Introduction
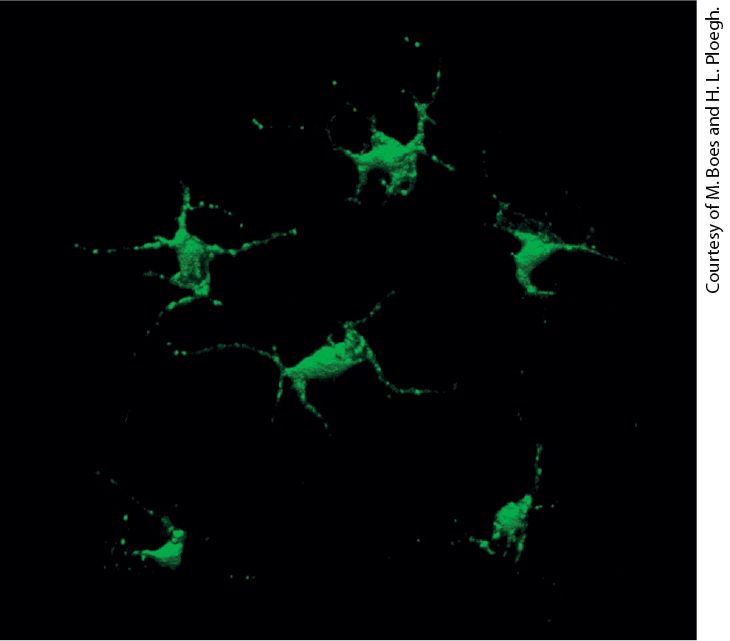
Chapter 24 Immunology Dendritic cells in the skin have class II MHC molecules on their surface. Those shown here were engineered to express a class II MHC–GFP fusion protein, which fluoresces green.
24.3 Generation of Antibody Diversity and B-Cell Development
24.5 T Cells, T-Cell Receptors, and T-Cell Development
24.6 Collaboration of Immune-System Cells in the Adaptive Response Metazoan animals and the cells within them are surrounded by a sea of pathogens — viruses, bacteria, and fungi — and have evolved a number of defenses to counter them, collectively termed the immune system. Immunity is a state of protection against the harmful effects of exposure to pathogens. Even insects such as Drosophila have an immune system with two types of defenses: (1) physical barriers and chemical defenses, and (2) cells that comprise an innate immune system that can recognize and destroy certain pathogens (Figure 24-1). Mechanical and chemical defenses operate continuously. Innate immune responses, which involve cells and molecules that are present in the body at all times, are rapidly activated (in minutes to hours). However, because they recognize molecules that are common to many foreign organisms, their ability to distinguish among different pathogens is somewhat limited.
FIGURE 24-1 The three layers of vertebrate immune defenses. Left: Mechanical defenses consist of epithelia and skin. Chemical defenses include the low pH of the gastric environment and antibacterial enzymes in tears. These barriers provide continuous protection against invaders. Pathogens must physically breach these defenses (step 1 ) to infect the host. Middle: Pathogens that have breached the mechanical and chemical defenses (step 2 ) are handled by cells and molecules of the innate immune system (blue). These include phagocytic cells (neutrophils, dendritic cells, macrophages) that engulf the invader and destroy it in lysosomes, natural killer (NK) cells, complement proteins, and certain interleukins (IL-1, IL-6). Innate defenses are activated within minutes to hours of infection. Right: In vertebrates, pathogens that are not cleared by the innate immune system are dealt with by the adaptive immune system (step 3 ), in particular B lymphocytes and their antibody products (Y-shaped icons), and several types of T cells and the many proteins they make that can destroy pathogens. Full activation of adaptive immunity requires days. The products of an innate response may potentiate an ensuing adaptive response (step 4 ). Likewise, the products of an adaptive immune response, including antibodies, may enhance innate immunity (step 5 ). Several cell types and secreted products are used in both the innate and adaptive immune systems and serve to connect these two layers of host defense. Description
The defenses against pathogens are depicted in 4 steps. Step 1: The skin acts as a mechanical defense. An illustration shows pathogens from which an arrow point at a cut in the mechanical defenses. Below the skin, chemicals give rise to chemical defenses against invading pathogens. Step 2: In the ensuing minutes to hours after infection, the innate immune system is active, involving macrophages, dendritic cells, neutrophils, and natural killer cells. Also, complement cascades of interleukins 1 and 6 occur. Arrows indicate that the products of the innate response can affect the adaptive immune response. Step 3: After days, the adaptive immune response, consisting mainly of killer T and B cells responds. An arrow indicates that products of adaptive immunity, such as antibodies can become part of the body’s innate immunity. In addition to these defense mechanisms, vertebrates have evolved a sophisticated immune system, the adaptive immune system, that can recognize and respond to many types of molecules not normally present in the body, including those that are parts of specific pathogens. The adaptive immune system generates secreted proteins termed antibodies, each of which binds to a specific foreign target molecule (see Figure 3-22). The adaptive immune system also generates many types of cells that attack and destroy pathogenic invaders. Adaptive immune responses take several days to develop fully and are highly specific; that is, they can distinguish between closely related pathogens based on very small molecular differences in the structures of their proteins or other polymers. The adaptive immune system makes adjustments to these threats over time, changing in response to the types and abundances of pathogens to which the host is exposed.
All pathogens have found ways to disarm the immune system or manipulate it to their own advantage, and host-pathogen interactions are an evolutionary work in progress. Virtually all pathogens have relatively short generation times compared with the hosts they infect and thus can evolve sophisticated countermeasures against their hosts’ immune system. This explains why, despite the evolution of remarkably sophisticated adaptive immune systems, pathogenic viruses, bacteria, and parasites continue to pose a threat to human populations. Seasonal outbreaks of influenza caused by new strains of influenza virus are just one example. These sophisticated defenses come at a price: an immune system capable of dealing with a massively diverse collection of rapidly evolving pathogens can sometimes mistake the host’s own tissues for pathogens and mount an attack against its own cells and tissues, a phenomenon called autoimmunity. Even so, we have learned to exploit the workings of the immune system to create vaccines that protect against a variety of infectious diseases. Vaccines are remarkably cost effective and have contributed to eliminating the scourge of many epidemics, such as outbreaks of smallpox. In this chapter, we deal mainly with the vertebrate immune system, with particular emphasis on those molecules, cell types, and pathways that uniquely distinguish the immune system from other types of cells and tissues. Four remarkable features characterize the vertebrate immune system. They are specificity, diversity, memory, and tolerance. Specificity is the immune system’s ability to distinguish between closely related substances. Diversity is the system’s capacity to specifically recognize an
astoundingly large number of different molecules. Memory is a host’s ability to recall previously experienced exposure to a foreign substance and more rapidly and effectively defend itself from that substance the next time it is encountered. Tolerance is the ability to avoid mounting an immune-system attack against the host’s own cells and tissues; that is, the ability to distinguish the body’s own tissues (“self”) from foreign materials including pathogens (“nonself”). Any material that can evoke an immune response is referred to as an antigen. As we shall see, the immune system achieves specificity and diversity by generating a large number of distinct proteins, such as antibodies and specific cell-surface receptors, each of which can bind very tightly to a target antigen such as a pathogenic molecule, but not to other, perhaps structurally very similar, molecules. Memory and tolerance depend on complex cellular systems. They are accomplished through the generation of a massively diverse set of cellsurface receptors that bind specific antigens. Cells that display these receptors have been “trained” so that they are largely unresponsive to self components (self-tolerant). From a practical perspective, the powers of the immune system can be exploited therapeutically. Today there is a multibillion-dollar market for monoclonal antibodies, which are used in the treatment of inflammatory conditions, autoimmune diseases, and cancer. The molecules that constitute the adaptive immune system — antibodies in particular — are also indispensable tools for the cell biologist, as we saw in Chapters 3 and
4. Antibodies allow visualization and isolation of the molecules they recognize with pinpoint precision. Their ability to do so has been invaluable in the accurate description of the components that make up the cell and its organelles and their localization, both in cells and in tissues. The technique of immunofluorescence, for example, is widely used by cell biologists to study cell morphology and behavior, while immunoblotting (Western blotting) has become an indispensable tool in the study of signal transduction. The ways in which these foreign materials are recognized and eliminated involve molecular and cell biological principles unique to the immune system. We begin this chapter with a brief sketch of the organization of the mammalian immune system, introducing the essential cell players in innate and adaptive immune responses and describing inflammation, a localized response to injury or infection that leads to the activation of immune-system cells and their recruitment to the affected site. In the next two sections, we discuss the structure and function of antibody (or immunoglobulin) molecules, which bind to specific molecular features on antigens, and how variability in antibody structure contributes to the recognition of specific antigens. The enormous diversity of antigens that can be recognized by the adaptive immune system finds its explanation in unique rearrangements of segments of DNA to generate antigen-specific receptors in B and T lymphocytes, commonly called B cells and T cells, which are the white blood cells that carry out antigen recognition. These gene rearrangements permit adaptation to a wide variety of pathogens by setting the specificity of antigen-binding receptors on lymphocytes; they also determine cell fate in the course of lymphocyte development.
Although the gene rearrangement mechanisms that give rise to antigenspecific receptors on B and T cells are very similar, the manner in which these receptors bind to (recognize) antigens is very different. The receptors on B cells — antibody molecules anchored in the plasma membrane — can interact with intact antigens directly, but the receptors on T cells cannot. Instead, as described in Section 24.4, the receptors on T cells recognize forms of antigen that are processed by target cells: small peptides and other small molecules that are displayed (or “presented”) on the surfaces of these cells by specialized cell-surface glycoproteins. These glycoproteins are encoded by genes in a region of the genome called the major histocompatibility complex (MHC). These MHC-encoded glycoproteins, also called MHC products, help determine the host’s ability to mount both T-cell and B-cell responses to antigens. Understanding these fundamental properties of the immune system has allowed us to answer a number of very practical questions: How can we best make antibodies that afford protection against an infectious agent? How can we raise antibodies to specific proteins we want to study in the laboratory? Knowledge of antigen processing and presentation thus informs both vaccine design to protect against infectious disease and the generation of tools essential for research. MHC-encoded glycoproteins also play a key role in an individual’s development of tolerance for his or her own antigens. We conclude the chapter with an integrated view of the immune response to a pathogen, highlighting the collaboration between different immune-system cells that is required for an effective immune response.
Pathogens Enter the Body Through Different Routes and Replicate at Different Sites
24.1 Overview of Host Defenses Because the immune system evolves in the presence of microbes, including pathogenic ones, we begin our overview of host defenses by examining where typical pathogens are found in a host and where they replicate. Then we introduce the basic concepts of innate and adaptive immunity, including some of the key cellular and molecular players. Pathogens Enter the Body Through Different Routes and Replicate at Different Sites Human skin has a surface area of some 20 square feet. The epithelial surfaces that line our airways, gastrointestinal tract, and urogenital tract present an even more formidable surface area of about 4000 square feet. All these surfaces are exposed on a daily basis to viruses, bacteria, and fungi in the environment. Some of these bacteria, called commensal bacteria, do not usually cause disease and in fact can be beneficial, helping to provide key nutrients or to maintain healthy skin. The microbiota also helps tune reactivity and composition of both the innate and adaptive immune systems via engagement of their receptors and the release of small molecules. It is thought that at any point in time, an adult human may be carrying as much as 3 pounds of microbes, against which most of us do not develop an overt inflammatory immune reaction. These
commensal microbes are not pathogenic as long as they remain on these outer surfaces of the body. If the normal barrier function of the epithelia that compose these surfaces is compromised, however, and these microbes enter the body, they can be pathogenic. Food-borne pathogens and sexually transmitted agents target the epithelia to which they are exposed. The sneeze of a flu-infected individual releases millions of virus particles in aerosolized form, ready for inhalation by a new host. Rupture of the skin, even if only by minor abrasions, or of the epithelial barriers that protect the underlying tissues provides an easy route of entry for pathogens, which then gain access to a rich source of nutrients (for bacteria) and to the cells required for replication (for viruses). Replication of viruses is confined strictly to the cytoplasm or nuclei of host cells, where viral protein synthesis and replication of the viral genetic material occur. Viruses can then spread to other cells either as free virus particles (virions) released from the initially infected cell or by direct transfer to an adjacent cell (cell-to-cell spreading). Many bacteria can replicate in the extracellular spaces of the body, but some are specialized to invade host cells and survive and reproduce within those cells. Such intracellular bacteria, such as Mycobacterium tuberculosis, the causative agent of tuberculosis, reside either in the membrane-delimited vesicles through which they enter cells by endocytosis or phagocytosis (see Figure 17-19) or in the cytoplasm if they escape from these vesicles. An effective host defense system, therefore, needs to be capable of eliminating not only extracellular viruses and bacteria, but also host cells that harbor pathogens.
Cells of the Innate and Adaptive Immune Systems Circulate Throughout the Body and Take Up Residence in Tissues and Lymph Nodes
Parasitic eukaryotes can also cause disease. Some of these parasites, such as the protozoans that cause sleeping sickness (trypanosomes) or malaria (Plasmodium species; see Figure 1-25), have very complex life cycles and have evolved complex countermeasures to avoid destruction by the host’s immune system. Cells of the Innate and Adaptive Immune Systems Circulate Throughout the Body and Take Up Residence in Tissues and Lymph Nodes The circulatory system (Figure 24-2) is responsible for moving blood throughout the body. Blood comprises cells (red and white blood cells, platelets) and liquid (plasma, which contains dissolved substances including proteins, ions, and small molecules). In addition to the hemoglobin-containing, oxygen-carrying erythrocytes (red blood cells) that compose the overwhelming majority of blood cells, the blood also contains leukocytes (white blood cells) and platelets (involved in blood clotting). Leukocytes encompass a variety of cell types, including lymphocytes (B and T cells), monocytes (precursors to the scavenger cells called macrophages), dendritic cells, neutrophils, and natural killer (NK) cells, all of which have distinct functions in the immune system. In contrast to erythrocytes, which never leave the circulation until they get old and die, leukocytes leave the circulation and enter target tissues to
help protect the body from invaders. The circulatory system moves leukocytes from the sites where they are generated (bone marrow, thymus, fetal liver) to the sites where they can be activated (lymph nodes, spleen), and then to the site of infection. Once leukocytes arrive at a given location, they may leave and re-enter the circulation in the course of their tasks.
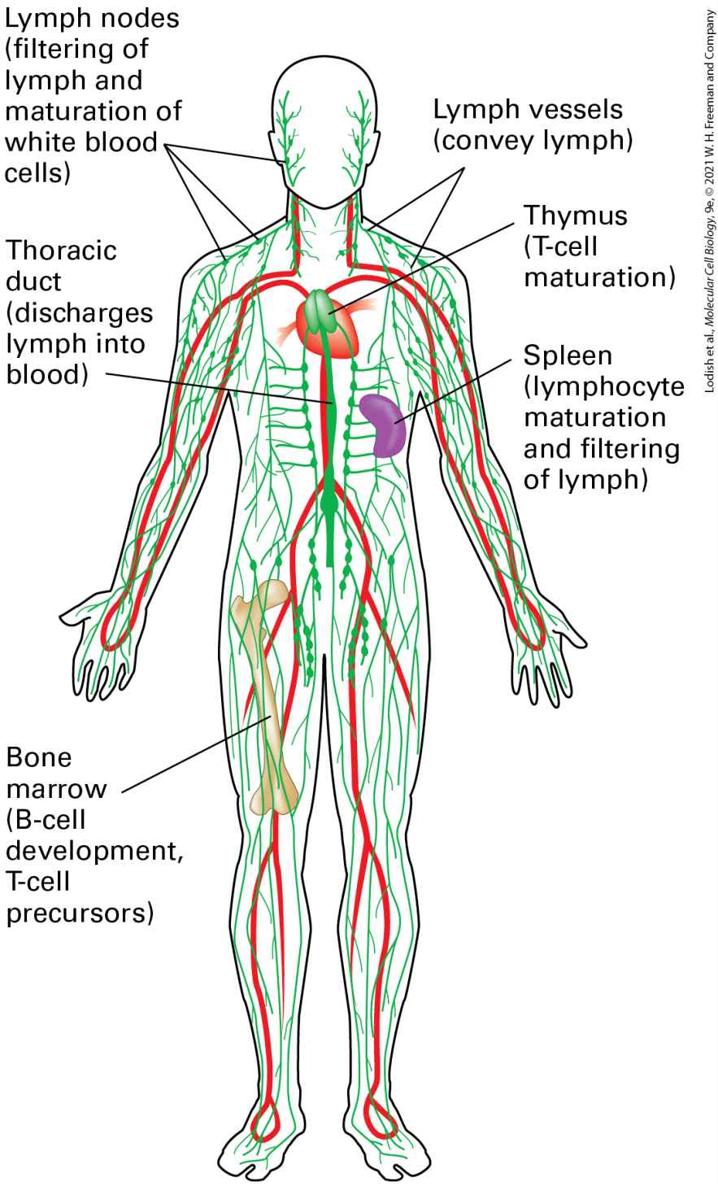
FIGURE 24-2 The circulatory and lymphatic systems. Positive arterial pressure exerted by the pumping heart is responsible for the movement of liquid from the circulatory system (red) into the interstitial spaces of the tissues, so that all cells of the body have access to nutrients and can dispose of waste. This interstitial fluid, whose volume is roughly three times that of all blood in the circulation, is returned to the circulation in the form of lymph, which passes through specialized anatomic structures called lymph nodes. The primary lymphoid organs, where lymphocytes are generated, are the bone marrow (B cells, T-cell precursors) and the thymus (T cells). The initiation of an immune response involves the secondary lymphoid organs (lymph nodes, spleen). Description The illustration also has the heart, spleen, and the femur bone highlighted. The lymph nodes (filtering of lymph and maturation of white blood cells) are labeled at the neck and shoulder region. A label at the lower left points to the thoracic duct in the center of the chest (discharges lymph into blood). At bottom left is a label pointing to the femur bone. The label reads: Bone marrow (B-cell development, T-cell precursors). On the right, at the neck, pointing to the neck and shoulder is a label that reads: lymph vessels (convey lymph). Below this is a label pointing to the heart area, reading: thymus (T-cell maturation). The last label on the right points to the spleen and reads: Spleen (lymphocyte maturation and filtering of lymph). The immune system, an interconnected system of vessels, organs, and cells, can be divided into primary and secondary lymphoid organs (see
Figure 24-2). Primary lymphoid organs are the sites at which lymphocytes — the subset of leukocytes that includes B and T cells — are generated and acquire their functional properties. These organs include the thymus, where T cells are generated from progenitors, and the bone marrow, where B cells are generated. Adaptive immune responses, which require functionally competent lymphocytes, are initiated in secondary lymphoid organs, which include lymph nodes and the spleen. All of the cells within
lymphoid organs are ultimately derived from hematopoietic stem cells (see Figure 22-18), generated initially in the fetal liver and subsequently in the bone marrow. The total number of lymphocytes in a young man is estimated to be . Roughly 15 percent of these cells are found in the spleen, 40 percent in the other secondary lymphoid organs (tonsils, lymph nodes), 10 percent in the thymus, and 10 percent in the bone marrow; the remainder circulate in the bloodstream or reside in other tissues. In normal circumstances, the pressure exerted by the pumping heart not only drives transport of the blood within blood vessels, but also forces cell-free liquid across blood vessel walls into the underlying tissue. This liquid delivers both nutrients and proteins, some of which carry out defensive functions. Its volume is up to three times the total blood volume. To maintain homeostasis, the fluid that leaves the circulation must ultimately return, and it does so in the form of lymph, via lymphatic vessels. At their most distal ends, lymphatic vessels are open to collect the interstitial fluid that bathes the cells in tissues. The lymphatic vessels merge into larger collecting vessels, which deliver lymph to lymph nodes (Figure 24-3). A lymph node consists of a capsule organized into areas that are defined by the cell types that inhabit them. Blood vessels entering a lymph node deliver B and T cells to it. The lymph that arrives in a lymph node carries soluble antigens and specialized cells that have encountered (“sampled”) antigens from the tissue drained by that particular afferent lymphatic vessel. In the lymph node, the cells and molecules required for the adaptive immune response interact, respond to the newly acquired
antigenic information, and then execute the necessary steps to rid the body of the pathogen (see Figure 24-3).
FIGURE 24-3 Initiation of the adaptive immune response in lymph nodes. Recognition of antigen by B and T cells (lymphocytes) located in lymph nodes initiates an adaptive immune response. T and B lymphocytes leave the circulation and take up residence in lymph nodes (step 1 ). Lymph carries antigen in two forms, soluble antigen and antigenladen dendritic cells; both are delivered to lymph nodes via afferent lymphatic vessels (steps 2 and 3 ). Dendritic cells, thus named because of their spine-like projections, are specialized cells of a type termed “antigen-presenting cells”; they degrade antigens into peptides or other small fragments and display them by means of MHC proteins on their surface, thus “presenting” antigens to T cells. Soluble antigen is recognized by B cells (step 4 ), and antigen-laden dendritic cells present antigen to T cells (step 5 ). Productive interactions between T and B cells (step 6 ) allow B cells to move into follicles and differentiate into plasma cells, which produce large amounts of secreted immunoglobulins
(antibodies). Efferent lymphatic vessels return lymph from the lymph node to the circulation. Description The cross section of the lymph node has a tube like structure labeled efferent lymphatic vessel. A U shaped blood vessel runs through the left side of the lymph node. The outer layer of the lymph node is labeled sinus. Two afferent lymphatic vessels are at the top. There are circular B cell follicles that line the inner membrane. The steps are as follows. Step 1: Mature T and B cells are delivered via the circulation and take up residence in lymph nodes. It points to an area lower center of node. Step 2: soluble antigen enters one of the afferent lymphatic vessels. Step 3: Antigen laden dendritic cell enters one of the afferent lymphatic vessels. Step 4: A B cell binds to soluble antigen and moves to follicle. Step 5: A T cell with an antigen-laden dendritic cell attached moving between the follicles and the blood vessel in lower left of the node. Activation of T cell by antigen-laden, activated dendritic cell; activated T cells may re-enter circulation. Step 6: A B cell follicle has B and T cells in it. Activated T cells interact with B cells, leading to B-cell differentiation and antibody production. Lymph nodes can be thought of as filters in which antigenic information gathered from distal sites throughout the body is collected and displayed to the immune system in a form suitable to evoke a response. All the relevant steps that lead to activation of a resting lymphocyte take place in lymphoid organs. Cells that have received proper instructions to become functionally active leave the lymph node via efferent lymphatic vessels that ultimately return lymph to the bloodstream. Such activated cells recirculate through the bloodstream and, now ready for action, may reach a location where they again leave the circulation in response to chemotactic cues, move into tissues, and seek out pathogenic invaders, destroy virus-infected cells, or produce the antibodies that recognize and tag the invaders for destruction.
Mechanical and Chemical Boundaries Form a First Layer of Defense Against Pathogens
The exit of lymphocytes and other leukocytes from the circulation, the recruitment of these cells to sites of infection, the processing of antigenic information, and the return of immune-system cells to the circulation are all carefully regulated processes that involve specific cell-adhesion events, chemotactic cues, and the crossing of endothelial barriers, as we will see later in this chapter. Having introduced the principal types of cells that form the immune system, we return to the first two lines of defense against foreign pathogens: physical barriers and chemical defenses, and an innate immune system. Mechanical and Chemical Boundaries Form a First Layer of Defense Against Pathogens As noted already, mechanical and chemical defenses form the first line of host defense against pathogens (see Figure 24-1). Mechanical defenses, which operate continuously, include skin, epithelia, and arthropod exoskeletons, all barriers that can be breached only by mechanical damage or through specific enzymatic attack. Chemical defenses include the low pH found in gastric secretions as well as enzymes such as lysozyme, found in tears and in intestinal secretions, that can attack microbes directly. The essential nature of mechanical defenses is immediately obvious in the case of burn victims. When the integrity of the skin (epidermis and
Innate Immunity Provides a Second Line of Defense
dermis; Figure 1-4) is compromised, the rich source of nutrients in the underlying tissues is exposed, and airborne bacteria or otherwise harmless commensal bacteria found on the skin can multiply unchecked, ultimately overwhelming the host. Viruses and bacteria have evolved strategies to breach the integrity of these physical barriers. For example, certain pathogenic bacteria (e.g., “flesh-eating bacteria,” which are highly pathogenic strains of Streptococcus) secrete collagenases that compromise the integrity of connective tissue and so facilitate access of the bacteria to underlying tissue. Innate Immunity Provides a Second Line of Defense The innate immune system is activated once the mechanical and chemical defenses have failed and the presence of an invader is sensed (see Figure 24-1). The innate immune system comprises cells and molecules that are always available for immediate response to pathogens. Phagocytes, cells that ingest and destroy pathogens (see Figure 17-19), have receptors on their surface that bind to molecules frequently present on pathogens but absent from normal body cells. Many yeasts and other fungi, for instance, have cell walls that contain polymers of the sugar mannose. Such polymers are not found on normal body proteins, and macrophages utilize a cell surface mannose receptor to bind many fungal pathogens. Macrophages are widespread throughout tissues and epithelia and can be recruited to sites of infection. Animals that lack an adaptive immune system, such as insects, rely exclusively on innate defenses to combat
infections. Likewise, plants rely exclusively on innate defenses and lack adaptive immunity altogether. Cells of the innate immune system, otherwise resembling lymphocytes, are now categorized as innate lymphoid cells (ILCs) and will be discussed later in this chapter. Phagocytes and Antigen-Presenting Cells The innate immune system includes macrophages, neutrophils, and dendritic cells. All of these cells are phagocytic and come equipped with other types of pathogen recognition receptors such as Toll-like receptors (TLRs; see Figure 24-37 for their molecular structure) and scavenger receptors on their cell surface. Like the mannose receptor, these receptors detect broad patterns of pathogen-specific markers, such as bacterial cellwall constituents or nucleic acids that contain unmethylated CpG or double-stranded RNA, and are thus key sensors for detecting the presence of bacteria or viruses. When these markers bind to TLRs, the cells produce effector molecules, including antimicrobial peptides. Dendritic cells and macrophages whose TLRs have detected pathogens also function as antigen-presenting cells (APCs) by processing and displaying foreign materials to antigen-specific T cells, thus bridging the innate and adaptive immune systems. The structure and function of TLRs and their role in activating dendritic cells are described in detail in Section 24.6. Inflammasomes and Non-TLR Nucleic Acid Sensors
Mammalian cells possess a family of proteins that are capable of recognizing all manner of nonself components and of perceiving danger signals. The molecules recognized by these proteins span a range from components of the bacterial cell wall to uric acid crystals, to heme degradation products, and even to asbestos and silica (Figure 24-4). Once recognized, these danger signals activate the assembly of a multiprotein complex called the inflammasome, which activates the effector proteins involved in inflammation. Proteins that make up the inflammasome contain modules that mediate interactions with adapter proteins that ultimately allow a physical connection with and activation of caspase-1, an enzyme that is critical in the production of cytokines that cause inflammation (a process described below). As we will see in Section 24.6, the inflammasome plays an important role in bridging the innate and adaptive immune response.
FIGURE 24-4 The NLRP3 inflammasome. The NLRP3 inflammasome activates caspase-1 only after receiving two signals. Signal 1 is provided by microbial antigens recognized via Toll-like receptors (TLRs) or by binding of endogenous cytokines such as TNF to the TNF receptor (TNFR). Signal 1 causes the up-regulation of NLRP3 (also called NALP3) and proIL-1β. Signal 2, which activates the NLRP3 inflammasome, can be provided by bacterial pore-forming toxins, by influenza virus M2 protein, by fungal particles via the kinase Syk (as shown for Candida albicans), or by cholera toxin. Cytosolic bacterial DNA can also activate the NLRP3 inflammasome, although the molecular details of this mechanism are not yet understood. Description Priming, signal 1: A microbial ligand is bound to T L R at the plasma membrane; there is a grape like structure that continues into the cytosol. The Endogenous cytokines (T N F) is present outside the plasma membrane. A T N F R molecule is bound to the plasma membrane as well. An arrow from the middle of the T L R and T N F R points at a D N A inside the nucleus. Gene transcription occurs which produces N A L P 3, which in
the presence of procaspase 1 yields N L R P 3 inflammasome which further yields caspase 1. Activation, signal 2: bacterial pore forming toxins, cholera toxin, candida albicans (yields S y k), cytosolic bacterial D N A, and influenza virus M 2 protein leads to the formation of N L R P 3 inflammasome which further yields caspase 1. The Complement System Another important component of the innate immune system is the complement system, a collection of serum proteins that can bind directly to microbial or fungal surfaces. This binding initiates a cascade of protease activation that culminates in, among other things, the formation of a membrane attack complex, a multiprotein complex that forms pores in the pathogen’s protective membrane (Figure 24-5). The cascade of complement activation is conceptually similar to the blood-clotting cascade, with amplification of the reaction at each successive stage of activation. At least three distinct pathways can activate the complement system. The classical pathway requires the presence of antibodies produced in the course of an adaptive immune response and bound to their antigens on the surface of the target microbe. How such antibodies are produced will be described in Section 24.3. This complement pathway represents an example of components of the innate immune system acting in concert with the adaptive immune system.
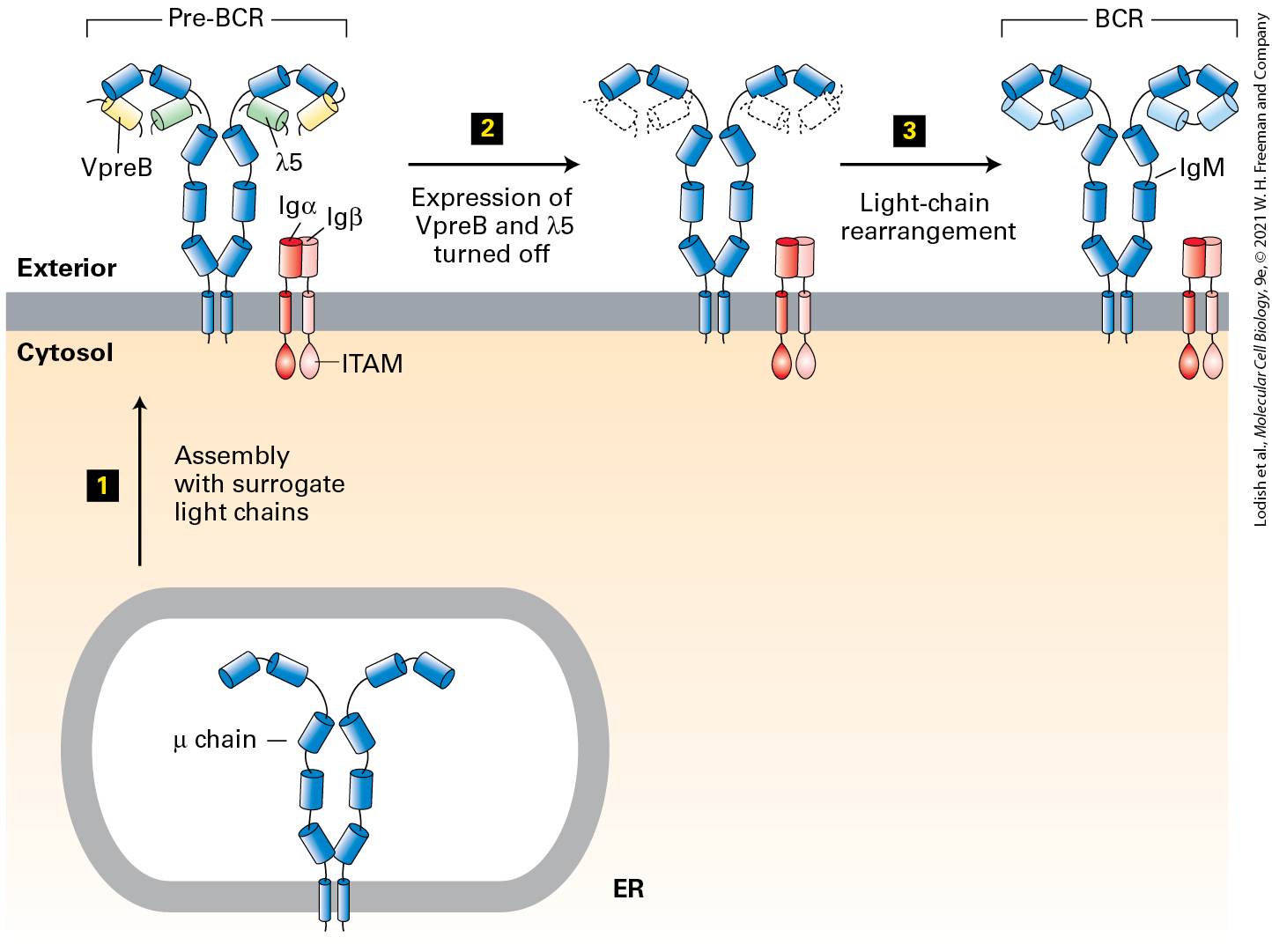
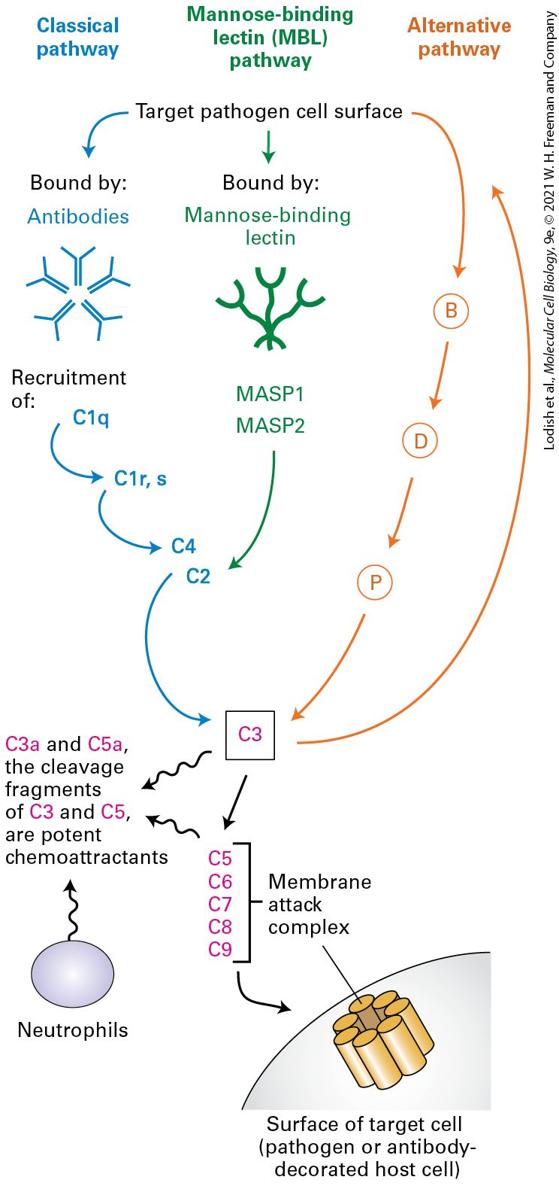
FIGURE 24-5 Three pathways of complement activation. The classical pathway involves the formation of antibody-antigen complexes. In the mannose-binding lectin pathway, mannose-rich structures found on the surfaces of many pathogens are recognized by mannose-binding lectin. The alternative pathway requires deposition of a special form of the serum protein C3, a major complement component, onto a microbial surface, upstream of which are factors B, D, and P. Each of the activation pathways is a cascade of proteases in which the downstream component is itself a protease. Amplification of activity occurs with each successive step. All three pathways converge on C3, which cleaves C5 and thus triggers formation of the membrane attack complex, leading to destruction of target cells. The small fragments of C3 and C5 generated in the course of complement activation are chemoattractants. They initiate inflammation by attracting neutrophils, phagocytic cells that can kill bacteria at short range or upon ingestion. Description All pathways end in C 3. In the classical pathway the target pathogen cell surface is bound by antibodies, which recruit C 1 q, C 1 r and C 1 s, C 4 which leads to C 3. In the mannose-binding lectin pathway (M B L), the target pathogen cell surface is bound by mannose-binding lectins, which recruit M A S P 1 and M A S P 2, which activate C 3 via C 2. The alternative pathway shows an arrow from the target pathogen cell surface that points at B, D, and P consecutively which finally leads to C 3. C 3 activates C 5 through C 9, which constitute the membrane attack complex on the surface of target cell (pathogen or antibody decorated host cell) The cleavage products of C 3 and C 5, C 3 a and C 5 a, are potent chemoattractants, and these attract neutrophils and other immune cells. In addition to the classical pathway of complement activation, pathogens that contain mannose-rich cell walls activate the complement cascade through the mannose-binding lectin pathway. (Note that this is distinct from the macrophage mannose receptor mentioned previously.) Mannosebinding lectin binds to distinctive groups of mannose sugars on the surface of the pathogen, which triggers activation of two mannose-binding lectin–
associated proteases, MASP-1 and MASP-2. Protease activation leads to activation of the downstream components of the complement cascade as shown in Figure 24-5. Finally, many microbial surfaces have physical and chemical properties, albeit incompletely understood, that result in activation of complement via the alternative pathway, an activation cascade that includes factors B, D, and P, all proteins found in plasma. The three pathways converge on the activation of complement protein C3. This protein is synthesized as a precursor that contains an internal, strained thioester linkage between a cysteine and a glutamate residue, requiring proteolytic conversion to become fully reactive. C3 is deposited only on antigen-antibody complexes that are nearby. Surfaces that are properly decorated with mannose-binding lectin or that receive C3 deposits via the alternative pathway are similarly targeted. This proximity restriction limits the effects of complement to nearby surfaces, avoiding an inappropriate attack on cells that do not display the targeted antigens. Regardless of the activation pathway, activated C3 unleashes the terminal components of the complement cascade, complement proteins C5 through C9, culminating in formation of the membrane attack complex, which inserts itself into almost any adjacent biological membrane and renders it permeable by forming a pore. The resulting loss of electrolytes and small solutes leads to lysis and death of the target cell. Whenever complement is activated, the membrane attack complex is formed and results in death of the cell onto which it is deposited. The direct microbe-killing (microbicidal) effect of a fully activated complement cascade is an important mechanism of host defense.
All three complement activation pathways also generate C3a and C5a cleavage fragments, which bind to G protein–coupled receptors and function to attract neutrophils and other cells involved in inflammation. In addition, phagocytic cells, such as macrophages, which recognize cells whose surfaces are covalently labeled with fragments from C3, ingest and destroy those cells. The complement cascade thus fulfills multiple roles in host defense: it can destroy the membranes that envelope a pathogen (bacteria, viruses); it covalently decorates the targeted pathogen so that it may be more readily ingested by phagocytic cells capable of killing the pathogen and presenting its contents to cells that will initiate an adaptive immune response. Finally, the act of complement activation yields signals to attract cells of the innate (neutrophils, macrophages, dendritic cells) and adaptive (lymphocytes) immune systems to the site of infection. These cues are called chemotactic signals. Natural Killer Cells In addition to bacterial and eukaryotic parasitic invaders, the innate immune system also defends against viruses. When the presence of a virus-infected cell is detected, still other cell types of the innate immune system become active, seeking out virus-infected target cells and killing them. For instance, when many types of cells (not just immune-system cells) are infected, they synthesize and secrete a class of proteins called type I interferons that act as intercellular signals, warning the immune system that an infection is present. The interferons are classified as
cytokines, small, secreted proteins that help regulate immune responses in a variety of ways. We will encounter other cytokines and discuss some of their receptors as the chapter progresses. Interferons activate natural killer (NK) cells, which belong to the group of innate lymphoid cells-1 (ILC1). These innate lymphoid cells will be discussed in Section 24.5. Activated NK cells help protect the body in several ways. First, they can kill host cells infected by a virus (hence the name “natural killer”), preventing those infected cells from making additional virus particles that would spread the infection. Second, NK cells secrete type II interferon γ which is essential for orchestrating many other aspects of antiviral defenses (Figure 24-6). Third, NK cells can kill target cells that have been decorated by antibodies. NK cells recognize their targets by means of several classes of surface receptors capable of yielding stimulatory (promoting cell killing) or inhibitory signals.
FIGURE 24-6 Natural killer cells. Natural killer (NK) cells or ILC1s are an important source of the cytokine interferon γ (IFN-γ), which is involved in antiviral defenses, and can kill virus-infected and cancerous cells directly by means of perforins. These pore-forming proteins allow access to the cytoplasm of the target cell by serine proteases called granzymes. Granzymes can also initiate apoptosis through activation of caspases (see
Chapter 22). Receptors on NK cells identify infected or stressed cells and stimulate the NK cell to kill them. Other receptors identify normal cells and inhibit NK cell activation. Description At the lower left, a normal cell is illustrated as a blue oval. A dotted line arrow from the normal cell labeled inhibits activation of N K cells points at an N K cell. The N K cell has three receptors of which one is negatively charged. The other two are positively charged. A dotted arrow from one of the positive receptors points towards stressed or
Inflammation Is a Complex Response to Injury That Encompasses Both Innate and Adaptive Immunity and Helps Destroy Pathogens
cancerous cell. A dotted arrow from the other positive receptor points towards virus infected cell. Both are labeled killing (perforins and granzymes). An arrow from the N K cell points at I N F gamma which leads to antiviral defense. Inflammation Is a Complex Response to Injury That Encompasses Both Innate and Adaptive Immunity and Helps Destroy Pathogens When a vascularized tissue (one that is supplied with blood vessels) is injured, the stereotypical response that follows is inflammation. The injury may be a consequence of physical or chemical processes, such as torn muscles, a simple paper cut, or infection with a pathogen. Inflammation, also called the inflammatory response, is characterized by four classic signs: redness, swelling, heat, and pain. These signs are caused by increased leakiness of blood vessels (vasodilation), attraction of immune-system cells to the site of damage, and the production of soluble mediators of inflammation, which are responsible for the sensation of heat and pain. Inflammation provides immediate protection through the activation of the cell types and soluble products that together mount the innate immune response and create a local environment conducive to the initiation of the adaptive immune response. If it is not properly controlled, however, inflammation can also be a major cause of tissue damage.
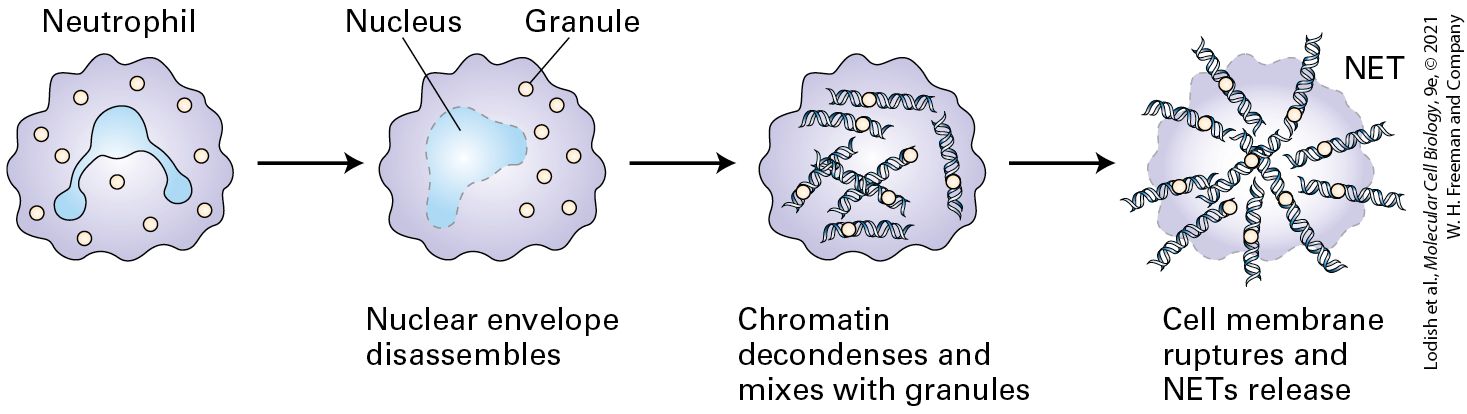
Figure 24-7 depicts the key players in the inflammatory response to bacterial pathogens and the subsequent initiation of an adaptive immune response. Tissue-resident dendritic cells sense the presence of pathogens via their TLRs and respond by releasing small soluble proteins such as cytokines and chemokines; the latter act as chemoattractants for immunesystem cells. Neutrophils leave the circulation and migrate to the site of injury or infection in response to the cytokines and chemokines produced there (see Figure 20-40). Neutrophils, which constitute almost half of all circulating leukocytes, are phagocytic (see Figure 17-19), directly ingesting and destroying pathogenic bacteria and fungi. Neutrophils can interact with a wide variety of pathogen-derived macromolecules via their TLRs. Engagement of these receptors, described in detail below, activates the neutrophils, which produce more cytokines and chemokines. The latter can attract more leukocytes — neutrophils, macrophages, and ultimately lymphocytes (T and B cells) — to the area to fight the infection. Activated neutrophils can also release bacteria-destroying enzymes (e.g., lysozyme and proteases) as well as small peptides with microbicidal activity, collectively called defensins. Activated neutrophils also turn on enzymes that generate the superoxide anion radical and other reactive oxygen species (see Section 12.4), which can kill microbes at short range. Neutrophils can also undergo a reaction referred to as NETosis. By committing suicide and releasing their nuclear DNA into the extracellular space, neutrophils throw out a fibrous net that can capture microbes and platelets (Figure 24-8). This reaction helps confine invaders to the site where neutrophil activation occurs. Another cell type that contributes to the inflammatory response is the tissue-resident mast cell. When activated by a variety of physical or chemical stimuli, mast cells release histamine,
a small molecule that binds to G protein–coupled histamine receptors. This binding leads to increased vascular permeability and thereby facilitates access to the tissue by plasma proteins (e.g., complement) that can act against the invading pathogen.
FIGURE 24-7 Interplay of innate and adaptive immune responses to a bacterial pathogen. Once a bacterium breaches the host’s mechanical and chemical defenses, the bacterium is exposed to components of the complement cascade, as well as to innate immune-system cells that confer immediate protection (step 1 ). Various inflammatory proteins induced by tissue damage contribute to a localized inflammatory response. Local destruction of the bacterium results in the release of bacterial antigens, which are delivered to the lymph nodes (step 2 ) via the afferent lymphatic vessels that drain the tissue. Dendritic cells acquire antigen at the site of infection, become migratory, and move to the lymph nodes, where they activate T cells (step 3 ). In the lymph nodes, antigen-stimulated T cells proliferate and acquire effector functions, including the ability to help B cells (step 4 ), some of which may move to the bone marrow and complete their differentiation into antibody-secreting plasma cells (step 5 ). In later stages of the immune response, activated T cells provide additional assistance to antigen-experienced B cells to yield plasma cells that secrete antigen-specific antibodies at a high rate (step 6 ). Antibodies produced as a consequence of the initial exposure to the bacterium act in synergy with complement to eliminate the infection (step 7 ), should it persist, or afford rapid protection in the case of re-exposure to the same pathogen. Description The mechanical defense of the body is represented by a brown line at the top. Bacteria enter via a cut in this defense line. The bacteria are engulfed into a dendritic cell. An N K cell and a neutrophil are in the vicinity. The dendritic cell and soluble antigens enter the lymph node to activate T cells. T cells proliferate. T cells activate B cells which enter into the bone marrow to be converted to a plasma cell with antibodies. The antibodies move towards the bacteria. A T cell from the lymph node moves towards the bacteria as well.
FIGURE 24-8 In NETosis neutrophils sacrifice themselves to facilitate capture of bacterial pathogens. Upon encounter with bacterial products such as lipopolysaccharides, neutrophils can undergo a form of cell death that results in the release of a network of nuclear DNA and associated molecules (a neutrophil extracellular trap or “NET”). NETs contain antimicrobial components and can kill bacteria extracellularly without reliance on phagocytosis. NETs can physically limit further spread of the pathogens. Description An illustration shows a neutrophil represented by an irregular round structure having a trilobed nucleus and granules. The nuclear envelop of the nucleus disassembles as the granules are still intact. Chromatin decondenses and mixes with granules. Cell membrane ruptures to release N E Ts which are represented by D N A strands bound to granules. A very important early response to infection or injury is the activation of a variety of plasma proteases, including the proteins of the complement cascade discussed above. As we have seen, the cleavage fragments produced during activation of these proteases attract neutrophils to the site of tissue damage (see Figure 24-5). They further induce production of cytokines such as interleukins 1 and 6 (IL-1 and IL-6), which cause inflammation. The recruitment of neutrophils also depends on an increase in vascular permeability, which is controlled in part by lipid-signaling molecules (e.g., prostaglandins and leukotrienes) that are derived from
Adaptive Immunity, the Third Line of Defense, Exhibits Specificity
phospholipids and fatty acids. All of these events occur rapidly, starting within minutes of injury. A failure to resolve the cause of this immediate response may result in chronic inflammation with ensuing tissue damage, in which cells of the adaptive immune system play an important role. When the pathogen burden at the site of tissue damage is high, it may exceed the capacity of innate defense mechanisms to deal with the infection. Moreover, some pathogens have acquired, in the course of evolution, tools to disable or bypass innate immune defenses. In such situations, the adaptive immune response is required to help control the infection. This response depends on specialized cells that are parts of both the adaptive and innate immune systems, including macrophages and dendritic cells, which are capable of ingesting and killing pathogens as well as presenting antigens to the adaptive immune system. Dendritic cells, in particular, can initiate an adaptive immune response by delivering newly acquired pathogen-derived antigens to secondary lymphoid organs (see Figure 24-7). Adaptive Immunity, the Third Line of Defense, Exhibits Specificity Adaptive immunity is the term reserved for the highly specific recognition of foreign substances by antigen-specific receptors, the full elaboration of which requires days or weeks after occurrence of the initial exposure. Lymphocytes bearing antigen-specific receptors are the key cells responsible for adaptive immunity. The B lymphocytes will differentiate
into plasma cells that produce a secreted version of their cell surface antigen receptors, proteins called immunoglobulins or antibodies. Immunoglobulins can neutralize (render inactive) not only bacterial toxins but also harmful agents such as viruses by binding directly to them in a manner that prevents the virus from attaching itself to host cells. The generation of neutralizing antibodies is the rationale underlying virtually all vaccination strategies. Vaccination is a form of active immunization that consists of deliberately exposing an individual to a foreign antigen to elicit protective immunity by generating an adaptive immune response (described below) and antibodies that bind the foreign antigen. In the same vein, antibodies raised against snake venoms can be administered to the victims of snake bites to protect them from intoxication, provided the administration occurs relatively soon after the bite. The antibodies bind to the toxic proteins in the venom, keeping them from binding to their targets in the host, and in so doing neutralize them. This procedure, called passive immunization, can save lives by instant neutralization of a noxious substance such as a toxin. Passive immunization is also used prophylactically to protect those who travel to areas where a disease such as viral hepatitis is endemic; administration of serum from immune individuals provides temporary protection against infection. Antibodies can thus have immediate protective effects. Given that today’s medical advances allow the survival of individuals whose immune systems are severely compromised (e.g., cancer patients receiving chemotherapy or radiation, transplant patients with a pharmacologically suppressed immune system, patients who suffer from AIDS, individuals with inborn deficiencies of the immune system), passive immunization
can be of immediate practical importance. The deliberate exposure of an animal such as a mouse or rabbit to a foreign substance (immunization) allows the production of antisera that specifically recognize that substance (the antigen). These antisera have become standard components of the cell biologist’s toolbox. KEY CONCEPTS OF SECTION 24.1 Overview of Host Defenses Mechanical and chemical defenses provide protection against most pathogens. This protection is immediate and continuous, yet possesses little specificity. Innate and adaptive immunity provide defenses against pathogens that breach the body’s mechanical or chemical boundaries (see Figure 24-1). The circulatory and lymphatic systems distribute the molecular and cellular players in innate and adaptive immunity throughout the body (see Figure 24-2). Innate immunity is mediated by the complement system (see Figure 24-5) and several types of leukocytes, including natural killer cells, neutrophils, and other phagocytic cells, such as macrophages and dendritic cells. The cells and molecules of the innate immune system are deployed rapidly (minutes to hours). Molecular patterns diagnostic of the presence of pathogens can be recognized by Toll-like and other receptors, but the specificity of recognition is modest, as these receptors are capable of recognizing rather broad sets of related molecules. Adaptive immunity is mediated by T and B lymphocytes. These cells require days for full activation and deployment, but they can distinguish between closely related antigens. This specificity of antigen recognition is the key distinguishing feature of adaptive immunity. Innate and adaptive immunity act in a mutually synergistic fashion. Inflammation, an early response to tissue injury or infection, involves a series of events that combines elements of innate and adaptive immunity (see Figure 24-7).
Immunoglobulins Have a Conserved Structure Consisting of Heavy and Light Chains
24.2 Immunoglobulins: Structure and Function Immunoglobulins (also called antibodies), produced by B cells, are the best understood of the molecules that confer adaptive immunity. An individual human has the capacity to make a limitless number of different antibodies, but any given specific antibody is typically made only when the individual has been exposed to the antigen (immunized) to which the antibody will bind specifically; hence antibody production is an adaptive immune response. In this section, we describe the structural organization of immunoglobulins, their diversity, and how they bind to antigens. The mechanisms that generate diverse antibodies are described in Section 24.3. Immunoglobulins Have a Conserved Structure Consisting of Heavy and Light Chains Immunoglobulins are abundant serum proteins that fall into several classes with distinct structural and functional properties. Immunoglobulins were identified as the class of serum proteins responsible for antibody activity when they were biochemically purified from serum isolated from immunized animals (called antiserum). They were purified based on their abilities to mediate the killing of microbes
and to bind directly to their corresponding, or cognate, antigens. Immunoglobulins of the most common class are composed of two identical heavy (H) chains, covalently attached to two identical light (L) chains (Figure 24-9; the various classes will be described in the next section). The typical immunoglobulin (sometimes abbreviated Ig) therefore has a twofold, symmetric structure, described as . One antibody molecule can usually bind to two antigen molecules (bivalent binding; see below). An exception to this basic architecture occurs in the immunoglobulins made by camelids (camels, llamas, vicuñas) and sharks. These animals can make immunoglobulins that are heavy-chain dimers and lack light chains.
FIGURE 24-9 The basic structure of an immunoglobulin molecule. Antibodies are serum proteins also known as immunoglobulins. They are twofold, symmetric structures composed of two identical heavy chains and two identical light chains. Fragmentation of antibodies with proteases yields fragments that retain antigen-binding capacity. The protease papain yields monovalent F(ab) fragments, and the protease pepsin yields bivalent fragments. The Fc fragment is unable to bind antigen, but this portion of the intact molecule has other functional properties. F(ab) fragments are now commonly made using recombinant DNA technology instead of relying on proteolytic digestion. Description The illustration shows an antibody which consists of two heavy chains (blue bars in a Y shape), and two light chains (light blue short rectangles along the arms of the Y shape), forming a Y-shaped protein. The two heavy chains are attached by disulfide bonds and carbohydrates. The light chains, present on the arms of the 'Y', the F (a b) units, are connected to the heavy chains by disulfide bonds. On digestion with papain, the two top units, the F (a b) units, are released from the tail of the Y, the F c unit. On digestion with pepsin, the F c tail is broken into multiple pieces, and the F (a b)2 bivalent head group is left intact. A biochemical approach was used to answer the question of how antibodies manage to distinguish among related molecules, that is, how one antibody can bind to its specific antigen but not to another, structurally very similar, molecule. Proteolytic enzymes were used to fragment immunoglobulins, which are rather large proteins (~150 kDa), to identify the regions in the protein that are directly involved in antigen binding (Figure 24-9). The protease papain yields fragments, called F(ab) for antigen binding fragment, that can bind a single antigen molecule (monovalent fragments), whereas the protease pepsin yields bivalent fragments, referred to as (F = fragment; ab = antibody) that exhibit
Multiple Immunoglobulin Isotypes Exist, Each with Different Functions
two binding sites, termed bivalent binding. These enzymes are used to convert intact immunoglobulin molecules into monovalent or bivalent reagents. Although F(ab) fragments are incapable of binding two antigens, a property often termed cross-linking when applied to antigens displayed at a cell surface, fragments can do so. Researchers frequently take advantage of this property to cross-link and thus activate surface receptors. Many receptors, such as the EGF receptor, dimerize upon engagement of ligand (ligand-induced dimerization), a prerequisite for full activation of downstream signaling cascades (see Chapter 16). Many receptors on immune-system cells behave in a similar fashion. The portion released upon papain digestion and incapable of antigen binding is called Fc because of its ease of crystallization (F = fragment; c = crystallizable). Multiple Immunoglobulin Isotypes Exist, Each with Different Functions Immunoglobulins can be divided into different classes, termed isotypes, based on their distinct biochemical properties. There are two light-chain isotypes, κ and λ. The heavy chains show more variation; in mammals, the major heavy-chain isotypes are μ, δ, γ, α and ε. These heavy chains can associate with either κ or λ light chains. Depending on the vertebrate species, further subdivisions occur within the α and γ isotypes, and fish possess an isotype that is not found in mammals. The fully assembled immunoglobulin (Ig) derives its name from the heavy chain: antibodies with μ heavy chains yield IgM; α chains, IgA; γ chains, IgG; δ chains, IgD; and ε chains, IgE. The general structures of the major Ig isotypes are
depicted in Figure 24-10. By means of the unique structural features of the Fc portions of their heavy chains, each of the different Ig isotypes carries out specialized functions.
FIGURE 24-10 Immunoglobulin isotypes. The different classes of immunoglobulins, called isotypes, may be distinguished biochemically and by immunological techniques. In mice and humans, there are two light-chain isotypes (κ and λ) and five heavy-chain isotypes (μ, δ, γ, ε, α). Each isotype defines a class of immunoglobulin based on the identity of the heavy chain. IgG, IgE, and IgD (not shown) are monomers with generally similar overall structures. IgM and IgA can occur in serum as pentamers and dimers, respectively, accompanied by an accessory subunit, the J chain, in covalent disulfide linkage. This volume-rendered depiction of the immunoglobulins highlights their modular design, with each barrel representing an individual Ig domain. Different isotypes have different functions. See Figure 24-13 for definitions of abbreviations. Description I g M: It has a pentameric structure, where five monomers are connected at the base of the F c tail by a circular J-chain. Each monomer is composed of I g domains labeled C mu 1 to C mu 4, V subscript H, C subscript L, and V subscript L. A text reads, pentameric I g M is stabilized by an additional polypeptide, the J chain. I g A is a dimer consisting of two monomers connected by a J-chain and the monomers comprise alpha
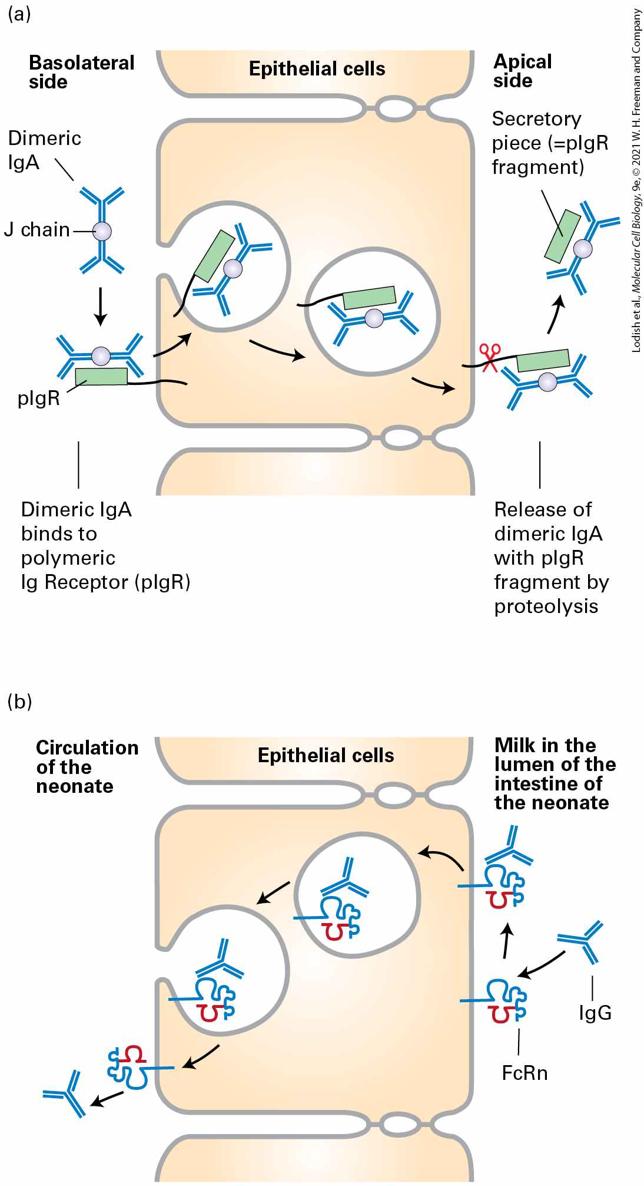
I g domains labeled C alpha 1 to 3, C subscript L, and V subscript L. I g E is a monomer comprised of epsilon domains labeled C epsilon 1 to 4, C subscript L, and V subscript L. I g G 1 is also a monomer composed of gamma I g domains namely C gamma 1 to 3, C subscript L, and V subscript L. The IgM molecule is secreted as a pentamer of chains, stabilized by disulfide bonds between the ends of the heavy chains and an additional chain, the J chain. In its pentameric form, IgM possesses 10 identical antigen-binding sites (2 for each ), which allow high-avidity interactions with surfaces that display the cognate antigen. Avidity is defined as the product of the strength of interactions (affinity) of the available individual binding sites and the number of such binding sites. Combining many low-affinity interactions can lead to a high-avidity interaction (as in Velcro). Upon its deposition on a surface that carries the cognate antigen, the pentameric IgM molecule assumes a conformation that is highly conducive to activation of the complement cascade; it thus is an effective means of damaging the membrane onto which it is adsorbed and onto which complement proteins are deposited as a consequence. The IgA molecule also interacts with the J chain, forming a dimer of molecules. Dimeric IgA can bind to an IgA receptor found on the basolateral plasma membrane of epithelial cells, where its binding triggers receptor-mediated endocytosis. Subsequently, the IgA receptor is cleaved, and the dimeric IgA, with a fragment of the receptor (the so-called “secretory piece”) still attached, is released from the apical side of the epithelial cell. This process, called transcytosis, is an effective means of delivering immunoglobulins from the basolateral side of an epithelium to
the apical side (Figure 24-11a). Tears and other secretions, especially in the gastrointestinal tract, are rich in IgA — grams of immunoglobulin are secreted from the blood into the lumen of the GI tract each day! — and so provide protection against environmental pathogens.
FIGURE 24-11 Transcytosis of IgA and IgG. (a) IgA, found in tears and in the secretions of various mucous membranes, must be transported across the epithelium. IgA binds to the polymeric IgA receptor on the basolateral surface of an epithelial cell and is endocytosed. As the resulting complex is transported across the epithelial monolayer, a portion of the receptor is cleaved, and the IgA, still bound to a portion of the receptor, the secretory piece, is released at the apical side. (b) Suckling rodents acquire Ig from their mother’s milk. At the apical surface of its intestinal epithelial cells, the newborn possesses the neonatal Fc receptor (FcRn), whose structure resembles that of class I MHC molecules (see Figure 2424). After this receptor binds to the Fc portion of IgG, transcytosis moves the acquired IgG to the basolateral side of the epithelium. In humans, the syncytial trophoblast in the placenta expresses FcRn and so mediates acquisition of IgG from the maternal circulation and its delivery to the fetus (transplacental transport). Description The illustration labeled (a) shows the transit of dimeric I g A immunoglobulins across epithelial cells from the basolateral side on the left to the apical side on the right. First, the dimeric I g A molecule binds to the polymeric I g receptor (p I g R), which is bound to the exterior basal surface of the cell. Then, the complex is endocytosed into the cell and the vesicle transits from the basolateral side to the apical membrane. Finally, on exocytosis at the apical side, the peptide anchoring p I g R to the membrane is cleaved, and the I g A dimer bound to the secretory piece is released. The illustration labeled (b) shows I g M antibodies in milk consumed by a neonate transferred into the circulation by transcytosis. First, I g G binds the F c R n receptor on the surface of the intestinal epithelial cells. Then endocytosis of the complex occurs. The vesicle is transported across the cell, and exocytosis occurs. On the circulatory side, the antibody is released from the F c R n complex into the blood. The IgG isotype is important for neutralization of virus particles. This isotype also helps prepare particulate antigens, such as viruses or larger fragments of bacteria, for acquisition by cells equipped with receptors specific for the Fc portion of the IgG molecule (see below).
Each Naive B Cell Produces a Unique Immunoglobulin
The immune system of the newborn mammal is immature, but protective antibodies are transferred from the mother to the newborn via the mother’s milk. The receptor responsible for capturing maternal IgG is the neonatal Fc receptor (FcRn), which in rodents is present on intestinal epithelial cells. Maternal IgG captured by an FcRn on the luminal side of the newborn’s intestinal tract is transcytosed across the gut epithelium and released into the infant’s circulation, generating passive immune protection of the infant rodent (Figure 24-11b). In humans, FcRn is found on fetal cells that contact the maternal circulation in the placenta. Transcytosis of IgG antibodies from the maternal circulation across the placenta delivers maternal antibodies to the fetus. These maternal antibodies will protect the newborn until its immune system is sufficiently mature to produce antibodies on its own. In adults, FcRn is also expressed on endothelial cells and helps control the turnover of IgG in the circulation as well as the delivery of IgG across the endothelial barrier and into underlying tissue. As we will see in Section 24.3, the IgM and IgD isotypes are expressed as membrane-bound receptors on newly generated B cells. Here the μ chains have an important role in B-cell development and activation. Each Naive B Cell Produces a Unique Immunoglobulin The clonal selection theory stipulates that each naive B lymphocyte (not yet having seen its specific antigen) expresses on its plasma membrane an
antigen-binding receptor of unique specificity. The receptor is an antibody extended at its C-terminus by a hydrophobic sequence that anchors the protein in the plasma membrane, as we discuss later (see Figure 24-19). When a lymphocyte encounters the antigen for which it is specific, clonal expansion occurs. This allows an amplification of the response, culminating in production and secretion by plasma cells of large amounts of this specific antibody (the same one made by the original precursor cell) (Figure 24-12). (Note that clonal expansion is rapid cell division of this single cell to form a group of cells — a clone — that all originate from this precursor cell.) The antigen-specific antibody is responsible for binding to the antigen and subsequently mediating the clearing of the antigen out of the body. In a typical immune response, the antigen that elicits the response is of complex composition: even the simplest virus contains several distinct proteins, and each protein may present to the immune system several molecularly distinct features that can be recognized independently of one another. Thus many individual lymphocytes respond to different parts of a given antigen and expand into independent clones in response to it, each producing its own antigenspecific receptor and antibody of unique structure and therefore with unique binding characteristics (affinity). Because each lymphocyte is endowed with a unique receptor and clonally expands in response to antigen, this response of multiple, independent B cell precursors is characterized as polyclonal.
FIGURE 24-12 Clonal selection. The clonal selection theory proposes the existence of a large set of B lymphocytes, each expressing on its surface a unique antigen-specific receptor (indicated by different colors). The receptor is an antibody extended at its C-terminus by a hydrophobic sequence that anchors it in the plasma membrane (see Figure 24-19). The antigen that fits with the receptor carried by a particular lymphocyte binds to it and stimulates that lymphocyte to expand clonally. From a modest number of antigenspecific cells, a large number of cells of the desired specificity may be generated. Many of these cells will differentiate into plasma cells, where a form of the antigen-specific receptor lacking the membrane anchor is secreted as an antibody. Note that, like secreted antibodies, the cell surface antigen-specific receptors each have two H and two L chains.
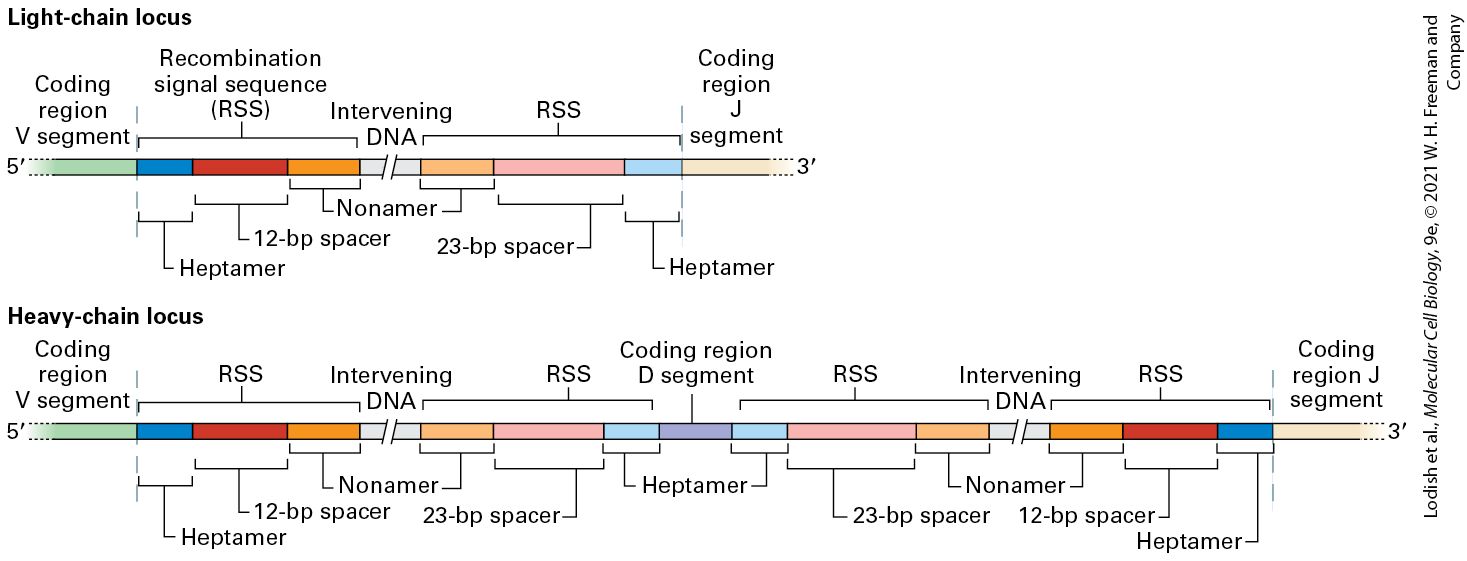
Description The illustration shows various antigen specific receptors on B cells, represented by different colors. It is titled activation of B cell. Clonal expansion of a B cell with an antigen specific receptor occurs to ultimately form 16 B cells which yield secreted antibodies. B-cell tumors, which represent malignant clonal expansions of individual B-lymphocytes, enabled the first molecular analysis of the processes that underlie the generation of antibody diversity. A key observation was that tumors derived from lymphocytes may produce large quantities of a single type of secreted immunoglobulin. Some of the light chains of these immunoglobulins are secreted in the urine of patients with such tumors. These light chains, called Bence-Jones proteins after their discoverers, can be readily purified and afforded the first target for a protein chemical analysis of immunoglobulins. Two key observations emerged from this work. First, no two independent B-cell tumors produced light chains with identical biochemical properties, suggesting that they were all unique in sequence. Second, the differences in amino acid sequence that distinguished one light chain from another were not randomly distributed but were clustered in a domain referred to as the variable region of the light chain, or . This domain comprises the N-terminal ~110 amino acids of the light chain. The remainder of the sequence is identical for the different light chains (provided they derive from the same isotype, either κ or λ) and is therefore referred to as the constant region, or . Immunoglobulins unique to each individual patient were subsequently purified from the patients’ serum. Sequencing of the
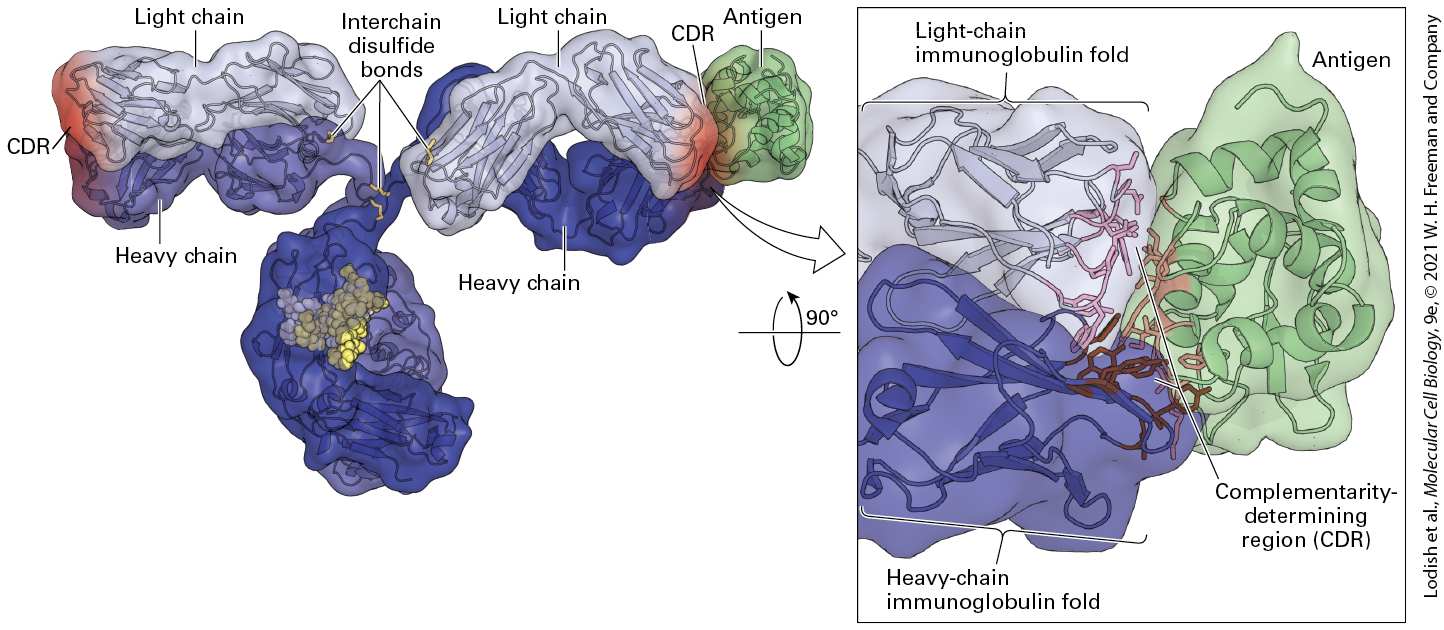
heavy chains from these preparations showed that the variable residues that distinguished one heavy chain from another were again concentrated in a well-demarcated domain, referred to as the variable region of the heavy chain, or . An alignment of variable-region sequences obtained from different light chains showed a nonrandom pattern of regions of variability, revealing three hypervariable regions — HV1, HV2, and HV3 — that are sandwiched between what are called framework regions (Figure 24-13a). (Similar alignments for the immunoglobulin heavy-chain sequences also yielded hypervariable regions.) In the properly folded three-dimensional structure of immunoglobulins, these hypervariable regions are in close proximity (Figures 24-13b and 24-14) and make contact with antigen. Thus that portion of an Ig molecule containing the hypervariable regions constitutes the antigen-binding site. For this reason, hypervariable regions are also referred to as complementarity-determining regions (CDRs).
FIGURE 24-13 Hypervariable regions and the immunoglobulin fold. (a) Amino acid variability varies with residue position in Ig light chains. Here the percentage of variableregion sequences with variant amino acids is plotted for each position in the sequence. Positions for which many different amino acid side chains are present are assigned a high variability index; those that are invariant among the sequences compared are assigned a
value of 0. This analysis reveals three regions of increased variability: hypervariability (HV) regions 1, 2, and 3; these regions are also called complementarity-determining regions (CDRs) because the surface topology these regions create are complementary to the surface of the antigen to which the immunoglobulin binds. (b) Volume-rendered depiction of fragment (right) and ribbon diagram of a typical Ig light-chain variable region with the positions of the hypervariable regions indicated in red (left). The hypervariable regions are found in the loops that connect the β strands and make contact with antigen. The β strands (rendered as arrows) make up two β sheets and constitute the framework region, which has a similar fold in all antibody domains. Each variable and constant domain has this characteristic three-dimensional structure, called the immunoglobulin fold. L = light chain; H = heavy chain; variable region; variable region; constant domains; constant region. Description The graph labeled (a) has its vertical axis representing variability, ranging from 0 to 100 in increments of 20. An upward vertical arrow is present on the left. The horizontal axis represents residue number ranging from 0 to 110, in increments of 10. For the majority of residues, the variability fluctuates around 10 percent. However, residues 25 to 35; H V 1 (C D R 1), 50 to 60, H V 2 (C D R 2), and 90 to 100, H V 3 (C D R 3) are highly variable, reaching variabilities of 80 percent; these residue positions correspond to the hypervariability regions 1, 2, and 3, respectively. The illustration labeled (b) shows the structure of an immunoglobulin monomer. The heavy chains are composed of three constant I g units and one variable I g unit. The light chains are composed of a constant unit and a variable unit. Both variable units of the heavy and light chains are located at the ends of the arms of the Y-shaped immunoglobulin. The ribbon diagram shows the arrangement of several beta-sheets in the light-chain variable domain. Connections between antiparallel beta-sheets are labeled as the hypervariability regions 1, 2, and 3. The hypervariability regions are also known as C D Rs - complementarity-determining regions.
FIGURE 24-14 Immunoglobulin structure. This model shows the three-dimensional structure of an immunoglobulin molecule complexed with hen egg-white lysozyme (a protein antigen) as determined by x-ray crystallography. [Data from E. A. Padlan et al., 1989, Proc. Nat’l Acad. Sci. USA 86:5938, PDB ID 1igt, 3hfm.] Description The illustration shows a characteristic y-shaped ribbon structure. The interchain disulfide bonds are highlighted. The light and heavy chains of the arms are labeled. At the end of the arms, the complementarity-determining region is colored red, and an antigen is bound to the C D R. An enlarged region shows the light chain immunoglobulin fold at the top, a heavy-chain immunoglobulin fold at the bottom and an antigen bonded to it. The C D R (Complementarity determining region) is present in the center of the three structures. The difficulty of encoding directly in the inherited genome (germ line) all the information necessary to generate the enormously diverse antibody repertoire (more than a million different antibody molecules in what we now know is a genome encoding about 20,000 independent genes) led to suggestions of unique genetic mechanisms to account for this diversity.
Immunoglobulin Domains Have a Characteristic Fold Composed of Two β Sheets Stabilized by a Disulfide Bond
Given the size of a typical antibody heavy chain and light chain (each heavy chain–light chain combination, if encoded as such, would require 2.5–3.5 kb of DNA, depending on the isotype), it is immediately obvious that encoding a set of antibody molecules of sufficient diversity to provide adequate protection against the wide array of pathogens and other foreign substances to which an organism is exposed would rapidly exhaust its DNA-coding capacity. We shall see that, indeed, unique mechanisms are at work to create an adequately diverse set of antibodies. Immunoglobulin Domains Have a Characteristic Fold Composed of Two β Sheets Stabilized by a Disulfide Bond Both the variable and constant domains of immunoglobulins fold into a compact, three-dimensional structure, an immunoglobulin (Ig) domain, composed exclusively of β sheets (see Figure 24-13b). A typical Ig domain contains two β sheets (one with three strands and one with four strands) held together like a sandwich by a disulfide bond. The residues that point inward are mostly hydrophobic and help stabilize this sandwich structure. The residues exposed to the aqueous environment show a greater frequency of polar and charged side chains. The spacing of the cysteine residues that make up the disulfide bond and a small number of strongly conserved residues characterize this evolutionarily ancient structural motif, termed the immunoglobulin fold. The basic immunoglobulin fold
is also found in numerous eukaryotic proteins that are not directly involved in antigen-specific recognition, including the Ig superfamily of cell-adhesion molecules, or Ig CAMs (see Chapter 20). The region on an antigen that makes contact with the corresponding antibody is called an epitope; the portion of the immunoglobulin that makes contact with antigen is the paratope. A protein antigen usually contains multiple epitopes, which are often exposed loops or surfaces on the protein and are thus able to bind to different antibody molecules. Each homogeneous antibody preparation derived from a clonal population of B cells recognizes a single molecularly defined epitope on the corresponding antigen. To determine the structure of an antibody complexed to its cognate epitope on an antigen, it is important to have a source of homogeneous immunoglobulin and of antigen in pure form (see Chapter 3). As we have seen, homogeneous immunoglobulins can be obtained from B-cell tumors, but in that case, the antigen for which the antibody is specific is usually not known. The breakthrough for generating homogeneous antibody preparations suitable for structural analysis was the development of techniques to obtain antibodies from hybridomas by use of a special selection medium. The creation of immortalized cell lines that produce antibodies of defined specificity, called monoclonal antibodies, has yielded essential tools for the cell biologist. Monoclonal antibodies are widely used not only for the specific detection of macromolecules, but also for detection and quantitation of drugs, drug metabolites, and even signaling molecules such as cAMP. Monoclonal antibodies can detect
An Immunoglobulin’s Constant Region Determines Its Functional Properties
proteins and their modifications (e.g., phosphorylation, nitrosylation, methylation, acetylation, etc.) as well as complex carbohydrates, (glyco)lipids, and nucleic acids and their modifications. They have therefore found widespread use in the laboratory as well as for diagnostic and therapeutic purposes. We now have detailed insights into the structure of a large number of monoclonal antibodies, each in a complex with its specific antigen. There are no hard-and-fast rules that describe these interactions, other than the usual rules of molecular complementarity of proteins with other molecules and macromolecules (see Chapter 3). The CDRs make the most important contributions to the antigen-antibody interface. The CDR3 of the region of the Ig heavy chain plays a particularly prominent role, as does the CDR3 of the region of the Ig light chain. An Immunoglobulin’s Constant Region Determines Its Functional Properties As we have seen, antibodies recognize antigen via their variable regions. Their constant regions determine which effector molecules they recruit to neutralize the pathogen. Antibodies attached to a viral or microbial surface can be recognized directly by cells that express receptors specific for the Fc portion of immunoglobulins. These Fc receptors (FcRs), which are specific for
individual classes and subclasses of immunoglobulins, display considerable structural and functional heterogeneity. By means of FcRdependent events, specialized phagocytic cells such as dendritic cells and macrophages can bind to antibody-decorated particles or cells and then ingest and destroy them. The decoration of an antigenic target with bound antibodies, or its covalent modification with complement components, is called opsonization. FcR-dependent events also allow some immunesystem cells (e.g., monocytes and natural killer cells) to directly engage target cells that display viral or other antigens to which antibodies are attached. This engagement may induce the immune-system cells to release toxic small molecules (e.g., reactive oxygen species) or the contents of cytotoxic granules, including perforins and granzymes. Perforins are proteins that can attach themselves to the surface of the engaged target cell and form pores in its membrane. These newly formed pores allow access by granzymes, proteases that initiate a sequence of events that will ultimately kill the target cell (see Figure 24-6). This process, called antibody-dependent cell-mediated cytotoxicity (ADCC), illustrates how cells of the innate immune system interact with, and benefit from, the products of the adaptive immune response. Antigen-antibody (immune) complexes of some immunoglobulin isotypes can initiate the classical pathway of complement activation (see Figure 24-5). IgM and IgG3 are particularly good at complement activation, but all IgG classes can in principle activate complement, whereas IgA and IgE are unable to do so. The large amounts of IgA found in the gut contribute to its barrier function by neutralizing gut-resident microbes.
KEY CONCEPTS OF SECTION 24.2 Immunoglobulins: Structure and Function Most immunoglobulins (antibodies) are composed of two identical heavy (H) chains and two identical light (L) chains . Each chain contains a variable (V) region and a constant (C) region. Proteolytic fragmentation yields monovalent F(ab) and bivalent fragments, which contain variable-region domains that retain antigen-binding capability (see Figure 24-9). The Fc fragment contains constantregion domains and determines their ability to activate complement components or bind to receptors specific for Fc regions expressed on leukocytes. Immunoglobulins are divided into classes based on the constant regions of their heavy chains (see Figure 24-10). In mammals, there are five major classes: IgM, IgD, IgG, IgA, and IgE; the corresponding heavy chains are referred to as μ, δ, γ, α and ε. There are two major classes of light chains, κ and λ, again characterized by the attributes of their constant regions. IgM and IgA can form higher order structures: IgM forms pentamers (five identical copies); and IgA forms dimers (two identical copies). Each individual B lymphocyte expresses an immunoglobulin of unique sequence and is therefore uniquely specific for a particular antigen. Upon recognition of antigen, only a B lymphocyte that bears a receptor specific for it will be activated and expand clonally (clonal selection) (see Figure 24-12). The antigen specificity of antibodies is conferred by their variable regions, which contain regions of high sequence variability, called hypervariable or complementarity-determining regions (see Figure 24-13a). These hypervariable regions are positioned at the tip of the variable region, where they can make specific contacts with the antigen for which a particular antibody is specific. The repeating immunoglobulin domains that make up immunoglobulin molecules have a characteristic three-dimensional structure, the immunoglobulin fold, which consists of two β sheets held together in a sandwich by a disulfide bond (see Figure 24-13b). The constant regions of the heavy chains endow antibodies with unique effector functions, such as the capacity to bind complement, the ability to be transported across epithelia, or the ability to interact with receptors specific for the Fc portion of immunoglobulins.
24.3 Generation of Antibody Diversity and B-Cell Development
24.3 Generation of Antibody Diversity and B-Cell Development Pathogens have short replication times, are quite diverse in their genetic makeup, and evolve quickly by mutation, generating enormous antigenic variation. An adequate defense must therefore be capable of mounting an equally diverse response. Antibodies provide the diversity required for successful host defense. The timing of the antibody response and its necessary adjustment to changes in the antigenic makeup of the pathogen pose unique demands on the organization and regulation of the adaptive immune system. A unique mechanism has evolved that allows not only virtually limitless variation in the set of antibodies that can be produced (called the repertoire), but also rapid improvement in the binding affinity of those antibodies to meet the demands posed by an ongoing viral or bacterial infection. Because optimal antibody production by B cells requires assistance from T cells, we will see below that the molecular mechanisms underlying lymphocyte diversity are fundamentally similar for B and T cells. B cells, which are responsible for antibody production, make use of a unique mechanism by which the genetic information required for synthesis of immunoglobulin heavy and light chains is stitched together from separate DNA sequences, or Ig gene segments, to create a functional transcription unit. The recombination mechanism that combines Ig gene
A Functional Light-Chain Gene Requires Assembly of V and J Gene Segments
segments itself dramatically expands the variability in sequence precisely at those sites where these genetic elements are joined together. This mechanism for generating a diverse array of antibodies is fundamentally different from meiotic recombination, which occurs only in germ cells, and from alternative splicing of exons (see Chapter 7). Because this recombination mechanism occurs in somatic cells but not in germ cells, it is known as somatic gene rearrangement or somatic recombination. This unusual recombination mechanism, unique to antigen-specific receptors on B and T lymphocytes, makes it possible to specify an enormously diverse set of receptors with minimal expenditure of DNA coding space. The discovery of somatic recombination is detailed in Classic Experiment 24.1. The ability to combine discrete genetic elements at will (combinatorial diversity), in addition to the generation of yet more sequence diversity in the encoded receptors by the underlying recombination mechanisms themselves, allows adaptive immune responses against a virtually limitless array of antigens, including molecules encoded by the host and chemical structures not naturally found in nature. Thus there are mechanisms at work not only to create this enormous diversity, but also to impose tolerance to curtail unwanted reactivity against self components. Neither mechanism is perfect: the adaptive immune system cannot generate receptors for all foreign substances. Furthermore, the unavoidable price we pay for how we generate B- and T-cell receptors is the likelihood of self-reactive receptors, which are implicated in autoimmunity.
A Functional Light-Chain Gene Requires Assembly of V and J Gene Segments Genes encoding intact immunoglobulins do not exist already assembled in the genome, ready for expression. Instead, the required gene segments are brought together and assembled in the course of B-cell development. The organization of the region of the genome containing the immunoglobulin genes is shown in Figure 24-15. In B cells, the DNA in this region is rearranged to generate assembled and fully functional immunoglobulinencoding genes in each B cell and its descendants. Although in the course of B-cell development the rearrangement of heavy-chain genes occurs before the rearrangement of light-chain genes, we discuss light-chain genes first, because of their less complex organization.
FIGURE 24-15 Overview of somatic gene rearrangement in immunoglobulin DNA. The stem cells that give rise to B cells contain multiple gene segments encoding portions of immunoglobulin heavy and light chains. During development of a B cell, somatic recombination of these gene segments yields functional light-chain genes (a) and heavychain genes (b). Each V gene segment carries its own promoter. Rearrangement brings an enhancer linked to or contained within the immunoglobulin locus close enough to the combined sequence to activate transcription. The light-chain variable region is encoded by two joined gene segments, and the heavy-chain variable region is encoded by three joined segments. Note that the chromosomal regions encoding immunoglobulins contain many more V, D, and J segments than those shown. In addition, the κ light-chain locus contains a single constant (C) segment, as shown, but the heavychain locus contains several distinct C segments (not shown) that encode the different immunoglobulin isotypes, as we detail later. Description The illustration labeled (a) shows a gene labeled gem line D N A consisting of several segments, which code, from 5-prime to 3-prime, for variable regions, for J segment, for
an enhancer, and for the constant domain. One variable region is marked. After rearrangement, this marked variable region has been brought into proximity to the region coding for the J segment. It is labeled rearranged D N A. The illustration labeled (b) before rearrangement, shows a gene consisting of, from 5prime to 3-prime, several variable domains, one of which is marked V, D-segments, one of which is marked, J segments, one of which is marked, an enhancer, and the constant segment. After rearrangement, the marked V, D, and J domains are close to each other. The immunoglobulin light-chain genes consist of clusters of V gene segments, followed downstream by a single C segment. Each V gene segment carries its own promoter sequence and encodes the bulk of the light-chain variable region, although a small piece of the nucleotide sequence encoding the light-chain variable region is missing from the V gene segment. This missing portion is provided by one of the multiple J segments located between the V segments and the single C segment in the unrearranged (or germ-line) κ light-chain locus (Figure 24-15a). (This J segment is a genetic element, not to be confused with the J chain, a polypeptide subunit of the pentameric IgM molecule and found also in association with IgA; see Figure 24-10.) In the course of B-cell development, commitment of a B-cell precursor to use a particular V gene segment — a random process — results in its physical juxtaposition with one of the J segments, again a random choice, to form an exon encoding the entire light-chain variable region (Figure 24-15a). This DNA rearrangement not only generates an intact and functional light-chain gene, but also places the promoter sequence of the rearranged gene within controlling distance of enhancer elements, located near the light-chain constant-region exon, that are required for its transcription. Only a fully
rearranged light-chain gene is transcribed and subsequently translated into protein. Recombination Signal Sequences Detailed DNA sequence analysis of the light-chain and heavy-chain regions revealed a conserved sequence element at the end of each V gene segment. This conserved element, called a recombination signal sequence (RSS), is composed of heptamer and nonamer sequences separated by a 23-bp spacer. At the end of each J segment, there is a similarly conserved RSS that contains a 12-bp spacer (Figure 24-16). The 12- and 23-bp spacers separate the conserved heptamer and nonamer sequences by one and two turns of the DNA helix, respectively.
FIGURE 24-16 DNA elements involved in V(D)J recombination in the immunoglobulin loci. Schematic representation of the immunoglobulin light-chain (top) and heavy-chain locus (bottom). At its end, each immunoglobulin V region coding sequence abuts a recombination signal, composed of a conserved heptamer (7 bp) and a conserved nonamer (9 bp) sequence, separated by a 12-bp spacer of variable sequence. The combination of heptamer, nonamer, and spacer is called a recombination signal sequence (RSS). Recombination requires juxtaposition of the gene segment that recombines with a second
gene segment with an RSS, which carries a heptamer and nonamer sequence that is complementary and antiparallel to the first RSS. For the second RSS, the spacer is 23-bp long, and of variable sequence. For the light-chain locus, the RSSs that flank the V and J segments are separated by intervening DNA. Recombination can occur only between elements that carry RSSs of different spacer length (the “12-23” rule). The heavy-chain locus comprises three sequence elements that can rearrange. The V region of the heavy chain carries an RSS at its end, the D segments are flanked on their and ends by an RSS, and the J segments carry an RSS at their end. The organization of the immunoglobulin heavy-chain locus thus enables rearrangements of D to J, and of V to DJ, but direct rearrangements of V to J are not allowed, as this would violate the 12-23 rule. Description The light chain locus runs from 5 prime to 3 prime. The following regions are labeled. Coding region V segment, recombination signal sequence (R S S) having a heptamer, 12 base pair spacer, and a nonamer, intervening D N A, R S S with a nonamer, 23 base pair spacer, and a heptamer, and a coding region J segment. The heavy chain locus has the following parts labeled running from 5 prime to 3 prime. Coding region V segment, R S S having a heptamer, 12 base pair spacer, and a nonamer, intervening D N A, R S S with a nonamer, 23 base pair spacer, and a heptamer, and a coding region D segment, heptamer, R S S with a heptamer, 23 base pair spacer, and a nonamer, intervening D N A, R S S with a nonamer, 12 base pair spacer, heptamer and a coding region J segment. Somatic recombination is catalyzed by two enzymes, the RAG1 and RAG2 recombinases, which are expressed only in lymphocytes. Thus these rearrangements do not occur in any other cells of the body. Juxtaposition of the two gene segments to be joined is stabilized by the RAG1/RAG2 complex (Figure 24-17). The structure of the RAG1/RAG2 complex juxtaposes the RSSs so that proper covalent joining of the immunoglobulin gene segments can occur, with concomitant release of the
intervening DNA, which is released as a covalently closed, circular DNA molecule.
FIGURE 24-17 RAG1/RAG2 structure. The RAG1 and RAG2 proteins (light and dark purple) are responsible for bringing together the immunoglobulin gene segments for rearrangement. RAG1/RAG2 complexes with the recombination signal sequences,
positioning the 12- and 23-bp spacer sequences to enable cleavage at the boundary of the coding sequence and the heptamer of the RSS. [Data from M.-S. Kim et al., 2015, Nature 518:507–511, PDB ID 4wwx; and F. F. Yin et al., 2009, Nat. Struct. Mol. Biol. 16:499–508, PDB ID 3gna.] Description The illustration shows D N A threaded through the R A G 1 and R A G 2 complex at the bottom of the structure. Along the left side, from bottom to top, the following areas are labeled: R A G 1, heptamer, R A G 1, 12-bp spacer, nonamer. Moving down the right side, from top to bottom, the following areas are labeled: 23-bp spacer, R A G 1, heptamer and R A G 2. The active site is present at the center of the R A G 2. The two coding segments on the D N A are labeled at the bottom. Defects in the synthesis of RAG proteins obliterate the possibility of somatic gene rearrangements. As described below, the rearrangement process is essential for B-cell development; consequently, RAG deficiency leads to the complete absence of B cells. People with defects in RAG gene function suffer from severe immunodeficiency. Targeted deletion of RAG genes in mice likewise leads to a complete defect in immunoglobulin (and T-cell receptor) gene rearrangement, resulting in a developmental block in the generation of B and T lymphocytes. Junctional Imprecision In addition to the random selection of V and J gene segments, processing of the intermediates created in the course of somatic recombination provides an additional means for expanding the variability of immunoglobulin sequences. This additional variability is created at the
Rearrangement of the Heavy-Chain Locus Involves V, D, and J Gene Segments
junction of the segments to be joined by random insertion or deletion of nucleotides to one of the cut ends before the V and J segments are joined, a process termed junctional imprecision. Whenever a V and a J segment recombine, the sequence and reading frame of the VJ product cannot be predicted; only one in three recombination reactions results in a reading frame that is compatible with light-chain synthesis. Light-chain diversity therefore arises not only from the combinatorial use of V and J gene segments, but also from junctional imprecision. Inspection of the three-dimensional structure of the light chain shows that the highly diverse joint generated as a consequence of junctional imprecision forms part of a loop — hypervariable region 3 (HV3) — that projects into the antigen-binding site and makes contact with antigen (see Figure 24-13b). Rearrangement of the Heavy-Chain Locus Involves V, D, and J Gene Segments The organization of the heavy-chain locus is more complex than that of the κ light-chain locus. The heavy-chain locus contains not only a large tandem array of V segments (each equipped with its own promoter) and multiple J segments, but also multiple D (diversity) segments (see Figure 24-15b). Somatic recombination of a V, a D, and a J segment generates a rearranged sequence encoding the heavy-chain variable region .
At the end of each V segment in heavy-chain DNA, there are conserved heptamer and nonamer sequences separated by spacer DNA, similar to the RSSs in light-chain DNA. These RSSs are also found in complementary and antiparallel configuration at the end and the end of each D segment (Figure 24-16). The J segments are similarly equipped at their end with the requisite RSS. The spacer lengths in these RSSs are such that D segments can join to J segments, and V segments to already rearranged DJ segments. However, neither direct V-to-J nor D-to-D joining is allowed, in compliance with the 12-23-bp spacer rule. Heavy-chain rearrangements proceed via the same mechanisms described above for light-chain rearrangements. In the course of B-cell development, the heavy-chain locus is always rearranged first, starting with D-J rearrangement. D-J rearrangement is followed by V-D-J rearrangement. In the course of the D-J and V-D-J rearrangements, an enzyme called terminal deoxynucleotidyl transferase (TdT) may add nucleotides to free OH ends of DNA in a templateindependent fashion. Up to a dozen or so nucleotides, called the N-region or N-nucleotides, may be added, generating additional sequence diversity at the junctions whenever D-J and V-D-J rearrangements occur (Figure 2418, step 7 ). As with rearrangements forming the light-chain gene, only one in three rearrangements yields the proper reading frame for the rearranged VDJ sequence. If the rearrangement yields a sequence encoding a functional protein, it is called productive. Although the heavy-chain locus is present on each of two homologous chromosomes, only one productive rearrangement is permitted, as we will see below.
FIGURE 24-18 The sequential steps in V(D)J recombination. Step 1 : Linear depiction of the immunoglobulin gene segments to be recombined. Each immunoglobulin gene segment carries a recombination signal sequence. For the V regions, at their end, for the J segments (light chain) at their end and for a rearranged DJ segment (heavy chain) also at the end. Step 2 : The RAG1/RAG2 recombinase makes a single strand cut to generate a free OH hydroxyl group. Step 3 : The OH group attacks the bottom strand to create a covalently closed hairpin. The intervening DNA, with an RSS at either end, is excised precisely at the boundaries of the RSS and yields a covalently closed “excision circle” (circularization step not shown in the figure). The gene segments that participate in rearrangement are held in close proximity by the RAG1/RAG2 recombinase and associated proteins. Step 4 : The covalently closed hairpins at the end of the coding sequences can be opened either symmetrically (left) or (step 5 ) asymmetrically (right). If the hairpin is opened asymmetrically (left), the unpaired nucleotides may be removed by nucleases to produce a “flush” end (not shown) or result in a short palindromic sequence that can be rendered flush by “filling in” the missing nucleotides (step 7 ); this adds nucleotides to where the actual junction will occur, thus creating additional variability in the final product of rearrangement. These nucleotides are called P nucleotides because of their
palindromic nature. Even if the hairpin opens symmetrically (step 4 , right), the enzyme terminal deoxynucleotidyl transferase (TdT) can add random nucleotides to the free OH in a template-independent manner (step 6 ), thus increasing sequence diversity at the joint. The target of TdT activity is confined to V, D, and J elements of the immunoglobulin heavychain locus. Once the ends of the coding sequences have been rendered flush (step 7 ), the coding ends are ligated together by nonhomologous end joining. Description 1: A double stranded D N A has three sections labeled from left to right: coding region of V segment, recombination signal sequences (R S S) and coding region of J segment (light chain) or D J segments (heavy chain). 2: A single strand break occurs at the start of R S S on the 5 prime to 3 prime strand and at the end of R S S on the 3 prime to 5 prime strand. 3: The ends at left and right connect to each other forming a hairpin shape and are labeled covalently closed hairpins at coding regions. A clean double strand break is labeled. 4: The right closed hairpin is labeled symmetric opening of hairpin. 5: The left closed hairpin is labeled asymmetric opening of hairpin. 6: The left hairpin breaks open at top and forms two unequal straight lines. The end is labeled palindromic overhang. The right hairpin breaks open. It has added nucleotides. A text reads, addition of N-region by terminal deoxynucleotidyl transferase (T d T). 7: The “filling in” of the overhangs occur. 8: These two straightened and amended strands are joined together, without the (R S S) part in between. An enhancer located downstream of the cluster of J segments and upstream of the constant-region segment activates transcription from the promoter at the end of the rearranged VDJ sequence (see Figure 24-15).
Somatic Hypermutation Allows the Generation and Selection of Antibodies with Improved Affinities
Splicing of the primary transcript produced from the rearranged heavychain gene generates a functional mRNA encoding the μ heavy chain. For both heavy-chain and light-chain genes, somatic recombination places the promoters upstream of the V segments within functional reach of the enhancers necessary to allow transcription, so that only rearranged VJ and VDJ sequences, and not the V segments that remain in the germ-line configuration, are transcribed. Somatic Hypermutation Allows the Generation and Selection of Antibodies with Improved Affinities In addition to somatic recombination and junctional imprecision, antigenactivated B cells can undergo an additional diversity-generating process called somatic hypermutation. Upon exposure to antigen and receipt of the proper signals, most of which are provided by T cells, expression of activation-induced deaminase (AID) is turned on. This enzyme deaminates cytosine residues, converting them to uracil. When a B cell that carries this lesion replicates its DNA, it may place an adenine on the complementary strand, thus generating a G-to-A transition (see Figure 514). Alternatively, the uracil may be excised by DNA glycosylase to yield an abasic site. Such abasic sites, when copied, give rise to an A, G, or C in the complementary DNA strand. That is, a C:G base pair can be replaced by somatic hypermutation with a T:A, A:T, or G:C base pair. Mutations thus accumulate with every successive round of B-cell division, yielding numerous mutations in the rearranged VJ and VDJ segments. Error-prone
filling by DNA polymerase of gaps created by nucleotide excision repair also contributes to somatic hypermutation. The process of somatic hypermutation occurs when lymphocytes reside in specialized microanatomic structures known as germinal centers. These structures, which arise within the follicles of secondary lymphoid organs upon immunization, consist of foci of thousands of rapidly proliferating and hypermutating B cells. In addition to B cells, germinal centers contain follicular dendritic cells, a cell type that serves as a depot for antigen that can be retrieved by B cells, and a small number of helper T cells specialized in providing selective signals that control B cells. Many of the somatic mutations induced by AID are deleterious, in that they reduce the affinity of the encoded antibody for an antigen, but some improve the encoded antibody’s affinity for an antigen. In a process analogous to Darwinian evolution, B cells carrying surface immunoglobulins with affinity-increasing mutations have a selective advantage in picking up antigen from follicular dendritic cells, which allows them to successfully compete for signals from the limiting number of helper T cells residing in the germinal center (described in some detail in Section 24.6). These signals trigger the proliferation, and thus the clonal selection, of B cells that have on their surface antibodies with higher affinity for the target antigen. This triggers further proliferation and additional mutations and differentiation into antibody-secreting plasma cells or memory B cells. The net result is the generation of a B-cell population whose antibodies, as a rule, show a higher affinity for the antigen.
B-Cell Development Requires Input from a Pre-B-Cell Receptor
In the course of an immune response, or upon repeated immunization, the adaptive immune response therefore exhibits affinity maturation — an increase in the average affinity of antibodies for an antigen as a function of time after antigen exposure — as the result of somatic hypermutation and selection. Antibodies produced following this phase of the adaptive immune response display affinities for antigen in the nanomolar (or better) range. For reasons that are not fully understood, but likely related to the chromatin structure of the rearranged immunoglobulin loci, the activity of AID is focused mainly on rearranged VJ and VDJ segments, and this targeting may therefore require active transcription. The entire process of somatic hypermutation is strictly antigen-dependent and shows an absolute requirement for interactions between the B cells and certain Tcell types. B-Cell Development Requires Input from a Pre-B-Cell Receptor B cells destined to make immunoglobulins must rearrange the necessary gene segments to assemble functional heavy-chain and light-chain genes. These rearrangements occur in a carefully ordered sequence during the development of a B cell, starting with heavy-chain rearrangements. Moreover, as noted above, the rearranged heavy-chain contains, near its C-terminus, a membrane-spanning segment that traverses the plasma membrane (Figure 24-19; also see Figure 24-20). This membrane-bound receptor first executes a cell-fate decision necessary to drive further B-cell development (and antibody synthesis) by permitting subsequent
rearrangement of the light-chain genes. Only a productive rearrangement that yields an in-frame VDJ combination can generate a complete μ heavy chain. The production of that μ chain serves as a signal to the B cell that it has successfully accomplished rearrangement and that no further rearrangements of the heavy-chain locus on the remaining gene copy are required. Recall that each lymphocyte precursor starts out with two immunoglobulin locus–bearing, homologous chromosomes in the germline (unrearranged) configuration. In accordance with clonal selection theory, which stipulates that each lymphocyte ought to come equipped with a single antigen-specific receptor, continued rearrangement would entail the risk of producing B cells with two different heavy chains, each with different specificity — an undesirable outcome.
FIGURE 24-19 Structure of the pre-B-cell receptor and its role in B-cell development. Successful rearrangement of V, D, and J heavy-chain gene segments allows synthesis of membrane-bound μ heavy chains in the endoplasmic reticulum (ER) of a pre-B cell. At this stage, no light-chain gene rearrangement has occurred. Newly made μ chains assemble with surrogate light chains, composed of λ5 and VpreB, and Igα/Igβ to yield the pre-B-cell receptor, pre-BCR (step 1 ). This receptor drives proliferation of those B cells that carry it. It also suppresses rearrangement of the heavy-chain locus on the other chromosome and so mediates allelic exclusion. In the course of proliferation, the synthesis of λ5 and VpreB is shut off (step 2 ), resulting in “dilution” of the available surrogate light chains and reduced expression of the pre-BCR. As a result, rearrangement of the light-chain loci can proceed (step 3 ). If this rearrangement is productive, the B cell can synthesize light chains and complete assembly of the B-cell receptor (BCR), which consists of a membrane-bound IgM and associated Igα and Igβ. The B cell is now responsive to antigen-specific stimulation. Description
The illustration shows a cell with its exterior and cytosol labeled. An oval E R is illustrated bonded to assembled heavy chains comprising mu domains. Step 1: Assembly with surrogate light chains: The heavy chain is moved to the exterior of the plasma membrane where it gets attached to the surrogate light chains labeled Vpre-B and lambda 5. I T A M is attached to the plasma membrane. It consists of I g alpha and I g beta. Step 2: Expression of V pre B and lambda 5 is turned off, and the surrogate light chains are lost from the heavy chains. Step 3: Light chain rearrangement occurs, and the light chains are added to the pre-B C R complex, forming the complete B cell receptor, which is labeled as I g M antibody. Successful rearrangement of V, D, and J segments in the heavy-chain locus thus allows the synthesis of a complete μ chain. B cells at this stage of development are called pre-B cells, as they have not yet completed assembly of a functional light-chain gene and therefore cannot engage in antigen recognition. As with other membrane-spanning proteins (see
Chapter 13), the μ chain is synthesized in the endoplasmic reticulum. It becomes part of a membrane-bound signaling receptor whose expression is essential for B-cell development to proceed in an orderly fashion. In pre-B cells, newly made μ chains form a complex with two so-called surrogate light chains, λ5 and VpreB (see Figure 24-19). The μ chain itself possesses a very short (three amino acids) cytosol-facing C-terminal tail and is therefore incapable of recruiting cytoplasmic components for the purpose of signal transduction. Instead, pre-B cells express two auxiliary transmembrane proteins, called Igα and Igβ, each of which carries in its cytoplasmic tail an immunoreceptor tyrosine-based activation motif, or
ITAM. The entire complex, including μ chain, λ5, VpreB, Igα, and Igβ, constitutes the pre-B-cell receptor (pre-BCR). Engagement of this receptor by (unknown) suitable signals results in recruitment and activation of a Src-family tyrosine kinase, which phosphorylates tyrosine residues in the ITAMs. In their phosphorylated form, ITAMs recruit other molecules essential for signal transduction (see below). Because no functional light chains are yet part of this receptor, it is presumed to be incapable of antigen recognition, as the antigen-binding site has contributions from both the heavy and light chains (see Figure 24-14) (except in camelids, which produce only H-chain antibodies). The pre-B-cell receptor has several important functions. First, it shuts off expression of the RAG recombinases, so that rearrangement of the other (allelic) heavy-chain locus cannot proceed. This phenomenon, called allelic exclusion, ensures that only one of the two available copies of the heavy-chain locus will be rearranged and thus expressed as a complete μ chain. Second, because of the association of the pre-B-cell receptor with Igα and Igβ, the receptor becomes a functional signal-transduction unit. Signals that emanate from the pre-BCR initiate proliferation of the pre-B cell to expand the numbers of those B cells that have undergone productive D-J and V-D-J recombination. In the course of this expansion, expression of the surrogate light chains, VpreB and λ5, subsides. The progressive dilution of VpreB and λ5 with every successive cell division results in insufficient fully assembled preBCR in the endoplasmic reticulum. As a consequence, the heavy chains are degraded (see Chapters 13 and 14) and the amount of pre-BCR
During an Adaptive Response, B Cells Switch from Making Membrane-Bound Ig to Making Secreted Ig
signaling decreases. This reduction in signaling allows re-initiation of expression of the RAG recombinases, which now target the κ or λ lightchain locus. A productive light-chain V-J rearrangement also shuts off rearrangement of the allelic locus (allelic exclusion). Upon completion of a successful V-J light-chain rearrangement, the B cell can make both μ heavy chains and κ or λ light chains and assemble them into a functional B-cell receptor (BCR), which can recognize antigen (see Figure 24-19). Once a B cell expresses a complete BCR on its cell surface, it can recognize antigen, and all subsequent steps in B-cell activation and differentiation require engagement with the antigen for which that BCR is specific. The BCR not only plays a role in driving B-cell proliferation upon a successful encounter with antigen, but also functions as a device for receptor-mediated endocytosis, an essential step that allows the B cell to process the acquired antigen and convert it into a signal that sends out a call for assistance by T lymphocytes; this antigen-presentation function of B cells is described in later sections. During an Adaptive Response, B Cells Switch from Making MembraneBound Ig to Making Secreted Ig As just described, the B-cell receptor, a membrane-bound IgM, provides a B cell with the ability to recognize a particular antigen, an event that triggers clonal selection and proliferation of that B cell, thus increasing the number of B cells specific for that antigen (see Figure 24-12).
However, key functions of immunoglobulins, such as neutralization of antigens or killing of bacteria, require that those products be released by the B cell so that they can accumulate in the extracellular environment and act at a distance from the site where they were produced. Whether to synthesize membrane-bound or secreted immunoglobulin is a choice made by the B cell during processing of the heavy-chain primary transcript. As shown in Figure 24-20, the μ locus contains two exons (TM1 and TM2) that together encode a C-terminal domain that spans the plasma membrane and anchors the IgM in it. One polyadenylation site is found upstream of these exons; a second polyadenylation site is present downstream. If the downstream poly(A) site is chosen, then further processing yields an mRNA that encodes the membrane-bound form of μ. (As described above, this choice is necessary for formation of the B-cell receptor, which includes membrane-bound IgM.) If the upstream poly(A) site is chosen, processing yields the secreted version of the μ chain. Similar arrangements are found for the other Ig constant-region gene segments (γ, α, ε), each of which can specify either a membrane-bound or a secreted heavy chain. The ability to switch between the membraneanchored and the secreted form of immunoglobulin heavy chains by alternative use of polyadenylation sites (not by alternative splicing) is so far unique to this family of gene products.
FIGURE 24-20 Synthesis of secreted and membrane IgM. The organization of the μ heavy-chain primary transcript is shown at the top: Cμ4 is the exon encoding the fourth μ constant-region domain; is a coding sequence unique for secreted IgM; TM1 and TM2 are exons that specify the transmembrane domain of the μ chain. Whether secreted or membrane-bound IgM is made depends on which poly(A) site is selected during processing of the primary transcript. (a) If the upstream poly(A) site is used, the resulting mRNA includes the entire Cμ4 exon and specifies the secreted form of the μ chain. (b) If the downstream poly(A) site is used, a splice donor site in the Cμ4 exon allows splicing to the transmembrane exons, yielding an mRNA that encodes the membrane-bound form of the μ chain. Similar mechanisms generate secreted and membrane-bound forms of other Ig isotypes. SS = signal sequence. Description The illustration shows from 5-prime to 3-prime, a heavy chain primary transcript consisting of S S, V D J, C mu 1, C mu 2, C mu 3, C mu 4, mu s, a poly-A site, T M 1, T M 2, and a final poly-A site. The C mu 4, mu s, poly-A site, T M 1 and T M 2 are highlighted while the remaining domains are grayed out. If polyadenylation at the upstream site occurs, the C mu 4 and mu S domains are expressed, forming secreted I g M. If polyadenylation at the downstream site occurs, splicing is carried out, resulting in the c mu 4, T M 1, and T M 2 domains being expressed, forming membrane I g M. The capacity to switch from the synthesis of exclusively membrane-bound immunoglobulin to the synthesis of secreted immunoglobulin is acquired
B Cells Can Switch the Isotype of Immunoglobulin They Make
by B cells in the course of their differentiation. Terminally differentiated B cells, called plasma cells, are devoted almost exclusively to the synthesis of secreted antibodies (see Figure 24-7). Each plasma cell synthesizes and secretes several thousand antibody molecules per second. It is this ramped-up production of secreted antibodies that underlies the effectiveness of the adaptive immune response in eliminating a pathogen and protecting against subsequent infection with the same pathogen. The protective value of antibodies is proportional to the concentration at which they are present in the circulation. Indeed, circulating antibody levels are often used as the key parameter to determine whether vaccination against a particular pathogen has been successful. The ability of plasma cells to secrete large amounts of immunoglobulins requires a massive expansion of the endoplasmic reticulum (ER), a hallmark of plasma cells. The unfolded-protein response (see Chapter 13) is initiated in B cells as an essential physiological mechanism to expand the ER and prepare the differentiating B cell for its future task as a highly active secretory cell. Interference with the unfolded-protein response abolishes the ability of B cells to turn into plasma cells. B Cells Can Switch the Isotype of Immunoglobulin They Make In the immunoglobulin heavy-chain locus, the exons that encode the constant segments of the μ chain lie immediately downstream of the rearranged VDJ exon (Figure 24-21, top). They are followed by exons that specify the constant segments of the δ chain. Transcription of a newly
rearranged immunoglobulin heavy-chain locus yields a single primary transcript that includes both the μ and δ constant regions. The splicing of this large transcript determines whether a μ chain or a δ chain will be produced. Downstream of the μ and δ exons are the exons that encode the constant segments of all the other heavy-chain isotypes. Upstream of each cluster of exons encoding one of the different isotypes (with the exception of the δ locus) is a repetitive sequence (switch region) that is recombination-prone, presumably because of its repetitive nature. Because each B cell necessarily starts out with surface IgM, recombination involving these sites, if it occurs, results in class switching from IgM to one of the other isotypes encoded downstream in the array of constantregion genes (Figure 24-21). The intervening DNA is deleted.
FIGURE 24-21 Class-switch recombination in the immunoglobulin heavy-chain locus. Class-switch recombination involves switch sites, which are repetitive sequences (colored circles) upstream of each of the heavy-chain constant-region genes. Recombination requires activation-induced deaminase (AID) as well as cytokines (e.g., IL-4) produced by certain helper T cells. Recombination eliminates the segment of DNA between the switch site upstream of μ exons and the constant region to which switching occurs. Class switching generates antibody molecules with the same specificity for antigen as that of the IgMbearing B cell that mounted the original response, but with different heavy-chain constant regions and therefore different effector functions.
Description An illustration shows a heavy chain locus. The gene consists of V, D, and J domains, a blue switch site, mu, delta, a gray switch site, gamma-three, a pink switch site, gamma1, a yellow switch site, gamma-2, an orange switch site, eta, a purple switch site, and alpha. Factors required for switching include C D 4 T cells, I L4, and A I D. An arrow from this gene points at I g G gamma 2 blood immunoglobulin which comprises of V, D, and J domains, a mixed blue-yellow switch site, gamma 2, orange switch site, eta, purple switch site, and alpha. Another arrow points at I g A, which is an immunoglobulin secreted across epithelia and comprises V, D, J, a mixed blue-purple switch site, and alpha. In the course of its differentiation, a B cell can switch Ig classes sequentially. Importantly, the light chain is not affected by this process, nor is the rearranged VDJ segment with which the B cell started out on this pathway. Class-switch recombination thus generates antibodies with different constant regions, but identical antigenic specificity because the variable region has not changed. Each immunoglobulin isotype is characterized by its own unique constant region. As discussed previously, these constant regions determine the functional properties of the various isotypes. Class-switch recombination is dependent on the activity of AID and on the presence of antigen as well as on helper T cells. Somatic hypermutation and class-switch recombination occur concurrently, and their combined effect allows fine-tuning of the adaptive immune response with respect to the affinity of the antibodies produced and the effector functions employed. KEY CONCEPTS OF SECTION 24.3
Generation of Antibody Diversity and B-Cell Development Functional antibody-encoding genes are generated by somatic rearrangement of multiple DNA segments at the heavy-chain and light-chain loci. These rearrangements involve V and J segments for immunoglobulin light chains and V, D, and J segments for immunoglobulin heavy chains (see Figure 24-15). Rearrangement of immunoglobulin gene segments is controlled by conserved recombination signal sequences (RSSs) composed of heptamers and nonamers separated by 12- or 23-bp spacers (see Figure 24-16). Only those segments that have spacers of different lengths can rearrange successfully: two segments to be joined must possess a 12- and a 23-bp spacer, not two of identical length. The molecular machinery that carries out the rearrangement process includes proteins made only in lymphocytes (recombinases RAG1 and RAG2). Antibody diversity is created by the random selection of Ig gene segments to be recombined and by the ability of the heavy and light chains produced from rearranged Ig genes to associate with many different light chains and heavy chains, respectively. Junctional imprecision generates additional antibody diversity at the joints of the gene segments brought together by somatic gene rearrangements. Further antibody diversity arises after B cells encounter antigen as a consequence of somatic hypermutation, which can lead to the selection and proliferation of B cells producing the highest affinity antibodies, a process termed affinity maturation. During B-cell development, heavy-chain genes are rearranged first, leading to expression of the pre-B-cell receptor. Subsequent rearrangement of light-chain genes results in assembly of an IgM membrane-bound B-cell receptor (see Figure 24-19). Only one of the allelic copies of the heavy-chain locus and of the light-chain locus is rearranged (allelic exclusion), ensuring that a B cell expresses Ig with a single antigenic specificity. Polyadenylation at different poly(A) sites in an Ig primary transcript determines whether the membrane-bound or secreted form of an antibody is produced (see
Figure 24-20). During an immune response, class switching allows B cells to adjust the class of antibody made and thus the effector functions of the immunoglobulins produced, while retaining the antibody’s specificity for antigen (see Figure 24-21).
24.4 The MHC and Antigen Presentation
24.4 The MHC and Antigen Presentation Antibodies can bind their target antigen without the involvement of other molecules; the presence of antigen and antibody is sufficient for their interaction. In the course of their differentiation, B cells receive essential assistance in the form of signals sent from T cells by a process that will be described in some detail below. This process, literally called T-cell help, is antigen-specific, and the T cells responsible for providing it are helper T cells. Although antibodies contribute to the elimination of bacterial and viral pathogens, it may also be necessary to destroy infected host cells that might serve as a source of new virus particles. This task is carried out by cytotoxic T cells. Both helper T cells and cytotoxic T cells make use of an antigen-specific T-cell receptor on its surface that, as we see in Section 24.5, is encoded by genes that are generated by mechanisms analogous to those used by B cells to generate immunoglobulin genes — including gene rearrangements. However, T cells recognize their cognate antigens in a manner very different from that used by B cells. The antigen-specific receptors on T cells recognize short segments of protein antigens but can do so only when these peptides are noncovalently bound to a glycoprotein complex present on the external surface of an antigen-presenting cell. Importantly, each T cell in an individual expresses many copies of a
The Killing Activity of Cytotoxic T Cells Is Antigen Specific and MHC Restricted
MHC proteins consist of two nonidentical single-pass transmembrane glycoproteins, both of which are encoded by the MHC. Description The illustration labeled (a) shows mouse M H C (H-2 complex). It consists of H-2 K, IA, I-E, H-2 D and 2 L genes. H-2 K, H-2 D, and 2 L are class one M H C proteins, while I-A and I-E are class two M H C proteins. The illustration labeled (b) shows human M H C (H L A complex). It consists of H L AD Q, H L A-D R, H D A-B, H L A-C, and H L A-A, the first two being class two M H C proteins and the remainder being class one M H C proteins. Interestingly, the human fetus may be considered a tissue graft in the mother: the fetus shares only half of its genetic material with the mother, the other half being contributed by the father. Antigens encoded by the paternal alleles may differ sufficiently from their maternal counterparts to elicit an immune response in the mother. Such a response can occur because in the course of pregnancy, fetal cells that slough off into the maternal circulation can stimulate the maternal immune system to mount an antibody response against the paternal antigens. We now know that these antibodies recognize proteins encoded by the human MHC. The fetus itself is spared rejection because of the specialized organization of the placenta, which prevents initiation of an immune response by the mother against fetal tissue. The Killing Activity of Cytotoxic T Cells Is Antigen Specific and MHC
Restricted Clearly MHC molecules did not evolve to prevent the exchange of surgical grafts. MHC molecules play an essential role in the recognition of virusinfected cells by a type of T cell termed a cytotoxic T cell, which is also called a cytolytic T lymphocyte (CTL). In virus-infected cells, MHC molecules bind to peptides derived from the viral proteins and display these fragments on the cell surface, where CTLs, charged with eliminating the infection, can recognize them. How such fragments of antigen are generated and displayed will be described below. CTLs that have receptors capable of recognizing a particular peptide-MHC complex unleash a payload of lethal molecules onto the infected target cells, destroying the target-cell membranes. The destruction of these target cells can be readily measured by the release of their cytoplasmic contents when they physically disintegrate. Thus CTL killing of infected host cells requires: (1) MHC presentation of antigenic peptides from the pathogen on the hostcell surface, (2) CTLs expressing antigen-MHC–specific T-cell receptors on their surface that can recognize the MHC-antigen complex, and (3) the activation of the CTL killing machinery once the T-cell receptors have bound to the MHC-antigen complex. Mice that have recovered from a particular viral infection are a ready source of CTLs that can recognize and kill target cells infected with the same virus. If CTLs are obtained from a mouse that has successfully cleared an infection with influenza virus, cytotoxic activity is observed against influenza-infected target cells, but not against uninfected controls
(Figures 24-23a and 24-23b, experiment 2 ). Moreover, the influenzaspecific CTLs will not kill target cells infected with a different virus, such as vesicular stomatitis virus (Figure 24-23b experiment 3 ). CTLs can even discriminate between closely related strains of influenza virus and can do so with pinpoint precision: differences of a single amino acid in the viral antigen may suffice to prevent recognition and killing by CTLs. These experiments show that CTLs are truly antigen specific and do not simply recognize some attribute that is shared by all virus-infected cells, regardless of the identity of the virus.
EXPERIMENTAL FIGURE 24-23 Chromium release assay allows direct demonstration of the cytotoxicity and specificity of cytotoxic T cells in a heterogeneous population of cells. (a) A suspension of spleen cells containing cytotoxic (killer) T cells is prepared from mice that have been exposed to a particular virus (e.g., influenza virus) and have cleared the infection. Target cells obtained from mice of the same strain are infected with the identical virus or left uninfected. After infection, cellular proteins are labeled nonspecifically by incubation of the target-cell suspension with . When the radiolabeled target cells are incubated with the suspension of cytotoxic T cells, the killing of infected target cells results in release of the -labeled proteins. Uninfected target cells are not killed and retain their radioactive contents. Lysis of cells by cytotoxic T cells can
therefore be readily detected and quantitated by measuring the radioactivity released into the supernatant. (b) Cytotoxic T cells (CTLs) harvested from mice that have been infected with influenza virus (X) can be tested against various target cells to determine the specificity of CTL-mediated killing. CTLs capable of lysing influenza virus–infected target cells ( 1 ) cannot kill uninfected cells ( 2 ) or cells infected with vesicular stomatitis virus (Y), for example ( 3 ). When these CTLs are tested on influenza virus–infected targets from a strain of mice that carries an altogether different MHC type (strain b, blue target cells), again no killing is observed ( 4 ). Cytotoxic T-cell activity is thus virus specific and restricted by the MHC. Description The illustration labeled (a) shows a mouse infected with influenza. The spleen is removed. From the spleen, killer T-cells are extracted. A target cell is labeled with chromium-51. The single cell suspension and labeled target cell is added in a test tube labeled control (uninfected). Another test tube has the virus infected target cell with killer T cells. No killing occurs in the control. Killing occurs in the other test tube. Both the test tubes are centrifuged. The supernatant from the second test tube is measured for chromium-51. The illustration labeled (b) shows a mouse. A text reads, infect mouse-a with influenza virus (x). An arrow points at the text, harvest T cells. At step 1, C T L-a, represented by a sphere, is infected with influenza virus (x) and a Target-a X is released. At step 2, C T L-a does not have influenza virus (x) in the target cell. At step 3, C T L-a is infected with V S V (Y) (a white shape is full of letter Ys) and labeled target-a Y. At step 4, C T L-a is infected with influenza virus (x) resulting in a target-b X cell. In the example in Figure 24-23a, we assumed that the CTLs harvested from an influenza-immune mouse are assayed on influenza-infected target cells derived from that same strain of mouse (strain a). However, if target cells from a completely unrelated strain of mouse (strain b, blue-colored target cell in Figure 24-23b experiment 4 ) are infected with influenza and used as targets, the CTLs from the strain a mouse are unable to kill the
T Cells with Different Functional Properties Are Guided by Two Distinct Classes of MHC Molecules
infected strain b target cells (Figure 24-23b, 1 vs. 4 ). It is therefore not sufficient that the antigen (an influenza-derived protein) is present; recognition of the antigen by CTLs is restricted by mouse strain-specific elements. Genetic mapping has shown that these restricting elements are encoded by genes in the MHC. Thus CTLs from a mouse strain that is immune to influenza will kill influenza-infected target cells from another mouse strain only if the two strains match at the MHC loci for the relevant MHC molecules. This phenomenon is therefore known as MHC restriction, and the MHC molecules involved are called restriction elements. T Cells with Different Functional Properties Are Guided by Two Distinct Classes of MHC Molecules The MHC encodes two types of glycoproteins essential for immune recognition, commonly called class I and class II MHC molecules. The genetic maps of mouse and human MHCs show the presence of several class I MHC genes and several class II MHC genes, although their arrangements differ between the two species (see Figure 24-22). In addition to class I and class II MHC molecules, the MHC encodes key components of the antigen-processing (e.g., proteolysis) and presentation machinery. Finally, the typical vertebrate MHC also encodes components of the complement cascade.
Both class I and class II MHC proteins are involved in presenting antigen to T cells, but they serve two broadly distinct functions. Class I MHC products present antigens to cytotoxic T cells, licensing them to destroy infected cells. Cytotoxic T cells are characterized by the expression of CD8, a surface glycoprotein that determines the ability of the T cells that carry it to interact with class I MHC products. Most, if not all, nucleated cells constitutively express class I MHC molecules, and many can support replication of viruses. Cytotoxic T cells thus recognize and kill infected cells of all body types via cell-surface class I MHC molecules that have bound to it (display) a virus-derived antigen (peptide). Class II MHC products are found exclusively on specialized antigenpresenting cells (APCs), also called professional APCs. These APCs present antigens bound to class II MHC molecules to a class of T lymphocytes called helper T cells. This presentation is the start of an adaptive immune response that also enables cytotoxic T cells to kill their targets and assists B cells in producing antigen-specific antibodies. B cells cannot undergo final differentiation into antibody-secreting plasma cells without assistance from helper T cells. Helper T cells express a surface glycoprotein called CD4 and interact with class II but not class I MHC molecules on target cells. The constitutive expression of class II MHC molecules is confined to professional APCs, which include B cells, dendritic cells, and macrophages. (Several other cell types, such as some epithelia, can be induced to express class II MHC molecules under specific circumstances, but we will not discuss them.) Again, the underlying cell biology that describes the expression, assembly, and mode
MHC Molecules Are Highly Polymorphic, Bind Peptide Antigens, and Interact with the T-Cell Receptor
of antigen presentation by class II MHC molecules fits this functional specialization rather neatly, as we shall see below. The two major groups of functionally distinct T lymphocytes — cytotoxic T cells and helper T cells — can thus be distinguished by the unique profile of membrane proteins displayed at their cell surface and by the MHC molecules they use as targets; these targets are often referred to as restriction elements. Cytotoxic T cells: CD8 marker; class I MHC restricted Helper T cells: CD4 marker; class II MHC restricted Both CD4 and CD8, along with many other proteins of the immune system, including B-cell and T-cell receptors and the polymeric IgA receptor, belong to the immunoglobulin (Ig) superfamily of proteins, all of which have one or more Ig domains. The molecular basis for the strict correlation between expression of CD8 and use of class I MHC molecules as the restriction element, or between expression of CD4 and use of class II MHC molecules as the restriction element, will become evident once the structure and mode of action of MHC molecules has been described. MHC Molecules Are Highly Polymorphic, Bind Peptide Antigens, and Interact with the T-Cell Receptor
Polymorphisms in the MHC Locus Are the Basis of Transplant Rejection Both class I and class II MHC molecules are highly polymorphic; that is, thousands of allelic variants, encoding similar proteins but with slightly different amino acid sequences, exist among individuals of the same species. The vertebrate immune system can respond to these allelic differences, and its ability to recognize allelic MHC variants is the underlying immunological cause for rejection of transplants that involve unrelated, genetically distinct individuals. The two classes of MHC molecules are structurally similar in many respects, as are their interactions with peptides and the T-cell receptor. MHC molecules are particularly important for recognizing self tissue and distinguishing it from nonself (and thus possibly pathogenic) substances. In general, except for close relatives, any two individuals have a very low chance of sharing the same MHC variants. Any inter-individual differences in MHC molecules in a graft recipient and donor will be recognized by the recipient’s immune system, which will treat the graft as foreign and eliminate it (graft rejection). The greater the similarity in the set of MHC alleles of a donor and a transplant recipient, the greater the chance that the transplant will be accepted. This is why surgeons look for an MHC-matched individual to donate an organ. If the tissue type (MHC alleles) of the donor does not exactly match that of the recipient, it is necessary to use drugs that suppress the immune responses of the recipient to prevent organ rejection.
The cell-biological mechanisms by which the immune system develops the capacity to distinguish self from nonself (or nonpathogenic from pathogenic) are complex, yet worth understanding. Understanding the molecular and cellular basis of immunity has enormous practical consequences for medicine and public health. We will therefore consider these molecular and cellular mechanisms in detail, beginning with the structures of the MHC proteins and the way by which peptides become bound to them. Class I MHC Molecules Class I MHC molecules, which belong to the Ig superfamily, consist of two polypeptide subunits. The larger subunit, for which there are multiple independent gene copies in the MHC region of mammalian genomes, is a type I membrane glycoprotein (see Figure 13-10). The smaller β2microglobulin subunit is not encoded by the MHC and corresponds in structure to an Ig domain. The larger subunits of class I MHC molecules in humans are encoded by the HLA-A, HLA-B, and HLA-C loci (see Figure 24-22). In the mouse, the larger subunits of class I MHC molecules are encoded by the H-2K, H-2D, and H-2L loci. The three-dimensional structure of a class I MHC molecule shows two Iglike domains (Figure 24-24a). These domains support an eight-stranded β sheet topped by two α helices. Jointly, the β sheet and the helices create a cleft, closed at both ends, in which a peptide binds. The mode of peptide binding by a class I MHC molecule requires that the peptide be about 8–10 amino acids long, so that the ends of the peptide can be tucked into
pockets that accommodate the charged amino and carboxyl groups at the termini of the peptide. Further, the peptide is anchored into the peptidebinding cleft by means of a small number of amino acid side chains, each of which is accommodated by a pocket in the MHC molecule that neatly fits that particular anchor amino acid residue (Figure 24-25a). On average, two such specificity pockets must be filled correctly to allow stable peptide binding, restricting binding to peptides with side chains that can fit into these pockets. In this manner, a given MHC molecule can accommodate a large number of peptides of diverse, yet circumscribed, sequence.
FIGURE 24-24 Three-dimensional structure of class I and class II MHC molecules. (a) Shown here is the structure of a class I MHC molecule with a bound antigenic (HA) peptide as determined by x-ray crystallography. The portion of a class I MHC molecule that binds a peptide consists of a β sheet composed of eight β strands and flanked by two α helices. The peptide-binding cleft is formed entirely from the MHC-encoded large subunit, which associates noncovalently with the small subunit (β2-microglobulin). (b) Class II MHC molecules are structurally similar to class I molecules but with several important distinctions. Both the α and β subunits of class II MHC molecules are MHC encoded and contribute to formation of the peptide-binding cleft. The peptide-binding cleft of class II MHC molecules accommodates a wider range of peptide sizes than that of class I molecules. Both class I and class II MHC proteins span the cell membrane, and each includes a transmembrane segment and a cytoplasmic tail (see Figures 24-21, 24-26, and 24-29). These are not included in the crystallographic analysis and are not shown in the figure. [Part (a) data from D. N. Garboczi, 1996, Nature 384:134, PDB ID 1ao7. Part (b) data from J. Hennecke, A. Carfi, and D. C. Wiley, 2000, EMBO J. 19:5611, PDB ID 1 fyt.] Description The illustration labeled (a) shows the side view, end view, and top view of class one M H C complex. The complex consists of the following parts: alpha helices, eight stranded beta sheet, I g like domains, and B 2 microglobulin. The top view shows the alpha helices. The illustration labeled (b) shows the side view, end view, and top view of class two M H C. The complex consists of multiple beta-sheet regions. The structure is very similar to that of the class one M H C complex. The top of the complex is the binding site for H A peptides.
FIGURE 24-25 Peptide binding and restriction in class I MHC. (a) Peptides that bind to class I molecules are on average 8–10 residues in length, require proper accommodation of the termini, and include two or three residues that are conserved (anchor residues). Amino acid residues in class I molecules that distinguish one allele from another (polymorphic residues) occur in and around the peptide-binding cleft. The polymorphic residues in the MHC affect both the specificity of peptide binding and interactions with T-cell receptors. Successful recognition of an antigenic peptide–MHC complex by a T-cell receptor requires a good fit among the receptor, peptide, and MHC molecule. (b) Steric clash and a lack of complementarity between anchor residues and the MHC molecule, and a similar clash between the T-cell receptor and the MHC molecule, prevent proper binding. Each T cell in an individual expresses one type of T-cell receptor, one that is thus restricted to binding specific peptide-MHC complexes. Description The illustration labeled (a) shows M H C a structure which has peptide binding specificity pockets represented by grooves. The peptide binding specificity pockets is bound to various groups of the peptide antigen. A T cell receptor is about to join the M H C a at the top. The M H C-peptide complex and the T-Cell receptor fit together well. (b) The M H C binding region of an allelic variant of an M H C molecule labeled M H C b has a slightly different peptide binding pockets shape. A lack of complementarity and steric clashes reduce the fit of the T-cell receptor with the M H C-antigen complex. Different anchor residue required. The polymorphic residues that distinguish one allelic MHC molecule from another are located mainly in and around the peptide-binding cleft. These residues therefore determine the architecture of the peptide-binding pocket and hence the specificity of peptide binding. Further, these polymorphic residues affect the surface of the MHC molecule that makes contact with the T-cell receptor. A T-cell receptor that can interact with one particular class I MHC allele will therefore, as a rule, not interact with unrelated
MHC molecules because of their different surface architectures (Figure 24-25b); this is the molecular basis of MHC restriction. The CD8 molecule on cytotoxic T cells functions as a co-receptor, binding to conserved portions of the class I MHC molecule. The presence of CD8 thus sets the class I MHC preference of any mature T cell that bears it. Class II MHC Molecules The two subunits (α and β) of class II MHC molecules are both type I membrane glycoproteins of the Ig superfamily. The typical mammalian MHC contains several loci that encode class II MHC molecules (see
Figure 24-22). Like the large subunit of class I molecules, both the α and β subunits of class II molecules show genetic polymorphism. The basic three-dimensional design of class II MHC molecules resembles that of class I MHC molecules. Two membrane-proximal Ig-like domains support a peptide-binding portion with a peptide-binding cleft (see Figure 24-24b). In class II MHC molecules, the α and β subunits contribute equally to the construction of the peptide-binding cleft. This cleft is open at both ends and thus supports the binding of peptides longer than those that bind to class I MHC molecules because the peptides can protrude from both ends of the cleft. The mode of peptide binding involves pockets that accommodate specific amino acid side chains as well as contacts between side chains of the MHC molecule and main-chain atoms of the bound peptide. As for class I MHC, class II MHC polymorphisms mainly affect residues in and around the peptide-binding cleft, so that peptidebinding specificity usually differs among different allelic products. A T-
cell receptor that interacts with a particular class II MHC molecule will not, as a rule, interact with a different class II MHC allelic variant, not only because of the difference in the peptide-binding specificity of the allelic MHC molecules, but also because of the polymorphisms that affect the residues that contact the T-cell receptor. Class I MHC is the basis for class II MHC restricted recognition of antigens. As we saw for class I MHC molecules and their CD8 co-receptor, the CD4 co-receptor recognizes conserved features on class II MHC molecules. Any mature T cell that bears the CD4 co-receptor uses class II MHC molecules for antigen recognition. As we will see below, class II MHC molecules evolved to present peptides generated predominantly in endosomes and lysosomes. Peptide binding to a class II MHC molecule takes place in those organelles, and class II MHC molecules are targeted specifically to those locations after their synthesis in the endoplasmic reticulum. This targeting is accomplished by means of a chaperone called the invariant chain, a type II membrane glycoprotein (see Figure 13-10). The invariant chain, termed Ii, plays a key role in the early stages of class II MHC biosynthesis by forming in the endoplasmic reticulum a trimeric structure onto which three newly made class II MHC αβ heterodimers assemble. The final assembly product thus consists of nine polypeptides: . The interaction between Ii and the αβ heterodimer involves a stretch of Ii, called the CLIP segment, which occupies the class II MHC peptide-binding cleft. Once the complex is assembled, the complex enters the secretory pathway and is diverted to endosomes and lysosomes at the trans-Golgi network (see Figure 14-2). The signals for this diversion are carried by the Ii cytoplasmic tail.
In Antigen Presentation, Protein Fragments Are Complexed with MHC Products and Posted to the Cell Surface
In Antigen Presentation, Protein Fragments Are Complexed with MHC Products and Posted to the Cell Surface The process by which foreign materials enter the immune system is the key step that determines the eventual outcome of an immune response. A successful adaptive immune response, which includes the production of antibodies and the generation of helper and cytotoxic T cells, absolutely requires the involvement of professional APCs. These cells are what acquire antigen, process it and then display it in a form that can be recognized by T cells. The pathway that converts antigen into a form suitable for T-cell recognition is referred to as antigen processing and presentation. The class I MHC pathway focuses predominantly on presentation of fragments of proteins synthesized by the cell itself (including pathogenencoded proteins found inside the infected cells), whereas the class II MHC pathway is centered on materials acquired from outside the APC. Recall that all nucleated cells express class I MHC products or can be induced to do so. This makes sense in view of the fact that a nucleated cell is capable of synthesizing nucleic acids and proteins and can thus in principle sustain replication of a viral pathogen. The ability to alert the immune system to the presence of an intracellular invader is inextricably linked to class I MHC-restricted antigen presentation. The distinction
The Class I MHC Pathway Presents Cytosolic Antigens
between the presentation of materials synthesized by an APC itself and the processing and presentation of antigen acquired from outside the cell is by no means absolute. Together, the class I and class II pathways of antigen processing and presentation sample all of the cellular and external compartments that need to be surveyed for the presence of pathogens. Antigen processing and presentation in both the class I and class II pathways may be divided into six discrete steps that are useful in comparing the two pathways: (1) acquisition of antigen, (2) tagging the antigen for destruction, (3) proteolysis, (4) delivery of peptides to MHC molecules, (5) binding of peptide to an MHC molecule, and (6) display of the peptide-loaded MHC molecule on the cell surface. Here we describe the molecular details of each pathway. The Class I MHC Pathway Presents Cytosolic Antigens
Figure 24-26 summarizes the six steps in the class I MHC pathway using a virus-infected cell as an example.
FIGURE 24-26 Class I MHC pathway of antigen processing and presentation. Step 1 : Acquisition of antigen is synonymous with the production of proteins with errors (e.g., premature termination, misincorporation) or the production of pathogen-derived proteins. Step 2 : Dysfunctional proteins are targeted for degradation by ubiquitinylation. Step 3 : Proteolysis is carried out by the proteasome. In cells exposed to interferon γ, the catalytically active β subunits of the proteasome are replaced by interferon-induced immune-specific β subunits. Step 4 : Peptides are delivered to the interior of the endoplasmic reticulum (ER) via the dimeric TAP peptide transporter. Step 5 : Each peptide is loaded onto a newly made class I MHC molecule within the peptide-loading complex.
Step 6 : The fully assembled class I MHC–peptide complex is transported to the cell surface via the secretory pathway. See text for details. Description An illustration shows a zoomed in view of a cell with the exterior, plasma membrane and cytosol labeled. The steps are as follows: Step 1: An illustration shows a ribosome attached to an m R N A strand. A cross is present on the proteins transcribed labeled error. A text reads, High error rate in translation targets dysfunctional proteins for U b addition and proteolysis. Step 2: A peptide epitope with an error from the ribosome is bonded to ubiquitin. Step 3: the peptide bonded to the U b enters the proteasome labeled beta 1, beta 2, and beta 5. The proteasome breaks down the peptide bonded to the U b so that the peptides and peptide epitope are separated. Step 4: The peptide epitope and fragments enter into the rough E R via a receptor. A T P is converted to A D P and phosphate in this step. Step 5: The E R membrane is attached a peptide loading complex which consists of T A P 1 and T A P 2, which is bound to tapasin which is further bound to a class 1 M H C. Class 1 M H C is derived from calnexin bound to calreticulin and E R p 57. Step 6: The amino peptidases cleave peptide epitope. The peptide epitope binds to class 1 M H C to be transported to the Golgi complex via vesicular transport from the cytosol to the plasma membrane. A text reads, peptide-loaded class 1 M H C complex is displayed on the cell surface. 1. Acquisition of Antigen: In the case of a viral infection, acquisition of antigen is usually synonymous with the infected state. Viruses rely on the host protein synthesis apparatus to generate new viral proteins. Protein synthesis, unlike DNA replication, is an error-prone process, in which a fraction of newly initiated polypeptide chains are terminated prematurely or suffer from other errors (e.g.,
misincorporation of amino acids, frameshifts, improper or delayed folding). These mistakes in protein synthesis affect the host cell’s own proteins and those specified by viral genomes equally. Such error-containing proteins must be rapidly removed so as not to clog up the cytoplasm, engage partner proteins in nonproductive interactions, or even act as dominant negative versions of a protein. Properly folded proteins may also sustain damage that leads to their unfolding, completely or in part, and necessitates their removal. These proteins are an important source of the peptides destined for presentation by class I MHC molecules. With the exception of a specialized process called cross-presentation (discussed below), the class I MHC pathway results in the formation of peptide-MHC complexes in which the peptides are derived from proteins synthesized by the class I MHC-bearing cell itself. 2. Targeting Antigen for Destruction: For the most part, polyubiquitinylation is responsible for targeting a protein for destruction (see Section 3.4). Polyubiquitinylation is a covalent modification that is tightly regulated. 3. Proteolysis: Polyubiquitinylated proteins are destroyed by proteolysis in proteasomes. The proteasome is a protease that engages its substrates and, without the release of intermediates, yields peptides in the size range of 3–20 amino acids as its final digestion products (see
Figure 3-32). During the course of an inflammatory response and in response to interferon γ, the three catalytically active β subunits (β1, β2, β5) of the proteasome can be replaced by three immune-specific subunits: β1i, β2i, and β5i, subunits that are encoded in the MHC region of the genome. The net result of this replacement is the generation of an immunoproteasome, the output (length of peptide
products) of which is matched to the requirements for peptide binding by class I MHC molecules. The immunoproteasome adjusts the average length of the peptides produced, as well as the sites at which cleavage occurs. Given the central role of the proteasome in the generation of the peptides presented by class I MHC molecules, proteasome inhibitors interfere potently with antigen processing via the class I MHC pathway. 4. Delivery of Peptides to Class I MHC Molecules: Protein synthesis, polyubiquitinylation, and proteasomal proteolysis all occur in the cytosol, whereas peptide binding by class I MHC molecules occurs in the lumen of the endoplasmic reticulum (ER). Thus peptides must cross the ER membrane to gain access to the peptide-binding sites of newly made class I molecules, a process mediated by the heterodimeric TAP complex, a member of the ABC superfamily of ATP-powered pumps (see Figure 11-15). The TAP complex binds peptides on the cytoplasmic face of the ER and, in a cycle that includes ATP binding and hydrolysis, translocates them into the ER. The specificity of the TAP complex is such that it can transport only a subset of all cytosolic peptides, primarily those in the length range of 5–10 amino acids, that are compatible with the circumscribed length of peptides that can fit into the class I MHC molecules. The mouse TAP complex shows a pronounced preference for peptides that with C-terminal leucine, valine, isoleucine, or methionine residues match the binding preference of class I MHC molecules. The genes encoding the TAP1 and TAP2 subunits composing the TAP complex are located in the MHC region. Peptidases in the cytosol and in the lumen of the ER may further modify the products of proteasomal proteolysis.
5. Binding of Peptides to Class I MHC Molecules: Within the ER, newly synthesized class I MHC molecules are part of a multiprotein complex referred to as the peptide-loading complex (Figure 24-27). This complex includes two chaperones (calnexin and calreticulin) and the oxidoreductase ERp57. Another chaperone (tapasin) interacts with both the TAP complex and the class I MHC molecule about to receive peptide. The physical proximity of TAP and the class I MHC molecule is maintained by tapasin. Once peptide loading onto the class I MHC molecule has occurred, a conformational change releases the loaded class I MHC molecule from the peptide-loading complex. This arrangement effectively ensures that only peptideloaded class I MHC molecules are released from the ER and then transported to and displayed at the cell surface. The overall efficiency of this pathway is such that approximately 4000 molecules of a given protein must be destroyed to generate a single MHC-peptide complex carrying a peptide from that particular polypeptide.
FIGURE 24-27 Class I MHC peptide-loading complex. The peptide-loading complex comprises not only the Class I MHC molecule that is about to receive a peptide, but also the TAP-associated protein tapasin, the chaperone calreticulin, and the oxidoreductase ERp57. Tapasin maintains the Class I MHC molecule in a peptidereceptive state and also provides a physical connection with the transporter associated with antigen presentation (TAP; not present in this structural diagram). Upon binding of peptide, the Class I MHC molecule is released from the peptide-loading complex and enters the secretory pathway to reach its destination, the cell surface. Description The illustration shows a bar where cytosol is labeled below and the E R lumen is labeled above. It has the following structures labeled clockwise from bottom left,
beta 2 m, calreticulin, tapasin, E R p 57, and M H C-1 h c. 6. Display of Class I MHC–Peptide Complexes at the Cell Surface: Once peptide loading is complete, the class I MHC–peptide complex is released from the peptide-loading complex and enters the constitutive secretory pathway (see Figure 14-2). Transfer from the Golgi to the cell surface is rapid and completes the biosynthetic pathway of a class I MHC–peptide complex. The entire sequence of events in the class I pathway occurs constitutively in all nucleated cells, all of which express class I MHC molecules and the other required proteins or can be induced to do so. As we have seen, exposure to cytokines such as interferon γ can induce immune-specific proteasomal subunits to generate immunoproteasomes with enhanced ability to produce the appropriate peptides for presentation by class I MHC molecules. In the absence of a viral infection, protein synthesis and proteolysis continuously generate a stream of peptides that are loaded onto class I MHC molecules. Healthy, normal cells therefore display on their surfaces a representative selection of peptides derived from their own proteins. There may be several thousand distinct MHC-peptide combinations displayed at the surface of a typical nucleated cell. The display of MHC–self-peptide complexes on the surfaces of normal, uninfected cells plays an essential role in the immune system. It is not until a virus makes its appearance that virus-derived peptides begin to make a contribution to the display of peptide-MHC complexes on cell surfaces.
As we noted above, a properly functioning immune system must be able to distinguish self (nonpathogenic) antigens from nonself (foreign, potentially pathogenic) antigens. The small organ called the thymus — located near the sternum at the level of the heart in humans — plays a critical role in controlling the ability of the immune system to identify self and nonself. Developing T cells in the thymus, referred to as thymocytes, calibrate their antigen-specific receptors to the sets of MHC-peptide complexes generated on thymic epithelial cells. The display of self peptides by self-MHC molecules in the thymus enables each developing T cell to learn which peptide-MHC combinations are self-derived and must therefore be ignored to avoid a self-destructive autoimmune reaction. Tcell development is thus driven by self-MHC molecules loaded with self peptides, a template on which a useful repertoire of T cells can be molded. Simply put, any T cell that bears a receptor that too strongly reacts with self-MHC–self-peptide complexes is potentially dangerous when it leaves the thymus, and it must be eliminated. This process of selection will be discussed below. An exception to the usual mode of antigen presentation that is nonetheless crucial in the development of cytotoxic T cells is cross-presentation. This term refers to the acquisition by dendritic cells of apoptotic cell remnants, complexes composed of antigen bound to antibody, and possibly other forms of antigen, by phagocytosis. By a pathway that has yet to be fully understood, these materials escape from phagosomal or endosomal compartments into the cytosol, where they are then handled according to the steps described above, resulting in fragments loaded onto class I MHC proteins. Endosomal or lysosomal proteolysis may likewise generate
The Class II MHC Pathway Presents Antigens Delivered to the Endocytic Pathway
peptides that find their way onto class I MHC proteins, either in the endocytic compartments themselves, or following delivery to the cytosol, where they may enter the canonical class I processing and presentation pathway. Dendritic cells are the most efficient at cross-presentation and so allow the loading of class I MHC molecules complexed with peptides that derive from cells other than the APC itself. The Class II MHC Pathway Presents Antigens Delivered to the Endocytic Pathway Although class I MHC and class II MHC molecules show a striking structural resemblance, the manner in which the two classes acquire peptide and their function in antigen recognition differ greatly. Whereas the primary function of class I MHC molecules is to guide CD8-bearing cytotoxic T cells to their target (usually infected) cells, class II MHC molecules serve to guide CD4-bearing helper T cells to the cells with which they interact, primarily professional APCs. Activated helper T cells provide protection not only by helping B cells to produce antibodies, but also by means of the complex sets of cytokines they produce, which activate phagocytic cells to clear pathogens or help set up an inflammatory response. As noted previously, class II MHC molecules are expressed primarily by professional APCs: dendritic cells and macrophages, which are phagocytic, and B cells, which are not. Hence the class II MHC pathway of
antigen processing and presentation generally occurs only in these cells. The steps in this pathway are depicted in Figure 24-28.
FIGURE 24-28 Class II MHC pathway of antigen processing and presentation. Step 1 : Particulate antigens are acquired by phagocytosis and nonparticulate antigens by pinocytosis or endocytosis. Step 2 : Exposure of antigen to the acidic and reducing environment of endosomes and lysosomes prepares the antigen for proteolysis. Step 3 : The antigen is broken down by various proteases in endosomal and lysosomal compartments. Step 4 : Class II MHC molecules, assembled in the ER from their subunits, are delivered to endosomal and lysosomal compartments by means of signals contained in the associated invariant (Ii) chain. This delivery targets late endosomes, lysosomes, and
early endosomes, ensuring that class II MHC molecules are exposed to the products of proteolytic breakdown of antigen along the entire endocytic pathway. The Ii polypeptide is degraded in this compartment, followed by removal of Ii remnants in exchange for other peptides destined for presentation. Step 5 : Peptide loading is accomplished with the assistance of DM, a class II MHC–like chaperone protein. Step 6 : Peptide-loaded class II MHC molecules are transported to the cell surface. See text for details. Description Macrophages and dendritic cells have a role in phagocytosis, pinocytosis, and receptormediated endocytosis. B cells have a role in B C R mediated endocytosis. The steps are as follows. Step 1 a: A microbe enters the cell via phagocytosis. Step 1 b: A microbe enters via pinocytosis. Step 1 c: A microbe enters via receptor-mediated endocytosis. Step 1 d: A microbe is bonded to an antigen on the plasma membrane. It enters the cell via B C R mediated endocytosis. Step 2: A the microbes enter into an oval lysosome where p H-dependent unfolding and reduction of S–S bonds occur. Step 3: Proteolysis by lysosomal peptidases: the microbes are cleaved to give peptides and peptide epitope. Step 4 a: Assembly of class 2 M H C occurs in the E R. Step 4 b: Class 2 M H C is transported to an endosome via the Golgi complex. Step 5: Peptide loading occurs in the endosome. The peptide epitope binds to class 2 M H C. Step 6: The peptide epitope and class 2 M H C complex is transported via a tubular endosome to the cell surface. A text reads, Peptide-loaded class 2 M H C complex displayed on the cell surface.
1. Acquisition of Antigen: In the class II MHC pathway, antigen is acquired by pinocytosis, phagocytosis, or receptor-mediated endocytosis. Pinocytosis, which is rather nonspecific, involves the delivery, by a process of membrane invagination and subsequent vesicle fusion, of a volume of extracellular fluid and the molecules dissolved therein. Phagocytosis, the ingestion of particulate materials such as bacteria, viruses, and remnants of dead cells, involves extensive remodeling of the actin-based cytoskeleton to accommodate the incoming particle. Although phagocytosis may be initiated by specific receptor-ligand interactions, these are not always required: even latex particles and other particulates such as glass beads can be ingested very efficiently by macrophages. Pathogens decorated (opsonized) by antibodies and certain complement components are targeted to macrophages and dendritic cells, which recognize them by means of cell-surface receptors for complement components or for the Fc portion of immunoglobulins. The cells then phagocytose them (Figure 24-29). Macrophages and dendritic cells also express several types of less-selective receptors (e.g., C-type lectins, Toll-like receptors, scavenger receptors) that recognize molecular patterns in both soluble and particulate antigens. These cells then internalize the bound antigens by receptor-mediated endocytosis. B cells, which are not phagocytic, can also acquire antigens by receptor-mediated endocytosis using their antigenspecific B-cell receptors (Figure 24-30). Finally, cytosolic antigens may enter the class II MHC pathway via autophagy (see Figure 1434).
FIGURE 24-29 Presentation of opsonized antigen by phagocytic cells. By means of Fc receptors such as FcγR displayed on their cell surface, specialized phagocytic cells such as macrophages or dendritic cells can bind and ingest pathogens that have been decorated with antibodies (opsonization). After digestion of the phagocytosed particle (e.g., immune complex, bacterium, virus), some of the peptides produced, including fragments of the pathogen (orange), are loaded onto class II MHC molecules (green). Class II MHC–peptide complexes displayed at the surface allow activation of T cells whose receptors are specific for these MHC-peptide combinations. Lipid antigens are delivered to the class I MHC–like molecule CD1 (pink), whose binding site is specialized to accommodate lipids. Certain pathogenderived peptides (purple) may be delivered to class I MHC products (blue) by means of cross-presentation. The molecular mechanisms that underlie cross-presentation remain to be clarified. Description The steps are as follows: Step 1: I g G-decorated bacterium binds to F c gamma R. An illustration shows a phagocytic cell (macrophage; dendritic cell). It has an F c receptor on its cell surface bonded to an antibody which is bonded to class 2 M H C restricted peptide and lipid antigens of a bacterium which is further bonded to an antibody. Step 2: Active F c gamma R stimulates phagocytosis. The bacterium is phagocytized. Step 3: Intracellular destruction of bacterium occur which leads to release of contents. Step 4: Presentation of bacterial antigens to T cells via class one crosspresentation and class 2 M H C. Lipid presentation via C D 1: An illustration shows a phagocyte with class 1 and 2 M H Cs which is bonded to bacterial antigens. C D 1 presents a lipid from the bacterium.
FIGURE 24-30 Antigen presentation by B cells. B cells bind antigen, even if present at low concentration, to their B-cell receptors, or surface Ig. The immune complex that results is internalized and then delivered to endosomal or lysosomal
compartments, where it is degraded. Peptides liberated from the immune complex, including fragments of the protein antigen, are displayed as class II MHC–peptide complexes at the cell surface. Helper T cells specific for the displayed complex can now provide help to the B cell, allowing it to proliferate and eventually form antibody-secreting plasma cells. This help is MHC restricted and antigen specific. Description The steps are as follows: Step 1: Surface I g captures antigen. An illustration shows a B cell with a surface antigen bind ro a circular structure. The circular antigen is made of a T cell epitope and a B cell epitope. Step 2: complex internalized. The antigen is internalized by the B cell. Step 3: Complex destroyed and T-cell epitope presented by class 2 M H C. Step 4: T cell provides help to B cell in antigen-specific fashion. An illustration shows a B cell with the destroyed complex contents and an M H C class 2 receptor presenting an antigen to the T C R of a T cell. 2. Targeting Antigen for Destruction: Proteolysis is required to convert intact protein antigens into peptides of a size suitable for binding to class II MHC molecules. Protein antigens are targeted for degradation by progressive unfolding, brought about by the drop in pH as proteins progress along the endocytic pathway. The pH of the extracellular environment is around pH 7.2, while in early endosomes it is between pH 6.5 and 5.5; in late endosomes and lysosomes the pH may drop to pH 4.5. ATP-powered V-class proton pumps in the endosomal and lysosomal membranes are responsible for this acidification (see
Figure 11-9). Proteins that are stable at neutral pH tend to unfold when they are exposed to extremes of pH through rupture of hydrogen
bonds and destabilization of salt bridges. Furthermore, the environment in the endosomal or lysosomal compartment is a reducing one, in which lysosomes attain a concentration of reducing equivalents in the millimolar range. Reduction of the disulfide bonds that stabilize many extracellular proteins can also be catalyzed by a thioreductase inducible by exposure to interferon γ. The combined action of low pH and reducing environment prepares the antigens for proteolysis. 3. Proteolysis: Degradation of proteins in the class II MHC pathway is carried out by a large set of lysosomal proteases, collectively referred to as cathepsins, which are either cysteine or aspartyl proteases. A wide range of peptide fragments is produced, including some that can bind to class II MHC molecules. The lysosomal proteases operate optimally at the acidic pH within lysosomes. Consequently, agents that inhibit the activity of the V-class proton pumps that maintain their acidification interfere with antigen processing, as do inhibitors of lysosomal proteases. 4. Encounter of Peptides with Class II MHC Molecules: Recall that most class II MHC molecules synthesized in the endoplasmic reticulum are directed to late endosomes. The peptides generated by proteolysis thus reside in the same intracellular space as the class II MHC molecules themselves — they do not have to cross a membrane, as is the case for peptides destined to bind to class I MHC molecules (see Figure 24-26). To allow peptides and class II MHC molecules to meet, the complex is transported from the endoplasmic reticulum via the secretory pathway to endosomal compartments.
5. Binding of Peptides to Class II Molecules: The complex delivered to endosomal compartments is incapable of binding peptide because the peptide-binding cleft in the class II molecule is occupied by the invariant chain (Ii). For the same reason, newly assembled complexes do not compete for class I MHC–destined peptides delivered to the ER via TAP: their peptide-binding site is already occupied by Ii. Recall that the ER is where both class I and class II MHC molecules are assembled. The presence of Ii in the nascent class II MHC complex ensures that class II MHC molecules do not bind peptide in the ER. The same proteases in endosomes and lysosomes that act on internalized antigens and degrade them into peptides also act on the complex, resulting in removal of the Ii molecule from the complex, with the exception of a small portion of Ii called the CLIP segment. Because it is firmly lodged in the class II MHC peptide-binding cleft, CLIP is resistant to proteolytic attack. The class II MHC molecules themselves are also resistant to unfolding and proteolytic attack under the harsh conditions that prevail in the endocytic pathway. Next the CLIP segment is removed from the αβ heterodimer by the chaperone DM, and the newly vacated peptide-binding cleft of the class II MHC molecule may now bind the peptides that are abundantly present in the endocytic pathway. Although the DM protein is MHC encoded and structurally very similar to class II molecules, it does not itself bind peptides. However, newly formed class II MHC–peptide complexes are themselves susceptible to further editing by DM, which may dislodge the peptide already bound, until the class II molecule acquires a peptide that binds so strongly that it cannot be removed by DM. The
resulting class II MHC–peptide complexes are extremely stable, with estimated half-lives far in excess of 24 hours. 6. Display of Class II MHC–Peptide Complexes at the Cell Surface: The newly generated class II MHC–peptide complexes are localized mostly in late endosomal compartments, which include multivesicular endosomes (or bodies) (see Figure 14-32). Endosomal compartments then elongate and ultimately deliver class II MHC– peptide complexes to the surface by membrane fusion. These events are tightly regulated: tubulation and delivery of class II MHC molecules to the surface are enhanced in dendritic cells and macrophages following their activation by signals generated in response to infection, such as bacterial lipopolysaccharide, which is detected by Toll-like receptors on the surfaces of these professional APCs, as well as inflammatory cytokines, such as interferon γ, produced by CD4-expressing helper T cells. For professional APCs, the above steps are constitutive — they happen all the time — but they can be modulated by exposure to microbial agents and cytokines. In addition to the pathways described here for class I and class II MHC products, there is a category of class I MHC–related molecules, the CD1 proteins, that are specialized in the presentation of lipid antigens. The structure of a CD1 molecule resembles that of a class I MHC molecule: a larger subunit complexed with β2-microglobulin. Many species of bacteria produce lipids whose chemical structures are not found in their mammalian hosts. These lipids can serve as antigens when presented by CD1 molecules (see Figure 24-29), to which they bind via a lipid-binding pocket that is conceptually similar to that of most MHC
molecules. Signals in the cytoplasmic tail of the larger CD1 subunit target these molecules to endosomal or lysosomal compartments, where loading with antigenic lipids occurs. The CD1-lipid complexes engage a relatively rare class of T cells, referred to as NKT cells, as well as γδ T cells, both described below. NKT cells fulfill an important role in cytokine production and help initiate and orchestrate adaptive immune responses via their cytokine outputs. Other proteins structurally related to Class I MHC products such as the MR1 protein can bind small metabolites and present them to a special subset of T lymphocytes called MAIT cells. KEY CONCEPTS OF SECTION 24.4 The MHC and Antigen Presentation The MHC, discovered as the genetic region responsible for acceptance or rejection of grafts, encodes many different proteins involved in the immune response. Two of these proteins, class I and class II MHC molecules, are highly polymorphic, occurring in many allelic variations (see Figure 24-22). The function of the class I and class II MHC proteins is to bind peptide antigens and display them on the surfaces of cells so that the antigen–MHC protein complex can interact with antigen-specific T-cell receptors on T cells. When an antigen–MHC protein complex on an antigen-presenting cell binds to its complementary T-cell receptor on a T cell, the T cell is activated to assume effector functions, such as the production of cytokines or the ability to kill a virus-infected cell. Class I MHC molecules are found on most nucleated cells, whereas the expression of class II MHC molecules is confined largely to professional APCs such as dendritic cells, macrophages, and B cells. The organization and structure of class I and class II MHC molecules is similar and includes a peptide-binding cleft that is specialized for binding a wide variety of peptides (see Figure 24-24). Different allelic variants of MHC molecules bind different sets of peptides because the differences that distinguish one allele from another include residues that define the architecture of the peptide-binding cleft (see Figure 24-25). Allelic variation also includes residues in the MHC molecule that directly contact the corresponding T-cell
receptor. Thus different allelic variants of an MHC molecule, even if they bind the identical peptide, do not usually react with the same T-cell receptor. This phenomenon is called MHC restriction. Peptide binding by class I and class II MHC molecules occurs in different intracellular compartments: class I molecules bind predominantly to cytosolic materials, whereas class II molecules bind to extracellular materials internalized by phagocytosis, pinocytosis, or receptor-mediated endocytosis. The process by which protein antigens are acquired, processed into peptides, and converted into surface-displayed MHC-peptide complexes is referred to as antigen processing and presentation. This process operates continuously in cells that express the relevant MHC molecules, yet can be modulated in the course of an immune response. Antigen processing and presentation can be divided into six discrete steps: (1) acquisition of antigen; (2) targeting of the antigen for destruction; (3) proteolysis; (4) encounter of peptides with MHC molecules; (5) binding of peptides to MHC molecules; and (6) display of the peptide-loaded MHC molecules on the cell surface (see Figures 24-25 and 24-27).
24.5 T Cells, T-Cell Receptors, and T-Cell Development
24.5 T Cells, T-Cell Receptors, and T-Cell Development T lymphocytes recognize antigen through specific interactions of their receptors with MHC molecules. The diverse, antigen-specific T-cell receptors made by individual T cells that are entrusted with this task are structurally related to the F(ab) portion of immunoglobulins. To generate a large repertoire of antigen-specific T-cell receptors, T cells rearrange the genes encoding the T-cell receptor subunits by mechanisms of somatic recombination essentially identical to those used by B cells to rearrange immunoglobulin genes. The development of T cells, like that of B cells, is strictly dependent on successful completion of these somatic gene rearrangements to yield a functional T-cell receptor. Genetic ablation of RAG activity eliminates all of the T cells as well as all of the antibodyproducing B cells. As noted previously, each T cell expresses a single type of T-cell receptor. In this section, we describe the receptor subunits that mediate antigen-specific recognition, how they pair up with membrane glycoproteins essential for signal transduction, and how these complexes recognize MHC-peptide combinations. As pointed out in the preceding section, an individual’s T cells recognize peptide antigens only when they are bound to the polymorphic MHC molecules present in that individual. In the course of T-cell development, T cells must learn the identity of these self MHC molecules and receive
The Structure of the T-Cell Receptor Resembles the F(ab) Portion of an Immunoglobulin
instructions about which MHC-peptide combinations to ignore, so as to avoid autoimmunity: the potentially catastrophic reactions of newly generated T cells with the individual’s own tissues. The Structure of the T-Cell Receptor Resembles the F(ab) Portion of an Immunoglobulin Much as B cells use the B-cell receptors on their surfaces to recognize antigens and generate intracellular signals that lead to clonal expansion, T cells depend on their T-cell receptors (TCRs) to initiate their participation in immune responses. T cells that have been activated via these antigen-specific receptors proliferate and acquire the capacity to kill antigen-bearing target cells (in the case of cytotoxic T cells) or to secrete cytokines that will assist B cells in their differentiation into antibodyproducing plasma cells (in the case of helper T cells). The TCR recognizes antigenic peptides bound to MHC molecules. The TCR is composed of two core glycoprotein subunits (Figure 24-31), each of which is encoded by a somatically rearranged gene. The receptor is usually composed of an α and a β subunit, but certain TCRs are alternatively composed of a γ and a δ subunit. The structure of these subunits is similar to that of the F(ab) portion of an immunoglobulin: at the N-terminal end is a variable region, followed by a constant region and a transmembrane segment. The cytoplasmic tails of the TCR subunits are short and do not directly interact with cytoplasmic signal transduction
molecules. Instead, the TCR associates with the CD3 complex, a set of membrane glycoproteins composed of γ, δ, ε, and ζ chains. The structure of this complex has been determined by cryoelectron microscopy (Figure 24-31b). The extracellular domains of the CD3 subunits are similar to immunoglobulin domains, and the cytoplasmic domain in each contains an ITAM (immunoreceptor tyrosine-based activation motif), which recruits adapter proteins upon phosphorylation of its tyrosine residues. The ζ chain is integrated into the CD3-TCR complex as a disulfide-bonded homodimer, and each ζ chain contains three ITAMs (see Figure 24-33).
FIGURE 24-31 Structure of the T-cell receptor and its co-receptors. (a) The antigenspecific T-cell receptor (TCR) is composed of two chains, the α and β subunits, which are produced by V-J and V-D-J recombination, respectively (see Figure 24-32). The α and β subunits must associate with the CD3 complex (not shown; see panel b and Figure 24-33) to allow the transduction of signals. The formation of a full TCRαβ–CD3 complex is required for surface expression. The TCR further associates with a co-receptor, CD8 (light blue) or CD4 (light green), which allows interaction with conserved features of class I MHC or class II MHC molecules, respectively, on antigen-presenting cells. (b) Structure of the extracellular portion of the TCR-CD3 complex as determined by cryoelectron microscopy. The subunits of the CD3 complex are color-coded. The extracellular portions of the T-cell receptor α(TCRα) and β(TCRβ) subunits as well as those of CD3γ,δ, and ε all possess the basic fold of immunoglobulin domains. The cytoplasmic portions of the CD3 complex subunits were not resolved by cryoelectron microscopy and are therefore absent from the figure. Unlike the CD3γ, δ, and ε subunits, the CD3ζ chain has a minimal extracellular domain. The intracellular portions of the CD3 complex contain the immunoreceptor tyrosine-based activation motifs (ITAMs) essential for signal transduction via the TCR-CD3 complex. [Part (b) republished with permission from Nature Publishing Group, from E. L. Reinherz, 2019, “The Structure of a T-Cell Mechanosensor,” Nature 573(7775):502–504; permission conveyed through Copyright Clearance Center, Inc.] Description The illustration labeled (a) shows the bottom section of two T cells. At the bottom of each T cell, a multicolor structure is present with the following labels, from top to bottom: Constant domains of T-cell receptor including alpha and beta units, T C R, variable domains of T-cell receptor, peptide bound to M H C, Class 1 M H C (on the left side)(C D 8), Class 2 M H C (on the right side) (C D 4). These are attached to two blue semicircles labeled A P C. The illustration labeled (b) shows the ribbon structure T C R embedded in the cell membrane. The cell exterior and the cytoplasm are labeled. Exteriorly, it has four areas labeled, clockwise from bottom left, F G loop, T C R beta, Variable domains, T C R alpha. At the cell membrane, the ribbons attach to 7 curled ribbons that are going
TCR Genes Are Rearranged in a Manner Similar to Immunoglobulin Genes
straight through the membrane. The labels on these ribbons, clockwise from top left to bottom are: C D 3 gamma, C D 3 delta, C D 3 epsilon, C D 3 zeta. TCR Genes Are Rearranged in a Manner Similar to Immunoglobulin Genes Virtually all antigen-specific receptors generated by somatic recombination contain a subunit that is the product of V-D-J recombination (e.g., Ig heavy chain; TCR β chain) and another that is the product of V-J recombination (e.g., Ig light chain; TCR α chain). The mechanism of V-D-J and V-J recombination for TCRs is essentially identical to that described for immunoglobulin genes and requires all the component proteins composing the nonhomologous end-joining machinery: RAG1, RAG2, Ku70, Ku80, the catalytic subunit of DNAdependent protein kinase, XRCC4, DNA ligase IV, and Artemis. Similarly recombination signal sequences (RSSs) are required (Figure 24-32).
FIGURE 24-32 Organization and recombination of TCR loci. The organization of TCR loci is in principle similar to that of immunoglobulin loci (see Figure 24-15). Left: The TCR β-chain locus includes a cluster of V segments, a cluster of D segments, and several J segments, downstream of which are two constant regions. The arrangement of the recombination signals is such that not only is D-J joining allowed, but also D-D joining followed by joining to a V segment. Direct V-J joining in the TCR β locus is not observed even though the arrangement of the recombination signal sequences would be consistent with such a possibility. Right: The TCR α-chain locus is composed of a cluster of V segments and a large number of J segments. SS = exon encoding signal sequence; Enh = enhancer.
Description T C R beta chain: The illustration shows a germ-line D N A for the T C R beta chain which consists of from 5 prime to 3 prime end S S, V-beta-1, S S, V-beta-n, D-beta-1, J- beta-1, C-beta-1, D-beta-2, J-beta 2, C-beta-2, and a beta-enhancer. After somatic recombination by D-J joining and then V-DJ joining, a shorter sequence comprising S S, V-beta-n, D-beta-1, J-beta, C-beta-1, J-beta-2, C-beta-2, and a beta enhancer is generated. Translation produces a primary R N A transcript, which then undergoes splicing. Finally, the messenger R N A is formed, consisting of S S, V-beta, D-beta, and J-beta segments in the variable portion and the constant C-beta-1 segment terminating in a poly-A tail. This m-R N A undergoes translation to form the beta chain with V and C regions. T C R alpha chain: The illustration shows a germ-line D N A for the T C R alpha-chain consists of S S, V-alpha-1, S S, V-alpha-n, J-alpha, C-alpha, and a beta-enhancer. Somatic recombination by V-J joining occurs, removing an S S segment and the Valpha-n segment, transcription forms the primary R N A transcript, which then undergoes splicing to form m-R N A comprising S S, V-alpha-1 and J-alpha in the variable portion and C-alpha segments in the constant region. Translation yields the alpha protein, which can then associate with the beta protein to form the assembled T C R molecule. A number of noteworthy features characterize the organization and rearrangement of the TCR loci. First, the organization of the RSSs is such that D-to-D rearrangements are allowed, unlike the case for Ig. Second, terminal deoxynucleotidyltransferase (TdT) is active at the time the TCR genes are rearranged, and therefore additional nucleotides, termed N nucleotides, can be present at the V-D and D-J junctions in all rearranged TCR genes. Third, in humans and mice, the TCR δ locus is embedded within the TCR α locus. This organization results in complete excision of the interposed δ locus when TCR α rearrangement occurs, so a choice of the TCR α locus for rearrangement precludes use of the δ locus, which is
Many of the Variable Residues of TCRs Are Encoded in the Junctions Between V, D, and J Gene Segments
lost by deletion. T cells that express the αβ receptor and those that express the γδ receptor are considered separate lineages with distinct functions. Among the T-cell γδ receptors are some capable of recognizing the CD1 molecule, which as we learned is specialized for the presentation of lipid antigens. The γδ T cells are programmed to home to distinct anatomic sites (e.g., the epithelium lining, the genital tract, or the skin) and probably play a role in host defense against pathogens commonly found at these sites. They may also have anti-tumor activity. Deficiencies in the key components of the recombination apparatus, such as the RAG recombinases, preclude rearrangement of TCR genes. As we have seen for B cells, development of lymphocytes is strictly dependent on the rearrangement of the antigen-receptor genes. A deficiency in either RAG1 or RAG2 thus prevents both B-cell and T-cell development. Mice with homozygous RAG gene knockouts are frequently used to assess the roles of B and T cells in physiological and pathophysiological processes. Many of the Variable Residues of TCRs Are Encoded in the Junctions Between V, D, and J Gene Segments The diversity created by somatic recombination of TCR genes is estimated to exceed unique receptors. Combinatorial use of different V, D, and J gene segments makes an important contribution to this diversity, as do the mechanisms of junctional imprecision and N-nucleotide addition already discussed for immunoglobulin gene rearrangements. The net result is a
degree of variability in the V regions that at least matches that of the immunoglobulins (see Figure 24-13). Indeed, each of the TCR’s variable regions includes three hypervariable regions (CDRs), equivalent to those in the BCR. Unlike immunoglobulin genes, however, the TCR genes do not undergo somatic hypermutation. Therefore, TCRs exhibit nothing equivalent to the affinity maturation of antibodies during the course of an immune response, nor is there the option of class-switch recombination or the use of alternative polyadenylation sites to create soluble and membrane-bound versions of the receptors. The crystal structures of a number of TCRs bound to class I MHC–peptide or class II MHC–peptide complexes have been determined. These structures show variation in how the TCR docks with the MHC-peptide complex, but the most extensive contacts in the somatically diverse CDR3 region are made with the central peptide-containing portion of the complex, with the germ line–encoded CDR1 and CDR2 contacting the α helices of the MHC molecules. Many of the TCRs for which a structure has been solved dock diagonally across the peptide-binding portion of the MHC-peptide complex. As a result, the TCR makes extensive contacts with the peptide as well as with the α helices of the MHC molecule to which it binds. The positions at which allelic MHC molecules differ from one another are frequently those residues that directly contact the TCR, thus precluding tight binding of unrelated allelic MHC products. Amino acid differences that distinguish one MHC allele from another also affect the architecture of the peptide-binding cleft. Even if the MHC residues that interact directly with the TCR were shared by two allelic
Signaling via Antigen-Specific Receptors Triggers Proliferation and Differentiation of T and B Cells
MHC molecules, their peptide-binding specificity would probably differ because of amino acid differences in the peptide-binding cleft. Consequently, the TCR contact residues provided by bound peptide, which are essential for stable interaction with a TCR, would be absent from the wrong MHC-peptide combination. A productive interaction with the TCR would then be unlikely to occur. As mentioned earlier, these molecular features are the underpinnings of MHC restriction. Signaling via Antigen-Specific Receptors Triggers Proliferation and Differentiation of T and B Cells The immune responses mediated by T cells and B cells are initiated once TCRs or BCRs bind their respective ligands and are thus activated. The ligands for TCRs are MHC-peptide complexes expressed on the surfaces of APCs. The ligands for BCRs are antigens that bind to the receptors without the need for MHC intervention or a presenting cell. TCR and BCR activation by their antigens is similar to the activation of signaling receptors like tyrosine kinase receptors that we have considered previously (see Chapter 16). In all cases, multiple phosphotyrosine signal transduction cascades are engaged. Several integral membrane proteins, as well as soluble cytosolic enzymes, participate in TCR and BCR signaling. In some cases, these membrane-associated proteins can be thought of as auxiliary subunits of the receptors. Examples of how such auxiliary proteins participate in signaling are shown in Figure 24-33. The cytosolic portions of the antigen-specific receptors do not protrude much beyond the
cytosolic leaflet of the plasma membrane, and are incapable of recruiting downstream signaling molecules. Instead, the antigen-specific receptors associate with auxiliary subunits that contain ITAMs. Engagement of the antigen-specific receptors by ligand initiates a series of receptor-proximal events: kinase activation, phosphorylation of one or two tyrosine residues in the ITAM sequence, and subsequent binding to these phosphotyrosines of SH2 domains of adapter proteins that serve as scaffolds for recruitment of other downstream signaling molecules.
FIGURE 24-33 Signal transduction from the T-cell receptor (TCR) and B-cell receptor (BCR). The signal transduction pathways used by the antigen-specific receptors of T cells (left) and B cells (right) are conceptually similar. The initial stages are depicted in this figure; downstream signaling events lead to changes in gene expression that result in proliferation and differentiation of the antigen-stimulated lymphocytes. See text for further discussion. Description An illustration shows a plasma membrane bonded to a T C R and a B C R. The exterior and cytosol are labeled. The T-cell receptor complex consists of several membraneanchored protein domains: a gamma, two etas, and the alpha and beta chains, which are anchored by zeta chains, a delta and the C D 4 complex. The cytoplasmic tails of these proteins are tyrosine-based activation motifs (also known as I T A Ms). The B-cell receptor complex consists of an I g immunoglobulin and I g-alpha and I g-beta complexes embedded in the membrane. I g-alpha and I g-beta also have cytosolic I T A Ms. The steps are as follows: Step 1: Binding of the M H C-peptide complex to the T-cell receptor complex activates the S r c kinases L c k, F-y-n, and L-y-n. Step 2: S r c kinases phosphorylate the cytosolic I T A Ms. L-c-k acts on the T C R whereas F-y-n and L-y-n act on the B C R. Step 3: The phosphorylated I T A Ms recruit non-S r c kinases via the S H 2 domains; these are Z A P-70 in the T C R and S-y-k in the B C R. Step 4: The activated non-S r c kinases recruit and phosphorylate multiple adapter proteins such as L-A-T in the T C R and S L P-65 in the B C R. Thus, signaling pathways are activated such as R a s, J n k, P K C, and N F-A T. One major downstream signal transduction pathway is outlined in Figure 24-33. Engagement of the antigen-specific receptors by ligand activates Src-family cytosolic tyrosine kinases (e.g., Lck in helper T cells; Lyn and
Fyn in B cells; Figure 24-33, step 1 ); these kinases are found in close proximity to or physically associated with the antigen-specific receptors. The active kinases phosphorylate tyrosine residues in the ITAMs (step 2 ). In their phosphorylated forms, the ITAMs bind to SH2 domains of non-Src-family tyrosine kinases (ZAP-70 in T cells, Syk in B cells), leading to activation of these kinases (step 3 ). The ITAMs bind other adapter proteins, which are activated by the non-Src-family tyrosine kinases. As with other phosphotyrosine-triggered signaling pathways (see
Chapter 16) this leads to activation of phosphoinositide-specific phospholipase Cγ and several PI-3 kinases. Subsequent downstream events parallel those described in Chapter 16 for signaling from receptor tyrosine kinases. Antigen-specific receptors on B and T cells are perhaps best characterized as modular receptor tyrosine kinases, with the ligand recognition units and kinase domains carried by separate molecules. Ultimately, signaling via these T cell and B cell antigen-specific receptors initiates transcription programs that determine the fate of the activated lymphocyte: proliferation and differentiation. T cells depend critically on the cytokine interleukin 2 (IL-2) for clonal expansion. Following antigen stimulation of a T cell, one of the first genes to be turned on is that for IL-2 (see Figure 16-19). The T cell responds to its own initial burst of IL-2 and proceeds to make more IL-2, an example of autocrine stimulation and part of a positive feedback loop. An important transcription factor required for the induction of IL-2 synthesis is the NF-AT protein (nuclear factor of activated T cells). This protein is sequestered in the cytoplasm in phosphorylated form and cannot enter the nucleus unless it is de-phosphorylated first. The phosphatase responsible
T Cells Capable of Recognizing MHC Molecules Develop Through a Process of Positive and Negative Selection
is calcineurin, a -activated enzyme. The initial rise in cytosolic leading to activation of calcineurin results from mobilization of ERresident stores triggered by hydrolysis of and the concomitant generation of (see Figure 15-28, steps 2 – 4 ). The immunosuppressant drug cyclosporine inhibits calcineurin activity through formation of a cyclosporine-cyclophilin complex, which binds and inhibits calcineurin. If de-phosphorylation of NF-AT is suppressed, NF-AT cannot enter the nucleus and induce transcription of the IL-2 gene. This precludes clonal expansion of antigen-stimulated T cells and so leads to immunosuppression, arguably the single most important intervention that contributes to the success of organ transplantation involving unrelated donors and recipients (individuals who are genetically different and therefore express different MHC products), referred to as allogeneic tissue transplantation. Although the success of transplantation varies with the organ used, the availability of strong immunosuppressants such as cyclosporine has expanded the possibilities of clinical transplantation enormously. T Cells Capable of Recognizing MHC Molecules Develop Through a Process of Positive and Negative Selection Rearrangement of the gene segments that are assembled to encode a functional T-cell receptor is a stochastic event, completed on the part of
the T cell without any prior knowledge of the MHC molecules with which its receptors must ultimately interact. As in somatic recombination of Ig heavy-chain loci in B cells, the first gene segments to be rearranged in the TCR β chain are the D and J elements; a V segment is then joined to the newly recombined DJ (see Figure 24-32). At this stage of T-cell development, productive rearrangement allows the synthesis of the TCR β chain, which is incorporated into the pre-TCR through association with a surrogate α subunit termed pre-Tα. This dimeric pre-TCR fulfills a function strictly analogous to that of the pre-BCR in B-cell development: it tells the T cell that it has successfully completed a productive β chain rearrangement, with no need for further rearrangements in the β chain gene on the homologous chromosome. The pre-TCR allows clonal expansion of the pre-T cells that successfully underwent TCR β chain rearrangement, and it imposes allelic exclusion to ensure that, as a rule, a single functional TCR β subunit is generated for a given T cell and its descendants. RAG expression subsides until the expansion phase of the pre-T cells is complete, after which it is re-initiated to allow rearrangement of the TCR α locus, ultimately leading to the generation of T cells with a fully assembled TCR containing a rearranged α and β subunit. Figure 24-34 illustrates the analogous steps in the development of T and B cells.
FIGURE 24-34 Comparison of T-cell and B-cell development. Cell fate decisions are executed by receptors containing either the newly rearranged μ chain (pre-BCR) or the newly rearranged β chain (pre-TCR). The pre-BCRs and pre-TCRs serve similar functions: signaling clonal expansion of cells that have successfully undergone rearrangement and allelic exclusion. This phase of lymphocyte development does not require antigen recognition. Both the pre-BCR and pre-TCR include subunits unique to each receptor type and absent from the antigen-specific receptors found on mature lymphocytes: VpreB and λ5 (orange, green) for the pre-BCR; pre-T α (blue) for the pre-TCR. Upon completion of the expansion phase, expression of the gene encoding the remaining subunit of the antigenspecific receptor begins: Ig light chain (light blue) for the BCR; TCR α chain (light red) for the TCR. Lymphocyte development and differentiation occur at distinct anatomic sites, and only fully assembled antigen-specific receptors (BCR, TCR) recognize antigen. Mature lymphocytes are strictly dependent on antigen recognition for their activation. Description The illustration, from left to right shows six stages of B cell and T cell development. The top half represents B cells, the bottom half represents T cells.
Stem cell: shows B and T lineage. Pro-lymphocyte: shows B and T cells. Prelymphocyte: B cell is bonded to a Y shaped antibody labeled pre-B C R on its surface. T cell is bonded to a Pre-T C R on its surface. Immature lymphocyte: B cell is bonded to a Y shaped antibody labeled B C R on its surface. T cell is bonded to a T C R on its surface. Mature lymphocyte: same as the previous stage. Differentiated effector lymphocyte: B cell and T cell release cytokines. A label at the bottom shows that the anatomic site for the first five stages is bone marrow or thymus and the last stage is in the periphery. A second label shows that the first four stages are antigen dependent, the fourth and fifth stages are labeled selfantigen, and the last stage is labeled foreign antigen. The last label shows that the first two stages are early maturation and expansion, the third stage is pre-antigen receptor expression. The fourth and fifth stages are where completion of antigen receptor, selection of receptor repertoire; differentiation occurs. The last two stages are where performance of effector functions occurs. How is the emerging repertoire of T cells, with its diverse set of pre-TCRs, further sorted so that the only surviving T cells are ones whose receptors have a productive interaction with self-MHC–peptide complexes? The random nature of the gene rearrangement process and the enormous variability engendered as a consequence produces a large and diverse set of TCRs. Yet the vast majority of this set of TCRs cannot interact productively with the host MHC products and are therefore useless. Thymocytes bearing useless receptors are not exported to the periphery: they wither on the vine and die. The immune system has developed selection processes to eliminate T cells that make TCRs incapable of a productive interaction with peptide complexes to the organism’s own MHC (self MHC). Selection in the thymus also removes T cells with TCRs that strongly interact with self-MHC–self-peptide complexes, because such T cells may be self-reactive, damaging normal healthy tissue
(autoimmunity). T cells whose TCRs recognize peptide-MHC complexes in the thymus with an affinity that falls between these two extremes receive survival signals and are positively selected. Some may by chance have the ability to bind strongly to an MHC protein complexed to a peptide derived from a foreign protein. Recall that antigen processing and presentation are constitutive processes, so that in the thymus, all selfMHC molecules are necessarily occupied with peptides derived from self proteins. These combinations of self peptides complexed to class I and class II MHC molecules constitute the substrate used by the set of newly generated T-cell receptors to determine what is self and ought to be ignored. The heterogeneity of peptide-MHC complexes presented by different cells in the thymus to T cells undergoing selection makes it highly probable that the T-cell receptor interprets signals not only by the strength and duration of the interaction with a particular peptide-MHC complex, but also in additive fashion, meaning the summation of the binding energies of the different MHC–self-peptide combinations it encounters. These help determine the outcome of the self–nonself selection. This process is called the avidity model of T-cell selection. Newly made T cells are killed by apoptosis only if the appropriate self antigen is represented adequately in the thymus in the form of MHCpeptide complexes. How does the immune system ensure that T cells generated in the thymus learn to ignore self antigens that are not normally expressed at that location? Proteins that are expressed in tissue-specific fashion or after the development of the thymus, such as insulin in the β
T Cells Commit to the CD4 or CD8 Lineage in the Thymus
cells of the pancreas or the components of the myelin sheath in the nervous system, fit this category. A factor called AIRE (autoimmune regulator) allows expression of such tissue-specific antigens in a subset of epithelial cells in the thymus. How AIRE accomplishes this is not known, but it is widely suspected that AIRE directly regulates the transcription of the relevant genes in the thymus and at select sites in secondary lymphoid organs. Defects in AIRE lead to a failure to express these tissue-specific antigens in the thymus. In individuals who do not express AIRE, developing T cells fail to receive the full set of instructions in the thymus that lead to the elimination of potentially self-reactive T cells. As a consequence, these individuals show a bewildering array of autoimmune responses, causing widespread tissue damage and disease. T Cells Commit to the CD4 or CD8 Lineage in the Thymus TCR gene rearrangement coincides with the acquisition of co-receptors. A key intermediate in T-cell development is a thymocyte that expresses both of the TCR co-receptors, CD4 and CD8, as well as a functional TCR-CD3 complex. These cells, called double-positive cells, are found only as developmental intermediates in the thymus. As the T cells mature, they lose either CD4 or CD8 to become single-positive cells. The choice of which co-receptor (CD4 or CD8) to express determines whether a T cell will recognize class I or class II MHC molecules. The question of how a cell is instructed to become a CD8 (class I MHC-restricted) T cell or a CD4 (class II MHC-restricted) T cell is not entirely settled, but
T Cells Require Two Types of Signals for Full Activation
we know that the transcription factors ThPOK and Runx3 play fundamental roles. ThPOK and Runx3 are regulated by TCR signaling. Cells that transiently express ThPOK will commit to the CD4 lineage and repress Runx3 expression. On the other hand, if ThPOK expression is not induced, Runx3 expression is high, and cells commit to the CD8 lineage. In mice, a loss-of-function mutation in the ThPOK gene abrogates CD4 Tcell development, and all thymocytes become CD8-expressing T cells. Another type of CD4 T cell also develops in the thymus, named natural (or thymically derived) regulatory T cells (Treg). Their function differs from that of CD4 helper T cells, as will be described below. The thymus also gives rise to other less numerous types of T cells, such as invariant natural killer T cells (iNKT) that express the NK cell marker NK1.1 and are selected based on their ability to bind the nonclassic MHC molecule CD1, which presents lipid antigens. Also generated are intraepithelial lymphocytes that will colonize the mucosal surfaces of the intestine. After the final stages of maturation, T cells of all types are exported to the peripheral lymphoid organs. T Cells Require Two Types of Signals for Full Activation All T cells require a signal via their TCR for activation, but that signal is not sufficient: the T cell also needs co-stimulatory signals that are present on an antigen-presenting cell. To perceive these co-stimulatory signals, T cells carry on their surface several additional receptors, of which the CD28
molecule is the best known example. It is these regulatory signals that are the basis of cancer immunotherapies using monoclonal antibodies that target certain of these signaling proteins: CTLA4, PD-1, and PD-L1. These therapies are discussed in Section 24.6. The CD28 protein on a T cell interacts with CD80 and CD86, two surface glycoproteins on the professional APC with which the T cell interacts. Expression of CD80 and CD86 increases when these APCs have themselves received the proper stimulatory signals, for example, by engagement of their Toll-like receptors (TLRs). The signals delivered to T cells via CD28 synergize with signals that emanate from the TCR when bound to its cognate self-MHC–peptide antigen complex, all of which are required for full T-cell activation (Figure 24-35, steps 1 – 3 ).
FIGURE 24-35 Signals involved in T-cell activation and its termination. The two-signal model of full T-cell activation: signal 1 is recognition of an MHC-peptide complex by the Tcell receptor (step 1 ), and signal 2 is recognition by CD28 of co-stimulatory molecules (CD80, CD86) on the surface of an antigen-presenting cell (step 2 ). Full activation, in turn, leads to increased expression of CTLA4 on the surface of T cells (step 3 ). CTLA4 binds CD80 and CD86, leading to inhibition of the T-cell response (step 4 ). Because the affinity of CTLA4 for CD80 and CD86 is greater than that of CD28, T-cell activation is eventually terminated. Note that after signal 1, if co-stimulation is not provided, the newly engaged T cell becomes unresponsive (anergic). Description The steps are as follows: Step 1: Signal via T C R (Signal 1): The illustration shows an A P C with an M H C presenting an antigen to the T C R of the T cell. An arrow inside the T cell from the T C R points at a text that reads signal 1. The T cell has four other receptors. Step 2: C D 28 on T cell interacts with C D 80, C D 86 on A P C (Signal 2): C T L A 4 is produced inside the T-cell. Step 3: Activation and proliferation of T cell: An autocrine loop occurs where I L-2 is secreted, stimulating the T-cell’s I L-2 receptors. Step 4: C T L A 4 is moved to the cell surface and competes with C D 28 for C D 80 and 86 binding. Once C T L A outcompetes C D 28, the signal is terminated. T cells, once activated, also express receptors that provide an attenuating or inhibitory signal upon recognition of these very same co-stimulatory molecules, providing negative feedback regulation. The CTLA4 protein, whose expression in T cells is induced only upon activation, competes with CD28 for binding of CD80 and CD86. Because the affinity of CTLA4 for the CD80 and C86 proteins is higher than that of CD28, the inhibitory
Cytotoxic T Cells Carry the CD8 Co-Receptor and Are Specialized for Killing
signals provided through CTLA4 will ultimately overwhelm the stimulatory signals coming via CD28. Co-stimulatory molecules can thus be stimulatory or — as was discovered later without adjusting the nomenclature — inhibitory, and they therefore provide an important means of controlling the activation status and duration of a T-cell response. Cytotoxic T Cells Carry the CD8 CoReceptor and Are Specialized for Killing As we have seen, cytotoxic T cells (CTLs) generally express on their surfaces the TCR co-receptor glycoprotein called CD8. These T cells kill target cells that display their cognate class I MHC–peptide combinations and do so with exquisite sensitivity: a single MHC-peptide complex suffices to allow a properly activated CTL to kill the target cell that bears it. The mechanism of killing by CTLs involves two classes of proteins that act synergistically: perforins and granzymes (Figure 24-36). Perforins exhibit homology to the terminal components of the complement cascade composing the membrane attack complex (see Figure 24-5). Like the attack complex, perforins attach to a membrane and form pores up to 20 nm across within the membrane. The pores cause electrolytes and other small solutes to leak from the cell, contributing to cell death. Granzymes are delivered to and are presumed to enter the target cell, probably via the
pores generated by perforin. Granzymes are serine proteases that activate caspases and so propel the target cell on a path of programmed cell death (apoptosis; see Chapter 22). Perforins and granzymes are packaged into cytotoxic granules, which are stored inside the cytotoxic T cell. Upon binding of the T-cell receptor to its cognate class I MHC–antigen complex, signal transduction from the TCR leads to release of the cytotoxic granules and their contents into the extracellular space that is formed between the cytotoxic T cell and the target cell, called the synaptic cleft. How the T cell avoids being killed upon release of granzymes and perforins into the synapse is unknown. Natural killer cells also exert cytotoxic activity and likewise rely on perforins and granzymes to kill their targets (see Figure 24-6).
T Cells Secrete an Array of Cytokines That Provide Signals to Other Immune-System Cells
FIGURE 24-36 Perforin- and granzyme-mediated cell killing by cytotoxic T cells. Upon recognition of a target cell (step 1 ), a cytotoxic T cell forms a tight antigen-specific contact with the target cell. Tight contact results in the formation of a synaptic cleft, into which the contents of cytotoxic granules, including perforins and granzymes, are released (step 2 ). Perforins form pores in the membranes onto which they adsorb, and granzymes are serine proteases that enter through the perforin pores (step 3 ). Perforins are believed to act not only at the surface of the target cell, but also at the surface of its endosomal compartments after the perforin molecules have been internalized from the cell surface (step 4 ). Once in the cytoplasm, the granzymes activate caspases, which initiate programmed cell death (step 5 ). Description The illustration shows a zoomed view of the synaptic cleft between a cytotoxic T cell and a target cell. The steps are as follows. Step 1: Class 1 M H C and antigen complex of the target cell and T C R of the cytotoxic T cell bind in the synaptic space between a target cell at the top, and a cytotoxic T cell at the bottom. Step 2: a lysosome of the T cell has cytotoxic granule (lysosome-related) with perforin (green cylinders) and granzymes (red circles) inside, being brought to the surface of the T cell to be released. Step 3: the perforins make a bundle and transport the granzymes into the target cell. The granzymes are bound to the Y shaped receptors of the target cell. Step 4 a: some perforins get accumulated at the groove formed at the target cell. Step 4 b: the perforins and granzymes are engulfed by target cell endosome. It leads to caspase activation. Step 5: cell death.
T Cells Secrete an Array of Cytokines That Provide Signals to Other Immune-System Cells Many lymphocytes and other cells in lymphoid tissues produce cytokines. These small secreted proteins control lymphocyte activity by binding to specific cytokine receptors on the surface of a lymphocyte and initiating a transcriptional program that allows the lymphocyte to either proliferate or differentiate into an effector cell ready to exert cytotoxic (cytotoxic T cells), helper (helper T cells), or antibody-secreting activity (B cells). Cytokines that are produced by or act primarily on leukocytes are called interleukins. More than 35 interleukins have been recognized and molecularly characterized. Interleukin receptors share structural similarity with each other and with receptors for erythropoietin; those interleukins whose structures are most closely related can be recognized by their cognate receptors. The interleukin-2 receptor (Figure 16-19b) is particularly well characterized. Interleukin 2 (IL-2), a T-cell growth factor, is one of the first cytokines produced when T cells are stimulated. IL-2 acts as an autocrine (self-acting) and paracrine (acting on neighboring cells) growth factor and drives clonal expansion of activated T cells. Interleukin 4 (IL-4), which is produced by helper T cells, induces activated B cells to proliferate and to undergo class-switch recombination and somatic hypermutation. Interleukin 7 (IL-7), produced by stromal cells in the bone marrow, is essential for development of T and B cells. Both IL-7 and IL-15 play a role in the maintenance of memory cells, which are
Helper T Cells Are Divided into Distinct Subsets Based on Their Cytokine Production and Expression of Surface Markers
antigen-experienced T cells that may be called upon when re-exposure to antigen occurs; these memory cells then rapidly proliferate and deal with the reinvading pathogens. The receptors for IL-2, IL-4, IL-7, and IL-15 all rely on a common subunit for signal transduction, the common γ chain , with α (IL-2, IL-15) and β subunits (IL-2, IL-4, IL-7, IL-15) providing ligand specificity (Figure 16-19b). Genetic defects in the result in nearly complete failure of lymphocyte development, illustrating the importance of these cytokines (and their receptors) not only during the effector phase of an immune response, but also in the course of lymphocyte development, where IL-7 in particular plays a key role. By knocking out the gene that encodes , immunodeficient mice can be generated that accept human tissues and tumors as grafts. Such mice have become very useful models to study tumor immunology and to create a setting in which a partial human immune system can be installed in mice, for example, to study HIV, which does not infect mouse lymphocytes. The mechanism of signal transduction by cytokine receptors through the JAK/STAT pathway is described in Chapter 16 (reviewed in Figure 1619a). Among the many genes under the control of interleukins and the STAT pathway are those that encode suppressors of cytokine signaling, or SOCS proteins. These proteins, which are themselves induced by cytokines, bind to the activated form of JAKs and target them for proteasomal degradation (see Figure 16-21b). Helper T Cells Are Divided into Distinct Subsets Based on Their
Cytokine Production and Expression of Surface Markers CD4-expressing T cells are helper T cells that provide assistance to B cells and guide their proliferation and differentiation into plasma cells. This function requires both the production and secretion of cytokines such as IL-4 as well as direct contact between the helper T cell and the B cell to which it provides help. A second class of helper T cell has as its major function secreting the cytokines that contribute to the establishment of an inflammatory environment. Multiple subtypes of such inflammatory T cells are categorized based on the spectrum of different cytokines they produce and their respective roles in regulating immune responses. Whereas all activated T cells can produce IL-2, other cytokines are produced only by particular helper T-cell subsets. Helper T cells classified as cells secrete interferon γ and tumor necrosis factor (TNF), which can activate macrophages and stimulate an inflammatory response. Referred to also as inflammatory T cells, cells nonetheless play an important role in antibody production, notably facilitating the production of complement-fixing antibodies such as IgG1 and IgG3. cells secrete IL-4 and IL-10, and through production of IL4, play an important role in B-cell responses that involve class switching to the IgG1 and IgE isotypes (see Section 24.3). Recall that in B cells, the induction of activation-induced deaminase (AID) prepares the B cell for
Innate Lymphoid Cells Regulate Inflammation and the Overall Immune Response
class-switch recombination and somatic hypermutation. This induction is a consequence of the precise mixture of cytokines produced by helper T cells and the binding of CD40, a surface membrane protein on the activated T cell, to a protein on the B-cell surface, called CD40 ligand or CD40L. Conventional helper T cells can also differentiate into cells, which produce IL-17, and into induced regulatory T cells (induced Tregs, distinct from the natural Tregs generated in the thymus). Both types of Treg cells attenuate immune responses by exerting a suppressive effect on other types of T cells. Natural Tregs restrain the activity of potentially selfreactive T cells and are important in maintaining peripheral tolerance (the absence of an immune response to self antigens), whereas induced Tregs are believed to regulate excessively strong immune responses against foreign antigens. cells are important in defense against bacteria (extracellular bacteria in particular) and also play a pathogenic role in autoimmune diseases. Innate Lymphoid Cells Regulate Inflammation and the Overall Immune Response The immune response against a pathogen exerts a selective pressure that allows the emergence of pathogen variants no longer recognized by antibodies or T cells. Many viruses with comparatively small genomes, such as the virus that causes influenza or human immunodeficiency virus,
replicate by means of error-prone polymerases that generate mutant descendants with amino acid substitutions, some of which escape immune recognition because the proper epitope is no longer expressed as a consequence of such mutations. Viruses with large genomes such as herpesviruses use other means of immune escape, for example, through down-regulation of Class I MHC products. This renders the infected cell refractory to killing by CD8 T cells. A similar situation applies to many cancerous cells, which often show starkly reduced expression of Class I MHC products. Natural killer (NK) cells can sense the absence of Class I MHC products because NK cells express receptors that recognize Class I MHC products and shut off NK cell function. A lack of Class I MHC products on the surface of a virus-infected cell or on a tumor cell relieves the inhibition mediated by such receptors and allows the NK cell to kill the target. Because of this, the combined action of CD8 T cells and NK cells affords a level of protection against virus-infected and tumor cells that is more effective than either cell type can confer on its own. Although NK cells were discovered several decades ago, they are now referred to as a member of the group of innate lymphoid cells or ILCs. Such cells resemble conventional lymphocytes in many of their functional properties such as the types of cytokine they can produce, but they do not make use of gene rearrangements to create the receptors characteristic of the cells of the adaptive immune system: the B and T cells. ILCs are important in regulating inflammation and in regulatory control of the immune response more generally. They are poised to rapidly secrete cytokines when activated and their preferred location at mucosal sites suggests a role in the control of various infections. The group 1 ILC
Leukocytes Move in Response to Chemotactic Cues Provided by Chemokines
lineage comprises ILCs such as NK cells that produce type 1 cytokines, notably interferon-γ and TNF. ILC2s express interleukin-5 (IL-5) and IL- 13, not unlike conventional CD4 T cells. ILC2s, together with CD4 T cells, protect against helminth (parasitic worms) infection. ILC3s were first defined as intestinal lymphoid cells that express the NK cell– activating receptor NKp46 but are otherwise quite distinct from NK cells (ILC1s). ILC3s express IL-17A and IL-22. Lymphoid tissue-inducer (LTi) cells are an ILC subset that appears to be closely related to ILC3s, but their exact relationship remains controversial. Together, LTi cells and ILC3s have been classified as group 3 ILCs. Leukocytes Move in Response to Chemotactic Cues Provided by Chemokines Interleukins tell lymphocytes what to do by eliciting a transcriptional program that allows lymphocytes to acquire specialized effector functions. Chemokines, on the other hand, tell leukocytes where to go. Many cells emit chemotactic cues in the form of chemokines. When tissue damage occurs, resident fibroblasts produce a chemokine, IL-8, that attracts neutrophils to the site of damage. The regulation of lymphocyte traffic within lymph nodes is essential for dendritic cells to attract T cells and for T cells and B cells to meet. These trafficking steps are all controlled by chemokines.
There are approximately 40 distinct chemokines and more than a dozen chemokine receptors. One chemokine may bind to more than one receptor, and a single receptor can bind several different chemokines. This flexibility creates the possibility of generating a combinatorial code of chemotactic cues of great complexity. This code is used to guide the navigation of leukocytes from where they are generated, in the bone marrow, into the bloodstream for transport to their target destination. Some chemokines direct lymphocytes to leave the circulation and take up residence in lymphoid organs. These migrations contribute to the population of lymphoid organs with the required sets of lymphocytes. Because these movements occur as part of normal lymphoid development, such chemokines are referred to as homeostatic chemokines. Those chemokines that serve the purpose of recruiting leukocytes to sites of inflammation and tissue damage are referred to as inflammatory chemokines. Chemokine receptors are G protein–coupled receptors that function as an essential component of the regulation of cell adhesion and cell migration. Leukocytes that travel through blood vessels do so at high speed and are exposed to high hydrodynamic shear forces. For a leukocyte to traverse the endothelium and take up residence in a lymph node or seek out a site of infection in tissue, it must first slow down, a process that requires interactions of glycoprotein surface receptors called selectins with their ligands on the surfaces of leukocytes, which are mostly carbohydrate in nature. If chemokines are adsorbed to the extracellular matrix and if the leukocyte possesses a receptor for those chemokines, activation of its
chemokine receptor elicits a signal that allows integrins carried by the leukocyte to undergo a conformational change. This change results in an increase in the affinity of the integrin for its ligand and causes firm arrest of the leukocyte. The leukocyte may now exit the blood vessel by a process known as extravasation (see Figure 20-40). KEY CONCEPTS OF SECTION 24.5 T Cells, T-Cell Receptors, and T-Cell Development The antigen-specific T-cell receptors are dimeric proteins consisting of α and β subunits or γ and δ subunits. T cells occur in at least two major classes defined by their expression of the glycoprotein co-receptors CD4 and CD8 (see Figure 24-31). Cells that use class I MHC molecules as the molecular guideposts for antigen recognition (restriction elements, in immunological parlance) carry CD8; those that use class II MHC molecules carry CD4. These classes of T cells are functionally distinct: CD8 T cells are cytotoxic T cells; CD4 T cells provide help to B cells and are an important source of cytokines. Genes encoding the TCR subunits are generated by somatic recombination of V and J segments (α chain) and of V, D, and J segments (β chain); their rearrangement obeys the same rules as does rearrangement of Ig genes in B cells (see Figure 24-32). Rearrangement of TCR genes occurs when the lymphocytes are present in the thymus and only in those cells destined to become T lymphocytes. A complete T-cell receptor includes not only the α and β subunits responsible for antigen and MHC recognition, but also the accessory subunits referred to as the CD3 complex, which is required for signal transduction. Each subunit of the CD3 complex carries in its cytoplasmic tail one or three ITAM domains; when phosphorylated, these ITAMs recruit adapter proteins involved in signal transduction (see Figure 24-33). In the course of T-cell development, the TCR β locus is rearranged first. If that locus encodes a functional β subunit, it is incorporated with the pre-Tα chain into a preTCR (see Figure 24-34). Like the pre-BCR, the pre-TCR mediates allelic exclusion, that is, the expression of a functionally rearranged T-cell receptor encoded by only one of the two alleles and proliferation of those cells that successfully underwent TCRβ rearrangement. Developing T cells that fail to recognize self-MHC molecules die for lack of survival signals. T cells that interact too strongly with self-peptide–self-MHC complexes
encountered during development are instructed to die (negative selection); those that have intermediate affinity for self-peptide–self-MHC complexes are allowed to mature (positive selection) and are exported from the thymus to the periphery. Innate lymphoid cells (ILCs) share properties with lymphocytes, notably the types of cytokines they release, without reliance on antigen-specific receptors, unlike T lymphocytes. T cells are instructed where to go (cell migration) through chemotactic signals in the form of chemokines. Receptors for chemokines are G protein–coupled receptors that show some promiscuity in terms of their binding of chemokines. The complexity of chemokine–chemokine receptor binding allows precise regulation of leukocyte traffic, both within lymphoid organs and in the periphery.
Toll-Like Receptors Perceive a Variety of Pathogen-Derived Macromolecular Patterns
24.6 Collaboration of ImmuneSystem Cells in the Adaptive Response An effective adaptive immune response requires the presence of B cells, T cells, and antigen presenting cells (APCs). For B cells to execute classswitch recombination and somatic hypermutation — prerequisites for production of high-affinity antibodies — they require help from activated T cells. These T cells, in turn, can be activated only by professional APCs such as dendritic cells. Dendritic cells sense the presence of pathogens through Toll-like receptors and other pattern-recognition receptors, such as the cell-surface mannose-binding protein and other related lectins that can recognize polysaccharides and carbohydrate determinants. The interplay between components of the innate and adaptive immune systems is therefore a very important aspect of adaptive immunity. This layered, interwoven nature of innate and adaptive immunity ensures a rapid early response of immediate protective value and primes the adaptive immune system for a specific response to any persisting pathogen. In this section, we describe how these various elements are activated and how the relevant cell types interact. Toll-Like Receptors Perceive a Variety of Pathogen-Derived Macromolecular
Patterns An important part of the innate immune system is its ability to immediately detect the presence of a microbial invader and respond to it. This response includes direct elimination of the invader, but it also prepares the mammalian host for a proper adaptive immune response, particularly through activation of professional APCs. These APCs are positioned throughout the epithelia (airways, gastrointestinal tract, genital tract), where contact with pathogens is most likely to occur. In the skin, a network of dendritic cells called Langerhans cells makes it virtually impossible for a pathogen that breaches this barrier to avoid contact with these professional APCs. Dendritic cells and other professional APCs detect the presence of bacteria and viruses through members of the Tolllike receptor (TLR) family. These proteins are named after the Drosophila protein Toll because of the structural and functional homology between them. Drosophila Toll was discovered because of its important role in dorsal/ventral patterning in the fruit fly, but related receptors are now recognized as capable of triggering an innate immune response in insects as well as in vertebrates. TLR Structure Toll itself and all TLRs possess a sickle-shaped extracellular domain, composed of leucine-rich repeats, that is involved in ligand recognition. The cytoplasmic portion of a TLR contains a domain responsible for the recruitment of adapter proteins to enable signal transduction. The
signaling pathways engaged by TLRs have many of the same components (and outcomes) as those used by receptors for the cytokine IL-1 (Figure 24-37; see also Figure 16-31).
FIGURE 24-37 Toll-like receptor activation. The extracellular portions of TLRs recognize ligands of diverse chemical nature (nucleic acids, lipopolysaccharides). The cytoplasmic portions of the TLRs, called TIR (BToll/IL-1β receptor homology) domains, associate with the adapter protein MyD88, present in six copies per complex, and recruit two types of
kinases, both members of the IRAK family. These complex interactions are maintained by TIR domains and death domains (DD) as shown in the figure. The assembled complex on the cytoplasmic side is referred to as the myddosome. As depicted in Figure 16-31, among the proteins bound to IRAK2 is TRAF6, an E3 ubiquitin ligase. Because of extensive TRAF6-TRAF6 interactions, hundreds of copies of this TLR complex become cross-linked together, forming a giant signalsome in the plasma membrane. [Republished with permission from Annual Reviews, from J. Y. Kang and J.-O. Lee, 2011, “Structural Biology of the Toll-Like Receptor Family,” Annu. Rev. Biochem. 80(1):917–941; permission conveyed through Copyright Clearance Center, Inc.] Description The illustration shows two toll-like receptors (T L R) domains in the extracellular space bonded by ligands. The cytoplasmic part contains several toll-interleukin-1-beta receptor homology domains (known as T-I-Rs). Interactions between the death domain and the T I R and death domain and the kinases are represented. The order of the T I Radaptor protein or kinase complexes is M y D 88, I R A K 4, and I R A K 2, moving from the membrane into the cytosol. The Drosophila Toll protein interacts with its ligand, Spaetzle, itself the product of a proteolytic conversion initiated by components of the cell walls of fungi that prey on Drosophila. In the fly, activation of Toll unleashes a signaling cascade that ultimately controls the transcription of genes that encode antimicrobial peptides. The activated receptor at the cell surface communicates with the transcriptional apparatus by means of a series of adapter proteins that activate downstream kinases interposed between Toll, the TLRs, and the transcription factors that are activated by them. A key step is the ubiquitin-dependent proteasomal degradation of the Cactus protein. Its removal allows the protein Dif to enter the nucleus and initiate transcription. This pathway is highly homologous in its
operation and structural composition to the NF-κB pathway in mammals (see Figure 16-30). Diversity of TLRs There are approximately a dozen mammalian TLRs that can be activated by various microbial products and are expressed by a variety of cell types; TLR function is crucial for the activation of dendritic cells and macrophages. Neutrophils also express TLRs. The microbial products recognized by TLRs include macromolecules found in the cell envelopes of bacteria, such as lipopolysaccharides, flagellins (subunits of bacterial flagella), and bacterial lipopeptides. Direct binding of at least some of these macromolecules to TLRs has been demonstrated in crystallographic analyses of the relevant complexes. The presence of distinct classes of microbial molecules is sensed by distinct TLRs: for example, TLR4 binds lipopolysaccharides; heterodimers of TLR1 and 2 and TLR2 and 6 bind lipopeptides; and TLR5 binds flagellin. Recognition of all bacterial envelope components by these TLRs occurs at the cell surface. A second set of TLRs — TLR3, TLR7, and TLR9 — sense the presence of pathogen-derived nucleic acids. They do so not at the cell surface but rather within the endosomal compartments where these receptors reside. Mammalian DNA is methylated at many CpG dinucleotides, whereas microbial DNA generally lacks this modification. TLR9 is activated by unmethylated, CpG-containing microbial DNA. Similarly, double-stranded RNA molecules, replication intermediates of RNA viruses, are present in these virus-infected cells and lead to activation of TLR3. Finally, TLR7
responds to the presence of certain single-stranded RNAs. Thus the full set of mammalian TLRs allows the recognition of a variety of macromolecules that are diagnostic for the presence of bacterial, viral, or fungal pathogens and parasites such as malaria. Inflammasome A variety of intracellular receptors for RNA and DNA have been described that recognize viral RNA and are structurally distinct from TLRs. The list of cytoplasmic receptors capable of recognizing DNA, both pathogenderived and host DNA-derived, continues to grow. Several of these receptors participate in the assembly of the inflammasome (Figure 24-38), whose major function is the conversion of the enzyme precursor procaspase-1 to the active caspase-1. Caspase-1 is a protease that converts pro-IL-1β into active IL-1β, a cytokine that elicits a strong inflammatory response. Mature IL-1β lacks a signal sequence and is released from producer cells via an unconventional secretory route that involves the formation of pores composed of gasdermin proteins (Figure 24-39). Gasdermins can be converted by caspases from their inactive precursors to the active form to yield pores of a size that allows passage of proteins. In so doing, these pores not only contribute to the release of IL-1β but also to certain forms of cell death called pyroptosis (“fiery death,” referring to the inflammation that occurs). The core components of inflammasomes are proteins with leucine-rich repeats, members of the neuronal inhibitors of apoptosis (NALP) family of proteins, and the NOD proteins, so named because of the presence of a nucleotide oligomerization domain. Ipaf-1, a protein related to the Apaf-1 molecule involved in apoptosis (see Chapter
22), recruits an adapter protein, ASC, to mediate complex formation with procaspase-1. Assembly of this multisubunit complex allows the conversion of procaspase-1 to active caspase-1 and of pro-IL-1β to IL-1β. Many seemingly unrelated substances can induce assembly and activation of an inflammasome, including silica, uric acid crystals, and asbestos particles. Accordingly, inhibition of the inflammasome signaling cascade, or blocking of the receptor for IL-1β, has shown therapeutic promise for a variety of inflammatory diseases.
FIGURE 24-38 The inflammasome. The inflammasome is a type of complex that senses the presence of cytoplasmic, pathogen-derived nucleic acids. It can also be activated by other danger signals including particulate matter such as uric acid crystals or asbestos. There are close to two dozen proteins that can participate in the formation of these complexes to yield inflammasomes of different composition, two of which are represented here. The fully assembled inflammasome activates caspase-1, the enzyme that converts proIL-1β into the active, cleaved cytokine IL-1β. NALP3, also referred to as NLRP = a member
of the protein family characterized by the presence of NACHT, LRR, and PYD domains; ASC = apoptosis-associated Speck-like protein containing a CARD (caspase recruitment domain). Description The illustration shows N A L P 3 inflammasome which forms from N A L P 3 on the presence of A S C, and procaspase-1. The I P A F inflammasome which forms from I P A F and procaspase-1. Both inflammasomes activate caspase-1, which converts prointerleukin-1-beta into interleukin-1-beta. They also both come together into a 6 petal like group called inflammasome.
FIGURE 24-39 Gasdermin pore structure. Structure of the gasdermin A3 pore determined by cryoelectron microscopy. Gasdermin subunits are converted by caspases upon activation of inflammasomes. The activated subunits self-assemble into a multimeric structure, shown here for 27 subunits of the gasdermin A3 complex in top and side views. The β barrel domain interacts with the fatty acyl chains of the lipid bilayer and spans the width of the membrane. The resulting pore allows the escape of cellular content, thus contributing to cell death. The large pores formed by an assembled gasdermin complex may
facilitate the release into the extracellular environment of intracellularly activated cytokines such as IL-1, a protein that lacks a signal sequence typical of secretory proteins. [Republished with permission from Springer Nature, from J. Ruan et al., 2018, “Cryo-EM Structure of the Gasdermin A3 Membrane Pore,” Nature, 557(7703):62–67; permission conveyed through Copyright Clearance Center, Inc.] Description The top of the illustration has the label 27-fold pore. This micro-drawing is a very detailed three dimensional ribbon diagram in the shape of a ring. The interior of the ring has a diameter of 180 angstroms. A label inside the ring notes 108 beta-strands. The measurement line at the right shows the outer diameter is 280 angstroms. A side view is shown with a depth of 70 angstroms. The top is labeled globular domain and the bottom is labeled beta-barrel domain. TLR Signaling Cascade As shown in Figure 24-37, engagement of mammalian TLRs leads to recruitment of the adapter protein MyD88, which in turn allows the binding and activation of IRAK (interleukin 1 receptor-associated kinase) proteins. After IRAK phosphorylates TNF-receptor–associated factor 6 (TRAF6), several downstream kinases come into play, leading to release of active NF-κB, a transcription factor, for translocation from the cytoplasm to the nucleus, where NF-κB activates various target genes (see Figure 1630). These target genes include those encoding the cytokines IL-1β and IL6, which as we saw contribute to inflammation, as well as the genes for TNF and IL-12. Expression of type I interferons, small proteins with antiviral effects, is also turned on in response to TLR signaling.
Engagement of Toll-Like Receptors Leads to Activation of Antigen-Presenting Cells
Cell responses to TLR signaling are quite diverse. For professional APCs, these responses include production of cytokines as well as the upregulation of co-stimulatory molecules, the surface proteins important for full activation of T cells that have yet to encounter antigen (referred to as “naive” T cells). TLR signaling allows dendritic cells to migrate from where they encounter a pathogen to the lymph nodes draining that area, where they can interact with naive lymphocytes. Not all activated TLRs evoke an identical response. In dendritic cells, each activated TLR controls production of a particular set of cytokines. For each engaged TLR, the combination of co-stimulatory molecules and the cytokine profile induced by TLR engagement creates a unique activated-dendriticcell phenotype. The identity of the microbial antigen encountered by a dendritic cell determines the pattern of the TLRs that will be activated. This pattern, in turn, shapes the differentiation pathways of activated dendritic cells, influencing the cytokines produced, the surface molecules displayed, and the chemotactic cues to which the dendritic cells respond. The mode of activation of a dendritic cell and the cytokines it produces in response create a unique local microenvironment in which T cells differentiate. Within this microenvironment, the neighboring T cells acquire the functional characteristics required to fight the infectious agent that led to engagement of the TLRs in the first place. Engagement of Toll-Like Receptors Leads to Activation of AntigenPresenting Cells
Professional APCs engage in continuous endocytosis, and in the absence of pathogens, they display at their surface class I and class II MHC molecules loaded with peptides derived from self proteins. In the presence of pathogens, the TLRs on these cells become activated, inducing the APCs to become motile. The cells detach from the surrounding extracellular matrix and start to migrate in the direction of the draining lymph node, following the directional cues provided by chemokines. An activated dendritic cell, for example, reduces its rate of antigen acquisition, up-regulates the activity of endosomal and lysosomal proteases, and increases the transfer of class II MHC–peptide complexes from the loading compartments to the cell surface. Finally, activated professional APCs up-regulate expression of the co-stimulatory molecules CD80 and CD86 (see Figure 24-35), which will allow these APCs to activate T cells more effectively. The initial contact of a professional APC with a pathogen thus results in its migration to the draining lymph node in a state that is fully capable of activating a naive T cell. Antigen is displayed in the form of peptide-MHC complexes, co-stimulatory molecules are abundantly present, and cytokines are secreted that assist in setting up the proper differentiation program for the T cells to be activated. Antigen-laden dendritic cells engage antigen-specific T cells, which respond by proliferating and differentiating. The cytokines produced in the course of this priming reaction determine whether a CD4-expressing T cell will polarize toward an inflammatory or a classic helper T cell phenotype. If engagement occurs via class I MHC molecules, a CD8-expressing T cell may develop from a precursor cytotoxic T cell into a fully active cytotoxic
Production of High-Affinity Antibodies Requires Collaboration Between B and T cells
T cell. Activated T cells are motile and move through the lymph node in search of B cells or enter the circulation to execute effector functions elsewhere in the body. Production of High-Affinity Antibodies Requires Collaboration Between B and T cells To generate the high-affinity antibodies that are necessary for tight binding to antigens and effective neutralization of pathogens, B cells require assistance from T cells. B-cell activation requires both a source of antigen to bind to the B-cell receptor (BCR) and the presence of activated antigen-specific T cells (Figure 24-40).
FIGURE 24-40 Collaboration between T and B cells is required to initiate the production of antibodies. Left: Activation of T cells by means of antigen-loaded, antigenpresenting dendritic cells (DCs). Right: Antigen acquisition by and subsequent activation of B cells to proliferate and differentiate into antibody-secreting plasma cells. Step 1 :
Professional antigen-presenting cells (dendritic cells, B cells) acquire antigen. Step 2 : Professional APCs internalize and degrade the antigen and present the resulting antigenderived peptides, bound to MHC proteins. T-cell activation occurs when dendritic cells present antigen to T cells with receptors that bind the antigen peptide-MHC complex. Step 3a : Activated T cells engage B cells that express a BCR (surface-bound immunoglobulin) for the antigen and that (like other APCs) present antigen to T cells through peptide-MHC complexes displayed on the surface of the B cell. Step 3b : T cells that are persistently activated initiate expression of the CD40 ligand (CD40L), a prerequisite for B cells becoming fully activated and turning on the enzymatic machinery (including AID) to initiate class-switch recombination and somatic hypermutation. Step 4a : A B cell that receives the appropriate instructions from CD4 helper T cells becomes an IgM-secreting plasma cell. Step 4b : A B cell that receives signals from activated CD4 helper T cells in the form of CD40-CD40L interactions and the appropriate cytokines can switch to other immunoglobulin isotypes and engage in somatic hypermutation, eventually differentiating into a plasma cell. Description The steps are as follows. Step 1: A dendritic cell requires an antigen and moves to the lymph node. B cells also acquire antigens. The antigens are represented by a sphere having T cell epitope (binds to M H C, recognized by T C R) and B cell epitope (binds to B C R). Step 2: The dendritic cell processes and presents the antigen to C D 4 T cells. Meanwhile, the B cell processes the antigen and displays it on its surface as part of the class two M H C-peptide complex. Step 3a: The T cell interacts with the B cells via the T C R-M H C interaction, activating the T cell. Step 4 a: The B cell is activated and becomes a plasma cell, secreting I g M. Step 3 b: The T cell interacts with a B cell via the T C R-M H C interaction and C D 40 L-C D 4 0 interaction, leading to the release of I L-4 from the B cell and increasing the production of activation-induced cytidine deaminase (also known as A I D).
Step 4 b. The B cell can undergo hypermutation, class-switch recombination, and produce high-affinity immunoglobulin. Soluble antigen reaches the lymph node from the periphery by transport through the afferent lymphatic vessels (see Figure 24-7). Bacterial growth is accompanied by the release of microbial products that can serve as antigens. If the infection is accompanied by local tissue destruction, activation of the complement cascade results in the killing of bacteria and the concomitant release of bacterial proteins, which are also delivered via the lymphatic vessels to the draining lymph node. Antigens covalently modified by proteins of the complement system are superior to their unmodified counterparts in the activation of B cells through engagement of complement receptors on those cells, which serve as co-receptors for the BCR. B cells that acquire antigen via their BCRs internalize the antibodyantigen complex by endocytosis and proteolytically degrade the antigen into peptides for presentation to T cells via the class II MHC pathway (see
Figure 24-28). B cells that have successfully engaged antigen thus convert it into a call for T-cell help in the form of a class II MHC–peptide complex expressed on the cell surface (Figure 24-40, step 2 ). Importantly, the sequence of amino acids on the antigen molecule recognized by the B-cell receptor (a membrane-anchored immunoglobulin) may be quite distinct from the peptide ultimately displayed on the B-cell surface in association with a class II MHC molecule. As long as the B-cell epitope on the antigen and the class II MHC–presented peptide — a T-cell epitope — are physically part of the same antigen, whether linked covalently or held
together through noncovalent interactions, successful B-cell differentiation and antibody production can be initiated by T-cell help. This concept of linked recognition — namely, the engagement of antigen by the B cell’s BCR and the display of antigen-derived fragments to T cells by class II MHC molecules — explains why there is a minimum size for molecules that can be used to successfully elicit a high-affinity antibody response, as we will see below. Such immunogenic molecules must fulfill several criteria: they must contain the epitope that binds to the B-cell receptor, they must undergo endocytosis and proteolysis, and a proteolyzed fragment of the protein must bind to the allelic class II MHC molecules available in order to be presented as a class II MHC–peptide complex, which serves as a call for T-cell help. Often investigators would like to generate an antibody (either polyclonal or monoclonal) that can recognize a short peptide fragment from a larger protein. Such antibodies can be used for a variety of experiments, including detection of a target molecule by immunofluorescence or immunoprecipitation. These antibodies are called anti-peptide antibodies. If the peptide alone is used as an immunogen [injected into an animal (e.g., a rabbit, goat, or mouse) to generate antibodies], it probably will not successfully induce robust antibody formation, even though there may be B cells with BCRs that can bind tightly to the peptide. The reason is that it is unlikely that those B cells will be able to generate a complex of a class II MHC with that same peptide and recruit helper T cells to drive proliferation and affinity
maturation. For this reason, synthetic peptides used to elicit antibodies are conjugated to carrier proteins to improve their immunogenicity; the carrier proteins serve as the source of peptides for presentation via class II MHC products. Only through recognition of such a class II MHC–peptide complex via its TCR can a T cell provide the help necessary for the B cell to run its complete course of differentiation leading to robust, highaffinity antibody production. This concept applies equally to B cells capable of recognizing particular modifications on proteins or peptides. Antibodies that recognize the phosphorylated form of a kinase are commonly raised by immunization of experimental animals with the phosphorylated peptide in question, conjugated to a carrier protein. An appropriately specific BCR recognizes the phosphorylated site on the peptide of interest, internalizes the phosphorylated peptide and carrier, and generates a complex set of peptides by endosomal proteolysis of the carrier protein. Among these peptides, there should be at least one that can bind to the class II MHC molecules carried by that B cell. If properly displayed at the surface of the B cell, this class II MHC–peptide complex becomes the call for T-cell help, which is provided by helper T cells equipped with receptors capable of recognizing the complex of class II MHC molecule and carrier-derived peptide. The helper T cell identifies, via its TCR, an antigen-experienced B cell by means of the class II MHC–peptide complex the B cell displays (Figure 24-40, step 3a ). The B cell also displays co-stimulatory molecules and receptors for cytokines produced by the activated T cell (e.g., IL-4). After
Vaccines Elicit Protective Immunity Against a Variety of Pathogens
interacting with T cells, these B cells proliferate. Some of them differentiate into plasma cells; others are set aside and become memory B cells. The first wave of antibodies they produce is always IgM (Figure 2440, step 4a ). Class switching to other isotypes and somatic hypermutation (necessary for the generation and selection of high-affinity antibodies) require the persistence of antigen or repeated exposure to antigen. In addition to cytokines, B cells require cell-cell contacts to initiate these processes. These contacts involve the CD40 protein on B cells and CD40L on T cells (Figure 24-40, step 3b ); these proteins are members of the TNF–TNF receptor family. Work on HIV suggests that extensive somatic hypermutation is a prerequisite for the generation of broadly neutralizing antibodies — antibodies that can neutralize a broad selection of highly variable HIV isolates. More insight into the control of somatic hypermutation will be required to understand the nature of the antigen(s) capable of eliciting such desirable antibodies as a prophylactic strategy. Vaccines Elicit Protective Immunity Against a Variety of Pathogens Arguably the most important practical application of immunological principles is the vaccine. Vaccines are materials that are designed to be innocuous but that can elicit an immune response for the purpose of providing protection against a challenge with the virulent version of a pathogen (Figure 24-41). It is not always known why vaccines are as
successful as they are, but in many cases, the ability to raise antibodies that can neutralize a pathogen (viruses) or that show microbicidal effects (bacteria) are good indicators of successful vaccination.
FIGURE 24-41 Time course of a viral infection. The initial antiviral response, seen when the number of infectious particles rises, includes activation of natural killer (NK) cells and production of type I interferons. These responses are part of the innate immune response. The production of antibodies and the activation of cytotoxic T cells (CTLs) follow, eventually clearing the infection. Re-exposure to the same virus leads to more rapid and more pronounced production of antibodies and to more rapid activation of cytotoxic T cells.
A successful vaccine induces an immune response similar in some respects to that following initial exposure to a pathogen, but without causing significant symptoms of disease. If a vaccinated person is subsequently exposed to the same pathogen, the adaptive immune system is primed to respond quickly and strongly. Description The graph plots days after viral infection on the horizontal-axis from 0 to 14 in intervals of one day. Several curves plot the changes in different components of the immune response. The virus titer rises rapidly in the first three days, peaks after five days, and then falls to zero after eleven days. Type 1 interferons and natural killer cells increase rapidly on infection, peaking after two days. The concentration of interferons falls to zero after four days, whereas natural killer cells reduce sharply after four days but remain in high concentration for a significant period afterward. Virus-specific C T Ls begin to rise after two days, reaching a peak at ten days after infection, and then falling sharply for a day before the decrease becomes more gradual. Antibody titers begin to increase after four days, reaching a maximum at ten days, and then falling away very gradually. On re-exposure to the virus, the virus titer increases rapidly, but falls within four days, the antibody titer increases rapidly, within two days, natural killer cells peak after three days, and virus-specific C T Ls rise sharply after four days. Several strategies can lead to a successful vaccine. Serial passage of a pathogen in tissue culture or from animal to animal often leads to attenuation; the molecular basis of attenuation is not well understood. Vaccines may be composed of live attenuated variants of more virulent pathogens. The attenuated version of the pathogen causes a mild form of the disease or causes no symptoms at all. However, by recruiting all the component parts of the adaptive immune system, such live attenuated vaccines can elicit protective levels of antibodies. These antibody levels may wane with advancing age because the lymphocytes responsible for immunological memory may have a finite life span, so repeated
immunizations (booster injections) are often required to maintain full protection. Live attenuated vaccines are in use against flu, measles, mumps, and tuberculosis. In the latter case, an attenuated strain of the mycobacterium that causes the disease is used (Bacille Calmette-Guerin; BCG). Although live attenuated poliovirus was used as a vaccine until recently, its use was discontinued because the risk of reemergence, by mutation, of more virulent strains of the poliovirus outweighed the benefit. Currently, killed poliovirus is used as the vaccine of choice in the United States and Europe, although live attenuated poliovirus vaccines continue to be used elsewhere. Vaccines based on the cowpox virus, a close relative of the human variola virus that causes smallpox, have been used successfully to eradicate smallpox; the first successful elimination of an infectious disease. Attempts to achieve a similar feat for polio are nearing completion, but socioeconomic and political factors or armed conflict often complicate the administration of vaccines, leading to reemergence of the disease, as seen recently in Asia. Spurious claims that vaccines may cause autism have led to skepticism among some people regarding the benefits of childhood vaccinations against serious childhood diseases such as rubella and mumps. The number of vaccinated individuals drops when parents choose not to vaccinate their children. As a consequence, the type of protection referred to as herd immunity wanes and exposes entire populations to the resurgence of diseases easily controlled by childhood vaccination. There is
no evidence that childhood vaccinations are in any way connected to the onset or severity of autism or related disorders. The other major type of vaccine is called a subunit vaccine. Rather than attenuated or killed pathogen, only one or several of the pathogen’s components are used to elicit immunity. In certain cases, this approach is sufficient to afford lasting protection against a challenge with the live, virulent source of the antigen used for vaccination. It has been successful in preventing infections with the hepatitis B virus. The commonly used flu vaccines are composed mainly of the envelope proteins neuraminidase and hemagglutinin (see Figure 3-10); these vaccines elicit neutralizing antibodies. For the vaccine against human papillomavirus HPV 16, a virus that causes cervical cancer, viruslike particles composed of the virus’s capsid structural proteins but devoid of the viral DNA are generated; these particles are noninfectious, yet in many respects mimic the intact virion. The HPV vaccine now licensed for use in humans is expected to reduce the incidence of cervical cancer in susceptible populations by perhaps as much as 80 percent, the first example of a vaccine that prevents a particular type of cancer.
The Immune System Defends Against Cancer
From a public health perspective, cheaply produced and widely distributed vaccines are formidable tools for preventing or even eradicating communicable diseases. Current efforts are aimed at producing vaccines against diseases for which no other suitable therapies are available (Ebola virus, coronaviruses such as the SARS-CoV-2 virus responsible for the 2020 pandemic) or where socioeconomic conditions have made the distribution of drugs problematic (malaria, HIV). With a more complete understanding of how the immune system operates, it should be possible to improve on the design of existing vaccines and extend these principles to diseases for which no successful vaccines are currently available. A remaining challenge is the massive genetic variation that pathogens can acquire: the error-prone reverse transcriptase of HIV introduces mutations with every successive cycle of viral replication, creating untold numbers of variants. Viable variants that carry such mutations may escape detection by the immune system. The design of effective vaccines, not only against HIV but also influenza virus, must therefore be focused on those structural elements that do not tolerate mutations and that can be seen by the adaptive immune system. The Immune System Defends Against Cancer
The immune system not only defends against the immediate consequences of infection with pathogens but may also help in warding off cancer. As we have seen, the adaptive immune system is purged of many self-reactive B and T cells by negative selection. Self-reactive lymphocytes that escape this process are usually silenced because they are not provided with the appropriate co-stimulatory signals. Conditions that lead to severe immunosuppression, such as a genetic lesion in the RAG somatic recombination machinery or immunodeficiency caused by infection with HIV, confer an increased risk of cancer, not only for cancers caused by transforming viruses but also for those elicited by carcinogens. This observation establishes a role for the immune response in keeping precancerous cells in check. Recall that B and T cells require activation not only via their antigenspecific receptors, but also by a second, co-stimulatory signal (e.g., binding of CD28 on T cells to CD80 or CD86 on APCs; see Figure 24-35). Withholding of this co-stimulatory signal silences, or anergizes, any selfreactive lymphocyte that escaped deletion in the course of T-cell selection. Because tumor cells are exceedingly similar to the progenitors that give rise to them (see Chapter 25), with only those few mutations (“driver mutations”) required to cause cancer, it is not immediately obvious how immune recognition aids in the eradication of (pre) malignant cells before they have chance to grow into larger tumors. Somatic Mutations Allow Immune Recognition of Tumor Cells
Nonetheless, somatic mutations in the developing tumor cell — even those that are adventitious and do not directly contribute to causing cancer — can create so-called neo-antigens that may be recognized by antigenspecific receptors on T cells. Chemical mutagens, as experienced by heavy smokers who expose their lungs to tobacco products, not only cause mutations in genes that then drive tumorigenesis, but also cause mutations in other genes (passenger mutations), providing a rich spectrum of altered gene products to which the developing immune system was never exposed. If there is no immune tolerance for these mutagen-induced neo-antigens, they may serve as targets for recognition by the host’s immune system. Often the deregulation of gene expression that accompanies a transformed phenotype results in re-expression of differentiation antigens characteristic of a much earlier developmental state. If these differentiation antigens were expressed at a stage of development when the immune system had not yet fully matured, immune tolerance for such differentiation antigens may not have been established. These antigens may therefore be targets for immune recognition. Finally, the levels of certain gene products may no longer be properly regulated in cancer cells and may begin to exceed a threshold required for immune recognition, notwithstanding the fact that they are proteins normally made by the host, albeit at much reduced levels. In summary, because cancer can be considered a disease caused first and foremost by mutations, whose effects are modified by epigenetic events (see Chapter 25), there is the potential for immune recognition of cancer cells. Furthermore, in much the same way that an immune response
against a virus or a bacterium can result in the outgrowth of variants that are no longer recognized by the immune system, selective pressure exerted by the immune system may also lead to variants of cancer cells that have lost expression of a possible tumor antigen. For example, many colon cancers show reduced levels, if not complete loss of expression, of class I MHC products, and are thus rendered invisible to cytotoxic T cells. Antibodies Against CTLA4, PD-1, and PD-L1 Are the Basis of Cancer Immunotherapy The tumor microenvironment is composed of stromal cells: fibroblasts and myeloid-derived cells, including macrophages. Lymphocytes are known to invade tumors, as do neutrophils. The interplay between tumor cells and the microenvironment in which these cells reside can create immunosuppressive conditions that preclude a successful anti-tumor immune response, even if the tumor cells themselves are sufficiently antigenically distinct to be recognized as such. Important players in establishing an immunosuppressive environment are molecules now referred to as immunological checkpoints, such as CTLA4, the expression of which on T cells increases as T cells undergo full activation and maturation. Normally, CTLA4 would play a role in terminating an immune response, but its expression on the surface of tumor-specific T cells would compromise their anti-tumor activity. Moreover, the thymus and peripheral lymphoid compartments produce Treg cells, which are capable of suppressing the activity of other T cells. An abundance of Treg cells would keep other T cells from attacking a tumor. By the same logic, these
Treg cells may keep potentially self-reactive T cells in check as a means of preventing the onset or reducing the severity of autoimmune disease. Two key inhibitors of immune responses are PD-1 on T cells and PD-L1 on T-cell targets. Binding of PD-L1 on a tumor cell to PD-1 on a T cell that recognizes a tumor neo-antigen inhibits T-cell functions, including its ability to kill the tumor cell. A spectacular breakthrough in the treatment of cancer is the use of antibodies that target the inhibitory T-cell proteins CTLA4 and PD-1 proteins as well as PD-L1, often present on certain tumor cells. Some 30 percent of patients with metastatic melanoma, refractory to other forms of therapy, respond to treatment with antibodies against CTLA4, PD-1, or PD-L1, which has resulted in complete remissions and even cures. Similar approaches are underway to treat different forms of lung and renal cancer. Treatment with anti-CTLA4 apparently broadens the repertoire of cytotoxic T cells capable of recognizing tumor antigens as well as suppressing the activity of Treg cells. Treatment with anti-PD-1 or anti PD-L1 enhances T-cell recognition of tumors. It is perhaps ironic that smokers with the heaviest exposure to tobacco products may benefit the most from these forms of treatment because of the high mutational load in their cancers. KEY CONCEPTS OF SECTION 24.6 Collaboration of Immune-System Cells in the Adaptive Response Antigen-presenting cells such as dendritic cells require activation by means of signals delivered to their Toll-like receptors and other pattern recognition receptors. These receptors are broadly specific for macromolecules produced by bacteria and viruses.
Their engagement activates the NF-κB signaling pathway, whose outputs include the synthesis of inflammatory cytokines (see Figure 24-37). Upon activation, dendritic cells become migratory and move to lymph nodes, ready for their encounter with T cells. Activation of dendritic cells also increases their display of MHC-peptide complexes and expression of co-stimulatory molecules required for initiation of a T-cell response. B cells require the assistance of T cells to execute their full differentiation program into plasma cells. Antigen-specific help is provided to B cells by activated T cells, which recognize class II MHC–peptide complexes on the surfaces of B cells. These B cells generate the relevant MHC-peptide complexes by internalizing antigen via BCRmediated endocytosis, followed by antigen processing and presentation via the class II MHC pathway (see Figure 24-40). In addition to cytokines produced by activated T cells, B cells require cell-cell contact to initiate somatic hypermutation and class-switch recombination. This contact involves CD40 on B cells and CD40L on T cells. Important applications of the immunological concept of collaboration between T and B cells include vaccines. The most common forms of vaccines are live attenuated viruses or bacteria, which can evoke a protective immune response without causing pathology, and subunit vaccines. The adaptive immune system can sometimes distinguish between normal cells and their cancerous counterparts. What complicates immune-system detection of cancer cells are the often relatively minor differences between normal and transformed cells. Immunological checkpoints dampen the activity of antigen-specific T cells under normal circumstances as a means of turning off or controlling an immune response.
Key Terms
End of Chapter Visit Achieve to access study tools and to learn more about the content in this chapter. Perspectives for the Future Classic Experiment 24.1: Two Genes Become One: Somatic Recombination of Immunoglobulin Genes Analyze the Data Chapter References Additional study tools, including videos, animations, and quizzes Key Terms antibody antigen antigen-presenting cell (APC) antigen processing and presentation autoimmunity B cell B-cell receptor (BCR) chemokine complement cytokine dendritic cell diversity
Review the Concepts
epitope Fc receptor (FcR) immunity immunoglobulin immunoglobulin fold inflammasome inflammation interleukin isotype lymphocyte macrophage major histocompatibility complex (MHC) memory monoclonal antibodies natural killer (NK) cell neutrophil opsonization paratope phagocyte T cell T-cell receptor (TCR) tolerance Toll-like receptor (TLR) transcytosis Review the Concepts
1. Describe the ways in which each of the following pathogens can disarm their host’s immune system or manipulate it to their own advantage: a. Pathogenic strains of Staphylococcus aureus, a Grampositive bacterium that replicates extracellularly and secretes cytotoxic products (toxins) b. Enveloped viruses such as HIV (see Section 5.7) 2. Trace the movement of leukocytes as they perform their functions throughout the body. 3. Identify the major mechanical and chemical defenses that protect internal tissues from microbial attack. 4. Compare and contrast the classical pathway of complement activation with the alternative pathway. 5. What is opsonization? What is the role of antibodies in this process? 6. In B cells, what mechanism ensures that only rearranged V genes are transcribed? 7. What prevents further rearrangement of immunoglobulin heavychain gene segments in a pre-B cell once a productive heavychain rearrangement has occurred? 8. How and why do B cells undergo a class switch from producing IgM antibodies to any of the other Ig isotypes? 9. What biochemical mechanism underlies affinity maturation of the antibody response? 10. Compare and contrast the structures of class I and class II MHC molecules. What kinds of cells express each class of MHC molecule? What are the functions of these cells?
11. Describe the six steps in antigen processing and presentation via the class I MHC pathway. 12. Describe the six steps in antigen processing and presentation via the class II MHC pathway. 13. What prevents self-reactive T cells from leaving the thymus? 14. Explain why T-cell–mediated autoimmune diseases are associated with particular alleles of class II MHC genes. 15. How are antigen-presenting cells and helper T cells involved in B-cell activation? 16. Outline the events in the innate and adaptive immune responses, from when a pathogen invades to clearance of the pathogen. 17. Define passive immunization and give an example. 18. How would you design a vaccine that protects against HIV infection without the possibility of infecting the patient? 19. The annual flu shot is composed of either live attenuated influenza virus or influenza subunits (the envelope proteins neuraminidase and hemagglutinin). How does the annual flu shot protect you against infection? 20. Design a laboratory protocol to develop a monoclonal or polyclonal antibody against a protein of interest (see Section 4.1). 21. Consider a person without any functioning plasma cells. What effects would this condition have on the person’s adaptive immune system? Innate immune system?