25 Microbial Infection and Pathogenesis
## Chapter 25 Microbial Infection and Pathogenesis
Killing Pathogens on Contact
The adherence of pathogenic bacteria to surfaces plays a crucial role in their ability to colonize areas of the body and cause disease. In addition to natural tissue surfaces in the body, such as tooth enamel and mucous membranes, pathogens can also adhere to artificial surfaces associated with implanted medical devices, including catheters, prosthetic heart valves, and artificial joints. The adherence of bacteria to these sites can lead to the formation of biofilms, which are notoriously difficult to treat using antibiotics.
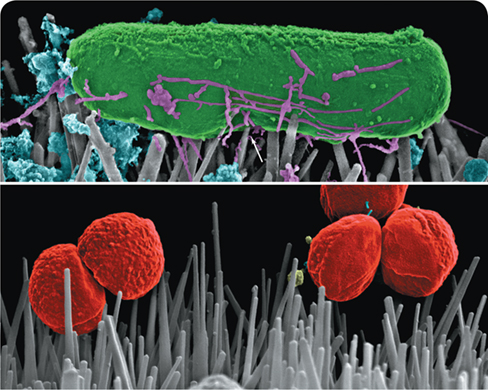
Because of the tendency of pathogenic bacteria to colonize artificial surfaces in the body, prosthetic joint infections (PJIs) are a constant threat in orthopedic medicine. Considering the ineffectiveness of antibiotics in treating these infections, successful treatment of PJIs often requires physical removal of the colonized biofilm from the prosthesis and surrounding tissues in a surgical procedure called debridement. To avoid the complications of treating infection, can we find ways to prevent the formation of biofilms that lead to PJIs in the first place?
Taking a lesson from nature itself, researchers are turning to the field of biomimetics to address these issues. Several animal and plant tissue surfaces exhibit antibacterial properties, including shark skin, insect wings, and lotus leaves. Such natural surfaces are composed of nanostructures that kill bacteria on contact, likely by disrupting the integrity of their cytoplasmic membranes. Borrowing this concept, antibacterial nanofabrication methods have centered on titanium dioxide (TiO2) as a potential substrate to mimic natural contact killing. Although TiO2 has shown limited potential for killing gram-positive cocci, such as Staphylococcus aureus (bottom photo), it has shown considerable promise in destroying gram-negative bacteria on contact, including Escherichia coli, in which the spiky nanostructures cause indentation and eventual puncture of the cells (arrow, top photo).
If artificial joints and other medical implants can be manufactured with surfaces having antibacterial nanostructures, PJIs and other biofilm-associated infections may become much easier to prevent and control—no antibiotics necessary.
Source: Jenkins, J., et al. 2018. Characterization of bactericidal titanium surfaces using electron microscopy. Microsc. Anal. (Am. Ed.). 32: 15.
I Human–Pathogen Interactions
To induce pathogenesis and begin to parasitize the host, pathogenic microbes must first adhere to host tissues and overcome immune defenses that seek to inhibit microbial colonization and invasion.
Humans are exposed to microorganisms of all sorts in their environment. Whether one is walking outdoors, sitting indoors, or participating in any type of physical activity, environmental microbes and humans interact. The human body is also a natural home to enormous numbers of microorganisms, as we saw in Chapter 24. Most of these are harmless, and in fact essential, with only a small percentage capable of causing disease. However, those that do, along with pathogens that are not part of the normal human microbiota, possess specific traits that underlie their pathogenic lifestyles. A major focus of this chapter will be a consideration of these specific traits and how they induce the diseased state.
25.1 Microbial Adherence
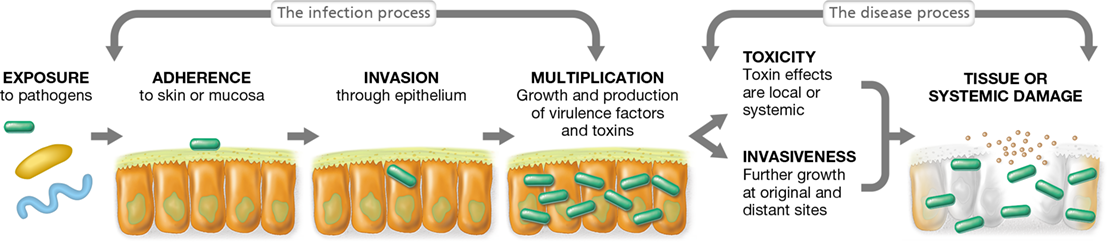
In the world of infectious diseases, the term infection is used to imply the growth of microorganisms on or in the host, whereas the term disease is reserved for actual tissue damage or injury that impairs host function. If a pathogen gains access to the specific tissues it infects, disease will occur only if it first adheres to those tissues, multiplies to yield many cells or viral particles, and then proceeds to damage tissues (or the entire organism) by the release of toxic or invasive substances (Figure 25.1). Adherence is the first step, and although adherence is required to initiate disease, it is not sufficient to initiate disease because the host has many innate defenses that can thwart infection; we consider these in Chapter 26.
Figure 25.1 Microbial pathogenesis.

Following exposure to a pathogenic microbe, a series of events leads to infection, and a further series of events results in disease.
Adherence Molecules
Pathogens typically adhere to epithelial cells through specific interactions between molecules on the pathogen and molecules on the host tissues. In addition, pathogens may adhere to each other, forming biofilms (Sections 4.9 and 20.4), with the biofilm itself adhering to specific tissues. In medical microbiology, adherence is the enhanced ability of a microorganism to attach to a cell or a surface. Pathogens gain access to host tissues by way of a portal of entry of one sort or another. These include mucous membranes, the skin surface, or under mucous membranes or the skin during penetration of these sites from puncture wounds, insect bites, cuts, or other abrasions. The portal of entry may be critical for the establishment of an infection because a pathogen that gains access to incompatible tissues is typically ineffective. For example, if cells of the bacterium Streptococcus pneumoniae are swallowed, they will be killed by the strong acidity of the stomach, whereas if the same cells reach the respiratory tract, they could trigger a fatal case of pneumonia.
Mastering Microbiology
Receptor molecules coating the surfaces of both the pathogen and cells of its host are often critical for adhering the pathogen to host tissues. Specific receptors can be important for the binding of any type of pathogenic microbe, including bacteria, viruses, and parasites (Figure 25.2). Receptors on pathogens have evolved to bind specifically to complementary molecules in the host, and the complementary nature of the receptors on pathogen and host cells alerts the pathogen that it has arrived on a suitable infection site. Receptors on the pathogen surface are called adhesins and are composed of glycoprotein or lipoprotein covalently bound to the outer layer of the cell (Figure 25.2b). In addition to extracellular matrices, such as the fibrinogen shown in Figure 25.2, host receptors for bacterial adhesins include cell surface glycoproteins or complex membrane lipids, such as gangliosides or globosides (both are sphingolipids containing sugars and other molecules).
Figure 25.2 Bacterial adherence.

A bacterial pathogen attaches specifically to host tissues by way of complementary receptors on the bacterial and host surfaces. (a) Cryogenic transmission electron micrograph of the gram-positive bacterium Staphylococcus aureus adhering to an extracellular matrix of fibrinogen, which the cells convert to a fibrin clot as a defense against host immune responses (see also Figure 25.12b). The S. aureus cells are about 0.75 μm in diameter. (b) Expanded diagram of S. aureus binding fibrinogen using the N2 and N3 domains of its SdrG adhesin protein.
Adherence Structures: Capsules, Fimbriae, Pili, and Flagella
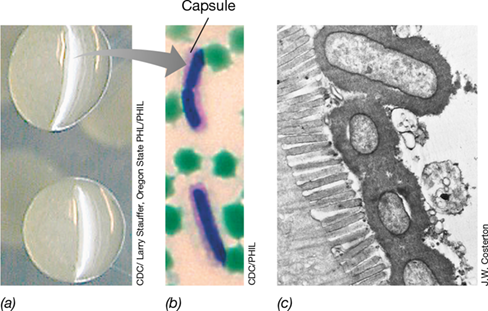
Some adhesins form part of an outer cell surface structure that may or may not be covalently linked to components of the cell wall. For example, some notable pathogenic bacteria form a capsule. In Bacillus anthracis (the bacterium that causes anthrax), the capsule is composed of polypeptide containing only the amino acid d-glutamate. The capsule of B. anthracis can be seen in cells by light microscopy, and the encapsulated cells form smooth, slimy colonies when grown on agar plates (Figure 25.3). The electron microscope can also clearly reveal bacterial capsules (Figure 25.3c). The capsule surface contains specific receptors that facilitate adherence to host tissues, but the inherently sticky nature of the capsule itself also assists in the overall attachment process. Although capsules are important for adherence of some pathogens to host tissues, many important pathogens, such as Vibrio cholerae, the causative agent of the disease cholera, lack them.
Figure 25.3 The bacterial capsule as a facilitator of pathogen attachment.

(a) Bacillus anthracis growing on an agar plate. The mucoid colonies of encapsulated cells are about 0.5 cm in diameter. (b) Light micrograph of cells of B. anthracis growing in horse blood and stained to show the capsule (dark pink surrounding blue cell). Cells are about 1 μm in diameter. (c) Cells of enteropathogenic Escherichia coli attached to the brush border of intestinal microvilli by way of a distinct capsule. The E. coli cells are about 0.5 μm in diameter.
Besides adherence, capsules are important for protecting pathogenic bacteria from host defenses. For example, the only known virulence factor for Streptococcus pneumoniae (bacterial pneumonia) is its polysaccharide capsule (Figure 25.4). Encapsulated strains of S. pneumoniae grow abundantly in the lungs where they initiate host responses that interfere with lung function, cause extensive host damage, and can cause death (Section 31.2). By contrast, nonencapsulated strains of S. pneumoniae are quickly and efficiently ingested and destroyed by phagocytes, white blood cells that ingest and kill bacteria by a process called phagocytosis (Sections 26.5, 26.6 and 26.7).
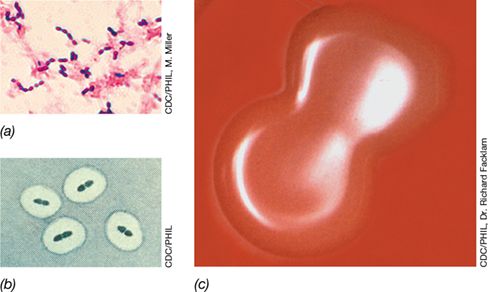
Figure 25.4 Capsules and colonies of *Streptococcus pneumoniae*.

(a) Gram stain of S. pneumoniae cells; capsules are not visible. (b) S. pneumoniae treated with anticapsular antibodies (Quellung reaction) that make the capsule visible. (c) Colonies of encapsulated S. pneumoniae cells grown on blood agar show a mucoid morphology with a sunken center. The colonies are about 2–3 mm in diameter and a single cell of S. pneumoniae is about 0.8 μm in diameter.
Many pathogens selectively adhere to particular types of cells through cell surface structures other than adhesins, capsules, or slime layers. For example, Neisseria gonorrhoeae, the pathogen that causes the sexually transmitted disease gonorrhea (Section 31.13), adheres specifically to mucosal epithelial cells in the genitourinary tract, eye, rectum, and throat; by contrast, other tissues are not infected. N. gonorrhoeae has a cell surface protein called Opa (opacity associated protein) that binds specifically to a host protein found only on the surface of epithelial cells of these body regions, allowing adherence of the pathogen to host cells. Likewise, influenza virus targets upper respiratory tract mucosal cells and attaches specifically to these and later to lung epithelial cells by way of the protein hemagglutinin present on the virus surface (Section 11.9 and Section 31.8).
Fimbriae and pili are bacterial cell surface protein structures (Section 2.6) that function in attachment (Figure 25.5). For instance, along with Opa, the pili of Neisseria gonorrhoeae play a key role in attachment to urogenital epithelia, and fimbriated strains of Escherichia coli are more frequent causes of urinary tract infections than strains lacking fimbriae. Among the best-characterized fimbriae are the type I fimbriae of enteric bacteria (Escherichia, Klebsiella, Salmonella, and Shigella), which are uniformly distributed on the surface of cells (Figure 25.5). Pili are typically longer and fewer in number than fimbriae, and in addition to attachment, some pili function in the bacterial genetic transfer process of conjugation (Section 9.8). Both pili and fimbriae function by specifically binding to host cell surface glycoproteins, thereby initiating adherence. Flagella may also facilitate adherence of bacterial cells to host cells (Figure 25.5), although their role is thought to be less important than that of fimbriae and pili.
Figure 25.5 Fimbriae.

Computer-generated image of a scanning electron micrograph of cells of Salmonella enterica (typhi ) showing the numerous thin fimbriae and the much thicker peritrichously arranged flagella. A single cell is about 1 μm in diameter.
Check Your Understanding
What event is required but not sufficient to cause an infectious disease?
Describe the molecules or structures that facilitate pathogen adherence to host tissues.
25.2 Colonization and Invasion
If a single pathogenic virus or cell attaches to its specific host tissue, it alone is insufficient to cause disease; the pathogen must establish residence there and multiply. Colonization, the growth of a microorganism after it has gained access to host tissues, begins at birth as a newborn child is naturally exposed to a suite of harmless (and in many cases necessary) bacteria and viruses that will be the infant’s initial normal microbiota (Chapter 24).
The human body is rich in organic nutrients and provides conditions of controlled pH, osmotic pressure, and temperature that are favorable for the growth of microorganisms. However, each body region, such as the skin, respiratory, gastrointestinal, and genitourinary tracts, differs chemically and physically from others, and thus provides a selective environment for the growth of certain microbes and not others. The result is that pathogens show rather rigid tissue specificities (Table 26.1), and this reality is often helpful in the diagnosis of microbial infections.
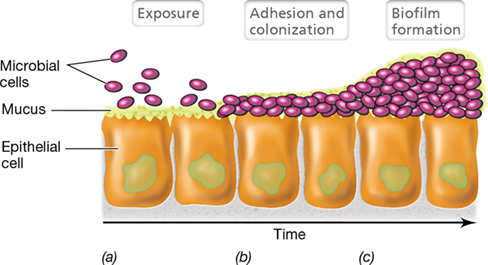
Colonization typically begins at sites in the mucous membranes (Figure 25.6). Mucous membranes are composed of epithelial cells, tightly packed cells that interface with the external environment. They are found throughout the body, lining the urogenital, respiratory, and gastrointestinal tracts. The epithelial cells in mucous membranes secrete mucus, a thick liquid secretion that contains water-soluble proteins and glycoproteins. Mucus retains moisture and naturally inhibits microbial attachment because most microbes are swept away by physical processes, such as swallowing or sneezing. Nevertheless, some microbes—both pathogens and nonpathogens—adhere to the epithelial surface and colonize. If these attached microbes are pathogens, it sets the stage for infection, invasion, and disease (Figure 25.1). Nonpathogenic microbes that penetrate mucous membranes include large numbers—potentially many billions—of innocuous bacteriophages that contribute to what has been called the “intrabody phageome.” Although poorly understood, the human phageome may play a role in modulating inflammation and other immune responses, which may have implications in controlling autoimmune diseases such as type 1 diabetes and Crohn’s disease (Section 28.1).
Figure 25.6 Bacterial interactions with mucous membranes.

(a) Loose association upon initial exposure. (b) Penetration of mucus leading to adhesion and colonization. (c) Further colonization leading to biofilm formation.
Growth of the Microbial Community: An Example from Human Dental Caries
Infection requires growth of the pathogen after it has attached to and colonized a surface (Figures 25.1 and 25.6), and the actual disease process may not be the result of a single type of microbe but of a community of interacting microorganisms. An example of this is found in the oral microbial disease dental caries (tooth decay), where attachment and infection have been well studied as models of these key events in the disease process.
Even on a freshly cleaned tooth surface, acidic glycoproteins from the saliva form a thin organic film several micrometers thick; this film provides an attachment site for bacterial cells, and oral streptococci quickly colonize it. These include in particular the two Streptococcus species most often implicated in tooth decay: S. sobrinus and S. mutans. Both of these organisms produce a capsule (Section 25.1). The S. sobrinus capsule contains adhesins specific for host salivary glycoproteins (**Figure 25.7a,*b***), whereas S. mutans resides in crevices and small fissures where it relies on dextran—a strongly adhesive exopolysaccharide—that it produces to secure cells to the tooth and gum surface (Figure 25.7c, d). Both S. sobrinus and S. mutans are lactic acid bacteria (Section 16.6) that ferment glucose to lactic acid, the agent that destroys tooth enamel. However, the trigger for decay activities is sucrose (table sugar), since it is sucrose that allows these species to produce the dextran exopolysaccharide and capsules necessary for attachment and colonization.
Figure 25.7 Cariogenic *Streptococcus* spp. and dental plaque.

(a) The low-magnification micrograph shows predominantly streptococcal cell morphology embedded in dental plaque. The species Streptococcus sobrinus (arrows) appears darker from a specific staining technique. (b) Higher-magnification micrograph showing a region in the plaque with S. sobrinus cells (dark, arrow). Note the extensive capsule (lighter grey area) surrounding the S. sobrinus cells. (c) Light micrograph of a Streptococcus mutans culture showing the characteristic cell chains of streptococci. (d) Scanning electron micrograph of the sticky dextran material that holds cells together in filaments. Individual cells of both S. sobrinus and S. mutans are about 1 μm in diameter.
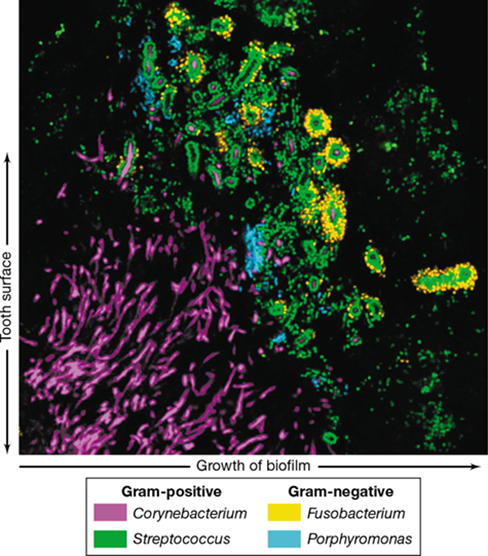
Extensive bacterial growth of these oral streptococci results in a thick biofilm called dental plaque (Figure 25.7). Using phylogenetic probes, it has been possible to more thoroughly explore the microbial diversity of dental plaque, and it is clear that the two Streptococcus species are not the entire story. Many other gram-positive and gram-negative Bacteria are present in plaque, including species of Corynebacterium, Porphyromonas, Leptotrichia, Neisseria, the filamentous anaerobe Fusobacterium, and many others (Figure 25.8). Moreover, the different species likely play specific structural and functional roles in mature dental plaque. This can be seen in FISH-stained (Section 19.5) sections of plaque, where filamentous streamers of cells of Corynebacterium attached to a thin biofilm on the tooth surface anchor cells of Streptococcus and other bacteria a short distance away from the tooth surface (Figure 25.8). Such an arrangement probably allows Streptococcus cells to extend out from the tooth surface into regions of the oral cavity where saliva, sugars, and other nutrients are more abundant.
Figure 25.8 Bacterial diversity of dental plaque.

Confocal micrograph of a FISH-stained (Section 19.5) section through human dental plaque using a suite of phylogenetic probes containing different fluorescent tags. Color matching to specific groups is shown below the photo. The tooth surface would be on the left with the filamentous Corynebacterium species (purple) containing attached streptococci (green) flaring out from an attachment site on the tooth surface.
Dental plaque is thus a complex mixed-culture biofilm composed of several different genera of Bacteria and their accumulated products. A few Archaea are also present in dental plaque, primarily methanogenic species such as Methanobrevibacter oralis. As dental plaque accumulates, the microbiota secrete locally high concentrations of lactic acid that decalcifies tooth enamel, resulting in dental caries. Tooth enamel is strongly calcified tissue, and the ability of microbes to invade this tissue plays a major role in the extent of dental caries and related, often more serious, oral pathologies, including gingivitis and periodontitis, bacterial diseases that erode the tooth-supporting gum and bone tissues.
Invasion and Systemic Infection
In the case of dental caries, the bacterial infection primarily resides on the tooth and gum surfaces. By contrast, in most infectious diseases, the pathogen must invade past the tissue surface in order to promote disease. Invasion is the ability of a pathogen to enter into host cells or tissues, spread, and cause disease. Some pathogens remain localized after initial entry, multiplying and invading at a single focus of infection, such as the boil that may arise from Staphylococcus skin infections (Section 31.9). However, sometimes the pathogens enter the bloodstream, from where they can travel to distant parts of the body. Depending on the pathogen and the condition of an individual and that individual’s immune system, the presence of bacteria in the blood can have mild or highly severe consequences.

The mere presence of bacteria in the blood is called bacteremia; this condition is typically self-limiting and asymptomatic, as the bacterial cells do not grow in the bloodstream, and the immune system quickly removes them. By contrast, in septicemia bacteria multiply in the bloodstream, and the organism spreads systemically from an initial focus and produces toxins or other poisonous substances. Septicemia usually begins as an infection in a specific organ such as the intestine, kidney, or lung, and then spreads rapidly throughout the body from there. Septicemia is typically associated with major symptoms and may lead to massive inflammation, culminating in septic shock (sepsis) and death. Viremia is the term used to describe viruses present in the bloodstream, and measles, a highly infectious disease in those not vaccinated (Section 31.6), is a good example of a systemic viremia.
A pathogen that causes disease in a given host can trigger mild or severe outcomes depending on its inherent capacity to elicit disease. We consider the principles that govern these capacities now with a focus on the important infectious disease concept called virulence.
Check Your Understanding
At what body sites do pathogens typically attach and colonize?
How does the capsule of Streptococcus mutans assist in the formation of dental caries?
Which is the more serious condition, bacteremia or septicemia, and why?
25.3 Pathogenicity, Virulence, and Virulence Attenuation
25.3 Pathogenicity, Virulence, and Virulence Attenuation
25.3 Pathogenicity, Virulence, and Virulence Attenuation
Unique properties of each pathogen contribute to its pathogenicity, the overall ability to cause disease. The measure of pathogenicity is called virulence, the relative ability of a pathogen to cause disease. Pathogenicity and virulence are not uniform properties of a given pathogen and can differ dramatically between different strains of the same bacterial species or virus. Highly virulent strains of a given pathogen tend to emerge every so often and when they do, they often trigger a particularly severe course of disease that is rapid and widespread, potentially resulting in an epidemic or pandemic (Chapter 30). The virulence of a given pathogen depends on a number of factors including its relative abilities to adhere, colonize, and invade (Figure 25.1), as well as its arsenal of virulence factors.
Virulence
Virulence is the net outcome of host–pathogen interactions, a dynamic relationship between the two organisms influenced by ever-changing conditions in the pathogen, the host, and the environment. Host damage in an infectious disease is mediated by virulence factors, toxic or destructive substances produced by the pathogen that directly or indirectly enhance invasiveness and host damage by facilitating and promoting infection. The second part of this chapter is devoted to major virulence factors.
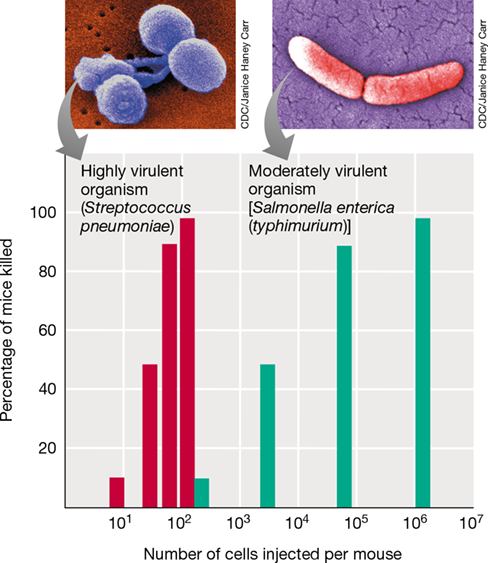
Virulence is a quantifiable entity, especially if a pathogen is lethal and an experimental animal model is available. For example, the LD50 (LD stands for “lethal dose”) is defined as the number of cells of a pathogen (or virions, for a viral pathogen) that kills 50% of the population of the host organism in a test group. Highly virulent pathogens frequently show little difference in the number of cells required to kill 100% of a test group as compared with the LD50. To illustrate this, recall the foundational work of the British microbiologist Frederick Griffith (Section 1.14 and Figure 1.38). Griffith worked with the gram-positive bacterium Streptococcus pneumoniae and discovered that strains of S. pneumoniae that contained a capsule (“smooth” strains because they formed smooth colonies on plates) were highly virulent for mice, whereas mutant derivatives lacking a capsule (“rough” strains) were not. The S. pneumoniae capsule is the primary virulence factor of this bacterium because it helps the bacterium evade immune surveillance. Griffith’s key discovery was that the smooth phenotype could be transferred to rough cells by treating rough cells with an extract from smooth cells (Figure 1.38). This was the first experimental example of transformation, a bacterial genetic transfer process (Section 9.6), and the transforming constituent in the extract was later shown (by other scientists) to be DNA.
Griffith’s choice of experimental organism was fortuitous because S. pneumoniae is both readily transformable and highly virulent for mice. Only a few cells of an encapsulated strain of S. pneumoniae can establish a fatal infection and kill all mice in a test population. As a result, the LD50 for S. pneumoniae in mice is not proportional to the number of cells delivered (Figure 25.9). By contrast, the number of cells of a less virulent pathogen, such as the gram-negative enteric bacterium Salmonella enterica (typhimurium), necessary to kill all of the mice in the test population is about 10,000-fold greater than the highly virulent S. pneumoniae cells, and the LD50 is proportionally related to the number of cells of the pathogen injected into the mice (Figure 25.9).
Figure 25.9 Microbial virulence.

Differences in microbial virulence demonstrated by the number of cells of Streptococcus pneumoniae (red bars) and Salmonella enterica (typhimurium) (green bars) required to kill mice. A colorized scanning electron micrograph of each bacterium is shown above its respective graph.
Mastering Microbiology
There are many examples of highly virulent human pathogens, especially among viruses. For example, some strains of the influenza virus are so highly virulent that only a few virions can initiate disease even though mortality rates are typically low. Ebola virus is also highly virulent, and the tiny inoculum necessary to initiate disease often results in a fatal infection. By contrast, the bacterial pathogen Vibrio cholerae (which causes cholera) is not especially virulent, as a large inoculum of this intestinal pathogen is necessary to initiate disease (Section 33.3).
Attenuation
The virulence of a pathogen can change. Attenuation is the decrease or loss of virulence of a pathogen (Figure 25.10). When pathogens are kept in laboratory culture rather than isolated from diseased animals, their virulence often decreases, or may be completely lost. Strains that have either a reduced virulence or are no longer virulent are said to be attenuated. Attenuation is thought to occur because nonvirulent or weakly virulent mutants grow faster than virulent strains in laboratory media where virulence has no selective advantage. After successive transfers in fresh media, such mutants are therefore selectively favored. However, if an attenuated culture is reinoculated into an animal, the organism may regain its original virulence, especially with continued in vivo passage as more-virulent strains are naturally selected. In some cases, though, the loss of virulence is permanent. For example, if a deletion mutation led to a major modification of a required receptor molecule (Figure 25.2b) or to the inability to produce a key virulence factor, such as the production of a toxin or invasive enzyme, then the mutant strain would be permanently attenuated.
Figure 25.10 Attenuation of virulence in vaccine production.

To produce the yellow fever vaccine, the isolated virus is attenuated by dozens of passages in hosts that are increasingly dissimilar to the original human host, with the final preparation occurring in embryonated chicken eggs. With each successive passage, the virus gradually becomes more adapted to its new host, while simultaneously becoming less virulent to the original human host. When introduced to a susceptible individual, the weakened viral strain elicits a protective immune response to yellow fever without causing symptoms of the disease itself.
Attenuated strains of various pathogens are valuable to clinical medicine because they are often used for the production of vaccines, especially viral vaccines. This principle was first demonstrated in the 1880s by Louis Pasteur with his remarkable development of the first rabies vaccine (Section 1.11 and Figure 1.32a), and since that time, attenuated viral strains have also been employed in the production of vaccines for measles, mumps, rubella, chicken pox/shingles, and yellow fever (Figure 25.10). Although attenuated viruses are “live” in the sense that, unlike “killed” strains, they are capable of replication and could in principle become virulent once again, properly attenuated virus vaccines (those free of any unattenuated virions) typically show greater efficacy and generate an overall stronger immune response than do killed virus vaccines.
Check Your Understanding
What are virulence factors? How can the LD50 test be used to define virulence of a pathogen?
What circumstances can contribute to attenuation of a pathogen?
25.4 Genetics of Virulence and the Compromised Host
25.4 Genetics of Virulence and the Compromised Host
25.4 Genetics of Virulence and the Compromised Host
The virulence of a bacterial pathogen and the eventual outcome of an infectious disease are the net result of genetic and physiological features of both the pathogen and the host. In the case of the host, a pathogen may infect a healthy, well-rested young adult or an individual compromised by an ailment, such as a physiological condition (old age, hospitalization, immune suppression), an ongoing infectious disease (for example, acquired immunodeficiency syndrome [AIDS] caused by the human immunodeficiency virus [HIV]), or a genetic disease (for example, cystic fibrosis). The outcome of infection—health or disease—may be very different in these different individuals, even if they are infected by the same strain of a viral or bacterial pathogen.
The virulence of a pathogen may be encoded by firmly entrenched chromosomal genes or by highly mobile genetic elements. For example, the gram-negative bacterium Bordetella pertussis, the causative agent of whooping cough (pertussis, Section 31.3), makes several toxins including pertussis toxin, a potent AB-type exotoxin (Section 25.6); collectively, these toxins trigger the symptoms of whooping cough. Other species of Bordetella do not make pertussis toxin, and the chromosomal gene encoding pertussis toxin does not readily move from B. pertussis to other species. But in contrast to B. pertussis, some bacterial pathogens routinely exchange genes encoding virulence factors with different bacterial species or even genera, and thus highly related versions of their most potent weapons may appear in several different pathogens. Salmonella is a well-studied example of the genetic transfer of virulence factors, and we focus on this bacterium now.
Virulence in *Salmonella*: Pathogenicity Islands and Plasmids
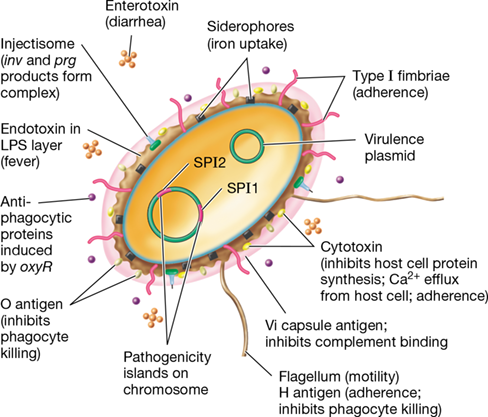
Salmonella species infect humans, leading to various gastrointestinal illnesses (Section 33.10), and they encode a large number of virulence factors that are important in disease. These include type I fimbriae (Section 25.1) to facilitate attachment of cells to gastrointestinal tissues; several different classes of exotoxins (Section 25.6); antiphagocytic proteins that block engulfment of bacterial cells by host phagocytes; proteins that promote survival if the bacterium does get phagocytosed; siderophores, organic molecules that bind iron tightly and, in pathogenic bacteria, allow the bacteria to outcompete host sequestration systems for iron; and endotoxin (Section 25.8). With the exception of endotoxin, many of these virulence factors are encoded by genes present on mobile DNA rather than on the cell’s chromosome (Figure 25.11).
Figure 25.11 Virulence factors in *Salmonella*.

Factors important for virulence and the development of pathogenesis in this gram-negative enteric pathogen are shown. Genes encoding many of the factors reside on the pathogenicity islands or plasmids.
Several genes that encode these virulence factors in Salmonella and related gram-negative pathogens, such as pathogenic strains of Escherichia coli (Section 33.11), are found clustered together on the chromosome as pathogenicity islands. Salmonella pathogenicity island 1 (SPI1) is a cluster of genes that encodes over 10 distinct proteins that promote virulence and invasion. One of these is invH, a gene encoding a surface adhesion protein (Section 25.1). Several inv genes encode proteins important for trafficking of virulence factors. For example, the InvJ regulator protein controls assembly of structural proteins InvG, PrgH, PrgI, PrgJ, and PrgK, which form a type III secretion system called the injectisome, an organelle in the bacterial envelope that allows for the direct transfer of virulence proteins into host cells through a needle-like assembly (Section 6.13 and Figures 6.43 and 6.44).
A second Salmonella pathogenicity island, SPI2, contains genes that are responsible for causing more systemic than localized disease and resistance to host defenses. In addition, several plasmid-encoded virulence factors, such as antibiotic resistance genes encoded on R plasmids (Section 6.2), can spread between Salmonella species and other genera of enteric bacteria. Pathogenicity islands and R plasmids allow for the facile and rapid transfer of virulence factors. It is thus not uncommon for genes encoding factors in one pathogen to be similar, if not identical, to those in another because of transfer of parts or all of the islands or plasmids between species by horizontal gene exchange (Chapter 9).
In the well-known opportunistic pathogen (see next subsection) Pseudomonas aeruginosa, pathogenicity islands can also contain genes encoding antibiotic resistance. As for Salmonella, many cases of multiple antibiotic resistance in P. aeruginosa are linked to plasmids. However, in some strains of P. aeruginosa, genomic islands containing transposable elements (Section 9.11) are present. By transposition these islands have “captured” multiple antibiotic resistance genes and can then disseminate them to other organisms; the genomic islands are present in P. aeruginosa in place of or in addition to resistance plasmids. Regardless of how they are encoded, these strains have become resistant to virtually all of the clinically useful antibiotics that have traditionally been used to control P. aeruginosa infections. This is a particularly serious problem for the compromised host and in the hospital environment, and we consider these issues now.
Mastering Microbiology
Art Activity: Figure 25.10 Virulence factors in Salmonella
The Compromised Host
Some individuals are simply more susceptible to infection than others for reasons that have little to do with the virulence of the pathogen. These so-called compromised hosts are individuals in which one or more mechanisms of resistance to disease are inactive and in whom the probability of infection is therefore increased. Many hospital patients with noninfectious diseases (for example, cancer and heart disease) acquire microbial infections more readily because they are compromised hosts. Such healthcare-associated infections (also called nosocomial infections; Section 29.2) affect up to 2 million individuals each year in the United States, with a nearly 5% mortality rate. Invasive medical procedures such as catheterization, hypodermic injection, spinal puncture, biopsy, and surgery may unintentionally introduce microorganisms into the patient. The stress of surgery and the anti-inflammatory drugs given to reduce pain and swelling can also reduce host resistance.
Some factors can compromise host resistance outside the hospital, including lifestyle choices that affect major organs of the body, such as intravenous drug usage, tobacco use, and excessive consumption of alcohol, or genetic diseases that eliminate parts of the immune system (Section 28.2). People that are physically compromised for any of a number of reasons may be more susceptible to infections, not only because they are physically weakened but also because their living conditions and lifestyle choices may put them in more continual contact with infectious agents. For example, infection with the human immunodeficiency virus (HIV) predisposes an individual to infections from opportunistic pathogens, microbes that cause disease only in the absence of normal host resistance. HIV causes AIDS by destroying a specific class of immune cell, the CD4 T lymphocytes (Section 31.15), which are key to an effective immune response. The reduction in CD4 T cells reduces immunity, and an opportunistic pathogen—one that does not cause disease in a healthy, uninfected host—can then cause serious disease or even death. Individuals with immunodeficiencies from underlying genetic causes rather than infection are also more susceptible to opportunistic infections because part of their immune system is either nonfunctional or suboptimal.
The outcome of an infectious disease thus depends on several factors, both host and pathogen related. Two individuals exposed to the same pathogen in the same way may well show different outcomes. But once an infection has proceeded to the actual stage of disease, the symptoms that appear are due to harmful products of the pathogens, and we turn our attention to these now.
Check Your Understanding
What major virulence factors are produced by Salmonella?
What is an opportunistic pathogen? What steps can a person take to help avoid opportunistic infections?
II Enzymes and Toxins of Pathogenesis
Many pathogenic microbes increase their competitiveness by synthesizing potent products—enzymes and toxins—that allow them to access nutrients in the host. These virulence factors may help the pathogen in initial invasion, be the final result of successful colonization, or be secreted into food or other nonliving substances later ingested by the host.
Bacterial pathogens damage host tissues (or the entire host) in two major ways: (1) by secreting tissue-destroying enzymes and (2) by secreting or shedding toxins that target specific host tissues or the entire host. In contrast to bacterial pathogens, most viral pathogens damage host tissues by lysing cells directly, although some viruses are nonlytic and instead introduce genes into host cells that may eventually harm the host (Section 5.7).
We turn our focus now to the enzymes and toxins of well-studied pathogenic bacteria, contrasting their potency and modes of action. Some of these virulence factors cause only minor disease symptoms, whereas others are among the most poisonous substances known.
25.5 Enzymes as Virulence Factors
Following adherence, colonization, and infection by a pathogen, invasiveness requires the breakdown of host tissues and access to nutrients released from host cells. In many classical bacterial pathogens, this is accomplished through the activity of enzymes that attack and destroy cells in one type of tissue or another (Table 25.1).
Table 25.1 Enzyme virulence factors of some well-known gram-positive bacterial pathogens

aThe activities of coagulase, hyaluronidase, and streptokinase are depicted in Figure 25.12.
Tissue-Destroying Enzymes
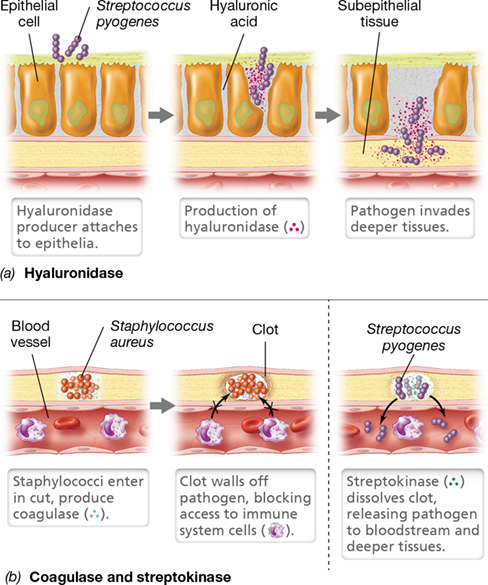
Many virulence factors are enzymes. For example, streptococci, staphylococci, and certain clostridia produce hyaluronidase (Table 25.1), an enzyme that promotes spreading of organisms in tissues by breaking down the polysaccharide hyaluronic acid. Among other functions, hyaluronic acid is a component of the extracellular matrix and functions as a type of “intercellular cement” in animal connective tissues, helping to maintain the organization of individual cells into tissues. The activity of hyaluronidase causes host cells to slough apart, allowing pathogens at an initial colonization site to spread between host cells to attack subsurface tissues (**Figure 25.12*a***). Similarly, the clostridia that cause gas gangrene produce collagenase, an enzyme that destroys collagen (a major protein of connective tissues in muscle and other body tissues); collagenase enables these bacteria to gain access to deeper host tissues and spread through the body. Recall that clostridia are anaerobes, and colonizing deeper tissues allows them to reach less oxic conditions and provides a ready source of nutrients from destroyed tissues (gangrene clostridia are typically proteolytic species, Section 16.8 and Section 32.9). Many pathogenic streptococci and staphylococci also produce proteases, nucleases, and lipases that degrade host proteins, nucleic acids, and lipids, respectively (Table 25.1).
Figure 25.12 Activity of some enzyme virulence factors.

(a) Hyaluronidase. (b) Coagulase (left) and streptokinase (right). Along with some other bacterial pathogens, virulent strains of Streptococcus pyogenes typically produce hyaluronidase and streptokinase, and virulent strains of Staphylococcus aureus typically produce coagulase.
Two virulence factors are enzymes that affect fibrin, the insoluble blood protein that triggers blood clots, but the activities of the enzymes yield opposing results (Figure 25.12b). Blood clotting is triggered by tissue injury and functions not only to stop blood loss but also, in the case of a bacterial infection, to isolate the pathogen, limiting the infection to a local region. Some pathogens counter this host protective mechanism by producing fibrinolytic enzymes, such as streptokinase produced by Streptococcus pyogenes. This bacterium is often associated with pus-forming wounds and secretes streptokinase to dissolve fibrin clots and make further invasion possible (Table 25.1, Figure 25.12b). Streptokinase specifically activates the host to produce plasmin, an enzyme that degrades fibrin blood clots. Because of this powerful activity, streptokinase also has a medically beneficial function. The protein is marketed as a pharmaceutical and administered intravenously to dissolve clots for conditions in which normal blood flow is blocked by blood clots, such as from heart attacks, deep vein thromboses, or embolisms.
Mastering Microbiology
Art Activity: Figure 25.11a Activity of some enzyme virulence factors
In contrast to the fibrin-destroying activity of streptokinase, some pathogens produce enzymes that actually promote the formation of fibrin clots. These clots protect the pathogen from host responses. For example, coagulase (Table 25.1), produced by virulent Staphylococcus aureus, converts fibrinogen to fibrin, resulting in the clotting of blood and the formation of fibrin surrounding the S. aureus cells; this blanket of fibrin protects the S. aureus cells from attack by cells of the host’s immune system (Figure 25.12b). The fibrin matrix produced as a result of coagulase activity may also account for the localized nature of many staphylococcal infections, as is typically seen in boils and pimples (Section 31.9). Coagulase-positive S. aureus strains are typically more virulent than coagulase-negative strains, a likely reflection of the former’s ability to evade innate immune responses such as phagocytosis (Chapter 26) and continue growth and tissue destruction for a longer period.
Enzyme Activities at the Host’s Mucosal Surface
Host mucosal surfaces are bathed in immune substances including enzymes such as lysozyme, an enzyme that cleaves the peptidoglycan of bacterial cells and promotes their osmotic lysis (Section 2.3). Virulence factors produced by the gram-positive bacterium Enterococcus faecalis—a major cause of bacteremia, surgical wound infections, and urinary tract infections—subvert the protective role of lysozyme by altering the structure of the bacterium’s peptidoglycan such that lysozyme can no longer recognize its substrate.
Antibodies are also present on mucosal surfaces, in particular a class of antibody called IgA (Section 27.3). These “secretory antibodies,” as they are called, help prevent pathogen adherence to host tissues (Section 25.1). However, certain pathogenic bacteria counter this protective role by producing enzymes that specifically cleave IgA (IgAases), rendering this host defense useless; Neisseria species such as N. gonorrhoeae (gonorrhea) and N. meningitidis (meningitis) are particularly notorious in this regard.
We thus see that pathogens can produce enzymes both as offensive weapons—to destroy host tissues—and as defensive weapons—to destroy or inactivate defensive weapons of the host. Both strategies accomplish a similar objective: The invasiveness of the pathogen is increased, and this allows it to ultimately extract more resources from its host.
Check Your Understanding
Identify host factors that limit or accelerate infection of a microorganism at selected local sites.
How do streptokinase and coagulase promote bacterial infection and invasion?
What is an IgAase, and why would a bacterial pathogen produce one?
25.6 AB-Type Exotoxins
Toxicity is the ability of an organism to cause disease by means of a toxin that inhibits host cell function or kills host cells (or the host itself). Exotoxins are toxic proteins secreted by the pathogen as it grows. These toxins travel from a site of infection and cause damage at distant sites. Some exotoxins are enterotoxins, toxic proteins whose site of action is the small intestine, generally causing secretion of fluid into the intestinal lumen, resulting in vomiting and diarrhea. Exotoxins fall into three categories in terms of their mechanism: AB toxins, cytolytic toxins, and superantigen toxins. In this section we consider the AB toxins, and in Section 25.7 we focus on cytolytic and superantigen toxins.
Mastering Microbiology
Art Activity: Figure 25.12 The activity of diphtheria toxin
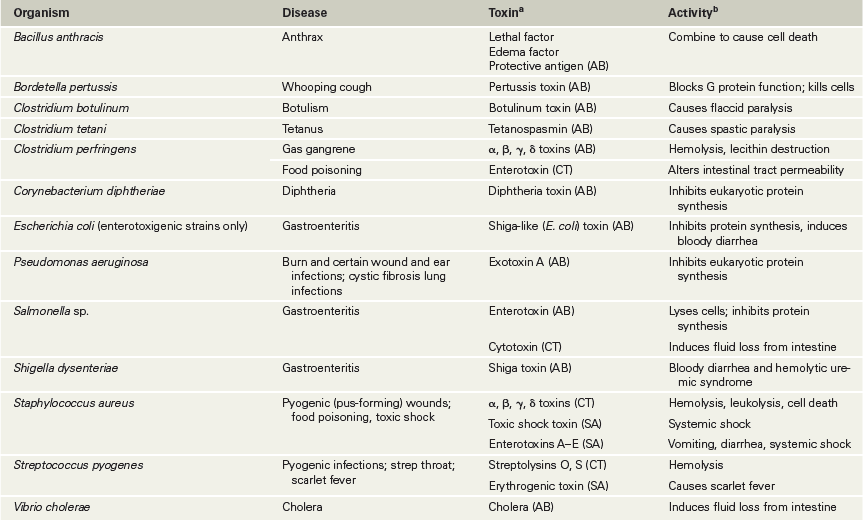
As the name implies, AB toxins consist of two subunits, A and B. The B component binds to a host cell surface molecule, facilitating the transfer of the A subunit across the cytoplasmic membrane, where it damages the cell. Some of the best-known and most potent exotoxins are AB toxins, including those expressed in diphtheria, tetanus, botulism, and cholera (Table 25.2).
Table 25.2 Some classic exotoxins and cytotoxins produced by human bacterial pathogens

bSee Figures 25.13–25.18 for the mode of action of some of these toxins.
Diphtheria Exotoxin: Blockage of Protein Synthesis
The diphtheria toxin produced by the aerobic gram-positive bacterium Corynebacterium diphtheriae is an AB toxin and an important virulence factor of the pathogen (Section 31.3). Diphtheria toxin inhibits protein synthesis in eukaryotic cells. Rats and mice are relatively resistant to diphtheria toxin, whereas humans are very susceptible, with only a single molecule of toxin sufficient to kill a cell. Diphtheria has a significant mortality rate, especially in the young, and death ensues from tissue destruction in vital organs such as the heart and liver as diphtheria toxin blocks protein synthesis.
Cells of C. diphtheriae secrete diphtheria toxin as a single polypeptide. One component of the toxin, subunit B, specifically binds to a host cell receptor protein on eukaryotic cells, the heparin-binding epidermal growth factor (Figure 25.13). After binding, proteolytic cleavage between subunit B and the remaining portion of the protein, subunit A, allows subunit A to move across the host cytoplasmic membrane into the cytoplasm. Here subunit A disrupts protein synthesis by blocking transfer of an amino acid from tRNA to growing polypeptide chains. Diphtheria toxin specifically inactivates elongation factor 2 (EF-2), a protein that functions in growth of the polypeptide chain, by catalyzing the attachment of adenosine diphosphate (ADP) ribose from NAD+. Following ADP-ribosylation, the activity of the modified EF-2 decreases dramatically, and protein synthesis stops (Figure 25.13).
Figure 25.13 The activity of diphtheria toxin.

Diphtheria toxin, an AB toxin produced by cells of Corynebacterium diphtheriae, binds to the host cytoplasmic membrane by way of its B subunit. Cleavage of the toxin allows the A subunit to enter the cell where it catalyzes ADP-ribosylation of elongation factor 2 (EF-2), a key factor in translation. The modified elongation factor no longer binds to the ribosome, resulting in the cessation of protein synthesis and host cell death. Inset photo: Light micrograph of Gram-stained cells of Corynebacterium diphtheriae. A single cell is about 0.75 μm in diameter.
Diphtheria toxin is not encoded by the bacterium but instead by a viral gene called tox present in the genome of the lysogenic bacteriophage β. Lysogenic phages are those whose genomes have become integrated into their host’s chromosome (Section 5.6). Toxigenic, pathogenic strains of C. diphtheriae are infected with phage β and hence produce the toxin. Nontoxigenic, nonpathogenic strains of C. diphtheriae can be converted to pathogenic strains by infection with phage β, a process called phage conversion (Section 9.7).
Exotoxin A of Pseudomonas aeruginosa functions similarly to diphtheria toxin, also modifying EF-2 by ADP-ribosylation. The enterotoxin produced by Shigella dysenteriae, called Shiga toxin, and the Shiga-like toxin produced by enteropathogenic Escherichia coli O157:H7 (Section 33.11) are also AB toxins (Table 25.2). Shiga and Shiga-like toxins target cells of the small intestine near where the pathogen colonized, shutting down protein synthesis. This leads to cell death, bloody diarrhea, and hemolytic uremic syndrome, a kidney disease that can trigger kidney failure, especially in children.
Neurological Exotoxins: Botulinum and Tetanus Toxins
Clostridium botulinum and Clostridium tetani are endospore-forming bacteria commonly found in soil and which cause the serious and potentially fatal diseases botulism and tetanus, respectively; both diseases are caused by the secretion of highly poisonous AB exotoxins that function as neurotoxins (Sections 32.9 and 33.9). Neither C. botulinum nor C. tetani is highly invasive, and therefore pathogenicity is almost exclusively due to neurotoxicity.
C. botulinum sometimes grows directly in the intestine, causing infant or wound botulism. Most frequently for adults, however, botulism results from cells of C. botulinum growing and producing toxin in improperly preserved foods, such as home-canned vegetables. Thus, infection and growth of the pathogen in the body are unnecessary. Botulinum toxins consist of seven related but distinct AB toxins, and of these, at least two are encoded on lysogenic bacteriophages specific for C. botulinum. Botulinum toxin and tetanus toxin both block the release of neurotransmitters that control muscle activities, but as we will see, the mode of action and symptoms of botulism and tetanus are quite distinct.
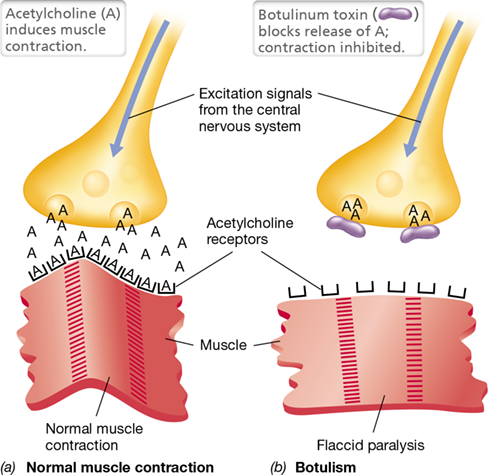
Normal transmission of a nerve impulse to a muscle cell at a neuromuscular junction requires interaction of the neurotransmitter acetylcholine with a muscle receptor (**Figure 25.14*a***). Botulinum toxins, which consist of both a heavy chain that binds the neuronal surface and a light-chain endopeptidase, prevent this interaction. After binding the motor end plate—the presynaptic membrane on the terminus of the stimulatory motor neuron—the toxin enters the cell by endocytosis. The heavy chain of the toxin then translocates the light chain across the vesicle membrane into the cytoplasm of the neuron. Depending upon which of the seven forms of botulinum toxin has entered the cell, the proteolytic light chain then cleaves any of several proteins that coordinate release of the neurotransmitter into the synaptic space, thus blocking the excitatory acetylcholine signal and preventing muscle contraction (Figure 25.14b). This is recognized as flaccid paralysis in a botulism victim, and it can lead to death by suffocation if the diaphragm muscles are severely affected.
Figure 25.14 The activity of botulinum toxin.

(a) Upon stimulation of peripheral and cranial nerves, acetylcholine (A) is normally released from vesicles at the neural side of the motor end plate. Acetylcholine then binds to specific receptors on the muscle, inducing contraction. (b) Botulinum toxin acts at the motor end plate to prevent release of acetylcholine from vesicles, resulting in a lack of stimulus to the muscle fibers, irreversible relaxation of the muscles, and flaccid paralysis.
Mastering Microbiology
Art Activity: Figure 25.15 The activity of cholera enterotoxin
Botulinum toxins are the most potent biological substances known; just one nanogram (10−9 g) of botulinum toxin is sufficient to kill a guinea pig. Thus, it is perhaps surprising that botulinum toxins have proved beneficial in the treatment of chronic pain. Because chronic pain, such as that associated with ongoing back and neck pain or frequent migraines, is often caused by neuropathy or stress-induced muscle tension, low-dose injections of dilute botulinum toxin (Botox®) can relax the affected tissues and effectively mitigate pain. This is especially useful considering that botulinum toxin treatments are not linked to the unwanted side effects that commonly accompany traditional drug treatments for these conditions.
In contrast to C. botulinum, C. tetani grows in the body in deep wounds that become anoxic, such as punctures. C. tetani cells rarely leave the wound where they were first introduced, growing relatively slowly at the wound site. On contact with the nervous system, tetanus toxin, called tetanospasmin, is transported through the motor neurons to the spinal cord, where it binds specifically to ganglioside lipids at the termini of the inhibitory interneurons (Figure 25.15). The inhibitory interneurons normally work by releasing an inhibitory neurotransmitter, typically the amino acid glycine, which binds to receptors on the motor neurons. Glycine from the inhibitory interneurons then stops the release of acetylcholine by the motor neurons and inhibits muscle contraction, allowing relaxation of the muscle fibers. However, if tetanus toxin blocks glycine release, the motor neurons cannot be inhibited, resulting in the continual release of acetylcholine and uncontrolled contraction of the muscle fibers (Figure 25.15b). Thus, in contrast to botulinum toxin, which prevents muscle contraction (Figure 25.14), tetanus toxin prevents muscle relaxation (Figure 25.15).
Figure 25.15 The activity of tetanus toxin.

(a) Muscle relaxation is normally induced by glycine (G) release from inhibitory interneurons. Glycine acts on the motor neurons to block excitation and release of acetylcholine (A) at the motor end plate. (b) Tetanus toxin binds to the interneuron to prevent release of glycine from vesicles, resulting in a lack of inhibitory signals to the motor neurons, constant release of acetylcholine to the muscle fibers, irreversible contraction of the muscles, and spastic paralysis. For the purpose of illustration, the inhibitory interneuron is shown near the motor end plate, but it is actually in the spinal cord.
The outcome in a case of tetanus is a twitching, spastic paralysis—the hallmark of tetanus (Section 32.9 and Figure 32.23b)—as affected muscles are constantly contracting. If the muscles of the mouth are involved, as is commonly the case, the prolonged contractions restrict the mouth’s movement, resulting in a condition called lockjaw. If the diaphragm is affected, its prolonged contraction may result in death due to asphyxiation.
Cholera Enterotoxin: Intestinal Distress
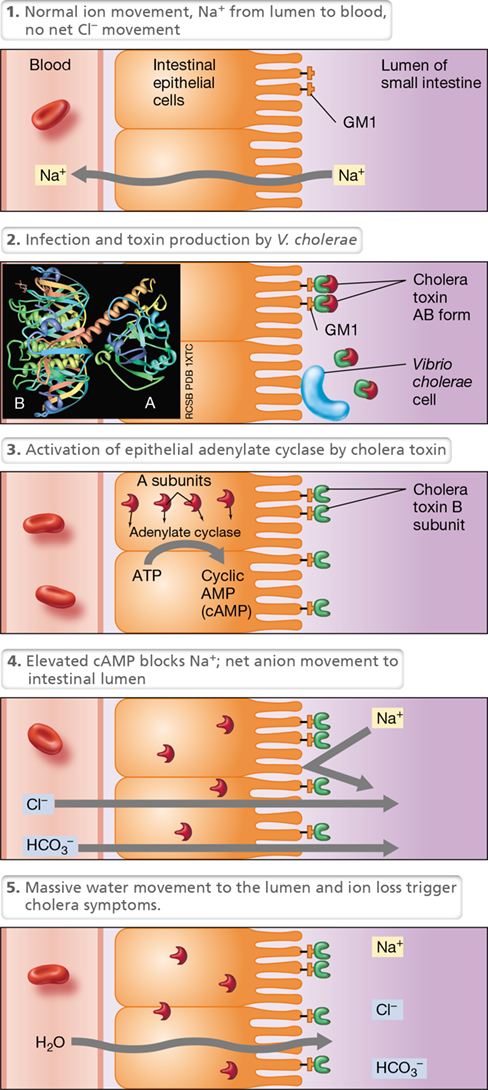
Cholera enterotoxin is an AB-type exotoxin produced by Vibrio cholerae, the causative agent of the waterborne disease cholera (Section 33.3). Cholera is characterized by massive fluid loss from the intestines, resulting in severe diarrhea, life-threatening dehydration, and electrolyte depletion (Figure 25.16). Cholera starts by ingestion of V. cholerae cells from food or water contaminated with human feces. The organism travels to the small intestine, where it colonizes and secretes cholera toxin. In the small intestine, the B subunit of cholera toxin, consisting of five identical monomers, binds specifically to GM1 ganglioside, a complex glycolipid found in the cytoplasmic membrane of intestinal epithelial cells (Figure 25.16).
Figure 25.16 The activity of cholera enterotoxin.

Cholera toxin is an AB enterotoxin that activates a second-messenger pathway, disrupting normal ion flow in the intestine, resulting in potentially life-threatening diarrhea. The thumbnail photo of the three-dimensional structure shows a side view of the toxin, with the separate cell-binding B subunit and the enzymatically active A subunit.
The B subunit targets cholera toxin specifically to receptors in the intestinal epithelium but has no toxicity itself; toxicity is a function of the A subunit, which crosses the cytoplasmic membrane and activates adenylate cyclase, the enzyme that converts ATP to cyclic adenosine monophosphate (cAMP). This molecule is a cyclic nucleotide (Figure 7.22) that mediates several regulatory systems in cells, including ionic balance. The increased cAMP induced by cholera enterotoxin blocks Na+ uptake by small intestine epithelial cells and induces secretion of chloride and bicarbonate (HCO3−) into the intestinal lumen. This change in ion concentrations leads to the secretion of large amounts of water; the rate of water loss into the small intestine is greater than the possible reabsorption of water by the large intestine, resulting in a large net fluid loss and watery diarrhea. If untreated, cholera victims can die within hours of the major onset. However, if lost fluids are replaced with an oral rehydration solution (the main treatment for cholera), the effects of cholera toxin can be neutralized, and a cholera victim can return to normal in just a few days.
A few other enterotoxins, most notably the Shiga-like toxin of enterotoxigenic strains of Escherichia coli (Section 33.11) and enterotoxins of Shigella and Salmonella, are also of the AB type (Table 25.2), and all of these function by inhibiting protein synthesis. This leads to major bouts of diarrhea, typically bloody and foul smelling, and severe dehydration. In addition, some cytolytic enterotoxins are known, and the powerful enterotoxins of Staphylococcus aureus (Table 25.2) are of the superantigen type. We consider these dangerous toxins now.
Check Your Understanding
Are bacterial growth and infection in the host necessary for the production of toxins? Explain and cite examples for your answer.
Why do botulism and tetanus show such opposing symptoms?
25.7 Cytolytic and Superantigen Exotoxins
The pathogenesis of exotoxins such as the cytolytic and superantigen toxins differs from those of the classical AB toxins. Cytolytic and superantigen exotoxins function by destroying host cells, such as from the activity of hemolysins, or by triggering a massive immune response, such as in the case of toxic shock exotoxin, the cause of toxic shock syndrome. As for other exotoxins, however, these proteins are produced by the pathogen to increase its invasiveness and release host cell resources that can benefit the pathogen.
Cytolytic Exotoxins
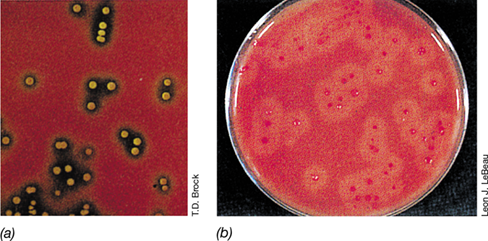
Cytotoxins (also called cytolytic exotoxins) are soluble proteins secreted by a variety of pathogens (Table 25.2). Cytotoxins damage the host cytoplasmic membrane, causing cell lysis and death. Because the lytic activity of these toxins is most easily observed in assays that use red blood cells (erythrocytes), the toxins are often called hemolysins (Table 25.2). However, hemolysins also lyse cells other than erythrocytes. The production of hemolysins can be demonstrated by streaking the pathogen on a blood agar plate (a rich medium containing 5% sterile blood). During growth of the colonies, hemolysin is released and lyses the surrounding red blood cells, releasing hemoglobin and creating a clear area, called a zone of hemolysis, around the growing colonies (**Figure 25.17*a***).
Figure 25.17 Hemolysis.

(a) Zones of hemolysis around colonies of Streptococcus pyogenes growing on a blood agar plate. (b) Activity of lecithinase, a phospholipase, around colonies of Clostridium perfringens growing on an agar medium containing egg yolk, a source of lecithin. Lecithinase dissolves the cytoplasmic membranes of red blood cells, producing cloudy zones of hemolysis around each colony.
Some hemolysins attack the phospholipid of the host cytoplasmic membrane. Because the phospholipid lecithin (phosphatidylcholine) is often used as a substrate, these enzymes are called lecithinases or phospholipases. An example is the α-toxin of Clostridium perfringens, a lecithinase that dissolves membrane lipids, resulting in cell lysis (Table 25.2, Figure 25.17b). Because the cytoplasmic membranes of all organisms contain phospholipids (Section 2.1), phospholipases can destroy bacterial as well as animal cell cytoplasmic membranes. In the case of C. perfringens, a major cause of gas gangrene, the activity of its lecithinase helps destroy tissues and release proteins that are then fermented by the bacterium in energy metabolism. Since C. perfringens lecithinase is secreted immediately after synthesis and is unable to reenter the cell, the enzyme does not affect the phospholipids in C. perfringens cells.
Some hemolysins, however, are not phospholipases. Streptolysin O, a hemolysin produced by streptococci, affects the sterols of the host cytoplasmic membrane. Leukocidins lyse white blood cells and thereby decrease the host immune response. Staphylococcal α-toxin (Figure 25.18 and Table 25.2) destroys both nucleated cells and erythrocytes. To do this, the seven α-toxin subunits first bind to the phospholipid bilayer. The subunits then combine to form into nonlytic heptamers, now associated with the membrane. Each heptamer then undergoes conformational changes to produce a membrane-spanning pore that releases cytoplasmic contents and allows the influx of extracellular materials, thus killing the cell (Figure 25.18).
Figure 25.18 Staphylococcal α-toxin.
Staphylococcal α-toxin is a pore-forming cytotoxin that is produced by growing Staphylococcus aureus cells. Released as monomers, seven identical protein subunits oligomerize in the cytoplasmic membrane of target cells to form a pore, releasing the contents of the cell. In red blood cells, hemolysis occurs, visually indicating cell lysis. The inset image on the top left shows the structure of α-toxin looking down through the pore; each of the seven identical subunits is shown in a different color. The inset photo on the top right is a scanning electron micrograph of S. aureus cells.
Superantigen Exotoxins
The gram-positive bacteria Staphylococcus aureus and Streptococcus pyogenes are major producers of exotoxin superantigens. We consider the mode of action of superantigens in Section 28.2 in the context of the immune response and focus here only on some basic disease symptoms.
Superantigen poisoning can be triggered by certain types of food poisoning (in particular, that caused by the enterotoxins of S. aureus), by toxic shock syndrome, or by pyrogenic fever (fever induced either from an internal source, such as small immune system proteins called cytokines, or from an external substance, such as endotoxin; see Section 25.8). Reactions to superantigen poisoning are severe and can even be fatal in some individuals, particularly those whose immune systems and overall health are weakened from cancer, drug treatments, HIV infection, or old age. Toxic shock syndrome (TSS) is the classic example of the systemic effects of a toxic superantigen and results from exposure to any of a series of exotoxins secreted during infection by certain strains of S. aureus or S. pyogenes (Sections 31.9 and 31.2, respectively).
S. aureus TSS commonly originates as a result of a localized rather than a generalized infection. By contrast, S. pyogenes TSS is typically the result of a systemic infection where bacteremia or septicemia (Section 25.2) is present, and tissue damage including extensive tissue necrosis occurs (Figure 31.10); as a result, mortality rates from S. pyogenes TSS are considerably higher than from S. aureus TSS. In both cases, however, the symptoms of TSS are triggered when the immune system recognizes the superantigen toxin, but rather than activating just a small subset of T lymphocytes (key cells in adaptive immunity, Chapter 27) as usually occurs in the adaptive immune response, a large proportion of the entire T lymphocyte pool becomes activated. It is the structure of the superantigen itself that leads to this overblown immune response, a result of which is widespread release of cell-signaling cytokines and systemic inflammation that leads to hypotension (low blood pressure), intestinal disruption, organ failure, and eventually systemic shock.
In a clinical diagnosis, either staphylococcal or streptococcal TSS is suspected when an individual has a fever of 39 °C or greater, systolic blood pressure of <90 mm Hg, and functional disruption of three or more organ systems, most often gastrointestinal, kidney, and liver. In severe cases of TSS, intensive care hospitalization may be needed with intravenous administration of antibiotics.
Check Your Understanding
Give an example of a cytolytic exotoxin and a superantigen exotoxin, as well as the bacteria that produce each.
How can activity of a hemolytic exotoxin be detected?
25.8 Endotoxins
Endotoxins are the toxic lipopolysaccharides found in the cell walls of most gram-negative Bacteria. Endotoxins are not proteins but are structural components of the gram-negative outer membrane (Section 2.4). In contrast to exotoxins, which are the secreted products of living cells, endotoxins are cell bound and released in toxic amounts only when the cells lyse. The basic properties of exotoxins and endotoxins are compared in Table 25.3.
Table 25.3 Properties of exotoxins and endotoxins

aA toxoid is a modified toxin that is no longer toxic but can still elicit an immune response against the toxin (Section 28.3).
Endotoxin Structure and Biology
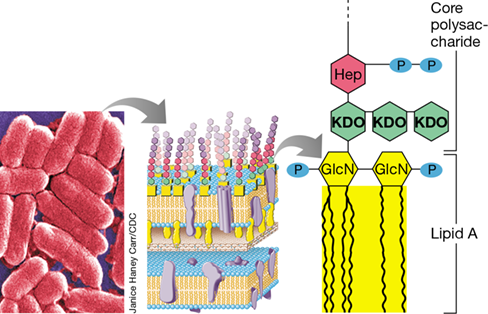
A major component of the gram-negative cell outer membrane is lipopolysaccharide (LPS) (Figures 2.12 and 2.13). LPS consists of three covalently linked subunits: the membrane-distal O-specific polysaccharide, a membrane-proximal core polysaccharide, and lipid A—a phosphoglycolipid and the membrane-anchoring portion of LPS (Figure 25.19). The lipid A portion of LPS is responsible for toxicity, whereas the polysaccharide fraction by itself is nontoxic. The polysaccharide functions to make the entire LPS complex soluble and immunogenic, and thus both the lipid and polysaccharide fractions must be delivered as a unit for toxicity to occur.
Figure 25.19 Endotoxin.

Left to right: Scanning electron micrograph of cells of the gram-negative bacterium Escherichia coli; structure of the gram-negative cell wall including the lipopolysaccharide (LPS) outer membrane; detailed structure of lipid A, the toxic portion of LPS, along with part of the core polysaccharide of LPS.
Endotoxins have been well studied in the bacteria Escherichia, Shigella, and especially in Salmonella, where they are another of the many virulence factors that contribute to pathogenesis (Figure 25.11). The biosynthesis pathway of the toxic component of endotoxin, lipid A, is known and is a highly conserved process among gram-negative bacteria. Nevertheless, not all lipid A is structurally the same, as the molecule can be modified using enzymes that catalyze postsynthesis modifications that control the presence, absence, or number of phosphate groups, and the chemistry and number of fatty acid side chains (Figure 25.19). These subtle but important alterations affect the properties of the LPS molecule and are a virulence strategy that certain pathogens have evolved to either evade recognition by the host immune system or increase the toxicity of the molecule. However, some lipid A alterations affect toxicity in a negative way. For example, the phosphate groups (Figure 25.19) are essential for binding lipid A to animal cell receptors, and although phosphate-free lipid A can evade immune surveillance, its toxicity is greatly reduced.
Endotoxins cause a variety of physiological effects. Fever is an almost universal result of endotoxin exposure because endotoxin stimulates host cells to release cytokines, soluble proteins secreted by certain cells of the immune system that function as endogenous pyrogens, proteins that affect the temperature-controlling center of the brain, causing fever. Cytokines released as a result of endotoxin exposure can also cause diarrhea, increased heart rate (tachycardia), a rapid decrease in the numbers of lymphocytes and platelets, and generalized inflammation (Section 26.8). Other physiological consequences of endotoxin exposure include activation of the complement cascade (complement is a series of immune system proteins, Section 26.9), which also triggers inflammation, and activation of the blood coagulation cascade, which can lead to blood clots and reduced blood flow. Large doses of endotoxin can cause death from hemorrhagic shock and kidney failure.
Although significant virulence factors, endotoxins are generally less toxic than most exotoxins and rarely cause symptoms that can lead to death in an otherwise healthy individual if exposure is from a gastrointestinal source (Table 25.3). For instance, in mice the LD50 (see Figure 25.9) for endotoxin is 200–400 micrograms per animal, whereas the LD50 for botulinum exotoxin is about 25 picograms, about 10 million times less. By contrast, intravenous administration of endotoxin, for example from a heavily contaminated intravenous solution, could have fatal consequences, and we discuss how such faulty solutions can be identified now.
*Limulus* Amoebocyte Lysate Assay for Endotoxin
Because endotoxins induce fever and can trigger other, more serious symptoms, pharmaceuticals such as injectable antibiotics and intravenous solutions must be free of endotoxin. An endotoxin assay of high sensitivity and specificity in widespread use employs lysates of amoebocytes from the horseshoe crab Limulus polyphemus (amoebocytes are mobile cells in the blood and fluids of invertebrates that are analogous to the white blood cells of vertebrates). Endotoxin specifically causes lysis of the amoebocytes (Figure 25.20). In the Limulus amoebocyte lysate (LAL) assay, Limulus amoebocyte extracts are mixed with the solution to be tested. If endotoxin is present, the amoebocyte extract forms a gel and precipitates, causing a change in turbidity. This reaction is measured quantitatively with a spectrophotometer and can detect as little as 10 picograms of LPS in a 1-ml sample.
Figure 25.20 *Limulus* amoebocyte assay for endotoxin.

(a) Normal amoebocytes from the horseshoe crab Limulus polyphemus. (b) Amoebocytes following exposure to bacterial lipopolysaccharide (LPS). LPS induces degranulation and lysis of the cells.
The LAL assay is also used to detect endotoxin in clinical samples such as serum or cerebrospinal fluid, and a positive result provides presumptive evidence for infection by gram-negative bacteria. Drinking water, water used for formulation of injectable drugs, and injectable aqueous solutions are routinely tested using the LAL assay to identify and eliminate endotoxin contamination from gram-negative bacteria. A commercially available assay uses horseshoe crab factor C made by recombinant DNA techniques (factor C is the key protein activated by endotoxin in the LAL assay). Rather than relying on amoebocytes collected from harvested horseshoe crabs, the recombinant protein is just as sensitive but less expensive, allows for a more standardized assay protocol, and is totally free of animal products.
Check Your Understanding
What part of the Escherichia coli cell contains endotoxin? Why do gram-positive bacteria not produce endotoxins?
Why is it necessary to test for endotoxin in water used for injectable drug preparations?
Chapter Review
Go to Mastering Microbiology for videos, animations, practice tests, and more.
I Human–Pathogen Interactions
25.1 If a pathogen gains access to the specific tissues it infects, disease will occur only if it first adheres to those tissues. Although adherence is required to initiate disease, it is not sufficient to initiate disease; colonization, invasion, and production of toxic substances are also required.
Q How is microbial adherence to host tissues facilitated by structures such as capsules and fimbriae?
25.2 Each body region differs chemically and physically from others, and thus provides a variety of selective environments for the growth of certain microbes but not others. Colonization of tissues by a pathogenic microbe followed by growth to form populations sufficient to trigger a biological effect is necessary before disease symptoms appear.
Q What common disease is a model for the study of bacterial attachment and colonization, and which species of bacteria cause the bulk of the damage in this disease?
25.3 The pathogenicity of a pathogen is a function of its virulence, its relative ability to cause disease. Virulence is a quantitative measure that can be assayed in terms of the number of cells (or virions, if a viral pathogen) required to infect or kill 50% of a given population—the LD50. Attenuated pathogens are strains with diminished virulence and are quite useful for preparing vaccines.
**Q What virulence factor, present in Streptococcus pneumoniae but absent from Salmonella enterica, makes S. pneumoniae so highly virulent for mice?**
25.4 The genetics and physiology of both the pathogen and the host affect the outcome of an infectious disease. In Salmonella species, chromosomal islands or conjugative plasmids are present that encode several virulence factors; these mobile genetic elements can quickly spread a suite of virulence factors to other gram-negative bacteria. Susceptibility to an infectious disease is increased in a compromised host.
Q What factors might diminish the ability of a host to fight off an infectious disease?
II Enzymes and Toxins of Pathogenesis
25.5 The virulence of a pathogen depends on the number and kinds of virulence factors it produces. Some pathogenic bacteria produce enzymes that function to either destroy host tissues or disarm host defenses. The activity of these enzymes releases nutrients to support growth of the pathogen and facilitate further invasiveness.
Q Give two reasons why a pathogen might benefit from the secretion of an enzyme that destroys host tissue integrity.
25.6 Exotoxins are toxic proteins and major virulence factors. Each exotoxin affects a specific host cell function. Enterotoxins are exotoxins that affect the small intestine. The clostridial botulinum and tetanus exotoxins are among the most poisonous substances known.
Q Assuming a person was poisoned with either botulinum toxin or tetanus toxin, how could the person’s physical state signal the type of poisoning that had occurred?
25.7 Cytotoxins and superantigens are toxic proteins that lyse host cells and trigger a massive immune response against host tissues, respectively. Hemolysis on a blood agar plate is a classic cytotoxic effect, whereas toxic shock syndrome, a potentially fatal condition, is one result of superantigen activity.
Q Distinguish between the mechanism of cytotoxins and AB toxins and provide one example of each.
25.8 Endotoxins are lipopolysaccharides derived from the outer membrane of gram-negative bacteria. Both the lipid and the polysaccharide components of endotoxin are necessary for toxicity. Symptoms of endotoxin poisoning include fever and intestinal distress.
Q Identify the structural features, origins, and major effects of endotoxins.
Application Questions
Coagulase is a virulence factor for Staphylococcus aureus that acts by causing clot formation at the site of S. aureus growth. Streptokinase is a virulence factor for Streptococcus pyogenes that acts by dissolving clots at the site of S. pyogenes growth. Reconcile these opposing strategies for enhancing pathogenicity.
Although mutants incapable of producing exotoxins are relatively easy to isolate, mutants incapable of producing endotoxins are much harder to isolate. From what you know of the structure and function of these types of toxins, explain the differences in mutant recovery.
Chapter Glossary
the enhanced ability of a microorganism to attach to a cell or surface Adhesins
glycoproteins or lipoproteins covalently bound to the outer layer of the pathogen that function in attachment to host tissues Attenuate
to decrease or eliminate the virulence of a pathogen or virus Bacteremia
a polysaccharide or protein outermost layer, usually rather slimy, present on some bacteria Colonization
the growth of a microorganism after it has gained access to host tissues Dental caries
tooth decay resulting from bacterial infection Dental plaque
a bacterial biofilm found on the teeth and consisting of cells embedded in a matrix of extracellular polymers and salivary products Disease
an injury to a host organism, caused by a pathogen or other factor, that is accompanied by specific signs and symptoms that affect host function Endotoxin
the lipopolysaccharide portion of the outer membrane of most gram-negative Bacteria, which is a toxin when solubilized Enterotoxin
a protein that is released extracellularly by a microorganism as it grows and that produces immediate damage to the small intestine of the host Exotoxin
a protein that is released extracellularly by a microorganism as it grows and that produces immediate damage to the host Infection
an event during which a microorganism not a member of the local microbiota is established and grows in a host, regardless of whether the host is harmed Invasion
the ability of a pathogen to enter into host cells or tissues, spread, and cause disease Mucous membrane
a layer of mucus-covered epithelial cells in the body that communicates with the external environment Mucus
a viscous liquid composed of mucin secreted by specialized epithelial cells that contains water-soluble glycoproteins and proteins that retain moisture and aid in resistance to microbial invasion on mucosal surfaces Opportunistic pathogen
an organism that causes disease only in the absence of normal host resistance Pathogen
the ability of a pathogen to cause disease Septicemia
the ability of an organism to cause disease by means of a preformed toxin that inhibits host cell function or kills host cells Virulence
the relative ability of a pathogen to cause disease Virulence factors
substances or strategies of a pathogen that indirectly or directly enhance invasiveness and host damage by facilitating and promoting infection