Introduction
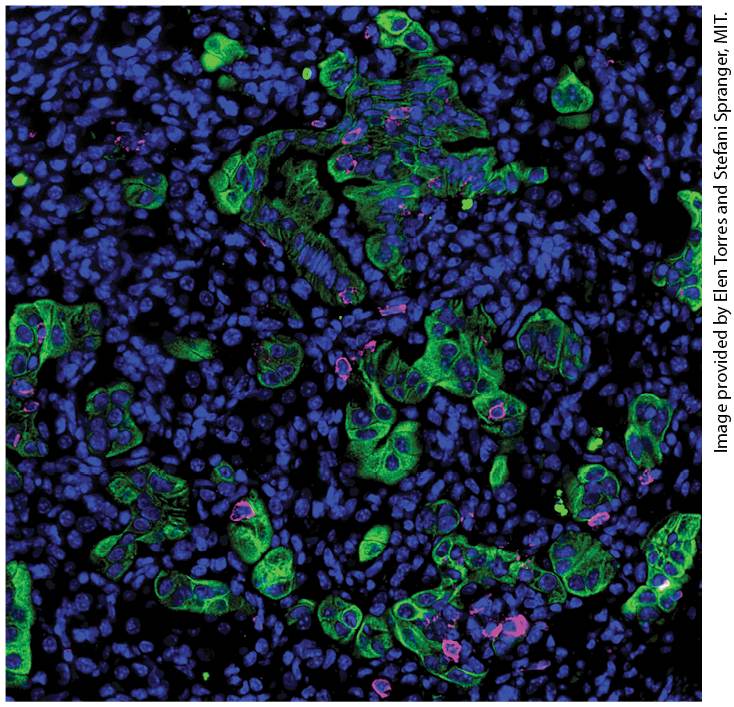
Chapter 25 Cancer Cells of the immune system often interact with cancer cells within the tumor microenvironment. This section of a lung adenocarcinoma shows all cell nuclei (blue), tumor cells (green) and T cells (magenta). Note the tendency of the T cells to be in contact with tumor cells.
25.3 Dysregulation of Cell Growth and Developmental Pathways Initiates Tumorigenesis
25.4 Evasion of Programmed Cell Death and Immune Surveillance Processes Cancer, described in the simplest terms, arises as a disease when normal somatic cells of the body acquire a set of mutations that cause them to escape from the orderly patterns of tissue development and to grow and spread through the body. Because there are many ways that loss of normal developmental regulation can lead to uncontrolled growth, cancer is actually many different diseases depending upon the origin of the cancer cells. Tumors derived from epithelial tissues such as endoderm (gut epithelium) or ectoderm (skin and neural epithelia) are classified as carcinomas, whereas tumors derived from mesodermal tissues (muscle, bone, cartilage and connective tissue precursors) are classified as sarcomas. Many tumors form solid masses, but tumors of the blood and bone marrow, such as the leukemias and lymphomas, can grow as individual cells circulating in the blood. The classification of cancer types is further subdivided according to the exact cell type of origin and sometimes even according to the types of mutations that have caused dysfunction in a particular cell type. There are about 200 types of cancer based on histological examination of tumor cells; the number of distinct
types of cancer that are recognized is increasing as differences between cancer cells can be discerned at a molecular level. Despite the wide variety of different types of cancer, all cancers are thought to arise by the same process, called oncogenesis or tumorigenesis, which begins with a single mutation in a somatic cell that eventually develops into a clone of cancer cells possessing many different oncogenic mutations. Although it is possible to imagine how one of the approximately dividing cells in the body could be struck by even a very rare mutational event, it seems inconceivable that a clone of cells could by chance alone acquire a whole set of highly specific mutations needed for all of the phenotypic properties of a cancer cell. The answer to this puzzle is that oncogenesis is the grim result of a version of natural selection in which cells that proliferate a bit more rapidly than their neighbors, or do not undergo apoptosis when they should, have greater opportunity to undergo further genetic changes. Most tumors develop slowly, acquiring additional oncogenic mutations by selection over a span of many years.
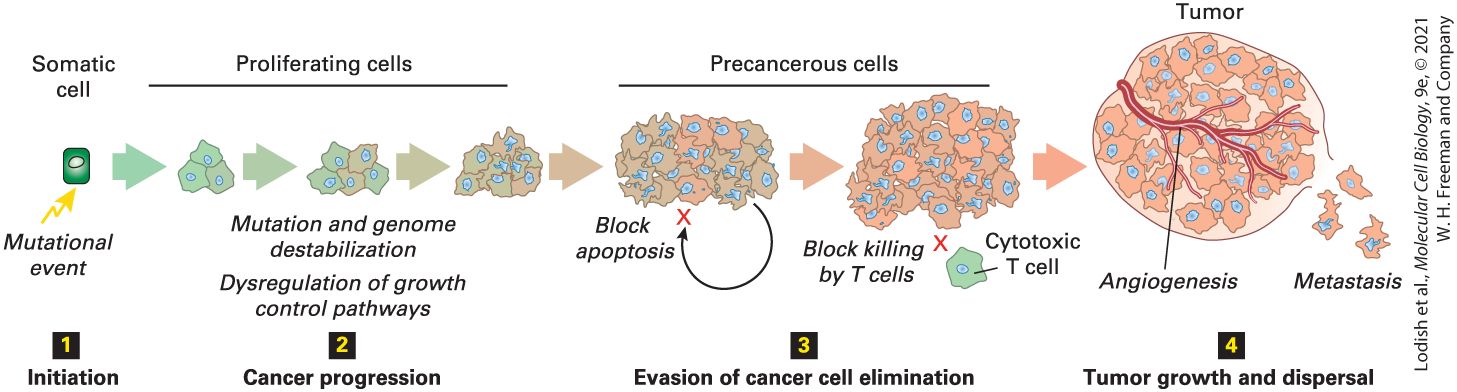
Figure 25-1 shows an outline for understanding how cancer progresses from an initiating mutation to a full tumor by the acquisition of successive mutations in a clone of cells successively acquiring oncogenic mutations. Usually, by the time a tumor is detected and its genome can be analyzed, so many mutations have accumulated that it is not possible to identify with certainty the original mutation that started a cell down the path to cancer. Nevertheless, two general types of mutations are known to be capable of initiating oncogenesis. The first type of initiating mutation may
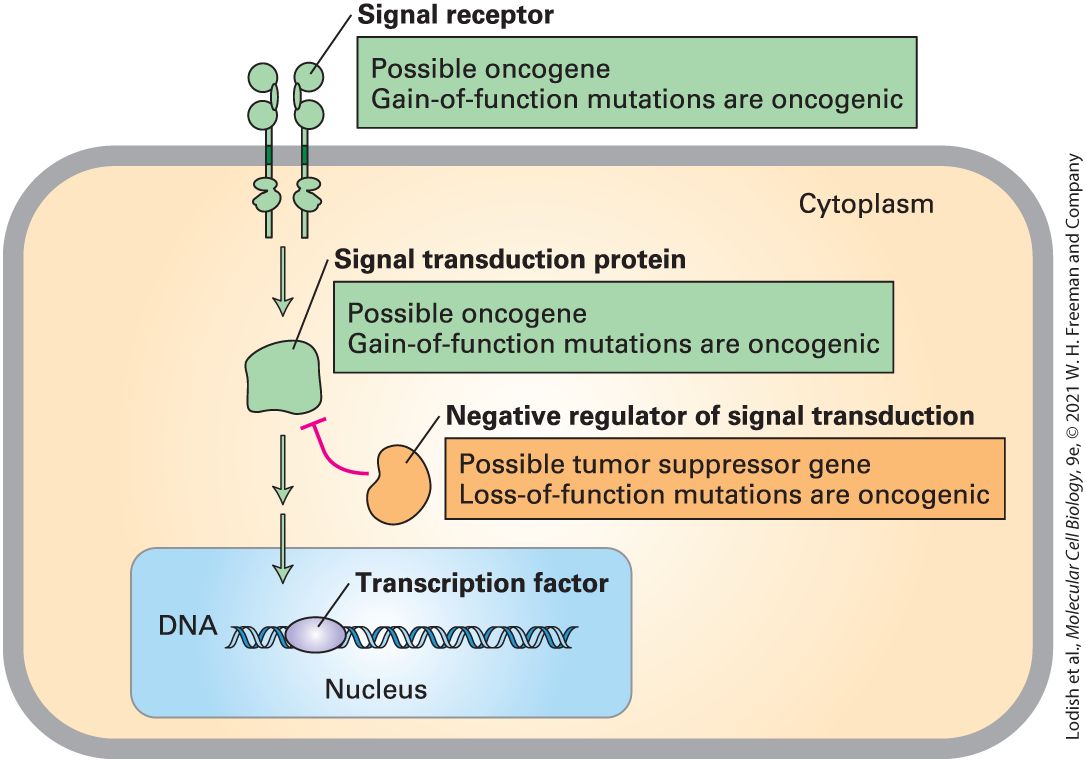
affect one of the many signal transduction pathways that regulate cell growth. Such mutations could either inappropriately activate a growthpromoting pathway or could inactivate a growth-inhibiting pathway. In either case, the mutant cells would grow more rapidly than their wellregulated neighbors and thus provide more opportunity for those cells to acquire additional oncogenic mutations. The second type of initiating mutation impairs the ability of the cell to repair damage to DNA caused by a mutagen or by an error in DNA replication. Such mutations could be in the DNA repair enzymes themselves or could prevent the arrest of the cell at one of the cell cycle checkpoints to allow the repair to take place (see
Chapter 20). As described in Chapter 5, DNA replication and cell division are necessary for damage to DNA to be converted to a mutation in the base sequence. This fundamental relationship between cell division and mutagenesis is one of the reasons why cells in actively dividing tissues are the most susceptible to acquiring oncogenic mutations and therefore generally the most prone to form tumors.
FIGURE 25-1 General framework for the evolutionary progression of cancer. The progression of cancer from a normal somatic cell to a full-grown tumor can be thought to take place in four stages. Step 1 : Initiation. Except in rare cases in which cancer is caused by a singular genetic event, such as infection by a tumor virus, we usually cannot pinpoint the initiating event but a somatic mutation is usually suspected. Step 2 : Cancer
progression. Precancerous cells acquire mutations that dysregulate growth-control pathways, causing inappropriate cell proliferation. In addition, they acquire mutations that cause genome instability and propensity to acquire yet more mutations, mainly by loss of the DNA damage checkpoint. Most cancers progress by a process of natural selection, sequentially acquiring multiple such mutations in different pathways. Step 3 : Evasion of cancer cell elimination. Precancerous cells are normally removed from the body by either apoptosis or immune-surveillance processes. To progress beyond this stage, tumor cells must acquire additional somatic mutations that allow them to evade these systems. Step 4 : Tumor growth and dispersal. Continued growth of a tumor requires a blood supply and tumor cells must acquire yet more changes that promote angiogenesis. To spread throughout the body by metastasis, cells from a solid tumor must acquire the ability to migrate from their original site and adhere at a new location in the body. Description The first step is labeled initiation. Above this label is a green rectangular cell labeled somatic cell. A yellow arrow points to the cell with a label mutational event. The second step is labeled cancer progression. An arrow that moves right from step 1 to 2 is labeled proliferating cells. The cell has become 3 cells, then five cells, and then many cells. The labels below read mutation and genome destabilization, and dysregulation of growth control pathways, and cancer progression. The third step is labeled evasion of cancer cell elimination. The next sideward arrow is labeled as precancerous cells. Now the group of cells is larger and a small curved arrow comes from the group out of the bottom area and around to the left and is labeled block apoptosis. The next group of cells is larger and there is a marked cell at the bottom, which is marked with a letter X. The label reads: block killing by T cells; cytotoxic T cell. The last step is labeled tumor growth and dispersal. The last group of cells is labeled tumor. The blood vessels wrap the tumor cells and the process is labeled angiogenesis. Small single cells are released at the bottom right with the label metastasis. The clonal descendants of an initial mutant are susceptible to acquiring additional oncogenic mutations of either a type that promotes cell proliferation or that inhibits DNA repair. These additional mutations in
turn increase the propensity for yet more oncogenic mutations. Each iteration of this cycle of mutation and selection for more rapidly growing cells produce cellular descendants exhibiting successively more uncontrolled growth and more defects in the genome. Tumors are usually detected only after they have reached in size, by which time their cells have undergone many cycles of error-prone replication and their genomes will carry many different single-base changes due to errors in replication and chromosome rearrangements due to erroneous repair of breaks in the DNA. The base sequence of cancer cell genomes usually reveals a profusion of mutations; it is often difficult to distinguish mutations that drive the oncogenic process from mutations that arose fortuitously but do not themselves contribute to oncogenic progression. Other oncogenic mutations enable cancer cells to evade the two major systems that cause removal of aberrant cells from the body. Mammalian cells are normally able to monitor the integrity of their genome, and if they suffer a major, irreparable aberration in genome structure will enter a cell death program (see Chapter 22) that will cause them to be eliminated before they can become cancerous. The precancerous cells that do survive to become tumors have lost one or more of the key elements of the programmed cell death pathway. Another class of oncogenic mutations often found in tumors has allowed the cancer cells to evade surveillance by T cells of the immune system. As a cancer cell accumulates an increasing burden of mutations in the genome, the proteins of the cell will increasingly carry amino acid substitutions, truncations, and domain rearrangements that will eventually cause a cancer cell to appear as
foreign to the immune system. Specific mutations in cancer cells allow them to avoid immune detection and destruction. The growth of a mass of rapidly dividing cells will reach a limit simply because the cells on the inside of the mass are restricted by diffusion for the availability of nutrients and oxygen. For a tumor to grow beyond the dimensions of several millimeters requires that the tumor cells be supplied with blood. Accordingly, a fourth type of oncogenic mutation found in solid tumors enables the tumor cells to signal for formation of blood vessels by secreting certain growth factors. In addition to the transformed cancer cells, tumors usually recruit numerous cells that are not mutated and that form differentiated structures in response to signals emanating from the cancer cells. Because tumors contain multiple cell types and structures, they more closely resemble developed organs than a clonal mass of cells. Finally, due to yet other oncogenic mutations, tumor cells often have acquired the ability to detach from the primary tumor, to enter circulating blood or lymphatic systems and adhere to a new site and form a secondary tumor in a different tissue. The capability of a tumor to spread throughout the body in this way is known as metastasis and is a hallmark of the deadliest forms of cancer. This basic outline of the origin, progression, growth, and spread of cancer shows why cancer is such a difficult disease to treat. Because cancer cells are derived from normal somatic cells, it is generally not possible to kill cancer cells selectively without to some extent killing normal cells as well. Most cancer therapy aims at killing rapidly dividing cells that cannot repair their DNA efficiently. For example, alkylating agents that cause
cross-links in DNA are among the most commonly used anticancer chemotherapeutic compounds that preferentially kill cells that are actively undergoing DNA replication but that have lost the ability to repair crosslinked and broken DNA. Even the most effective anticancer compounds cause tremendous collateral damage to actively dividing, normal cells, such as the dividing cells in the hair follicles, bone marrow, and immune system, causing hair loss, anemia, and susceptibility to infections. Another avenue to cancer therapy is to reverse the effects of oncogenic mutations by, for example, inhibiting the product of an overexpressed oncogene. This type of targeted therapy is effective at treating rare forms of cancer that arise because of a single oncogenic event. As we will see, the vast majority of cancers have acquired multiple oncogenic mutations in different pathways for cell growth, DNA repair, and programmed cell death and there are no prospects yet for a magic bullet that can simultaneously reverse the dysregulation of multiple pathways. Currently, our understanding of how cancer develops at the cellular level offers the most promise for early diagnosis and prevention of the disease. All of the known oncogenic mutations as well as their physiological consequences can be thought of as molecular markers that could allow early detection of the presence of precancerous cells. In the early stages of progression of the disease, precancerous cells are likely to be the most vulnerable to correction by a drug targeted to a specific pathway or by elimination by heightened immune surveillance. We know that most cancers develop in stages over a long period of time; detection of the first steps of oncogenesis should afford significant opportunities to intervene.
In this chapter, we first introduce the properties of tumor cells, illustrating how the genome, cellular metabolism, regulation of growth and proliferation, and morphology all can be altered in cancer. We will also see how the interaction of cancer cells with their environment enables development of large tumors and the spread of cancer by metastasis. We then discuss the genetic and genomic basis of cancer, how cancer can originate with inherited mutations or by somatic mutations caused by carcinogens and how the breakdown of genome maintenance functions contributes to tumorigenesis. Next we consider the general types of genetic changes affecting both growth-promoting and growth-inhibiting processes that can result in excess cell proliferation. We conclude the chapter with a discussion of how cancer cells acquire mutations that allow them to escape from programmed cell death and immune surveillance. We will end with promising new ways to treat mature tumors based on activation of the immune system to recognize and destroy cancer cells, known as cancer immunotherapy.
25.1 How Tumor Cells Differ from Normal Cells
25.1 How Tumor Cells Differ from Normal Cells Before examining the genetic basis of cancer in detail, let’s consider the general properties of tumor cells that distinguish them from normal cells. The change from a normal cell to a cancer cell commonly involves multiple steps, each one adding properties that make cells more likely to grow into a tumor. The genetic changes that underlie oncogenesis alter several fundamental properties of cells, allowing those cells to evade normal growth controls, modulating their tissue microenvironment, and ultimately conferring the full cancer phenotype (see Figure 25-1). Cancer cells acquire a drive to proliferate that does not require an external inducing signal. They fail to sense signals that restrict cell division, and they survive when they should die. They often change their attachment to surrounding cells or to the extracellular matrix, breaking loose to move away from their tissue of origin. Solid tumors characteristically outgrow their initial blood supply and become hypoxic (oxygen starved), so to grow to more than a small size, tumors must obtain an additional source of blood flow. They often do so by inducing the growth of blood vessels into the tumor. As cancer progresses, further adaptations are acquired by selection allowing growth and invasion of surrounding tissues, often spreading to distant sites in the body.
The Genetic Makeup of Most Cancer Cells Is Dramatically Altered
In this section, we describe the characteristics of cancer cells. We first discuss the changes in the cancer cell’s genetic makeup that affect virtually all cellular functions, allowing the cancer cell to escape proliferation regulation and acquire the ability to divide indefinitely. We then see how the genetic changes in a tumor cell and its interactions with its environment facilitate its escape from the constraints of the tissue it was once a part of and allow it to invade neighboring tissues and colonize distant sites in the body. The Genetic Makeup of Most Cancer Cells Is Dramatically Altered At the turn of the twentieth century, David von Hansemann and Theodor Boveri first documented what we now know to be an almost universal feature of cancer cells: their entire genetic makeup differs dramatically from that of normal cells. Chromosomes from cancer cells stained to be visible in the light microscope often show a highly altered karyotype and large chromosomal amplifications and deletions, translocations, and aberrant numbers of chromosomes — generally too many, a condition known as aneuploidy. Typical cancer cells exhibit whole chromosome or chromosome arm gains and losses involving a quarter of their genome (Figure 25-2). The advent of efficient next generation DNA-sequencing methods has allowed complete cancer genome sequences to be obtained from thousands of different tumors. In addition to gross chromosomal abnormalities, sequencing reveals single-base mutations and small, local amplifications and deletions often affecting about 10 percent of the cancer
cell’s genome (Figure 25-3). Perhaps the most surprising result revealed by the sequencing of cancers is the high degree of variation in mutation rates across different cancers. Mutations are rare in pediatric cancers, with substitution rates as low as a few hundred base changes per genome, but those rates may be as high as 500,000 base changes per genome in mutagen-induced cancers such as certain lung cancers and melanomas. As we will see in Section 25.2, the frequency and type of genomic mutations in a given tumor can often be traced to the presence of mutations in DNA repair pathways. These genetic changes affect virtually all aspects of cellular homeostasis, proliferation, tissue organization, and migratory properties as well as survival and proliferation at foreign sites in the body.
FIGURE 25-2 Cancers often have highly abnormal karyotypes. Image of chromosomes obtained from an SW403 colorectal adenocarcinoma cell line. Chromosomes are arranged and color coded according to labeling with probes specific for each of the 24 chromosomes. Two characteristics of the cancer cells are evident. First, a normal karyoptype would contain exactly two copies of each chromosome and the cancer cells contain one or two extra copies of many of the chromosomes. Second, many chromosomes are composites of pieces from different chromosomes, which result from translocation events. [From W. M. Abdel-Rahman et al., 2001, “Spectral Karyotyping Suggests Additional Subsets of Colorectal Cancers Characterized by Pattern of Chromosome Rearrangement.”
Proc. Nat’l Acad. Sci. USA 98(5):2538–2543, Fig. 3c. Copyright (2001) National Academy of Sciences, U.S.A.] Description The illustration shows 22 groups of chromosomes with two, three, or four copies of chromosomes in each group. The X and Y-chromosomes show three and no copies of chromosomes, respectively.
Uncontrolled Proliferation Is a Universal Trait of Cancer
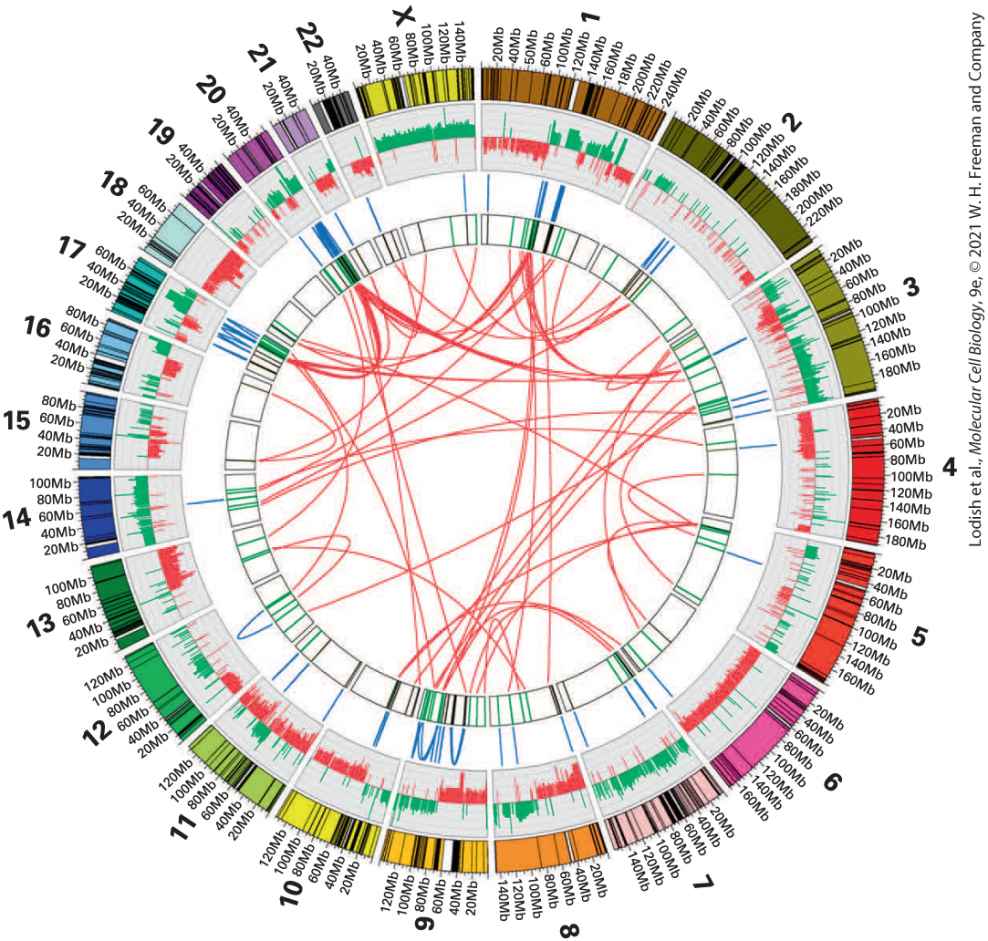
FIGURE 25-3 Cancer genome sequencing reveals thousands of sequence alterations. The complete genome sequence of the MCF-7 adenocarcinoma breast cancer cell line displayed as a Circos plot. The 23 chromosomes are arranged around the outside ring. In the next ring, DNA copy number is represented by increases (green) or decreases (red) from the normal 2n (diploid) number of copies, showing that most of the MCF-7 genome has an abnormal copy number. Intrachromosomal and interchromosomal rearrangements are represented respectively by blue and red arcs. A total of 157 chromosomal breakpoints were found. [Oliver A. Hampton et al., 2009, Genome Res. 19:167–177; Copyright © 2009 by Cold Spring Harbor Laboratory Press.] Description This Circos plot shows the numbered chromosomes labeled around the outside of the circle, with each numbered chromosome from 1 to 22 and X with each having several choices of sequence under it. Multiple lines are drawn through the center of the circle to illustrate possible combinations. Uncontrolled Proliferation Is a Universal Trait of Cancer In normal tissues, cell proliferation is a highly controlled process. Growthpromoting factors are released in a highly controlled fashion to ensure that target cells proliferate only as needed to replenish a tissue. A universal feature of cancer cells is that they have acquired oncogenic mutations that allow them to escape these tight controls and to proliferate continuously and indefinitely. Here we will briefly introduce three of the most common oncogenic mutations as illustrative examples, and in Sections 25.3 and
25.4, we will develop a more comprehensive picture of the ways that different mutations can enable cancer cell proliferation. The Ras protein described in Chapter 16 acts a GTPase switch in many of the signaling pathways that receive growth stimulatory signals via a receptor tyrosine kinase. Ras activates the MAP kinase signal transduction pathway that in turn activates many transcription factors regulating aspects of cell proliferation. The first oncogenic mutation to be described is a point mutation in the Ras gene that causes constitutive activation of the MAP kinase pathway. This pathway normally is only activated when a growth factor is present in the cell’s environment. Thus precancerous cells that carry an oncogenic Ras mutation proliferate as if they are continually receiving a growth signal. Most human tumors have oncogenic mutations in p53 or in proteins that regulate p53 activity. Normally, p53 plays a major role in maintaining the integrity of the genome. As described in Chapter 19, when DNA damage is first detected in a cell, p53 is responsible for causing cell cycle arrest before M phase, giving repair enzymes sufficient time to correct the damage before the cell cycle continues. If the DNA damage cannot be repaired, p53 will eventually activate apoptosis to ensure that cells with a compromised genome are eliminated from the body. Precancerous cells that have insufficient p53 activity continue to divide even when their DNA is badly damaged. Such unchecked cell divisions destabilize the genome and greatly accelerate the acquisition of additional oncogenic mutations.
Cellular Housekeeping Functions Are Fundamentally Altered in Cancer Cells
A third example of oncogenic mutations that allows uncontrolled cell proliferation commonly found in tumors are DNA rearrangements that cause increased expression of telomerase, the enzyme responsible for adding telomeres, short tandem DNA sequences, to the ends of linear chromosomes. As discussed in Chapter 7, telomeres become shorter every time a chromosome is replicated. Most human somatic cells produce only a small amount of telomerase and uncontrolled proliferation would eventually lead to extensive shortening of telomeres, which is recognized by the cell as a double-strand break that consequently triggers cell cycle arrest and apoptosis. Tumor cells overcome this fate by up-regulating telomerase expression to produce a state of replicative immortality. Cellular Housekeeping Functions Are Fundamentally Altered in Cancer Cells Cancer cells can often be distinguished from normal cells by microscopic examination. They are usually less well differentiated than normal cells. Cancer cells frequently exhibit the characteristics of non-cancerous rapidly growing cells: a high nucleus-to-cytoplasm ratio, prominent nucleoli, an increased frequency of mitotic cells, and relatively little specialized structure. Tumor cells differ from normal cells not only in their appearance, but in their entire protein composition. Losses and gains of whole chromosomes or chromosome parts are characteristic of cancer cells and have a profound
effect on the protein composition of the cell and hence on many cellular functions. Imbalances in the composition of multiprotein particles cause many proteins to remain only partially folded; this in turn induces a stress response, similar to that caused by raising the cell to an elevated temperature (see Chapter 21), aimed at offsetting these protein imbalances. As a result, cancer cells rely heavily on protein-folding chaperones and degradation mechanisms, including proteasomes, for their survival as a direct result of their dramatically altered chromosome composition. Another hallmark of cancer cells is their dependence on an energygenerating pathway that resting cells only use if they are deprived of oxygen. Most normal differentiated cells rely on relatively efficient mitochondrial oxidative phosphorylation to satisfy their energy needs. Cells metabolize glucose to carbon dioxide by oxidation of pyruvate through the tricarboxylic acid (TCA) cycle in the mitochondria (see
Chapter 12). Only under anaerobic conditions do normal resting cells undergo anaerobic glycolysis and produce large amounts of lactate. Most cancer cells, however, rely on glycolysis for energy production irrespective of whether oxygen levels are high or low (Figure 25-4). The use of glycolysis to produce energy even in the presence of oxygen, called aerobic glycolysis, was first discovered in cancer cells by the biochemist Otto Warburg and is therefore called the Warburg effect. Aerobic glycolysis generates only 2 ATP molecules per molecule of glucose and appears to be much less efficient than oxidative phosphorylation, which generates up to 36 molecules of ATP per molecule of glucose. However, by not using the TCA cycle for generating energy, proliferating cells can
instead utilize the molecules in the TCA cycle and their precursors to synthesize nucleotides, amino acids, and lipids. This rewiring of glucose metabolism diverts the flow of metabolic intermediates away from ATP synthesis and toward the production of cellular building blocks, a balance more favorable for rapid cell growth.
FIGURE 25-4 Energy production in cancer cells by aerobic glycolysis. In the presence of oxygen, nonproliferating (differentiated) cells metabolize glucose into pyruvate via glycolysis. Pyruvate is then transported into mitochondria, where it is fed into the TCA cycle. Oxygen is required as the final electron acceptor during oxidative phosphorylation. Thus when oxygen is limiting, cells metabolize pyruvate into lactate, allowing glycolysis to continue by cycling NADH back to . Cancer cells and proliferating cells convert
Cancer Cells Exhibit Altered Cell-Cell Interactions to Form Heterogeneous Organs
most glucose to lactate regardless of whether oxygen is present or not. The production of lactate in the presence of oxygen is called aerobic glycolysis. [Data from M. G. Vander Heiden, L. C. Cantley, and C. B. Thompson, 2009, Science 324:1029.] Description In differentiated tissue, glucose is converted to pyruvate and then, in the presence of oxygen, to carbon dioxide by oxidative phosphorylation. In the absence of oxygen, lactate is produced. Oxidative phosphorylation yields thirty-six moles of A T P per mole of glucose; in contrast, anaerobic glycolysis only yields 2 moles of A T P per mole of glucose. In proliferative or tumor tissue, in both the absence and presence of glucose, pyruvate is converted to lactate. Aerobic glycolysis or Warburg effect yields four moles of A T P per mole of glucose. Not only do cancer cells rewire their metabolic pathways, but they also produce increased levels of some metabolites and metabolic by-products. These so-called onco-metabolites directly or indirectly alter patterns of gene expression, including genes involved in blood vessel formation. Cancer Cells Exhibit Altered Cell-Cell Interactions to Form Heterogeneous Organs Many human cell types grow in well-ordered sheets. Cells do not continue to divide and grow outside the plane of the sheet because of a phenomenon of contact inhibition, which causes arrest in the phase of the cell cycle when a cell is completely surrounded by other cells. Precancerous cells
have lost contact inhibition, are less adherent and more rounded than normal cells, and will continue to divide as a three-dimensional cluster of cells (a focus) that can be recognized under a microscope. Adhesion molecules such as E-cadherin, cell polarity factors, actin cytoskeleton regulators, and the Hippo pathway (see Chapter 21) all function in mediating cell cycle arrest when cell-cell contacts are established. However, the exact mechanisms whereby this occurs, and how these pathways are disrupted in cancer, remain to be worked out. Not all tumors are made up of uniform cells, even if they originated from a single initiating cell. In some types of tumors, for example, only certain tumor cells, called cancer stem cells, are capable of seeding a new tumor. Within these tumors, some cells cease dividing, while others can continue cancerous growth. The latter, of course, are the most dangerous and the most important to destroy with anticancer treatments. Cancer stem cells are thought to give rise to some cells with high replicative capabilities and others with more limited replicative potential. The origins of these cancer stem cells are not clear. In some cancers, a normal tissue stem cell may give rise to the cancer stem cells. In others, dedifferentiation of terminally differentiated cells to form progenitor cells may give rise to cancer stem cells. Irrespective of their origin, cancer stem cells share gene expression signatures with normal tissue stem cells, leading to their designation as stem cell-like cells. The immediate environment of a tumor — the tumor microenvironment — contributes to the heterogeneity of cells within the tumor, influencing the behavior of the cancer stem cells and the tumor cells in general. Some
Tumor Growth Requires Formation of New Blood Vessels
neighboring cells may be more conducive to tumor growth than others. The importance of the tumor microenvironment extends to one of the most common environmental influences on a tumor cell: inflammatory cells. It is now widely accepted that cells of the immune system interact with the tumor. As we detail in Section 25.4, cytotoxic T lymphocytes and natural killer cells surround and often migrate into the tumor, where they can inhibit tumor formation. Mice deficient in these and other components of the immune system are more prone to carcinogen-induced tumors than normal mice. These findings lead to the idea that the immune system eliminates cancer cells. Current information about how cancer cells escape this immune surveillance is discussed in Section 25.4. More and more evidence is mounting that immune-system cells can also have tumorigenic properties. It has been known for a long time that cancers frequently arise at sites of injury or chronic infection. It is estimated that up to 20 percent of cancers are linked to chronic infection. For example, persistent infection of the stomach by the bacterium Helicobacter pylori is associated with gastric cancer. Crohn’s disease, an inflammatory autoimmune disease that affects the intestines, is associated with colon cancer. Infection with hepatitis B or C viruses increases the risk of a form of liver cancer, hepatocellular carcinoma. Immune-system cells migrate to sites of injury or infection and produce growth factors, thereby stimulating tumor cell proliferation. They also produce factors to induce the growth of blood vessels, which — as we will discuss next — is an essential aspect of tumor growth and dissemination to distant sites.
Tumor Growth Requires Formation of New Blood Vessels Tumors must recruit new blood vessels in order to grow to a large size. In the absence of a blood supply, a tumor can grow into a mass of about cells, roughly a sphere 2 mm in diameter. At this point, division of cells on the outside of the tumor mass is balanced by death of cells in the center from an inadequate supply of nutrients. Such growth-limited tumors, unless they secrete hormones, cause few problems. However, most tumors induce the formation of new blood vessels that invade the tumor and nourish it, a process called angiogenesis. This complex process requires several discrete steps: degradation of the basement membrane that surrounds a nearby capillary, migration of endothelial cells lining the capillary into the tumor, division of these endothelial cells, and formation of a new basement membrane around the newly elongated capillary. Many tumors produce growth factors that stimulate angiogenesis; other tumors induce surrounding normal cells to synthesize and secrete such factors. Basic fibroblast growth factor (β-FGF), transforming growth factor α (TGF-α), and vascular endothelial growth factor (VEGF), which are secreted by many tumors, all have angiogenic properties. New blood vessels allow the tumor to increase in size and thus increase the probability that additional harmful mutations will occur. The presence of an adjacent blood vessel also facilitates the process of metastasis.
Invasion and Metastasis Are Late Stages of Tumorigenesis
The VEGF receptors, which are tyrosine kinases, regulate several aspects of blood vessel growth, such as endothelial cell survival and growth, endothelial cell migration, and vessel wall permeability. VEGF expression can be induced by oncogenes and by hypoxia. As detailed in Chapter 21, the hypoxia signal is mediated by hypoxia-inducible factor 1α (HIF-1α), a transcription factor that is activated in low-oxygen conditions and that binds to and induces transcription of the VEGF gene and about 30 other genes, including many enzymes in the glycolytic pathway. An enhancement of glycolysis can in turn stimulate the growth of many cancer cells. HIF-1α activity is controlled by an oxygen sensor composed of a prolyl hydroxylase that is active at normal levels but inactive when deprived of . Hydroxylation of HIF-1α causes ubiquitinylation and degradation of the transcription factor, a process that is blocked when is low. Mutations in genes encoding a subunit of the ubiquitin ligase that degrades HIF-1α at ambient oxygen levels trigger certain kidney tumors. Compounds that inhibit angiogenesis have excited much interest as potential therapeutic agents, but their success in the clinic has thus far been limited. Invasion and Metastasis Are Late Stages of Tumorigenesis Tumors arise with great frequency, especially in older individuals, but most pose little risk to their host because they are small and localized. We call such tumors benign; an example is a wart, a benign skin tumor. The cells composing benign tumors closely resemble and may function like
normal cells. The cell-adhesion molecules that hold tissues together keep benign tumor cells, like normal cells, localized to the tissues where they originate. A fibrous capsule usually delineates the extent of a benign tumor and makes it an easy target for a surgeon. Benign tumors become serious medical problems only if their sheer bulk interferes with normal functions or if they secrete excess amounts of biologically active substances, such as hormones. For example, acromegaly, the overgrowth of head, hands, and feet, can occur when a benign pituitary tumor causes overproduction of growth hormone. In contrast to benign tumor cells, malignant tumor cells are able to invade nearby tissue, spreading and seeding additional tumors while the cells continue to proliferate (Figure 25-5). This ability is a major characteristic that differentiates malignant tumors from benign ones. Some malignant tumors, such as those in the ovary or breast, remain localized and encapsulated, at least for a time. When these tumors progress, however, the cells invade surrounding tissues and form metastases (Figure 25-6a).
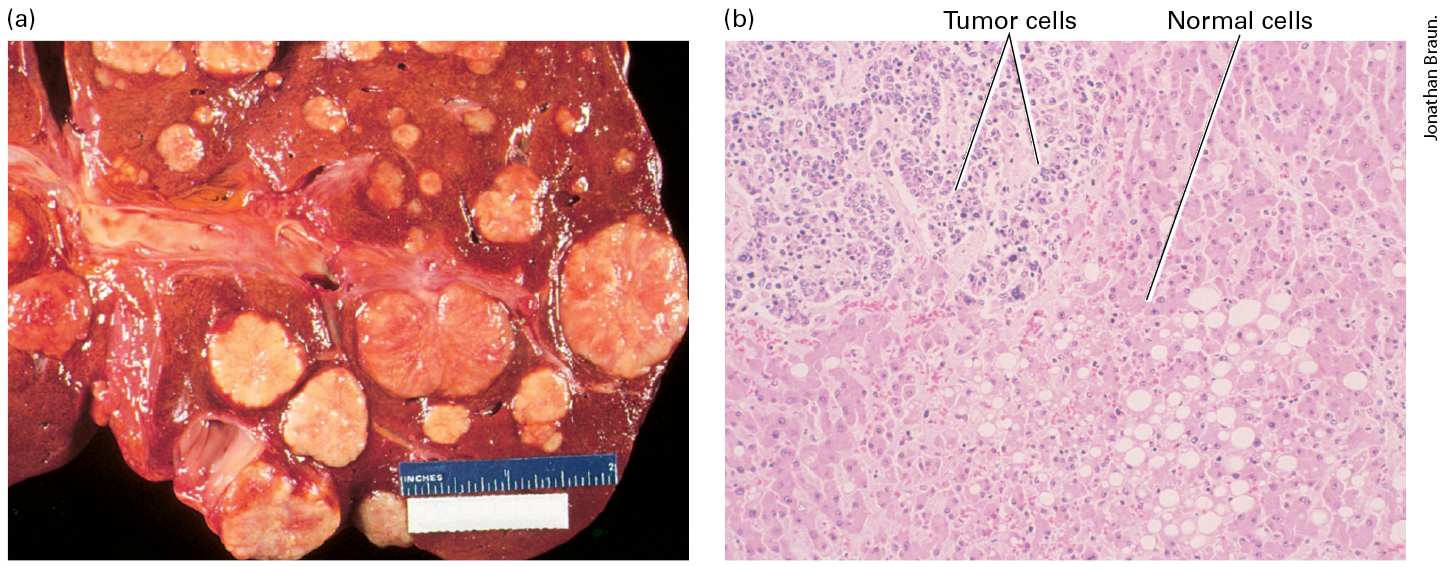
FIGURE 25-5 Gross and microscopic views of a tumor invading normal liver tissue. (a) The gross morphology of a human liver in which a metastatic lung tumor is growing. The white protrusions on the surface of the liver are the tumor masses. (b) A light micrograph of a section of the tumor in (a), showing areas of small, dark-staining tumor cells invading a region of larger, light-staining, normal liver cells.
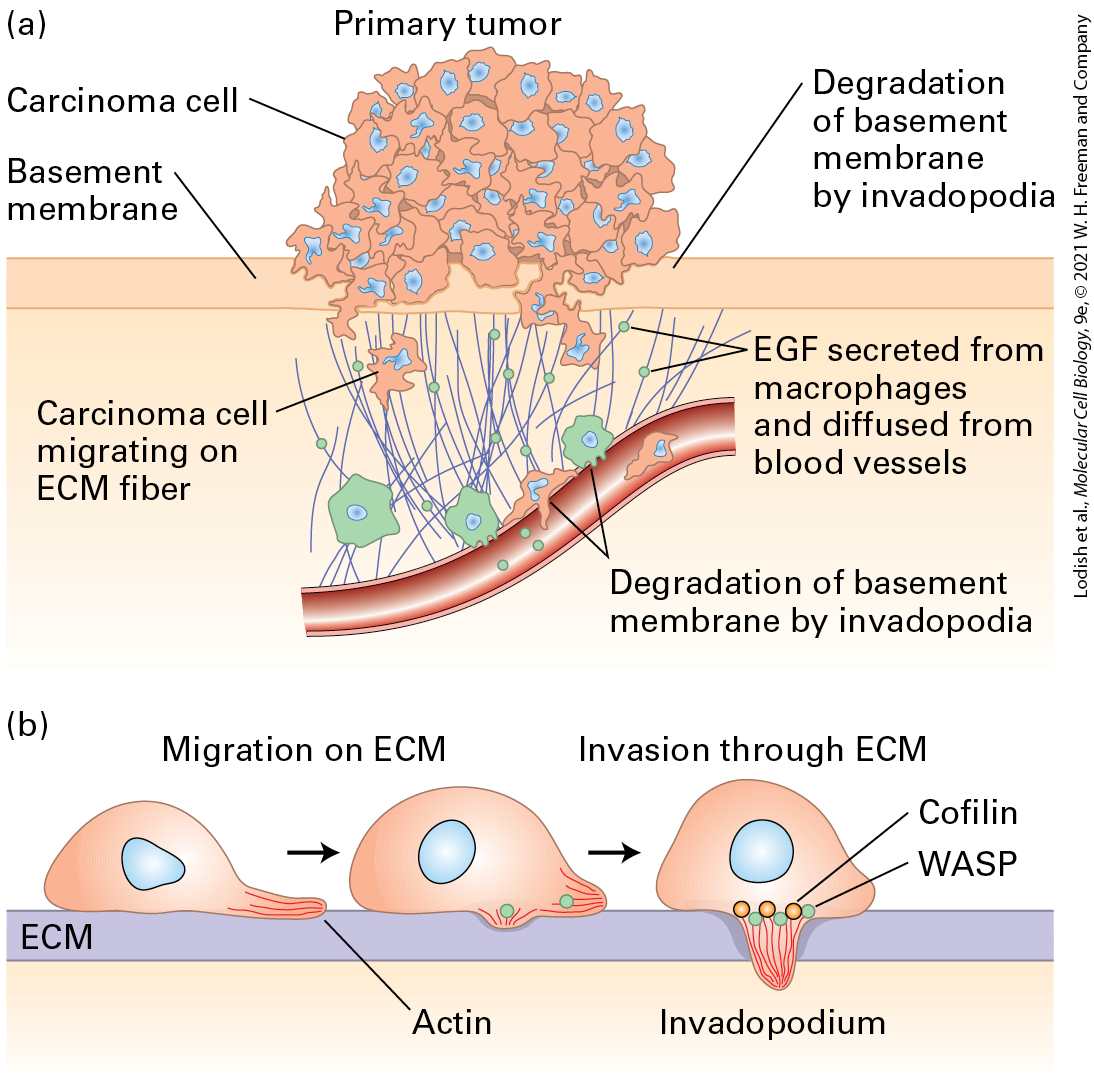
FIGURE 25-6 Metastasis. (a) First steps in metastasis, using breast carcinoma cells as an example. Cancer cells leave the main tumor and attack the basement membrane, using extracellular matrix (ECM) fibers to reach the blood vessels. The cancer cells can be
attracted by signals such as epidermal growth factor (EGF), which can be secreted by macrophages. At the blood vessels, they penetrate the layer of endothelial cells that forms the vessel walls and enter the bloodstream. (b) Carcinoma cells penetrate the extracellular matrix and blood vessel wall by extending invadopodia (actin-rich protrusions of the plasma membrane), which produce matrix metalloproteases and other proteases to open up a path. [Data from H. Yamaguchi, J. Wyckoff, and J. Condeelis, 2005, Curr. Opin. Cell Biol. 17:559.] Description The illustration labeled (a) shows a primary tumor mass with carcinoma cells. The tumor is on the surface of the basement membrane. The basement membrane is degraded by the invadopodia of the cancer cells. This is represented by small cancer cells moving through the membrane. On breaking through the basement membrane, the carcinoma cells migrate along extracellular matrix fibers. The carcinoma cells are attracted to blood vessels by the epidermal growth factor, which is released from macrophages and diffuses from blood cells. The illustration labeled (b) shows a carcinoma cell that extends invadopodia (looks like a foot being extended by an amoeba), filled with actin filaments along with the fiber, which allows migration in the extracellular matrix. Extension of the invadopodia into the extracellular matrix allows the cells to penetrate tissue. Proteins such as cofilin and W A S P help to disassemble the extracellular matrix. Normal cells are restricted to their place in an organ or tissue by cell-cell adhesion and by physical barriers such as the basement membrane, which underlies layers of epithelial cells and also surrounds the endothelial cells of blood vessels (see Chapter 20 and Figure 1-26). In contrast, cancer cells have acquired the ability to penetrate basement membranes using a cell protrusion called an invadopodium and to migrate to distant sites in the body (Figure 25-6b). A developmental process known as the epithelial-tomesenchymal transition (EMT) is thought to play a crucial role during
the process of metastasis in some cancers. During normal development, the conversion of epithelial cells into mesenchymal cells is a step in the formation of some organs and tissues. An EMT requires distinct changes in patterns of gene expression and results in fundamental changes in cell morphology, such as loss of cell-cell adhesion, loss of cell polarity, and the acquisition of migratory and invasive properties. During metastasis, the EMT regulatory pathways are thought to be activated at the invasive front of a tumor, producing single migratory cells. As the basement membrane disintegrates, some tumor cells enter the bloodstream, but fewer than 1 in 10,000 cells that escape the primary tumor survive to colonize another tissue and form a secondary, metastatic tumor. Much of preventative medicine is currently focused on developing methods to identify the rare tumor cells that circulate in the bloodstream. The ability to capture these circulating tumor cells would not only provide a powerful and noninvasive tool for the early detection of cancer, but also their analysis could provide insights into the nature of the disease and inform treatment. In order to produce metastases, tumor cells must not only enter the bloodstream, but also adhere to the lining of the blood vessel in a new location and migrate through it into the underlying tissue, in a process called extravasation (see Chapter 20). In order to seed a metastasis at a distant site, the tumor cells must not only disseminate, but also adapt to a foreign tissue environment. At least initially, metastatic tumor cells may not be well adapted to their new environment, but they are thought to evolve to survive and thrive in a foreign context. Little is known about the
molecular pathways that facilitate this adaptation, but mounting evidence suggests that some environments are more conductive to cancer cell colonization than others. Because metastasis is the most common reason for morbidity associated with cancer, much effort is being put into understanding which tumors will become metastatic and how metastasis occurs. Traditionally, the properties of tumor and normal cells have been assessed using microscopic tools, and the prognosis for many tumors could be determined, within certain limits, from their histology. However, the appearance of cells alone has limited information content, and better ways to discern the properties of cells are desirable, both to understand tumorigenesis and to arrive at meaningful and accurate decisions about prognosis and therapy. The advent of methods to assess a tumor’s patterns of RNA, protein, lipid, and metabolite production is allowing for a more detailed examination of tumor properties. Not surprisingly, primary tumors are often distinguishable from metastatic tumors by the RNAs and proteins that they produce. Analyses of global patterns of gene expression (described in Chapter 6) are now routinely used to predict patient outcomes and to determine the best course of treatment for many types of cancers. They will soon become the standard in determining treatment options. KEY CONCEPTS OF SECTION 25.1 How Tumor Cells Differ from Normal Cells
The genomes of most cancer cells acquire thousands of mutations and other genetic alterations that include point mutations, deletions, amplifications, chromosomal rearrangements and whole chromosome gains and losses. The changes in genetic makeup of cancer cells include oncogenic mutations that affect a wide variety of cellular functions. Oncogenic mutations in growth control pathways, cell cycle regulation, and in telomere maintenance are examples of how cancer cells acquire the ability to proliferate indefinitely. Cancer cells often exhibit altered metabolism, such as a switch of glucose metabolism to anaerobic glycolysis. Cancer cells lose contact inhibition, which allows them to grow in a mass outside the confines of a normal cell layer. Tumors are complex organs composed of different cell types that interact with their environment to obtain a maximal growth advantage. Tumors require angiogenesis, the formation of new blood vessels, in order to grow to a large mass. Cancer cells sometimes invade surrounding tissues, often breaking through the basement membranes that define the boundaries of tissues and spreading through the body to establish secondary areas of growth, a process called metastasis. Metastatic tumor cells acquire migratory properties in a process called the epithelialto-mesenchymal transition.
25.2 Genetic and Genomic Basis of Cancer
25.2 Genetic and Genomic Basis of Cancer Cancer can be considered to be a genetic disease in two different ways. Considering the mechanism of cancer formation, we know that tumors arise from somatic cells that have acquired a collection of mutations that give them the ability to proliferate when their neighbors cannot. In this sense, somatic mutations are the cause of cancer. When we consider the tumors themselves, we can see that they typically have acquired thousands of point mutations and genomic rearrangements; in this sense, mutations are a consequence of cancer. These contrasting relationships can be reconciled by viewing cancer progression as a form of Darwinian evolution acting on the growth of clones of cancer cells with cycles of mutation and selection. In this view, early in the process of cancer formation, the genome of a precancerous cell becomes highly mutable and the cells and its descendants begin to acquire random mutations. Although the vast majority of these mutations are not in genes and have little or no phenotypic effect on the cells that harbor them, a small fraction will have functional consequences. Those mutations that enable the precancerous cells to grow more rapidly, to avoid being eliminated, or to spread through the body will be selected for and will become part of the lineage that forms a tumor. Oncogenic mutations are those that are selected because they are causal drivers of cancer progression. Most modern cancer biology
Carcinogens Induce Cancer by Damaging DNA
research aims at identifying oncogenic mutations and understanding their underlying mechanism. In this section, we first consider the major causes of somatic mutations in cancer — these are environmental DNA-damaging agents known as carcinogens and either inherited or somatic mutations that prevent DNA damage from being repaired with high fidelity. We then consider the spectrum of mutations found in cancer genomes which include a small fraction of oncogenic driver mutations. Next we consider the major types of oncogenic mutations that include virus-borne onncogenes, and gain-offunction mutations and loss-of-function mutations in the genome. Finally, we consider oncogenic mutations that exert their effect through general changes in gene expression via chromatin structure or microRNA expression. Carcinogens Induce Cancer by Damaging DNA The ability of chemical carcinogens to induce somatic-cell mutations results from the DNA damage they cause as well as the errors introduced into DNA that result from the mechanisms to repair that damage. The strongest evidence that carcinogens act through mutagenesis comes from the observation that there is a strong correlation between the ability of a compound to cause mutations in DNA of cultured cells and to transform cells and induce cancer in animal models.
Some Carcinogens Have Been Linked to Specific Cancers
Although substances identified as chemical carcinogens have a broad range of chemical structures, they can be classified into two general categories. Direct-acting carcinogens, of which there are only a few, are highly reactive compounds that can react with and modify nucleotide bases in DNA so as to distort the normal pattern of base pairing. If the modified nucleotides are not repaired, they allow an incorrect nucleotide to be incorporated during replication. This class of carcinogens includes ethylmethane sulfonate (EMS), dimethyl sulfate (DMS), and nitrogen mustards. In contrast, indirect-acting carcinogens are relatively unreactive compounds that become modified to more reactive forms by cellular cytochrome P-450 enzymes. P-450 enzymes normally function to add electrophilic centers, such as OH groups, to nonpolar foreign chemicals in order to solubilize them so that they can be excreted from the body. For some compounds, modification by cytochrome P-450 increases the reactivity of the compound with DNA converting it into a mutagen and a carcinogen. Some Carcinogens Have Been Linked to Specific Cancers Since the earliest awareness of cancer as a disease intrinsic to the cells of the body, it became clear that at least some cancers resulted from the action of environmental poisons. As early as the eighteenth century, chimney sweeps’ exposure to soot was associated with a high incidence of
scrotal cancer, and the use of tobacco as snuff was associated with nasal cancer. Environmental chemicals were originally associated with cancer through experimental studies in animals. The classic test for a carcinogen is to repeatedly paint a test substance on the back of a mouse and look for development of local or systemic tumors in the animal. Such assays led to the purification of a pure chemical carcinogen, benzo(a)pyrene, from coal tar in 1933. Although chemical carcinogens are believed to be risk factors for many human cancers, a direct link to specific cancers has been established in only a few cases, the most important being lung cancer and the other cancers that are associated with smoking (of the larynx, pharynx, stomach, liver, pancreas, bladder, cervix, and more). Epidemiological studies first indicated that cigarette smoking was the major cause of lung cancer. The chemical benzo(a)pyrene, found in cigarette smoke as well as in coal tar, undergoes metabolic activation by cytochrome P-450 in the lungs (Figure 25-7) to form a potent mutagen that mainly causes conversion of cytosine (C) to adenine (A) bases, a transversion mutation. When applied to cultured bronchial epithelial cells, activated benzo(a)pyrene induces transversion mutations at many loci. Benzo(a)pyrene thus leaves a specific fingerprint in the DNA of cancer cells from heavy smokers. Cigarette smoke is known to contain more than 60 different carcinogens, and the overall profile of mutations in lung cancer appear as a complex pattern of overlapping signatures.
FIGURE 25-7 Enzymatic processing of benzo(a)pyrene to a more potent mutagen and carcinogen. Liver enzymes, particularly P-450 enzymes, modify benzo(a)pyrene in a series of reactions, producing 7,8-diol-9,10-epoxide, a highly potent mutagenic species that reacts with DNA primarily at the atom of a guanine (G) base. The resulting adduct, , causes polymerase to insert an A rather than a C opposite the modified G base. The next time the DNA is replicated, a T will be inserted opposite the A, and the mutation will be complete. Horizontal arrows indicate alterations toward greater potency, while vertical arrows indicate changes in the direction of reduced toxicity. The large “O” symbol represents the rest of the multi-ring structure shown in the complete benzo(a)pyrene molecule at the left. Description Benzo-(a)-pyrene is composed of five fused benzene rings. Oxidation of benzo-(a)- pyrene results in the formation of benzo-(a)-pyrene-7, 8-epoxide. Further oxidation leads to benzo-(a)-pyrene-7, 8-diol, then benzo-(a)-pyrene-7, 8-diol-9, 10-epoxide. At this point, the epoxide is highly mutagenic which is responsible for cancer development forms an adduct with guanine: the (+)-trans-anti-B-(a)-P-N superscript 2-d G adduct. Alternatively, the epoxide can be reduced to benzo-(a)-pyrene tetraol, or the D N A damaged by adduct formation can be repaired.
Familial Syndromes That Cause Loss of DNA Repair Can Lead to Cancer
In addition to chemical mutagens, electromagnetic radiation of sufficient energy to damage DNA is also carcinogenic. X-rays and gamma rays are of a category of radiation known as ionizing radiation because they have sufficient energy to strip electrons from atoms, generating ions. When ionizing radiation hits DNA it has sufficient energy to break a strand of DNA. X-rays have been known to cause mutations in Drosophila since the classic experiments of Hermann J. Muller were published in 1927. A connection between ionizing radiation and cancer was first indicated by the high incidence of cancer in the mouths of “radium girls” who worked in watch factories and ingested radium from licking the paintbrushes used to paint luminous dials. A causal relationship was solidified from a largescale study from atomic bomb survivors who were shown to have an increased incidence of cancer, principally leukemia, in proportion to their exposure to radiation from the bomb blast. Because the repair of DNA breaks is often error prone, ionizing radiation causes a very broad spectrum of mutations. UV light does not have the energy to break DNA strands but can interact with adjacent pyrimidine residues to create chemically altered DNA that is repaired by the nucleotide excision repair pathway (see Figure 5-18). The predominant signature of melanoma, a cancer caused by exposure to sunlight, is a preponderance of cytosine (C) to thymine (T) bases conversions, consistent with the known mutagenic effect of UV light. Familial Syndromes That Cause Loss of DNA Repair Can Lead to Cancer
Even without exposure to any external carcinogens or mutagens, normal biological processes generate a large amount of DNA damage. That damage is due to depurination reactions, to alkylation reactions, and to the generation of reactive species such as oxygen radicals, all of which alter DNA. It has been estimated that in every cell, more than 20,000 alterations to the DNA occur each day from reactive oxygen species and depurination. The vast majority of this damage is correctly repaired by the high-fidelity DNA-repair systems that are described in Chapter 5. As we will see, the large number of mutations found in most cancers results from defects in one or more of these repair systems. One well-recognized way for DNA repair to fail is by familial inheritance of a defect in one of the DNA-repair pathways. Table 25-1 shows the best understood familial syndromes that result from defects in DNA-repair pathways and their corresponding effects on an increase in the propensity to develop cancer. For example, people who inherit the disease xeroderma pigmentosum (XP) have a 1000-fold increase in the propensity to develop skin cancer. Seven of the eight known XP genes have been identified as components of the nucleotide excision-repair machinery, and the connection to skin cancer is clear since repair of DNA damage caused by UV light primarily requires nucleotide excision repair. Hereditary nonpolyposis colorectal cancer (HNPCC, also known as Lynch syndrome) causes a greatly increased propensity to develop colon cancers. HNPCC genes encode components of the mismatch-repair system; in the absence of mismatch repair, colon cancer progresses from benign polyps to fullfledged tumors much more rapidly than usual, presumably because the precancerous cells acquire oncogenic mutations at an increased rate.
Defects in mismatch repair should in theory increase the probability of oncogenic mutations in all cells in the body. The specificity of HNPCC for colon cancer is not well understood.
TABLE 25-1 • Some Human Hereditary Diseases and Cancers Associated with DNA-Repair Defects Disease DNA-Repair System Affected Sensitivity Cancer Susceptibility Symptoms Hereditary nonpolyposis colorectal cancer DNA mismatch repair UV irradiation, chemical mutagens Colon, ovary Early development of tumors Xeroderma pigmentosum Nucleotide excision repair UV irradiation, point mutations Skin carcinomas, melanomas Skin and eye photosensitivity, keratoses Bloom’s syndrome Repair of double-strand breaks by homologous recombination Mild alkylating agents Carcinomas, leukemias, lymphomas Photosensitivity, facial telangiectases, chromosome alterations Fanconi anemia Repair of double-strand breaks by homologous recombination DNA crosslinking agents, reactive oxidant chemicals Acute myeloid leukemia, squamouscell carcinomas Developmental abnormalities including infertility and deformities of the skeleton, anemia Hereditary Repair of Breast and Breast and
deficiency double-strand breaks by homologous recombination ovarian cancer ovarian cancer SOURCE: Modified from A. Kornberg and T. Baker, 1992, DNA Replication, 2d ed., W. H. Freeman and Company, p. 788; J. H. J. Hoeijmakers, 2001, Nature 411:366; and L. H. Thompson and D. Schild, 2002, Mutat. Res. 509:49. Double-strand breaks are particularly severe lesions because incorrect rejoining of double strands of DNA can lead to gross chromosomal rearrangements and translocations, such as those that produce a hybrid gene or bring a growth regulatory gene under the control of a different promoter or enhancer. Often the repair of such damage depends on using the homologous chromosome as a template (see Figure 5-21). The B and T cells of the immune system are particularly susceptible to DNA rearrangements caused by double-strand breaks created during rearrangement of their immunoglobulin or T-cell receptor genes, which explains the frequent involvement of these loci in leukemias and lymphomas. BRCA1 and BRCA2, genes implicated in human breast and ovarian cancers, encode important components of DNA-break repair systems. As we saw in Chapter 5, broken DNA ends can normally be repaired by recombination with the homologous chromosome, which is usually a high-fidelity process, returning the broken chromosome to the wild-type sequence. However, if homologous recombination fails, the broken ends can be repaired by nonhomologous end joining (NHEJ), which is highly mutagenic. Cells lacking either of the BRCA functions are unable
Somatic Mutations in the DNA Damage Response Pathway Are Oncogenic
to repair broken DNA strands by homologous recombination and they have no alternative but to repair by NHEJ, which introduces an insertion, a deletion, or a chromosome rearrangement at the site of the break. Somatic Mutations in the DNA Damage Response Pathway Are Oncogenic Inherited predispositions for a defect in DNA repair account for only a small fraction of tumors, yet the vast majority of cancer cells exhibit a much greater propensity for genome instability and a higher rate of mutagenesis than normal somatic cells. This propensity of cancer cells to acquire new mutations at a high rate is the result of somatic mutations acquired during cancer progression that compromise efficient DNA repair. The majority of these repair-defective somatic mutations do not directly affect the repair pathways themselves but alter the ways that cells respond to DNA damage. We saw in Chapter 19 that a general response to DNA damage of all dividing eukaryotic cells is to arrest the cell cycle to give the repair systems time to repair the damage before entry into mitosis. In brief outline, serine kinases ATM or ATR are recruited to sites of DNA damage and become activated (Figure 19-34). The activated kinases then signal the presence of DNA damage by phosphorylating p53 on a serine residue in the N-terminus of the protein. This phosphorylation causes the protein to evade ubiquitin-mediated degradation, leading to a marked stabilization of
p53 that activates transcription of the gene encoding p21, which in turn binds to and inhibits mammalian CDK2, CDK1, and CDK4/6 complexes (Figure 19-34). The net outcome of this signaling process is for cells with damaged DNA to activate cell cycle checkpoints causing an arrest in the cell cycle, allowing time for DNA repair to be completed before chromosome segregation in mitosis. Once DNA repair has been completed, p53 is degraded and the cell cycle will recommence to complete mitosis. If for some reason the DNA damage cannot be repaired, the cell will be arrested permanently and become senescent. This pathway that enables DNA-repair enzymes to operate effectively appears to be the aspect of DNA repair that is most vulnerable to somatic mutation. Most, if not all, human tumors have mutations either in p53 itself or in other proteins that regulate p53 activity. When the p53, , and S phase checkpoint control does not operate properly, damaged DNA can replicate, generating mutations and DNA rearrangements that are passed on to daughter cells and make their transformation into metastatic cells more likely. For example, loss of p53 function leads to a hundredfold or greater increase in the frequency of gene amplification. At the same time, loss of p53 function limits the duration of arrest, allowing cells with damaged DNA to enter mitosis prematurely. The activity of p53 is not limited to inducing cell cycle arrest, and in Section 25.4 we will describe the role of p53 in inducing apoptosis. The fact that cancer cells usually contain one or more defective DNA-repair systems provides an Achilles heel that allows tumors to be
Cancer Genome Sequencing Reveals an Enormous Diversity of Somatic Mutations
killed by chemicals that intentionally damage DNA. For example, traditional anticancer chemotherapeutic agents like cis-platinum cause DNA cross-links that tumor cells cannot efficiently repair, while drugs like doxorubicin and etoposide, which inhibit DNA topoisomerases, cause large numbers of DNA breaks during S phase, blocking entry into mitosis and resulting in tumor cell death. This loss of normal DNA repair, combined with the rapid proliferation of tumor cells, explains why commonly used chemotherapy drugs preferentially kill cancer cells, but do not kill the normal nonproliferating cells in the body. An exception to this are the rapidly proliferating normal cells in the bone marrow and immune system, which are also damaged to some extent by these drugs. This explains why cancer patients develop anemia and are susceptible to infections during chemotherapy treatment. Cancer Genome Sequencing Reveals an Enormous Diversity of Somatic Mutations The advent of next-generation DNA-sequencing technology has enabled rapid sequencing of thousands of cancer genomes and compilation of the vast array of somatic mutations in cancer cells. The large collection of cancer genome sequences shown in Figure 25-8 shows the complexity and variation in the types and numbers of different somatic mutations found in tumors. Normal somatic cells acquire an average of about one mutation per cell division. Given the total accumulation of mutations in cancer cells, the mutation rate during cancer progression is at least 1000
times greater than normal. Despite the enormous diversity of somatic mutations in cancer, a number of useful generalizations can be made.
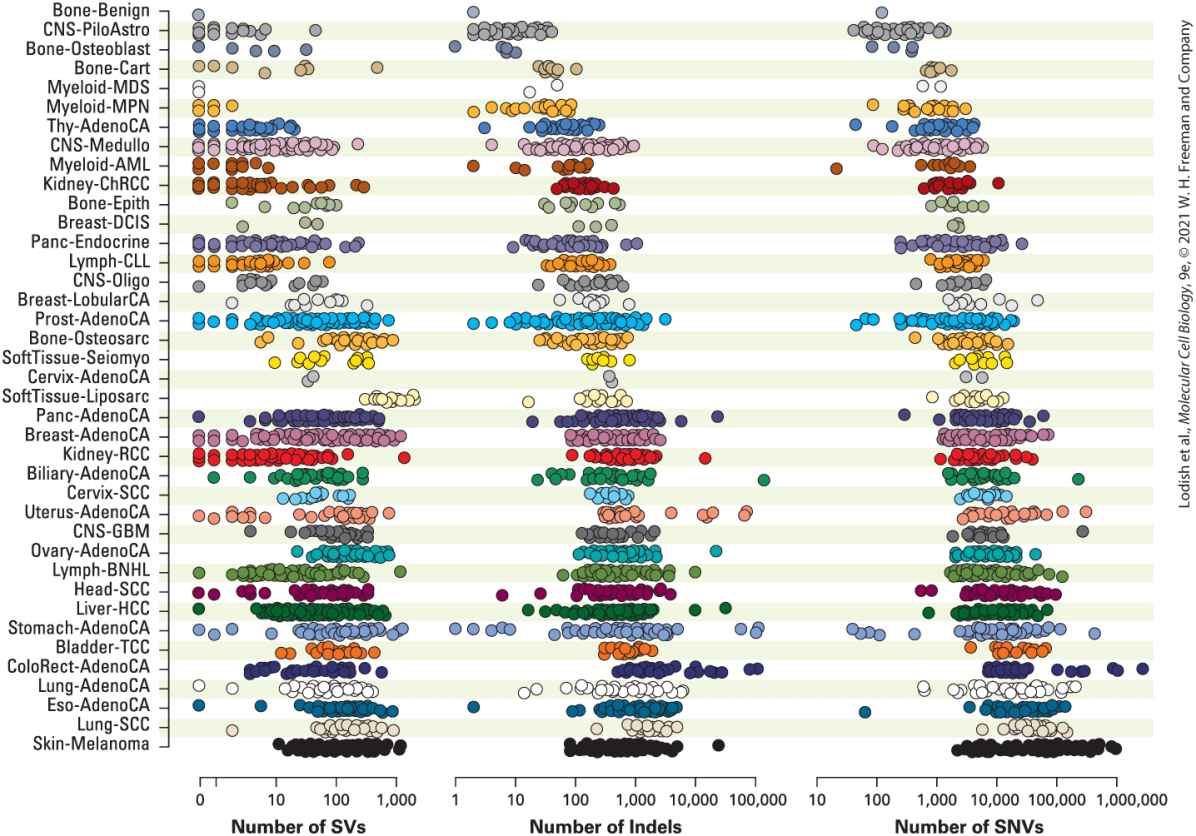
FIGURE 25-8 Sequenced cancer genomes carry a large number and wide variety of somatic mutations. A compendium of 2658 cancer genome sequences compared to matching normal somatic cells. The sequences are grouped into 38 tumor types and each dot specifies the number of mutations in a single tumor categorized as single nucleotide variants (SNVs), short insertions or deletions (Indels), or structural variants (SVs). [P. J. Campbell et al., 2020, “Pan-Cancer Analysis of Whole Genomes,” Nature 578:82–93, https://doi.org/10.1038/s41586-020-1969-6, (Creative Commons Attribution 4.0 International License).] Description The chart shows 38 normal somatic cell names in a column and next to each cell names are three groups of dots. The first group labeled number of structural variants has a
Oncogenes Were Discovered by Their Association with Tumor Viruses
scale range from 0 to 1000. The second group labeled number of indels has a scale range from 1 to 100,000. The third group labeled number of single nucleotide variants and has a scale range from 10 to 1,000,000. The number of dots next to each somatic cell type gets larger as the column moves to the bottom. 1. Somatic mutations can be grouped into three general types: single nucleotide variants (SNVs, or point mutations); insertions or deletions (Indels); or structural variants (SVs), which include chromosomal rearrangements, duplications, and copy number variations. 2. Although a typical tumor might contain 5000 SNVs, 500 Indels, and 50 SVs, there is enormous variation in number from one tumor to another even for the same type of cancer. 3. The cancers that have the greatest number and variation in mutations arise from tissues that have been exposed to mutagens: melanoma from sunlight and lung tumors from heavy smokers. 4. The types of mutations can reflect the underlying mechanism for increased mutagenesis. For example, breast tumors from women carrying a BRCA1 mutation tend to have a greater proportion of Indels and SVs than sporadic breast tumors. This is consistent with the expectation that a BRCA1 defect would lead to error-prone repair of DNA breaks, leading to Indels and SVs. Oncogenes Were Discovered by Their Association with Tumor Viruses Now that we have seen some of the ways that cancer cells acquire a large number of random somatic mutations, we turn to the problem of
identifying the small fraction of these mutations that have functional consequences and have contributed to cancer progression. With the modern capability of analyzing entire genomes, we now know that a typical tumor may have approximately five known driver mutations that contribute to the uncontrolled cell proliferation, avoidance of programmed cell death, and unstable genome that are hallmarks of cancer cells. Finding a few driver mutations among tens of thousands of somatic mutations has only been possible recently; the first known genetic drivers of oncogenesis were revealed in cases in which a single powerful driver was activated through an unusual genetic event. The genes that caused the oncogenic transformation are known as oncogenes. Pioneering studies by Peyton Rous beginning in 1911 led to the initial recognition that a virus could cause cancer when injected into a suitable host animal. Many years later, molecular biologists showed that his Rous sarcoma virus (RSV) is a retrovirus whose RNA genome is reversetranscribed into DNA, which is then incorporated into the host-cell genome (see Figure 5-44). In addition to the normal genes present in all retroviruses, oncogenic transforming viruses such as RSV contain an oncogene — in the case of RSV, the v-src gene. Subsequent studies with mutant forms of RSV demonstrated that only the v-src gene, not the other viral genes, was required for cancer induction. In the late 1970s, scientists were surprised to find that normal cells from chickens and other species contain a gene that is closely related to the RSV v-src gene. This normal cellular gene, a proto-oncogene, is commonly distinguished from the viral gene by the prefix c for “cellular”
Single Oncogenic Drivers Can Be Activated by Chromosome Rearrangements
(c-SRC). The product of this gene, c-Src, is a cytosolic protein tyrosine kinase which participates in many signal transduction pathways. RSV and other oncogenic transforming viruses are thought to have arisen by incorporating a normal host cellular proto-oncogene into their genome. Subsequent mutations in the incorporated gene then converted it into a dominantly acting oncogene, encoding a constitutively active kinase that is able to transform host cells even in the presence of the normal c-SRC proto-oncogene. When this phenomenon was first discovered, it was startling to find that these dangerous viruses were turning the hosts’ own genes against them. In other cases, retroviruses can cause cancer not by carrying an oncogene but by integrating into the host-cell DNA near a cellular proto-oncogene and activating its expression. For example, in the cells from tumors caused by avian leukosis virus (ALV), the retroviral DNA is inserted near the MYC gene. These cells overproduce MYC protein, which causes abnormally rapid proliferation of cells, initiating the cancer progression. Additional subsequent mutations have to occur before a full-fledged tumor becomes evident. Single Oncogenic Drivers Can Be Activated by Chromosome Rearrangements In the 1960s, researchers first realized that some cancers harbor characteristic chromosome rearrangements that could be detected by light
microscopy. Chronic myelogenous leukemia (CML), a common leukemia in humans, was found to be associated with the Philadelphia chromosome (Figure 25-9a), which is generated by a translocation between chromosomes 22 and 9. The two chromosomes exchange their terminal regions, which leads to a characteristic alteration in the size of chromosome 22. Subsequent cloning and analysis of the DNA at the breakpoint of this translocation showed that a new fusion protein, called BCR-ABL, is generated. This hybrid protein kinase phosphorylates proteins that the wild-type ABL kinase normally does not phosphorylate, thereby inappropriately activating many intracellular signal-transducing proteins. If this translocation occurs in a hematopoietic cell in the bone marrow, the activity of the chimeric BCR-ABL oncogene results in the initial phase of CML, characterized by an expansion in the number of white blood cells.
FIGURE 25-9 BCR-ABL protein kinase. (a) Origin of the Philadelphia chromosome from a translocation of the tips of chromosomes 9 and 22 and the oncogenic fusion protein formed by that translocation. (b) The BCR-ABL fusion protein is a constitutively active kinase that phosphorylates multiple signal-transducing proteins. Imatinib binds near the active site of BCR-ABL and stabilizes the inactive form that lacks kinase activity. (c) Imatinib bound to the BCR-ABL active site. [Data from B. Nagar et al., 2002, Cancer Res. 62:4236, PDB ID 1iep.] Description In the illustration labeled (a), chromosome 9 (represented as a tall bubble letter X) contains the A B L gene, and chromosome 22 (represented as a short U-shaped structure with appendages at the top) contains the B C R gene. Translocation results in the transfer of the tips of these chromosomes and the formation of the d e r (9) and d e r (22) chromosomes. The d e r (22) chromosome, also known as the Philadelphia chromosome, contains the B C R and A B L genes where the A B L gene has been attached to the breakpoint in the B C R gene. Transcription and translation of the B C R-A B L gene result in the B C R-A B L fusion protein, which has a functional active site. The illustration labeled (b) shows two ways that the B C R-A B L fusion protein can act. The top example shows the B C R-A B L as a pink structure with a blue structure entering at its side. The blue structure is labeled substrate, for example, J A K 2, S T A T 5. A right arrow points to the B C R-A B L with the blue substrate separated, now with a yellow phosphate circle at its tip. The label reads, substrate activated by phosphorylation. The lower example starts with the B C R-A B L with the gray structure attached. The gray structure is labeled imatinib binds to active site and inhibits substrate binding. The blue substrate is separated. A right arrow leads to a label; tumor cell cannot proliferate. The illustration labeled (c) shows the three-dimensional ribbon model of the B C R-A B L fusion protein with imatinib attached close to the center.
The discovery of the Philadelphia chromosome and the critical oncogene it creates, BCR-ABL, combined with the discovery of the molecular action of the ABL protein, together have led to a powerful new therapy for CML. After a painstaking screening of compounds that can inhibit the enzymatic activity of ABL kinase, a highly specific inhibitor named imatinib (Gleevec) was identified as a possible treatment for CML. Imatinib, which binds directly to the active site of the ABL kinase and holds it in an inactive conformation to inhibit its kinase activity, is lethal to CML cells while sparing normal cells (see Figure 25-9b, c). After clinical trials showing that imatinib is remarkably effective in treating CML despite some side effects, it was approved by the FDA in 2001 as the first cancer drug targeted to a signal-transducing protein unique to tumor cells. Imatinib inhibits several other tyrosine kinases that are implicated in different cancers and has been successful in trials for treating those diseases, including forms of gastrointestinal tumors, as well. There are 90 functional protein tyrosine kinases encoded in the human genome, and drugs that inhibit several of these are proving useful in treating other types of cancers. The development of imatinib represents one of the great successes of an approach to cancer therapy known as targeted therapy. In its general outline, the idea behind targeted therapy is to find the oncogenic drivers for a particular type of cancer and then to design a drug that reverses the effect of the oncogenic mutation. The case of targeting BCR-ABL that is the oncogenic driver of CML demonstrates both the power of this method and also reveals its limitations. CML is unusual in that it forms as the result of a single oncogenic driver whose effect can be reversed to cure the
Inherited Predisposition for Cancer Enabled Identification of Some Oncogenic Drivers
disease. As we will see, the majority of cancers result from multiple driver mutations; reversing the effect of any one of them would not cure the disease. In addition, we have seen that most cancers have a high mutation rate and thus would rapidly acquire mutations that confer resistance to a specific enzyme inhibitor. Indeed, CML tumors eventually acquire mutations in the gene encoding the BCR-ABL fusion protein that prevent binding of imatinib. Researchers have identified molecules that specifically inhibit some of these mutant BCR-ABL kinases; these are used as a second-line therapy for CML. The twin challenges of multiple pathways affected and generation of drug resistance through mutation are features of the underlying genetic complexity of most cancers and remain the most significant obstacles to development of targeted therapies. Inherited Predisposition for Cancer Enabled Identification of Some Oncogenic Drivers Another way that individual genes responsible for driving progression were identified was through genetic analysis of syndromes that have a hereditary predisposition to certain cancers. For most of these syndromes, individuals inherit from a parent a loss of function mutation in one allele of the gene; then a somatic mutation of the second allele of the gene initiates cancer progression. A classic inherited cancer syndrome is retinoblastoma, which is caused by loss of function of RB. As discussed in
Chapter 19, the protein encoded by RB regulates cell cycle entry.
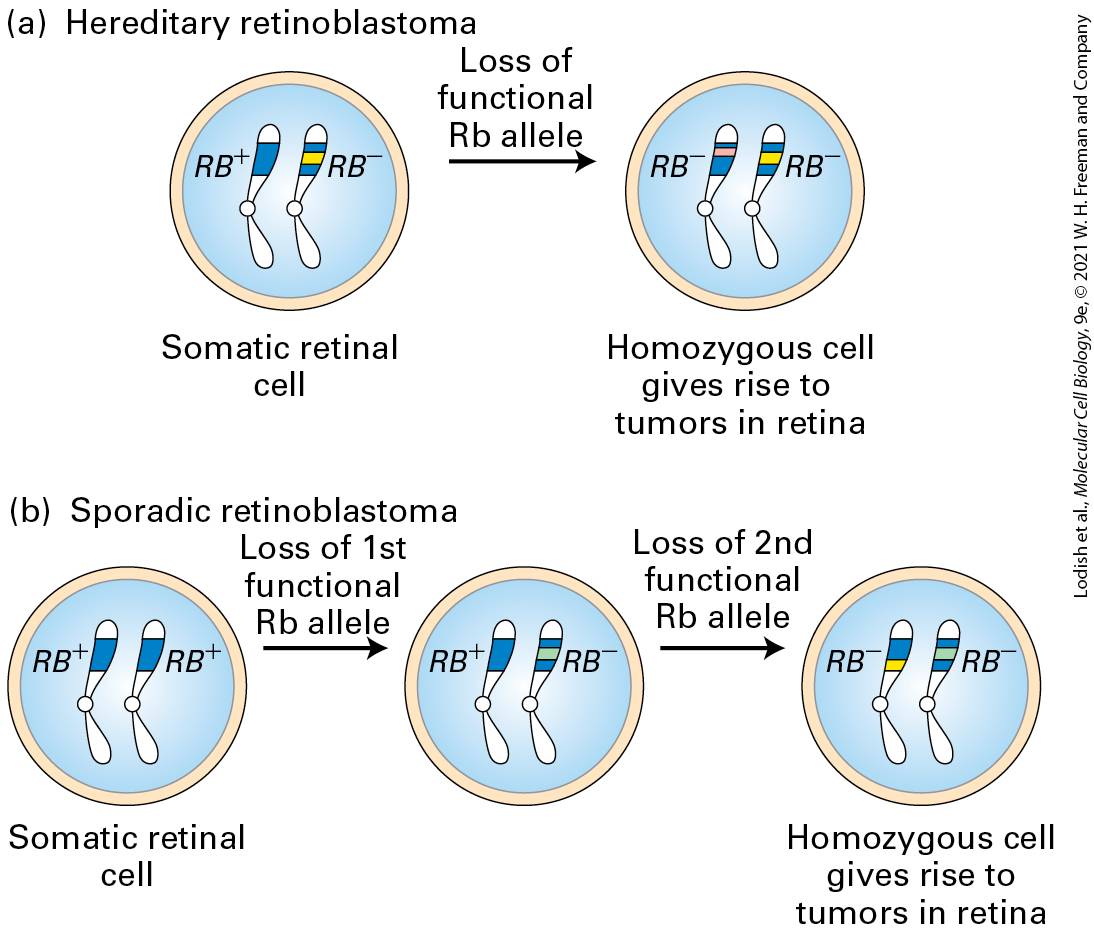
Children with hereditary retinoblastoma inherit one defective copy of the RB gene, sometimes seen as a small deletion on one of the two copies of chromosome 13. These children develop multiple retinal tumors early in life and generally in both eyes. The loss or inactivation of the normal RB gene on the other chromosome is an essential step in tumor formation, giving rise to a cell that produces no functional Rb protein (Figure 2510a). Individuals with sporadic retinoblastoma, in contrast, inherit two normal RB alleles, each of which has undergone a loss-of-function somatic mutation in a single retinal cell (Figure 25-10b). Because losing both copies of the RB gene in a somatic cell is far less likely than losing one, sporadic retinoblastoma in individuals with two normal copies of the RB gene is very rare and usually affects only one eye.
FIGURE 25-10 Role of spontaneous somatic mutation in retinoblastoma. This disease is marked by retinal tumors that arise from cells carrying two mutant alleles. (a) In hereditary (familial) retinoblastoma, a child inherits a normal allele from one parent and a mutant allele from the other parent. When the second normal allele is lost in a heterozygous somatic retinal cell, a cell is generated that lacks any RB gene function. (b) In sporadic retinoblastoma, a child inherits two normal alleles. Two separate RB loss events must occur in a particular retinal cell to produce a cell lacking all RB function. Description In the illustration labeled (a), the somatic retinal cell contains two chromosomes, one R B plus, and the other R B minus. A right arrow labeled loss of functional R b allele from the somatic retinal cell leads to a homozygous R B minus cell that gives rise to retinal tumors. In the illustration labeled (b), the somatic retinal cell contains
homozygous R B plus cell. A right arrow labeled loss of first functional R b allele from the somatic retinal cell leads to a heterozygous R B plus and R B minus cell. A right arrow labeled loss of second functional R b allele from the heterozygous R B cell leads to a homozygous R B minus cell that gives rise to retinal tumors. If retinal tumors are removed before they become malignant, children with hereditary retinoblastoma often survive until adulthood and produce children, but they are at an increased risk of developing other types of tumors later in life. Because their germ cells contain one normal and one mutant RB allele, these individuals will, on average, pass on the mutant allele to half their children and the normal allele to the other half. Children who inherit the normal allele are normal if their other parent has two normal RB alleles. However, those who inherit the mutant allele have the same enhanced predisposition to develop retinal tumors as their affected parent, even though they inherit a normal RB allele from their other, normal parent. Thus the tendency to develop retinoblastoma is inherited as a dominant trait: one mutant copy is sufficient to predispose a person to develop the cancer. As we will see, many human tumors (not just retinal tumors) contain mutant RB alleles or mutations affecting other components of the RB pathway; most of these tumors arise as the result of somatic mutations. Although hereditary retinoblastoma cases number about 100 per year in the United States, about 100,000 other cancer cases each year involve RB mutations acquired in somatic cells.
Oncogenic Driver Mutations Have Been Identified in Many Genes
Estimates vary, but hereditary cancers (cancers that arise due in part to an inherited version of a gene) are thought to constitute about 10 percent of human cancers. Further work tracing the contributions of human genes seems likely to increase the percentage. It is important to remember, however, that the inherited germ-line mutation alone is not sufficient to cause tumor development. Not only must the inherited normal allele be lost or inactivated, but also mutations affecting other genes must occur for cancer to develop. Thus a person with a recessive tumor suppressor gene mutation can be exceptionally susceptible to environmental mutagens, such as radiation. Mutation in only one copy of a tumor suppressor gene itself typically does not cause cancer because the remaining normal allele prevents aberrant growth. However, the subsequent loss or inactivation of the remaining normal allele in a somatic cell, referred to as loss of heterozygosity (LOH) causes cancer to develop. Three mechanisms exist that can cause the loss of the normal allele. First, the normal allele can become inactive due to a de novo inactivating mutation or deletion. Second, chromosome missegregation can cause loss of the chromosome carrying the normal allele. Neither mechanism is particularly common. By far the most frequent mechanism for LOH is mitotic recombination between a chromatid bearing the normal allele and a homologous chromatid bearing a mutant allele. Subsequent chromosome segregation can generate a daughter cell that is homozygous for the mutant tumor-suppressor allele.
Oncogenic Driver Mutations Can Be Identified by Comparing Cancer Genomes
Oncogenic Driver Mutations Have Been Identified in Many Genes After discovery of the first oncogenes carried by tumor viruses, in the ensuing 40 years dozens of different genes that can carry oncogenic driver mutations have been identified. These genes were typically identified by studying the unique genetic markers of a particular tumor sample or transformed cell line. The greatest advances have come from studying tumor types, like pediatric cancers and rare types of adult cancer, where the total number of genetic changes is small. Most oncogenic drivers were first identified by one of the following six approaches (we have already seen examples of the first five): 1. Identification of an oncogene associated with a tumor-causing virus. 2. Identification of a gene activated to cause cancer by insertion of a retrovirus. 3. Identification of a gene at the breakpoint in a chromosome rearrangement associated with cancer. 4. Mapping of a gene that causes a familial predisposition to cancer. 5. Identification of a gene that, when activated or up-regulated, can cause cell transformation. 6. Identification of a gene specifically up-regulated in cancer cells. Oncogenic Driver Mutations Can Be Identified by Comparing Cancer Genomes
All of the oncogenic driver mutations in a given tumor should be discoverable by comparing the genomic sequence of the tumor with that of the sequence of a normal tissue-matched somatic cell. The difficulty with this approach is that for most tumors, out of tens of thousands of somatic mutations only a few will be oncogenic driver mutations. However, when a large number of different cancer genome sequences are compared, patterns emerge that allow driver mutations to be identified among the much larger number of adventitious somatic mutations in tumor cells. The most obvious pattern would be to look for mutations in the same gene appearing across multiple tumors of the same type. This method is based on the concept that cancer cells of the same type have similar morphology, metabolism, and regulatory and gene expression networks and therefore are likely to be the result of similar sets of oncogenic driver mutations. More sophisticated analysis of the sequence information will identify common mutations of the same type across tumors. For example, mutations in RB tend to be deletions that cause a loss of gene function, whereas mutations in Ras are usually point mutations that have a gain of function. This type of analysis, applied to 2500 different tumor sequences from different types of cancer, yields a set of common oncogenic drivers shown in Figure 25-11. The average number of oncogenic driver mutations found in these tumors was five and the most common driver gene mutated is p53 (TP53), which was found in 77 percent of the tumors. Although at first inspection the number and variety of oncogenic drivers can be bewildering, almost all of these genes had previously been associated with cancer progression and their basic cellular functions are known. Moreover,
many of the oncogenic driver genes operate in a relatively small number of regulatory pathways and cellular processes, as shown in Table 25-2. In Section 25.3, we describe in greater detail the function of oncogenic drivers that participate in cell growth control pathways; in Section 25.4, we consider oncogenic drivers that prevent programmed cell death and allow evasion of immune surveillance.
FIGURE 25-11 Oncogenic driver mutations identified in sequenced cancer genomes. A compendium of 2583 cancer genome sequences from 26 different tumor types were searched for shared oncogenic driver mutations. The columns of the table represent different types of cancer and the rows represent the most prevalent driver mutations in order of frequency. The numbers in the table represent the number of tumors of a given type found with a given driver mutation. The colored bars represent the proportion of driver mutations of a given type. The most common types of mutations are single nucleotide
Oncogenic Drivers Can Be Gain-of-Function or Loss-of-Function Mutations
variants in the coding sequence (yellow), which can be either loss-of-function mutations as in the case of TP53 or gain-of-function mutations as in the case of KRAS. Common loss-offunction mutations are structural variants which lead to a deleted or truncated gene (red) and less common intron splicing mutants (light red). Common gain-of-functions mutations are structural variants that lead to an amplified gene or a gene fusion (green) and less commonly promoter mutations (light green). Tumor suppressor genes (labeled red) predominantly carry loss-of function oncogenic driver mutations, whereas oncogenes (labeled green) carry gain-of function oncogenic driver mutations. [Data from ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, 2020, Nature 258:82.] Description The chart is organized so that the most common cancers are at the top, sorted into columns labeled by the normal cells they occur in. The total number of patients that were studied for each row is at the right. Between this total and the individual columns is a bar color-coded by the type of single nucleotide variants and the structural variants of each cancer type. Next to the total number of patients is a column that shows the name of genes involved in each row. Oncogenic Drivers Can Be Gain-ofFunction or Loss-of-Function Mutations In the simplest terms, gene function can be altered by a mutation in one of two ways. A gene can be partially or completely inactivated by a mutation to cause a loss of function, or a gene can possibly acquire from a mutation increased activity or a new activity that could be considered a gain of function. It is usually possible to deduce from a cancer genome sequence,
whether an oncogenic mutation causes a loss of function or a gain of function and thus to obtain important information about the mechanism by which the mutation drives cancer progression. Typical loss-of-function mutations: 1. A deletion of all or part of a gene. 2. A point mutation in the coding sequence that introduce a stop codon or a frameshift. Typical gain-of-function mutations: 1. Duplication or amplification of the gene leading to overproduction of the encoded protein. 2. A point mutation that results in a hyperactive or constitutively active protein product. 3. A promoter mutation that results in increased expression of the protein product. 4. A DNA rearrangement that places the gene under the control of an enhancer or promoter that results in increased or inappropriate expression of the protein product. Genes for which the oncogenic forms arise because of a loss-of-function mutation are usually known as tumor suppressor genes. RB is the prototypical tumor suppressor gene and we have already seen that retinoblastoma arises because of loss-of-function mutations in both copies of the RB gene. We could also deduce that RB was a tumor suppressor gene from the cancer genome sequences in Figure 25-11, since most of the
Tumor Suppressor Genes and Oncogenes Often Operate in the Same Pathway
oncogenic mutations in RB are deletions. Similarly, it is evident that p53 is a tumor suppressor gene since about half of the oncogenic mutations in the TP53 gene are deletions. Genes for which the oncogenic forms arise because of a gain-of-function mutation are usually known as oncogenes. For example, the oncogenic mutation in the RAS gene is due to a single base change that permanently activates the Ras protein in the active GTP-bound state and inappropriately activates the MAP kinase pathway, generating an excessive or uncontrolled proliferation-promoting signal. Almost all of the RAS mutations identified in tumors are of this type. In contrast, the MYC oncogene typically acquires a gain of function by amplification of the MYC gene, which increases the abundance of the protein product. Tumor Suppressor Genes and Oncogenes Often Operate in the Same Pathway Most of the common oncogenic mutations shown in Figure 25-11 affect pathways that regulate growth control or cell cycle progression. These pathways contain some elements that act positively, but they usually contain negatively acting elements as well (Figure 25-12). For pathways that promote cell growth and cancer progression, gain-of-function oncogenic mutations can occur in pathway elements that act positively, whereas loss-of-function mutations may occur in pathway elements that act negatively. Thus both oncogenes and tumor suppressor genes can be
found that function in the same pathway. However, most tumors usually have only one oncogenic mutation that activates a given pathway. For example, many tumors have oncogenic mutations in Ras or Raf (see Figure 25-11), but very few have mutations in both genes since these proteins operate in the same signal transduction pathway (see Figure 16-13 and
FIGURE 25-12 A typical cell growth pathway showing how gain-of-function or loss-of- function mutations in the same pathway can drive cancer progression. A typical pathway is shown that activates cell proliferation by gene transcription and initiation of cell cycle progression in response to an extracellular mitogenic signal. If proteins that act positively in this pathway, such as the receptor protein or an intracellular signaling protein, can become oncogenes by acquiring a gain-of-function mutation. In contrast, a negative regulator of the pathway may activate the pathway because of a loss-of-function mutation. In such a case the corresponding gene for the negative regulator would be a tumor suppressor gene.
Description The illustration shows a large rectangle with a cell membrane. The signal receptor is between the exterior cell surface and the cytoplasm. A downward arrow moves to a green structure, labeled signal transduction protein. A text box next to this structure reads, possible oncogene, gain-of-function mutations are oncogenic. Downward arrows from the signal transduction protein point toward the nucleus of the cell that has a D N A helix with a transcription factor. Another possibility is presented by an orange structure next to the signal transduction protein, which is labeled negative regulator of signal transduction. A text box next to this structure reads, possible tumor suppressor gene, loss-of-function mutations are oncogenic.
TABLE 25-2 • Common Types of Mutations That Drive Cancer Progression Cellular Process Protein (Human Gene) Type of Mutation (Gain of Function or Loss of Function) Activation of a mitogenic growth pathway (e.g., the MAP kinase pathway) Ras (KRAS) Raf (BRAF) Nf1 (NF1) Activating coding (GoF) Activating coding and promoter (GoF) Mostly deletion (LoF) Inactivation of a growth inhibitory pathway (e.g., TGFβ pathway) Smad4 (SMAD4) Mostly deletion (LoF) Activation of cell cycle initiation p16 (CDKN2A) Rb (RB) Mostly deletion (LoF) Mostly deletion (LoF) Gene amplification (GoF)
MicroRNAs Can Promote and Inhibit Tumorigenesis
Cyclin D (CCND1) Sustaining chromosome replication by elongation of telomeres Telomerase (TERT) Promoter (GoF) Loss of DNA damage checkpoint p53 (TP53) ATM (ATM) Deletion and coding (LoF) Deletion and coding (LoF) Avoidance of apoptosis p53 (TP53) Bcl2 (MCL1) PI-3 kinase (PIK3CA)
(PTEN) Deletion and coding (LoF) Gene amplification (GoF) Activating coding (GoF) Mostly deletion (LoF) Change in chromatin state by SWI/SNF
(ARID1A) Mostly deletion (LoF) Activation of nuclear transcription factors MYC (MYC) β-catenin (CTNNB1) Gene amplification (GoF) Activating coding (GoF)
MicroRNAs Can Promote and Inhibit Tumorigenesis In the last two decades, a new class of oncogenic factors has emerged. Noncoding RNAs (RNAs that do not encode proteins), especially microRNAs (miRNAs), play a critical role in tumorigenesis. As discussed in Chapter 9, generation of miRNAs typically involves the transcription of a precursor RNA that, through a number of processing steps, is trimmed down to a mature miRNA that is 20–22 nucleotides long. The mature miRNA usually base-pairs with the untranslated region (UTR) of its target RNA and inhibits its translation or sometimes causes its degradation (Chapter 9). To date, more than 1500 miRNAs have been identified in humans and have been implicated in the regulation of as many as 30 percent of the cell’s mRNAs, with fundamental roles in cell proliferation, differentiation, and apoptosis. A number of miRNAs have also been shown to function as tumor suppressor genes or oncogenes. The first known role for miRNAs in tumorigenesis was revealed by the analysis of chromosomal region 13q14.3. This genomic region is found deleted in most cases of chronic lymphocytic leukemia (CLL), prostate cancer, and pituitary adenomas. Characterization of the disease-causing deletion showed the absence of two distinct miRNAs encoded in the deleted region. Deletions of the homologous miRNAs in a mouse strain caused the mice to develop CLL, showing that these miRNAs act as tumor suppressors. The two miRNAs appear to repress translation of certain cell proliferation proteins; in the absence, proliferation of B cells is increased.
Epigenetic Changes Can Contribute to Tumorigenesis
Similarly, the let-7 family of miRNAs has been implicated in lung, colon, breast, and ovarian cancer. Let-7 miRNAs repress the translation of Ras mRNA. Thus in the absence of the miRNAs, Ras is constitutively overproduced, contributing to tumorigenesis. Let-7 miRNAs have other targets as well, such as the oncogenic transcription factor MYC, which we will discuss in detail in the next section. A general theme that emerges in the study of miRNAs in cancer is that each miRNA has multiple targets and therefore ample opportunities to contribute to tumorigenesis. Other miRNAs have also been found to be overexpressed in cancer. Of particular interest is miR-21, which is overexpressed in most solid tumors, including glioblastomas and breast, lung, pancreatic, and colon tumors. This miRNA targets several tumor suppressor genes, among them the gene encoding the PTEN phosphatase, which normally inhibits the kinase signal transduction pathway (Chapter 16). Much more needs to be learned about how miRNAs contribute to tumorigenesis, but it is clear that through their ability to regulate many different genes, they can influence disease progression in more than one way. Epigenetic Changes Can Contribute to Tumorigenesis We have just seen how mutations can be oncogenic by causing a loss of function of a tumor suppressor gene or a gain of function in an oncogene. As we saw in Chapter 9, gene expression can either decrease or increase according to the chromatin state. Genes with global effects on chromatin
state through changes in DNA methylation, as well as changes in the activity of histone-modifying enzymes or chromatin-remodeling complexes, are now recognized as major drivers of tumorigenesis. DNA methylation occurs at cytosines of CpG islands, which are found largely in promoters of genes. Methylation of these Cs leads to repression of the promoters. A large fraction of colorectal cancers are characterized by DNA hypermethylation. DNA hypomethylation is also a hallmark of cancer. The promoters of many genes involved in cancer are hypomethylated, and expression of the genes under their control is therefore increased. For example, 25 percent of acute myeloid leukemias are characterized by DNA hypomethylation that is due to inactivating mutations in an enzyme that catalyzes the methylation of CpG dinucleotides. Genes encoding chromatin modifiers and regulators have also emerged as drivers of tumorigenesis. Systematic, whole genome sequencing of many tumor types has revealed highly recurrent alterations in approximately 40 genes encoding epigenetic regulators. Recurrent mutations were found in genes encoding enzymes that modify histones or proteins that bind to these post-translational modifications of histones. Genes encoding histone methyl transferases, histone demethylases, and histone acetyl transferases have all been found mutated in a wide variety of tumors. Interestingly, tumors typically harbor only a single mutated allele of a gene encoding a chromatin-modifying enzyme, indicating that these mutations are haploinsufficient. Presumably, losing both alleles would kill the cell, but having
only one functional allele alters the extent of histone modification and thus the expression of target genes sufficiently to promote tumorigenesis. Central among the chromatin-remodeling factors implicated in cancer are the SWI/SNF complexes. These large and diverse multiprotein complexes, which have an ATP-dependent helicase at their core, often control histone modification and chromatin remodeling (see Chapter 8). For example, they can cause changes in the positions or structures of nucleosomes, making genes accessible or inaccessible to DNA-binding proteins that control transcription. If a target gene is normally activated or repressed by SWI/SNF-mediated chromatin changes, mutations in the genes encoding SWI or SNF proteins will cause changes in the expression of that gene. Studies with transgenic mice suggest that SWI/SNF normally plays a role in repressing the expression of E2F genes, thereby inhibiting progression through the cell cycle. Thus loss of SWI/SNF function can lead to overgrowth and perhaps cancer. Recent evidence from humans and mice has strongly implicated the SNF5 gene in cancer. The SNF5 protein is a core member of the SWI/SNF complex. In humans, inactivating somatic SNF5 mutations cause rhabdoid tumors, which most commonly form in the kidney, and an inherited (familial) disposition to form brain and other tumors. Subsequent studies have found genes encoding various BAF proteins, which are also subunits of the SWI/SNF complex, to be mutated in 40 percent of renal cancers, 50 percent of ovarian cancers, and a high fraction of liver and bladder cancers. In summary, epigenetic misregulation has emerged as a major contributor to tumorigenesis. In hindsight, this notion is probably not
surprising, given that epigenetic regulation offers the opportunity to change the expression of many factors and regulatory pathways simultaneously. KEY CONCEPTS OF SECTION 25.2 Genetic and Genomic Basis of Cancer Oncogenic mutations can result from DNA-copying errors or through the effect of environmental mutagens such as DNA-alkylating agents, UV light, and ionizing radiation. Mutagens that can cause cancer are known as carcinogens. Some carcinogens such as benzo(a)pyrene must first be activated by cytochrome P-450 enzymes. A major driver of mutagenesis in cancer cells is through the loss of normal DNArepair processes. Inherited defects in DNA-repair processes, such as mutations in the BRCA1 gene, are associated with an increased susceptibility for certain cancers. The most common somatic mutations that increase mutagenesis in cancer cells cause loss of the DNA damage checkpoint. Most such mutations are in the tumor suppressor gene p53. Sequencing of cancer genomes typically reveals thousands of mutations, which include single nucleotide variants (SNVs), insertions or deletions (Indels), or structural variants (SVs) that include chromosomal rearrangements, duplications, and copy-number variations. The first cancer-causing oncogenes were identified by their association with tumor viruses. Cellular oncogenic driver mutations have been identified at the break points of chromosome rearrangements, by mapping genes responsible for familial cancer syndromes, and by assays for transformation of tissue-culture cells. The first tumor suppressor gene to be identified, RB, is mutated in retinoblastoma and many other tumors. Inheritance of a single mutant allele of RB greatly increases the probability that a cancer will develop, because a somatic cell readily lose the functional copy of RB by loss of heterozygosity (LOH) by mutation or deletion of the normal allele, chromosome mis-segregation, or mitotic recombination. Comparison of large numbers of genome sequences from the same tumor type allows oncogenic driver mutations to be distinguished from the many adventitious mutations present in cancer genomes.
Many oncogenic driver genes cause cell proliferation by activating growth-promoting pathways or by inhibiting growth-inhibiting and apoptotic pathways. A gene that becomes an oncogenic driver as a result of a gain-of-function mutation is known as an oncogene. A gene that becomes an oncogenic driver as a result of a loss-of-function mutation is known as a tumor suppressor gene. Micro-RNAs (miRNAs) can promote or inhibit tumorigenesis by affecting the expression of multiple oncoproteins or tumor suppressor proteins. Mutations affecting epigenetic regulators such as histone-modifying enzymes or chromatin remodelers are associated with a variety of tumors.
Receptor Mutations Can Cause Proliferation in the Absence of External Growth Factors
25.3 Dysregulation of Cell Growth and Developmental Pathways Initiates Tumorigenesis Uncontrolled cellular proliferation is the central hallmark of cancer. We have already seen that many of the known oncogenic drivers of cancer are in genes involved in the pathways that keep growth of normal tissues in check by a balance of growth-promoting and growth-inhibiting signals. In this section, we examine these pathways in greater detail, with an emphasis on the mechanism by which oncogenic mutations exert their effects. Receptor Mutations Can Cause Proliferation in the Absence of External Growth Factors Oncogenes encoding cell-surface receptors that transduce growthpromoting signals have been associated with several types of cancer. A major class of these receptors, known as receptor tyrosine kinases (RTKs), have intrinsic protein tyrosine kinase activity in their cytosolic domains, which is quiescent until activated. As detailed in Chapter 16, ligand binding to the external domains of RTKs leads to their dimerization and
activation of their kinase activity, initiating an intracellular signaling pathway that ultimately promotes proliferation. In some cases, a point mutation changes a wild-type RTK into one that dimerizes and is constitutively active even in the absence of ligand. For instance, a single point mutation converts the normal human EGF receptor 2 (HER2) into the NEU oncoprotein (“NEU” for its first known role, in neuroblastoma) (Figure 25-13, left). Similarly, human tumors called multiple endocrine neoplasia type 2 produce a constitutively active dimeric glia-derived neurotrophic factor (GDNF) receptor that results from a point mutation in the extracellular domain. The GDNF receptor and the HER2 receptor are both protein tyrosine kinases, so by the signal transduction pathways detailed in Chapter 16, the constitutively active forms excessively activate their downstream target proteins, which ultimately promote cell cycle progression, cell survival and proliferation. In other cases, deletion of much of the extracellular ligand-binding domain produces a constitutively active oncogenic receptor. For example, deletion of the extracellular domain of the normal EGF receptor (Figure 25-13, right) converts it to the dimeric ErbB oncoprotein (from erythroblastosis virus, in which a viral version of the altered gene was first identified). Mutations leading to overproduction of a normal RTK can also be oncogenic. For instance, many human breast cancers overproduce a normal HER2 receptor because of amplification of its encoding gene. As a result, the cells are stimulated to proliferate in the presence of very low concentrations of EGF and related hormones, concentrations too low to stimulate proliferation of normal cells (see Chapter 16).
FIGURE 25-13 Effects of oncogenic mutations in proto-oncogenes that encode cellsurface receptors. Left: A mutation that alters a single amino acid (valine to glutamine) in
Many Oncogenic Mutations Constitutively Activate Signal-Transducing Proteins
the transmembrane region of the HER2 receptor causes dimerization of the receptor, even in the absence of the normal EGF-related ligand, transforming it into the oncoprotein NEU, a constitutively active protein tyrosine kinase. Right: A deletion that causes loss of the extracellular ligand-binding domain in the EGF receptor leads, for unknown reasons, to constitutive activation of the kinase activity of the resulting oncoprotein, ErbB. Description The illustration at the top left titled proto-oncogene receptor proteins shows two orange structures labeled H E R 2 receptor. The plasma membrane is with half of this protein in the exterior and the other half down in the cytosol. A black line curled in the center of the protein that is inside the membrane is labeled Valine. A downward arrow has two labels: on the left, (Valine to Glutamine), on the right oncogenic mutations. Below the arrow, the orange structures have moved close together and the label glutamine is in the membrane. In the cytosol, the phosphate molecules represented in yellow circles are added to the orange structure. At the top right, a set of two blue structures in the exterior are labeled E G F receptor. The blue structures are in the cytosol and labeled inactive receptor tyrosine kinase. The downward arrow also shares the oncogenic mutations label and has the label deletion at its right. At the bottom, the blue receptors have joined in the cytosol. A label at the top reads, E r b B oncoprotein. A label pointing to the joined area reads, constitutively active protein-tyrosine kinase. At the bottom of both diagrams is a label that reads, Ligand-independent receptor oncoproteins. Many Oncogenic Mutations Constitutively Activate SignalTransducing Proteins A large number of oncogenes are derived from proto-oncogenes whose encoded proteins are components or regulators of signal transduction
pathways — most prominent among them the Ras pathway. As we saw in
Chapter 16, Ras is a key component in the transduction of signals from activated receptors to a cascade of protein kinases. In the first part of this pathway, a signal from an activated RTK is carried via two adapter proteins to Ras, converting it to the active GTP-bound form (see Figure 16-10). In the second part of the pathway, activated Ras transmits the signal via two intermediate protein kinases to MAP kinase. The activated MAP kinase then phosphorylates a number of transcription factors that induce synthesis of important growth and proliferation proteins (see Figures 16-13 and 16-14). Virtually every component of this RTK/Ras/MAP kinase signaling cascade has been identified as an oncogene or tumor suppressor gene (Figure 25-14).
FIGURE 25-14 RTK/Ras/MAP kinase pathway components are frequently mutated in cancer. Oncogenic mutations that activate the RTK/Ras/MAP kinase pathway have been identified in many human cancers. Most components, highlighted in green, activate the pathway and oncogenic mutations cause a gain of function. In contrast, NF1, highlighted in
red, normally inactivates the pathway and the oncogenic mutations cause a loss of function of both copies of the gene. Description The illustration shows the cell membrane as a thin gray line at the top of the diagram. Above the membrane, a circle labeled growth hormone enters a green gateway shape that goes through the membrane. Attached to this gateway, in the cytosol, are 3 small green ovals labeled: S h c, G r b 2, and S o s. The P G r b 2 is to a larger green structure labeled R a s. To the right, there are two arrows, one going opposite the other. Below them is a red rectangle labeled N F 1. The R a s structure is displayed again after the arrows, now with a green rectangle labeled N-terminal regulatory domain, then another R a f with 3 yellow phosphate circles on it. A gray structure labeled M E K attaches to R a f and separates it from the membrane. Another gray structure is added and labeled M A P kinase. Two phosphorus yellow circles are added to the R a f structure and a downward arrow leads it into the nucleus. The structure attaches to the D N A helix with transcription factors and the D N A shows a gray rectangle labeled transcription. A legend box indicates that green structures are proto-oncogenes and the red structure is a tumor suppressor gene. Among the best studied oncogenes are the genes themselves, which were the first nonviral oncogenes to be recognized. Any one of a number of changes in Ras can lead to its uncontrolled and therefore dominant activity. In particular, if a point mutation substitutes any other amino acid for the glycine at position 12 in the Ras sequence, the normal protein is converted into a constitutively active oncoprotein (see Chapter 16). This single mutation reduces the protein’s GTPase activity, thus maintaining Ras in the active GTP-bound state. Activating Ras mutations short-circuit the first part of the RTK pathway, making upstream activation triggered by ligand binding to the receptor unnecessary. Constitutively active Ras oncoproteins are produced by many types of human tumors, including
Growth Control Pathways Ultimately Regulate Initiation of the Cell Cycle
bladder, colon, mammary, skin, and lung carcinomas, neuroblastomas, and leukemias. Constitutive Ras activation can also arise from loss-of-function mutations in a GTPase-activating protein (GAP). The normal function of a GAP is to accelerate hydrolysis of GTP and thus the conversion of active GTP-bound Ras to inactive GDP-bound Ras (see Figure 3-35). Loss-of-function mutations in the Ras-GAP protein, the product of the NF1 gene, leads to sustained activation of downstream signal-transducing proteins. The relationship between RAS, an oncogene, and NF1, a tumor suppressor gene acting in the same pathway, is a good example of the regulatory circuit shown in Figure 25-12. NF1 was first discovered as the underlying cause of the familial cancer syndrome neurofibromatosis. Individuals who have inherited a single mutant NF1 allele then develop neurofibromas, a benign tumor of the sheath cells that surround nerves, caused by loss of both alleles through LOH. Oncogenes encoding other altered components of the RTK/Ras/MAP kinase pathway have also been identified (see Figure 25-14). For example, constitutively active forms of Raf have been identified in approximately 50 percent of melanomas. As in the case of constitutively active forms of Ras, these mutant Raf forms no longer require regulatory signals coming from the cell surface and signal continuously for cell growth and proliferation.
Growth Control Pathways Ultimately Regulate Initiation of the Cell Cycle Growth stimulatory pathways such as the RTK/Ras/MAP kinase pathway ultimately have two outputs: transcription of a set of genes required for cell growth and activation of the cell cycle to initiate a new round of cell division. During the cell cycle, once a cell progresses past a certain point in , called the restriction point, it becomes irreversibly committed to entering S phase and replicating its DNA. Cyclin Ds, cyclin-dependent kinases (CDKs), and the Rb protein are all elements of the control system that regulates passage through the restriction point. Many of these proteins that regulate cell cycle initiation are targets for oncogenic mutations. The pathway that controls entry into the cell cycle is estimated to be misregulated in approximately 80 percent of human cancers. At the heart of this pathway are cyclin D–CDK4/6 complexes and the transcriptional repressor Rb (see Figure 19-16). The expression of cyclin D genes is induced by many extracellular growth factors, or mitogens. These cyclins assemble with a partner, CDK4 or CDK6, to generate catalytically active cyclin-CDK complexes whose kinase activity promotes progression through . Mitogen withdrawal prior to passage through the restriction point leads to accumulation of two CDK inhibitors. As described in
Chapter 19, these two proteins, p15 and p16, bind to cyclin D–CDK4/6 complexes and inhibit their activity, thereby causing arrest.
Most tumors contain an oncogenic mutation that causes the overproduction or loss of one of the components of the pathway that controls entry into S phase, so that the cells are propelled into S phase in the absence of the proper extracellular growth signals. For example, elevated levels of cyclin D1, one of the three cyclin Ds, are found in many human cancers. One mechanism that results in overproduction of cyclin D is translocation. In certain tumors of antibody-producing B lymphocytes, the cyclin D1 gene is translocated such that its transcription is under the control of an antibody-gene enhancer, causing elevated cyclin D1 production throughout the cell cycle irrespective of extracellular signals. That cyclin D1 can function as an oncoprotein was shown by studies with transgenic mice in which the cyclin D1 gene was placed under the control of an enhancer specific for mammary duct cells. Initially, the duct cells underwent hyperproliferation, and eventually breast tumors developed in these transgenic mice. A second mechanism that can lead to overproduction of cyclin D is gene amplification. Amplification of the cyclin D1 gene and concomitant overproduction of the cyclin D1 protein is common in human breast cancers; the extra cyclin D1 helps to drive cells through the cell cycle. We have already seen that inactivating mutations in both RB alleles lead to childhood retinoblastoma, a relatively rare type of cancer. However, lossof-function mutations in the RB gene are also found in the more common cancers that arise later in life (see Figure 25-11). These tissues, unlike retinal tissue, probably produce other proteins (e.g., p107 and p130, both structurally related to RB) whose function is redundant with that of RB, and thus RB is not so critical for preventing cancer in these tissues. In the
retina, however, regulation of cell cycle entry appears to rely exclusively on the Rb protein, which is why patients heterozygous for the RB gene first develop tumors in this tissue. Rb function can be eliminated not only by inactivating mutations, but also by the binding of an inhibitory protein, designated E7, that is encoded by human papillomavirus (HPV), another nasty viral trick to create virus-producing tissue. At present, this binding is known to occur only in cervical and oropharyngeal cancers. The proteins that function as cyclin-CDK inhibitors are also targets for oncogenic mutations. In particular, loss-of-function mutations in p16 (CDKN2A) that prevent it from inhibiting cyclin D–CDK4/6 kinase activity are among the most common oncogenic drivers in several cancers (see Figure 25-11 and Table 25-2). Loss of p16 mimics overproduction of cyclin Ds and thus p16 normally acts as a tumor suppressor. Although the p16 tumor suppressor gene is deleted in some human cancers, the p16 sequence is normal in others. In some of these latter cancers (e.g., lung cancer), the p16 gene, or genes encoding other functionally related proteins, is inactivated by hypermethylation of its promoter region, which prevents its transcription. What promotes this change in the methylation of p16 is not known, but it prevents production of this important cell cycle control protein. The locus encoding p16 is highly unusual in that it encodes no less than three tumor suppressor genes, which makes it the most vulnerable locus in the human genome to oncogenic changes. In addition to harboring the p16encoding gene, CDKN2A, it has the CDKN2B locus immediately upstream, which encodes p15, another cyclin D–CDK4/6 inhibitor (Figure 25-15).
Inappropriate Production of Nuclear Transcription Factors Can Induce Transformation
The locus also encodes a key activator of the tumor suppressor p53. This protein, p14ARF (p19ARF in the mouse), is encoded by an exon upstream of the first CDKN2A exon and shares its exon 2 and exon 3 with CDKN2A. As we will see in Section 25.4, this protein controls the stability of p53. Thus mutations in this locus can simultaneously affect the two major tumor-suppressor pathways in the cell, the Rb and p53 pathways.
FIGURE 25-15 The p15-ARF-p16 locus encodes three tumor suppressor genes. Exons are designated as E. The two p15 exons (orange) are located upstream of the ARF locus. ARF (blue) is encoded by a unique E1β exon but shares exons E2 and E3 with p16 (green). ARF encodes a p53 activator. [Data from C. J. Sherr, 2006, Nat. Rev. Cancer 6:663–673.] Description In the illustration, Exons 1 and 2 (orange rectangles) are separated by about 4 kilobase at the left of the D N A line. Downstream, the exon-1-beta (blue rectangle) is located, and further downstream of this, exon 1-alpha (green rectangle), followed by exons 2 and 3 (green rectangles). The orange rectangle area at the left has the label p 15. The blue rectangle has the label P 14 A R F and the green rectangles are labeled p 16.
Inappropriate Production of Nuclear Transcription Factors Can Induce Transformation Mutations that create oncogenes or inactivate tumor suppressor genes eventually cause broad changes in gene expression. These changes can be measured by comparing the amounts of different mRNAs produced in normal cells and in tumor cells. Since the most direct effect on gene expression is exerted by transcription factors, it is not surprising that many oncogenes encode transcription factors. Two such transcription factors with a clear role in tumorigenesis are the FOS and MYC proteins, which stimulate transcription of genes encoding proteins that promote progression through the phase of the cell cycle and the -to-S transition. We discuss the deregulation of these proteins below. JUN and FOS were initially identified in transforming retroviruses and later found to be overexpressed in some human tumors. The JUN and FOS proto-oncogenes encode proteins that sometimes associate to form a heterodimeric transcription factor, called AP1, that binds to a sequence found in promoters and enhancers of many genes (see Figure 8-26a and
Chapter 16). These proteins function as oncoproteins by activating the transcription of key genes that encode growth-promoting proteins and by inhibiting the transcription of growth-repressing genes. Many nuclear proto-oncogene proteins are produced when normal cells are stimulated to grow, indicating their direct role in growth control. For
example, platelet-derived growth factor (PDGF) treatment of quiescent mouse 3T3 cells induces an approximately 50-fold increase in the production of the transcription factors FOS as well as MYC, the products of the FOS and MYC proto-oncogenes. Initially, there is a transient rise of FOS and later a more prolonged rise of MYC (Figure 25-16). The levels of both proteins decline within a few hours, a regulatory effect that may, in normal cells, help to avoid cancer. EXPERIMENTAL FIGURE 25-16 Addition of serum to quiescent 3T3 cells yields a marked increase in the activity of two proto-oncogene products, FOS and MYC. Serum contains factors such as platelet-derived growth factor (PDGF) that stimulate the growth of quiescent cells. One of the earliest effects of growth factors is to induce expression of FOS and MYC, whose encoded proteins are transcription factors. [Data from M. E. Greenberg and E. B. Ziff, 1984, Nature 311:433.]
Description In the graph, the horizontal axis plots time in minutes and hours ranging from 0 to 90 minutes in increments of 30 minutes and then from 2 to 10 hours in increments of two hours. The vertical axis plots relative activity ranging from 0 to 15 in increments of 10. An arrow indicates that serum is added at time zero. The purple curve and blue curve represents F O S and M YC, respectively. The F O S curve rises rapidly on the addition of serum and reaches a maximum of 15 after about 20 minutes. Then the curve falls rapidly to about 2 after 60 minutes and zero after 90 minutes. The M Y C curve rises slowly to 7 after an hour, then gradually falling to a relative activity of 3 after four hours and leveling off at 2 after ten hours. The oncogenic forms of FOS and MYC are due to gain-of-function mutations. In normal cells, the mRNAs for these genes and the proteins they encode are intrinsically unstable and degrade rapidly after the genes are transcribed. Some of the genetic changes that turn FOS from a normal gene into an oncogene involve deletions of the sequences that normally make the FOS mRNA and protein short-lived. Conversion of the MYC proto-oncogene into an oncogene can occur by different mechanisms. In cells of the human tumor known as Burkitt’s lymphoma, the MYC gene is translocated to a site near the heavy-chain antibody genes, which are normally active in antibody-producing white blood cells (Figure 25-17). The MYC translocation is a rare aberration of the normal DNA rearrangements that occur during maturation of antibody-producing cells. The translocated MYC gene, now regulated by the antibody-gene enhancer, is continually highly expressed, causing the cell to become cancerous. Localized amplification of a segment of DNA containing the MYC gene, which occurs in several human tumors, also causes inappropriately high production of the otherwise normal MYC protein. This mechanism of
oncogenic activation is similar to the formation of BCR-ABL oncogene by a translocation that produces the Philadelphia chromosome.
FIGURE 25-17 Chromosomal translocation in Burkitt’s lymphoma. As a result of a translocation between chromosomes 8 and 14, the MYC gene is placed adjacent to the gene for part of the antibody heavy chain , leading to overproduction of the MYC transcription factor in lymphocytes and hence their growth into a lymphoma. Description The illustration shows two chromosomes, at the left is a chromosome labeled 8 with M Y C gene and at the right is a chromosome labeled 14 with a heavy chain constant domain (C H) and heavy chain variable domain (V H). The translocation leads to the exchange of the bottom tips. Chromosome 8 has V H of chromosome 14 and labeled 8 q minus. Chromosome 14 has C H and M Y C gene of chromosome 8 and labeled 14 q plus. The MYC gene encodes a basic helix-loop-helix protein that acts as part of a set of interacting proteins that can dimerize in various combinations, bind to DNA, and coordinately regulate the transcription of target genes. Other members of this protein set include MAD, MAX, and MNT. MAX
Aberrations in Signaling Pathways That Control Development Are Associated with Many Cancers
can heterodimerize with MYC, MAD, and MNT. MYC-MAX dimers regulate genes that control proliferation, such as cyclins. MAD proteins inhibit MYC proteins, which has led to an interest in using MAD proteins, or drugs that stimulate MAD proteins, to rein in excessive MYC activity that contributes to tumor formation. MYC protein complexes affect transcription by recruiting chromatin-modifying complexes containing histone acetyl transferases (which usually stimulate transcription; see
Chapter 8) to MYC target genes. MAD and MNT work with the SIN3 corepressor protein to bring in histone deacetylases that help to block transcription. Together, all these proteins form a regulatory network that employs protein-protein association, variations in DNA binding, and transcriptional regulation to control cell proliferation. Overproduction of MYC protein tips the scales in favor of cell growth and division. Aberrations in Signaling Pathways That Control Development Are Associated with Many Cancers During normal development, secreted signals such as Hedgehog (Hh), Wnt, and TGF-β are used to direct cells to particular developmental fates, which may include the property of rapid cell cycling. The effects of such signals must be regulated so that growth is limited to the right time and place. Among the mechanisms available for reining in the effects of these powerful developmental signals are inducible intracellular antagonists, receptor blockers, and competing signals. Mutations that prevent such
restraining mechanisms from operating are likely to be oncogenic, causing inappropriate or cancerous growth. The Hedgehog signaling pathway, which is used repeatedly during development to control differentiation, is a good example of a signaling pathway implicated in cancer induction. In the skin and cerebellum, one of the human Hh proteins, Sonic Hedgehog, stimulates cell division by binding to and inactivating a membrane protein called Patched1 (PTC1) (see Figure 16-29). Loss-of-function mutations in PTC1 permit cell proliferation in the absence of an Hh signal; thus PTC1 is a tumor suppressor gene. People who inherit a defective copy of PTC1 have a propensity to develop skin and brain cancer; either can occur when the remaining PTC1 allele is lost through the mechanism of LOH that we saw for RB and NF1 cancer syndromes. Spontaneous mutations in both copies of this gene have also been observed in sporadic cases of these cancers. Mutations in other genes in the Hh signaling pathway are also associated with cancer. Some such mutations create oncogenes that turn on Hh target genes inappropriately; others are recessive mutations that affect negative regulators such as PTC1. Many of the signaling pathways described in Chapters 16 and 20 also play roles in controlling embryonic development and cell proliferation in adult tissues. In recent years, mutations affecting components of most of these signaling pathways have been linked to cancer. Indeed, once one gene in a developmental pathway has been linked to a type of human cancer, knowledge of that pathway gleaned from model organisms such as worms, flies, or mice allows focused investigations of the possible involvement of
additional pathway genes in other cases of the cancer. For example, APC, a gene that is mutated early on in colon cancer, is now known to be part of the Wnt signaling pathway (see Chapter 16). That knowledge, in turn, led to the discovery of β-catenin mutations in colon cancer. Mutations in tumor-suppressor developmental genes promote tumor formation in tissues where the affected gene normally acts to restrain growth. For example, transforming growth factor β (TGF-β), despite its name, primarily acts to inhibit proliferation of many cell types, including most epithelial and immune-system cells. Binding of TGF-β to its receptor activates cytosolic Smad transcription factors (see Figure 16-24). After translocating to the nucleus, Smads can promote expression of the gene encoding p15, an inhibitor of cyclin-dependent kinase 4 (CDK4), which causes cells to arrest in . TGF-β signaling also promotes expression of genes encoding extracellular matrix proteins and plasminogen activator inhibitor 1 (PAI-1), which reduces the plasmin-catalyzed degradation of the matrix. Loss-of-function mutations in either TGF-β receptors, as noted above, or in Smads thus promote cell proliferation and probably contribute to the invasiveness and metastasis of tumor cells (Figure 25-18). Such mutations have in fact been found in a variety of human cancers. For example, deletion of the Smad4 gene occurs in many human pancreatic cancers; retinoblastoma and colon cancer cells lack functional TGF-β receptors and therefore are unresponsive to TGF-β growth inhibition. Originally Smad4 was called DPC (deleted in pancreatic carcinoma).
Experimental Reconstruction of the Multi-Hit Model for Cancer
FIGURE 25-18 Effect of loss of TGF-β signaling. Binding of TGF-β, an anti-growth factor, to its receptor causes activation of Smad transcription factors. In the absence of effective TGF-β signaling owing to either a receptor mutation or a Smad mutation, cell proliferation and invasion of the surrounding extracellular matrix increase. See X. Hua et al., 1998, Genes Dev. 12:3084. Description In the illustration, type two and type one receptors are embedded in the cell membrane. Docking of T G F-beta with the receptors leads to activation of the cytosolic tails, resulting in activation of S M A D 3 and its combination with S M A D 4. The formation of the S M A D 3-S M A D 4 complex results in the activation of the p 15 promoter and P A I-1 promoter in the nucleus and increased transcription of P 15, a cell-cycle inhibitor, and P A I-1, a gene that inhibits E C M-protease production, genes. Loss of function mutations in the receptor block the T G F-beta signal. Loss of function mutation in S M A D's also blocks the T G F-beta signal. Both of these results in decreased production of p 15, resulting in increased proliferation, and decreased production of P A I-1, allows increased extracellular matrix degradation and, hence, metastasis. Experimental Reconstruction of the Multi-Hit Model for Cancer As we have seen from the sequencing of cancer genomes, a typical tumor may have acquired thousands of mutations, five of which have been selected for because they are drivers of oncogenesis; many of which are genes we just discussed that regulate cell growth and division. As the data in Figure 25-19 show, the incidence of many types of human cancer increases drastically with age, indicating that it can take decades for the
required multiple mutations to occur. This model for the slow progression of cancer raises fundamental questions about the sequence of events needed to lead to a tumor. In what sequential order do the driver mutations occur? Do different driver mutations act synergistically? Answers to these questions are necessary to complete the picture of how cancer forms and will be important for strategies for early diagnosis and cancer prevention.
FIGURE 25-19 The incidence of human cancers increases as a function of age. The marked increase in the incidence of cancer with age is consistent with the multi-hit model of cancer induction. Note that the logarithm of annual incidence is plotted versus the logarithm of age. [Data from B. Vogelstein and K. W. Kinzler, 1993, Trends Genet. 9:138–141.]
Description In the graph, the horizontal axis plots age in years ranging from 20 to 80 in increments of 10. The vertical axis plots annual incidence of cancer per one-hundred thousand males ranging from 0.1 to 500 in logarithmic intervals. Each plot is linear and rises with age. Skin cancer has an incidence of 1 at age 23 and rises to 200 at age 80; esophageal cancer, one at age 43, rising to 50 at age 80; stomach cancer, one at age 28, rising to 300 at age 80; rectal cancer, 1 at age 33, rising to 150 at age 80; pancreatic cancer, 1 at age 37, rising to 100 at age 80; and prostate cancer, 1 at age 48, rising to almost 500 at age 80. All values are approximate. Experimental reconstruction of a multi-hit progression has been carried out with transgenic mice, which have shown that a variety of combinations of oncogenes can cooperate in causing cancer. For example, mice have been made that carry either the mutant dominant oncogene or the MYC proto-oncogene, in each case under the control of a mammary-cellspecific promoter/enhancer. This promoter is induced by endogenous hormone levels and tissue-specific regulators, leading to overexpression of MYC or in breast tissue. As noted, the MYC protein is a transcription factor that induces expression of many genes required for the transition from the to the S phase of the cell cycle. Heightened transcription of MYC in these mice mimics previously identified oncogenic mutations that increase MYC transcription, converting the proto-oncogene into an oncogene. By itself, the MYC transgene causes tumors only after 100 days, and then in only a few mice; clearly only a minute fraction of the mammary cells that overproduce the MYC protein actually become malignant. Production of the mutant protein alone causes tumors earlier, but still slowly and
with about 50 percent efficiency over 150 days. When the MYC and overexpressing transgenic mice are crossed, however, all mammary cells in their offspring overproduce both MYC and . In these mice, tumors arise much more rapidly, and all animals succumb to cancer (Figure 25-20). Such experiments emphasize the synergistic effects of multiple oncogenes. They also suggest that the long latency of tumor formation, seen even in the double-transgenic mice, is due to the need to acquire still more mutations. EXPERIMENTAL FIGURE 25-20 The kinetics of tumor appearance in female mice carrying either one or two oncogenic transgenes shows the cooperative nature of multiple mutations in cancer induction. Each of the transgenes was driven by the mouse mammary tumor virus (MMTV) breast-specific promoter. The hormonal stimulation
The Succession of Oncogenic Mutations Can Be Traced in Colon Cancers
associated with pregnancy activates the MMTV promoter and hence the overexpression of the transgenes in mammary tissue. The graph shows the time course of tumorigenesis in mice carrying either MYC or transgenes as well as in the progeny of a cross of MYC carriers with carriers, which contain both transgenes. The results clearly demonstrate the cooperative effects of multiple mutations in cancer induction. See E. Sinn et al., 1987, Cell 49:465. Description In the graph, the horizontal axis plots age in days ranging from 0 to 200 in increments of 50. The vertical axis plots tumor-free mice in percentage ranging from 0 to 100 in increments of 20. A purple curve represents M Y C, a pink curve represents R a s-v 12, and a black curve represents M Y C plus R a s-v 12. All curves start at 100. The M Y C curve is flat until 100 days and then falls slowly to 80 percent after almost 200 days. The R a s-v 12 curve begins to fall after about 40 days and then falls slowly to 60 percent after almost 200 days. The curve for M Y C plus R a s-v 12 begins to fall after 40 days and then drops sharply to 40 percent after 50 days. After 50 days, the curve drops more slowly, reaching zero after 150 days. The Succession of Oncogenic Mutations Can Be Traced in Colon Cancers A direct way to reconstruct the progression of a tumor is to take sequential samples from a slowly developing tumor. Surgeons can obtain fairly pure samples of many human cancers, but since the tumor is observed at only one time, its exact stage of progression cannot be easily determined. An exception is colon cancer, which evolves through distinct, wellcharacterized morphological stages. Its intermediate stages — polyps,
benign adenomas, and carcinomas — can be isolated by a surgeon during colonoscopies, allowing mutations that occur in each of these stages to be identified. Numerous studies have shown that colon cancer arises from a series of mutations that commonly occur in a well-defined order, providing strong support for the multi-hit model (Figure 25-21).
FIGURE 25-21 The development and metastasis of human colorectal cancer and its genetic basis. A mutation in the APC tumor suppressor gene in a single epithelial cell causes the cell to divide (although surrounding epithelial cells do not), forming a mass of localized benign tumor cells, called a polyp. Subsequent mutations lead to expression of a constitutively active Ras protein and loss of the tumor suppressor gene p53. These mutations, together with additional genetic changes yet to be identified, generate a malignant cell. The cell continues to divide, and its progeny invade the basement membrane that surrounds the tissue, but do not penetrate the basement membrane of capillaries (bottom left). Some tumor cells spread into blood vessels that will distribute them to other sites in the body (bottom right). Additional mutations permit the tumor cells to exit from the blood vessels and proliferate at distant sites. See B. Vogelstein and K. W. Kinzler, 1993, Trends Genet. 9:138–141. Description The illustration shows the physical signs from top to bottom. Step 1. Loss of the A P C tumor-suppressor gene on chromosome five. Small growths, polyps, begin to grow at the site of the mutation on the colon wall. Step 2. A benign, precancerous tumor grows. Step 3. Activation of the K-r a s oncogene on chromosome 12. The growth of the tumor continues, becoming a class two adenoma. At this stage, the tumor is still benign. Step 4. As the tumor grows, it changes from class two to class three tumor. At this stage, the tumor is still benign. Step 5. The loss of the p 53 tumor-suppressor gene on chromosome 17, resulting in a change from benign to malignant carcinoma. At this stage, various other genetic changes can occur, and the cancer cells can metastasize, spreading to other parts of the body. A schematic shows how the cell in the polyp invade the normal colon epithelial cells, penetrating the basement membrane, invading the blood vessels, and allowing metastasis. Insight into the progression of colon cancer first came from the study of inherited predispositions to colon cancer such as familial adenomatous polyposis (FAP). Mutations in the Wnt signaling pathway have been identified in many of these syndromes, and it is now believed that
deregulation of Wnt signaling results in formation of polyps (precancerous growths) on the inside of the colon wall — not only in people with inherited polyposis syndromes, but also in people afflicted with sporadic (noninherited) forms of colon cancer. The APC (adenomatous polyposis coli) protein is a negative regulator of Wnt signaling (see Figure 16-26), which promotes cell cycle entry by activating expression of the MYC gene. The absence of functional APC protein thus leads to inappropriate production of MYC, and cells homozygous for APC mutations proliferate at a rate higher than normal and form polyps. Loss-of-function mutations in the APC gene are the most frequent mutations found in early stages of colon cancer. Most of the cells in a polyp contain the same one or two mutations in the APC gene that result in its loss or inactivation, indicating that they are clones of the cell in which the original mutation occurred. Thus APC is a tumor suppressor gene, and both alleles of the APC gene must carry an inactivating mutation for polyps to form because cells with one wild-type APC allele express enough APC protein to function normally. If one of the cells in a polyp undergoes another mutation, this time an activating mutation of the ras gene, its progeny divide in an even more uncontrolled fashion, forming a larger adenoma. Inactivation of the p53 gene follows and results in the gradual loss of normal regulation and the consequent formation of a malignant carcinoma (see Figure 25-21). The p53 protein is a tumor suppressor that halts progression through the cell cycle in response to DNA damage and other stresses and can induce apoptosis (see Section 25.4). While the three “hits” listed here are certainly crucial parts of the picture, there are likely to be additional
contributing genetic events. Not every colon cancer, however, acquires all the later mutations or acquires them in the order depicted in Figure 25-21. Thus different combinations of mutations may result in the same phenotype. DNA from different human colon carcinomas generally contains mutations in all three genes mentioned here — loss-of-function mutations in the tumor suppressors APC and p53 and an activating (gain-of-function) mutation in the oncogene K-RAS (one of the RAS family of genes) — establishing that multiple mutations in the same cell are needed for the cancer to form. Some of these mutations appear to confer growth advantages at an early stage of tumor development, whereas other mutations promote the later stages, including invasion and metastasis, which are required for the malignant phenotype. For the majority of tumors that are only biopsied or removed at a late stage in cancer development there are now sequencing-based methods that allow some of the events in cancer progression to be reconstructed. The idea is to gain information from multiple individual cells from the tumor to create a lineage map for the tumor as a whole. To see how this analysis works, imagine a simple case for a tumor that arose as the result of two oncogenic driver mutations. At the time that the tumor is biopsied, all of the cells should have the first driver mutation that occurred, whereas only some of the tumor cells would have the second. This method will allow the order of appearance to be determined for any two oncogenic driver mutations as long as at least a few cells in the tumor can be found that lack one of the mutations. This method for reconstructing cell lineages within a
Cancer Development Can Be Studied in Animal Models
tumor is now feasible because of advances in next generation sequencing that enable genomic sequence information to be obtained from a single tumor cell. Cancer Development Can Be Studied in Animal Models Genetically engineered mice have also provided tremendous insights into the steps of tumor initiation and progression. Using mouse models to study cancer is not always straightforward, however. Many tumor suppressor genes serve essential functions during normal mouse development, so mice lacking both copies of these genes are not viable. The essential functions of these genes during early embryogenesis preclude the study of their role in tumor progression. To circumvent this problem, researchers have begun to employ conditional knock-in and knockout strategies that allow for the targeted activation or inactivation of a gene in a certain tissue or at a certain stage of development. In the conditional mouse model, an allele of a particular oncogene or tumor suppressor gene is wild type until activated or inactivated with exogenous chemicals or viruses in a tissue- or time-specific manner. At the heart of these conditional systems are the Cre and FLP recombinases. These recombinases facilitate homologous recombination between loxP and FRT sites, respectively (Figure 25-22; see also Figure 6-40). When the recombinases are under the control of a tissue-specific promoter, recombination occurs only in the tissue that produces the recombinase.
The recombinase method can be used in two ways. First, the recombinase target sites may flank an exon. Upon induction of the recombinase, that exon is lost and the gene is inactivated (Figure 25-22a). This method is especially useful for inactivating tumor suppressor genes in a tissuespecific manner. Second, expression of an oncogene can be controlled by introducing into the oncogene an additional exon that contains a stop codon, which makes the gene nonfunctional. However, if the additional exon is flanked by recombinase target sites, the oncogene will be expressed upon induction of the recombinase (Figure 25-22b). Using this system, researchers have examined the role of oncogenic forms of Ras in the mouse and have, using a conditional oncogenic ras allele, created a mouse model of human lung cancer.
FIGURE 25-22 Conditional mouse models of cancer. In the inactivating system (a), an exon of interest is flanked by two loxP or FRT sites as shown. Expression of the Cre or FLP recombinase leads to homologous recombination between the two loxP and FRT sites, respectively. This recombination leads to excision of the exon, rendering the gene nonfunctional. In the activating system (b), an additional exon with a stop codon is introduced into the gene of interest, making the gene nonfunctional. This exon is flanked by
Molecular Cell Biology Is Changing How Cancer Is Diagnosed and Treated
loxP or FRT sites. When Cre or FLP recombinase is induced, the stop codon–containing exon is recombined out, and the gene of interest is expressed. Description The illustration labeled (a) titled inactivating system shows a D N A and it carries the label, functional. At left is a green rectangle labeled Exon 1 followed by a small square labeled I o x P or F R T. A red rectangle is next, labeled Exon 2, which is also followed by a small square with the same label. At the last is a green rectangle, labeled Exon 3. A downward arrow points from Exon 2 and is labeled C r e or F L P recombinase. The arrow points to Exon 1, a small square with I o x P label or F R T, and Exon 3, without Exon 2 and this structure carries the label, nonfunctional. The illustration labeled (b) shows the same set up of exons and small squares as the illustration (a) and labeled nonfunctional. The first green rectangle is labeled Exon 1, the red rectangle is labeled exon with no number, and above this is a label stop. The last green rectangle is labeled Exon 2. A downward arrow carries the same label as the illustration (a) and leads to Exon 1, a small square with I o x P or F R T label, and Exon 2. Above, Exon 2 is a label G 12 V. The structure carries the label functional. Molecular Cell Biology Is Changing How Cancer Is Diagnosed and Treated The identification of oncogenic driver mutations, such as mutations in genes that regulate cell growth and development pathways, has not only provided us with a molecular understanding of how cancer arises and progresses, but has also revolutionized the way cancers are diagnosed and treated. Historically, tumors were characterized and treated based on the organ in which they arose and the histologic characteristics of the tumor
cells when they were viewed under the microscope. We now know that similarly appearing tumors may behave very differently and can be better separated into specific tumor subsets by examining their genomic changes. This type of characterization, which is extensively used in breast and brain cancer diagnosis, has significant implications for treatment. Each difference between the cancer cells and normal cells provides a new opportunity to identify a specific drug or treatment that kills only the cancer cells or at least stops their uncontrolled growth. Thus knowledge of the molecular cell biology of a tumor is critical information that can be exploited by researchers to develop anticancer treatments that more precisely target cancer cells. Breast cancer provides a good example of how molecular cell biology techniques have affected treatments, both curative and palliative. Breast cancer is the second most frequent cause of women’s cancer deaths. Breast cancers are often diagnosed during routine mammogram (x-ray) examinations. Typically, a very small biopsy is taken with a needle to confirm the diagnosis and is tested with antibodies to determine whether a high level of estrogen or progesterone receptors is present. These steroid receptors are capable of stimulating tumor growth and are sometimes expressed at high levels in breast cancer cells. If either receptor is present, it is exploited in the treatment. A drug called tamoxifen, which inhibits the estrogen receptor (see Figure 8-26), can be used to deprive the tumor cells of this growth-stimulating hormone. The biopsy is also tested for amplification of the proto-oncogene HER2/NEU, which, as we saw in
Chapter 16, encodes the human EGF receptor HER2. A monoclonal
antibody specific for HER2 has been a strikingly successful new treatment for the subset of breast cancers that overproduce HER2. HER2 antibody injected into the blood recognizes HER2 and causes it to be internalized, selectively killing the cancer cells without any apparent effect on normal breast (and other) cells that produce only moderate amounts of HER2. More recently, covalent conjugates of HER2 monoclonal antibodies with a potent cytotoxic drug became available to target these agents into HER2overproducing tumor cells, enhancing the killing effects of these monoclonal antibodies. These advanced antibody conjugates are used as a second line of defense for tumors that have resisted more conventional treatments. Breast cancer is still initially treated with a combination of surgery, radiation therapy, and chemotherapy. The first step is surgical resection (removal) of the tumor and examination of lymph nodes for evidence of metastatic disease. The subsequent treatment typically involves 8 weeks of chemotherapy with several different types of agents and 6 weeks of radiation. These harsh treatments are designed to kill the dividing cancer cells; however, they also cause a variety of side effects, including suppression of blood cell production, hair loss, nausea, and neuropathy. These effects can reduce the strength of the immune system, risking infection, and cause weakness due to poor oxygen supply. To help offset these effects, patients are given the cytokine G-CSF to promote the formation of neutrophils (a type of white blood cell that fights bacterial and fungal infections) and erythropoietin (Epo) to stimulate red blood cell formation (see Chapter 16). Even with this aggressive treatment, about a third of women will nevertheless succumb to cancer over the next 10
years. This risk can be reduced by 15 percent by hormone-blocking treatment such as tamoxifen and by another 10 percent by treatment with antibodies against the HER2/NEU oncoprotein. In aggregate, advanced molecular methods can reduce morbidity from breast cancer by as much as 25 percent. KEY CONCEPTS OF SECTION 25.3 Dysregulation of Cell Growth and Developmental Pathways Initiates Tumorigenesis Mutations that permit receptors for growth factors to dimerize in the absence of their normal ligands lead to constitutive receptor activity. Overproduction of growth-factor receptors can have the same effect and lead to abnormal cell proliferation. Most tumor cells produce constitutively active forms of one or more intracellular signal-transducing proteins, causing growth-promoting signaling in the absence of normal growth factors. A feature of the proliferation of most cancers is unregulated passage through the restriction point of the cell cycle. This can be caused by gain-of-function mutations in cyclin D1 or loss-of-function mutations in p16 or RB. Inappropriate production of nuclear transcription factors such as FOS, JUN, and MYC can induce transformation. In Burkitt’s lymphoma cells, MYC is translocated close to an antibody gene, leading to overproduction of MYC. Loss of signaling by TGF-β, a negative growth regulator, promotes cell proliferation and development of malignancy. A typical tumor carries about five oncogenic mutations in genes of cell growth and development pathways that are acquired over many years. A major unsolved problem in understanding how cancer progresses will depend on reconstruction of the sequential order of these mutational events. Colon cancer develops through distinct, identifiable morphological stages that can be traced to specific oncogenic mutations. Genome sequencing of multiple individual cells in a tumor can enable the lineages of cells with specific oncogenic mutations to be reconstructed.
25.4 Evasion of Programmed Cell Death and Immune Surveillance Processes
25.4 Evasion of Programmed Cell Death and Immune Surveillance Processes So far in this chapter, we have seen that early stages of the progression of cancer involve two types of oncogenic mutations: those discussed in Section 25.2 that compromise DNA repair and lead to destabilization of the genome, and those discussed in Section 25.3 that activate growthpromoting pathways and inactivate growth-inhibiting pathways. In addition to these mutations that drive oncogenesis, many cells that have reached this precancerous stage also carry a significant burden of adventitious mutations, chromosomal rearrangements, and other defects in chromosomal DNA. Cells that carry significant chromosomal abnormalities or irreparable damage to their DNA will enter the apoptotic pathway and be targeted for programmed cell death. Cells that carry many point mutations in protein-coding genes will express mutant proteins that may cause them to be recognized and targeted for destruction by cells of the immune system. Although we have little direct visibility into what happens to small numbers of precancerous cells when they do arise, it is likely that the vast majority of them are efficiently eliminated by one or both of these mechanisms. For this reason, the tumor cells that we eventually do observe owe their existence to having acquired mutations that allowed them to evade both apoptosis and immune surveillance.
Oncogenic Driver Mutations Enable Cancer Cells to Evade Apoptosis
The acquisition by most cancer cells of mutations that block the apoptotic pathway and of other mutations that suppress an immune response to them is a powerful example of how cancer progression results from an evolutionary process of mutation and selection. We will see in this section some of the kinds of oncogenic mutations that allow tumor cells that carry a high mutational load to survive. At the end of the section, we will highlight some of the promising new therapies that are based on reactivating the immune system to target cancer cells. Oncogenic Driver Mutations Enable Cancer Cells to Evade Apoptosis As we saw in Chapter 22, programmed cell death, or apoptosis, plays an important role in development. For example, during mammalian limb formation apoptosis is responsible for sculpting the digits out of a paddleshaped limb bud. In addition, apoptosis serves an editing function to maintain the organization of mature tissues by removing abnormal excesses of cells not needed for development of a working organ and by removing cells that have been compromised by errors in mitosis or DNA damage (see Chapter 22). For some cells, apoptosis appears to be the outcome by default, and signals from the PI 3-kinase/AKT pathway are required to ensure cell survival (see Figure 22-42). A major class of oncogenic mutations blocks apoptosis, allowing tumor cells to continue to proliferate inappropriately. For example, chronic lymphocytic leukemia (CLL) occurs because cells survive when they
p53 Can Activate Either the DNA Damage Checkpoint or Apoptosis in Response to DNA Damage
should not. The cells accumulate slowly, and most are not actively dividing, but they do not die. CLL cells have chromosomal translocations that activate the BCL2 gene, a critical blocker of apoptosis (see Figure 2242). The resultant inappropriate overproduction of the BCL2 protein prevents normal apoptosis and allows survival of these tumor cells. CLL tumors are therefore attributable to a failure of cell death. Another dozen or so proto-oncogenes that are normally involved in negatively regulating apoptosis have been found to be mutated to become oncogenes. Overproduction of their encoded proteins prevents apoptosis even when death is needed to stop cancer cells from growing. p53 Can Activate Either the DNA Damage Checkpoint or Apoptosis in Response to DNA Damage We have already seen that the p53 protein is a central player in tumorigenesis, and it is thought that most, if not all, human tumors have mutations either in p53 itself or in proteins that regulate p53 activity. In Section 25.2, we saw that p53 has a major part in causing an arrest in the cell cycle in response to DNA damage. Because cells carrying oncogenic p53 mutations do not arrest to give enough time for the DNA damage to be repaired, these mutants have unstable genomes and accumulate somatic mutations. p53 mutant cells also do not respond to extensive DNA damage by entering the apoptotic pathway and thus can continue to proliferate even after they have sustained damage that would cause a normal cell to be removed by programmed cell death.
Although the oncogenic form of p53 is due to a loss of function, it is nevertheless possible for certain missense mutations in only one of the two p53 alleles in a cell to abrogate almost all p53 DNA activity. This behavior comes from the fact that the active form of p53 is tetramer of four identical subunits and mixed complexes that contain at least one defective subunit have reduced ability to activate transcription. Precancerous cells with one missense mutant of p53 are probably selected for because of a proliferative growth advantage. However, the loss of function is incomplete and in order to proliferate more rapidly tumor cells often lose the remaining functional allele. This unusual feature of positive selection for a first and then a second mutation in the p53 gene may explain why tumor cells so frequently carry oncogenic driver mutations in p53. We do not yet understand why p53 causes cell cycle arrest under some circumstances and apoptosis under others, but both responses can occur as a result of p53 being stabilized in response to DNA damage. Unlike other cell cycle proteins, p53 is present at very low levels in normal cells because it is extremely unstable and rapidly degraded. The activity of p53 is normally kept low by a protein called MDM2. When MDM2 is bound to p53, it inhibits the transcription-activating ability of p53 and at the same time, because it has E3 ubiquitin ligase activity, catalyzes the ubiquitinylation of p53, thus targeting it for proteasomal degradation. After DNA damage, ATM or ATR are recruited to the sites of damage and their serine kinase activity is activated. Phosphorylation of p53 on a serine residue in the N-terminus of the protein by ATM or DNA-PK displaces bound MDM2 from p53, thereby stabilizing it (Figure 25-23). The
stabilized p53 activates transcription of the gene encoding p21, which binds to and inhibits mammalian CDK2, CDK1, and CDK4/6 complexes (see Figure 19-34).
FIGURE 25-23 Arrest in in response to DNA damage. The kinase activity of ATM, ATR, and DNA-PK is activated in response to DNA damage due to various stresses (e.g., UV irradiation, heat). Activated ATM and ATR then trigger two principal pathways leading to arrest in . Step 1 : Phosphorylation of p53 stabilizes it, permitting p53-activated expression of genes encoding proteins that cause arrest in , promote apoptosis, or participate in DNA repair. Step 2 : The second pathway is another way of controlling the pool of p53. The MDM2 protein in its active form can form a complex with p53, inhibiting its transcription-factor activity and causing p53 ubiquitinylation and subsequent proteasomal degradation. ATM and DNA-PK phosphorylate MDM2 to inactivate it, causing increased stabilization of p53. In addition, MDM2 levels in the cytoplasm are controlled by ATR and p14ARF (p19ARF in the mouse), sequesters it in the nucleolus, where it cannot
access p53. The p14ARF gene is induced by high levels of mitogenic signaling, which are frequently observed in cells carrying oncogenic mutations in growth factor signaling pathways. The human MDM2 gene is frequently amplified in sarcomas, which presumably causes excessive inactivation of p53. Similarly, p14ARF is also found mutated in some cancers. Description The illustration shows the damaged D N A that activates A T M. The response mechanism is described in two steps. Step 1. The downward arrow leads to a pink oval labeled p 53 with two phosphate yellow circles attached. Another downward arrow, labeled transcription activation points to p 53, which is attached to a blue rectangle with transcription factors. Three downward arrows come from this structure. Left goes to apoptosis, center goes to sustained G 1 and G 2 arrest, and right goes to DNA repair. Step 2. An angled arrow from A T M points to a green oval labeled M D M 2 with one phosphate yellow circle. Above this oval is another oval labeled M D M 2 with a pink p 53 oval. The p 53 has a side arrow to the right leading to proteasomal degradation and tiny pink dots. The left M D M 2 is labeled inactive, and the right M D M 2 oval has a blue rectangle attached, which has the label p 14 A R F, which is labeled oncogenic signaling. These blue ovals have downward arrows both leading to single arrow, the bottom of which is labeled pool of p 53 protein increases. Stabilized p53 also can induce expression of proteins such as Bax and PUMA that bind to and inhibit Bcl2 and thus induce apoptosis (see Figure 22-42). When cells suffer extensive DNA damage, the p53-induced expression of pro-apoptotic proteins leads to their quick elimination. While apoptosis may seem like a drastic response to DNA damage, it prevents proliferation of cells that are likely to accumulate multiple mutations. When p53 function is lost, apoptosis cannot be induced, and the accumulation of additional mutations required for cancer to develop and progress becomes more likely.
The Immune System Is a Second Line of Defense Against Cancer Formation
Apoptosis can be triggered in response to a number of intrinsic and extrinsic signals. For example, we will see in the next section that cytotoxic T cells can kill a target by inducing apoptosis in the targeted cell. The Immune System Is a Second Line of Defense Against Cancer Formation We have seen that tumor cells carry thousands of mutations, including point mutations, Indels, and chromosomal structural variants (see Figure 25-8). For a typical tumor with 5000 mutations, on average about 50 of these would lie in protein-coding sequences and thus may give rise to a protein product that was altered in some way. For tumors that arise from exposure to mutagens, such as melanomas (UV light) and lung carcinomas (tobacco smoke) the number of mutations may be hundredfold greater than this. These altered proteins are known as neo-antigens and would be expected to cause the tumor cell to be recognized and eliminated by the immune system in much the same way as cells that contain foreign proteins induced by infection with a pathogen such as a virus or bacterium specifically targeted for destruction. Most precancerous cells bearing neo-antigens are thought to be efficiently eliminated by immune surveillance. Evidence for this comes from studies of immunodeficient mouse mutants, which are more likely to acquire carcinogen-induced cancer than wild-type mice. This raises the crucial question of how did the tumors that we do see escape immune
The Tumor Microenvironment and Immunoediting Limit the Ability of the Immune System to Detect and Kill Established Tumors
surveillance? In the following section, we discuss how the local environment within the tumor suppresses the immune system, and how monoclonal antibodies that recognize molecules enriched on the surface of tumor cells can redirect certain immune system cells to kill the tumor cell. Finally, we discuss newer ways of genetically engineering T cells of the immune system so they express on their surface monoclonal antibodies that enable these cells to recognize and kill many types of tumor cells. The Tumor Microenvironment and Immunoediting Limit the Ability of the Immune System to Detect and Kill Established Tumors As solid tumors develop, they recruit a variety of other cell types in addition to the cancerous cells themselves. These cells, which include fibroblasts from the organ in which the tumor develops, as well as a wide variety of different types of cells from the immune system, transform the tumor into a type of tissue, rather than simply a clone of isolated cancer cells. The non-cancer cells that surround the malignant tumor cells comprise what is called the tumor microenvironment. Cancer cells influence the behavior of the fibroblasts and immune cells in the microenvironment, and conversely, cells in the tumor microenvironment influence the way the cancer cells evolve, particularly with respect to immune recognition.
During the very early stages of tumor development, inflammatory immune cells including neutrophils, macrophages, and dendritic cells respond to the presence of these abnormal tumor cells by releasing cytokine molecules that inhibit tumor cell proliferation and angiogenesis and send signals to activate various types of T cells. A particularly important class of T cells involved in killing tumor cells are called cytotoxic T cells; they are distinguished by the presence of the CD8 protein on their surface (see
Chapter 24). To understand the mechanisms by which cytolytic T cells potentially can kill tumor cells and why this often does not occur, we must review some of the salient points by which T cells in general, and cytotoxic T cells in particular, recognize foreign molecules. Cytotoxic T Cells Target Tumor Cells As detailed in Chapter 24, T cells recognize not intact proteins but peptide fragments of these proteins of about 10 amino acids in length. Cells constantly degrade representative samples of all cellular proteins — whether they are made in the cell or brought in by pathogens — to peptides of this size, which are then bound to one of several two-chain glycoprotein complexes called MHC molecules. These MHC-peptide complexes are then transported to the cell surface where they can be detected by passing T cells. In the case of peptides derived from mutant proteins that are displayed on the surface of the tumor cells themselves, these are generally bound to class I MHC molecules (see Figure 24-26). Because most tumor cells, like all nucleated cells in the body, express MHC class I molecules on their surface, cytotoxic T cells can directly recognize tumor cells expressing neo-antigens. However, only a small
fraction of neo-antigens appear to be detected by T cells as foreign. Therefore, only tumors with a high mutational burden, such as melanomas, reach the threshold for recognition as a suitable target for destruction by immune surveillance. Engagement of tumor cells bearing foreign tumor antigens on their MHC class I molecules by cytotoxic T cells can result in killing of the tumor cells by two different mechanisms. Some activated cytotoxic T cells release granules containing the proteins perforin and granzyme-B. Perforin and granzymes enter a target cell through endocytosis, and perforins punch holes in the endocytic vesicle membrane, allowing granzymes to enter the cytoplasm. Granzymes are serine proteases and cleave and activate apoptotic molecules within the target cells, causing programmed cell death (see Figure 24-36). Other cytotoxic T cells express on their surface FasL, a natural ligand molecule related to TNF. When FasL on the cytotoxic T cell binds to its receptor, the Fas protein on the tumor cells, an apoptotic signaling pathway in the tumor cells becomes activated, which also leads to tumor cell death. During early stages of solid tumor development, these processes effectively eliminate many of the incipient tumor cells. As a consequence, the developing tumor and the immune system reach balance where the tumor does not grow but is not completely eliminated. Tumor Cells Escape from Killing by Cytotoxic T Cells
Even as growth plateaus, tumor cells continue to divide in a steady state balance with immune-mediated killing. Eventually, the tumor cells may acquire additional oncogenic mutations that allow them to escape the immune system and to begin to proliferate again. This immune evasion can occur through a variety of mechanisms. In some cases, the tumor cells send signals that change the types of immune cells that are recruited into their microenvironment, so that pro-inflammatory macrophages and neutrophils are replaced with anti-inflammatory, wound-healing neutrophils and macrophages, which secrete a different set of cytokines that fail to recruit T cells into the tumor. In other cases, the tumor downregulates the expression of class I MHC proteins, rendering the tumors invisible to cytotoxic T cells. Sometimes the tumor cells up-regulate the expression of anti-apoptotic proteins like BCL2 that blunt the killing effects of granzymes, perforins, and FasL. Two other mechanisms that tumor cells use to survive involve a direct attack by tumor cells on the signaling mechanisms that T cells use to kill them. In order for cytotoxic T cells to become licensed to kill cells bearing foreign antigens, they require a second co-stimulatory activation signal in addition to recognition of peptide fragments of tumor antigens presented on class I MHC molecules (see Figure 24-35 for more detail). This second check before killing probably exists to prevent cytotoxic T cells from accidently killing normal cells and causing autoimmune injury to normal tissues. One of the best understood co-stimulatory signals comes from CD28, a cell-surface protein on T cells that binds to either of two cellsurface proteins that are on the surface of antigen-presenting cells, CD80 and CD86 (Figure 25-24, step 3 ). Only when T cells see MHC-bound
foreign tumor antigens and receive a signal through CD28 do they become activated for cytokine production and T-cell–mediated killing. Within a day after activation, T cells up-regulate a different cell-surface protein, CTLA-4, which competes with CD28 for binding to CD80 and CD86 and sends a negative signal that inhibits T-cell responses (Figure 25-24, step 4 ). This is important because it restrains the extent of immune activation in order to limit inflammatory tissue damage. Because of the upregulation of CTLA-4 on anti-tumor cytotoxic T cells, they become selfinhibited in killing tumor cells.
FIGURE 25-24 Overview of T-cell signaling. Activation of a T cell is initiated by the T-cell receptor (TCR), which contains one α and one β chain, each of which differs slightly in sequence in different T cells of an individual. Each αβ TCR binds to a single or group of
related target peptides bound (“presented”) on the surface of an MHC protein on an antigen presenting cell (step 1 ). TCRs also contain signaling ζ chains as well as γ, δ, and ε chains. Binding of the TCR to a MHC-peptide complex triggers activation of the ZAP70 protein tyrosine kinase attached to the cytosolic domain of the ζ chains (step 2 ); in a cytotoxic T cell this triggers a signal transduction pathway (signal 1) essential for activation of the ability of the T cell to kill the antigen-presenting cell. Activation of the T cell also requires a second signal, triggered by binding of one of several proteins on the antigen-presenting cell, predominantly CD80 or CD86, to the CD28 protein on the surface of the T cell (step 3 ), triggering activation of a second signal transduction pathway that together with the first leads to activation of the T cell. Among the proteins induced during T-cell activation is CTLA-4; binding of CTLA-4 to CD80 or CD86 on the antigen-presenting cell (step 4 ) activates another signaling pathway leading to inactivation of the T cell. If the antigenpresenting cell expresses on its surface either the PD-L1 or the PD-L2 protein, these can bind to the PD-1 protein on the T cell (step 5 ) and trigger a signaling pathway that induces apoptosis of the T cell, preventing it from killing the antigen-presenting cell. A second T-cell surface receptor protein that acts as a negative regulator of T-cell function is PD-1 (programmed death-1). PD-1 has a structure similar to CTLA-4 and when activated negatively regulates T-cell function by inducing apoptosis of activated T cells. The two ligands of PD-1, PDL1 and PD-L2, are in the same protein family as CD80 and CD86 and are present on the surface of antigen-presenting cells; the ligands thus induce apoptosis of activated cytotoxic T cells, preventing inappropriate killing of antigen-presenting cells by T cells (Figure 25-24, step 5 ). Many tumor cells overexpress PD-L1 and thus escape death from cytotoxic T cells by in effect masquerading as antigen-presenting cells. Both the CTLA-4 pathway and the PD-1/PD-L1 signaling system are called immune checkpoint pathways because they normally keep the immune system in check in order to prevent the development of
Activation of the Immune System Presents a Major Opening for Cancer Therapy
autoimmune diseases. Both pathways are co-opted by tumor cells in order to suppress anti-tumor immunity. This entire process in which tumor cells are initially eliminated by the immune system, reach a steady state plateau, and then eventually escape from immune control by a variety of molecular mechanisms is referred to as immunoediting, since the final tumor outgrowths have been shaped by its response to the innate and adaptive immune system. Activation of the Immune System Presents a Major Opening for Cancer Therapy As we have seen, the immune system has the capacity, largely based on activation of cytotoxic T cells, to recognize and eliminate precancerous cells, especially those that express abundant neo-antigens. We have also seen that cancer cells that are able to escape immune surveillance and continue to proliferate to form tumors have acquired the capacity to suppress killing by cytotoxic T cells. One avenue for cancer therapy is the reactivation or reengineering of the immune system to recognize and eliminate mature tumors. Here we describe two of the most promising approaches for cancer immunotherapy. Monoclonal Antibodies Against Immune Checkpoint Molecules to Treat Cancers
One very successful method is to use antibodies that block CTLA4 from binding to CD80 and CD86 and thus prevent cytotoxic T cells from receiving inhibitory signals from CTLA-4. A CTLA-4-blocking antibody, ipilimumab, was approved for the treatment of advanced melanoma. It and similar monoclonal antibodies that block immune checkpoint activation have proven very effective in treating cancers such as melanomas that have a large mutational burden and thus have many neo-antigens recognized by many different T cells. Monoclonal antibodies directed against either PD-L1 or PD-1 are also effective in treating several types of advanced cancers, particularly those tumors that contain many mutant proteins such as melanomas and certain lung cancers. However, when administered alone, monoclonals against CTLA-4, PD-1, or PD-L1 are effective in treating only a fraction of patients with most cancers. Increasingly, combination therapies, in which these monoclonals are combined with other antibodies and drugs are increasing the efficacy of this type of cancer immunotherapy and may eventually allow even some terminal cancer patients to survive. Engineering T Cells to Recognize Tumor Antigens We have just seen that cytotoxic T cells, when reactivated by inhibition of the immune checkpoint, have the capacity to recognize and kill some tumors with a large burden of mutations. However, most tumors, probably because they express an insufficient number of neo-antigens, do not respond to monoclonal antibodies to checkpoint inhibitors such as CTLA4 or PD-1. An alternative approach is to genetically engineer T cells in the laboratory that can directly attack tumor cells bearing a marker protein on
their surface and then infuse them into patients where they can perform their tumor-killing function. These T cells contain specially engineered Tcell receptors, called chimeric antigen receptors, and the modified cells are called CAR T cells. To understand how they work, we need to first review signaling pathways that connect T-cell receptor binding to antitumor activity. The cytosolic domains of the a and b subunits of the T-cell receptor do not directly activate a signal transduction pathway. Rather, binding of the extracellular TCR domain to a MHC-peptide complex triggers activation of ZAP70 protein tyrosine kinases attached to the cytosolic domains of the two adjacent ζ chains (see Figure 25-24, bottom left). The idea behind CARs was to engineer the T-cell receptor to trigger activation of ZAP70 in response to the presence of a tumor-specific antigen rather than the MHCpeptide complex. The first generation of CARs accomplished this by employing the antigen-binding and domains of a monoclonal antibody that binds to a protein enriched on the surface of a tumor cell (e.g., CD19 on a B-cell tumor) linked to the cytosolic domain of the ζ subunit through a transmembrane segment. T cells expressing such a chimeric receptor do activate ZAP70 when they encounter a tumor cell expressing the CD19 protein on its surface (Figure 25-25a), but they fail to kill the tumor cell. Realizing that this failure could be due to the absence of crucial co-stimulatory signals (signal 2 in Figure 25-24), researchers designed chimeric antigen receptors in which the cytosolic domain contained signal-transducing segments from the cytosolic domain of the ζ subunit as well as from one or more of the co-stimulatory receptors such as CD28 or 4-1BB (see Figure 25-24). In clinical trials, such CARs
targeting CD19 were expressed using lentiviral vectors in normal T cells isolated from a large number of patients with several types of human Bcell tumors (Figure 25-25b). Retransfusion of these engineered CAR T cells back into these patients in many cases led to complete remission of the cancer. Currently, researchers are developing CAR T cells expressing CARs targeting a number of proteins enriched on the surface of many types of human tumors. These are being tested against a number of malignancies for which there are presently no cures.
FIGURE 25-25 Three generations of CAR T cells. (a) Chimeric antibodies (CARs) expressed in T cells. The first generation CAR T cells to treat B-cell malignancies expressed
on the T-cell surface of a chimeric protein containing the and transmembrane domain and the cytosolic domain of the ζ subunit of the T-cell receptor (second panel). Recall that binding of the TCR to a peptide MHC complex normally triggers activation of the ZAP70 protein tyrosine kinase attached to the cytosolic domain of the ζ chains. Secondgeneration CARs contained, in addition, the cytosolic signaling domains from one of the co-stimulatory receptors that form the 4-1BB protein or the CD28 protein (see Figure 2524). Subsequent CARs incorporated both of these co-stimulatory domains and signaling domains from other proteins as well. These co-stimulatory domains activate multiple signaling pathways that trigger rapid and extensive activation of T-cell killing of target tumor cells. (b) Generation of CAR T cells. A cDNA encoding a chimeric antibody receptor is cloned into a lentiviral vector — similar to the retrovirus vector depicted in Figure 6-35 except derived from the human HIV retrovirus. Leukapheresis, a procedure to extract T cells from a patient with a B-cell malignancy, is performed, and the T cells are infected in culture with the lentivirus vector. After culture to allow integration of the viral DNA into the genome and expression of the chimeric antibody, the T cells are transfused back to the patient. The production of CAR T cells normally takes 17–21 days. Description The illustration labeled (a) shows a cell membrane with a double oval structure in the exterior. The left oval is labeled V H segment of anti-C D 19 monoclonal antibody. The right oval is labeled V L segment of anti-C D 19 monoclonal antibody. This pair of ovals is presented 4 times as changes happen at the bottom of the structure inside the cytosol. First-generation C A R shows the attachment of a green rounded rectangle to the antibody in the membrane. The green rectangle is labeled T-cell activation domain. In the second diagram, an orange rectangle is added to the green one and labeled C D 28 co-stimulatory domain. In the third diagram, a second antibody has a blue rectangle labeled 4-1 B B co-stimulatory domain. These two diagrams are labeled as secondgeneration C A R's. The last diagram shows all three rectangles joined together and labeled third-generation C A R. The illustration labeled (b) starts with a human outline. An arrow from the structure in the human body points to a group of 5 T-cells. Above and to the right is a virus with receptors around it labeled retroviral transduction with anti-C D 19 C A R. An arrow shows that this virus goes into the T-cells. An arrow coming away from the T-cells to the right leads to three blue cells labeled C A R T cells. An arrow shows the structure
from the illustration (a) is the C A R part of these cells. The blue cells have an arrow pointing to an I V bag labeled anti-C D 19 C A R T-cell infusion which is connected to another human outline. Into the other arm of the human outline is the pink I V bag labeled Preconditioning chemotherapy. Complications of Cancer Immunotherapy One caveat to all of these immune-activating treatments — both monoclonal antibodies against immune checkpoint molecules and CAR T cells — is that they unleash the full force of the immune system not only against the tumor, but often against the adjacent normal tissues that are not cancerous. Consequently, a major side effect of these therapies is a cytokine storm that leaves the patient with potentially devastating tissue damage. Successful application of cancer immunotherapy will require finding a balance between activation of the T cells to recognize tumors with other treatments that are less injurious of normal tissues. KEY CONCEPTS OF SECTION 25.4 Evasion of Programmed Cell Death and Immune Surveillance Processes Precancerous cells often have sustained DNA damage and are normally eliminated by programmed cell death or apoptosis. Oncogenic driver mutations that suppress apoptosis allow continued tumor cell proliferation even in the presence of sustained DNA damage. The most common driver oncogenic driver mutations are in p53, a transcription factor that activates the expression of genes for cell cycle arrest or apoptosis. Cancer cells that have loss of function mutations in p53 continue to proliferate damage to their genomes and thus have a propensity to accumulate mutations. Immune cells in the tumor microenvironment detect mutant proteins and shape the developing tumor through a three-step process involving tumor cell elimination,
equilibrium between the tumor cells and the immune system, and late escape of tumor cells from immune control. Cytotoxic T cells recognize fragments of mutant proteins presented by MHC proteins on the surface of tumor cells. However, the killing activity of the T cells is usually abated in part because of activation of inhibitory receptors on the T-cell surface, such as CTLA4 and PD-1. Monoclonal antibodies to CTLA4 and PD-1 and to PD-L1, the ligand of PD-1, prevent activation of these inhibitory receptors and enhance the ability of the T cells to kill the tumor cells. These monoclonals are used to treat cancers such as melanomas in which many mutations have accumulated. CAR T cells are normal T cells in which lentivirus vectors have been used to introduce a chimeric protein, a CAR, containing the and domains of a monoclonal antibody targeting a protein on the surface of a tumor cell, a transmembrane segment, and cytosolic domains from the TCR ζ submit and from any of several co-stimulatory receptors. CAR T cells are efficacious in killing many types of human tumors and in curing the cancer.
Key Terms
End of Chapter Visit Achieve to access study tools and to learn more about the content in this chapter: Perspectives for the Future Classic Experiment 25.1: An Experiment That Led to the Identification of the RAS Oncogene Analyze the Data Additional study tools, including videos, animations, and quizzes Key Terms angiogenesis benign Burkitt’s lymphoma cancer stem cell carcinogen carcinoma CAR T cells epithelial-to-mesenchymal transition (EMT) immune checkpoint pathways immunoediting leukemia loss of heterozygosity (LOH) lymphomas
Review the Concepts
malignant metastasis multi-hit model mutagen oncogene oncogenesis p53 protein Philadelphia chromosome proto-oncogene Ras protein Rb protein retrovirus sarcoma telomerase transformation tumorigenesis tumor microenvironment tumor suppressor gene Warburg effect Review the Concepts 1. Despite differences in origin, cancer cells have several features in common that differentiate them from normal cells. Describe these. 2. What characteristics distinguish benign from malignant tumors?
3. Which important characteristic of tumor cells did Otto Warburg discover? 4. Because of oxygen and nutrient requirements, cells in a tissue must reside within 100 μm of a blood vessel. Based on this information, explain why many malignant tumors often possess gain-of-function mutations in one of the following genes: βFGF, TGF-α, and VEGF. 5. Ninety percent of cancer deaths are caused by metastatic rather than primary tumors. Define metastasis. Explain the rationale for the use of the cancer treatment agent batimastat, an inhibitor of matrix metalloproteases and of the plasminogen activator receptor. 6. What is the importance of the EMT during metastasis? 7. What hypothesis explains the observations that incidence of human cancers increases exponentially with age? Give an example of data that confirm the hypothesis. 8. Distinguish between proto-oncogenes and tumor suppressor genes. To become cancer promoting, do proto-oncogenes and tumor-suppressor genes undergo gain-of-function or loss-of- function mutations? Classify the following genes as protooncogenes or tumor-suppressor genes: p53, ras, BCL-2, JUN, MDM2, and p16. 9. Describe how mutations in genome maintenance factors promote tumorigenesis. Why would inactivation of a mismatch repair gene cause colon cancer? 10. Hereditary retinoblastoma generally affects children in both eyes, while spontaneous retinoblastoma usually occurs during adulthood only in one eye. Explain the genetic basis for the
epidemiological distinction between these two forms of retinoblastoma. Explain the apparent paradox: loss-of-function mutations in tumor-suppressor genes act recessively, yet hereditary retinoblastoma is inherited as an autosomal dominant. 11. Explain the concept of loss of heterozygosity (LOH). Why do most cancer cells exhibit LOH of one or more genes? How does failure of the spindle assembly checkpoint lead to loss of heterozygosity? 12. Many malignant tumors are characterized by the activation of one or more growth-factor receptors. What is the catalytic activity associated with transmembrane growth-factor receptors such as the EGF receptor? Describe how a point mutation that converts a valine to glutamine within the transmembrane region of the HER2 receptor leads to activation of the relevant growthfactor receptor. 13. Describe the common signal transduction event that is perturbed by cancer-promoting mutations in the genes encoding RAS and NF1. Why are mutations in RAS more commonly found in cancers than mutations in NF1? 14. Describe the mutational event that produces the MYC oncogene in Burkitt’s lymphoma. Why does the particular mechanism for generating oncogenic MYC result in a lymphoma rather than another type of cancer? Describe another mechanism for generating oncogenic MYC. 15. Pancreatic cancers often possess loss-of-function mutations in the gene that encodes the Smad4 protein. How does this mutation promote the loss of growth inhibition and highly metastatic phenotype seen in pancreatic tumors?
16. Why are mutations in the p16-arf-p15 locus so dangerous? 17. Explain how epigenetic changes can contribute to tumorigenesis. 18. Several strains of human papilloma virus (HPV) can cause cervical cancer. How does the viral E7 protein contribute to host cell transformation? 19. Loss of p53 function occurs in the majority of human tumors. Name two ways in which loss of p53 function contributes to a malignant phenotype. Explain how the carcinogen benzo(a)pyrene can cause loss of p53 function. 20. The active form of p53 is a tetramer of four identical subunits. However, a mutant version of just one subunit can inactivate the entire tetramer. Explain how this dominant negative interaction increases the probability that mutations in p53 are selected for during cancer progression. 21. Explain why only about 1 percent of mutations in a tumor cell have an effect on a cellular protein and thus may cause production of a neo-antigen. 22. CAR T cells are genetically engineered to kill cancer cells by introduction of a chimeric protein composed of the antigenbinding domain of a monoclonal antibody linked to the cytosolic ζ subunit of the T-cell receptor. Explain the function in targeting and killing cancer cells of the antibody part and the T-cell receptor part of the chimeric protein.