13 Microbial Evolution and Genome Dynamics
## Chapter 13 Microbial Evolution and Genome Dynamics
I Early Earth and the Origin and Diversification of Life
Exploring Viral Genesis
Unlike cells, viruses cannot perform metabolic reactions or conserve energy, yet viruses thrive in a world filled with cells. Viruses can evolve with frightening speed, and a tremendous diversity of viruses exist. Indeed, it is likely that every cellular organism can be a host for several different viruses.
While we have learned much about viruses, we know little about their evolution. Most viral genomes have few genes—indeed, the smallest have as few as three—and as a result, it is difficult to reconstruct the evolutionary history of all viruses as has been done for cells. Hence, we can only speculate about viral origins. All viruses might have diverged from one ancestral virus that appeared soon after the origin of cellular life, some 3.8 to 4.3 billion years ago. However, another possibility is that viruses have multiple origins, appearing spontaneously again and again over the history of life.
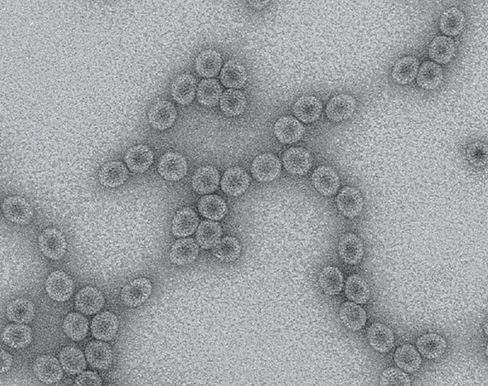
To address these hypotheses, we must understand mechanisms that promote viral genesis. One important step in viral genesis is the origin of the viral capsid. All capsids are formed by self-assembly of one or a few proteins into a hollow molecular structure. Similar structures already exist in cells. For example, consider flagella and carboxysomes; both are hollow structures formed by self-assembling proteins. Another example is the enzyme lumazine synthase produced by the bacterium Aquifex aeolicus. Copies of this protein spontaneously assemble into capsid-like protein shells, and, with a few simple modifications in the laboratory, can be altered to package their own mRNA, resulting in virus-like nucleocapsids containing an RNA genome (see photo, capsids are 31 nm in diameter).
This discovery suggests that it could be surprisingly easy for viruses to originate spontaneously by mutation of cellular proteins that captured bits of nucleic acid. Thus, it is indeed possible that the viral diversity we observe today has many different origins.
Source: Terasaka, N., et al. 2018. Laboratory evolution of virus-like nucleocapsids from nonviral protein cages. Proc. Natl. Acad. Sci. 115: 5432.
I: Early Earth and the Origin and Diversification of Life
I: Early Earth and the Origin and Diversification of Life
I Early Earth and the Origin and Diversification of Life
**Microbes first appeared on Earth about 4 billion years ago and proceeded to colonize every habitable environment and oxygenate the planet. Cells of Bacteria and Archaea combined in some way to give rise to eukaryotic cells, and the evolutionary path that all cells have traveled is recorded in the nucleotide sequences of their DNA.**
Evolution is a process that unifies all branches of biology because all living things undergo change over time and all have descended from the same universal ancestor. In this chapter, we tackle the topic of microbial evolution and describe methods for unraveling evolutionary relationships between microorganisms. We will also see how microbial evolution informs microbial systematics, a system we will use to describe relationships among microorganisms as we explore the diversity of the microbial world in the following five chapters.
In these first sections, we consider the possible conditions under which life arose, the earliest evidence for cellular life, and its divergence into three evolutionary lineages: Bacteria, Archaea, and Eukarya. Although much about these events and processes remains speculative, geological and molecular evidence has combined to build a plausible scenario for the earliest events in the evolution of life and for the fundamental impacts that microbes have had on the history of our Earth.
13.1 Formation and Early History of Earth
The Earth of 4 billion years ago (bya) would be foreign and inhospitable to human eyes, but this sterile wasteland of blasted rock and boiling seas was the incubator from which all life sprang. Since few fossils exist to tell the story of the early Earth, most of what we know is inferred from chemical and isotopic analyses of ancient rocks and minerals. However, we will see in this chapter that we can infer ancient evolutionary events from living organisms by comparing their molecular structures and sequences to determine their common history of descent. The story of life begins not long after the dawn of our solar system with the formation of Earth itself.
Origin of Earth
Earth formed about 4.5 bya (Figure 13.1), based on analyses of slowly decaying radioactive isotopes. Our planet and the other planets of our solar system arose from materials making up a disk-shaped nebular cloud of dust and gases released by the supernova of a massive old star. As a new star—our sun—formed within this cloud, it began to compact, undergo nuclear fusion, and release large amounts of energy in the form of heat and light. Materials left in the nebular cloud began to clump and fuse due to collisions and gravitational attraction, forming tiny aggregates that gradually grew larger to form clumps that eventually coalesced into planets. Energy released in this process heated the emerging Earth as it formed, as did energy released by radioactive decay within the condensing materials, forming a planet Earth of fiery hot magma. As Earth cooled over time, a metallic core, rocky mantle, and a lower-density, thin surface crust formed.
Figure 13.1 Major landmarks in biological evolution, Earth’s changing atmospheric chemistry, and microbial metabolic diversification.

The oldest date for the origin of life is fixed by the time of Earth’s origin, and the minimum time for the origin of oxygenic photosynthesis is fixed by the Great Oxidation Event, about 2.4 billion years ago (bya). Note how the oxygenation of the atmosphere from cyanobacterial metabolism was a gradual process, occurring over a period of about 2 billion years. Compare this figure with the introduction to the antiquity of life on Earth shown in Figure 1.10.
The inhospitable conditions of early Earth, characterized by a molten surface under intense bombardment by asteroids and other objects from space, persisted for up to 500 million years. The intense heat and the absence of liquid water indicate that early Earth was completely devoid of life. Furthermore, the early Earth was anoxic, and the absence of O2 means that Earth lacked an ozone layer, allowing intense ultraviolet (UV) light to sterilize its surface. Water on Earth originated from volcanic outgassing of the planet’s interior and from innumerable collisions with icy comets and asteroids. Given Earth’s heat at the time, this water would have been present only as water vapor. No rocks dating to the origin of Earth have yet been discovered, presumably because they have undergone geological metamorphosis. However, ancient crystals of the mineral zircon (ZrSiO4) have been discovered that were formed on the early Earth, and these materials have given us a glimpse of conditions on Earth prior to the origin of life.
Liquid water is a requirement for life, and its eventual presence on Earth made possible the origin of life. Analyses of ancient zircon crystals, including impurities trapped in these crystals and their oxygen isotope ratios (we discuss the use of isotopic analyses as indications of living processes in Section 19.10), provide evidence for a solid crust and liquid water on Earth as early as 4.3 bya (Figure 13.1). Additional evidence of water comes from ancient sedimentary rocks, since sedimentary rocks are only formed under water. Among the oldest surviving sedimentary rocks are those found in southwestern Greenland, dating to 3.86 bya. These ancient rocks indicate the presence of early oceans. These rocks also contain the fossilized remains of cells (Figure 13.2), as well as carbon isotope ratios that provide evidence for life. Furthermore, graphite inclusions having carbon isotope ratios of biogenic origin are found in zircon minerals that are between 3.7 and 4.1 bya. These lines of evidence show that liquid water appeared prior to 4 bya and soon thereafter the first life forms appeared.
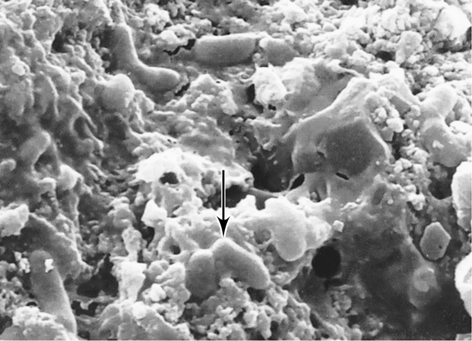
Figure 13.2 Ancient microbial life.
Scanning electron micrograph of microfossil bacteria from 3.45-billion-year-old rocks of the Barberton Greenstone Belt, South Africa. Note the rod-shaped bacteria (arrow) attached to particles of mineral matter. The cells are about 0.7 μm in diameter.
Origin of Cellular Life
The origin of life on Earth remains the greatest of mysteries, obscured by the depths of time. Few rocks survive unaltered to testify about this period of Earth’s history. Experimental evidence indicates that organic molecules such as RNA nucleotides, amino acids, and lipids can form spontaneously under conditions that were present on the early Earth, providing the preconditions needed for the first living systems. However, conditions on Earth’s surface more than 4.1 bya, in particular the extremely hot temperatures and high levels of ultraviolet radiation, were likely hostile to the origin of life as we know it.
Mastering Microbiology
Art Activity: Figure 13.3 Submarine mounds and their possible link to life origins
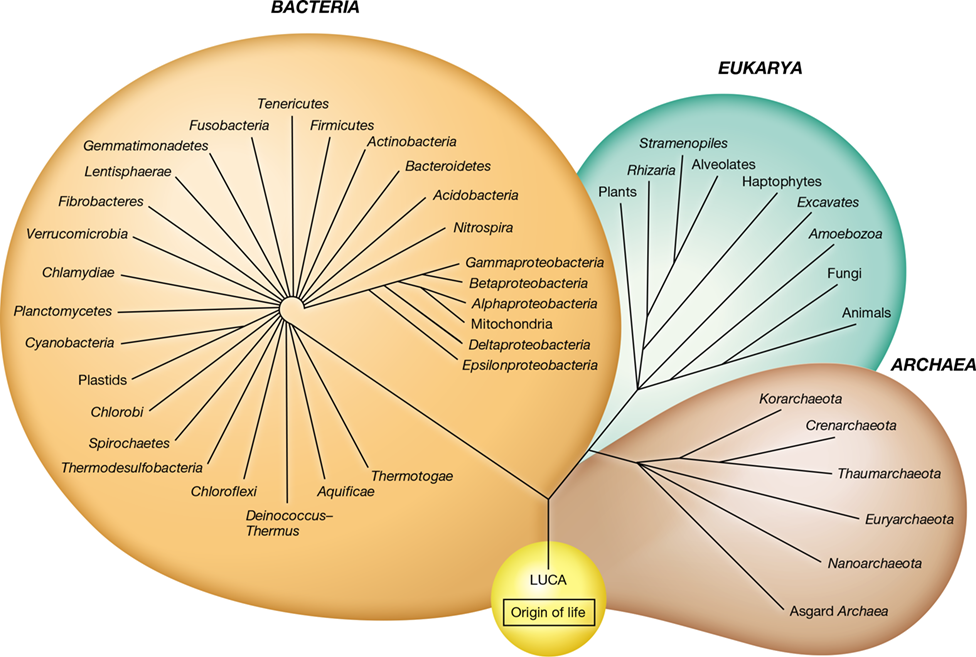
One hypothesis holds that life may have originated at hydrothermal systems on the ocean floor (Figure 13.3). Deep on the ocean floor, protected from the lethal UV radiation at Earth’s surface, conditions would have been less hostile and more stable than on land. A steady and abundant supply of energy in the form of reduced inorganic compounds—hydrogen (H2), hydrogen sulfide (H2S), and elemental sulfur (S0), for example—would have been available at these hydrothermal systems. The unique geochemistry of these sites can support the abiotic production of molecules critical for the emergence of life, such as amino acids, lipids, sugars, and nucleotides. Furthermore, minerals that form in these systems can produce compartmentalized structures that may have been necessary for conserving energy prior to the emergence of biological membranes. Whether on the seafloor or elsewhere, some form of prebiotic chemistry must have facilitated the development of the first self-replicating systems, the precursors to cellular life.
Figure 13.3 Submarine mounds and their possible link to the origin of life.


Deep-sea hydrothermal vents are a likely site for the origin of life on Earth. They are stable environments conducive to life and they provide a source of chemical energy, precursors required for the formation of biological molecules, and mineral pores that can serve as compartments to house pre-cellular biochemical reactions. (a) Precursor molecules form abiotically and accumulate within mineral pores. (b) Chemical gradients that form within mineral pores provide a source of energy that drives the replication of pre-cellular biological molecules. (c) Membranes eventually take over the role of mineral compartments, leading to the formation of the first cells. (d) Photo of an actual hydrothermal mound. Hot, mineral-rich hydrothermal fluid mixes with cooler, more oxidized ocean water, forming precipitates of Fe and S compounds, clays, silicates, and carbonates. These mineral precipitates form pores that could have served as energy-rich compartments to facilitate the evolution of pre-cellular forms of life.
Anna-Louise Reysenbach and Woods Hole Oceanographic Institution
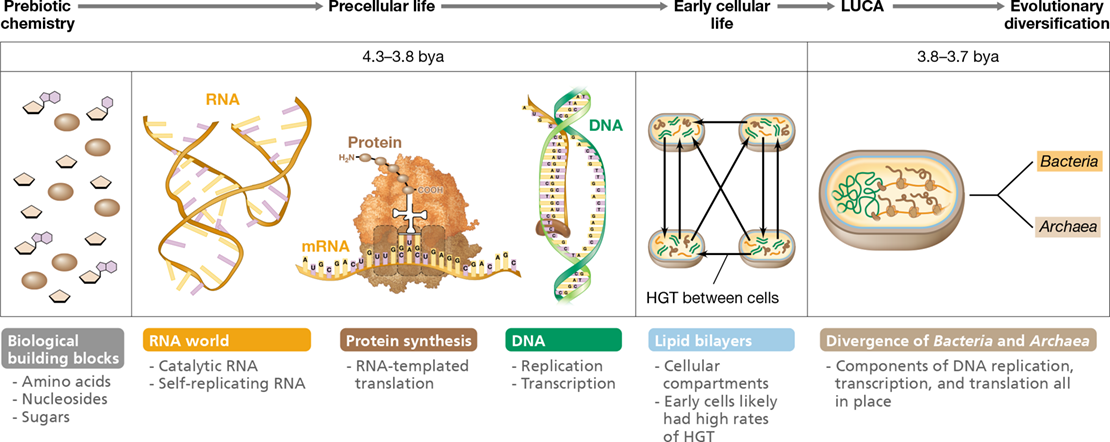
Molecules of RNA were likely a central component of the first self-replicating systems, and it is possible that life began in an RNA (rather than DNA) world (Figure 13.4). RNA molecules can perform diverse biochemical functions. For example, RNA molecules are the backbone of certain molecules essential to all cells, such as ATP (used in energy transfer), NADH (used in electron transfer), and coenzyme A (used in many biosynthetic reactions) (Chapter 3). RNA can also bind small molecules (such as nucleotides and amino acids), can catalyze certain simple biochemical reactions, and can be the template for its own synthesis. In addition, RNA is able to catalyze protein synthesis through the activities of ribosomal RNAs, tRNAs, and mRNAs (Section 6.10). Hence, it is not unreasonable to imagine that the earliest forms of life relied exclusively on RNAs and had little or no need for DNA or proteins.
Figure 13.4 Events hypothesized to precede the origin of cellular life.

The earliest self-replicating biological systems may have been based on catalytic RNA. At some point RNA enzymes evolved the capability to synthesize proteins, and proteins became the main catalytic molecules. Conversion from RNA- to DNA-based genomes required the evolution of DNA and of RNA polymerases. The lipid bilayer is the site of electron transport, and the evolution of this structure was likely important for energy conservation, in addition to containing and protecting biomolecules. The last universal common ancestor (LUCA), which preceded the divergence of Bacteria and Archaea (see Figure 13.9), was a cellular organism that had a lipid bilayer and used DNA, RNA, and protein. Horizontal gene transfer (HGT) may have allowed rapid transfer of beneficial genes among early forms of life.
Eventually, however, proteins synthesized by RNA molecules replaced the catalytic role of RNAs in primitive cells (proteins can catalyze many reactions that RNA cannot) and at some later point, DNA, a molecule that is inherently more stable than RNA and therefore a better repository of genetic (coding) information, assumed the role of the genome and became the template for RNA synthesis (Figure 13.4). The earliest cellular forms of life likely possessed elements of this three-part system of DNA, RNA, and protein, in addition to a membrane system capable of conserving energy and preventing cell leakage. The last universal common ancestor (LUCA) of all extant life likely existed at 3.8–3.7 bya, the point at which Bacteria and Archaea diverged and life began to diversify into the cellular forms we would recognize today.
The Last Universal Common Ancestor
Following the origin of cells, microbial life likely experienced a long period of metabolic diversification, exploiting the various energy resources available on Earth. For much of Earth’s history the planet, including all of its oceans, was anoxic (Figure 13.1). Thus, the energy-generating metabolism of primitive cells would have been exclusively anaerobic. Furthermore, organic matter was largely absent and so early cells must have been chemolithotrophic, using substances such as H2 and H2S spewed out of volcanoes and thermal springs as electron donors. H2 is also produced abiotically when pyrrhotite (FeS), an abundant volcanic mineral on the early earth, reacts with H2S to yield pyrite (FeS2) and H2. Regardless of the source of H2, this simple molecule is thought to have been the basis of a primitive energy metabolism based on the proton motive force (Figure 13.5). Elemental sulfur (S0), also abundant on the early Earth, was likely one of the first acceptors of electrons from H2 oxidation; the reduction of S0 to yield H2S is exergonic and can be accomplished by a single enzyme (Figure 13.5). Because organic carbon was scarce on the early Earth, these primitive chemolithotrophs likely used CO2 and N2 as major sources of C and N for biosynthesis (see autotrophy and nitrogen fixation, Chapter 3).
Figure 13.5 A possible energy-conserving scheme for primitive cells.

The earliest cells could have generated energy by using H2 as electron donor and S0 as electron acceptor, both of which are abundant in hydrothermal systems and are also emitted from volcanoes. H2 can also be formed from an abiotic reaction between FeS and H2S, both of which were common on the early Earth. Such reactions could have allowed early cells to colonize diverse habitats.
Mastering Microbiology
Art Activity: Figure 13.4 Events hypothesized to precede the origin of cellular life
By analyzing the gene content of genomes from living organisms across the tree of life, we can identify those genes that were most likely present in the earliest cells. A total of just 355 genes trace back to the last universal ancestor. These genes indicate that LUCA was an anaerobic organism that lived in a high-temperature environment rich in sulfur and iron and used H2 as a source of energy and CO2 as a source of carbon. These traits suggest that LUCA could have lived in a hydrothermal environment (Figure 13.3). Unsurprisingly, these cells had all the genetic machinery required for DNA replication, transcription, and translation, as well as ATP synthase. These early cells had the capacity to metabolize H2 and CO2, likely producing either acetate (acetogens, Section 14.14) or methane (methanogens, Section 14.15) as their waste products. These early forms of chemolithotrophic metabolism along with the reduction of S0 (Figure 13.5), all driven by H2 as electron donor, would likely have supported the production of large amounts of organic compounds from autotrophic CO2 fixation. Over time, these organic materials would have accumulated and provided the conditions needed for the evolution of chemoorganotrophic bacteria with diverse metabolic strategies to conserve energy from the oxidation of organic compounds.
Eventually Earth became a highly oxic planet, with an atmosphere containing the high levels of O2 that we breathe today (Figure 13.1). Microbes catalyzed this key geochemical transition in Earth’s history, and we explore this topic now.
Check Your Understanding
What characteristics would have made the surface of Earth inhospitable to the formation of life 4.5 billion years ago?
How do we know when oceans were first present on Earth? Why is the presence of oceans significant to the origins and diversification of life?
What lines of reasoning support the hypothesis that the first self-replicating systems were based on RNA molecules?
13.2 Photosynthesis and the Oxidation of Earth
The evolution of photosynthesis was a biological breakthrough that radically changed the chemistry of Earth. Phototrophic organisms use energy from the sun to oxidize molecules such as H2S, S0, Fe2+, or H2O and to synthesize complex organic molecules from carbon dioxide or simple organic molecules (Sections 14.2 and 14.3). Over time, the products of photosynthesis accumulated in the biosphere, stimulating the further diversification of microbial life. Earth’s first phototrophs were anoxygenic (cells that do not produce O2, Sections 14.5 and 15.4, 15.5, 15.6 and 15.7), but from these evolved the Cyanobacteria, the earliest O2-producing (oxygenic) phototrophs (Figure 13.1, Section 15.3).
Fossilized microbial formations called stromatolites can be found in rocks that are 3.5 bya, providing some of the earliest conclusive evidence of life on Earth (Figure 13.6). Stromatolites, or “layered rocks,” are formed when microbes cause the deposition of carbonate or silicate minerals that promote fossilization (we discuss microbial mats in Section 20.5). Stromatolites were diverse and common on Earth between 2.8 and 1 bya but declined dramatically in abundance over the last billion years. Stromatolites are largely gone from Earth today, and yet modern examples of this ancient microbial ecosystem can still be found in certain shallow marine basins (Figure 13.6c, e) or in hot springs (Figure 13.6d). Phototrophic bacteria, such as cyanobacteria (Section 15.3) and anoxygenic phototrophs, play a central role in the formation of modern stromatolites. Ancient stromatolites contain microfossils that appear remarkably similar to modern species of phototrophic bacteria (**Figure 13.7*a***). Hence, the earliest phototrophic organisms may have evolved in the Bacteria more than 3.5 bya, giving rise to the stromatolites we observe in the fossil record.
Figure 13.6 Ancient and modern stromatolites.

(a) The oldest known stromatolite, found in a rock about 3.5 billion years old from the Warrawoona Group in Western Australia. Shown is a vertical section through the laminated structure preserved in the rock. Arrows point to the laminated layers. (b) Stromatolites of conical shape from 1.6-billion-year-old dolomite rock from northern Australia. (c) Modern stromatolites, Darby Island, Bahamas. The large stromatolite in the foreground is about 1 m in diameter. (d) Modern stromatolites composed of thermophilic cyanobacteria growing in a thermal pool in Yellowstone National Park, USA. Each structure is about 2 cm high. (e) Modern stromatolites from Shark Bay, Australia. Individual structures are 0.5–1 m in diameter.
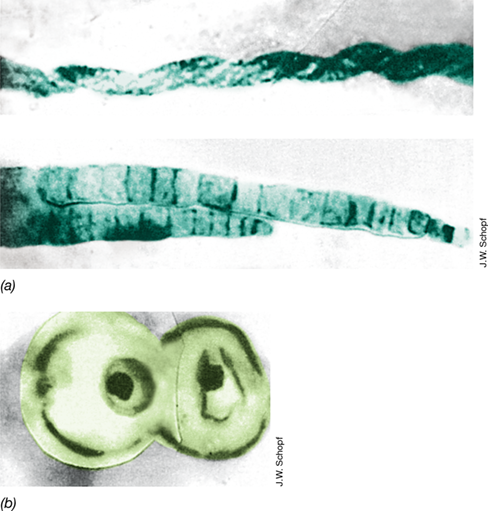
Figure 13.7 More recent fossil bacteria and eukaryotes.

(a) One-billion-year-old microfossils from central Australia that resemble modern filamentous cyanobacteria. Cell diameters, 5–7 μm. (b) Microfossils of eukaryotic cells from the same rock formation. The cellular structure is similar to that of certain modern green algae, such as Chlorella species. Cell diameter, about 15 μm. Color was added to make cell form more apparent.
The first phototrophs were anoxygenic and likely used H2S as electron donor for CO2 fixation, generating elemental sulfur (S0) as a waste product (Section 14.5). How could the first phototrophs have evolved at a time when life existed mostly near hydrothermal systems? A clue came from the recent discovery of anoxygenic phototrophs living at hydrothermal vents in the complete darkness of the deep ocean. These phototrophs actually carry out photosynthesis using infrared radiation generated by the heat of hydrothermal vents. Likewise, the first photosynthetic organisms likely lived in the dark, at hydrothermal vents where H2S and infrared radiation were abundant. Diversification of anoxygenic phototrophs led to species that were able to use a range of electron donors including Fe2+, which was abundant throughout Earth’s early oceans. The ability to use Fe2+ as an electron donor likely allowed early phototrophs to escape from hydrothermal systems and colonize shallow regions of Earth’s early oceans where light was abundant but where overlying water still provided protection from UV radiation.
The ability to use solar radiation as an energy source allowed phototrophs to diversify extensively. By 2.5–3.3 bya, the cyanobacterial lineage evolved a photosystem capable of oxygenic photosynthesis (Section 14.6) in which H2O supplanted H2S as the reductant for CO2, thereby generating O2 as a waste product. About a billion years later, eukaryotic oxygenic phototrophs appeared and can be seen in the microfossil record (Figure 13.7b). The origin of oxygenic photosynthesis and the rise of O2 in Earth’s atmosphere (Figure 13.1) caused the greatest change ever in the history of our biosphere and set the stage for the evolution of even newer and more complex forms of life that evolved to exploit the energy available from O2 respiration. We look at the evidence for and consequences of Earth’s oxidative transformation now.
The Rise of Oxygen: Banded Iron Formations
In the absence of O2, most of Earth’s iron would have been present in reduced forms (Fe0 and Fe2+), and large amounts of iron were dissolved in Earth’s anoxic oceans. Molecular and chemical evidence indicates that oxygenic photosynthesis first appeared on Earth several hundred million years before significant levels of O2 appeared in the atmosphere. However, the O2 that cyanobacteria produced could not accumulate in the atmosphere because it reacted spontaneously with the reduced iron minerals in the oceans to make iron oxides. By 2.4 bya, O2 levels had risen to one part per million, a tiny amount by present-day standards, but enough to initiate what has come to be called the Great Oxidation Event (Figure 13.1).
The metabolism of cyanobacteria yielded O2 that oxidized reduced minerals containing Fe2+ to iron oxides containing Fe3+. These iron oxide minerals became a prominent marker in the geological record. Iron oxides are poorly soluble in water and would have precipitated in the oceans, raining down onto the seafloor and forming sedimentary structures we see today as banded iron formations (Figure 13.8), laminated sedimentary rocks formed in deposits of iron- and silica-rich materials. In addition to cyanobacteria, anoxygenic phototrophs that oxidize Fe2+ likewise contributed to the formation of these banded iron formations. Much of the iron in rocks of Precambrian origin (>0.5 bya, see Figure 13.1) exists in banded iron formations, and today these minerals are mined as a major source of iron ore. During this span of Earth history, lasting more than 1.5 billion years, the presence of precipitating iron minerals could have caused the oceans to appear brown, or black, or even red rather than the blue color we know today. Only after the abundant Fe2+ on Earth was oxidized could O2 begin to accumulate in the atmosphere, and not until 600–900 million years ago did atmospheric O2 reach present-day levels (∼21%, Figure 13.1).
Figure 13.8 Banded iron formations.
An exposed cliff made of sedimentary rock about 10 m in height in Western Australia contains layers of iron oxides (arrows) interspersed with layers containing iron silicates and other silica materials. The iron oxides contain ferric iron (Fe3+) produced from ferrous iron (Fe2+) primarily by the oxygen released by cyanobacterial photosynthesis.
As O2 accumulated on Earth, the atmosphere gradually changed from anoxic to oxic. Species of Bacteria and Archaea unable to adapt to this change were increasingly restricted to anoxic habitats because of the toxicity of O2 and because it chemically oxidized the reduced substances upon which their metabolisms depend. However, the oxic atmosphere also created conditions for the evolution of various new metabolic schemes, such as sulfide oxidation, nitrification, and the various other aerobic chemolithotrophic processes described in Chapter 14. Microbes that evolved the capacity to respire O2 gained a tremendous energetic advantage because of the high reduction potential of the O2/H2O couple (Section 3.2 and Figure 3.4), and with more energy at their disposal, aerobes could reproduce far more rapidly than anaerobes.
Mastering Microbiology
Art Activity: Figure 13.9 The universal phylogenetic tree as supported by comparative SSU rRNA gene sequence analysis
The Ozone Shield
An important consequence of O2 for the evolution of life was the formation of ozone (O3). The sun bathes Earth in intense amounts of ultraviolet (UV) radiation, which is lethal to cells and can cause severe DNA damage (Section 9.4). When O2 is subject to UV radiation from the sun, it is converted to ozone, which strongly absorbs UV radiation in wavelengths up to 300 nm. The conversion of O2 to O3 creates an ozone shield, a barrier that protects the surface of Earth from much of the UV radiation from the sun. Prior to the generation of the ozone shield, the punishing UV irradiation from the sun would have made Earth’s surface fairly inhospitable for life, restricting life to environments that provided protection from UV radiation, such as in the oceans or in the subsurface. However, as Earth developed an ozone shield, organisms could range over the terrestrial surface of Earth, exploiting new habitats and evolving ever-greater diversity. Figure 13.1 summarizes some landmarks in biological evolution and Earth’s atmospheric chemistry as Earth transitioned from an anoxic to a highly oxic planet.
Microscopic examination of ancient rocks has revealed timelines for the origin of life and major changes in Earth’s geochemistry. But what about modern organisms—how can we unravel their evolutionary relationships? We consider next how this is possible.
Check Your Understanding
Why is the origin of cyanobacteria considered a critical step in evolution?
What caused the formation of banded iron formations?
What lines of evidence indicate that microbial life was present on Earth 3.5 billion years ago?
13.3 Living Fossils: DNA Records the History of Life
13.3 Living Fossils: DNA Records the History of Life
13.3 Living Fossils: DNA Records the History of Life
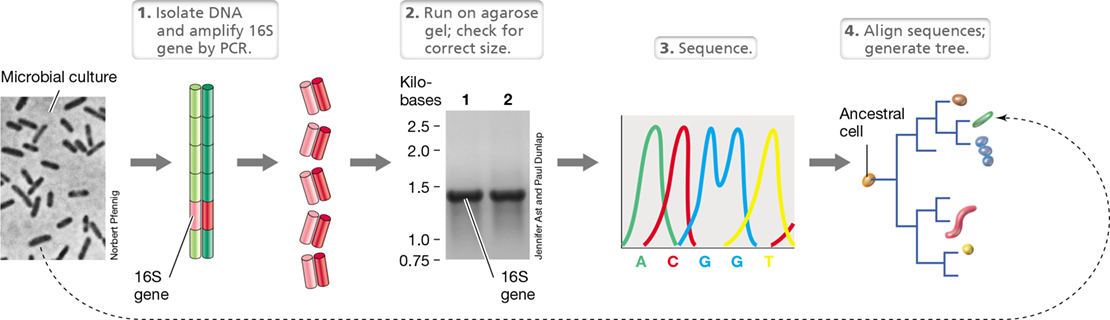
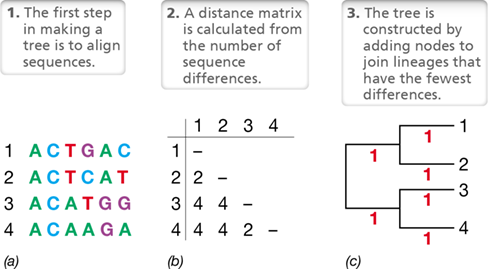
The evolutionary origins of microorganisms remained a mystery until the discovery that certain molecular sequences are a record of evolutionary history (Section 1.15). Mutations occur at random and accumulate over time, causing heritable changes in the sequence of DNA (Chapter 9) that drive the evolutionary process. Hence, organisms that share a recent ancestor have similar DNA sequences, and organisms that are more distantly related have DNA sequences that are more dissimilar. This means we can reconstruct the evolutionary history, or phylogeny, of a set of related DNA sequences by analyzing similarities in their nucleotide sequences (see Section 13.11).
The Universal Tree of Life
Carl Woese was the first to construct a universal tree of life that he inferred from nucleotide sequence similarity in the ribosomal RNA (rRNA) genes of diverse organisms (Section 1.15). The universal tree of life (Figure 13.9) is a genealogy of life on Earth. It depicts the evolutionary history of all cells and clearly reveals the three-domain concept in which all cells can be classified as Bacteria, Archaea, or Eukarya. The root of the universal tree represents a point in time when the common ancestor of all life on Earth today, the last universal common ancestor, LUCA (Figures 13.4 and 13.9), was alive. The universal tree of life shows that the first living things were microorganisms, and that microbes were the dominant life form for most of the history of life on Earth.
Figure 13.9 Schematic universal phylogenetic tree as supported by comparative analysis of small subunit (SSU) rRNA gene sequences.

Only a few key organisms or lineages are shown in each domain. The branch lengths in this tree are arbitrary, and nodes have been collapsed to reflect phylogenetic uncertainty. At least 84 phyla of Bacteria have now been identified, although many of these have not yet been cultured. LUCA, last universal common ancestor (Figure 13.4). The procedures used to generate phylogenetic trees are discussed in Section 13.11.
Genomics (Chapter 10) supports the three-domain concept through phylogenetic analysis of most genes central to cellular function. For example, at least 60 genes (including rRNA genes) are shared by nearly all modern cells, and these genes were likely present in the universal ancestor. Most of these genes encode core functions in transcription, translation, and DNA replication. Across these conserved genes, those of Eukarya show greater sequence similarity to those of Archaea than to those of Bacteria. These and other data support the conclusion that the Bacteria and Archaea diverged prior to the origin of eukaryotic cells (Figure 13.9).
Horizontal Transfer and the Tree of Life
The manner in which the three domains were established remains a topic of debate. It is clear that the three domains represent the major evolutionary cell lineages that exist on Earth, and also clear that Bacteria and Archaea diverged prior to the divergence of Eukarya from Archaea. However, there are many examples of genes shared by any two of the three domains. One hypothesis is that early in the history of life, before the primary domains had diverged, horizontal gene transfer (Section 13.9 and Chapter 9) was extensive. During this time, any genes that provided a strong benefit likely were transferred rapidly among early forms of life (Figure 13.4). As the domains continued to diverge over time, barriers to unrestricted horizontal gene transfer evolved in order to maintain genomic stability. As a result, the previously promiscuous cell populations began to diverge into the primary lines of evolutionary descent we recognize today (Figures 13.4 and 13.9). However, the ability of genes to move horizontally between different microbial species complicates our understanding of microbial phylogeny, a concept we will return to later in the chapter.
It seems likely that the domains Bacteria and Archaea had already diverged by about 3.7 billion years ago (Figure 13.1). Following this, there was a further bifurcation, somewhere between 2.7 and 1.5 billion years ago, at which time the Eukarya diverged from Archaea to form a distinct domain. As each domain continued to evolve, certain traits became fixed within each group, giving rise to a multitude of genetic, physiological, and structural differences (Chapters 2, 9, 10, and 14, 15, 16, 17 and 18). After nearly 4 billion years of microbial evolution, we see the grand result: three domains of cellular life that are each evolutionarily distinct and yet share certain features that reveal their common descent from a universal cellular ancestor.
Before we turn our attention to the evolutionary process per se, we focus briefly on the origin of the eukaryotic cell and eventually the domain Eukarya, a phylogenetically distinct group (Figure 13.9) whose cell structure contrasts with the prokaryotic cell structure of the Bacteria and Archaea (Chapter 2). The major properties and phylogentic diversity of microbial eukaryotes are discussed in Chapter 18.
Check Your Understanding
What kinds of evidence support the three-domain concept of life?
What is LUCA and what are some of its characteristics?
13.4 Endosymbiotic Origin of Eukaryotes
The evolution of the eukaryotic cell marked a major milestone in cellular evolution that paved the way for the evolution of complex multicellular organisms. Here we consider the origin of the Eukarya and show how eukaryotes are genetic chimeras containing genes from both Archaea and Bacteria.
The Last Eukaryotic Common Ancestor and Its Genetic Makeup
For around two billion years, about half the time life has existed on Earth, the Earth was anoxic and all living things had prokaryotic cell structure. The origin of the eukaryotic cell and the evolution of multicellularity roughly coincide with the oxygenation of Earth’s atmosphere, and this event may have facilitated eukaryote evolution. While the exact origins of the eukaryotic cell remain unclear, the oldest microfossils that have recognizable nuclei are about 2 billion years old. Multicellular and increasingly complex microfossils of algae are evident from 1.9 to 1.4 billion years ago (Figure 13.7b). By 0.6 billion years ago, with O2 near present-day levels, large multicellular organisms, the Ediacaran fauna, were present in the sea. In a relatively short time geologically speaking, multicellular eukaryotes diversified into the ancestors of modern-day algae, plants, fungi, and animals during what has been coined the “Cambrian explosion” (Figure 13.1).
The exact pathway by which the eukaryotic cell emerged remains a major unresolved question in evolution; however, it is clear that the modern eukaryotic cell is a genetic chimera, containing genes from both Bacteria and Archaea. By analyzing the genomes of living organisms across the tree of life (Figure 13.9), we can identify those genes that were most likely present in the last eukaryotic common ancestor (LECA), the ultimate ancestor of all eukaryotic organisms.
A total of about 4000 genes trace back to LECA, and this organism had all the characteristic features of eukaryotic cells. The latter included a nucleus, mitochondria that respired O2, organelles, a complex cytoskeleton, spliceosomal machinery and genes with introns (Chapter 6), and other eukaryotic characteristics (Sections 2.13, 2.14 and 2.15). More than 70% of these 4000 genes exist only in Eukarya, which clearly reveals the Eukarya to be a unique domain of life unambiguously differentiated from the Bacteria and Archaea. However, LECA shares its remaining genes with the Bacteria and Archaea. Of these genes, about two-thirds of them derive from Bacteria, whereas the remaining third are from Archaea. The genes of bacterial origin encode mostly metabolic functions, while the genes of archaeal origin encode the information-processing machinery of the cell. For example, eukaryotic cells share with Archaea molecular features of transcription and translation (Chapter 6), while features eukaryotes share with Bacteria include their ester-linked membrane lipids (Section 2.1) and glycolytic pathway (Section 3.6).
Endosymbiotic Theory
A well-supported explanation for the origin of organelles in the eukaryotic cell is the endosymbiotic theory, in which at least two scenarios have been proposed (Figure 13.10). This theory states that the mitochondrion of modern-day eukaryotes arose from the stable incorporation of a bacterium capable of aerobic respiration into the cytoplasm of an early eukaryotic cell. Endosymbiotic mitochondria would have been beneficial to these early eukaryotic cells because they increased the cell’s respiratory capacity, and thus these early mitochondria-containing cells proceeded to become the ancestors of all living Eukarya. Virtually all eukaryotic cells have mitochondria (Section 2.14), though these structures were subsequently lost in certain lineages of anaerobic microbial eukaryotes (Chapter 18).
Figure 13.10 Endosymbiotic models for the origin of the eukaryotic cell.

(a) The serial endosymbiosis hypothesis proposes that the eukaryotic ancestor diverged from the archaeal line and possessed a nucleus and other features of eukaryotic cells prior to endosymbiosis with the bacterial ancestor of the mitochondrion. A later endosymbiosis with the cyanobacterial ancestor of the chloroplast gave rise to the eukaryotic ancestor of all plants and all other photosynthetic eukaryotes. (b) The symbiogenesis hypothesis proposes that the eukaryotic cell evolved from a symbiotic relationship between Bacteria and Archaea. The bacterial partner was eventually engulfed by its archaeal partner and evolved over time into the mitochondrion. The nucleus and other features of the eukaryotic cell evolved after establishment of endosymbiosis. A later endosymbiosis with the cyanobacterial ancestor of the chloroplast gave rise to the eukaryotic ancestor of all plants and all other photosynthetic eukaryotes. Note the position of the mitochondrion and plastids (chloroplasts are a type of plastid) on the universal phylogenetic tree (Figure 13.9).
A second endosymbiotic event, in an important lineage of the Eukarya, also had a major impact on the evolution of life. Chloroplasts arose, after the acquisition of mitochondria, from the stable incorporation of a cyanobacterium-like cell into the cytoplasm of a eukaryotic lineage, and this endosymbiotic event triggered the origin of photosynthesis within Eukarya (Figure 13.10). All phototrophic eukaryotes, including plants and algae, have descended from the lineage of cells that acquired endosymbiotic chloroplasts. Atmospheric oxygen is intimately associated with the endosymbiotic origins of organelles, being consumed by the ancestor of the mitochondrion and being produced by the ancestor of the chloroplast. These endosymbiotic events diversified the metabolism of early eukaryotic cells, with mitochondria providing aerobic respiration and chloroplasts providing the ability to exploit sunlight for energy. Moreover, the endosymbiotic origin of organelles set the stage for the diversification of Eukarya into the forms we know today.
Several lines of evidence support the endosymbiotic theory. The earliest evidence that suggested an endosymbiotic origin of organelles was that chloroplasts and mitochondria are about the same size as bacteria and they replicate independently within the cytoplasm of the eukaryotic cell. However, the endosymbiotic theory rests largely on evidence derived from molecular biology. First, mitochondria and chloroplasts contain their own genomes composed of bacterial genes. These genomes are circular like those of Bacteria and encode proteins of the respiratory chain (mitochondrion) and photosynthetic apparatus (chloroplast) as well as ribosomal RNAs and transfer RNAs. Second, both mitochondria and chloroplasts contain bacterial ribosomes. The 16S rRNA gene sequences of mitochondria and chloroplasts are also characteristic of those from Bacteria, and sequence analyses place the ancestor of mitochondria in the phylum Proteobacteria, and the ancestor of chloroplasts in the phylum Cyanobacteria (Figure 13.9). (Phyla are the major lineages of organisms within the Bacteria, Archaea, and Eukarya; see Section 13.12 and Table 13.1.) Third, the same antibiotics that inhibit ribosome function in free-living Bacteria inhibit ribosome function in these organelles. Fourth, the eukaryotic nucleus contains genes derived from Bacteria. Genomic sequences of eukaryotic cells have revealed genes that encode functions specific to mitochondria and chloroplasts. Moreover, because these gene sequences more closely resemble those of Bacteria than those of Archaea or Eukarya, it is concluded that these genes were translocated to the nucleus from ancestors of mitochondria and chloroplasts during the initial stages of endosymbiosis.
Formation of the Eukaryotic Cell
It is generally agreed that the acquisition of mitochondria was the critical event that led to the origin of the eukaryotic cell and establishment of the domain Eukarya. It is also thought that most of the bacterial genes found in eukaryotic genomes were transferred from the genome of the mitochondrial ancestor. However, the phylogenetic nature of the cell that acquired the mitochondrion remains a matter of debate; but even here, molecular evidence is paving the way to an answer.
The fact that the genes encoding DNA replication, transcription, and translation in Eukarya are derived from Archaea is generally used to support the hypothesis that the Archaea are ancestral to Eukarya. The recent discovery of Lokiarchaeota—a new phylum of Archaea—has provided fresh insights into the origin of Eukarya. Lokiarchaeota were discovered through metagenomic analyses (Section 10.7) of microbial communities that inhabit deep marine sediments near a hydrothermal vent system known as Loki’s Castle (Figure 13.11), located along the Mid-Atlantic Ridge between Greenland and Norway. The genomes of Lokiarchaeota contain a number of eukaryotic features that suggest the presence of a eukaryotic-like cytoskeleton (Section 2.15) and the ability to synthesize intracellular membranes, traits that may have facilitated stable integration of an endosymbiont. Because of these strong genetic links to eukaryotes, it is thought that Lokiarchaeota may be close ancestors of the cell line that ultimately gave rise to the Eukarya.
Figure 13.11 Habitat of *Lokiarchaeota* and the origin of the *Eukarya*.
Lokiarchaeota were discovered in a metagenomic survey of marine sediments near a hydrothermal vent system known as Loki’s Castle (shown in photo). These Archaea share certain key similarities with Eukarya and might be the closest known relatives of the last eukaryotic common ancestor.
The exact series of events that led to the eukaryotic cell is unknown, but two major pathways have been proposed (Figure 13.10). In the serial endosymbiosis hypothesis, eukaryotes arose from a complex nucleus-bearing cell line that diverged from the Archaea and later acquired mitochondria and chloroplasts by endosymbiosis (Figure 13.10a). Endosymbiosis is posited to have occurred when this cell line engulfed or was invaded by a bacterial cell that, rather than being destroyed, managed to survive and replicate as an endosymbiont within the cytoplasm of the host cell. According to this hypothesis, eukaryotic genes that resemble those of Bacteria were acquired in gene transfers from the endosymbiont to the nuclear genome.
A second hypothesis for the formation of the eukaryotic cell is called symbiogenesis and proposes that the eukaryotic cell arose from a symbiotic relationship between cells of Bacteria and Archaea that ultimately resulted in engulfment of the bacterial partner to form mitochondria (Figure 13.10b). One version of symbiogenesis proposes that the eukaryotic cell arose from an association between a H2-producing species of Bacteria and a H2-consuming species of Archaea. This “hydrogen hypothesis” proposes that the bacterial cell was a facultative aerobe and a chemoorganotroph that could grow by aerobic respiration or by syntrophic production of H2 (Section 14.22). The archaeal partner was an anaerobic chemolithotroph that required H2 for growth. These symbiotic partners could have coevolved for an extended period prior to bacterial engulfment and formal development of the mitochondria.
The origin of the nucleus was critical to the evolution of the eukaryotic cell, yet it remains unclear whether the nucleus appeared before or after the endosymbiotic origin of mitochondria. One hypothesis for the origin of the nucleus is that its formation is associated with the evolution of RNA processing in Eukarya. In contrast to the genes of Bacteria and Archaea, eukaryotic genes frequently contain introns (Section 6.6). For proper gene expression to occur in Eukarya, RNA splicing must remove introns prior to translation. Eukaryotic cells have a molecular complex called the spliceosome that performs RNA splicing and operates in the cell nucleus. Hence, the nuclear membrane may have evolved in Eukarya as a mechanism to separate spliceosomes (enzymes that can cut RNA) in the nucleus from ribosomes (major sources of RNA) in the cytoplasm.
Check Your Understanding
What evidence supports the idea that the mitochondrion and chloroplast were once free-living members of the domain Bacteria?
In what ways are modern eukaryotes a combination of attributes of Bacteria and Archaea?
Describe the different hypotheses for the formation of the eukaryotic cell.
13.5 Viral Evolution
The tree of life does not contain viruses, and for good reason: Even though viruses show some of the properties of cells, viruses are not cells (Chapters 5 and 11). Among other cellular deficiencies, viruses lack ribosomes (and hence, ribosomal RNA), so it has been impossible to place viruses on the universal tree of life (Section 13.3 and Figure 13.9). Nevertheless, the question of when viruses first appeared on Earth and what their relationship is to cells remains an intriguing question and one to which several hypotheses have been proposed.
All known viruses require a host cell for their replication, and this leads to the natural conclusion that viruses evolved sometime after cells appeared on Earth. However, viruses have very few genes, they evolve rapidly, and their genes are only shared among closely related viruses. Hence there are no universally conserved viral genes. As a result, it is impossible to generate a tree of life that contains viruses, or even a universal phylogenetic tree of viruses (a phylogenetic tree of a group of viruses that share a common gene is possible, however, Figure 11.5b). Indeed, it is possible that viruses have multiple origins and that different types of viruses have evolved independently over the history of life.
Viruses as Selfish DNA
Mobile DNA elements such as plasmids and transposons are well known (see Section 13.9 and Chapter 9). These elements have been called “selfish DNA” because they replicate independently of the host chromosome, contain genes that enable their transfer between cells, have discrete host ranges, and make no contribution to an organism’s fitness (and may even be harmful). Ultimately, viruses are not much different from these selfish DNA elements. For example, many viruses reside indefinitely in the host’s cytoplasm or integrated into its genome and do not kill the host cell immediately or at all. The main differences between viruses and mobile DNA are that viruses typically have higher rates of replication, often resulting in the death of the host cell, and viral DNA is encased in a protein capsid that facilitates transmission between cells. There exists a diversity of viral capsid proteins, however, and so it is likely that different viral families have different evolutionary origins.
One clue as to viral origins comes from the discovery of gene transfer agents. Gene transfer agents are bacteriophage-like particles that transfer genes between cells (Section 9.7). Gene transfer agents facilitate a type of horizontal gene transfer and are encoded by genes present on the chromosome. These genes produce a capsid within which is packaged a piece of chromosomal DNA. This capsid allows for gene transfer between cells, a trait that can be beneficial for cell populations. Gene transfer agents share ancestry with viruses, but the nature of this relationship remains disputed. One possibility is that viruses appeared first and gene transfer agents evolved from defective bacteriophages. But it is also possible that gene transfer agents appeared first and viruses evolved when some of these agents gained the ability to package the genes necessary for their own production and escaped from cellular control (**Figure 13.12*a***). It is clear that different gene transfer agents evolved independently in various lineages of bacteria and that these agents are very ancient. Hence, viruses may have evolved on multiple occasions as rogue mobile genetic elements that escaped from host control by exploiting preexisting genetic mechanisms for DNA replication and transmission (see MicrobiologyNow on page 392).
Figure 13.12 Hypothetical schemes for viral origins.

(a) Gene transfer agents are mobile genetic elements that facilitate horizontal gene transfer by packaging host DNA and exporting it in a capsid. Random mutations in the DNA encoding gene transfer agents could have resulted in their escape from host control, leading to replication and transmission of infective agents that evolved into viruses. (b) Prior to the evolution of modern cells, during the RNA world (Figure 13.4), RNA-based life forms might have been highly interdependent. Following the origin of DNA, these RNA-based organisms could have evolved further dependency to the point that their replication could only take place within DNA-based cells, thereby resulting in the evolution of viruses.
Viruses as a Vestige of Pre-Cellular Life
Structural features shared among viral proteins have been suggested as a method to probe viral evolution. While no genes or proteins are shared among all viruses, certain structural domains, those parts of proteins that have discrete conserved functions (Section 6.7), have been found in a diverse array of viruses. It is possible that the analysis of these conserved structural domains will provide new insights into how viruses first appeared on Earth.
Analyses of conserved structural domains shared among viral proteins suggests an ancient origin of viruses. Such analyses provide evidence that the earliest viruses had RNA genomes and may have existed prior to the origin of cellular life (Figure 13.12b). Some form of self-replicating RNA must have existed in the “RNA world” prior to the origin of cells (Section 13.1). Viruses may have originated from within this RNA world and taken an alternate evolutionary path from that of cellular life. Some of this pre-cellular RNA may have adapted to the appearance of cells by becoming obligate parasites. Under this scenario, viruses are a holdover from the pre-cellular days of life and have been infecting cells ever since the origin of cellular life itself.
Check Your Understanding
Why is it difficult to place viruses on the universal tree of life?
Explain the hypothesis that viruses have evolved many different times from mobile DNA elements.
Explain the hypothesis that viruses first evolved prior to the appearance of cellular forms of life.
II Mechanisms of Microbial Evolution
Evolution modifies a microbe’s genome over long time periods through mutations and genetic recombination. Organisms can also express new properties quickly when they receive genes from other cells. The ability of microbes to grow rapidly allows microbiologists to document evolutionary events in the laboratory.
While many of the basic principles of evolution are conserved across all domains of life, certain aspects of microbial evolution are uncommon in plants and animals. For example, Bacteria and Archaea are generally haploid and asexual, they have several mechanisms for horizontal gene transfer that result in the asymmetrical exchange of genetic material uncoupled from reproduction, and their genomes can be remarkably heterogeneous and highly dynamic. Despite these differences, we can study microbial evolution and explore the evolutionary history of life by directly comparing the genomes of living organisms. In this part of the chapter, we consider the processes that cause the diversification of microbial lineages and how these forces affect the evolution of microbial genomes.
13.6 The Evolutionary Process
In its simplest form, evolution is a change in allele frequencies in a population of organisms over time, resulting in descent with modification. Alleles are alternate versions of a given gene. New alleles and totally new genes arise as a result of mutation and recombination, and changes in allele frequencies can occur through a variety of processes. We see here how these simple mechanisms give rise to the origin and divergence of microbial species.
Origins of Genetic Diversity
As we have seen, mutations are random changes in DNA sequence that accumulate over time, and they are a fundamental source of the natural variation that drives the evolutionary process. Most mutations are neutral or deleterious, though some can be beneficial. Mutations take several forms including substitutions, deletions, insertions, and duplications (Chapter 9). Gene duplication events produce a redundant copy of a gene that can be modified by further mutation without losing the function encoded by the original gene (Section 13.8). Hence, duplications allow for the diversification of gene function.
Recombination is a process by which segments of DNA are broken and rejoined to create new combinations of genetic material (Section 9.5). Recombination can cause reassortment of genetic material already present in a genome and is also required for the integration of DNA acquired through horizontal gene transfer. Recombination can be broadly classified as either homologous or nonhomologous. Homologous recombination requires short segments of highly similar DNA sequence flanking the region of DNA being transferred. By contrast, nonhomologous recombination is mediated by several mechanisms that share in common the fact that they do not require high levels of sequence similarity to initiate successful DNA integration.
Selection and Genetic Drift
New alleles and new genes result when mutation and recombination cause variation in gene sequences. Evolution occurs when different alleles change in frequency in a population over a span of many generations. Evolutionary biologists have described many different mechanisms that can govern this evolutionary process, but chief among them are the forces of selection and genetic drift.
Evolutionary selection is defined on the basis of fitness, the ability of an organism to produce progeny and contribute to the genetic makeup of future generations. Most mutations are neutral with respect to fitness and they have no effect on the cell, as a result of the degeneracy of the genetic code (Section 6.9). These mutations generally accumulate in DNA over time. Some mutations are deleterious, and these decrease the fitness of an organism by disrupting gene function. Deleterious mutations are generally purged from populations over time by natural selection. Some mutations can be beneficial, increasing the fitness of an organism, and these mutations are favored by natural selection, increasing in frequency in a population over time. An example of a beneficial mutation would be a mutation that induces antibiotic resistance in a pathogenic bacterium infecting a person undergoing antibiotic therapy. It is important to remember that all mutations occur by chance; the selective nature of the environment does not cause adaptive mutations but simply selects for the growth and reproduction of those organisms that have incurred mutations that provide a fitness advantage.
While Darwin proposed natural selection as the mechanism by which gene frequencies change over time, evolutionary change can occur through mechanisms other than selection. A chief example is genetic drift (Figure 13.13), a random process that can cause gene frequencies to change over time, resulting in evolution in the absence of natural selection. Genetic drift occurs because some members of a population will have more offspring than others simply as a result of chance. Over time, these chance events can result in evolutionary change in the absence of selection. Genetic drift is most powerful in small populations and in populations that experience frequent “bottleneck” events. The latter occur when a population experiences a severe reduction in population size followed by regrowth from the cells that remain. For example, genetic drift can be very important in the evolution of pathogens because each new infection is caused by a small number of cells colonizing a new host. Hence, pathogen populations can change rapidly as a result of random genetic drift (Figure 13.13).
Figure 13.13 Genetic drift.

Genetic drift is a random process that can cause gene frequencies in a population to change over time, causing evolution without natural selection. In this example, a population containing four different bacterial genotypes (indicated by different colors), each at equal frequency, is present in the ancestor tube. Four cells at random are then transferred to each of three new tubes and the cells allowed to grow to fill each tube. There is no difference in fitness between the cells and so they grow equally. Cells taken at random are then transferred in two successive rounds. Striking differences in genotype frequencies between the populations are observed after only three rounds of transfers.
Origins of Microbial Species
Microbial species can be very old and can encompass a wide variety of individual strains having diverse traits (we consider the microbial species concept in Section 13.12). The accumulation of mutations over time can be used to explore the evolutionary origins of species. Sequence changes due to mutation can be used as a molecular clock to estimate the time since a species has diverged from its closest relatives. Major assumptions of the molecular clock approach are that nucleotide changes accumulate in a sequence in proportion to time, that such changes are generally neutral and do not interfere with gene function, and that they are random. Molecular clock estimates are most reliable when they can be calibrated with evidence from the geological record. The most well documented of these have used obligate bacterial symbionts of insects (Section 23.7) for which the insect host provides a fossil record to accurately date evolutionary events.
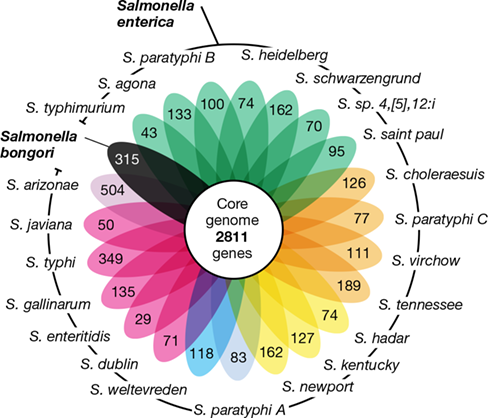
Molecular clock estimates make it possible to estimate the divergence of microbial species based on dissimilarity in their gene sequences. For example, molecular clock estimates indicate that the closely related species Escherichia coli and Salmonella enterica (typhimurium), which have 2.8% dissimilarity in their 16S rRNA gene sequences, last shared a common ancestor some 100–140 million years ago. Likewise, it is estimated that two well-characterized strains of E. coli, the harmless strain K-12 and the foodborne pathogenic strain O157:H7 (Section 33.11), diverged about 4.5 million years ago. Many microbial species, like E. coli, can encompass an enormous number of different strains possessing a diversity of traits that collectively define the species. Molecular clock estimates reveal that microbial speciation takes a very long time. However, we will see in the next section that while speciation can take a long time, strains of a species can acquire new traits rapidly.
Check Your Understanding
What are the different processes that give rise to genetic variation?
What is the difference between selection and genetic drift, and how do they promote evolutionary change?
13.7 Experimental Evolution
Experimental evolution is a field that employs laboratory experiments with microorganisms to investigate evolutionary processes. Such evolution experiments are enabled by the rapid growth of bacteria and the ability to preserve them indefinitely by freezing. The latter makes it possible to maintain a living “fossil record” of ancestral organisms that can be thawed later and grown to compare with evolved strains. Microorganisms typically form large populations and can reproduce quickly, producing a new generation in as few as 20 minutes for some species, and thus evolutionary events in microbial populations can often be observed in the laboratory on relatively short time scales. Here we consider two examples of rapid evolutionary change in bacteria, one involving the rapid loss of a characteristic trait in Rhodobacter, and one involving the acquisition of a new trait in Escherichia coli.
Loss of Function
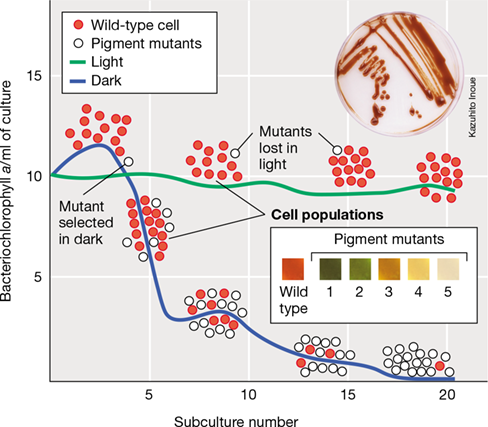
Rhodobacter is a phototrophic purple bacterium that carries out anoxygenic photosynthesis (Section 14.5) in illuminated anoxic environments in nature. When cultured anaerobically, the cells synthesize photopigments in both the presence and the absence of light because pigment synthesis is regulated by O2 availability and not by light. In the light these pigments participate in photosynthetic reactions that lead to ATP synthesis, but in darkness, the pigments provide no benefit to the cell. Random mutations, by chance, occasionally generate Rhodobacter cells that produce reduced levels of photopigments. In nature, the ability to carry out photosynthesis provides a fitness benefit and wild-type cells outcompete such pigment-deficient mutants. However, when cultured in constant darkness, wild-type cells have no such advantage. In darkness, mutants that produce reduced levels of photopigments gain the advantage because they do not waste energy and resources synthesizing photopigments that provide them no benefit. As a result, photopigment mutants can grow at a faster rate and quickly take over Rhodobacter cultures grown in darkness because under these conditions, the mutants have greater fitness than wild-type cells (Figure 13.14). Mutations affecting photosynthesis occur at the same rate in the light as in the dark, but in the light the selection for the wild-type phototrophic phenotype is so strong that photopigment mutants are quickly lost from the population.
Figure 13.14 Survival of the fittest and natural selection in a population of phototrophic purple bacteria.

Serial subculture of the purple bacterium Rhodobacter capsulatus in the light (green line) provides a benefit to wild-type phototrophic strains and they outcompete nonphototrophic mutants. In the dark (blue line), however, phototrophy provides no benefits and nonphototrophic mutants quickly outcompete wild-type cells still making bacteriochlorophyll and carotenoids. Photos: top, plate culture showing colonies of phototrophic cells of R. capsulatus; bottom, close-up photos showing the color of colonies of wild-type cells and five pigment mutants (1–5) obtained during serial dark subculture. Wild-type cells are reddish-brown from their assortment of carotenoid pigments. The color of mutant colonies reflects the absence (or reduced synthesis) of one or more carotenoids. Mutant strain 5 lacked bacteriochlorophyll and was no longer able to grow phototrophically. Mutant strains 1–4 could grow phototrophically but at reduced growth rates from the wild type. Data adapted from Madigan, M.T., et al. 1982. J. Bacteriol. 150: 1422.
Gain of Function
The E. coli long-term evolution experiment (LTEE), which has been running since 1988, has tracked the evolution of 12 parallel lines of E. coli through more than 60,000 generations. The E. coli LTEE cultures have been grown aerobically on a minimal medium with glucose as a sole source of carbon and energy. E. coli is typically propagated in a rich medium that contains an excess of nutrients, and so the minimal glucose medium used in the LTEE represents a new adaptive environment in which E. coli can evolve over time.
In the LTEE, both the ancestor and the evolved lines were genetically engineered to contain a neutral marker that made their colonies either red or white. The marker made it possible to measure the fitness of evolved strains relative to the ancestor by putting them into competition with one another (**Figure 13.15*a***). Genome sequencing during the experiment revealed that mutations accumulated randomly over time in the evolved lines. However, the relative fitness of the evolved lines on minimal glucose medium increased dramatically over the first 500 generations as a result of selection acting on mutations beneficial in this new environment (Figure 13.15b). The fitness of the evolved lines continued to increase, albeit at a reduced rate, as a result of further selection over the course of the experiment. Most remarkably, after 31,500 generations, one of the evolved lines obtained the ability to use citrate as an energy source (Figure 13.15c). Citrate was present as a pH buffer in the growth medium and was not considered a potential carbon source for E. coli, because the inability to grow aerobically on citrate is a diagnostic trait used for identifying E. coli. However, the random accumulation of mutations in this one evolved line modified preexisting genes in such a way as to allow for the evolution of a new adaptive trait. The diverged strains can now exploit a new resource that was unavailable to the ancestral population. Since they can now catabolize both citrate and glucose, these cells grow to higher cell density than the ancestor (Figure 13.15c). The fact that only one of the 12 parallel lines evolved the ability to grow on citrate demonstrates the chance nature of evolution.
Figure 13.15 Long-term evolution of *Escherichia coli*.

(a) In the E. coli long-term evolution experiment (LTEE), ancestral and derived lines differ in a mutation that affects their ability to use arabinose, allowing them to be differentiated by their colony color when grown on tetrazolium arabinose agar. (b) Competition experiments between evolved and ancestral strains show that relative fitness in minimal glucose media increases dramatically for evolved lines. (c) The ability to use citrate aerobically evolved in one of 12 LTEE lines. Cells growing on minimal and citrate allowed the mutant cell line to reach significantly higher cell densities. Relative fitness is a measure of the growth rate of the evolved strain to that of the ancestral strain. Note that the mutation rate in the E. coli cultures is constant and that mutations accumulate randomly over time.
The transitions shown in these experiments remind us of how quickly evolutionary pressures can shift even major properties (such as metabolic strategies) of a microbial cell population. In the case of Rhodobacter (Figure 13.14) a mutation that is deleterious in the wild provides a selective advantage when the organism is grown in the laboratory in a continuously dark environment. Under this new condition, evolution causes Rhodobacter to lose unneeded metabolic machinery. In the case of E. coli (Figure 13.15), the accumulation of random mutations set the stage for genetic diversity in a population. That is, natural variation caused by spontaneous mutation generated a new trait—the ability to use citrate—and since the environment in which the cells were grown happened to contain citrate, this mutation provided a selective advantage to those cells. In the absence of citrate, these mutations would still occur at the same rate. However, in the absence of a selective benefit, cells able to use citrate would likely disappear from the population over time.
Check Your Understanding
In the experiment of Figure 13.14, why did the cell population in the dark lose its pigments?
Why did only one of twelve Escherichia coli populations in the LTEE eventually evolve the ability to grow on citrate? What does this tell us about the process of evolution?
In the LTEE, if the rate of mutation in E. coli remains constant, then why does the fitness of E. coli improve rapidly upon initial addition to a new environment and then more slowly in later generations (as in Figure 13.15)?
13.8 Gene Families, Duplications, and Deletions
The duplication and deletion of genetic information within the genome plays a major role in microbial evolution. Genomes vary dramatically in size (Chapter 10), and duplication and deletion mutations are a major force that governs the size of microbial genomes. In addition, these mutations govern the gene content of the genome, allowing for removal of nonessential genes and the expansion of gene function. In order to examine the impact of deletions and duplications on the evolution of microbial genomes, we must first classify genes based on their shared ancestry so that we can compare the presence and absence of genes across genomes.
Duplications and Gene Families
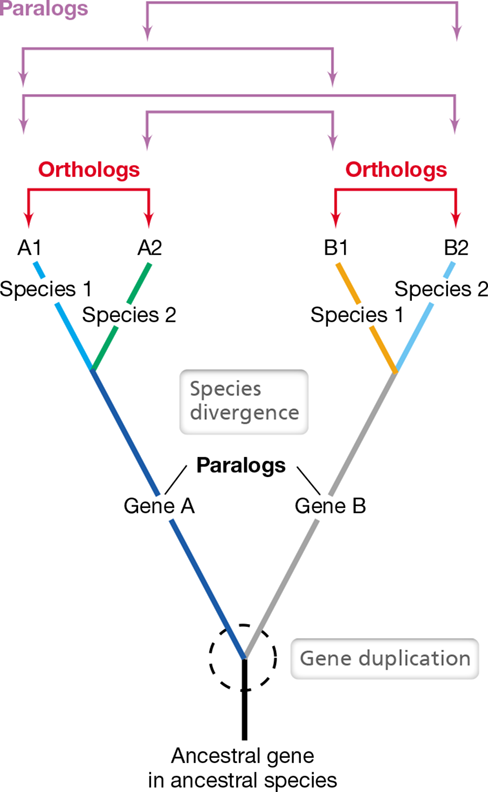
A set of genes that have all descended from a single ancestral gene present in a shared ancestor are homologs (Figure 13.16). Homologous genes tend to have similar nucleotide sequences, and we can infer homology based on nucleotide similarity. Genes that are highly similar in sequence often have the same function, and homologous genes that share the same function are orthologs. However, not all homologous genes have the same function. The accumulation of mutations over time can eventually cause genes to evolve new functions. Genes that are homologous but have different functions are paralogs. Over time a single ancestral gene sequence can diverge to have many different functions in many different organisms. A group of homologous genes is called a gene family, and gene families can contain diverse paralogs that all share a common evolutionary origin.
Figure 13.16 Homologs.
This family tree depicts the evolution of a gene family composed of homologous genes (orthologs and paralogs), which means that all have evolved from a single ancestral gene present in an ancestral species. A gene duplication occurs in the ancestral species, allowing the redundant gene copy, gene B, to evolve a new function within the genome of the ancestral species (see Figure 13.17a). Over time, the ancestral species evolves into two new species whose gene sequences have diverged as a result of evolution. Genes A1 and A2 have the same functions and they have evolved from the same ancestral gene, and so they are orthologs, as are B1 and B2. Genes that have evolved from the same ancestral gene but have different functions are paralogs.
Gene duplication is thought to be a major driving force behind the evolution of gene families and the organisms that contain them (**Figure 13.17*a***). A duplication mutation creates a redundant copy of a gene sequence in the genome. The original gene copy retains its function, making it possible for the second copy to accumulate mutations freely without a loss of gene function in the cell. In this way, evolution can “experiment” with one copy of the gene. Genomic analyses have revealed many examples of protein-encoding genes that were clearly derived from gene duplication. Figure 13.17b shows this for the enzyme RuBisCO, a key enzyme of autotrophic metabolism (Section 3.12): An ancestral RuBisCO gene gave rise to paralogous enzymes with different but related catalytic activities.
Figure 13.17 Evolution by gene duplication.

(a) The principle of gene duplication. After duplication, the “spare” copy of a gene is free to evolve to encode a new function. (b) The large subunit of the enzyme RuBisCO, which fixes CO2 during photosynthesis, is part of a gene family that contains many different genes. Different orthologous forms of RuBisCO (forms I, II, and III), which all retain the same function, are present in different microbial groups. However, RuBisCO is in turn derived from an ancestral gene (black bars) of unknown function that was duplicated to produce paralogous genes encoding an enzyme in methionine metabolism (yellow bar) and several genes whose function is still unknown (purple bars). RLP, RuBisCO-like protein.
Gene Deletions in Microbial Genomes
Gene deletions play an important role in microbial genome dynamics. It is a common misconception that evolution inevitably causes organisms to increase in complexity over time. In reality, evolution is both a give and a take proposition. Fitness changes are completely dependent on the environment, and fitness in some environments can actually be improved by deletion of genetic material.
It is common in prokaryotic cells for nonessential and nonfunctional materials to be deleted over evolutionary time. Deletions occur with far greater frequency than insertions or duplications in microbial genomes. This bias toward deletions is the force that maintains the small size of many microbial genomes and the reason why the genomes of Bacteria and Archaea are tightly packed with genes and contain relatively few noncoding sequences (Section 10.3). Selection is the main force that counters the effect of deletions, preserving those genes that provide a fitness benefit to the cell.
Deletions are also thought to be a driving force for the tiny genomes observed in many obligate intracellular symbionts and intracellular pathogens (Section 10.3 and Section 23.7). The genomes of these bacteria are highly streamlined because many metabolites are available in the cytoplasm of their host organisms. Hence deletion mutations that remove biosynthetic genes might have little effect on the fitness of an intracellular pathogen if those biosynthetic functions are provided by the host. Over time, deletion mutations eliminate redundant functions. Insect endosymbionts have carried this theme to an extreme and show the smallest of all cellular genomes (Section 10.3, Table 10.1, and Figure 10.8). The same process is ultimately responsible for the minute genomes found in mitochondria and chloroplasts (Section 10.4).
Deletion Mutations and the Evolution of Interdependence in Microbial Communities
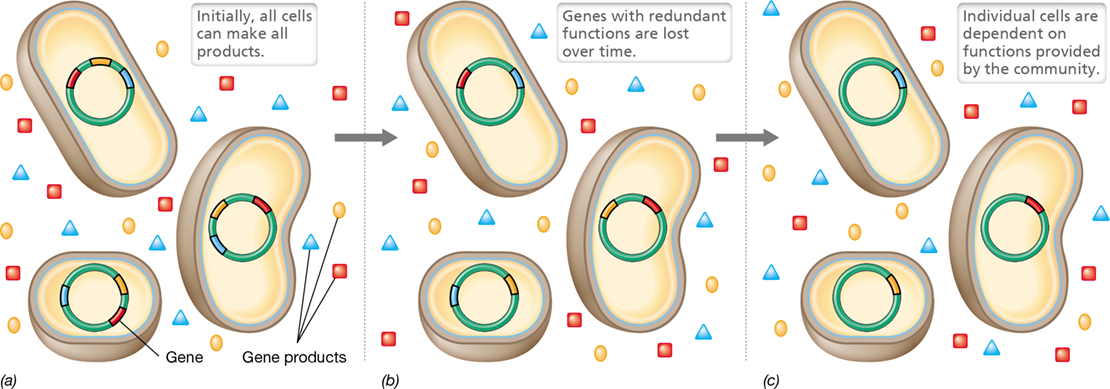
A deletion mutation that prevents an organism from producing an essential nutrient such as a vitamin when growing in pure culture would be a lethal event. But in nature, microorganisms grow in microbial communities, and in such a setting, a deletion mutation that prevents an organism from producing an essential nutrient may not be lethal if the organism can obtain the nutrient from some other organism in the environment (Figure 13.18).
Figure 13.18 The evolution of interdependence in microbial communities.

(a) Three species in a community each have three different genes that make extracellular products that benefit the whole community (a gene and its product are shown in the same color). (b) Over time, random mutations cause functions to be lost from the genomes. (c) As long as some members of the community continue to make each product, there will be no fitness cost when a single species loses a single gene. Over time, the three species become mutually dependent.
All genes impose some cost on the cell, and organisms living in a microbial community can delete a gene to avoid this cost if other community members provide the gene’s function. Such deletions can increase an organism’s fitness but promote the interdependency of members of the microbial community. Organisms having such “loss-of-function” mutations will remain competitive as long as they remain within the community but may be unable to grow if separated from the community in which they coevolved. This sharing of resources in natural communities explains the observation that many bacteria grown in pure culture require one or more growth factors. For example, regardless of where they are isolated from, all strains of the phototrophic purple bacterium Rhodospirillum rubrum require the vitamin biotin when grown in pure culture, while all strains of Rhodobacter capsulatus (see Figure 13.14) require thiamine. In nature, these two phototrophs could coexist in the same microbial community if each phototroph (or other microbes in the community) secreted a small amount of the vitamin that the other phototroph required. Such gene deletions result in metabolic interdependencies within a microbial community (Figure 13.18), making deletions one of the factors that govern the microbial composition of a microbial community.
Many microbes can grow in pure culture without any growth factor additions. Such organisms preserve all essential functions and bear the costs of maintaining all gene functions; this puts them at a disadvantage relative to mutually dependent competitors when the competition takes place within a complex microbial community. However, cells that maintain their ability to grow independently earn a fitness bonus in that, unlike their mutually dependent competitors, they retain the option of dispersing to new habitats and growing there independently.
Check Your Understanding
In what way can a gene deletion cause an increase in the fitness of an organism?
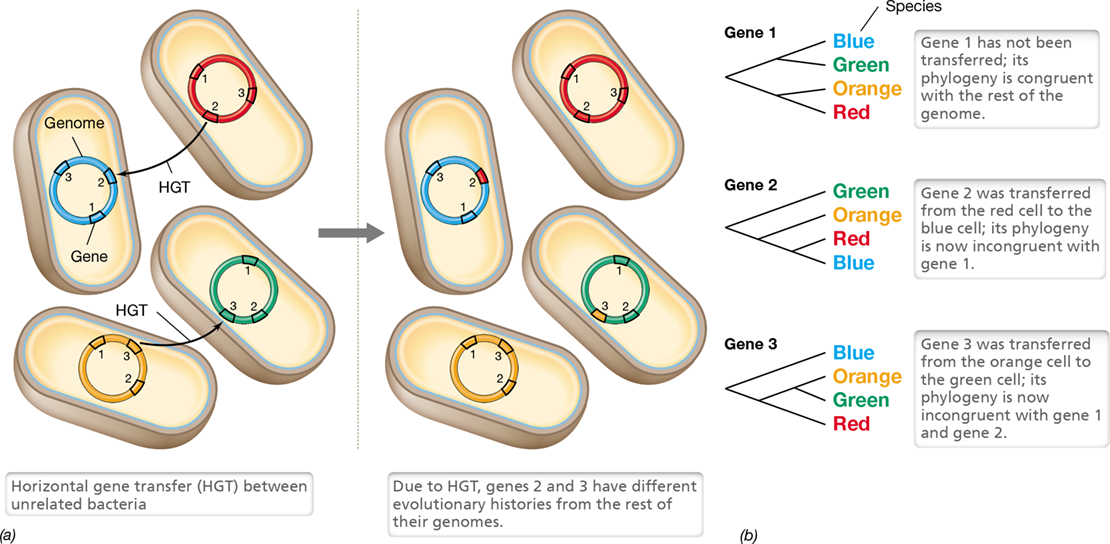
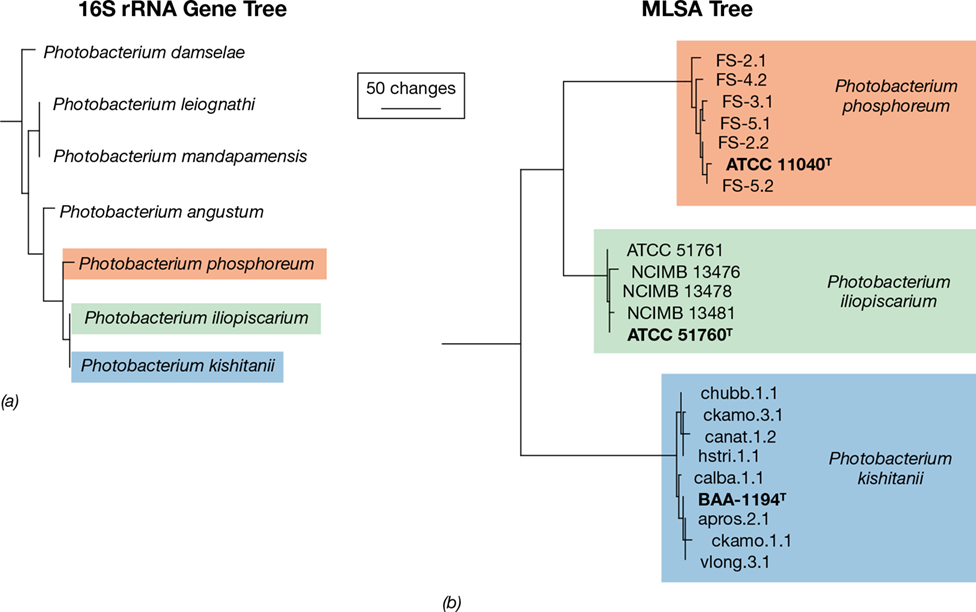
13.9 Horizontal Gene Transfer
Microorganisms can exchange DNA between cells through the process of horizontal gene transfer (Figure 13.19), and this process has a tremendous impact on microbial evolution. We often think of evolution as a vertical process that takes place along the branches of a phylogenetic tree starting with an ancestor at the tree’s root and ending with descendants at its topmost branches. However, horizontal gene transfer allows the transfer of DNA between distant branches of the tree. At least three mechanisms of horizontal gene transfer exist: transformation, transduction, and conjugation (Figure 13.19 and Chapter 9).
Figure 13.19 Vertical versus horizontal gene transfer.

Vertical gene transfer occurs when cells divide. Horizontal gene transfer occurs when a donor cell contributes DNA to a recipient cell. In Bacteria and Archaea, horizontal transfer occurs through one of three mechanisms: transformation, transduction, and conjugation.
Horizontal gene transfer can be thought of as a type of recombination in which DNA from the donor is passed to the recipient and becomes part of the recipient’s genome. Horizontal transfer is unlike sexual recombination because it is (1) unidirectional, with DNA passing from donor to recipient, (2) asymmetrical, in that only a small amount of DNA is usually transferred, and (3) not constrained by species boundaries because the donor and recipient can be distantly related.
Horizontal gene transfer can have a range of consequences depending on the mechanism of recombination used to incorporate the DNA into the genome. The effect of horizontal transfer on the genome is strictly additive when the newly acquired DNA is inserted by nonhomologous recombination. However, when the newly acquired DNA is incorporated by homologous recombination, the result can be gene conversion, in which the recipient’s copy of a gene is replaced by the donor’s copy.
Like most mutations, most genes acquired by horizontal gene transfer can be expected to be neutral or deleterious to the cell. Hence, horizontally acquired genes that do not convey a fitness benefit are deleted from the genome over time. In contrast, those genes that do confer a fitness benefit are likely to persist within the genome. Horizontal gene transfer allows a cell to “explore” a tremendous diversity of genetic information, and newly acquired genes can change over time as a result of further mutation and recombination. As a result, horizontal gene transfer is an effective means for microbes to acquire new functions and to evolve new traits.
The Mobilome
Horizontal gene transfer is facilitated by the mobilome, the sum total of all mobile genetic elements in a genome. Mobile genetic elements are discrete segments of DNA that have genes encoding their own replication, recombination, and transmission. Mobile genetic elements include plasmids, prophages (integrated viral genomes), insertion sequences, transposons, and integrons (Chapter 9). Although the mobilome is not essential to the cell, it can nevertheless play an important role in genome evolution by shuffling genes within and between genomes.
Insertion sequences are simple mobile elements that consist of a transposase gene flanked by inverted repeats (Section 9.11). Transposase is an enzyme that recognizes the inverted repeats and catalyzes nonhomologous recombination, allowing the insertion sequence to jump between different segments of DNA through a “cut and paste” mechanism. If multiple insertion sequences are present in a genome, these elements can generate chromosomal rearrangements such as deletions, inversions, or translocations (Figure 13.20). In addition, insertion sequences shared between a chromosome and a plasmid can facilitate the exchange of genes between these molecules.
Figure 13.20 Mobile elements promote genome evolution.

Mobile genetic elements facilitate the transfer of DNA from one organism to another, thus adding new genes to the recipient genome. The most common mobile genetic elements are plasmids, bacteriophages, and transposons. Mobile genetic elements can also facilitate chromosomal rearrangements. For example, transposase can cause deletions and inversions of host DNA next to sites of transposon insertion.
Transposons are mobile genetic elements that are larger than insertion sequences but similar in function, having both terminal inverted repeats and a transposase gene (Section 9.11). Transposons, however, contain multiple genes that can have a diversity of functions unrelated to transposition. Hence, transposons can move a wide variety of genes between different segments of DNA. Furthermore, some transposons contain genes that cause conjugative transfer, thereby promoting horizontal transfer between cells by conjugation. Because of this tendency, transposable elements are a strong driver of genome evolution.
Integrons also facilitate recombination and DNA exchange. Integrons contain an integrase gene flanked by a promoter and a recombination site that contains direct repeats. Integrons typically contain a gene cassette consisting of one or more genes flanked by a series of corresponding recombination sites. The integron promoter drives the expression of genes within the cassette. The integrase catalyzes site-specific recombination of DNA at the integron recombination site. Hence, DNA sequences flanked by corresponding recombination sites can be inserted or excised from the integron gene cassette. Integrons are often found on other mobile elements such as plasmids or transposons. This mechanism can facilitate the rapid acquisition and rearrangement of new genes.
Detecting Horizontal Gene Flow
The rate of horizontal gene transfer per cell per generation is quite low, but the effect of horizontal gene transfer on genome composition is integrated over the entire evolutionary history of an organism dating back to the last universal common ancestor. As a consequence, the vast majority of genes in a microbial genome have been subject to horizontal gene transfer at some point in their evolutionary history. However, detecting horizontally transferred genes and unraveling their evolutionary history can be a challenge.
A variety of methods are used to identify horizontally transferred genes, but these methods typically take one of two different approaches: statistical analysis of sequence composition, or phylogenetic analysis. Microbial genomes often have distinguishing nucleotide sequence characteristics, much like a fingerprint, that can be used in their identification. These differences in sequence composition can be used to identify segments of DNA acquired from other organisms. One example is to compute the percentage of G+C (guanine–cytosine) base pairs in the genome (genomic GC content, Figure 10.8). In this case, horizontal gene transfer is signaled if the percentage of G+C base pairs in a segment of DNA differs significantly from that of the rest of the genome. In addition, genomes also contain DNA corresponding to each of the 64 possible codons at different frequencies, and these codon-use frequencies (codon bias, Section 10.2) can also be used to identify horizontally acquired genes. A variety of statistical tests can be applied to look for such differences in sequence composition across the genome.
Horizontally acquired genes can also be detected through phylogenetic analysis by making phylogenetic trees for different genes in the genome (Section 13.11). If a gene has a different pattern of ancestry from the rest of the genes in the genome, that is an indication that it was horizontally transferred. Horizontally transferred genes are most easily detected when they are transferred between highly dissimilar organisms and more difficult to detect when transferred between close relatives.
We move on now from the effects of horizontal transfers of individual genes or sets of genes to consider the evolution of the entire genome.
Check Your Understanding
What is one way that you can identify genes acquired by horizontal gene transfer?
List the three major mechanisms that mediate horizontal gene transfer between cells.
How is an integron different from an insertion sequence? Which would be more likely to cause a major genome rearrangement?
13.10 The Evolution of Microbial Genomes
Comparative genomics makes use of whole genome sequencing technologies (Chapter 10) to analyze differences in gene content and DNA sequence composition across entire sets of genomes. Comparative analyses have revealed microbial genomes to be highly diverse and dynamic. A microbial species is composed of many individual strains that differ in gene composition. In the case of microbial genomes, those differences in gene composition can be substantial. Figure 13.21 illustrates the diverse nature of microbial genomes within a single species of the genus Salmonella. All strains of S. enterica—an important human pathogen (Section 33.10)—whose genomes have been sequenced share 2811 genes, but each strain of this species also has a unique set of strain-specific genes. The genomes of many microbial species, like S. enterica, are evolving constantly as diverse strains gain genes by horizontal gene transfer and lose genes as a result of deletions.
Figure 13.21 The *Salmonella enterica* pan genome includes a large number of strain-specific genes.

The species Salmonella enterica contains many different strains that differ in various traits including their pathogenicity. While these strains all share the same 2811 core genes, each individual has some number of strain-specific genes. Salmonella bongori is a species distinct from S. enterica.
Data from Jacobsen, A., R.S. Hendriksen, F.M. Aaresturp, D.W. Ussery, and C. Friis. 2011. The Salmonella enterica pan-genome. Microb. Ecol. 62: 487.
From genomic studies of several strains of a single species, it is now clear that the genome of a microbial species is really composed of two segments: the core genome, consisting of genes shared by all individual strains of a species, and the pan genome, consisting of the core genome plus genes that are not shared by all strains of a species (**Figure 13.22*a***). As we have seen, horizontal gene transfer of mobile genetic elements (Section 13.9) is widespread. The existence of the pan genome shows that microbes are constantly sampling genes from other organisms in the environments in which they live. Consequently, there can be major differences in genome size, gene content, and functional traits (virulence, symbiosis, biodegradation, and the like) between individual strains within a single microbial species.
Figure 13.22 The core and pan genome concept.

Microbial genomes are dynamic and heterogeneous. The first three genomes sequenced from different strains of Escherichia coli were found to have only 39% of their genes in common. The core genome is considered the set of genes that are shared by all members of a species (darkest green in part a), while the pan genome is the totality of all genes found in the different strains of a species (all the genes in genomes 1–3 in part a). The size of the core and pan genome can vary between species. In E. coli the core genome is composed of approximately 1976 genes (b). The size of the pan genome in E. coli is not fixed, as each different strain has a unique complement of genes acquired from horizontal gene transfers. Data adapted from Touchon, M., et al. 2009. PLoS Genetics 5 (1): e1000344.
The Dynamic Nature of the *Escherichia coli* Genome
The dynamic nature of microbial genomes was revealed in dramatic fashion when the first genomes were sequenced from multiple strains of a single species. For example, genome sequencing of Escherichia coli strain K-12 and two pathogenic strains showed that only 39% of their genes were shared among all three genomes (Figure 13.22). The three genomes varied in size by more than a million base pairs and each contained a unique and diverse complement of genes acquired through horizontal gene transfer.
E. coli genomes have on average 4721 genes, with individual strains having as few as 4068 or as many as 5379 total genes. The core genome consists of only 1976 genes present in all strains, accounting for less than half the genes present in the average E. coli genome. The size of the core genome can be expected to decrease as the evolutionary distance of strains increases.
The number of unique genes observed in E. coli, and the size of its pan genome, continues to increase indefinitely as each new strain of E. coli has its genome sequenced. For example, a survey of genomes from 20 E. coli strains revealed a total of 17,838 unique genes (Figure 13.22b). Subtracting the contribution from the core genome, this indicates that as many as 15,862 genes are not shared by all strains. A great many of these genes have clearly been obtained from horizontal gene transfers rather than through vertical patterns of inheritance. Patterns of gene exchange appear to be governed by phylogenetic distance, with rates of gene exchange declining as the phylogenetic distance increases between genomes. In the core genome of E. coli, for example, it has been found that most horizontal gene transfer takes place between close relatives and occurs by homologous recombination of DNA segments of 50 to 500 base pairs in length.
Chromosomal Islands
Comparative genomic analyses have revealed that in addition to a gene or two, entire genetic pathways can be acquired as a result of horizontal gene transfer. These so-called chromosomal islands (also called genomic islands) contain clusters of genes for specialized functions (Figure 13.23). Several lines of evidence indicate that chromosomal islands derive from horizontal gene transfer. First, chromosomal islands are often flanked by inverted repeats, implying that the whole region was inserted into the chromosome by transposition (Section 13.9). Second, the base composition and codon bias in chromosomal islands often differ significantly from those of the genome proper. Third, chromosomal islands often belong to the pan genome, being found in some but not all strains of a species. In addition, some chromosomal islands carry an integrase gene, suggesting similarity with mobile elements such as conjugative transposons (Section 13.9). Finally, in a few cases, the transfer of chromosomal islands has even been demonstrated experimentally in the laboratory.
Figure 13.23 Chromosomal islands and pathogenicity islands in *Escherichia coli.*

Genome comparisons of nonpathogenic E. coli strain K-12 and pathogenic E. coli strains 536 and 073 (both of which cause urinary tract infections) reveal that many differences in gene content are associated with chromosomal islands (CI) and pathogenicity islands (PAI). The inner ring represents genome position (in base pairs). The middle ring shows the frequency with which G+C base pairs occur across the genome, and anomalous regions are colored red. The outer ring compares gene content between genomes: green, genes common to all strains; red, genes present only in pathogenic strains; blue, genes found only in strain 536; orange, genes of strain 536 present in a different location in strain 073. Note the correlation between chromosomal islands and skewed GC content. Some very small inserts are deleted for clarity. Prophage, DNA from a temperate bacteriophage.
Data adapted from Brzuszkiewicz, E., et al. 2006. Proc. Natl. Acad. Sci. (USA) 103: 12879.
Chromosomal islands that encode virulence factors, molecules that facilitate disease and are found in pathogenic bacteria (Chapter 25), are called pathogenicity islands. The pathogenicity islands of uropathogenic strains of E. coli have been particularly well studied (Figure 13.23). Only a few strains of E. coli cause bladder and kidney infections, but those that do—the uropathogenic strains—contain pathogenicity islands that encode a variety of virulence factors including adhesins that facilitate binding to host tissues and a capsule that helps cells evade immune surveillance. Pathogenicity islands are also present in certain pathogenic strains of the gram-positive bacterium Staphylococcus aureus, which can cause serious skin infections (Section 31.9). These pathogenicity islands can be moved between cells by transduction, a major mechanism of horizontal gene transfer (Section 9.7).
Chromosomal islands in pathogenic bacteria have drawn the most attention from microbiologists because in many cases, genes encoding disease-associated functions have been linked to an island. However, chromosomal islands are also known whose genes encode proteins that degrade pollutants such as aromatic hydrocarbons and herbicides (Section 22.4). In addition, many of the genes essential for the symbiotic relationship of species of the nitrogen-fixing bacterium Rhizobium with the root nodules of plants (Section 23.4) are carried in chromosomal islands. Perhaps the most unique chromosomal island is the magnetosome island of the bacterium Magnetospirillum; this DNA fragment carries genes that encode the formation of magnetosomes, intracellular magnetic particles that orient the cell in a magnetic field and influence the direction of its movement (Section 2.12).
In the final part of this chapter we consider how phylogenetics has revolutionized microbial systematics and has begun to crystallize our concept of a microbial species.
Check Your Understanding
What is the difference between core genome and pan genome of a given species?
What is a chromosomal island and how can one be identified?
What factors likely influence the size of microbial genomes?
III Microbial Phylogeny and Systematics
Microbial systematics blends phenotypic and genotypic information about microbes to yield a working blueprint of how the microbial world evolved and how it is organized. Phylogenetic trees are an important part of this blueprint and are used to show the evolutionary connections between microbes and to describe new taxa.
Systematics is the study of the diversity of organisms and their relationships. It links phylogeny with taxonomy, the science in which organisms are characterized, named, and classified according to defined criteria. Taxonomists describe microorganisms using a polyphasic approach that evaluates phenotypic, genotypic, and phylogenetic information. Phenotypic analyses examine the morphological, metabolic, physiological, and chemical characteristics of the cell. Genotypic analyses consider characteristics of the genome. These two analyses are used to categorize organisms based on similarities and are complemented by phylogenetic analyses, which seek to place organisms within an evolutionary framework using molecular sequence data (see Figure 13.9). We begin by exploring how molecular sequences can provide phylogenetic insight.
13.11 Molecular Phylogeny: Making Sense of Molecular Sequences
13.11 Molecular Phylogeny: Making Sense of Molecular Sequences
13.11 Molecular Phylogeny: Making Sense of Molecular Sequences
Molecular sequences provide a record of past evolutionary events and can be used to build phylogenetic trees, which are diagrams that depict evolutionary history. As we have seen in this chapter, mutations accumulate in DNA sequences over time. These mutations occur at random and are a major source of the natural variation that makes evolution possible (Section 13.6). Hence, the number of nucleotide sequence differences between any two homologous genes will be a function of the number of mutations that have accumulated since they shared a common ancestor. As a result, differences in DNA sequences can be used to infer evolutionary relationships, and the technology for doing this is well developed.
Obtaining DNA Sequences
Obtaining DNA from a microorganism is relatively easy if the organism can be cultivated in isolation in the laboratory. Once the cultivar’s genomic DNA is isolated, either the genome can be sequenced directly or the genomic DNA can be used to amplify one or more specific genes, using the polymerase chain reaction (PCR, Section 12.1) and primers specific for the gene(s) of interest. Advances in DNA sequencing technology (Section 10.2) have made genome sequencing a standard tool employed in analyses of microbial phylogeny. However, sequence analysis of small subunit (SSU) ribosomal RNA (rRNA) genes (Figure 13.24), which encode the rRNA molecule found within the small subunit of the ribosome (Section 6.10), remains a cornerstone of molecular phylogeny in microbiology because SSU rRNA genes are highly conserved, present in all cellular organisms, and easily sequenced and analyzed (see Figure 13.9).
Figure 13.24 Ribosomal RNA (rRNA).
Primary and secondary structure of 16S rRNA from Escherichia coli (Bacteria). The 16S rRNA from Archaea is similar in secondary structure (folding) to that of Bacteria but has numerous differences in primary structure (sequence). The molecule is composed of conserved and variable regions (V1–V9). The approximate positions of the variable regions are indicated in color. Inset: the 70S ribosome of Bacteria is composed of 30S and 50S subunits;16S rRNA is part of the 30S subunit whereas 5S and 23S rRNAs are parts of the 50S subunit.
PCR primers exist for many highly conserved genes, such as the SSU rRNA gene, and can be designed in such a way as to have different levels of phylogenetic specificity. That is, primers can be designed that target distinct species (or all species) within a genus, all genera within a phylum, or all phyla within a domain, and there are even “universal” PCR primers that will amplify the SSU rRNA gene from any organism. PCR products are visualized by agarose gel electrophoresis, excised from the gel, extracted and purified from the agarose, and then sequenced, often using the same oligonucleotides as primers for the sequencing reactions (Figure 13.25). Instead of starting with a microbial culture, it is also possible to amplify SSU rRNA genes from DNA extracted directly from an environmental sample or to sequence environmental DNA directly without amplification (metagenomics, Section 10.7 and Section 19.6). These latter approaches are widely used to characterize microorganisms that are difficult or not yet possible to grow in laboratory culture. Once sequences are obtained, they must be aligned and analyzed, issues we turn to now.
Mastering Microbiology
Art Activity: Figure 13.16 PCR amplification of the 16S rRNA gene
Figure 13.25 PCR amplification of the 16S rRNA gene from an unknown microbial culture.

Following DNA isolation, primers complementary to the ends of the 16S rRNA (Figure 13.24) are used to PCR-amplify the 16S rRNA gene from the genomic DNA of the bacterium. The PCR product is then run on an agarose gel (lane 1) versus a standard (lane 2). The bands of amplified DNA are approximately 1465 nucleotides in length (positions that DNA of different lengths would run in the gel are indicated at the left). The band from the unknown culture is then excised from the gel, purified, and sequenced. The sequence is then aligned with 16S rRNA sequences from other organisms and a phylogenetic tree constructed.
Sequence Alignment
Phylogeny can be inferred only from genes that have homology, that is, genes that have been inherited from a common ancestor. Thus, homology is a binary trait; sequences are either homologous or they are not. The concept of homology is often confused with that of sequence similarity. The latter is a continuous trait defined as a percentage of nucleotide positions shared between any two sequences. Sequence similarity is used to infer homology, but a similarity value can be calculated between any two sequences regardless of their function or evolutionary relationship. Thus, the terms similarity and homology are not interchangeable. Genes that have homology can be either orthologs, if they have the same function and originate from a single ancestral gene in a common ancestor, or paralogs, if they have evolved to have different functions as a result of gene duplication (Section 13.8 and Figures 13.16 and 13.17). Phylogenetic analyses typically focus on analyses of orthologous genes because of their common phylogeny and function.
Phylogenetic analyses estimate evolutionary changes from the number of sequence differences across a set of homologous nucleotide positions. Some mutations introduce nucleotide insertions or deletions, and these cause gene sequences to differ in length, making it necessary to align nucleotide positions prior to phylogenetic analysis of gene sequences. The purpose of sequence alignment is to add gaps to molecular sequences in order to establish positional homology; that is, to be sure that each position in the sequence was inherited from a common ancestor of all organisms under consideration (Figure 13.26). Proper sequence alignment is critical to phylogenetic analysis because the assignment of mismatches and gaps caused by deletions is in effect an explicit hypothesis of how the sequences have diverged from a common ancestral sequence.
Figure 13.26 Alignment of DNA sequences.
(a) Sequences for a hypothetical region of a gene are shown for three species before alignment and after alignment. A sequence alignment should display homologous positions in vertical columns. Sequence alignment is achieved by adding gaps, indicated by hyphens, to maximize local sequence similarity between the species in the alignment. (b) The distance matrices show the number of sequence differences that would be inferred for each species pair both before and after alignment.
Phylogenetic Trees: Composition and Construction
A phylogenetic tree is a diagram that depicts the evolutionary history of the organisms on the tree and bears some resemblance to a family tree. Most microbes have not left fossils and so their ancestors are unknown, but ancestral relationships can be inferred from the DNA sequences of organisms that are alive today. Organisms that share a recent ancestor are likely to share other characteristics as well, and thus phylogenetic trees allow us to make predictions about an organism’s characteristics. Phylogenetic trees are also of great use in taxonomy and species identification, as we will discuss later (Section 13.12).
A phylogenetic tree is composed of nodes and branches (Figure 13.27). The tips of the branches in a phylogenetic tree represent species that exist today. The nodes represent a past stage of evolution where an ancestor diverged into two new lineages. The branch length represents the number of changes that have occurred along that branch. In a phylogenetic tree, only the position of nodes and the branch lengths are informative; rotation around nodes has no effect on the tree’s topology (Figure 13.27).
Figure 13.27 Phylogenetic trees and their interpretation.

Three equivalent versions of the same phylogenetic tree are shown. The only difference between the trees is that their nodes have been rotated at the points indicated by red circles. The vertical position of species is different between the trees, but the pattern of ancestry (the nodes shared by each species) remains unchanged.
There is only one correct phylogenetic tree that most accurately depicts the evolutionary history of a group of gene sequences, but inferring this tree from sequence data can be a challenging task. The complexity of the problem is revealed by considering the total number of different trees that can be formed from a random set of sequences. For example, only three possible trees can be drawn for any four arbitrary sequences. But if the number of sequences is doubled to eight, now 10,395 trees are possible. This complexity continues to expand exponentially such that 2×10182 different trees can be drawn to represent 100 arbitrary sequences. Phylogenetic analysis uses molecular sequence data in an attempt to identify the one correct tree that accurately represents the evolutionary history of a set of sequences.
A variety of methods are available for inferring phylogenetic trees from molecular sequence data. The structure of a phylogenetic tree is inferred by applying either an algorithm or some set of optimality criteria. An algorithm is a programmed series of steps designed to construct a single tree (Figure 13.28). Algorithms used to build phylogenetic trees include the unweighted pair group method with arithmetic mean (UPGMA) and the neighbor-joining method. Alternatively, phylogenetic methods that employ optimality criteria include parsimony, maximum likelihood, and Bayesian analyses. These latter methods evaluate many possible trees and select the one tree that has the best optimality score, that is, they select the tree that best fits the sequence data given a discrete model of molecular evolution. Optimality scores are calculated on the basis of evolutionary models that describe how molecular sequences change over time. For example, evolutionary models can account for variation in substitution rates and base frequencies between sequence positions.
Figure 13.28 Building phylogenetic trees.

The number of nucleotide differences between gene sequences can be used to build a phylogenetic tree. In the sequence alignment (a) we can count the number of differences between each pair of sequences to build a distance matrix (b). This distance matrix can be used to build a tree (c) where the cumulative lengths of the horizontal branches (labeled with a red “1”) between any two species in the tree are proportional to the number of nucleotide differences between these species.
Limitations of Phylogenetic Trees
Molecular phylogeny provides powerful insights into evolutionary history, but it is important to consider the limitations of building and interpreting phylogenetic trees. For example, it can be difficult to choose the true tree based on available sequence data if several different trees fit the data equally well. Bootstrapping, a statistical method in which phylogenetic information is resampled at random, is an approach used to deal with uncertainty in phylogenetic trees. Bootstrap values indicate the percentage of the time that a given node in a phylogenetic tree is supported by the sequence data. High bootstrap values indicate that a node in the tree is likely to be correct, while low bootstrap values indicate that the placement of a node cannot be accurately determined given the available sequence data.
Homoplasy, also known as convergent evolution, occurs when organisms share a trait that was not inherited from a common ancestor. An example is the evolution of wings in insects and birds. These traits evolved separately and do not indicate that a winged ancestor was shared among insects and birds. Homoplasy occurs in molecular sequences as well, when similar sequence positions result from recurrent mutation rather than inheritance from a common ancestor (**Figure 13.29*a***). The problem of homoplasy in molecular phylogeny then increases in proportion to evolutionary time (Figure 13.29b). As a result of homoplasy, the reconstruction of accurate phylogenetic trees gets more difficult when sequence divergence between organisms is very high.
Figure 13.29 The problem of homoplasy due to recurrent mutation.

It is possible for recurrent mutation to obscure the true number of mutations that have occurred since a pair of sequences have shared a common ancestor. (a) Two series of mutations during the evolution of a gene sequence are compared. On the left side, the number of mutations is equal to the number observed between species 1 and 4. However, if there is recurrent mutation (right side), the number of mutations observed between species 1 and 4 can be less than the number that actually occurred. (b) The likelihood of recurrent mutation increases as more and more mutations accumulate over time.
Mastering Microbiology
Art Activity: Figure 13.20 The problem of homoplasy due to recurrent mutation
The prevalence of horizontal gene transfer (Section 13.9 and Chapter 9) also creates complications when considering the evolutionary history of microorganisms. When the sequence of a gene is used to infer the phylogeny of an organism, it must be assumed that the gene is inherited in a vertical fashion—from mother to daughter—throughout the evolutionary history of the organism. The horizontal exchange of genes between unrelated organisms violates this assumption (Figure 13.30). Hence, it is important to consider the difference between a gene phylogeny, which depicts the evolutionary history of an individual gene, and an organismal phylogeny, which depicts the evolutionary history of the cell.
Figure 13.30 Horizontal gene transfer.
The horizontal transfer of a gene will cause it to have a different evolutionary history from the rest of the genome. (a) Genes are transferred horizontally between distantly related microorganisms. Colors are used to match microorganisms with their genomes. (b) As a result of the horizontal transfer events in part a, different phylogenetic trees for gene 1, gene 2, and gene 3 are obtained. Only the gene tree for gene 1, which was not transferred, remains congruent with the organismal phylogeny.
In general, genes encoding SSU rRNAs appear to be transferred horizontally at very low frequencies, and rRNA gene phylogenies agree largely with those prepared from other genes that encode genetic informational functions. Thus, SSU rRNA gene sequences are generally considered to provide an accurate record of organismal phylogeny. Nevertheless, many microbial genomes contain genes that have been acquired by horizontal gene transfer at some point in their evolutionary history, and this has important implications for microbial evolution that cannot be adequately assessed by analyzing only a single gene, even one as phylogenetically informative as that encoding SSU rRNA.
Check Your Understanding
How are DNA sequences obtained for phylogenetic analysis?
Why is sequence alignment critical to phylogenetic analysis?
13.12 Microbial Systematics
Microbial systematics is the branch of microbiology concerned with naming and classifying microorganisms, describing their traits, and investigating their evolutionary histories. Nomenclature—the naming of organisms—follows the binomial system devised by the Swedish medical doctor and botanist Carl Linnaeus in which species are given genus names and species epithets. The names are Latin or Latinized Greek derivations in which the genus name is a noun and the species epithet an adjective describing some key property of the organism or the habitat from which the organism was obtained. Occasionally, a species epithet will be derived from the last name of a scientist who has made major contributions to the study of a particular group of organisms.
Taxonomy is the classification of organisms into groups of similar organisms. Individual microbial strains are classified into species, species classified into genera, genera into families, families into orders, orders into classes, classes into phyla, and phyla into domains (Table 13.1). By classifying organisms into groups and naming them, we order the natural microbial world and make it possible to communicate effectively about all aspects of particular organisms, including their behavior, ecology, physiology, pathogenesis, and evolutionary relationships.
Table 13.1 Taxonomic hierarchy for the purple sulfur bacterium *Allochromatium warmingii*

Species are the fundamental units of biological diversity, and how we distinguish and classify species in microbiology greatly affects our ability to explain and assess the diversity of the microbial world. For the purpose of microbial systematics, a microbial species is a taxonomic category that defines a population of individuals that (1) is monophyletic (descended from a common ancestor, see next subsection), (2) is genomically coherent, (3) is phenotypically coherent, and (4) can be clearly differentiated from other species. The description of microbial species employs a polyphasic approach, using many different methods in combination, to identify, describe, and name species of Bacteria and Archaea. In this section we describe methods commonly used for characterizing species of Bacteria and Archaea.
Phylogenetic Analyses
A microbial species should be monophyletic, that is, the strains composing the species should all share a recent common ancestor to the exclusion of other species. The phylogenetic relationships of microorganisms are typically determined by analyzing their SSU rRNA gene sequences (Section 13.11). Phylogenetic trees will show that the SSU rRNA gene sequences from strains of a distinct species will all form a coherent group that shares a recent common ancestor to the exclusion of strains of any other species. In addition, SSU rRNA genes from strains of the same species typically have greater than 98.6% sequence similarity, whereas SSU rRNA genes from different species typically have less than 97% sequence similarity (Figure 13.31).
Figure 13.31 Classification of bacterial and archaeal species.

Species identification is facilitated by performing pairwise comparison of 16S rRNA similarity and average nucleotide identity (ANI) between strains. Strains with >98.6% 16S rRNA similarity and >96% ANI typically belong to the same species. Strains with 97–98.6% 16S rRNA similarity and 93–96% ANI possibly belong to the same species, but further tests are needed to confirm identity. Strains with <97% 16S rRNA similarity and <93% ANI are highly unlikely to belong to the same species.
SSU rRNA gene sequence similarity can also be useful in classification at higher taxonomic ranks. While there are no established cutoffs or formal criteria used to define higher taxonomic ranks, taxonomic ranks above species generally correlate with SSU rRNA gene sequence similarity. For example, the median SSU rRNA gene sequence similarity between strains of the same group is approximately 95% at the genus level, 92% for family, 89% for order, 86% for class, and 83% for phylum.
Multilocus sequence analysis (MLSA) can be used in addition to SSU rRNA gene sequence analysis to determine phylogenetic relationships. SSU rRNA gene sequences are highly conserved, and while they provide valuable phylogenetic information, they are not always useful for distinguishing closely related species. By contrast, other highly conserved genes, such as recA, which encodes a recombinase protein (Sections 9.4 and 9.5 and Figures 9.10 and 9.12), and gyrB, which encodes DNA gyrase (Sections 6.1 and 6.4 and Figures 6.4 and 6.16), can be useful for distinguishing bacteria at the species level, and techniques are available to examine them collectively. The DNA sequences of protein-encoding genes accumulate mutations more rapidly than rRNA genes and thus their sequences can often distinguish prokaryotic species that cannot be resolved by rRNA gene sequence analyses alone (Figure 13.32). MLSA multigene analyses generally employ a minimum of four genes but can include more than a thousand genes if genome data are available. These genes can be analyzed independently, or they can be linked into a single alignment and analyzed as a unit to produce a single phylogenetic tree (Figure 13.32).
Figure 13.32 Multilocus sequence analysis (MLSA).

Phylogenies are shown for species in the genus Photobacterium. (a) 16S rRNA gene tree, showing the species to be poorly resolved. (b) MLSA based on the combined analysis of the 16S rRNA gene and gyrB and luxABFE genes in 21 isolates of three Photobacterium species. MLSA clearly resolves the strains into three distinct species, P. phosphoreum, P. iliopiscarium, and P. kishitanii. The scale bar indicates the branch length equal to a total of 50 nucleotide changes. The type strain of each species is listed in bold. (All abbreviations are part of strain designations.) Phylogenetic analyses courtesy of Tory Hendy and Paul V. Dunlap, University of Michigan.
Genomic Analyses
The use of entire genomes for the identification and description of bacteria is becoming increasingly common as DNA sequencing capacities improve and costs decline (Section 10.2). A wide range of sequence analyses are performed on whole genomes, thereby providing insights into microbial physiology and microbial evolution. Comparative analyses of gene content (presence or absence of genes), synteny (the order of genes in the genome), and genome G+C content (Section 13.9) provide powerful insights into evolutionary relationships between strains. Whole genome sequences can also be used for metabolic reconstruction to determine the potential physiological characteristics of a given strain.
Several methods in comparative genomics and population genomics have been developed for use in systematic analyses. In particular, metrics of overall genome relatedness can be calculated by determining sequence similarity across the entire genome. Average nucleotide identity (ANI) is the most commonly used metric for estimating overall genome relatedness between a pair of organisms. To do this, a computer breaks a genome sequence into fragments about 1000 bp in length. Each fragment is then aligned to its orthologous sequence in a second genome and the average nucleotide identity between the two genomes calculated. Genomes from strains of the same species typically have greater than 96% ANI, and genomes from different species typically have less than 93% ANI (Figure 13.31).
Phenotypic Analyses
The observable characteristics—the phenotype—of a bacterium provide many traits that can be used to differentiate species. Typically, several phenotypic traits are determined routinely when describing a new microorganism. These phenotypic results are then compared to the phenotypes of organisms that have been described previously. The phenotypic traits that are determined will depend on the type of organism being described. For example, in applied situations, such as clinical diagnostic microbiology, where timely identification is important, a well-defined subset of traits can be tested to rapidly discriminate between different types of microorganisms. Table 13.2 lists general categories and examples of some phenotypic traits used in identifications and species descriptions.
Table 13.2 Some phenotypic characteristics of taxonomic value

Phenotypic characteristics of a given organism can be highly dependent on growth conditions, and the phenotype of an organism observed in the laboratory may not be the same as that expressed in the natural environment. Thus, care must be taken in using phenotypic characteristics in systematic analyses. In addition, the value of different phenotypic characteristics for reaching a firm systematics conclusion can vary with the taxonomic groups being examined.
The Description of New Species
When a newly discovered strain of Bacteria or Archaea is thought to be unique, a decision must be made as to whether it is sufficiently different from established taxa to be described as a new taxon. The creation of new taxa of Bacteria and Archaea must follow the rules described in the International Code of Nomenclature of Prokaryotes (the Prokaryotic Code). To achieve formal validation of taxonomic standing as a new genus or species, a detailed description of the organism’s characteristics and distinguishing traits, along with its proposed name, must be published in a specialized peer-reviewed journal, and viable cultures of the organism must be deposited in at least two international culture collections. Most new taxa are described in the International Journal of Systematic and Evolutionary Microbiology (IJSEM), the publication of record for the nomenclature of Bacteria and Archaea. Two websites provide listings of valid, approved bacterial names: List of Prokaryotic Names with Standing in Nomenclature (http://www.bacterio.net), and Prokaryotic Nomenclature Up-to-Date (http://www.dsmz.de).
Microbial culture collections play an important role in protecting microbial biodiversity, just as museums do in preserving plant and animal specimens for future study. Culture collections store microorganisms as viable cultures, typically frozen or in a freeze-dried state, and distribute cultures to qualified scientists who wish to study them. These storage methods maintain the cells indefinitely in a living state and prevent genetic changes that might occur if the organisms were continually subcultured. Culture collections also serve as repositories for type strains. When a new species of bacteria is described in a scientific journal, a strain is designated as the type strain and serves as the nomenclatural type of the taxon for future taxonomic comparison with other strains of that species. Major international culture collections exist in the United States (ATCC), Germany (DSMZ), Japan (JCM Riken), and several other countries. Each collection has a website linked to its acronym from which its holdings can be searched.
It is possible to use molecular and genomic techniques (Chapters 10 and 19) to characterize the phenotypic and genotypic characteristics of microorganisms without the need for the cultivation of an isolated strain. However, in the absence of an isolate that can be deposited in two international culture collections, it is not yet possible to validly name a new species of microorganism under the Prokaryotic Code. However, when an organism is well characterized but not yet obtained in pure culture, a provisional taxonomic name can be applied. The moniker Candidatus is appended to candidate taxonomic ranks. For example, “Candidatus Pelagibacter ubique” is a globally widespread and well-characterized marine bacterium that is difficult to grow in laboratory media (Section 20.12) and so has not been formally named under the Prokaryotic Code. By contrast, the bacterium “Candidatus Heliomonas lunata” can be grown in laboratory culture but the culture is not pure, so this bacterium also retains Candidatus status.
The classification system most widely accepted by microbiologists is that of Bergey’s Manual of Systematics of Archaea and Bacteria, a major taxonomic treatment of Bacteria and Archaea that has served microbiologists since 1923 as a compendium of information on all recognized prokaryotic species. Each chapter, written by experts, contains tables, photos, figures, and other systematic information useful for identification purposes. A second major source describing the physiology, ecology, phylogeny, enrichment, isolation, and cultivation of Bacteria and Archaea is The Prokaryotes. This work is available online by subscription through university libraries. Collectively, Bergey’s Manual and The Prokaryotes offer microbiologists both the concepts and the details of the biology of Bacteria and Archaea as we know them today and are the primary resources for microbiologists characterizing newly isolated organisms.
How Many Species of *Bacteria* and *Archaea* Exist?
The result of nearly 4 billion years of evolution is the microbial world we see today (Figure 13.9). Microbial taxonomists agree that no firm estimate of the number of bacterial and archaeal species can be given at present. However, they also agree that in the final analysis, this number will be very large. The diversity of bacterial and archaeal species on Earth is unquestionably higher than that of all plant and animal species combined, and the total species numbers of Bacteria and Archaea are likely several orders of magnitude higher than the 14,000 species that have been characterized to date.
Every environment on Earth contains a diverse community of microorganisms. For example, analyses of environmental SSU rRNA gene sequences (Section 19.6) suggest that over 14,000 different species can coexist in a single gram of soil! Since 1977, more than 3.3 million SSU rRNA sequences have been deposited in international databases such as GenBank and used to characterize the vast diversity of the microbial world. These SSU rRNA sequences encompass more than 225,000 operational taxonomic units (OTUs), species-like units of diversity which are defined at a 97% sequence similarity threshold (Figure 13.31). The Ribosomal Database Project (RDP; http://rdp.cme.msu.edu) contains an ever-growing collection of these sequences and provides computational programs for their analysis and for the construction of phylogenetic trees.
While we do not yet know the extent of biodiversity of microbial life, we do know that nearly all plant and animal species have microbiomes (Chapter 23) that contain countless numbers of unique microbes. Thus, microorganisms are not only the oldest but also unquestionably the most diverse forms of life on our planet. We will see this diversity in action over the next five chapters.
Check Your Understanding
What are some key criteria used to determine whether two strains belong to the same species?
What is average nucleotide identity and how is it calculated?
What is the process for describing and naming a new species of bacteria?
Chapter Review
Go to Mastering Microbiology for videos, animations, practice tests, and more.
I Early Earth and the Origin and Diversification of Life
13.1 Planet Earth is about 4.5 billion years old. After its formation, Earth was hot and sterile. Gradual cooling of the planet allowed formation of liquid water, a requirement for the origin of life. The earliest evidence of life comes from isotopic analysis of ancient sedimentary rocks and zircon minerals indicating that life was present on Earth by 3.7–4.1 billion years ago.
Q What is LUCA, and what is a plausible explanation for the origin of cellular life?
13.2 In rocks 3.5 billion years old or younger, microbial formations called stromatolites are abundant and show extensive microbial diversification. The evolution of oxygenic photosynthesis caused O2 to accumulate 2.4 billion years ago, eventually leading to the formation of banded iron formations, the ozone shield, and an oxygenated atmosphere, which set the stage for rapid diversification of metabolic types and the evolution of multicellularity.
Q Why was the origin of cyanobacteria of such importance to the evolution of life on Earth?
13.3 Ribosomal RNA genes have been used to construct a universal tree of life revealing that life on Earth evolved in three major directions, forming the domains Bacteria, Archaea, and Eukarya. The universal tree of life shows that the domains Bacteria and Archaea diverged billions of years ago, and that Eukarya split from Archaea later in the history of life.
Q What evidence supports the classification of life into three domains?
13.4 The eukaryotic cell developed from endosymbiotic events. The modern eukaryotic cell is a chimera with genes and characteristics from both Bacteria and Archaea. 16S rRNA sequence analyses indicate the ancestors of mitochondria are found in the phylum Proteobacteria and those of chloroplasts are found in the phylum Cyanobacteria.
Q What is the endosymbiotic theory for the origin of mitochondria and chloroplasts? What evidence supports this theory?
13.5 Viruses lack universally conserved genes and so cannot be placed on the universal tree of life. Viruses might have evolved many times from rogue mobile genetic elements such as gene transfer agents, or they might have a single origin as remnants of pre-cellular life from the RNA world.
Q All viruses protect their nucleic acid using a capsid shell composed of protein. Provide a hypothesis to explain the evolution of capsid proteins.
II Mechanisms of Microbial Evolution
13.6 Evolution is defined as a change in allele frequencies in a population of organisms over time, resulting in descent with modification. New alleles are created through the processes of mutation and recombination. Mutations occur at random and most mutations are neutral or deleterious, but some are beneficial. Natural selection and genetic drift are two mechanisms that cause allele frequencies to change in a population over time.
Q What is fitness? To what degree does fitness depend on the environment in which organisms live?
13.7 Experimental evolution uses microbial systems to investigate evolution in the laboratory. Mutations occur at random, but those that improve fitness will increase in frequency in a population of cells. Environmental conditions dictate whether mutations cause a change in fitness. Mutations may cause a gain, loss, change, or no change in function.
**Q Do Rhodobacter pigment mutations occur at the same frequency in the light and in the dark? In the LTEE, do you expect that citrate-using mutants would still have occurred if citrate had never been present in the growth medium?**
13.8 A gene family contains a group of homologs that have all descended from the same ancestral gene. Homologs are orthologs if they have same function or paralogs if they have evolved different functions. Paralogs often evolve as a result of gene duplication. Deletion mutations are common in microbial genomes, and they cause the rapid elimination of genetic material that is unnecessary or redundant in function.
Q Explain how deletion mutations can increase fitness. Explain how duplication mutations can promote the evolution of new gene functions.
13.9 Horizontal gene transfer allows genes to be exchanged between cells irrespective of species boundaries. The mobilome is the portion of the genome that contains mobile genetic elements that facilitate horizontal gene exchange and chromosomal rearrangements. The mobilome includes plasmids, viruses, transposons, integrons, and insertion sequences.
Q Explain how horizontally transferred genes can be detected in a genome.
13.10 Microbial genomes are dynamic, and genome size and gene content can vary considerably between strains of a species. The core genome is defined as the set of all genes shared by a species, while the pan genome is defined as the core genome plus genes whose presence varies among strains of a species. Many bacteria contain chromosomal islands, large clusters of genes acquired by horizontal gene transfer, that encode specialized functions such as virulence (pathogenicity islands).
Q What are some processes that influence the content of the pan genome?
III Microbial Phylogeny and Systematics
13.11 Molecular sequences accumulate random mutations over time, and molecular phylogenetic analysis examines differences in molecular sequences to determine the evolutionary history of life. A phylogenetic tree is a diagram that depicts the evolutionary history of a set of genes or organisms.
Q What is the difference between a gene tree and an organismal tree?
13.12 Systematics is the study of the diversity and relationships of living organisms. Species of Bacteria and Archaea are classified using phylogenetic, genomic, and phenotypic information. Strains of the same species are monophyletic, typically have >98.6% SSU rRNA sequence similarity and >96% average nucleotide identity across their genomes, and are expected to share a coherent set of phenotypic traits.
Q Is it possible to provide a formal name for a microorganism that has not been cultivated in isolation? How might you attempt to describe and name such a species?
Application Questions
Compare and contrast the physical and chemical conditions on Earth at the time life first arose with conditions today. From a physiological standpoint, discuss at least two reasons why animals could not have existed on early Earth. In what ways has microbial metabolism altered Earth’s biosphere? How might life on Earth be different if oxygenic photosynthesis had not evolved?
For the following sequences, construct the phylogenetic tree that best depicts their evolutionary relationships (assume that the sequences are aligned).
Imagine that you have been given several bacterial strains from various countries around the world and that all the strains are thought to cause the same gastrointestinal disease. What methods could you use to test whether the different strains are actually members of the same species?
Chapter Glossary
a domain of life consisting of microorganisms that have prokaryotic cell structure and are distinct from Bacteria Average nucleotide identity (ANI)
a genome-wide measure of genetic similarity between two microorganisms based on aligning all orthologous gene pairs across the two genomes and determining the percentage of matching nucleotides (see also orthologs) Bacteria
a domain of life consisting of microorganisms that have prokaryotic cell structure and are distinct from Archaea Banded iron formation
iron oxide–rich ancient sedimentary rocks containing zones of oxidized iron (Fe3+) formed by oxidation of Fe2+ Binomial system
the system devised by the Swedish scientist Carl Linnaeus for naming living organisms in which an organism is given a genus name and a species epithet Chromosomal island
a chromosome region of foreign origin containing a cluster of genes that work together to confer upon the cell a particular trait such as virulence or symbiosis Core genome
those genes shared by all strains of a species Endosymbiotic theory
the idea that mitochondria and chloroplasts originated from Bacteria Eukarya
a domain of life consisting of organisms that have eukaryotic cell structure; includes algae, protists, fungi, slime molds, plants, and animals Evolution
a change over time in gene sequence and frequency within a population of organisms, resulting in descent with modification Evolutionary selection
a process that results in a change in allele frequencies in a population when individuals that are favored in a given environment are able to produce more offspring and make a greater contribution to the gene content of future generations Fitness
the capacity of an organism to survive and reproduce in a given environment as compared to that of competing organisms Gene family
genes related in sequence to each other because of common evolutionary origin Genetic drift
a process that results in a change in allele frequencies in a population as a result of random changes in the number of offspring from each individual over time Homologs
genes that were inherited from a shared ancestor; includes both orthologs and paralogs Homoplasy
when two organisms have the same trait as a result of recurrent mutation or convergent evolution Horizontal gene transfer
the unidirectional transfer of genes between cells through a process uncoupled from reproduction Mobilome
the mobile genetic elements in a genome Molecular clock
a DNA sequence, such as a gene for rRNA, that can be used as a comparative temporal measure of evolutionary divergence Monophyletic
in phylogeny, a group descended from a single shared ancestor Multilocus sequence analysis (MLSA)
phylogenetic analysis of microorganisms performed using multiple different genes Mutation
a heritable change in the base sequence of the genome of an organism Orthologs
genes that are homologous (that is, they are descended from a common ancestor) and share the same function (see also paralogs) Pan genome
the totality of the genes present in the different strains of a species Paralogs
genes that are homologous (that is, they are descended from a common ancestor) but which have diverged to have different functions; typically, paralogous genes are the result of gene duplication (see also orthologs) Pathogenicity island
a bacterial chromosome region of foreign origin that contains clustered genes for virulence Phenotype
the observable characteristics of an organism Phylogenetic tree
a diagram that depicts the evolutionary history of an organism; consists of nodes and branches Phylogeny
a major lineage of organisms in one of the three domains of life Recombination
a resorting or rearrangement of DNA fragments resulting in a new sequence Ribosomal RNA (rRNA)
the types of RNA found in the ribosome; some participate actively in protein synthesis Sequence alignment
the insertion of gaps into a set of molecular sequences organized in rows so that homologous positions are organized in vertical columns. Alignment is necessary prior to phylogenetic analysis because deletion and insertion mutations cause variations in the length of molecular sequences 16S rRNA
the type of SSU rRNA found in Bacteria and Archaea; its eukaryotic counterpart is 18S rRNA (see also SSU rRNA) Species
defined in microbiology as a collection of strains that all share the same major properties and differ in one or more significant properties from other collections of strains; defined phylogenetically as a monophyletic, exclusive group based on DNA sequence SSU rRNA
(small subunit rRNA) the rRNA molecule found in the small subunit of the ribosome, comprised of 16S rRNA, which is present in the 30S ribosomal subunit of Bacteria and Archaea, or its orthologous counterpart 18S rRNA, which is present in the 40S ribosomal subunit of Eukarya. SSU rRNA genes are conserved in all forms of cellular life, and this gene is often used in phylogenetic analysis of microorganisms Stromatolite
a laminated microbial mat that can become fossilized and is typically built from layers of filamentous microorganisms Systematics
the study of the diversity of organisms and their relationships; includes taxonomy and phylogeny Taxonomy
the science of identification, classification, and nomenclature Type strain
the strain chosen to represent the nomenclatural type of a species; type strains are deposited in microbial culture collections and are used to represent the typical characteristics of their species Universal tree of life
a phylogenetic tree that shows the positions of representatives of all domains of cellular life