mechanism that brings about this loss of cohesins from chromosomes is conserved as well. A protease known as separase cleaves the cohesin subunit Scc1 (Figure 19-28), breaking the protein circles linking sister chromatids. Once this link is broken, anaphase begins as poleward force exerted on the kinetochores moves the split sister chromatids toward opposite spindle poles.
FIGURE 19-28 Regulation of separase through phosphorylation and ubiquitin-mediated degradation of an inhibitor establishes the timing of cohesin cleavage. Separase is inhibited before anaphase by the binding of the protein securin. Mitotic CDKs also inhibit separase by phosphorylating it. When all the kinetochores have properly attached to spindle microtubules and the spindle apparatus is properly assembled and oriented, the Cdc20 specificity factor associated with APC/C directs it to ubiquitinylate securin and mitotic cyclins. Following securin degradation and a decrease in mitotic CDK activity, separase cleaves the Scc1 subunit, breaking the cohesin circles and allowing sister chromatids to be pulled apart by the spindle apparatus that is pulling them toward opposite spindle poles. Description The illustration shows chromatids around which there is a cohesin ring with the S c c 1 region labeled. A forward arrow points at chromatids around which there is a broken cohesin ring as S c c 1 is cleaved by an oval structure labeled separase. Above the arrow separase bound to a phosphate group is attached to securin. This complex by the action of A P C slash C and C d c 20 gets disassembled to leave separase to cleave the cohesin ring. Securin attached to three ubiquitin molecules exits the reaction. Cohesin cleavage was discovered in budding yeast. Insight into the identity of the protein that was responsible for the cleavage of cohesin came from analysis of previously identified yeast mutants that failed to segregate chromosomes during anaphase. A mutant form of the gene encoding Esp1 — what we now call separase — failed to produce the cleavage fragment. Subsequent analyses revealed that separase is a
APC/C Activates Separase Through Securin Ubiquitinylation
protease and that cleavage of cohesin by separase is essential for chromosome segregation. Given the irreversible nature of Scc1 cleavage, it is absolutely essential that separase activity be tightly controlled. Next we discuss its regulation. APC/C Activates Separase Through Securin Ubiquitinylation Prior to anaphase, the protein securin binds to and inhibits separase (see
Figure 19-28). Once all kinetochores have attached to spindle microtubules in the correct bi-oriented manner, the APC/C ubiquitinprotein ligase, directed by specificity factor Cdc20 (known as ) (see Figure 19-14), ubiquitinylates securin. The polyubiquitinylated securin is rapidly degraded by proteasomes, thus releasing separase. is phosphorylated and activated in prophase by mitotic CDK phosphorylation. We will see in Section 19.7, however, that phosphorylated is inhibited by a checkpoint pathway — the spindle assembly checkpoint pathway — that ensures anaphase is not initiated until all of the chromosomes have achieved proper attachment to the mitotic apparatus (i.e., are bi-oriented). Cdc20 is inhibited until every kinetochore has attached to microtubules and proper tension has been applied to the kinetochores of all sister chromatids. In vertebrate cells, separase itself is also negatively regulated by CDK phosphorylation during prophase and metaphase. Separase becomes active and triggers
Mitotic CDK Inactivation and Protein De-phosphorylation Triggers Exit from Mitosis
chromosome segregation when -mediated protein degrades because mitotic CDK activity begins to decline at the metaphase-anaphase transition. Once cohesins are cleaved, anaphase chromosome movement ensues. As discussed in Chapter 18, chromosome segregation is mediated by microtubule depolymerization and motor proteins as the spindle poles move away from each other. Decline in mitotic CDK activity is important for these anaphase chromosome movements. If mitotic CDK inactivation is inhibited, anaphase does occur, but it is abnormal. De-phosphorylation of a number of microtubule-associated proteins that affect microtubule dynamics appears to be important for this process. In budding yeast, this de-phosphorylation is brought about by the protein phosphatase Cdc14, which, as we will see, plays an essential role in the final cell cycle stage, exit from mitosis. Mitotic CDK Inactivation and Protein De-phosphorylation Triggers Exit from Mitosis Anaphase spindle elongation and the events associated with exit from mitosis — mitotic spindle disassembly, chromosome decondensation, and nuclear envelope re-formation — require the de-phosphorylation of CDK substrates. In other words, the phosphorylation events that triggered all of the different mitotic events need to be undone for the cell to revert to the state.
De-phosphorylation of mitotic CDK substrates involves two steps. First, the mitotic CDKs themselves have to be inactivated. In most organisms, mitotic CDK inactivation is triggered by -mediated degradation of mitotic cyclins. However, in budding yeast, only about 50 percent of mitotic cyclins are degraded by . An additional pathway called the mitotic exit network plays a particularly important role in shutting off mitotic cyclins in yeast by controlling the activation of the Cdc14 phosphatase through a GTPase pathway called the mitotic exit network. Activation of Cdc14 at anaphase de-phosphorylates Cdh1, allowing formation , which then degrades the remaining mitotic cyclins. In addition, Cdc14 promotes the re-accumulation of the CDK inhibitor Sic1, which inhibits any residual mitotic CDK activity, similarly to how it inhibits phase cyclins in previous stages of the cell cycle (Section 19-4). This process leads to exit from mitosis. Phosphatase activity is also essential for exit from mitosis in vertebrate cells, although they do not appear to possess a mitotic exit network like that found in budding yeast. Ultimately, reversal of mitotic CDK phosphorylation changes the activities of many proteins back to their interphase states. De-phosphorylation of condensins and other chromatin-associated proteins leads to decondensation of mitotic chromosomes in telophase. De-phosphorylated inner nuclear membrane proteins are thought to bind to chromatin once again. As a result, multiple projections of regions of the ER membrane containing these proteins are thought to associate with the surfaces of the decondensing chromosomes and then fuse with one another, directed by an unknown mechanism to form a continuous double membrane around each
chromosome (Figure 19-29). De-phosphorylation of nuclear pore subcomplexes allows them to reassemble into complete NPCs traversing the inner and outer membranes soon after fusion of the ER projections. Ran⋅GTP, required for driving most nuclear import and export (see
Chapter 13), stimulates both fusion of the ER projections to form daughter nuclear envelopes and assembly of NPCs (Figure 19-29). The Ran⋅GTP concentration is highest in the vicinity of the decondensing chromosomes because the Ran-guanine nucleotide–exchange factor (Ran-GEF) is bound to chromatin. Consequently, membrane fusion is stimulated at the surfaces of decondensing chromosomes. Sheets of nuclear membrane with inserted NPCs then fuse with one another to form one nuclear membrane around all of the chromosomes.
Cytokinesis Creates Two Daughter Cells
FIGURE 19-29 Model for reassembly of the nuclear envelope during telophase. Extensions of the endoplasmic reticulum (ER) associate with each decondensing chromosome and then fuse with one another, forming a double membrane around the chromosome. De-phosphorylated nuclear pore subcomplexes reassemble into nuclear pores, forming individual mini-nuclei called karyomeres. The enclosed chromosome further decondenses, and subsequent fusion of the nuclear envelopes of all the karyomeres at each spindle pole forms a single nucleus containing a full set of chromosomes. NPC: nuclear pore complex. See B. Burke and J. Ellenberg, 2002, Nat. Rev. Mol. Cell Biol. 3:487–497. Description The illustration shows a part of the nucleus labeled chromatin. The nucleus is enclosed by connected tubular structures labeled E R which is present in the cytosol. The ends of this tubular structure are attached to the nucleus. A downward arrow labeled membrane recruitment points at a part of the nucleus which is now attached to E R where its ends have splayed to attach to the nucleus. Arrows indicate that the ends of the E R start to grow. A downward arrow labeled membrane fusion and N P C assembly points at a part of the nucleus now fully surrounded by the outer and inner nuclear membrane and N P C. net like structures labeled nuclear lamin assembly lines the inner nuclear membrane. Cytokinesis Creates Two Daughter Cells When chromosome segregation is completed, the cytoplasm and organelles are distributed between the two future daughter cells. This process is called cytokinesis. With the exception of higher plants, the division of the cell is brought about by a contractile ring made of actin and the actin motor myosin (see Figure 17-34). During cytokinesis, the ring contracts in a manner similar to muscle contraction, pulling the membrane
inward and eventually closing the connection between the two daughter cells (Figure 19-30).
FIGURE 19-30 Control of RhoA regulates cytokinesis. (a) The Centralspindlin complex (green) recruits and activates the RhoGEF Ect2 (orange) at the central spindle during anaphase. These colocalize with and activate RhoA (red) on the cell cortex opposite the central spindle, generating an actomyosin-based contractile ring that creates a cleavage furrow and divides the cell in half. (b) Molecular pathway of RhoA activation and control of cleavage furrow formation by phosphorylation. [Information from A. Basant and M. Glotzer, 2018. Curr. Biol. 28:R570-R580.] Description The illustration labeled (a) shows a cell in cytokinesis. Centromeres are present at the poles attached to microtubules which pull the chromatids apart to move to the poles. At the center of this divided cell, two arrows point at structures present lining the center.
The illustration labeled (b) shows a flowchart which begins at the top with the numbers 14-3-3. A downward line has an addition of aurora B and moves to the label centralspindlin (M K L P 1 slash C y k 4). A downward arrow moves and shows addition of C D K 1 and P l k 1 and moves to a label E c t 2 (R h o A·G E F). A downward arrow leads to a reversible set of two arrows. Left label reads R h o A-G D P, and right label reads R h o A-G T P. The R h o A-G T P continues with downward arrows to the label F-actin and myosin. Another downward arrow moves to the label cytokinetic furrow. Cytokinesis must be coordinated with other cell cycle events in space and time. For cell division to produce two daughter cells each containing half of the parent cell’s cytoplasmic content and exactly half of the genetic content, the division plane must be properly spatially positioned. Cytokinesis must also be temporally coordinated with completion of mitosis. In what follows, we explore both these aspects of cytokinesis regulation. In animal cells, the contractile ring forms during anaphase and must be placed exactly in the middle of the anaphase spindle to ensure that each daughter cell receives half of the genetic material. Contractile ring assembly requires the small GTPase RhoA (see Section 17.7). Regions of active RhoA form on the cortical surface of the cell opposite the central part of the spindle at sites where the contractile ring will form (Figure 1930a). Active GTP-bound RhoA both stimulates formin-mediated F-actin assembly and promotes the action of myosin II (see Figure 17-42). Recall that in yeast, small GTPases play a critical role in forming the mitotic exit network, which is absent in animal cells. Instead, small GTPases in animal
cells are required for cytokinesis through a process that is tightly controlled by mitotic protein kinases (Figure 19-30b). RhoA itself is activated by a conserved Rho guanine nucleotide–exchange factor (GEF) called Ect2, which is itself tightly regulated by a multiprotein complex called Centralspindlin. Centralspindlin is a heterotetramer composed of two copies of the kinesin-6 motor protein MKLP1 and a dimer of a Rho GTPase-activating protein (GAP). As one can guess from its name, most of the Centralspindlin localizes to the midzone of the anaphase spindle, where the MKLP1 components form oligomers that bundle spindle microtubules and organize the central spindle in preparation for cell abscission at the end of cytokinesis. A variable amount of Centralspindlin, however, localizes to the cell cortex adjacent to the central spindle, together with the RhoA activator Ect2 and RhoA (Figure 19-30a). MKLP1 is inhibited by the phosphoserine/threonine-binding protein 14-3-3 (see Figure 19-30b). This ensures that the spindle midzone microtubules do not compact until the end of anaphase. Similarly, the RhoA activator Ect2 is auto-inhibited during prior stages of mitosis, but it contains two phospho-binding BRCT domains that are important for its activation. During anaphase, when cyclin B–CDK1 activity is extinguished but Aurora and Polo kinases are still active, Aurora B phosphorylates MKLP1 and displaces the 14-3-3 inhibitor, allowing MKLP1 to acquire catalytic motor activity and compact the spindle midzone. At the same time, Plk1 phosphorylates the Rho-GAP component, recruiting the BRCT domains of Ect2 to form an active Centralspindlin-Ect2 complex. This catalyzes the GTP-dependent
activation of RhoA, leading to cleavage furrow formation and division of the cytoplasm to form two completely separate cells. This concludes our discussion of the molecular events of cell division. As we have seen, cyclin-dependent kinases, phosphoserine/threonine-binding domains, and ubiquitin-mediated protein degradation are at the center of its control (Figure 19-31). In Section 19.7, we discuss the mechanisms that ensure that a cell cycle stage is not initiated until the previous one has been completed and that each cell cycle step occurs accurately.
FIGURE 19-31 Fundamental processes in the eukaryotic cell cycle. A synopsis of major events that regulate progression through the cell cycle. See text for details.
Description The illustration shows a cycle with arrows labeled S, G subscript 2, M, and early G subscript 1 and mid-late G subscript 1. The cycle starts just before the S phase. D N A prereplication complexes assemble at origins. G subscript 1 slash S phase C D Ks inactivate c d h 1, G subscript 1 slash S phase C D Ks activate expression of S phase cyclin C D Ks components, and G subscript 1 slash S phase C D Ks phosphorylate S phase inhibitors. S phase C D Ks are attached to an inhibitor. Then, in the S phase, S C F-proteasomes degrade phosphorylated S phase C D K inhibitors, and D N A replication occurs. S phase C D Ks activate prereplication complexes. Entering G subscript 2, c d c 25 phosphatase activates mitotic C D Ks, which activate early mitotic events. In metaphase, A P C slash C proteasome degrades securin, leading to anaphase. Then, phosphatases activate c d h 1, and A P C slash C proteasome degrades mitotic cyclins, leading to telophase and cytokinesis. KEY CONCEPTS OF SECTION 19.6 The Mitotic Spindle, Chromosome Segregation, and Exit from Mitosis Cleavage of cohesin by separase induces chromosome segregation during anaphase. At the onset of anaphase, APC/C is directed by Cdc20 to ubiquitinylate securin, which is subsequently degraded by proteasomes. The degradation of securin, along with phosphorylation by Plk1, activates separase (see Figure 19-28). Exit from mitosis is triggered by mitotic CDK inactivation mainly brought about by mitotic cyclin degradation. Exit from mitosis requires the activity of protein phosphatases such as Cdc14 to remove mitotic phosphates from many different proteins, permitting mitotic spindle disassembly, decondensation of chromosomes, and reassembly of the nuclear envelope. Cytokinesis finalizes cell fission and must be coordinated with the site of nuclear division. In animal cells, this coordination involves the small GTPase RhoA and is regulated by Plk1, Aurora B, and phospho-binding domains (see Figure 19-30).
19.7 Surveillance Mechanisms in Cell Cycle Regulation
19.7 Surveillance Mechanisms in Cell Cycle Regulation Surveillance mechanisms known as checkpoint pathways operate to ensure that the next cell cycle event is not initiated until the previous one has been completed. Checkpoint pathways consist of sensors that monitor a particular cellular event, a signaling cascade that initiates the response, and an effector that halts cell cycle progression and activates repair pathways when necessary until the defect is corrected. If the defect cannot be repaired, the checkpoint pathway induces apoptosis. Cell cycle events monitored by checkpoint pathways include growth, DNA replication, DNA damage, kinetochore attachment to the mitotic spindle, and positioning of the spindle within the cell. These pathways are responsible for the extraordinarily high fidelity of cell division, ensuring that each daughter cell receives the correct number of accurately replicated chromosomes. Checkpoint pathways function through protein phosphorylation, targeting proteins to bind to phosphoserine/threoninebinding domains that control the activities of the cyclin-CDKs. This control involves a variety of mechanisms: blocking synthesis and increasing degradation of cyclins, phosphorylating CDKs at inhibitory sites, sequestering CDK activators away from the CDKs, stabilizing CDK inhibitors (CKIs) that inactivate cyclin-CDK complexes, and inactivating the APC/C ubiquitin-protein ligase.
The DNA Damage Response System Halts Cell Cycle Progression and Recruits DNA Repair Machinery When DNA Is Compromised
The DNA Damage Response System Halts Cell Cycle Progression and Recruits DNA Repair Machinery When DNA Is Compromised The complete and accurate duplication of the genetic material is essential for cell division. If cells enter mitosis when DNA is incompletely replicated or otherwise damaged, genetic changes occur. In many instances, those changes lead to cell death, but as we will see in Chapter 25, they can also lead to genetic alterations that result in loss of control over cell growth and division and, eventually, cancer. This risk is underscored by the finding that many proteins involved in sensing DNA damage and its repair are frequently found mutated in human cancers. The enzymes that replicate DNA are accurate, but not enough to ensure complete accuracy during DNA synthesis. Furthermore, environmental insults such as x-rays and UV light can cause DNA damage, and this damage must be repaired before a cell enters mitosis. Cells possess a DNA damage response system that senses many different types of DNA damage and responds by activating repair pathways and halting cell cycle progression until the damage has been repaired. Cell cycle arrest can occur in , S phase, or , depending on whether DNA damage occurred before cell cycle entry or during DNA replication. In multicellular organisms, when the DNA damage is severe, cells forego repair and instead initiate either apoptosis (“programmed cell death”), a mechanism that is discussed
in detail in Chapter 22, or they senesce, becoming large, flattened cells that have permanently stopped dividing. DNA damage exists in many different forms and degrees of severity. A break of the DNA helix, known as a double-strand break, is perhaps the most severe form of damage because such a lesion would almost certainly lead to DNA loss if mitosis ensued in its presence. This is because the severed piece of DNA that was not attached to the centromere of the chromosome, where the kinetochore assembles, would not be reliably separated into the two daughter cells. More subtle defects include singlestrand breaks, structural changes in nucleotides, and DNA mismatches. For our discussion here, it is important to note that cells have sensors for all these different types of damage. These sensors scan the genome and, when they detect a lesion, assemble signaling and repair factors at the site of the lesion. Central to the detection of these different types of lesions is a pair of homologous protein kinases called ATM (for ataxia telangiectasia mutated) and ATR (for ataxia telangiectasia and Rad3-related protein). In addition to ATM and ATR, a related protein kinase called DNA-PK (for DNA-dependent protein kinase) is also critical for DNA damage signaling and repair. All three protein kinases are recruited to sites of DNA damage through specific protein complexes that form on the chromatin at the site of the damage. They then initiate the sequential recruitment of adapter proteins and DNA repair proteins near the site of the DNA lesion. In addition, they activate another set of downstream effector protein
kinases called Chk1, Chk2, and MK2 that establish and maintain cell cycle checkpoints that stop progression through the cell cycle (Figure 19-32).
FIGURE 19-32 An overview of the DNA damage response system. The protein kinases DNA-PK, ATM, and ATR are activated at sites of damaged DNA. ATR responds to a variety of DNA damage — most likely to the single-stranded DNA that exists either as a result of the damage itself or as a result of repair. ATM and DNA-PK are specifically activated by double-strand breaks. Because double-strand breaks are converted into single-stranded DNA as a part of the repair process, they also, albeit indirectly (as depicted by a dashed line), activate ATR. These kinases phosphorylate additional proteins at the DNA lesion
resulting in the recruitment and activation of DNA repair machinery. ATM and ATR, once activated by DNA damage, activate a triad of related protein kinases, Chk1, Chk2, and MK2. These kinases cause cell cycle arrest by phosphorylating and thus inhibiting Cdc25 and also by stabilizing the transcription factor p53, which induces the transcription of the CDK inhibitor CKI p21. If the DNA damage is severe, p21 can cause the cells to senesce or p53 can induce apoptosis. Description Double-strand breaks activate D N A-P K, A T M, and A T R. Stalled replication forks, D N A mismatches, and nucleotide damage activates A T R. D N A-P K, A T M, and A T R take part in D N A repair. A T M activates C H K 2, and A T R activates C H K 2, C H K 1, and M K 2 leading to repair. Inhibition of c d c 25 activates C D Ks. Activation of P 5 3, results in cell cycle arrest by p 2 1 or apoptosis. P 2 1 can activate senescence and inhibit C D Ks. ATR, ATM, and DNA-PK recognize different types of DNA damage. ATM and DNA-PK respond primarily to double-strand breaks. ATR recognizes diverse types of DNA damage, such as stalled replication forks, DNA mismatches, damaged nucleotides, and single-strand breaks. ATR recognizes these diverse types of damage because all of them contain some amount of single-stranded DNA, either as part of the damage itself or because repair enzymes create single-stranded DNA as part of the repair process. ATR binding to single-stranded DNA is mediated through an ATR-interacting protein called ATRIP and results in increased ATR protein kinase activity. ATR then phosphorylates adapter proteins whose function is to recruit DNA repair proteins and help activate the Chk1 and MK2 kinases that stop progression through the cell cycle until the damage is repaired.
Both DNA-PK and ATM recognize double-strand DNA breaks. Two subunits of DNA-PK, called Ku70/80, form a ringlike structure that encircles the free DNA end. In contrast, ATM is directly recruited to the DNA ends by a protein complex known as the MRN complex (Figure 1933, step 1 ). Binding of DNA-PK and ATM to the broken ends helps to hold them together. ATM also activates Chk2 and MK2 to stop progression through the cell cycle until repair is complete. In addition, DNA-PK, ATM, and ATR phosphorylate a variant histone called H2AX within the chromatin near the damage site (Figure 19-33, step 2 ). Phosphorylated H2AX acts as a landing pad for recruiting additional protein complexes that control DNA repair in a manner that is conceptually similar to other aspects of cell cycle progression and control described in previous sections. That is, by using additional protein phosphorylations, recruiting additional proteins through phospho-binding domains, and marking proteins at the damage site, including H2AX using ubiquitin protein ligases (Figure 19-33, step 3 ), as well as other protein modifications like methylation. The end result is the recruitment of two proteins that determine what specific type of DNA repair occurs at the break. One type of repair is homologous recombination, as described in Chapter 5. This involves the creation of single-stranded overhangs that in turn recruit and activate ATR and its effectors, further enhancing the DNA damage response. Repair by this process requires the presence of a homologous chromosome and so is restricted to late S and phases of the cell cycle. This type of repair requires a protein called BRCA1 (Figure 19-33, step 4 ). In contrast, recruitment of another protein called 53BP1 to the DNA break (Figure 19-33, step 5 ) suppresses homologous recombination and promotes an alternative repair pathway known as nonhomologous end
joining. Here the two broken ends of the DNA are simply glued back together, sometimes with deletion of some of the DNA bases at the break joining site; hence this type of repair can be mutagenic. This end joining process requires the catalytic activity of DNA-PK. Since no homologous DNA sequence is required, nonhomologous end joining is the primary mechanism of repair for cells in and is also the dominant mechanism of repair in S and , even though homologous DNA from chromosome replication is available. Other than cell cycle stage, we do not know what controls which repair pathway is used at a particular break.
FIGURE 19-33 Activation of cell cycle checkpoints and recruitment of repair machinery at DNA double-strand breaks. The activation and recruitment process depends on a concerted series of protein phosphorylations and ubiquitin modifications, together with recruitment of additional proteins through phosphoserine/threonine-binding domains and ubiquitin-binding modules. Step 1 : The ends of a double-strand break are recognized by the MRN complex, which activates ATM. The MRN complex also has nuclease activity that helps process the broken end for repair. Alternatively, the free double-stranded DNA end can bind to Ku70/80 (not shown) to activate DNA-PK. Step 2 : ATM, ATR, or DNA-PK phosphorylates the variant histone H2AX within nucleosomes located close to the break. This recruits additional proteins that recognize phospho-H2AX through their phosphobinding domains and are themselves then phosphorylated. Step 3 : One of the recruited phospho-binding proteins ubiquitinates H2AX. This allows H2AX to now bind to a ubiquitin-binding protein complex. Step 4 : Phosphorylation of the ubiquitin-binding complex recruits BRCA1 through its phospho-binding BRCT domains, which promotes homologous recombination. Step 5 : Alternatively, methylated histones on histone H4 near the break recruit 53BP1 through its methyl lysine-binding domain. Together with the catalytic activity of DNA-PK (not shown), this suppresses DNA end resection at the break, blocking homologous recombination and promoting nonhomologous end joining. Description The illustration has 5 steps, starting at Step 1 and moving counterclockwise to step 5. Step 1 is at the end of a D N A strand which is attached to a structure labeled M R N. Step 2 shows a comma-shaped structure labeled A T M-A T R, D N A-P K, with red arrows moving to three places. The first two arrows move to a blue structure and add phosphate yellow circles to it at step 3. The third red arrow from step 2 moves to step 4. A phosphate is added to a green structure labeled B r c a 1 which has an arrow from it labeled homologous recombination. To the right of the green structure, a blue and purple structure is present attached to two ubiquitin units. This is attached to a cylinder shaped structure labeled H 2 A X. The H 2 A X is a part of a group of four cylinders. The other three are labeled H 3, H 4, and H 2 B. Attached to the other end of the D N A strand and H 4 are 2 blue circles labeled M e, which are then attached to a light blue structure labeled 5 3-B P 1. This structure has an arrow to the left labeled nonhomologous end-joining.
The identification of BRCA1 as a molecule involved in DNA repair emerged from genetic studies of breast-cancer prone families in whom the development of breast tumors showed a Mendelian-like pattern of inheritance. The genomic locus associated with this trait was localized to a single gene on chromosome 17 by Mary-Claire King and colleagues and designated BReast Cancer Associated-1 (BRCA1). The gene was subsequently cloned by several academic and industrial research teams and shown to be mutated in the patients with breast cancer. Subsequent work by many labs implicated the BRCA1 protein as a key component of the homologous DNA repair pathway. Cancer-associated mutations in BRCA1 disrupt its DNA repair function. Downstream from ATR and ATM, the kinases Chk1, Chk2, and MK2 halt the cell cycle (see Figure 19-32). These protein kinases inhibit Cdc25 family members by phosphorylating them on sites that are distinct from the CDK-activating phosphorylation sites. These damage-induced phosphorylation sites target Cdc25 proteins to bind to various phosphoserine/threonine-binding proteins (Figure 19-34). When the DNA damage occurs during , phosphorylation of Cdc25A targets it for binding to an F-box–containing ubiquitin-protein ligase (see Figure 1914), targeting Cdc25A for proteasomal degradation. Loss of Cdc25A inhibits phase CDKs and S phase CDKs (see Figure 19-34). As a result, these kinases cannot initiate DNA replication. When the DNA damage occurs during S phase or in , phosphorylation of Cdc25B and Cdc25C by Chk1/2 and MK2 targets them for binding to 14-3-3 proteins. This reduces the catalytic activity of Cdc25B and Cdc25C and sequesters
them, preventing them from binding to and activating mitotic CDKs, which results in arrest in (see Figures 19-34 and 19-21).
FIGURE 19-34 Overview of DNA damage checkpoint controls in the cell cycle. In response to DNA damage, the ATM or ATR protein kinases (ATM/R) inhibit Cdc25 family members via the Chk1/2 and MK2 protein kinases. They also activate p53, which induces production of the CKI p21. During , the p53-p21 pathway inhibits CDKs. In S and replication stress (slow DNA replication fork movement or DNA replication fork collapse), activates the ATR-Chk1 protein kinase cascade, while DNA double-strand breaks activate the ATM-Chk2 protein kinase cascade. Chk1 and Chk2, together with MK2, phosphorylates and inactivates Cdc25B and C, blocking activation of mitotic CDKs and entry into mitosis. During and S, the DNA damage checkpoint pathways also
phosphorylate Cdc25A, targeting it for -mediated degradation, thereby inhibiting phase CDKs and S phase CDKs and blocking entry into or passage through S phase. Red symbols indicate pathways that inhibit progression through the cell cycle. Description The circular pattern of arrows is labeled, moving clockwise from top right, G subscript 1, S phase, G subscript 2, and M. In the G subscript 1 area, there are two flowcharts. At left is one that starts with D N A damage (black) with arrows downward through three steps in red: A T M slash R, p 53, p 21, then G 1 C D Ks( in black). The second flowchart starts with D N A damage (black) and shows one downward arrow in red labeled A T M slash R. This step has a side arrow to C h k 1 slash 2, M K 2 (red) which goes downward to S C F superscript beta T r C P (red) to C d c 25 A (black). Back at A T M slash R, there are two downward arrows in red: p 53, p 21. Both arms of this flowchart end at G 1 slash S phase and S phase C D Ks. In the S phase area the flow chart starts at the bottom with replication stress (black) two red upward arrows read: A T R, C h k 1. Then C d c 25 A (black) moves up to S phase C D Ks, which moves up to S phase. Back at C h k 1, a red branch goes off to the left with the label late replication (black). In the M phase, one flowchart starts at left with the title ongoing D N A replication, with 2 downward arrows labeled A T R (red) and C h k 1 14-3-3 (red). The flowchart to the right starts with D N A damage, then moves down to A T M slash R (red). This branches off into two columns. The left column shows, moving downward: C h k 1 slash 2, M K 2 (Red) C d c 2 5 C B slash C (black) and Mitotic C D Ks (red). The right column shows, moving downward, p 53(red), p 21(red) and mitotic C D Ks (red). Even in the absence of DNA damage, active DNA replication also inhibits entry into mitosis. ATR activates Chk1 and MK2 to inhibit Cdc25B/C until all replication forks complete DNA replication and disassemble. This mechanism makes the initiation of mitosis dependent on the completion of chromosome replication. Finally, cells also sense DNA replication stress that results in stalling or slowing of replication forks. Such stress triggers
activation of the ATR-Chk1 checkpoint pathway and results in downregulation of S phase CDK activity and prevents the firing of latereplicating origins. In addition, ATR also stabilizes the stalled fork so that resumption of DNA replication can ensue once the replication stress is resolved. Chk1-, Chk2-, and MK2-mediated inhibition of Cdc25 phosphatases is not the only means of inhibiting cell cycle progression when there is DNA damage or incomplete replication. DNA damage also leads to the activation of p53, a transcription factor that induces expression of the gene encoding the CDK inhibitor p21. p21 binds to and inhibits all metazoan cyclin-CDK complexes. As a result, cells are arrested in and (see
Figure 19-34). The transcription factor p53 is known as a tumor suppressor because its normal function is to limit cell proliferation in the face of DNA damage. This protein is extremely unstable and generally does not accumulate to high enough levels to stimulate transcription under normal conditions. The instability of p53 results from its ubiquitinylation by a ubiquitin-protein ligase called Mdm2 and its subsequent proteasomal degradation. Rapid degradation of p53 is inhibited by ATM and ATR, which phosphorylate p53 at a site that interferes with Mdm2 binding. This and other modifications of p53 in response to DNA damage greatly increase its ability to activate transcription of specific genes that help the cell cope with DNA damage, such as p21.
The Spindle Assembly Checkpoint Pathway Prevents Chromosome Segregation Until Chromosomes Are Accurately Attached to the Mitotic Spindle
Under some circumstances, such as when DNA damage is extensive, p53 also activates expression of genes that lead to apoptosis, the process of programmed cell death (see Figure 19-32). In metazoans, the p53 response leading to induce apoptosis (see Chapter 22) evolved presumably to prevent the accumulation of multiple mutations that might convert a normal cell into a cancer cell. The dual role of p53 in both cell cycle arrest and induction of apoptosis may account for the observation that nearly all cancer cells have mutations in the p53 gene or in the pathways that stabilize p53 in response to DNA damage (see Chapter 25). The consequences of mutations in p53, ATM, and Chk2 provide dramatic examples of the significance of cell cycle checkpoint pathways to the health of a multicellular organism. The Spindle Assembly Checkpoint Pathway Prevents Chromosome Segregation Until Chromosomes Are Accurately Attached to the Mitotic Spindle The spindle assembly checkpoint pathway is a global surveillance mechanism that ensures that all of the chromosomes are properly attached to spindle microtubules through amphitelic attachments before entry into anaphase occurs. If even a single kinetochore is unattached or not under tension, then anaphase is inhibited, since any attempt to pull the sister chromatids apart in the presence of such a defect would almost certainly
lead to chromosome loss. As we saw in Section 19.5, the spindle assembly checkpoint pathway monitors for kinetochores that are not attached to spindle microtubules. Microtubule-kinetochore interactions that are under insufficient tension are destabilized by the protein kinase Aurora B, which phosphorylates the microtubule-binding component of the kinetochore, the Ndc80 complex. This leads to the loss of stable kinetochore-microtubule interactions that are then recognized by the spindle assembly checkpoint pathway. In this manner, the kinase activity of Aurora B at the level of individual chromosomes and the spindle assembly checkpoint pathway that surveys attachments of all of the chromosomes collaborate during every cell cycle to accurately ensure that every single pair of sister chromatids is attached to the mitotic spindle in the correct, bi-oriented manner. Components of the spindle assembly checkpoint pathway bind to unoccupied microtubule binding sites at kinetochores and create an anaphase inhibitory signal that ultimately inhibits . Recall that is the ubiquitin ligase that triggers cyclin B degradation, and the ubiquitinylation and subsequent degradation of securin, triggering the activation of separase to degrade cohesins, and entry into anaphase (see
Figure 19-28). When a kinetochore is not attached to microtubules, the outer kinetochore component Knl1 is phosphorylated by the spindle assembly checkpoint pathway protein kinase Mps1 (Figure 19-35). This phosphorylation, in turn, recruits other checkpoint pathway components to the unattached kinetochore. Critical for the shutting down of is the recruitment and activation of the Mad1-Mad2 complex by unattached kinetochores. Importantly, the activated Mad1-Mad2 complex
can convert inactive (“open”) Mad2 molecules in the cytoplasm to an active (“closed”) conformation that is capable of binding to and inhibiting . Mad2 bound to recruits additional checkpoint factors to the complex to form the mitotic checkpoint complex (MCC). The MCC then prevents from recognizing and ubiquitinylating substrates. This spindle assembly checkpoint pathway enables a single unattached kinetochore to inhibit all cellular Cdc20, and thus anaphase, until the kinetochore becomes properly associated with spindle microtubules.
FIGURE 19-35 The spindle assembly checkpoint pathway. The spindle assembly checkpoint pathway is active until every single kinetochore has attached properly to spindle microtubules. Step 1 : When a kinetochore is unattached, the outer kinetochore component Knl1 is phosphorylated by the checkpoint kinase Mps1. Phosphorylated Knl1 then binds the checkpoint protein kinase Bub1-Bub3 and the checkpoint protein BubR1. These three proteins, in turn, recruit the Mad1-Mad2 complex to the kinetochore. The Mad1-Mad2 complex bound to a kinetochore adopts the active closed form (shown as Mad2-C; step 2 ) and has the ability to convert inactive open Mad2 (Mad2-O) in the cytoplasm into active closed Mad2 that is able to bind and inhibit it. Complete inhibition of requires the recruitment of the checkpoint factors Bub1-Bub3 and BubR1
into the complex. Together, these proteins form the mitotic checkpoint complex (MCC) that prevents from recognizing and ubiquitinylating its substrates. Silencing of the spindle assembly checkpoint pathway occurs once all kinetochores have attached to microtubules in a tension-generating manner. Protein phosphatase 1 then de-phosphorylates Knl1, thereby eliminating checkpoint protein binding sites at kinetochores. In addition, disassembles the MCC, allowing to degrade mitotic cyclins and securin to trigger anaphase. [Data from E. A. Foley and T. M. Kapoor, 2013, Nat. Rev. Mol. Cell Biol. 14:25–37; and A. Mussachio, 2015, Curr. Biol. 25:R1002–R1018.] Description The illustration has the following sequence: 1.An unbound kinetochore is attached to a phosphorylated K n l 1, which is further attached to b u b 3 checkpoint kinase. 2. This complex catalyzes the formation of M A D-2 A, which binds to active A P C-C complexes in the cytoplasm, to inhibit them. Once all chromosomes have attached to the mitotic spindle in the correct amphitelic manner, the spindle assembly checkpoint pathway must be silenced to allow to degrade securin and initiate anaphase. Silencing of the spindle assembly checkpoint pathway occurs through multiple mechanisms. Protein phosphatase 1 de-phosphorylates Knl1, thereby eliminating checkpoint protein-binding sites at kinetochores. In addition, a protein known as disassembles the MCC, thereby activating and initiating anaphase (Figure 19-35). The spindle assembly checkpoint pathway is essential for viability in mice, highlighting the importance of this quality-control pathway during every cell division. If anaphase is initiated before both
kinetochores of a replicated chromosome become attached to microtubules from opposite spindle poles, chromosomes are mis-segregated in a process called nondisjunction. The resulting condition, in which cells have either lost or gained whole chromosomes, is called aneuploidy. Aneuploidy has profound effects on health and fitness. It leads to the misregulation of genes and can contribute to the development of cancer. When nondisjunction occurs during the meiotic division that generates a human egg or sperm, trisomy (gain of a chromosome) or monosomy (loss of a chromosome) can be the result. As we will see in Section 19.8, meiosis is especially prone to nondisjunction, which can lead to spontaneous abortions or Down syndrome. KEY CONCEPTS OF SECTION 19.7 Surveillance Mechanisms in Cell Cycle Regulation Surveillance mechanisms known as checkpoint pathways establish dependencies among cell cycle events and ensure that the next cell cycle event does not occur prior to the completion of a preceding event. Checkpoint pathways consist of sensors that monitor a particular cellular event, a signaling pathway, and an effector that halts cell cycle progression and activates repair pathways when necessary. These pathways make extensive use of protein kinases, phospho-binding domains, and ubiquitin-protein ligases. Cells are able to detect and respond to a wide variety of DNA damage, and their response depends on what stage of the cell cycle cells are in (see Figure 19-34). In response to DNA damage, three related protein kinases, DNA-PK, ATM, and ATR, are recruited to the damage site, where they activate signaling pathways that lead to cell cycle arrest, repair, and under some circumstances, apoptosis. ATM and ATR establish cell cycle checkpoints through the actions of three effector checkpoint kinases Chk1, Chk2, and MK2 (see Figure 19-32). The spindle assembly checkpoint pathway, which prevents premature initiation of anaphase, utilizes Mad2 and other proteins to regulate , which targets securin and mitotic cyclins for ubiquitinylation (see Figure 19-35).
19.8 Meiosis: A Special Type of Cell Division
19.8 Meiosis: A Special Type of Cell Division In nearly all diploid eukaryotes, meiosis is the form of nuclear division that generates haploid germ cells — eggs or sperm. An egg or sperm from one individual can then fuse with a germ cell from another individual to generate a diploid zygote that develops into a new individual. Meiosis is a fundamental aspect of the evolution of eukaryotes because it results in the reassortment of the chromosome sets received from an individual’s two parents. Chromosome reassortment and homologous recombination between parent DNA molecules during meiosis guarantee that each haploid germ cell will receive a unique combination of alleles that is distinct from each parent and from every other haploid germ cell formed. Meiosis, like mitosis, is preceded by a , S, and phase. The mechanisms of meiosis are analogous to those of mitosis. However, several features of meiosis generate genetically diverse haploid cells (see
Figure 6-3). In the mitotic cell cycle, each S phase is followed by chromosome segregation and cell division. In contrast, during meiotic cell division, one round of DNA replication is followed by two consecutive chromosome segregation phases. This leads to the formation of haploid daughter cells. During the two divisions, maternal and paternal chromosomes are shuffled so that the daughter cells are different in genetic makeup from the parent cell. In this section, we discuss the
Extracellular and Intracellular Cues Regulate Germ Cell Formation
similarities between mitosis and meiosis and the meiosis-specific mechanisms that transform the canonical mitotic cell cycle machinery so that it leads to the formation of haploid daughter cells. Extracellular and Intracellular Cues Regulate Germ Cell Formation Identifying signals triggering entry into meiosis in metazoans is an active area of research. We know that extracellular signals induce a transcriptional program, which produces meiosis-specific cell cycle factors that bring about the meiotic cell divisions. This modification of the cell cycle goes hand in hand with a program that induces characteristic features of gametes, such as the development of a flagellum in sperm or the production of a stress-resistant cell wall in fungi. One of the extracellular signals inducing entry into meiosis in mammals is retinoic acid. Binding of retinoic acid to the retinoic acid receptor initiates activation of many different developmental processes (see Figure 9-43). The cellular targets of this hormone and how it functions to specify the meiotic fate remain to be discovered. The molecular mechanisms underlying the decision to enter meiosis are well understood in S. cerevisiae. The decision to enter the meiotic divisions is made in . Depletion of nitrogen and carbon sources induces diploid cells to undergo meiosis instead of mitosis, yielding haploid spores (see Figure 1-24). During the meiotic divisions, budding is repressed. Pre-meiotic S phase and the two meiotic divisions thus occur
Several Features Distinguish Meiosis from Mitosis
within the confines of the parent cell. Spore walls are produced around the four meiotic products. Recall that budding and the initiation of DNA replication are induced by phase CDKs. Their expression needs to be inhibited to prevent budding. Nutritional starvation represses expression of phase cyclins, thereby inhibiting budding. However, DNA replication also relies on phase CDKs. How can pre-meiotic DNA replication occur in the absence of phase CDKs? The sporulationspecific protein kinase Ime2 takes over the role of phase CDKs in promoting DNA replication. Ime2 promotes (1) phosphorylation of the APC/C specificity factor Cdh1, inactivating it so that S phase and M phase cyclins can accumulate (see Figure 19-17); (2) phosphorylation of transcription factors to induce expression of genes whose products are required for S phase, including DNA polymerases and S phase cyclins (see
Figure 19-18); and (3) phosphorylation of the S phase CDK inhibitor Sic1, leading to release of active S phase CDKs and the onset of pre-meiotic DNA replication. Several Features Distinguish Meiosis from Mitosis The meiotic divisions differ in several ways from mitotic divisions. These differences are summarized in Figure 19-36. The two consecutive cycles of cell division are termed meiosis I and meiosis II (Figure 19-37). Meiosis II resembles mitosis in that sister chromatids are segregated. However, meiosis I is very different. During this division, homologous chromosomes — the chromosome inherited from your mother and the
same chromosome inherited from your father — are segregated. This unusual chromosome segregation requires three meiosis-specific modifications to the chromosome segregation machinery. Next we discuss these modifications and explain why they are needed.
FIGURE 19-36 Comparison of the main features of mitosis and meiosis. See text for details. Description Row 1: In mitosis, one cell division results in two daughter cells. An arrow from a circular parent cell points at two circular daughter cells. In meiosis, two cell divisions
result in four products of meiosis. A circular meiocyte divides to two cells which further divide to become four cells labeled products of meiosis. Row 2: In mitosis, chromosome number per nucleus is maintained (example, for a diploid cell). A cell labeled 2 n divides into two cells labeled 2 n. In meiosis, chromosome number is halved in the products of meiosis. A cell labeled 2 n divides into two cells which further divide to form four cells all labeled n. Row 3: In mitosis, one pre-mitotic S phase occurs per cell division, whereas in meiosis, one pre-meiotic S-phase occurs for both cell divisions. Two graphs plot D N A per nucleus versus cell stages respectively. Row 4: In mitosis, normally, no pairing of homologous chromosomes occurs in prophase. Two separate chromosomes are depicted. A chromatid is labeled. In meiosis, full synapsis of homologous chromosomes in prophase occurs. A chromatid from two chromosomes combines to become bivalent. Row 5: In mitosis, normally no recombination occurs in prophase. In meiosis, at least one recombination between nonsister chromatids occurs. A chiasma is formed between two nonsister chromosomes. Row 6 and 7: In mitosis, bi-oriented sister kinetochores form and there is loss of cohesion between sister chromatid arms during prophase. Two chromosomes have an line passing through their centromeres. In meiosis, co-orientation of sister kinetochores in meiosis one occurs, and the cohesion between sister chromatid arms is maintained. Two chromosomes with chiasma between them have two lines each extending from their centromeres. Row 8: In mitosis, the centromeres divide at anaphase, whereas in meiosis, no division occurs in anaphase 1 but division does occur in anaphase 2. Below row 8: in mitosis, the daughter cells' genotypes are identical to the parents, whereas variations occur in meiosis. One more row: In addition, cells undergoing mitosis can be diploid or haploid, whereas in meiosis, cells are diploid or multiples thereof.
FIGURE 19-37 Meiosis. Pre-meiotic cells have two copies of each chromosome (2n), one derived from the paternal parent and one from the maternal parent. For simplicity, the paternal and maternal homologs of only one chromosome are diagrammed. Step 1 : All chromosomes are replicated during S phase before the first meiotic division, giving a 4n chromosomal complement. Cohesin complexes (not shown) link the sister chromatids composing each replicated chromosome along their full lengths. Step 2 : As chromosomes condense during the first meiotic prophase, the replicated homologs pair and undergo homologous recombination, leading to at least one crossover event. At metaphase I, shown here, both chromatids of one chromosome associate with microtubules emanating from one spindle pole, but each member of a homologous chromosome pair associates with microtubules emanating from opposite poles. Step 3 : During anaphase of meiosis I, the homologous chromosomes, each consisting of two chromatids, are pulled to opposite spindle poles, and the chiasmata are dissolved as the recombination between homologous chromosomes is completed. Step 4 : Cytokinesis yields two daughter cells (now 2n), which enter meiosis II without undergoing DNA replication. At metaphase of meiosis II, shown here, the sister chromatids associate with spindle microtubules from opposite spindle poles, as they do in mitosis. Steps 5 and 6 : Segregation of sister chromatids to opposite spindle poles during anaphase of meiosis II, followed by cytokinesis, generates haploid gametes (1n) containing one copy of each chromosome. Micrographs on the left show meiotic metaphase I and metaphase II in developing gametes from Lilium (lily) ovules. Chromosomes are aligned at the metaphase plate. Description The illustration has the following sequence. A premeiosis cell with 2 n chromosomes is present, one containing the paternal homolog and the other containing the maternal homolog. Meiosis 1: 1. Chromosomes are replicated, leading to 4 n chromosomes. 2. Recombination and synapsis between homologs occurs in prophase. Metaphase 1 has bivalent chromosomes attached to the spindle fibers. An electron micrograph shows the same. 3. Anaphase 1 starts, and the chromosomes move to the poles of the cell. Meiosis 2: 4. Cell division occurs and the daughter cells enter metaphase 2. An electron micrograph shows a cell with chromosomes lined at the center. 5. Anaphase two occurs, and the chromosomes in the daughter cells are pulled to the poles of the cells. 6. Four gamete cells are produced, each with 1 n chromosome.
The tension-based sensing mechanism responsible for accurately attaching chromosomes to the spindle during mitosis is also responsible for segregating chromosomes during meiosis I. Thus homologous chromosomes must be linked so that this tension-based mechanism can function. Homologous recombination between homologous chromosomes creates these linkages (see Figure 19-36). The molecular mechanisms of homologous recombination are discussed in detail in Chapter 4. Here we restrict our discussion to the importance of homologous recombination to successful meiotic divisions. In and prophase of meiosis I, the two replicated chromatids of each chromosome are linked together by cohesin complexes along the full length of the chromosome arms, just as they are following DNA replication in a mitotic cell cycle. In prophase of meiosis I, homologous chromosomes (i.e., the maternal and paternal chromosome 1, the maternal and paternal chromosome 2, etc.) pair with each other and undergo homologous recombination. The pairing of homologous chromosomes with each other, a unique feature of meiosis, is called synapsis, and is mediated by a proteinaceous complex known as the synaptonemal complex (SC) that initially forms at specific points on the chromosomes and spreads all along their lengths. This pairing precisely aligns the genes of homologous chromatids adjacent to one another in order to support homologous recombination. Significantly, at least one recombination event must occur between a maternal and a paternal chromosome. Crossing over of chromatids produced by recombination occurs at large multiprotein assemblies, called recombination nodules, and can be observed microscopically in the first meiotic prophase and metaphase as
Recombination and a Meiosis-Specific Cohesin Subunit Are Necessary for the Specialized Chromosome Segregation in Meiosis I
structures called chiasmata (singular, chiasma). Homologous chromosomes are linked through chiasmata by a meiotic-specific cohesin called Rec8 and are called bivalents (see Figure 19-36). During the later part of prophase, the synaptonemal complex is disassembled as the chromosomes condense until the homologous chromosomes are only attached at the chiasmata and adjacent cohesin molecules, which now provide the resistance to the pulling force exerted by microtubules on the metaphase I spindle (see Figure 19-37). At the meiotic metaphase to anaphase transition, Rec8 is cleaved by separase, much like how the mitotic cohesin Scc1 is cleaved by separase during mitosis, allowing the homologous chromosome pairs to separate. Recombination between homologous chromosomes that occurs in prophase of meiosis I has two functional consequences: first, it contributes to genetic diversity among individuals of a species by ensuring new combinations of alleles in different individuals; second, it connects homologous chromosomes during meiosis I metaphase. The homologs, now connected through at least one chiasma, must align on the spindle in metaphase of meiosis I so that maternal and paternal chromosomes segregate away from each other during anaphase of meiosis I. Recombination and a Meiosis-Specific Cohesin Subunit Are Necessary for the Specialized Chromosome Segregation in Meiosis I
As we have seen, in metaphase of meiosis I, both sister chromatids in one (replicated) chromosome associate with microtubules emanating from the same spindle pole, rather than from opposite poles as they do in mitosis (Figure 19-38). Two physical links between homologous chromosomes resist the pulling force of the spindle until anaphase: (1) the chiasmata that result from crossing over between chromatids, and (2) cohesins distal to the crossover point (see Figure 19-39b, top). Evidence for the linking function of recombination during meiosis comes from the observation that when recombination is blocked by mutations in proteins essential for the process, chromosomes segregate randomly during meiosis I; that is, homologous chromosomes do not necessarily segregate to opposite spindle poles.
FIGURE 19-38 Chiasmata and cohesins distal to them link homologous chromosomes in meiosis I metaphase. Connections between chromosomes during meiosis I are most easily visualized in organisms with acrocentric centromeres, such as the grasshopper. The kinetochores at the centromeres of sister chromatids attach to spindle microtubules emanating from the same spindle pole, with the kinetochores of the maternal (red) and paternal (blue) chromosomes attaching to spindle microtubules from opposite spindle poles. The maternal and paternal chromosomes are attached to each other by chiasmata, which are formed by recombination between them and by the cohesion between sister chromatid arms that persists until metaphase I. Note that elimination of cohesion between sister chromatid arms by separase cleavage of the meiotic cohesin Rec8 is all that is required for the homologous chromosomes to separate at anaphase. See L. V. Paliulis and R. B. Nicklas, 2000, J. Cell Biol. 150:1223–1232; and S. B. C. Buonomo et al., 2000, Cell 103:387–398.
Description The illustration shows circular centrioles are at top and bottom poles with spindle microtubules extending from it attaching to pairs of chromatids. The chiasma is present between the pairs of chromatids on the left center and right side. Co-oriented sister kinetochores are labeled at the top left of a chromosome.
FIGURE 19-39 Cohesin function during mitosis and meiosis. (a) During mitosis, sister chromatids generated by DNA replication in S phase are initially linked by cohesin complexes along the length of the chromatids. During chromosome condensation, cohesin complexes (yellow) become restricted to the region of the centromere at metaphase. Shugoshin (purple) recruits PP2A to centromeres, where they antagonize Polo kinase and Aurora B, preventing the dissociation of cohesins from centromeric regions. Dissociation of
Shugoshin from centromeres and activation of separase lead to removal of cohesins at the centromere. Sister chromatids now separate, marking the onset of anaphase. (b) In prophase of meiosis I, maternal and paternal chromatids establish linkages between each other by homologous recombination. By metaphase I, the chromatids of each replicated chromosome are cross-linked by cohesin complexes along their full length. Rec8, a meiosis-specific homolog of Scc1, is cleaved along chromosome arms but not around the centromere, allowing homologous chromosome pairs to segregate to daughter cells. Centromeric Rec8 is protected from cleavage by PP2A, recruited to centromeric regions by the PP2A regulator Shugoshin (shown in purple). By metaphase II, the Shugoshin-PP2A complex dissociates from chromosomes. Cohesins can now be cleaved during meiosis II, allowing sister chromatids to segregate. See F. Uhlmann, 2001, Curr. Opin. Cell Biol. 13:754–761. Description The illustration labeled (a) titled mitosis shows a cell in metaphase where the centromeres are attached to microtubules holding a chromosome bound to cohesin rings, kinetochores and shugoshin-P P 2 A. With the action of separase the chromosome bound complexes disintegrate and the chromosome splits. The anaphase stage starts with the two chromatids formed moving towards the poles with the help of microtubules. The illustration labeled (b) titled meiosis shows a cell in metaphase 1 where the centromeres are attached to microtubules holding a pair of chromosome bound to cohesin rings, kinetochores and shugoshin-P P 2 A. R e c 8 is labeled. With the action of separase the chromosome bound complexes disintegrate and the chromosomes split. The anaphase 1 stage starts with the two chromosomes formed moving towards the poles with the help of microtubules. Below, a cell shows metaphase 2 where the centromeres are attached to microtubules holding a chromosome bound to R e c 8. With the action of separase the chromosome bound complexes disintegrate and the chromosome splits. The anaphase 2 stage starts with the two chromatids formed moving towards the poles with the help of microtubules.
At the onset of meiotic anaphase I, cohesins between chromosome arms are cleaved by separase. This cleavage is required for homologous chromosomes to segregate. If cohesins were not lost from chromosome arms, the recombined sister chromatids would be torn apart during anaphase I. The maintenance of centromeric cohesion during meiosis I is necessary for the proper segregation of sister chromatids during meiosis II. Studies in many organisms have shown that a specialized cohesin subunit, Rec8, is necessary for the stepwise loss of cohesins from chromosomes during meiosis. Expressed only during meiosis, Rec8 is homologous to Scc1, the cohesin subunit that closes the cohesin ring in the cohesin complex of mitotic cells (see Figure 19-20). Immunolocalization experiments revealed that during early anaphase of meiosis I, Rec8 is lost from chromosome arms but is retained at centromeres. During early anaphase of meiosis II, centromeric Rec8 is cleaved by separase, so the sister chromatids can segregate, as they do in mitosis (see Figure 19-39b, bottom). Consequently, understanding the regulation of Rec8-cohesin complex cleavage is central to understanding chromosome segregation in meiosis I. Cohesin removal differs for meiosis I because when Rec8 replaces Scc1 in the cohesin complex, the complex does not dissociate in prophase when it is phosphorylated. The meiotic cohesin complex can be removed from chromatin only by the action of separase. To be cleaved by separase, Rec8, unlike Scc1, requires phosphorylation by several protein kinases. During meiosis I, the centromere-specific isoform of PP2A targeted to
centromeric chromatin by Shugoshin prevents this phosphorylation. The PP2A targeting factor and PP2A then dissociate from chromosomes by metaphase II, allowing separase to cleave Rec8. Meiosis I is much more error-prone than mitosis. It is estimated that 10 percent of all conceptions in humans are aneuploid. These aneuploidies are largely caused by chromosome nondisjunction in meiosis I. When recombination fails to take place between homologous chromosomes, or when the chiasmata are too close to the end of the chromosomes, either no tension or too little tension is exerted on kinetochore–microtubule attachments, and they are destabilized, leading to nondisjunction. As a result, eggs or sperm receive too few or too many chromosomes. All monosomies (lack of a chromosome) and most trisomies (gain of a chromosome) result in embryonic lethality or death shortly after birth. Only trisomy 21, also known as Down syndrome, results in viable offspring, but it causes developmental abnormalities and intellectual disabilities. Nondisjunction in meiosis I increases dramatically with maternal age. Whereas fewer than 0.1 percent of babies born to women under the age of 30 have Down syndrome, this rate increases to 3.5 percent at age 45. This increase in chromosome nondisjunction with maternal age is not specific to chromosome 21. The incidence of trisomy in clinically recognized pregnancies is less than 5 percent in women under 30 but is as high as 35 percent in women over the age of 42 (Figure 19-40). The reason for this dramatic increase in nondisjunction with maternal age lies in the biology of female meiosis in vertebrates. In all vertebrates, pre-meiotic DNA
replication and recombination occur in the female embryo. The oocytes then become arrested in of meiosis I until the female reaches sexual maturity, which in humans is between the ages of 12 and 16. It is at this time that the first oocyte enters meiosis and progresses to the second meiotic metaphase, where it arrests and awaits fertilization (see Figure 195). The oocytes that enter the meiotic divisions in a 40-year-old woman have been arrested in for 40 years, during which time cohesins that hold the homologous chromosomes together during this very long phase can deteriorate, causing the homologous chromosomes to dissociate from each other. This can cause the homologous chromosomes to missegregate, leading to the formation of aneuploid eggs.
FIGURE 19-40 Fetal aneuploidy increases with maternal age. The percentage of trisomic embryos identified among clinically recognized pregnancies is shown as a function of maternal age. [Data from T. Hassold and P. Hunt, 2001, Nat. Rev. Genet. 2:280–291.] Description The vertical axis of the graph represents incidence of trisomy (percentage of clinically recognized pregnancies) ranging from 0 to 35 percent in increments of 5. The
Co-orienting Sister Kinetochores Is Critical for Meiosis I Chromosome Segregation
horizontal axis represents maternal age in years ranging from 15 to 40 in increments of 5. A curve shows low rates of trisomy, about 2.5 percent between 15 and 25. However, from 20, there is a slight rising trend, and by 35, the incidence has increased to 10 percent. By 40, the incidence as more than tripled to 35 percent. Co-orienting Sister Kinetochores Is Critical for Meiosis I Chromosome Segregation At metaphase in mitosis and meiosis II, sister kinetochores attach to spindle microtubules emanating from opposite spindle poles; the kinetochores are said to be bi-oriented. This is essential for segregation of sister chromatids to different daughter cells. In contrast, at meiosis I metaphase, sister kinetochores attach to spindle microtubules emanating from the same spindle pole; these sister kinetochores are said to be cooriented (see Figure 19-38). Obviously, attachment of sister kinetochores to the proper microtubules in meiosis I and II is critical for correct meiotic segregation of chromosomes. Proteins required for meiosis I sister kinetochore co-orientation were first identified in S. cerevisiae. In this organism, a single microtubule attaches to each kinetochore. We now know that a protein complex known as the monopolin complex associates with sister kinetochores during meiosis I and fuses them into a single kinetochore unit, to which one microtubule attaches. In all other organisms, kinetochores attach to multiple microtubules. In vertebrates, Rec8-containing cohesins are essential for
sister kinetochore co-orientation. These meiosis-specific cohesins impose a rigid kinetochore structure, restricting the movement of sister kinetochores and thereby favoring their attachment to microtubules from the same spindle pole. Correct attachment of meiosis I chromosomes is mediated by a tensionbased mechanism, as it is during mitosis and meiosis II. During meiotic metaphase I, kinetochore-associated microtubules are under tension (even though the co-oriented kinetochores of sister chromatids attach to microtubules coming from the same spindle pole) because chiasmata generated by recombination between homologous chromosomes and the cohesins distal to the chiasmata prevent them from being pulled to the poles (see Figure 19-38). Since kinetochore-microtubule attachments are unstable in the absence of tension (due to Aurora B–mediated phosphorylation), kinetochores that attach to microtubules from the wrong spindle release those microtubules, which enables them to bind microtubules again until attachments are made that generate tension. As in mitosis, once tension is generated, microtubule attachment to the kinetochores is stabilized. KEY CONCEPTS OF SECTION 19.8 Meiosis: A Special Type of Cell Division Meiosis is a specialized type of cell division in which meiosis-specific gene products modulate the mitotic cell division program (see Figure 19-36). The meiotic division comprises one cycle of chromosome replication followed by two cycles of cell division to produce haploid germ cells from a diploid pre-meiotic cell. During meiosis I, homologous chromosomes are segregated; during meiosis II, sister chromatids separate.
Specialized environmental conditions induce a developmental program that leads to the meiotic divisions. During prophase of meiosis I, homologous chromosomes pair with each other and undergo recombination. At least one recombination event must occur between the chromatids of each pair of homologous chromosomes for proper chromosome segregation (see Figure 19-37). Chiasmata and cohesins distal to them are responsible for holding the homologous chromosomes together during prophase and metaphase of meiosis I (see Figure 1938). At the onset of anaphase of meiosis I, cohesins on chromosome arms are phosphorylated and, as a result, cleaved by separase, but cohesins in the region of the centromere are protected from phosphorylation and cleavage. This protection is brought about by a meiosis-specific cohesin subunit and a protein phosphatase that associates with centromeres. As a result, the sister chromatids remain linked to each other during segregation in meiosis I (see Figure 19-39). Cleavage of centromeric cohesins during anaphase of meiosis II allows individual chromatids to segregate into germ cells (see Figure 19-39). Meiotic cohesins facilitate the attachment of sister kinetochores to microtubules emanating from the same pole during meiosis I.
Key Terms
End of Chapter Key Terms amphitelic attachment anaphase-promoting complex (cyclosome; APC/C) aneuploidy ataxia telangiectasia and Rad3-related protein (ATR) ataxia telangiectasia mutated (ATM) Aurora kinase bi-oriented Cdc14 phosphatase Cdc25B phosphatase Cdc25C phosphatase CDK-activating kinase (CAK) centriole centromere centrosome (spindle pole body) centrosome disjunction checkpoint pathway chromatid cohesin condensin co-oriented critical cell size cytokinesis
DNA-PK (DNA-dependent protein kinase) E2F transcription factor (E2F) effector cyclin-CDK phase cyclin-CDK kinetochore meiosis merotelic attachment mitogen mitotic cyclin-CDK mitotic spindle monopolin complex monotelic attachment nondisjunction nucleoporin p53 S phase cyclin-CDKs SCF securin sensor separase signaling cascade sister chromatid resolution synaptonemal complex (SC) syntelic attachment temperature-sensitive mutant Wee1
Review the Concepts
Review the Concepts 1. What cellular mechanism(s) ensure that passage through the cell cycle is unidirectional and irreversible? What molecular machinery underlies these mechanism(s)? 2. What types of experimental strategies do researchers employ to study cell cycle progression? How do genetic and biochemical approaches to this topic differ? 3. Tim Hunt shared the 2001 Nobel Prize in Physiology or Medicine for his work in the discovery and characterization of cyclin proteins in eggs and embryos. Describe the experimental steps that led him to his discovery of cyclins. 4. What physiological differences between S. pombe and S. cerevisiae make them useful yet complementary tools for studying the molecular mechanisms involved in cell cycle regulation and control? 5. In Xenopus, one of the substrates of mitotic CDKs is the phosphatase Cdc25. When phosphorylated by mitotic CDKs, Cdc25 is activated. What is the substrate of Cdc25? How does this information help to explain the rapid rise in mitotic CDK activity as cells enter mitosis? 6. Explain how CDK activity is modulated by the following proteins: (a) cyclin, (b) CAK, (c) Wee1, (d) p21. 7. Explain the role of CDK inhibitors. If cyclin-CDK complexes are necessary to allow regulated progression through the eukaryotic cell cycle, what would be the physiological rationale for CDK inhibitors?
8. Cancer cells typically lose cell cycle entry control. Explain how the following mutations, which are found in some cancer cells, lead to a bypass of these controls: (a) overexpression of cyclin D, (b) loss of Rb function, (c) loss of p16 function, (d) hyperactive E2F. 9. The Rb protein has been called the “master brake” of the cell cycle. Describe how the Rb protein acts as a cell cycle brake. How is the brake released in mid to late to allow the cell to proceed to S phase? 10. A common feature of cell cycle regulation is that the events of one phase ensure progression to a subsequent phase. In S. cerevisiae, and phase CDKs promote S phase entry. Name two ways in which they promote the activation of S phase. 11. For S phase to be completed in a timely manner, DNA replication must be initiated from multiple origins in eukaryotes. In S. cerevisiae, what role do S phase CDKs and DDKs play to ensure that the entire genome is replicated once and only once per cell cycle? 12. In 2001, the Nobel Prize in Physiology or Medicine was awarded to three cell cycle scientists. Paul Nurse was recognized for his studies with the fission yeast S. pombe, in particular for the discovery and characterization of the gene. What did the characterization of the gene tell us about cell cycle control? 13. What role do phosphoserine/threonine-binding proteins play in controlling entry from into S phase and entry from into M phase?
14. Describe how cells know whether sister kinetochores are properly attached to the mitotic spindle. 15. Describe the series of events by which APC/C promotes the separation of sister chromatids at anaphase. 16. Describe how cytokinesis is regulated so that cell cleavage only occurs after chromosome separation. 17. Leland Hartwell, the third recipient of the 2001 Nobel Prize in Physiology or Medicine, was acknowledged for his characterization of cell cycle checkpoint pathways in the budding yeast S. cerevisiae. What is a cell cycle checkpoint pathway? When during the cell cycle do checkpoint pathways function? How do cell cycle checkpoint pathways help to preserve the genome? 18. What role do tumor suppressors, including p53, play in mediating cell cycle arrest for cells with DNA damage? 19. Individuals with the hereditary disorder ataxia telangiectasia suffer from neurodegeneration, immunodeficiency, and an increased incidence of cancer. The genetic basis for ataxia telangiectasia is a loss-of-function mutation in the gene encoding ATM (ATM; ataxia telangiectasia mutated). Besides p53, what other substrate is phosphorylated by ATM? How does the phosphorylation of this substrate lead to inactivation of CDKs to enforce cell cycle arrest? 20. Overall, meiosis and mitosis are analogous processes involving many of the same proteins. However, some proteins function uniquely in each of these cell-division events. Explain the meiosis-specific function of the following: (a) Ime2, (b) Rec8, (c) monopolin.
21. Explain why the incidence of Down syndrome increases with maternal age.