24 Microbial Symbioses with Humans
I Structure and Function of the Healthy Adult Gastrointestinal and Oral Microbiomes
II Urogenital Tract and Skin Microbiomes and the Human Viral Microbiome
III From Birth to Death: Development of the Human Microbiome
IV Disorders Attributed to the Human Microbiome
One of the Most Abundant Viruses on Earth Discovered First in the Human Viral Microbiome
Past discoveries of novel viruses have required that the host they naturally infect first be identified and isolated. However, since most bacterial and archaeal diversity is still uncultured, new viral discoveries were few until next-generation sequencing allowed virus-like particles collected from the environment to be characterized directly. Today, viral metagenomics is the primary method of viral discovery and has revealed a vast reservoir of novel genetic diversity, most of which has no precedent in known viruses.
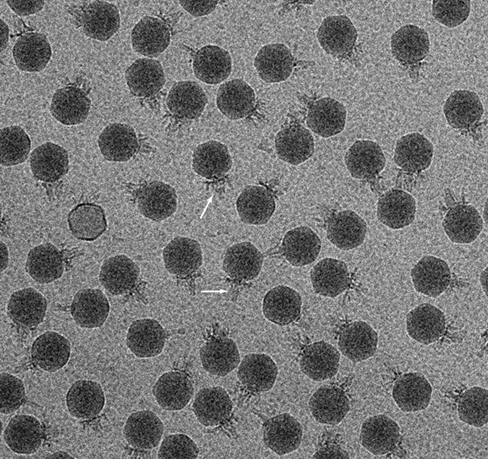
One of the most remarkable findings from metagenomic viral studies thus far has been the discovery of a human gut–associated bacterial virus (bacteriophage) that comprises up to 90% of viral genomic sequences found there. The virus was named crAssphage for the cross-assembly method used to reconstruct its roughly 100-kbp circular DNA genome from metagenomic sequences obtained from different subjects. Genomic sequences from crAssphage were found to match more than one-fifth of all viral genomic sequences recovered from marine, terrestrial, and animal sources. Thus, crAssphage-like viruses are probably one of the most abundant biological entities on Earth! But a remaining mystery was “Whom do crAssphage-like viruses infect?”
The hunt was on using the standard method of screening cultured bacteria for infection by crAssphage. By screening 54 gut species for infection by phage-enriched fecal filtrates, the first crAssphage virus was isolated on the gut bacterium Bacteroides intestinalis; the phage has an icosahedral capsid about 80 nm in diameter (photo) and a short tail (arrows).The crAssphage-like viruses are now known to comprise a large viral family predicted to infect many species of Bacteroidetes in diverse environments.
Since changes in the gut virome are associated with human gastrointestinal abnormalities such as inflammatory bowel disease and malnutrition, we can expect that the isolation of new bacteriophages discovered through metagenomics will reap important rewards for human health.
Source: Shkoporov, A.N., Khokhlova, E.V., Fitzgerald, C.B., et al. 2018. ϕ CrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 9: 4781.
I: Structure and Function of the Healthy Adult Gastrointestinal and Oral Microbiomes
I: Structure and Function of the Healthy Adult Gastrointestinal and Oral Microbiomes
I Structure and Function of the Healthy Adult Gastrointestinal and Oral Microbiomes
Our bodies host tremendous numbers of microorganisms distributed among different body sites, together comprising the human microbiome. Colonization begins at birth, and the assembly, composition, and activities of our gastrointestinal and oral microbiomes in particular greatly influence our health and predisposition to disease.
The human body contains huge numbers of microbes—our microbiome—that our very existence depends on. We begin our consideration of this microbial menagerie with an overview of the human microbiome and then consider the microbiota—the types of organisms present in specific regions of the body.
The clinical benefits of knowing a person’s microbiome seem promising and include the development of biomarkers for predicting predisposition to specific diseases, the design of therapies increasing or decreasing the activities of selected microbial species in particular body sites, personalized drug therapies, and tailor-made probiotics (Section 24.12). However, there is a caveat. Not only is a person affected by his or her microbiota, but a person’s microbiota also responds to his or her activities, health, and diet. Thus, cause and effect relationships are often not immediately obvious and can sometimes be difficult to sort out.
Microbiome studies to date have revealed an incredible diversity of microbial life in and on the human body and have signaled several likely connections between the microbial composition of a body site and the health status of the person. However, at this point most microbial associations with health or disease states are only correlations or are based on animal models; a firm causal relationship between a person’s microbiome and health status has been well established in only a few instances.
24.1 Overview of the Human Microbiome
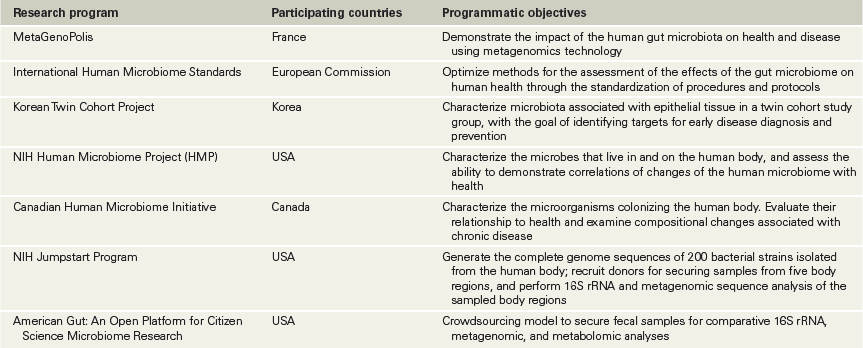
The sites of the human body inhabited by microorganisms include the mouth, nasal cavities, throat, stomach, intestines, urogenital tracts, and skin (Figure 24.1). It is estimated that the microbes in the human microbiome number around 1013 cells, which is about the same as the number of human cells in a single person. Together, the human body as the host and its associated microbes constitute a host–microbiome supraorganism. For example, the gut microbial community in the healthy human was once considered to consist of merely commensal microbes, but we now know that gut microbes are critical to development of the immune system, overall health later in life, and predisposition to disease. This recognition has stimulated the formation of several national and international research programs devoted to examining the human microbiome in great detail (Table 24.1). Collectively, these studies have shown that on the one hand the microbial diversity between individuals is so great that no one microbial species is present in all individuals, but on the other hand, particular microbial groups typically dominate.
Figure 24.1 Microbial habitats of the human body.

Primary body sites characterized in ongoing studies of the human microbiome (see Table 24.1).
Table 24.1 Major human microbiome research programs

Methodologies for Probing the Human Microbiome
Because the vast majority of microorganisms cannot yet be readily cultured or enumerated using growth-dependent approaches, it was the development of advanced nucleic acid sequencing methods (Chapters 10 and 19) that first offered the means to initiate a robust census of microbial diversity and abundance patterns in humans. The current “megasequencing era” has allowed microbiologists to quickly compare the microbiomes of different individuals and to begin to resolve the dynamic relationship between the microbiome and host history, genetics, health status, and diet.
This focus on molecular sequences in microbiome studies does not diminish the importance of cultivation in the study of the human microbiome. Indeed, since sequence-based surveys have shown that many human-associated microorganisms have not yet been isolated, there has been a renaissance in efforts to bring those microorganisms into culture, and the development of successful culture conditions has been guided by the results of metagenomic sequencing and the other omics tools of the microbial ecologist (Section 19.8). Using the omics toolbox, longitudinal studies of “at risk” human study groups have explored how the composition of the human microbiome, often determined early in life, affects such major diseases as obesity, diabetes, and inflammatory bowel disease. The results clearly show microbial links to these conditions.
A Snapshot of the Human Microbiome
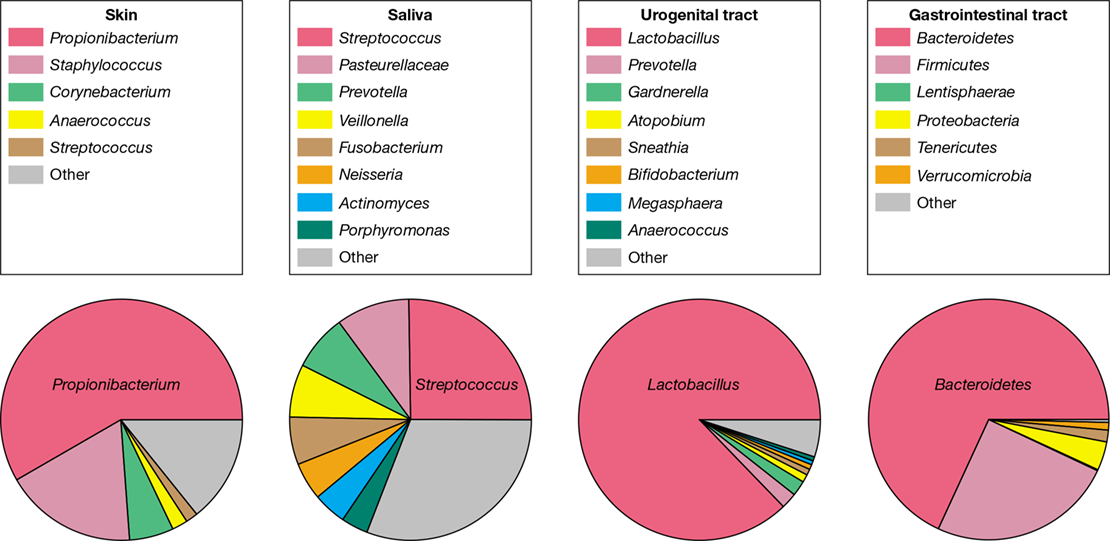
An overview of the bacterial composition of the four heavily colonized human body sites (skin, oral, urogenital, and gastrointestinal), as defined by higher taxonomic levels, is shown in Figure 24.2. In each site, one group of bacteria tends to dominate, and at each site, the dominant group is different. For example, the diversity of gram-positive bacteria is greatest on the surface of the skin and the urogenital tract while the diversity of gram-negative bacteria is greatest in the gastrointestinal tract. The following sections in this chapter will examine each of these sites at a higher resolution to more fully describe the diversity of microbes inhabiting the human body. Later sections will address the functional significance of the microbiome and how it might be modified for health benefit.
Figure 24.2 Overview of major microbial populations in the body sites sampled by human microbiome projects.

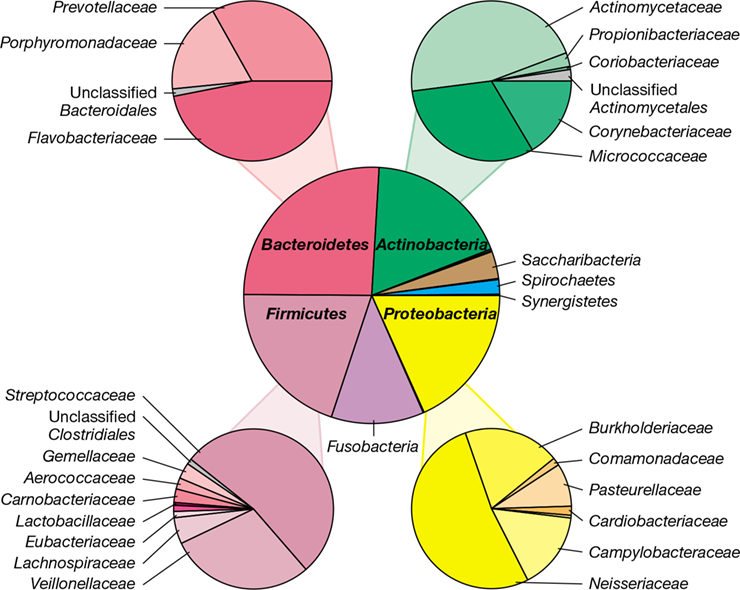
Diversity among body sites was evaluated by 16S rRNA gene sequence analysis (Section 13.11). Note that each body site tends to be dominated by one population type: skin by Propionibacterium species; oral by Streptococcus species; urogenital by Lactobacillus species; and gastrointestinal by Bacteroides species.
A remarkable feature that emerged early in human microbiome studies is the individuality and resilience of the different microbial communities. For example, an adultlike gut microbiome develops in an individual within three years of birth and shows a species composition unique to that individual that is retained throughout life. An individual’s microbiome also has tremendous plasticity, responding quickly to disturbances such as changes in diet, antibiotic therapy, or cohabitation with people and pets. However, following the removal of the disturbance, an individual’s unique microbiome tends to reemerge.
As we will explore in later sections, differences in the strains or even species of microbes in people’s microbiomes likely influence their response to therapies, such as fecal transplants designed to alter their microbiota for health benefit (Section 24.11). Similarities in the microbiomes of different individuals are more evident at higher bacterial taxonomic levels (such as phyla, families, and to some extent genera) rather than at the species and strain levels.
We start our tour of the human microbiome with a focus on the microbial communities that inhabit the gut, the human body site most heavily colonized by microbes and a site where the absence or presence of specific microbes may play a major role in an individual’s overall health or lack thereof.
Check Your Understanding
Which major body sites do microbes most heavily colonize?
What methods have been used to assess the human microbiome?
Why might knowing our microbiome and how it functions be useful?
24.2 Gastrointestinal Microbiota
The human gastrointestinal tract consists of the stomach, small intestine, and large intestine (Figure 24.3), and is responsible for the digestion of food and the absorption of nutrients; many important nutrients are also produced by the indigenous microbiota. Starting with the stomach, the human digestive tract is a long, folded tube of nutrients mixed with microbes, primarily species of Bacteria. The host and its gut microorganisms share the easily digestible nutrients. The nutrients are moved through this tube, encountering ever-changing microbial communities and abundance (Figure 24.3).
Figure 24.3 The human gastrointestinal tract and major members of its microbiota.

These taxa are representative of microorganisms found in healthy adults. Not every individual harbors all of these microorganisms.
The human gastrointestinal tract has about 400 m2 of surface area and is home to a total of about 1013 microbial cells. In the human duodenum, ingested food passed down from the stomach is blended with bile, bicarbonate, and digestive enzymes. About 1–4 h after ingestion, food reaches the gut (the large intestine, with near-neutral pH) and total bacterial numbers have increased from about 104/g (in the stomach) to 108/g (in the small intestine) to 1011−1012 per g (in the large intestine) (Figure 24.3).
The Stomach and Small Intestine
The stomach was long thought to be either sterile or only minimally populated until the discovery in 1983 that the highly acid-tolerant bacterium Helicobacter pylori colonizes the stomachs of about 50% of all humans. This discovery prompted a closer inspection of the microbiology of the human stomach through both cultivation and 16S rRNA sequence analyses. The stomach is now recognized as the home of a vibrant bacterial community with hundreds of phylotypes distributed between the gastric lumen (pH 1–2) and the mucus layer of the wall (pH 6–7). Although low pH prevents bacterial overgrowth, the healthy human stomach holds a core microbiome that is distinct from the transient passage of ingested oral populations and is dominated by Bacteroidetes (Prevotella), Firmicutes (Streptococcus, Veillonella, Lactobacillus), Actinobacteria (Rothia, Propionibacterium), Fusobacteria, and Proteobacteria (Haemophilus, Methylobacterium).
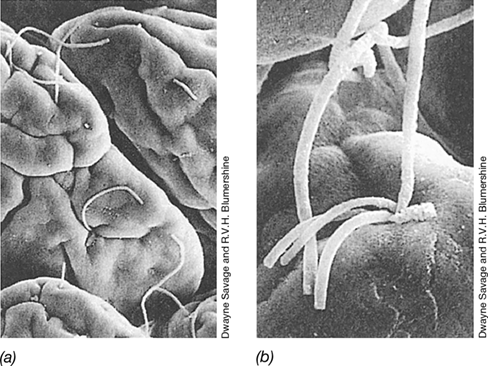
Firmicutes, Bacteroidetes, and Actinobacteria dominate gastric fluid samples, whereas Firmicutes and Proteobacteria are more abundant in gastric mucosal samples. When present, H. pylori accounts for the vast majority of stomach microbial biomass and can cause upper gastrointestinal tract disease (Section 31.10). Distal to the stomach, the intestinal tract consists of the small and large intestines, each of which is divided into different anatomical segments and supports a characteristic microbiota (Figure 24.3). The small intestine has three distinct chambers, the duodenum and the ileum connected by the jejunum. The duodenum, adjacent to the stomach, is fairly acidic and its microbiota typically resembles that of the stomach. From the duodenum to the ileum, the pH gradually becomes less acidic and bacterial numbers increase. In the lower ileum, cell numbers of 105−107/gram of intestinal contents are common, even though the environment becomes progressively more anoxic. Fusiform (spindle-shaped) anaerobic bacteria are typically present, with one end attached to the intestinal wall (Figure 24.4). Whereas the colonic microbiota (next subsection) is largely supported by the degradation of complex indigestible carbohydrates, microbial residents of the small intestine must compete with the host for the rapid uptake of small carbohydrates and therefore consist of bacteria well-adapted to “feast or famine” conditions.
Figure 24.4 Microbiota in the small intestine.

Scanning electron micrographs of the microbial community on epithelial cells in the mouse ileum. (a) An overview at low magnification shows long, filamentous fusiform bacteria on epithelial cells in the mouse ileum (part of the small intestine, see Figure 24.19). (b) Higher magnification shows several filaments attached at a single depression. The attachment is at the end of the filaments. Individual cells are 10–15 μm long.
Structure, Cell Numbers, and Retention Times in the Large Intestine
The ileum empties into the cecum, the beginning of the large intestine. The colon makes up the rest of the large intestine. In the colon, Bacteria are present in enormous numbers and Archaea (primarily methanogens) can be present, too, depending on the individual. The colon is essentially a fermentation vessel, with the microbiota using nutrients derived from the digestion of food (Figure 24.3). Facultative aerobes such as Escherichia coli are present but in much smaller numbers than other bacteria; instead, obligate anaerobes predominate. The facultative aerobes consume traces of oxygen that make it to the colon, rendering the large intestine strictly anoxic. Anoxia promotes growth of obligate anaerobes such as Clostridium and Bacteroides species; indeed, Bacteroidetes and gram-positive Bacteria account for more than 99% of all prokaryotic cells.
The total number of obligate anaerobes in the colon is enormous. Bacteria are by far the dominant population—bacterial cell counts of 1010 to 1011/gram of fecal contents are typical. During the passage of food through the gastrointestinal tract, water is absorbed from the digested material, which gradually becomes more concentrated and is converted to feces (Figure 24.5). Bacteria compose about one-third of the weight of fecal matter. Organisms inhabiting the lumen of the large intestine are continuously displaced downward by the flow of material, and bacteria that are lost are continuously replaced by new growth, similar to a continuous culture system (Section 4.8). The time needed for passage of material through the human gastrointestinal tract is about 24 h, and the growth rate of bacteria in the lumen averages one to two doublings per day.
Figure 24.5 Different microenvironments in the large intestine.
The inner mucin layer, produced by and contacting the gut mucosa, is partly oxygenated but generally free of bacteria. The sparsely populated outer mucus layer is adjacent to the heavily colonized anoxic lumen, which receives undigested food particles from the small intestine.
A person sheds a total of about 1013 bacterial cells each day in feces. Most of those organisms are restricted to the lumen of the large intestine. Production of mucin (a thick liquid secretion containing water-soluble proteins and glycoproteins) by goblet cells (a specialized class of epithelial cells) in the intestinal epithelium forms a protective layer (inner mucus layer) immediately adjacent to the intestinal epithelium (Figure 24.5). This inner mucus layer, unlike the outer mucus layer, is rarely colonized by bacteria. Goblet cells also produce various antimicrobial peptides that help prevent microbial contact with the underlying epithelium.
Bacterial Diversity of the Large Intestine
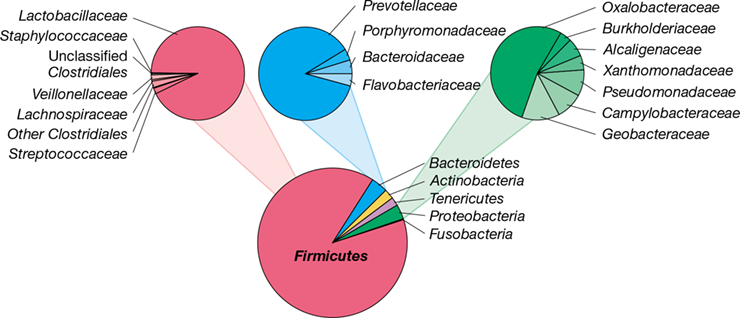
Figure 24.6 is a molecular snapshot of bacterial diversity in the human colon as determined by 16S rRNA gene sequence analysis of feces. Two major families of Firmicutes (Lachnospiraceae and Ruminococcaceae) dominate gram-positive bacterial diversity in the gut and are important in the digestion of polysaccharide polymers in plant fiber, such as cellulose and pectin; these are depolymerized and the sugars fermented in the large intestine. Ruminococcus species play an important role in the digestion of starch and resistant starch, a form of starch common in seeds and whole grains that is not digested in the small intestine. Since fermentation of resistant starch in the colon generates beneficial short-chain fatty acids (see Section 24.9), there is increasing interest in using resistant starch as a food additive, called a prebiotic (see Section 24.12), to encourage the growth of ruminococci and other fermenting species.
Figure 24.6 Bacterial diversity of feces.


The results are pooled analyses of approximately 1 million sequences (from 184 samples) of the V1–V3 region of the 16S rRNA gene (Figure 13.24). Members of the Bacteroidetes and Firmicutes dominate the normal microbiota of the large intestine. Many of these groups are discussed in Chapters 15 and 16. Data assembled and analyzed by Nícolas Pinel.
Gut Bacteroidetes largely consist of different species of the genus Bacteroides, organisms that also specialize in the degradation of polysaccharides, although some species are proteolytic. When dietary polysaccharides are not available, or in low abundance, glycolytic Bacteroides use host glycans and glycoproteins derived from the mucin layer (Figure 24.5).
In contrast to the rather limited phylum-level gut bacterial diversity, gut species diversity is enormous. For instance, one study of diversity in human fecal samples (based on the analysis of millions of 16S rRNA sequences from a large sample of people) identified between 3500 and 35,000 bacterial “species,” depending on exactly how one defines a bacterial species (Section 13.12). A relatively small number of these, several hundred or so, are shared by most individuals. Archaea (represented by a phylotype closely related to the methanogen Methanobrevibacter smithii), yeasts, fungi, and protists are either absent or compose only a minor part of the human gut community (compare this with the rumen, Section 23.16). When eukaryotic microbes are present, they are usually yeasts of the genus Candida (Sections 34.1 and 34.2).
Although there is high variability from person to person in gut community composition at the species level, any particular individual’s community is relatively stable over long periods. In a longitudinal study of healthy adults, about 65% of strains persisted for at least 5 years and most strains likely remain residents for decades, perhaps even for the lifetime of an individual. Three broad gut communities (called enterotypes) have been described, differing primarily by the enrichment of one microbial group in each enterotype. Enterotype 1 is enriched in Bacteroides, enterotype 2 is enriched in Prevotella, and enterotype 3 is enriched in Ruminococcus (Figure 24.6). The enterotypes are functionally distinct, differing in capacity for producing vitamins and energy-rich metabolites that can be used by the host. Evidence also exists that a person’s enterotype influences the response to diet and drug therapy and may contribute to health or disease status in yet unknown ways.
Products of Intestinal Microbiota
Some products of gut microbial metabolism are relatively simple compounds (Table 24.2), such as the volatile fatty acids generated by microbial fermentation of plant material, as also occurs in the rumen of herbivores (Figure 23.46). Other products generated by the activities of fermentative bacteria and methanogens include H2, CO2, and CH4 (similar to fermentation products of the rumen) and several other substances and nutrients (Table 24.2).
Table 24.2 Biochemical/metabolic contributions of intestinal microorganisms

aCapacity for amino acid biosynthesis inferred from the identification of biochemical pathways encoded in gut metagenomic sequences (Sections 10.7 and 19.8).
Gut microbes also produce vitamins B12 and K and some amino acids humans are unable to make (so-called essential amino acids, such as lysine). These essential compounds are not synthesized by humans (and vitamin B12 is not present in plants) but are made by the gut microbiota and absorbed from the colon. In addition, steroids, produced in the liver and released into the intestine from the gallbladder as bile acids, are modified in the intestine by the microbiota; the modified bioactive steroid compounds are then absorbed from the gut.
Many other microbial metabolites or transformation products that can be generated in the gut have significant influence on host physiology. These include post-translationally modified peptides such as the lantibiotics and bacteriocins (Table 24.3), substances that function to help secure colonization of the producing organism by inhibiting organisms closely related to the producer. Gut bacteria can also synthesize high levels (>100 mg/day in some cases) of metabolites derived from the reduction of amino acids. These include substances such as tryptamine, a tryptophan metabolite thought to function as a neurotransmitter that signals the enteric nervous system, and 4-ethylphenylsulfate, shown to affect behavior in mice (Table 24.3). These examples suggest that an animal’s microbiome and its nervous system—called the gut–brain axis—are likely connected (see Explore the Microbial World, “The Gut–Brain Axis”).
Table 24.3 Small bioactive molecules produced by bacteria in the large intestine

aRibosomally synthesized and post-translationally modified peptides.
bThese small molecules promote colonization by normal microbiota.
The Gut Microbiome and “Educating” the Immune System
The gut microbiome has far-reaching effects on the long-term health of a human. For example, the immune system does not properly develop in the absence of microbial stimulation, and exposure to a variety of microorganisms soon after birth is essential for developing immune tolerance of beneficial microorganisms (our normal microbiota) and recognizing pathogens as foreign. Indeed, the result of excessive hygiene in an infant’s development may be a poorly trained immune system that is more likely to attack beneficial organisms with an inflammatory response. Such an immune status is thought to promote autoimmune conditions such as allergies, asthma, and inflammatory bowel diseases later in life.
Much of our understanding of how the immune system is trained to accept the normal microbiota comes from the study of mice (Section 24.7). For example, a key factor contributing to the successful colonization of Bacteroides fragilis, part of the normal microbiota of the mouse gut, is production of a “symbiosis factor” called polysaccharide A (Table 24.3). Polysaccharide A is a diffusible oligosaccharide derived from the B. fragilis outer membrane (Section 2.4) that signals the host’s immune system to promote the tolerance needed for successful colonization by this bacterium. Tolerance is associated with the capacity of polysaccharide A to induce the production of cytokines (Section 26.5) in the host that suppress inflammation or activate specific immune cells. Clostridium species also suppress inflammation and promote tolerance using a similar mechanism. Besides promoting its own colonization, B. fragilis has also been shown to protect mice from colitis caused by a pathogenic bacterium, Helicobacter hepaticus. In experimental animals colonized with B. fragilis mutants unable to express polysaccharide A, H. hepaticus colonizes the gut and elicits inflammatory bowel disease, whereas control animals colonized by wild-type B. fragilis do not.
The gut is a complex structure that hosts a diversity of microbes, and these examples of the “dialogue” between gut microbiota and host indicate the importance of exposing infants to a variety of microbes early in life in order to develop a healthy immune system that can readily distinguish “friend from foe.” The identification of common and variably distributed biologically active compounds produced by the human microbiota is a rapidly emerging research area of great significance and will be critical to predicting future health outcomes and developing therapies for human pathologies linked to the gastrointestinal microbiome.
Check Your Understanding
How does the general metabolism of microorganisms colonizing the small and large intestines differ and why?
What is an enterotype, and what data indicate that it a stable feature of an individual?
24.3 Oral Cavity and Airways
Mucous membranes throughout the body support the growth of a normal microbiota that helps prevent infection by pathogenic microorganisms. Mucous membranes consist of epithelial cells, tightly packed cells that interface with the external environment, and are found throughout the body, lining the urogenital, respiratory, and gastrointestinal tracts (Figure 24.5). Mucous membranes secrete mucin, forming the mucus layer that retains moisture and inhibits microbial attachment; invaders are usually swept away by physical processes like swallowing or sneezing, but some microorganisms adhere to the epithelial surface and colonize it. Here we discuss two mucosal environments and their resident microbes. In Chapter 25 we explore the mechanism of specific attachment of microbes to mucous membranes leading to growth and development of the normal microbiota or, alternatively, to microbial disease.
Oral Microbial Diversity
The complex mixture of nutrients present in the mouth is supplied by saliva (containing proteins, glycoproteins, amino acids, peptides, and vitamins) and the ingestion of food. This nourishes a diverse community comprised of both aerobes and anaerobes. However, saliva itself is not a good microbial growth medium because saliva contains antibacterial substances. These include lysozyme, an enzyme that cleaves glycosidic linkages in peptidoglycan of the bacterial cell wall, weakening the wall and causing cell lysis. Another enzyme, lactoperoxidase, found in both milk and saliva, kills bacteria by a reaction that generates singlet oxygen (a toxic form of oxygen, Section 4.16). Despite the activity of these antibacterial substances, food particles and cell debris provide high concentrations of nutrients near surfaces such as the teeth and gums, creating favorable conditions for extensive local microbial growth that can contribute to tissue damage and disease.
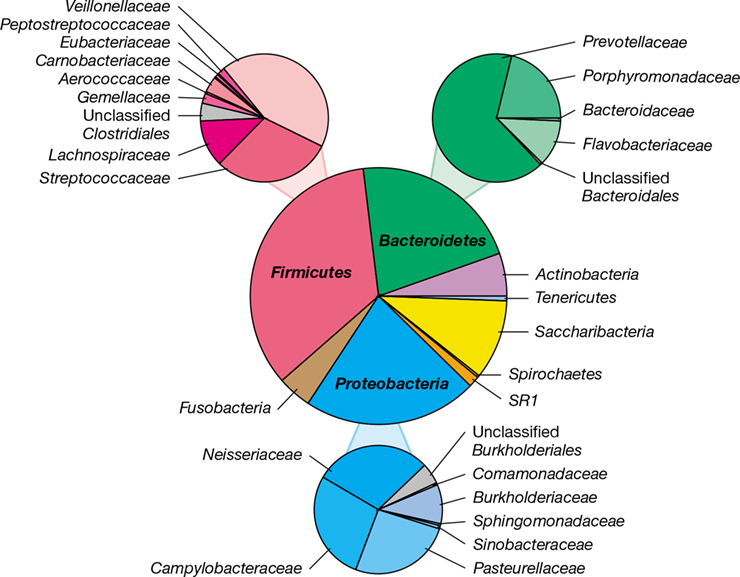
The oral cavity provides a variety of microbial habitats, each colonized by species that grow primarily as biofilms (Sections 4.9, 8.10, and 20.4). The microbiota in saliva consists of microorganisms shedding from multiple sites within the oral cavity and provides an overview of oral microbial diversity (Figure 24.7). The oral microbiome is essentially as diverse as that of the gut, but human populations share greater proportions of common taxa for the mouth than for the gut. Almost all of the microorganisms inhabiting the healthy oral cavity can be assigned to seven bacterial phyla: Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, Fusobacteria, Saccharibacteria, and Spirochaetes.
Figure 24.7 Bacterial diversity of saliva.

The results are pooled analyses of approximately 750,000 sequences (from 159 samples) of the V1–V3 region of the 16S rRNA gene (Figure 13.24). Note the lower fraction of anaerobes affiliated with Fusobacteria and Spirochaetes relative to the distribution of taxa observed in subgingival plaque (Figure 24.11). Many of these groups are discussed in Chapters 15 and 16. Data assembled and analyzed by Nícolas Pinel.
At least 750 species of aerobic and anaerobic microbes, including a minor representation of methanogenic Archaea and yeast, reside in the human oral cavity, distributed among teeth, tissue surfaces, and saliva. Most of these microorganisms have facultatively aerobic metabolisms, but some, such as Bacteroidetes and Spirochaetes, are obligately anaerobic. A few have strictly aerobic metabolisms, such as species of Neisseria, Campylobacter, and Haemophilus (a member of the Pasteurellaceae) in the Proteobacteria phylum. The most abundant genera are Firmicutes, major degraders of polysaccharides derived from the host or diet. Among Firmicutes, Streptococcus (∼20%) and the obligately anaerobic Veillonella (∼9%) are most abundant, although their relative numbers vary greatly as a function of oral cleanliness. Acid-producing, carbohydrate-fermenting genera physiologically and morphologically similar to the streptococci are also common Firmicutes, including species of Abiotrophia (Aerococcaceae), Gemella (Gemellaceae), and Granulicatella (Carnobacteriaceae).
Nanosynbacter lyticus is the first described species of the phylum Saccharibacteria (Figure 24.7) and is unique among oral bacteria (Figure 24.8). N. lyticus is a member of the ultrasmall (200–300 nm) bacterial radiation first observed in the terrestrial subsurface (Section 20.8 and Explore the Microbial World, “Tiny Cells” in Chapter 1). This diminutive bacterium has a highly reduced genome and lacks the ability to synthesize any amino acid, vitamin, or cell wall precursor. It exists as an obligate epiparasite, drawing nutrients from other oral bacteria by physical attaching to them. Lactate, a major metabolic product of oral streptococci, seems to be an important carbon and energy source for N. lyticus, as assessed from genomic analysis of cultured N. lyticus cells grown with oral actinobacteria as host (Figure 24.8). A similar symbiotic relationship occurs in the Archaea where the tiny parasitic and hyperthermophilic Nanoarchaeum equitans grows attached to the surface of cells of Ignicoccus (Section 17.6 and Figure 17.17).
Figure 24.8 Nanosynbacter lyticus.

The epiparasite N. lyticus is a species of the phylum Saccharibacteria. The tight physical association between N. lyticus cells (arrows) and its host Actinomyces odontolyticus is shown in (a) a transmission electron micrograph of a thin section, and (b) fluorescence in situ hybridization (FISH, Section 19.5) imaging of intact cells (red N. lyticus cells attached to white Actinomyces filaments). An N. lyticus cell is about 250 nm in diameter.
Ryan Hunter, Batbileg Bor, Xuesong He, and Jeffrey McLean
Oral Microenvironments and Their Microbiota
Bacteria found in the mouth during the first year of human life (when teeth are absent) are predominantly aerotolerant anaerobes such as streptococci and lactobacilli, and a few aerobes. When the teeth appear, the newly created surfaces are rapidly colonized by aerotolerant and obligate anaerobes that are specifically adapted to growth in biofilms on the surfaces of the teeth and in the gingival crevices (Figure 24.9). The formation of biofilms on teeth (dental plaque) is an ordered process of microbial assembly, where order is determined by highly specific binding between cell surface adhesins of one species and specific receptors on another species. Streptococcus and Actinomyces are the pioneer colonizers of a freshly cleaned tooth surface, followed by successive adherence by species of Veillonella, Prevotella, Fusobacterium, and other genera. The highly ordered structure can be revealed using fluorescence microscopy to visualize the layers of different genera in plaque (Figure 24.10). Close physical proximity may serve the function of metabolic cooperation. For example, Veillonella and Streptococcus establish a close physical and metabolic association in which the Veillonella consumes the lactate produced by streptococcal fermentation of carbohydrates.
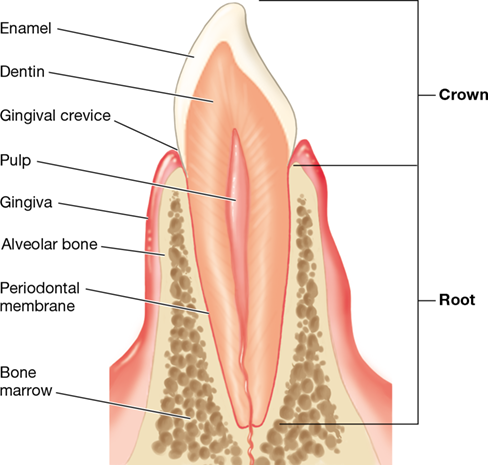
Figure 24.9 Section through a tooth.

The diagram shows the tooth architecture and the surrounding tissues that anchor the tooth in the gum.
Figure 24.10 Structured microbial community of dental plaque.

Organization of different bacterial genera in a thin section through human dental plaque visualized by fluorescence in situ hybridization (FISH, Section 19.5). The micrograph shows streptococci (stained green) located primarily at the periphery of the plaque beyond several other genera that combine to form a scaffold emerging from the tooth surface. These include early-colonizing Corynebacterium spp. (purple) near the tooth surface, covered by later-colonizing fusobacteria (Fusobacterium, yellow; Leptotrichia, blue-green), and capped by species of Capnocytophaga (red), Streptococcus (green), and Haemophilus (orange) in this highly organized assembly.
Most of these bacteria contribute to the health of the host by keeping pathogenic bacteria in check and preventing them from gaining access to mucosal surfaces. Tooth decay, gum inflammation, and periodontal disease are among the most visible manifestations of a breakdown in these generally stable mutualisms. We discuss the microorganisms associated with the hard tooth surface and their contribution to the formation of dental plaque and dental caries in Section 25.2.
There is significant site variation in the diversity and specificity of colonization by different oral bacterial species. For example, there are differences in abundant microbial taxa associated with subgingival plaque (plaque that forms under the gums) (Figure 24.11) compared with that found in saliva (Figure 24.7). Compare Figure 24.7 and Figure 24.11 and note the greater representation by Actinobacteria and Spirochaetes in the subgingival plaque. Different species of Corynebacterium (a genus of the Actinobacteria, Section 16.10) demonstrate significant site specificity. For instance, Corynebacterium matruchotii is almost exclusively found in the supragingival plaque, whereas Corynebacterium argentoratense occurs mostly in the saliva. Lautropia mirabilis (Proteobacteria) selectively colonizes the supragingival plaque, whereas the spirochete Treponema socranskii is found mostly in subgingival plaque, presumably because this site provides the low-oxygen environment needed for microaerobic growth of this bacterium. The distribution of Firmicutes, Proteobacteria, and Bacteroidetes is similar between the oropharynx (see Figure 24.12) and saliva. The hard palate (roof of the mouth) harbors a much lower diversity of microbiota than does the gingival plaque, possibly as a result of constant shedding of epithelial cells and the shear forces associated with chewing and swallowing.
Figure 24.11 Bacterial diversity of subgingival plaque.

The results are pooled analyses of approximately one million sequences (from 183 samples) of the V1–V3 region of the 16S rRNA gene (Figure 13.24). Compare the fractional distribution of different bacterial taxa with that observed in saliva (Figure 24.7), noting the higher representation of anaerobic fusobacteria and spirochete populations in the oxygen-limited gingival crevice. Many of these groups are discussed in Chapters 15 and 16. Data assembled and analyzed by Nícolas Pinel.
Microenvironments of the Respiratory Tract
The anatomy of the respiratory tract is shown in Figure 24.12 and Figure 24.1. In the upper respiratory tract (including the sinuses, nasal and oral cavities, and throat—pharynx, tonsils, and larynx), microbes live bathed in secretions from the mucous membranes. Bacteria continually enter the upper respiratory tract from the air during breathing, but most are trapped in the mucus of the nasal and oral passages and expelled with nasal secretions or swallowed and then killed by the acid in the stomach. However, a few microbes colonize respiratory mucosal surfaces in all individuals; those most commonly present are species of staphylococci, streptococci, diphtheroid bacilli, and gram-negative cocci.
Figure 24.12 The respiratory tract.

In healthy individuals the upper respiratory tract has a large variety and number of microorganisms. By contrast, the lower respiratory tract in a healthy person has few microorganisms.
Mastering Microbiology
Occasionally, potential pathogens such as Staphylococcus aureus and Streptococcus pneumoniae are part of the normal microbiota in the nasopharynx of healthy individuals. These individuals are carriers of the pathogens but do not normally develop disease, presumably because the other resident microorganisms compete successfully for nutritional and metabolic resources and limit pathogen attachment, colonization, or activities. The innate immune system (Chapter 26) and components of the adaptive immune system such as secreted antibodies (Chapter 27) are particularly active at mucosal surfaces and inhibit growth and invasion by potential pathogens.
The lower respiratory tract (trachea, bronchi, and lungs, Figure 24.12) has no naturally resident microbiota in healthy adults, but does harbor very low numbers of bacteria (primarily Prevotella, Veillonella, and Streptococcus) that are introduced from the upper respiratory tract during normal breathing. Dust particles, which are fairly large, settle in the upper respiratory tract. As the air passes into the lower respiratory tract, the flow rate decreases, and organisms settle onto the walls of the respiratory passages. The walls of the entire respiratory tract are lined with ciliated epithelial cells, and the cilia, beating upward, push bacteria and other particulate matter toward the upper respiratory tract where they are then expelled in saliva and nasal secretions or are swallowed. Only particles smaller than about 10 μm in diameter reach the lungs. Nevertheless, these include some pathogenic microbes, most notably certain bacteria and viruses that cause pneumonia (inflammation of the lungs, Sections 31.1, 31.2, and 31.8).
We now leave the oral cavity and explore the microbial diversity of other potentially heavily colonized body sites, the urogenital system and the surface of the skin.
Check Your Understanding
Compare the microbial microenvironments in the oral cavity in newborns and adults.
Identify the major microbes that predominate in the adult oral cavity by taxa and metabolic requirements.
Why is the lower respiratory tract typically microbe-free?
II: Urogenital Tract and Skin Microbiomes and the Human Viral Microbiome
II: Urogenital Tract and Skin Microbiomes and the Human Viral Microbiome
II Urogenital Tract and Skin Microbiomes and the Human Viral Microbiome
A resilient lactobacilli community colonizes the healthy vagina and maintains a weakly acidic environment. Different oily, moist, and dry microenvironments of healthy skin select for different distributions of primarily gram-positive bacteria. The human viral microbiome (virome) consists of both animal viruses and bacteriophages.
24.4 Urogenital Tracts and Their Microbes
In the urogenital tracts of healthy adults (Figure 24.13), the kidneys and bladder are believed to be sterile; however, facultatively aerobic gram-negative bacteria colonize epithelial cells lining the distal urethra, and the bladder of even healthy individuals may harbor a resident microbial community similar in composition to that of the urethra. Potential pathogens such as Escherichia coli and Proteus mirabilis, normally present in small numbers in the body or in the local environment, can multiply in the urethra and cause disease if conditions such as changes in pH occur. Such organisms are a frequent cause of urinary tract infections, especially in females. Proteus is especially notorious as a urinary tract pathogen. This bacterium is a strong urease producer; it generates ammonia from urea and uses the ammonia as a nitrogen source. Ammonia also causes urine pH to become quite alkaline, and this can trigger other urinary tract conditions such as the formation of kidney stones.
Figure 24.13 Microbial growth in the urogenital tracts.

(a) The urogenital tracts of the human female and male, showing regions (red) where microorganisms often grow. The upper regions of the urogenital tracts of both males and females are sterile in healthy individuals. (b) Gram stain of Lactobacillus acidophilus, the predominant organism in the vagina of women between the onset of puberty and the end of menopause. The individual gram-positive rods are 3–4 μm long. Species of Lactobacillus are anaerobic bacteria that ferment glucose and other sugars primarily to lactic acid as a fermentation product (Section 16.6).
The vagina of the adult female is weakly acidic (pH ∼5) and contains significant amounts of glycogen. Lactobacillus acidophilus, a resident organism in the vagina, ferments the polysaccharide glycogen, producing lactic acid that maintains a local acidic environment (Figure 24.13b). Other organisms, such as species of the yeasts Torulopsis and Candida, various streptococci, and E. coli, may also be present. Before puberty, L. acidophilus is absent, the female vagina is neutral in pH and does not produce glycogen, and the microbiota consists predominantly of staphylococci, streptococci, corynebacteria, and E. coli. After menopause, glycogen production ceases, the pH rises, and the microbiota again resembles that found before puberty.
Metagenomic 16S rRNA sequence analyses have confirmed earlier culture-based observations that the vaginal microbial community is less complex at the genus level than the oral or gut communities. A healthy vaginal microenvironment is dominated by lactobacilli (Figures 24.1 and 24.13b) and vaginosis (major changes in the balance of microbes in the vagina) is characterized by increased bacterial diversity, elevated pH, and a vaginal discharge. But even in the healthy adult female, vaginal microbiota consists of over 100 bacterial genera that group into several primarily gram-positive bacterial families (Figure 24.14).
Figure 24.14 Vaginal bacterial diversity.

The results are pooled analyses of approximately 600,000 sequences (from 86 samples) of the V1–V3 region of the 16S rRNA gene (Figure 13.24). Note the dominant fractional representation by Lactobacillus species and related Firmicutes, as compared to the diversity found at other body sites. Many of these groups are covered in Chapters 15 and 16. Data assembled and analyzed by Nícolas Pinel.
There are at least five types of “normal vaginal communities” containing different compositions of Lactobacillus spp., the dominant gram-positive organisms in the vagina. Four types are defined by dominance by one of L. crispatus, L. iners, L. reuteri, or L. jensenii (see also Figure 24.27), while the fifth is a more heterogeneous type characterized by higher overall diversity and a greater proportion of other strict anaerobes relative to the lactobacilli. Although all vaginal community types are associated with an acidic pH, pH varies with community type. The L. crispatus type shows the lowest average pH (∼4.0), whereas the heterogeneous type shows the highest average pH (∼5.3).
Thus, unlike the picture of microbial diversity we have seen in the gut or the oral cavity, the microbiota in the vagina of the healthy female is overwhelmingly dominated by lactobacilli (Figure 24.14). In contrast to the vagina, studies of the penis microbiota are fewer, but the general picture shows that the bacterial diversity of the penis is similar to that in the vagina, the patterns being especially so in sexual partners. Species of Lactobacillus, Streptococcus, and Sneathia are commonly found in both the male and female urethra. However, the microbiota on the circumcised versus the uncircumcised penis can be quite different, and bacterial abundance on the uncircumcised penis is typically much greater as well.
Check Your Understanding
What is the importance of vaginal Lactobacillus in healthy adult women?
What variable feature of the vagina is most closely associated with different community compositions of Lactobacillus species?
24.5 The Skin and Its Microbes
The skin is a complex human organ functioning primarily to prevent loss of moisture and restrict the entry of pathogens. An average adult human has about two square meters (2 m2) of skin surface that varies greatly in chemical composition and moisture content. Skin also provides an environment for part of the human microbiome. The skin microbiota consists of a rich community of microorganisms that associates intimately with the host’s hormonal, nervous, and immunological systems.
There are approximately 1 million resident bacteria per square centimeter of skin, for a total of about 1010 skin microorganisms covering the average adult. Although these numbers are much lower than the oral and gut communities, molecular analyses have shown that the skin harbors a diverse microbial community of bacteria and fungi (primarily yeast) that varies significantly with location on the body as a function of the diversity of habitats. These habitats consist of microenvironments of varying temperature, pH, moisture, sebum content (sebum is the oily secretions of the sebaceous glands), and surface characteristics. One distinct set of microenvironments includes moist skin areas such as the inside of the nostril, the armpit, and the umbilicus. Moist skin is separated by only a few centimeters from dry microenvironments such as the forearms and the palms of the hands. A third microenvironment consists of areas with high concentrations of sebaceous glands such as those by the side of the nose, the back of the scalp, and the upper chest and back. In addition to these site-specific differences, sweat is high in salt and other antimicrobial substances such as free fatty acids and antimicrobial peptides and thus plays a role in controlling diversity.
Mastering Microbiology
Art Activity: Figure 24.13a Normal skin microbiota
Microbial Diversity of Skin Microenvironments
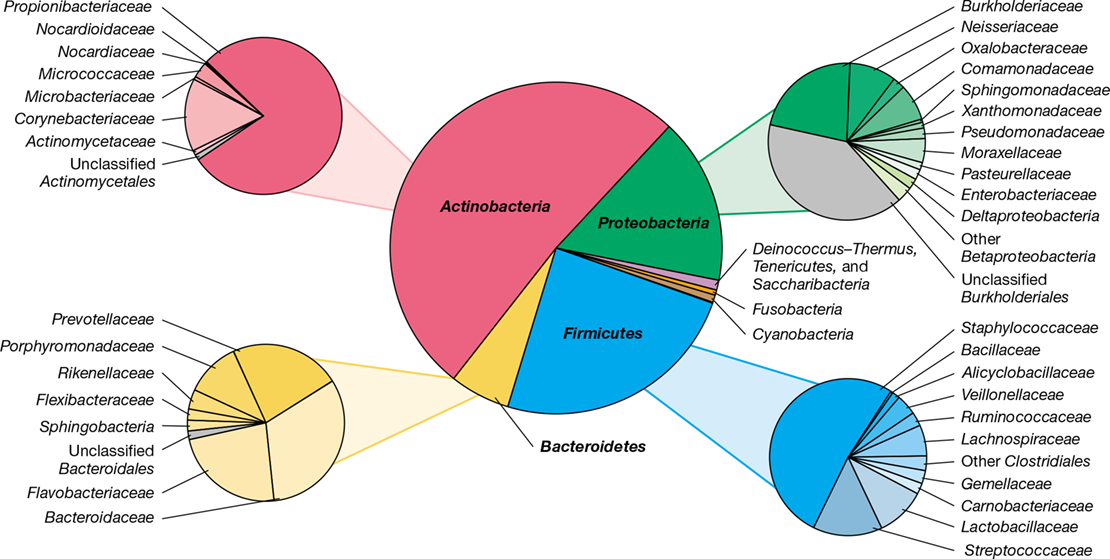
A 16S rRNA sequencing comparison of 20 diverse skin sites categorized as moist, dry, or oily revealed tremendous diversity and variation among sites and individuals and also showed some common patterns (Figure 24.15). Collectively, nearly 20 bacterial phyla were detected, but most phylotypes affiliated with one of four groups: Actinobacteria (∼52%), Firmicutes (∼24%), Proteobacteria (∼16%), and Bacteroidetes (∼6%). Over 200 different genera were identified, but species of three genera, Corynebacterium and Propionibacterium (both Actinobacteria) and Staphylococcus (Firmicutes) typically dominated the observed phylotypes (Figure 24.15b).
Figure 24.15 Overview of the skin microbiota.

(a) Analysis of the skin microbiome from 10 healthy human volunteers detected 19 bacterial phyla. Four phyla were predominant. (b) Composite populations of Bacteria from the same volunteers, divided according to sebaceous, moist, and dry skin microenvironments. Data adapted from Grice, E.A., et al. 2009. Science 324: 1190.
Each skin microenvironment showed a unique microbiota. Moist sites are dominated by corynebacteria and staphylococci, while drier sites support a mixed population dominated by Betaproteobacteria, corynebacteria, and Flavobacteriales. Species of Propionibacterium predominate in sebaceous areas (Figure 24.15b). For example, colonization of the follicular sebaceous gland system by Propionibacterium acnes is promoted by its ability to hydrolyze triglycerides present in sebum, resulting in release of free fatty acids that promote adherence of this bacterium, which sometimes causes disease (acne, Section 24.10).
A higher-resolution molecular diversity study of 400 different body sites of one male and one female subject revealed 850 distinct species, as bacterial species are currently defined (Section 13.12). The results of a high-resolution analysis of a single dry skin site (the inside of the elbow) are shown in Figure 24.16. As shown by the more general studies (Figure 24.15a), the most common microbial phyla were Actinobacteria, Firmicutes, Proteobacteria, and Bacteroidetes, but in this work, the breakdown of each phylum into family-level taxa shows a significant hidden diversity within each major group. Although staphylococci, propionibacteria, and Betaproteobacteria dominate, many other groups are also present (Figure 24.16).
Figure 24.16 Skin bacterial diversity at the inside of the elbow.

Note that Propionibacterium species comprise the vast majority of the Actinobacteria, and that Staphyloccocus species dominate skin-associated Firmicutes. The results are pooled analyses of approximately 80,000 sequences (from 123 samples) of the V1–V3 region of the 16S rRNA gene (Figure 13.24). Many of these groups are discussed in Chapters 15 and 16. Data assembled and analyzed by Nícolas Pinel.
The abundance of specific bacterial taxa can also be visualized in a “heat map” diagram (Section 10.10 and Figure 10.28) showing the major locations of different taxa on the skin. An example is shown in Figure 24.17 where it can be seen that Propionibacterium tends to localize on sebaceous regions (head, face, upper back, and upper chest), whereas species of Staphylococcus and Corynebacterium are more prevalent on less exposed regions, such as the groin, armpit, and toe web—areas higher in temperature and moisture content (Figure 24.17).
Figure 24.17 Distributions of *Staphylococcus, Propionibacterium*, and *Corynebacterium* on the human skin.

Heat maps of microbial species distributions at 400 different body sites inferred from 16S rRNA gene sequences from the skin of male (top) and female (bottom) subjects (in scale bar, red indicates higher relative abundance and blue lower abundance). Propionibacterium species tend to localize on sebaceous regions (head, face, upper back, and upper chest); Corynebacterium species are most common on the head, groin, and toes; and Staphylococcus species are most abundant on the foot.
Other Aspects of the Skin Microbiome
Eukaryotic microbes and Archaea are also present on the skin. The yeast Malassezia is the most common fungus found on the skin, and at least five different species of this yeast are typically present on the skin of healthy individuals. In an individual with a weakened immune system, for example, someone who has HIV/AIDS or whose normal microbiota has been compromised, Candida and other potentially pathogenic fungi can also colonize the skin and cause serious (even fatal) infections (fungal pathogens are discussed in Chapter 34). A remarkable finding emerging from 16S rRNA gene surveys of skin is that ammonia-oxidizing Archaea (Section 17.5) can comprise as much as 4% of the skin microbiota in some individuals, presumably sustained by ammonia present in the sweat of more physically active individuals.
Environmental and host factors influence the composition of the normal skin microbiota. For example, the weather may cause an increase in skin temperature and moisture, which increases the abundance of the skin microbiota. The age of the host also has an effect; young children have a more varied skin microbiota and carry more potentially pathogenic gram-negative Bacteria than do adults. Personal hygiene also greatly influences the resident skin microbiota; individuals with poor hygiene typically have higher microbial population densities on their skin. And finally, many microorganisms that would otherwise colonize skin cannot survive there simply because of its low moisture content and presence of antimicrobial fatty acids. Thus, the skin is a natural barrier to microbial colonization (Figure 26.2) while at the same time supporting a diverse array of normal microbiota.
As yet, there have been no significant longitudinal studies of early colonization and succession of the skin microbiome as have been conducted for the gut microbiome (Section 24.8). However, it is well recognized that the skin microbiome undergoes a major transition associated with sexual maturation. The skin of young children is dominated by Streptococcus spp., Betaproteobacteria, and Gammaproteobacteria. By contrast, these taxa are largely absent from the skin of postadolescent young adults; in this group the skin is characteristically dominated by species of Propionibacterium and Corynebacterium, as we have seen (Figures 24.15 and 24.17). And finally, encounters with man’s best friend can contribute to the human skin microbiota. Studies have shown that adult dog owners have more skin microbes in common with their own dogs than with other dogs. This demonstrates that close and regular contact between two distinctly different species of animal can result in a major sharing of their microbiomes. In contrast to the skin microbiota, microbes in the mouths and guts of canines differ quite distinctly from those of their owners.
Check Your Understanding
Compare the populations of microorganisms in the three major skin microenvironments.
Describe the properties of microorganisms that grow well on the skin.
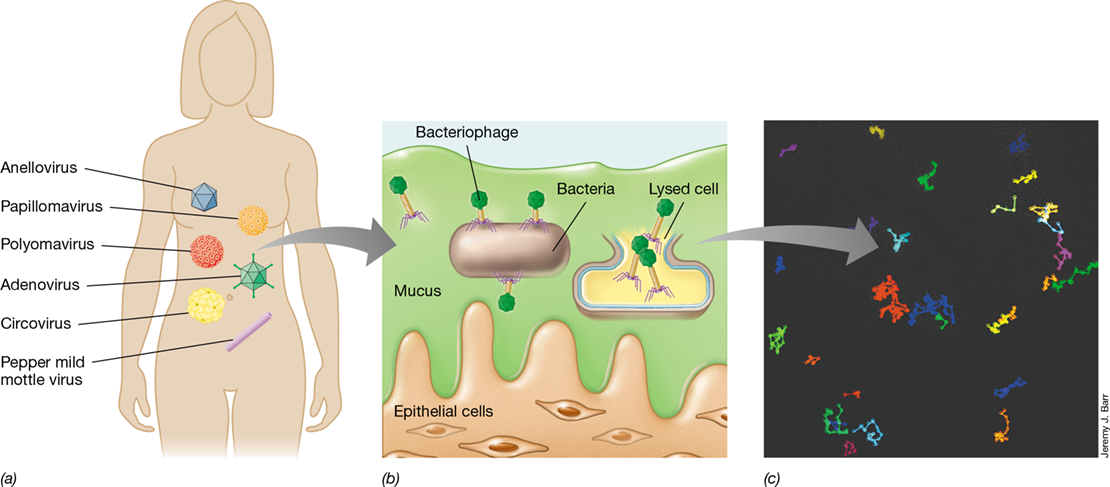
24.6 The Human Virome
In Chapter 5 we highlighted the infection process of animal viruses, and we described the general morphology and genomic structure of common animal viruses in Chapter 11 (Figures 11.2 and 11.3). Animal viruses differ from bacteriophages and archaeal viruses in that an animal cell takes up the entire virion instead of just the viral genome. There are also more than two lifestyles for animal viruses, including virulent infection, latent infection, persistent infection, and cellular transformation (Figure 5.22). A healthy human is teeming with viruses—not just animal viruses but also bacteriophages and possibly even some plant viruses—and we examine this suite of viruses now.
The Human Body and the Virome
While profiling the viruses of the human body has been hampered by limitations in cell culturing, the power of metagenomics has not only allowed for the human microbiome to be characterized (Section 24.1), but also the human virome. The human virome encompasses the entire population of viruses present in and on the human body (Figure 24.18). And, as for the human microbiome, the human virome is both unique to an individual and relatively stable over long periods.
Figure 24.18 The human virome.

(a) Common viruses of eukaryotes found in the virome of a healthy human. (b) Bacteriophages within the mucosa of the human respiratory and gastrointestinal tracts protect epithelial cells from the invasion of bacterial pathogens. (c) Movement of T4 bacteriophages in mucus. Tracks represent movement of fluorescently labeled individual bacteriophage virions within a microfluidic chip containing 1% mucin to simulate the mucus environment shown in part b.
Depending on the individual, animal viruses of the human virome include those that cause severe diseases such as hepatitis and severe acute respiratory syndrome, those that cause milder acute infections such as influenza and viruses of the common cold, and those that are completely benign. Other common animal viruses of the human virome cause latent infections, such as the herpesviruses, in particular human cytomegalovirus, present in most human adults (the biology of these viruses is covered in Chapter 11 and the disease syndromes themselves in Chapters 31, 32 and 33).
Viruses that contain DNA genomes dominate the viromes of healthy humans. Areas of the human body in which the virome has been assessed are the nose, skin, mouth, and gastrointestinal tract (from fecal samples). Animal viruses commonly detected in these areas include the single-stranded DNA anelloviruses and circoviruses and the double-stranded DNA adenoviruses, polyomaviruses, and papillomaviruses (Figure 24.18a). Anelloviruses are nonenveloped viruses that establish persistent infections in the body early in life with no known connection to disease. Circoviruses possess some of the smallest of all viral genomes and are commonly found in poultry and pigs, suggesting that their presence in the human gastrointestinal tract may originate from these food sources.
Adenoviruses are common respiratory viruses that have been detected in children with fevers but are also found in the nose and upper respiratory tract of healthy humans. Similarly, polyomaviruses are commonly found in healthy individuals, but in immunocompromised individuals they can also cause urinary tract infections and a type of brain disorder called leukoencephalopathy. Several different papillomaviruses are also common in the human virome, especially in the skin and saliva. Papillomaviruses are nonenveloped double-stranded DNA viruses (Baltimore class I, Figure 11.2) that replicate on skin and in mucosal epithelia of virtually all mammals. Most papillomavirus infections are asymptomatic; however, the human papillomavirus (HPV) causes persistent infections that can develop into skin warts, and certain strains of HPV can cause premalignant lesions in the female reproductive tract that can lead to cervical cancer later in life. Nearly all cervical cancers are attributed to HPV, and this virus also causes many cancers of the vagina, vulva, penis, anus, and oropharynx.
Besides these common animal viruses, most human viromes also contain viruses that infect plants, such as the pepper mild mottle virus (Figure 24.18a). These viruses are undoubtedly transmitted to humans from foods and pass through the intestines. While the host specificity of these viruses is for plants, it is thought that their presence in humans may trigger inflammation, which may lead to some of the symptoms susceptible individuals have to spicy foods or certain plant products. Another possible interaction between the human virome and the immune system is the prevalence of human endogenous retrovirus (HERV) elements in human chromosomes; HERVs are remnants of retroviral genes that have been incorporated into and constitute 5–8% of the human genome. Although most HERVs are thought to be harmless, connections have been proposed between certain HERVs and the autoimmune disorders rheumatoid arthritis and systemic lupus erythematosus (Section 28.1), and with other afflictions such as inflammatory bowel (Crohn’s) disease and multiple sclerosis.
Bacteriophages and the Human Virome
While every human body contains a unique mixture of different animal viruses, the most abundant viruses in all body sites are not animal viruses but instead are bacteriophages. The large intestine is a hotbed for such viruses, as this organ contains an abundance of prokaryotic cells and virions (numbering about 1011 and 109, respectively, per gram of feces). However, unlike the marine virosphere (Section 20.13), for example, in which bacteriophage virions greatly outnumber bacteria, the virion to microbe ratio (VMR) in the gut is much lower and could be as low as one virus for every 100 bacteria. As with the animal viruses of the human virome, DNA bacteriophages dominate, and many of these viruses are thought to benefit bacterial cells by transferring genes encoding antibiotic resistance or enzymes for specialized metabolisms through the genetic transfer processes of transduction (Section 9.7) and lysogeny (Section 5.6).
A unique feature of the gut virome is that most bacteriophages are in a lysogenic state, reproducing in concert with their host bacterium and therefore persisting for prolonged periods. Observations of bacteriophage diversity within an individual have shown it to be remarkably stable, with 80–95% of bacteriophages retained for over 2 years, and likely for much longer. Persistence and stability likely derive from the stability of the bacterial community (see Section 24.8) and a preference for the bacteriophage to enter into a stable lysogenic state. Thus, although genetic transfers from gut bacteriophages to their hosts likely help the gut microbiota adapt to changing nutrient conditions and stabilize it from the stresses of antibiotic treatment, the mechanisms and frequency of exchange may be quite different in the gut than in open environmental systems.
Metagenomic surveys of DNA recovered from virions separated from feces have shown that between 75% and 99% of bacteriophages in the gut microbiome are not closely related to known viral genomes. Thus, matching bacteriophage genomes with the bacterial hosts they infect is a daunting challenge. Even so, important progress has been made in identifying novel bacteriophages by matching CRISPR sequences (Section 9.12) between phage and bacterial genomes to search for links in a virus–host association. Using this strategy, the isolation of a major group of novel tailed bacteriophages (called crAssphage) present in at least half of the human population was achieved by recovering the bacteriophage on cultures of the Bacteroides species predicted by metagenomic sequencing to serve as its host (see MicrobiologyNow).
Function of the Human Virome
Why are there so many bacteriophages in and on us? Probably because our bodies are home to such enormous levels of bacteria. To our benefit, phages likely play a protective role in human health. Bacteriophages of the human virome can be a first line of defense against certain pathogens, especially within mucosal surfaces where bacteriophages accumulate. It has been estimated that 20 times more bacteriophages than bacteria exist in the mucosa of our lungs and intestines. The bacteriophages present in mucous linings are anchored to sugar residues produced by mucosal cells (Figure 24.18b). Here bacteriophages presumably scavenge invading pathogens and kill them before they can cross the mucous barrier. Thus, phages within the mucus layer can be considered to have a symbiotic relationship with the human host and provide a form of host-independent immunity. Using fluorescently labeled T4 phages, movement of the phages through a model mucus layer can actually be tracked microscopically in the laboratory (Figure 24.18c).
Besides killing or conferring antibiotic resistance to bacteria within the microbiome, the bacteriophage component of the virome can also enhance the pathogenicity of certain bacteria. An example of this is the lysogenic bacteriophage CTXϕ and its host, the bacterium Vibrio cholerae (the bacterium that causes cholera). It is the phage genome rather than the host genome that actually encodes the cholera toxin that induces disease symptoms (Section 33.3), and cells of V. cholerae that are not CTXϕ lysogens are nonpathogenic. This phage–bacterium link is also seen in the toxin-coregulated pilus, a structure essential for V. cholerae cells to attach to intestinal cells. Genes encoding the pilus are part of a viral genome that is integrated into the host’s genome (a prophage, Section 5.6) and is only present in pathogenic strains of V. cholerae.
With recognition of the potentially huge impact on human health by the virome coupled with the continually improving field of metagenomics, we can expect to have a much clearer understanding of the role bacteriophages play in health and disease in the near future. Indeed, a handful of therapeutic bacteriophage treatments have already been developed for humans and shown to be effective (see MicrobiologyNow at the beginning of Chapter 5 and the MicrobiologyNow).
We now turn our attention to how the human microbiome first develops and the importance to human health of early exposure to the microbial world.
Check Your Understanding
What accounts for the remarkable stability of the gut virome?
How do bacteriophages benefit and harm human hosts?
III: From Birth to Death: Development of the Human Microbiome
III: From Birth to Death: Development of the Human Microbiome
III From Birth to Death: Development of the Human Microbiome
Different body sites of the newborn are rapidly colonized, receiving microbes from the mother and the local environment. A mature gut community takes about three years to develop and is greatly influenced by early life events. Each individual harbors a unique microbiome but one that can be altered by diet and aging.
At birth, a baby is exposed to both maternal and environmental microorganisms. These early encounters determine the composition of microorganisms that first colonize different body sites. In this part of the chapter we focus primarily on colonization of the gut, the body site harboring the largest part of the human microbiome, and consider factors that govern early colonization and subsequent successional events of a community now recognized to be critical to general health and the education of the immune system.
24.7 Human Study Groups and Animal Models
Establishing relationships between the composition of a person’s microbiome and its contribution to health and disease has been complicated by problems surrounding sampling, the uncertainty about the contribution of the host’s genetic background, and the limitations of controlling diet and other contributing lifestyle factors. Nevertheless, several human study groups and animal model experiments have revealed some of the basic principles and given microbiome studies a starting point.
Human Microbiome Study Groups
Most functional understanding of the human microbiome has been based on surveys of selected study groups (Table 24.1). For example, one of the most ambitious early studies was the Human Microbiome Project (HMP) funded by the U.S. National Institutes of Health. This study collected samples from 242 individuals (all American medical students in good health), sampling different body sites (15 to 18 sites depending on gender) at one to three time points, and then evaluated bacterial diversity based on 16S rRNA gene sequencing and limited metagenomic analyses (Figures 24.1 and 24.2).
The objective of the HMP was to develop baseline information about what constituted a “healthy” microbiome. Although the HMP generated a huge amount of data, the study group represented only a small fraction of human diversity. This limitation was clearly revealed in the Global Gut Project, which examined three distinct populations (U.S. citizens, Malawians, and a group of indigenous peoples from Venezuela) and different age groups within those populations. The gut microbiomes of the two non-U.S. populations were distinct from those of the U.S. individuals, showing that the HMP greatly underestimated the potential for variation in gut microbiomes across nationalities. These studies also suffered from a lack of appropriate metadata; for example, detailed information about dietary habits (vegetarian, vegan, omnivorous) and how much fiber or protein was ingested daily. Because there is still very limited understanding of the influence of specific environmental variables such as lifestyle, diet, gender, and genetics on microbiome structure and function, new and ongoing studies (e.g., the American Gut Project) are obtaining these valuable metadata at the time of sampling. The overarching goal is to develop robust correlations between the microbiome, host genetics, diet, lifestyle, health, and pathologies thought to have a microbial connection, in particular, heart disease, cancer, stroke, diabetes, and obesity.
The Mouse Model
Even if appropriate metadata are available, a major limitation of the human microbiome studies to date is establishing causality, something that is only possible with highly controlled animal studies. Hence the mouse has become the major model animal system for linking cause and effect in the gut microbiome.
Although the mouse and human digestive systems have some significant differences (Figure 24.19), the mouse (and to a more limited extent, the rat) has been the workhorse of experimental microbiome studies. Compared to humans, mice have a relatively larger colon and cecum, which are needed to extract nutrients derived primarily from the fermentation of plant materials. For example, the average ratio of the length of the murine small intestine to its colon is about 2.5 whereas in humans it is about 7. In mice, fermentation of plant material is compartmentalized in the cecum (Section 23.14), while in humans this fermentation takes place in the colon. Nevertheless, mice have several experimental advantages, including the availability of well-defined genetic lines, low maintenance costs, and short life cycle. This has allowed researchers to explore the importance of the host genetic background through selective gene knockouts, the manipulation of gut microbiota composition using germ-free mice (raised free of all microbes), tests of the influence of a strictly controlled diet, assessing the consequences of antibiotic treatment, and exploring the transfer of physiological traits through fecal transplantation. These studies have clearly associated the composition of the mouse microbiome to different pathologies, including obesity and inflammatory bowel disease, as we will discuss in Section 24.9.
Figure 24.19 Anatomy of the mouse and human intestinal tracts.

The mouse (a) and human (b) intestinal tracts have significant structural differences that are associated with differences in the composition of the microbiota of mice and humans despite similarities in their physiologies.
Unfortunately, because of anatomical differences (Figure 24.19), mouse studies are not directly applicable to humans, and these anatomical differences may in part account for observed differences in the relative abundance of dominant bacterial genera in mouse and human guts. For example, the genera Prevotella, Faecalibacterium (Firmicutes), and Ruminococcus are in high abundance in the human gut (Figure 24.6), whereas the genera Lactobacillus, Alistipes, and Turicibacter are more abundant in the mouse gut. Thus, although the mouse model gives us much experimental latitude not available in gut microbiome studies of humans, actual results from the two systems are not directly comparable. However, despite these differences in bacterial composition, experimenters have glimpsed much useful information from the study of animal models that has accelerated our understanding of the human gut microbiome (for example, see Section 24.9 and Figure 24.25).
We now follow development of the human gut microbiota from birth through adulthood, comparing and contrasting the major organisms seen in healthy humans. An overview of the mature and highly stable gut microbiota was presented in Figures 24.2, 24.3, and 24.6.
Check Your Understanding
In hindsight, which aspects of the HMP were well controlled for and which were not?
Why has it been so difficult to associate human disease, or health, with changes in the gut microbial community?
What are some of the major differences between the mouse and human gastrointestinal systems?
24.8 Colonization, Succession, and Stability of the Gut Microbiota
24.8 Colonization, Succession, and Stability of the Gut Microbiota
24.8 Colonization, Succession, and Stability of the Gut Microbiota
Colonization of an initially sterile gut begins immediately after birth; a succession of microbial populations replace each other in turn until a relatively stable, adult microbial community is established. Colonization begins when the infant first encounters microbes by passage through the mother’s vagina. The newborn is quickly inoculated by bacteria present in breast milk and skin, and later through contact with visitors and household pets.
The structure of the infant’s gut community in the first year of life is highly dynamic and greatly influenced by early life events. In particular, birth by vaginal delivery versus cesarean section has a profound influence on early community structure. It is also now known that these early colonization events are critical to the microbe–immune system “cross talk” essential to the development of a well-functioning immune system. Disruption of a normal colonization sequence, such as may result from early-life treatment with antibiotics, is correlated with the development in later life of allergy and asthma, as well as type 1 diabetes and obesity. As we will review in Section 24.12, modulation of the developing gut community by early feeding of beneficial probiotic microorganisms is increasingly viewed as a means to avoid diseases associated with the early disturbance of the normal gut colonization sequence.
Microbial Community Structure and Function in the First Three Years of Life
The young infant gut community is dominated by bifidobacteria—fermentative anaerobes of the bacterial class Actinobacteria (Section 16.10)—and does not reach an adultlike composition until about age 3. The newborn’s relatively simple community progressively evolves into a more complex and adultlike composition in these first three years. The first year is a period of tremendous fluctuation in community composition; the second year is a transitional phase and is followed by maturation to a stable, adultlike composition during year three.
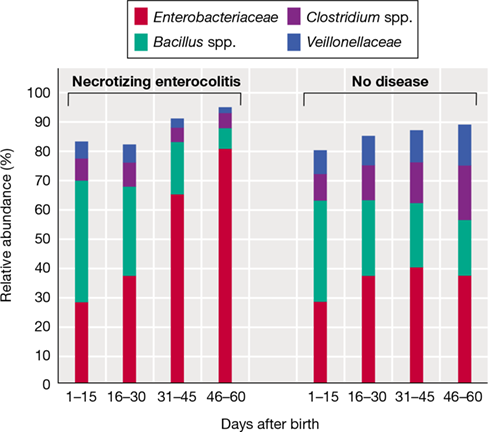
Early microbial colonizers are an important source of amino acids and vitamins. Microbial genes encoding the synthesis of vitamins K2 (menaquinone), B6 (pyridoxal), and B7 (biotin) are elevated in the newborn’s microbiota. As the gut microbiome matures, there is a greater prevalence of microbial genes encoding synthesis of the vitamins B1 (thiamine), B5 (pantothenate), and B12 (cobalamin). The immature gut community contains a large number of species affiliated with facultatively aerobic Enterobacteriaceae (Figure 24.20). The presence of bacterial genera such as Escherichia and Enterococcus, both facultative aerobes, in the newborn gut is also indicative of a more aerobic state of the early gut system and a greater role for the citric acid cycle and respiration in microbial energy production in the neonate.
Figure 24.20 Maturation of the infant gut community.

Normal maturation of the breastfed infant’s gut bacterial community during the first three years of life. Bifidobacterium and Bacteroides species are important early colonizers. As the gut community matures with termination of breastfeeding, species of Firmicutes (Lachnospiraceae and Ruminococcaceae) and Bacteroidaceae increase in relative abundance while Enterobacteriaceae decrease to very low levels.
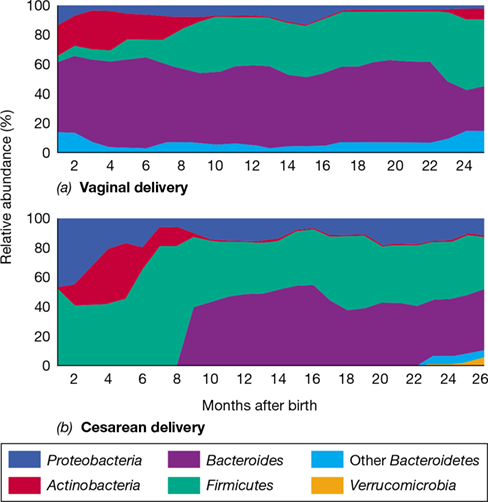
Major factors controlling the early assembly of the gut microbiome following birth are whether birth was vaginal or by cesarean section (C-section) (Figure 24.21) and whether initial nutrition came from breast milk or from formula. A vaginally delivered infant is colonized by a gut microbiota similar to that of the mother, suggesting direct transfer from mother to neonate during passage through the birth canal and subsequent intimate contact with the mother. In contrast, the gut microbiota of a child delivered by C-section is significantly different from that of the mother. Approximately 70% of babies delivered vaginally develop a gut microbiome similar to their mothers, including species of Escherichia and Bilophila (Proteobacteria), Bifidobacterium (Actinobacteria), Enterococcus (Firmicutes), and Bacteroides. By contrast, only about 40% of the species match those of the mother in the gut of a C-section neonate (Figure 24.21). The latter tends to be enriched in Staphylococcus and Streptococcus species (Firmicutes), and Veillonella species, indicating that skin and oral microbes, respectively, as well as environmental populations, were early colonizers. Bacteroides species are either less prevalent or totally missing in the infants delivered by C-section (Figure 24.21b). The difference in the gut microbiomes of babies born vaginally or by C-section is significantly less by 12 months of age, but the C-section gut microbial community at 8 months remains distinctive and more variable in composition than that of infants born vaginally (Figure 24.21).
Figure 24.21 Impact of birth mode on the young infant’s gut community.

Early colonization and temporal changes in the relative abundance of major microbial groups in children delivered (a) vaginally or (b) by cesarean section. With vaginal delivery, species of Bacteroides are present from an early age and Proteobacteria only poorly colonize. Bacteroides species are undetectable during the first 8–9 months of life following delivery by cesarean section, and a higher initial colonization by Proteobacteria persists throughout early life.
The Microbiology of Breastfeeding versus Formula
Infants that have been breastfed develop a different gut microbiome than those who have not. Breastfeeding is associated with elevated numbers of fermentative Bifidobacterium, Lactobacillus, and Staphylococcus species. Viable populations of Bifidobacterium and Lactobacillus species are present in breast milk, and Staphylococcus species colonize the areolar skin. Thus, these species are transferred directly from mother to infant. Staphylococcus epidermidis is an opportunistic colonizer, essentially absent in formula-fed infants and an organism whose numbers decrease over time following cessation of breastfeeding.
The enrichment of Bifidobacterium species, in particular the species B. longum, is related to the composition of human milk. Breast milk contains a complex mixture of unusual oligosaccharides that most gut microbes (other than Bifidobacterium species) and humans are unable to digest; for example, the sugar monosialyl-lacto-N-neohexaose I (Figure 24.22). B. longum subspecies infantis is unique in having an exceptionally large repertoire of glycosidases and oligosaccharide transporters and grows well on this and other human milk oligosaccharides. These characteristics have also made this strain an important probiotic for treating enterocolitis in premature infants (see Section 24.12). With cessation of breast milk feeding and maturation of the infant gut microbiome, B. longum subspecies infantis is displaced by other strains of B. longum that can use both mammalian and plant-derived oligosaccharides. However, even with the introduction of solid food, the gut communities of infants that continue to receive some breast milk are less diverse than in those no longer getting breast milk. Thus, maturation of the gut microbiome is largely driven by the removal of breast milk from the diet—which generally occurs between 9 and 12 months—rather than by the introduction of solid foods.
Figure 24.22 Example of an oligosaccharide found in human breast milk.

This and other short polysaccharides selectively nourish and enrich for desirable microbial populations, such as Bifidobacterium and Lactobacillus species, in the infant’s developing gut microbial community.
At 4 months there remains a clear difference between children who have received exclusively breast milk compared with those given formula milk. Formula-fed infants tend to have elevated numbers of Clostridioides difficile—a potentially serious pathogen (Section 30.7 and Figure 30.12)—as well as other potential pathogens such as Citrobacter and Enterobacter, whereas the guts of children that remain on breast milk at 12 months continue to be dominated by Bifidobacterium and Lactobacillus. The abundance of B. longum in breastfed infants also leads to the production of short-chain fatty acids; this creates an environment favoring the growth of the normal microbiota important for “educating” the immune system. In addition, since the structures of human milk oligosaccharides mimic carbohydrates lining the infant gut, they function to suppress pathogenic bacterial infection by blocking the pathogens’ receptors for attachment. Therefore, in addition to providing nutrition to the newborn, breast milk selects for a specific group of earlier colonizing normal microbiota important to the overall health of the child and proper development of its immune system.
Termination of breastfeeding results in a shift of the community toward a more adultlike composition, as reflected by enrichment in Bacteroides, Bilophila, Roseburia, Clostridium, Anaerostipes, and Eikenella. Many of these later-arriving genera are efficient degraders of dietary fibers and complex carbohydrates, consistent with a transition to more solid foods following the cessation of breastfeeding; they also produce short-chain fatty acids. The increased abundance of Bacteroides thetaiotaomicron, a bacterium encoding a diverse set of glycan-degrading enzymes, is specifically associated with the increase in pectin in solid foods. Although the development of a fully adultlike gut microbial community varies a bit from child to child, the average is three years.
Stability of the Adult Microbiome and Transitions with Age
Although metagenomic diversity studies have identified several thousand gut bacterial species, the number of species in any given individual is closer to 200, a number that remains stable for long periods. The most stable of these species are typically affiliated with the Bacteroidetes and Actinobacteria. Although Actinobacteria, such as Bifidobacterium spp., comprise a rather small part of the adult gut microbiota compared to the Bacteroidetes, specific species tend to associate with individuals over long periods of time. By contrast, species of Firmicutes and Proteobacteria appear to be less stable members of the gut community. The species composition of the gut of an individual tends to reflect that of family members, and likely was established early in life. Thus, the early colonizers—including microbes acquired from parents or siblings—in large part determine the metabolic and immunological properties of the adult microbiome and may be an important factor in adult health and predisposition to disease.
As we age, so does our microbiome. The most well-established change is the age-related alteration in the relative proportions of Firmicutes and Bacteroidetes, with an increasing proportion of Bacteroidetes seen in the elderly as compared with the higher proportions of Firmicutes in young adults. Also observed with age are significant decreases in bifidobacteria and certain clostridial species. It also appears that the old age–related inability to perform routine daily activities, a measure of frailty, is correlated with decreased diversity in the gut microbiota. Consistent with this observation, elderly individuals living at home or in supportive communities have a more diverse gut community compared with those living in residential care facilities, such as nursing homes. Thus, maintaining a diverse gut microbiota is likely one of many links to health for the aged and is possibly even a positive effector of longevity.
With this overview of changes in the human microbiome with time, we move on to consider some startling discoveries about human diseases linked to unfavorable changes in the human gut microbiota. We then end this chapter by looking at some therapies for dealing with these problems.
Check Your Understanding
What factors contribute to early colonization of the newborn’s gut community?
How do microorganisms in the infant and adult gut community contribute differently to vitamin and amino acid requirements?
What factor(s) are most important in the transition from an immature to a mature gut microbial community?
IV Disorders Attributed to the Human Microbiome
Many major human health issues including obesity and inflammatory bowel disease correlate with disturbances of the microbiome. Maintenance of gut and general health depends on the continuous production of short-chain fatty acids by microbial fermentation of dietary fiber.
Alteration of the structure and activity of the human microbiome is associated with a variety of pathologies, including obesity, type 2 (non-insulin-dependent) diabetes, asthma, atopic dermatitis, liver disease, colorectal cancer, kidney stones, psoriasis, tooth decay (dental caries), and periodontitis. As we learn more about the relationship between the human microbiome and health and disease, therapeutic intervention may well be possible. This might include promoting the growth of protective beneficial bacteria, inhibiting the growth of specific microbes (or specific assemblages of microbes) that compromise health, fecal transplants to introduce a desirable gut microbiota, and developing appropriate behavioral interventions, such as diet modification.
24.9 Syndromes Linked to the Gut Microbiota
A healthy gut community is loosely defined as constituting a relatively stable and resilient community associated with health of the human host. A gut microbial community demonstrating stability and resilience is said to be in a state of homeostasis. Variations from the homeostatic state may or may not trigger disease. We will first consider the healthy state and then explore some cases where alterations in the gut microbiome are strongly correlated with specific disease syndromes.
Diet and Gut Health
A specific definition of healthy homeostasis based on species composition has not been possible because individuals differ significantly in their specific gut community makeup, and community makeup shifts with diet. Thus, a healthy gut microbiota is increasingly being defined in dietary, metabolic, and immunological terms. Diet is a critical determinant of homeostasis. In particular, the short-chain fatty acids (SCFAs; primarily butyrate, propionate, and acetate) produced by fermentation of fiber by the gut microbiota serve a central role in the development and maintenance of a healthy gut. Not only are SCFAs an important energy source for intestinal cells, but they also function as signaling molecules to regulate cellular metabolism and immune surveillance (Figure 24.23). Binding of SCFAs to specific intestinal cell surface receptors regulates cellular metabolism, increases the secretion of antimicrobial peptides known as defensins (Section 26.2), elevates production of hormones affecting energy recovery and sensations of hunger, and has a dampening effect on the inflammatory response through the regulatory T cell (Treg) signaling system (Figure 24.23 and Section 27.8).
Figure 24.23 Role of short-chain fatty acids in intestinal health.

Short-chain fatty acids (SCFAs) produced by microbial fermentation of fiber and dietary polysaccharides modulate the metabolic activity of colonocytes (see Figure 24.5) lining the intestinal epithelium. Butyrate activates the PPAR-γ regulatory protein, which then accelerates beta (β)-oxidation of fatty acids and the synthesis of tight junction proteins between cells and concurrently depresses colonocyte glycolysis. SCFAs also signal to regulatory T cells (Treg), suppressing the potential inflammatory response to normal gut microbiota. Together these shifts in metabolic activities reduce oxygen leakage into the lumen and maintain the integrity of the epithelial barrier.
Among SCFAs, butyrate in particular serves a critical function in the control of immune surveillance and the metabolism of colonocytes. Colonocytes are the absorptive epithelial cells of the large intestine (Figure 24.5). Butyrate activates a specific regulatory protein of colonocytes called PPAR-γ that accelerates mitochondrial beta-oxidation of fatty acids (Section 14.23). This oxygen-consuming process helps to maintain an anoxic environment near the epithelial surface that is favorable to the obligately anaerobic bacteria that produce butyrate (Figure 24.23). If butyrate-producing microbes are depressed by, for example, a dramatic diet change or as a result of antibiotic treatment, colonocyte metabolism is shifted toward glucose fermentation and lower oxygen consumption. This results in greater passage of oxygen through the intestinal mucosal surface and a proliferation of facultatively aerobic bacteria (such as species of Enterobacteriaceae) that can invoke an inflammatory response. Since SCFA signaling also promotes tight junction formation (Figure 24.23), it is thought that a depression of PPAR-γ activity contributes to the development of leaky gut syndrome in humans. Disruption of tight junctions leads to a greater exposure to gut microbiota, which in turn elicits an inflammatory response. Such a disruption of healthy gut function is called dysbiosis, typified by increased numbers of facultative aerobes, such as species of Enterobacteriaceae, and a state of chronic inflammation. These physiological changes are also thought to play important roles in the development of inflammatory bowel disease and obesity, and we turn our attention to these now.
Inflammatory Bowel Disease
The gut microbiota plays important roles in shaping the immune system in infancy; during this time, the normal microbiota and their products interact with immune cells to initiate and maintain host tolerance. The failure to develop tolerance to the normal microbiota early in life is associated with different immune-mediated diseases, including allergies and chronic inflammation of the gut such as inflammatory bowel disease (IBD).
It is widely accepted that IBD is not caused by a specific pathogenic microbe but rather an imbalance, or dysbiosis, between the immune system and the normal gut microbiota. The observation that antibiotic use in early life increases the risk of IBD points to the importance of the development of a normal gut microbiota in “educating” the immune system to differentiate between the normal microbiota and invading pathogens. For example, relative to healthy children, children with IBD have higher levels of species of Veillonella, Prevotella, Lactobacillus, and Parasporobacterium and lower levels of species of Bifidobacterium and Verrucomicrobium.
There is also evidence from mouse studies that once developed, IBD is transmissible. Fostering or co-caging healthy mice with IBD-predisposed mice was sufficient to cause IBD development in the healthy mice and was correlated with the transfer of the enteric bacterial species Klebsiella pneumoniae and Proteus mirabilis from the IBD mice to the healthy mice. In humans, metagenomic analyses of healthy subjects and patients with IBD shows that the gut microbiota of IBD patients shares fewer genes in common with healthy subjects, relative to the number of genes shared among healthy subjects. The microbial community of IBD patients also tends to have significantly reduced functional capacity, as reflected by a reduction in the number of nonredundant (functionally unique) genes relative to subjects that do not have IBD (Figure 24.24).
Figure 24.24 Reduced functional capacity of the gut microbiome of patients with inflammatory bowel disease.

Metagenomic analysis of human gut microbiota in healthy subjects and patients with inflammatory bowel disease (IBD) revealed a tendency toward fewer nonredundant bacterial genes in patients with IBD.
The mechanism of IBD formation is unclear but may be the result of disruption of mucosal barrier integrity (leaky gut) by a gut pathogen or a toxic insult, permitting commensal bacteria to interact with and activate the adaptive immune system (Chapter 27). This stimulates the proliferation and differentiation of T cells into commensal-specific effector cells that can persist in the intestine long after resolution of the infection. For example, the IBD syndromes of Crohn’s disease and ulcerative colitis are known to be associated with a T cell response to intestinal commensal bacteria.
There is also a strong correlation between IBD and diet. With the typical Western diet rich in animal protein, carbohydrates are first fermented in the upper colon (Figure 24.3). As digesta move further along the colon, protein and amino acids are then fermented, generating potentially harmful metabolites such as ammonia, phenols, trimethylamine (TMA), and hydrogen sulfide (H2S) that have been implicated in promoting IBD as well as playing a role in colon cancer. Animal studies have shown that these compounds can promote the development of a leaky gut and gut inflammation, as well as cardiovascular disease (CVD). TMA is converted to trimethylamine oxide (TMAO) in the liver, and TMAO contributes to coronary thrombosis by increasing blood platelet hyperresponsiveness. A high concentration of TMA accelerates atherosclerosis (clogging and stiffening of the arteries) development in mice and is correlated with a higher incidence of CVD in humans. In contrast, a diet rich in plant-based foods (high fiber) works against development of these pathologies, highlighting the importance of a fiber-rich diet and carbohydrate-based fermentation in the healthy gut. Finally, IBD does not always affect identical twins; this observation argues against a strong genetic connection to IBD and supports the hypothesis that the environment and diet are the major triggers of this intestinal condition.
The Role of the Gut Microbiota in Obesity: Mouse Models
Obesity is a significant health risk that contributes to secondary health issues such as high blood pressure, cardiovascular disease, and type 2 diabetes. More than 600 million adults and 100 million children worldwide are obese (defined as a body mass index of >30), and because of this, several studies have focused on possible correlations between the gut microbiome and obesity.
Initial evidence linking the gut microbiota to host fat accumulation in humans came from studies using germ-free (microbe-free) mice. In these experiments, normal mice had 40% more total body fat than those raised under germ-free conditions, although both mouse populations were fed the same amount and type of food. After germ-free mice were inoculated with fecal material from a normal mouse (a fecal transplant, see later), they developed a gut microbiota and their total body fat increased, although there had been no changes in food intake or energy expenditure.
Experimental colonization of germ-free mice with individual microbial species or microbial communities have demonstrated that colonization triggers the expression of host genes for glucose uptake and lipid absorption and transport in the ileum. This indicates that there is likely a link between gut microbial composition and the ability of the host to harvest energy from its diet, ultimately contributing to obesity. This has been demonstrated in experiments with mice. Mice that are genetically obese have microbial gut communities that differ from those of normal mice, with 50% fewer Bacteroidetes, a proportional increase in Firmicutes, and a greater number of methanogenic Archaea. It has been hypothesized that methanogens increase the efficiency of microbial conversion of fermentable substrates to volatile fatty acids (VFAs) by consuming molecular hydrogen (H2), as also occurs in the rumen (Section 23.16); this makes more VFAs available for absorption by the mouse. In the non-obese mouse, by contrast, fewer H2-consuming methanogens are present, and the buildup of H2 functions as a partial inhibitor of microbial fermentation. This favors growth of Bacteroidetes species, making fewer VFAs from Firmicutes fermentation available for the mouse (**Figure 24.25*a***). What triggers this difference in the methanogen content of lean versus obese mice is not well understood. However, a similar scenario but involving different microbes also appears to be a factor in human obesity (see next subsection).
Figure 24.25 Fermentation in the colon of lean and obese mice and transfer of an obese condition by fecal transplant.

(a) The greater abundance of methanogens in the gut of obese mice is thought to promote fermentation by consuming H2. (b) Transplanting fecal material from the gut contents of a paired identical human twin study group (one twin was obese and the other lean) to germ-free mice showed that the obese twin microbiota made the mouse obese. Conversely, transfer of gut contents from the lean twin did not contribute to an obese phenotype. Part b adapted from Ridaura, V.K., et al. 2013. Science 341 doi:10.1126/science.1241214.
Mastering Microbiology
Art Activity: Figure 24.19 Differences in gut microbial communities between lean and obese mice
The importance of the gut community to a predisposition to obesity in mice has also been demonstrated by fecal transplant studies, transplanting a small sample of the gut contents from a set of paired human twins (one obese and one lean) into germ-free mice (see Section 24.11 for more coverage of fecal transplants). Although the recipient mice were fed identical high-fiber diets, the mice receiving fecal contents from the obese twin gained significantly more weight than the mice receiving fecal contents from the lean twin (Figure 24.25b). This is direct evidence that a lean or obese body type can be altered by changes in the gut microbiota, even when the microbiota originates from a different species. Put another way, specific but widely distributed gut microbes exist that can control an animal’s metabolism to yield a lean or obese body type.
The Gut Microbiota and Human Obesity
Animal model inferences have been more difficult to demonstrate with human subjects, since strict control of diet and host genotype is not feasible and gut microbiota manipulation is more difficult to achieve. Although studies of humans could not confirm the Bacteroidetes–Firmicutes relationship established in mice, they have shown that obese humans are more likely to harbor species of Prevotella and methanogenic Archaea than are lean humans, suggesting that a version of the mouse model (Figure 24.25a) is likely applicable to humans. In humans, the methanogens are proposed to remove H2 produced by Prevotella, facilitating fermentation by Prevotella and increasing nutrients to the host. This model is also supported by the study of germ-free mice colonized with Bacteroides thetaiotaomicron (having a metabolism similar to Prevotella) and the methanogen Methanobrevibacter smithii. Relative to controls containing just one of these species, co-colonized mice have a higher number of total gut bacteria, higher acetate levels in the intestinal lumen and blood, and greater body fat.
This relatively simple explanation for the cause of obesity is insufficient to fully explain microbial contributions to obesity. For example, the gut microbiota in lean mice produce greater amounts of the fatty acids propionate and butyrate and actually digest more of the plant fiber than do the microbiota of obese mice. Similarly, although a fiber-rich diet increases the amount of material available for fermentation in the human large intestine, such a diet also reduces the risk of obesity. As we now know, this may be an attribute of healthy gut homeostasis (Figure 24.23) where binding of SCFAs to specific intestinal cell surface receptors elevates production of hormones affecting energy recovery and sensations of hunger. Similarly, obesity in humans is also associated with a variety of metabolic complications, including low-grade inflammation exacerbated by reduced production of SCFAs from a diet low in fiber. These factors tend to complicate the simple microbiota models of obesity proposed to date.
An influence of host physiology on the gut community is also suggested by changes during pregnancy in humans. The period between the first and third trimesters of pregnancy is associated with a decrease in gut microbial diversity and enrichment in the gut community of species of Proteobacteria and Actinobacteria. These changes are associated with the increased body fat and insulin insensitivity that develop later in gestation. A simple interpretation of these findings is that a pregnant woman’s body in some way manipulates her gut microbiome as part of its preparations for a greater demand on stored energy reserves. If true, this once again underscores the complex interplay between the gut microbiota and multiple host variables (genetic, physiological, behavioral, and environmental) that may be contributing factors in the development of human obesity and associated metabolic disorders.
Check Your Understanding
What is dysbiosis? How might this condition lead to inflammatory bowel disease?
How are higher numbers of gut Prevotella species linked to obesity in humans?
Why are the gut microbiota results obtained in mice more difficult to confirm through human studies?
24.10 Syndromes Linked to the Oral, Skin, and Vaginal Microbiota
24.10 Syndromes Linked to the Oral, Skin, and Vaginal Microbiota
24.10 Syndromes Linked to the Oral, Skin, and Vaginal Microbiota
Imbalances in the normal microbiota have been linked to human health issues other than those affecting the gut. These include in particular diseases of the oral cavity, skin, and vagina, and we consider some of these important disease centers here.
Dental Caries and Periodontitis
Dental caries and periodontal diseases are among the most common chronic human maladies. We consider dental caries in Chapter 25, where we examine how bacteria attach to solid surfaces. The mechanisms by which oral bacteria stick to the teeth and gums has been used as a major model system for bacterial attachment to solid surfaces. The attached cells eventually form a diverse microbial community in dental plaque and this leads to dental disease from the generation of lactic acid by fermentation (Section 25.2 and Figures 25.7 and 25.8). Although Streptococcus mutans is a major contributor to dental caries, colonization of tooth surfaces (Figure 24.26) with organisms other than S. mutans can also lead to caries. In the absence of S. mutans, other acidogenic and acid-tolerant species can initiate caries.
Figure 24.26 Colonization of tooth surfaces.

(a) The colonies are growing on a model tooth surface inserted into the mouth for 6 h. (b) Higher magnification of the preparation in part a. Note the diverse morphology of the organisms present and the slime layer (arrows) holding the organisms together.
Species of the bacterial genera Streptococcus, Granulicatella, and Actinomyces are typically elevated in children who develop severe early dental caries, whereas caries-free children have a higher relative abundance of Aestuariimicrobium. In older children, caries is associated with decreasing bacterial diversity and increasing prevalence of Porphyromonas and Prevotella species. Similarly, dental plaque is more microbially complex in healthy subjects than in those with caries. Chronic periodontitis (inflammation and loss of tissue and bone that supports the teeth) is also associated with decreased microbial diversity, although no single bacterial phylotype seems to be directly associated with disease.
Collectively, these observations suggest that no single pathogen leads to the formation of dental caries or periodontal disease, but rather that community-wide changes in microbial composition trigger the diseases. Periodontal disease is particularly serious because it is thought to contribute to several debilitating systemic conditions, including cardiovascular disease, diabetes, pneumonia, and arthritis. Although the etiology of these systemic pathologies is unclear, they appear to be associated at least in part with dysbiosis of the oral cavity similar to that observed in dysbiosis of the gut microbiota in inflammatory bowel disease (Section 24.9).
Acne Vulgaris
The interrelationship of species of the bacterial genus Propionibacterium with the innate and adaptive arms of immunity (Chapters 26 and 27, respectively) has been examined extensively because of the association of this bacterium with acne vulgaris, usually just called acne, a cutaneous inflammatory disease of the skin that affects more than 85% of adolescents worldwide. However, the suspected causative agent of acne, P. acnes, is also a member—and a dominant member—of the normal cutaneous microbiota (Figures 24.2 and 24.15–24.17) and thus is also present in individuals without inflammatory disease.
Although Propionibacterium species have the capacity to elicit an inflammatory response, this alone is insufficient to demonstrate a causal association with disease. A better understanding of the cause(s) of acne will likely emerge from a better understanding of variation among strains of P. acnes. Notably, experiments using metagenomic and multilocus sequence analysis (Section 13.12) suggest that a specific strain of P. acnes in susceptible individuals may trigger the acne condition, whereas a much greater diversity of strains is observed among individuals having good skin health. Because of the scarcity of nutrients available for the skin microbiota, even minor differences in nutritional requirements between strains may favor the development of one or another P. acnes strain. If true, this might lead to therapies for acne using probiotics or prebiotics (see Section 24.12) that foster the growth of harmless strains or the growth of other species that displace P. acnes through competition for space and nutrients. For example, a lotion could be applied to the skin that contains a nonpathogenic strain or a nutrient selective for protective strains as a means to prevent or limit colonization by pathogenic strains.
Vaginal Conditions
Dysbiosis of the normal vaginal microbiota (Section 24.4 and Figure 24.14) is associated with either an inflammatory infection (vaginitis) or a less severe clinical form (vaginosis) that may be asymptomatic or associated with malodor and discharges. Common types of vaginitis result from the overgrowth of species of the yeast Candida (candidiasis) or from infection by the sexually transmitted protozoan Trichomonas vaginalis, both discussed in Chapter 34. Diagnosis of the less highly inflammatory vaginosis in a clinical research lab is commonly based on a microscopic scoring of a Gram-stained vaginal smear. However, culture-independent molecular methods (Chapter 19) are increasingly being employed to more precisely define vaginal communities associated with health, disease, and predisposition to disease, including neonatal infections, miscarriage, pre-term birth, and increased susceptibility to HIV and sexually transmitted infections.
Longitudinal studies of individual women over multiple months have revealed that the vaginal microbial communities of some women are highly dynamic, changing in composition over relatively short time periods, whereas the communities of others are relatively stable (Figure 24.27). Most vaginal microbial communities appear to be stable, displaying resilience (the capacity to return to the pre-disturbance community structure following disruption, such as menstruation) or resistance (the capacity to resist community disruption in response to disturbance). For example, a low pH–tolerant, Lactobacillus crispatus–dominated vaginal community is relatively stable (Figure 24.27), showing either resilience (subject 1) or resistance (subject 2). By contrast, other vaginal communities are more prone to converting to a vaginosis form. For example, the healthy L. crispatus–dominated community can convert to a healthy L. iners–dominated community before returning to L. crispatus (Figure 24.27). However, although both the L. crispatus and L. iners communities are associated with vaginal health, an L. iners–dominated community is more likely to transition to vaginosis. Vaginal bacterial diversity also waxes and wanes over shorter time periods, such as a woman’s menstrual cycle. Higher bacterial diversity is observed during days of menstruation than in intervening days, with diversity following a cycle inversely related to that of the estrogen hormone estradiol.
Figure 24.27 Resilience of the normal vaginal microbiota in two subjects.

Both vaginal communities were dominated by Lactobacillus crispatus and demonstrated resilience to perturbation, most notably the disruption associated with menstruation (indicated by horizontal red bars) in subject 1.
As for most ongoing human microbiome research, molecular microbial community analyses of the vagina have revealed connections between the microbial diversity of the vagina, vaginal health, and the susceptibility to vaginal diseases. However, we do not yet fully understand how the vaginal community is established and maintained or how vaginal dysbiosis develops and resolves. Nevertheless, if some community types are associated with clinical symptoms or are associated with higher risk for vaginal disorders, then early intervention may be important to maintaining women’s health and the health of a mother and baby in particular.
Check Your Understanding
What observations indicate that dental caries are not due solely to Streptococcus mutans?
Why might it be possible to have a high abundance of Propionibacterium acnes without developing acne vulgaris?
What are some clinical advantages of a community-structure-based evaluation of vaginal health?
V Modulation of the Human Microbiome
Gut dysbiosis can be resolved by introducing microbes from a healthy gut by fecal transplant. Selective modulation of the gut community can be accomplished by the use of probiotics and prebiotics. Early intervention with such therapies can reduce the incidence of serious intestinal disease in infants.
One important basic-science goal of human microbiome studies is to understand how the microbial composition and activity of human-associated microbes promotes health or predisposes the body to a variety of health disorders. Here we look at how the introduction of specific microbes or even transplants of the entire gut community can be used to promote health.
24.11 Antibiotics and the Human Microbiome
Antibiotics are naturally produced antimicrobial substances whose efficacy varies against different bacteria (Chapter 28). However, when an antibiotic is taken orally it kills or inhibits to at least some extent the normal microbiota as well as the targeted pathogen(s). Antibiotic treatment can lead to a significant loss of the normal gut microbiota (Section 24.2). When antibiotic therapy ends, the normal intestinal microbiota is usually, but not always, reestablished spontaneously in adults.
Use of antibiotics during the first few months of life is a particular problem for the developing normal microbiota. This disruption can affect normal development of the immune system and predispose the infant to later autoimmune disorders, such as IBD and allergies (Section 24.9). Research has also shown that early disruption of the gut microbiota influences host energy metabolism. For example, antibiotic exposure during the first 6 months of life is associated with increased weight gain in infants between 10 and 38 months of age compared to those not receiving antibiotics. With growing concerns about the frequency of childhood obesity leading to adult obesity along with the growing number of childhood autoimmune disorders, it is clearly important to avoid, if possible, disruption of the normal development of the human gut microbiota early in life.
*Clostridioides difficile* Infections
Sometimes antibiotic-resistant opportunistic pathogens become established in young children or the elderly following antibiotic treatment that disrupts the normal microbiota. A particularly problematic complication of antibiotic therapy is infection with toxigenic Clostridioides difficile (Section 30.7) and the inability to resolve infections with repeated follow-on administrations of antibiotics (Figure 24.28). There is a strong association between antimicrobial therapy and the subsequent development of C. difficile infection, and the risk of infection is greater if C. difficile is resistant to the antimicrobial agents used in therapy.
Figure 24.28 Antibiotic treatment increases the risk of *Clostridioides difficile* infection.

During antibiotic therapy the displacement of sensitive gut populations results in a significant loss of community diversity, allowing antibiotic-resistant populations such as toxigenic C. difficile to increase in abundance. When antibiotic therapy ends, C. difficile may be displaced (solid green line) by reestablishment of a normal gut microbiota, or alternatively it may persist (dotted line), causing intestinal disease.
In those infected with C. difficile, symptoms vary from a mild diarrhea to severe abdominal pain and fever. The most severe complications are inflammatory lesions and bowel perforation; as a result of these, septic shock (Section 25.2) and death are possible. In the past, if intravenous fluids and antibiotics did not resolve the infection, then surgical removal of the colon was a last-resort lifesaving measure. However, the prognosis for recovery from a C. difficile infection today has been dramatically improved by a novel nondrug and nonsurgical therapy: the infusion of fecal material from the gut contents of a healthy donor into the gut of a C. difficile–infected patient—more commonly called a fecal transplant. In most cases such treatment has been shown to restore a healthy colon in patients suffering recurring C. difficile infections, and we examine this procedure now.
Fecal Transplants
The goal of a fecal transplant is to reintroduce normal microbiota into the gut of someone with an intestinal disease of bacterial origin and have the transplanted microbial community become established and exclude the disease-causing organisms. Although it has taken the medical community some time to adopt this therapy, the dramatic improvement in the rate of resolution from C. difficile infection by fecal transplant from a healthy donor has prompted widespread adoption of this procedure.
As we have seen, once established, C. difficile is a particularly difficult pathogen to eradicate (Figure 24.28). However, the percentage of otherwise nonresponsive patients cured of C. difficile infection without relapse is nearly 90% with fecal transplant therapy versus only about 25% with standard antibiotic treatment. It has also been shown that fecal transplants can alleviate some forms of metabolic syndrome, a condition that is characterized by elevated blood pressure and glucose levels, excess belly fat, and abnormal cholesterol levels and that is a frequent precursor of type 2 (insulin-nonresponsive) diabetes. Metabolic syndrome patients receiving a fecal transplant from a lean donor had significantly increased insulin sensitivity, and also showed increased fecal levels of butyrate, greater overall gut microbial diversity, and increased abundance of the gut bacterium Roseburia intestinalis, thought to be a beneficial microbe because of its butyrate production (Figure 24.23).
Fecal transplants require that the donor be healthy and free of infection. And, because feces are considered a body fluid, screening and testing of the donor is essential. Potential donors must complete a screening questionnaire similar to that required for donating blood, and any prospective donors who have risk factors for HIV (AIDS) or hepatitis infections or any history of gastrointestinal disease, autoimmune disease, or cancer are screened out. Prospective fecal donors must also undergo blood tests for a suite of pathogens, and their feces are tested for fecal bacterial pathogens and parasites. If everything checks out, samples of their feces are frozen in small vials and then thawed and used when needed. Fecal transplant recipients typically receive the transplant via a saline solution during colonoscopy to ensure that the transplanted feces actually reach the upper colon and that the colon has previously been cleansed to remove as much of the previous microbial community as possible.
Mastering Microbiology
Art Activity: Figure 24.23 Antibiotic treatment increases the risk of Clostridium difficile infection
Although a fecal transplant may seem somewhat crude and an essentially uncontrolled or nonprecise therapy, studies are accumulating that show that the appropriate manipulation of the gut microbiota can have significant health benefits and that gut microbiota transplants can be maintained for long periods. More generally, the success of fecal transplants has shown that manipulation of the human microbiome for improved health is feasible and that with greater understanding of these complex communities, more targeted intervention may be possible, as we consider in the next section.
Check Your Understanding
Why do healthy adults usually not contract Clostridioides difficile infections?
Why is it unsafe for a fecal transplant recipient to receive feces from an unscreened donor?
24.12 Probiotics, Prebiotics, and Synbiotics
We close this chapter with a brief discussion of how ingestion of living microbial cultures (probiotics), particular nutrients typically derived from plants (prebiotics), or a combination of probiotic and prebiotic (called synbiotic) supplements could be used to treat disease and promote health by modulating the gut microbial community.
Probiotics
The United Nations Food and Agriculture Organization and the World Health Organization define a probiotic as a “live microorganism which, when administered in adequate amounts, confers a health benefit on the host.” Species of Bifidobacterium and Lactobacillus bacteria are the most commonly discussed probiotics. They are usually delivered to the gastrointestinal system by ingestion of a fermented milk product, such as yogurt (Figure 24.29), with the hope of suppressing gut or urogenital disturbances.
Figure 24.29 Examples of some probiotic foods and supplements widely available worldwide.

Some probiotic products also contain prebiotics (specific polysaccharides obtained from various plants and other sources).
Most physicians and scientists agree that ingestion of probiotic foodstuffs probably has limited therapeutic value. Nevertheless, human microbiome research has spurred a renaissance in probiotics research and investment, especially for the development of effective and more targeted probiotics. The remarkable success of fecal transplants (Section 24.11) points to the therapeutic potential of modification of the gut microbiome. However, although fecal transplants have proven very effective, there is minimal understanding of the mechanisms behind their successes and also why they do not work in a limited number of cases.
Molecular analyses of Clostridioides difficile infections have highlighted the importance of a mechanistic understanding for the development of effective probiotic therapies. Resistance to C. difficile infection following antibiotic therapy (Figure 24.28) in both mice and humans is correlated with the presence of a Clostridium species, C. scindens. Administration of this species to mice infected with C. difficile has been shown to be an effective probiotic therapy; suppression has been linked to the ability of C. scindens to convert cholic acid (found in bile) into deoxycholic acid, a growth inhibitor of C. difficile. In similar studies, recovery from diarrhea caused by Vibrio cholerae (cholera, Section 33.3) has been associated with an increased abundance of the bacterium Ruminococcus obeum. Mouse studies demonstrated that production of a quorum-sensing autoinducer (AI-2) (Section 7.7) by Ruminococcus suppressed expression of V. cholerae colonization factors and effectively prevented the cholera bacterium from establishing an infection.
The promise of effective probiotic therapies suggested from mouse studies have been supported in several human studies. For example, the fortuitous ingestion of a Bacillus strain present in a rural Thai environment was shown to prevent colonization by pathogenic Staphylococcus aureus. Similar to the mechanism of action observed in R. obeum, the Bacillus prevents colonization by interfering with the S. aureus quorum-sensing signaling system, ultimately preventing transcription of genes encoding virulence (Figure 24.30). This particular Bacillus strain could thus be marketed as an effective probiotic to eliminate intestinal as well as nasal S. aureus colonization. This is important because treatment of S. aureus infections has become increasingly difficult due to the prevalence of multi-antibiotic-resistant strains, such as methicillin-resistant S. aureus (MRSA; Section 31.9). Thus, an effective probiotic therapy that targets a specific pathogen and avoids the disruptive impact of broad-spectrum antibiotics on the human microbiome (see Section 24.11) would be a powerful addition to clinical medicine.
Figure 24.30 Probiotic inhibition of *Staphylococcus aureus* intestinal colonization.

Some intestinal Bacillus species prevent colonization of the opportunistic pathogen S. aureus by producing fengycin, an inhibitor of quorum sensing that is structurally similar to the autoinducing peptide (AIP) made by S. aureus. Fengycin binding to the ArgC sensor kinase (Figure 7.20) inhibits AIP binding but does not induce the signal transduction required for virulence gene expression and colonization of the gut by S. aureus.
Probiotics have also been shown effective in reducing the incidence of necrotizing enterocolitis (NEC), a severe inflammatory injury to the intestines of low-birthweight premature infants. NEC affects as many as 10% of premature infants, and about a quarter of those affected will die. Molecular characterization by 16S rRNA gene sequencing (Section 19.6) of the developing gut communities in these infants showed that during the first 60 days of life, species of Veillonellaceae (Negativicutes) and Clostridia increased in infants not developing NEC but were displaced by increasing numbers of Enterobacteriaceae species in those developing NEC (Figure 24.31). Thus, NEC is a type of dysbiosis, the disruption of the normal sequence of gut colonization.
Figure 24.31 Dysbiosis contributing to necrotizing enterocolitis of premature infants.

Inflammation and necrosis of the infant’s intestine is associated with an overgrowth of enteric bacteria, displacing protective species such as those of the Veillonellaceae and the genus Clostridium.
The most effective preventive treatment of NEC has been the probiotic feeding of commercially available species of Bifidobacterium and Lactobacillus; early treatment with these bacteria reduced the risk of death by 25%, likely by competing for space and resources with pathogenic enteric bacteria. From these promising results, current research is focused on identifying the most effective species and strains in hopes of more precisely targeting probiotic treatment of NEC and getting the gut microbiome of premature infants back on a normal track (Section 24.8).
Prebiotics and Synbiotics
Probiotics should not be confused with prebiotics. The prebiotic approach to developing a healthy normal microbiota promotes the ingestion of certain nutrients as microbial growth stimulants with the idea that they will nurture particular bacterial species already present in the gut and known to be associated with a healthy colon. Prebiotics are typically carbohydrates that are indigestible by the body but are excellent carbon and energy sources for certain fermentative gut bacteria. Fructooligosaccharides, polymers of the sugar fructose present in many vegetables, are thought to stimulate desirable gut microbes, and the complex polysaccharide inulin is widely promoted commercially as a major prebiotic.
Some probiotic formulations (such as certain yogurts) contain prebiotics as well to supply both beneficial aspects in a single treatment; these combined formulations are called synbiotics. The prebiotic in a synbiotic is added to promote the growth and colonization of the probiotic organism. The most dramatic success of synbiotics has been in the treatment of sepsis in young children. Sepsis in newborns, a systemic infection originating from an inflammation of the gut similar to NEC, is a major cause of child illness and death in the developing world, causing over half a million deaths a year. A synbiotic containing Lactobacillus plantarum and fructooligosaccharide given orally to newborns for 7 days beginning 2 days after birth reduced the risk of death from sepsis by 40% in a trial in India. This again demonstrates the importance of early intervention in gut colonization for preventing dysbiosis with its complications of possible sepsis and later life health problems.
The remarkable success of these treatment examples offers tremendous hope for future development of effective and inexpensive therapeutic agents for the prevention of disease and improved human health. The most successful advances to date have come from careful scientific studies based on our steadily improving understanding of the developing and mature microbiome and its relationship with its human host.
Check Your Understanding
What is a probiotic? How does it differ from a prebiotic and a synbiotic?
Why do you think fecal transplants have stimulated renewed research into the development of probiotic and prebiotic therapies?
Explain how probiotics based on interference of quorum sensing can suppress infection.
The Gut–Brain Axis
Interactions between the gut microbiota and the host brain and general nervous system (called the gut–brain axis) have been gaining attention because of possible contributions to behavioral disorders. We know that there is a clear relationship between gut dysbiosis and pathologies such as inflammatory bowel disease and obesity (Section 24.9). However, gut dysbiosis is also associated with autism, a general term for a spectrum of behavioral disorders (together called autism spectrum disorder) that can emerge in the first 36 months of life, causing substantial impairments in social interaction and communication and marked by repetitive behaviors and unusual interests.
Human autism has been attributed to a combination of genetic and environmental factors. An environmental risk factor for a child developing autism and other behavioral disorders is maternal immune activation (MIA), which involves elevated levels of inflammatory factors in the blood, placenta, and amniotic fluid during pregnancy and can be caused, for example, by viral infection. Although the relationship between MIA during pregnancy and impaired development of the fetal central nervous system is complex, mouse models suggest that some features of autism can be alleviated by correcting associated defects in the gut microbial community.
The offspring of mice in which MIA has been induced display autism-like behaviors, such as communication defects and anxiety (**Figure 1a,*b***). Those behavioral changes are associated with a loss of intestinal integrity, a shift in the composition of gut clostridial and bacteroidal populations reminiscent of changes observed in autistic humans, and a 46-fold increase in serum levels of the chemical 4-ethylphenylsulfate (Figure 1d). This compound is made from the amino acid tyrosine by certain gut microbiota. When administered alone, it induces anxiety-like behavior in normal mice.1 However, a remarkable finding is the ability to ameliorate defects in communication, anxiety, 4-ethylphenylsulfate levels, and gut permeability by feeding the affected mice a human gut commensal bacterium, either Bacteroides fragilis or Bacteroides thetaiotaomicron (Figure 1c). It is thought that these organisms displace the 4-ethylphenylsulfate producers and return gut chemistry to normal.
Figure 1 Influence of gut microbiota on behavior.

Mice offspring of mothers with maternal immune activation (MIA) show autistic-like behavior, marked by (a) a fear of exploring the center of the test field and (b) reduced vocalization. (c) Feeding the affected offspring the human commensal bacterium Bacteroides fragilis ameliorates these behavioral abnormalities. (d) Certain gut microbiota can convert the amino acid tryptophan into 4-ethylphenylsulfate, the neuroactive compound thought to trigger the mouse autistic-like behaviors shown in a and b. Modified from Hsiao, E.Y., et al. 2013. Cell 155: 1451.
These studies offer an example of the importance of the gut microbiota–brain connection in behavior and point to the possibility of modifying the gut community, such as might be possible with targeted probiotic therapy (Section 24.12), to alleviate symptoms in human neurodevelopmental disorders such as autism. Furthermore, amelioration of aberrant behavior by reducing serum levels of the gut microbiota–derived metabolite 4-ethylphenylsulfate to normal levels suggests that other neurodevelopmental illnesses may also be linked to the accumulation of microbial metabolites in serum by an unbalanced gut microbiota. However, one must be cautious about overinterpreting this study because the science thus far is only at the early stages of associating the gut microbiota with behavioral disorders.
1 Hsiao, E.Y., S.W. McBride, S. Hsien, et al. 2013. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155: 1451.
Chapter Review
Go to Mastering Microbiology for videos, animations, practice tests, and more.
I Structure and Function of the Healthy Adult Gastrointestinal and Oral Microbiomes
24.1 The human body is colonized by a diverse assemblage of microorganisms in numbers that approximately equal the total number of cells in the human body. Different body sites provide distinctive habitats that influence the types of microorganisms inhabiting each site. Collectively, these microbes constitute the human microbiome, now recognized to be intimately associated with human health and disease.
Q What are some of the possible future benefits of studies of the human microbiome?
24.2 The human gastrointestinal tract is composed of many segments that differ dramatically in their nutritional and pH characteristics and support quite distinct bacterial populations.
Q How do microbial diversity and abundance vary along the length of the gastrointestinal tract?
24.3 Distinctive groups of microorganisms colonize the mucous membranes and hard surfaces of the mouth and are constantly shed in saliva. The different microhabitats of the mouth (hard and soft surfaces, gingival crevice) sustain a high diversity of both aerobic and anaerobic populations.
Q Where would you expect to find the highest numbers of spirochetes in the mouth?
II Urogenital Tract and Skin Microbiomes and the Human Viral Microbiome
24.4 With the exception of the vagina and distal urethra, the male and female urogenital tracts harbor no or few microorganisms. The healthy vaginal microbial community is dominated by Lactobacillus species, which suppress the growth of other populations by lowering the vaginal pH to about 5 through fermenting glycogen to lactic acid.
**Q How does the composition of different species of Lactobacillus in the vaginal community impact predisposition to infection?**
24.5 The skin is a complex human organ covering about 2 square meters of the body and hosting about 1010 microorganisms in the healthy adult. Microbial community composition varies among the diversity of skin sites, generally categorized as moist, dry, or oily. The most common skin microorganisms are bacteria of the phyla Actinobacteria and Firmicutes.
**Q What characteristic of Propionibacterium species accounts for their colonization of the sebaceous gland system?**
24.6 The human virome consists of the vast diversity of animal and bacterial viruses that colonize the body and its microbiome. The greatest diversity and abundance of viruses is found in the gut, consisting of many novel bacteriophages that infect that complex microbial community. Unlike the environmental virosphere, where phage virions greatly exceed microbial cell numbers, most gut phages are lysogenic and have unknown impact on the gut microbial ecology.
Q What accounts for the low ratio of virions to bacteria in the gut compared to the open environment, such as in the ocean (Section 20.13)?
III From Birth to Death: Development of the Human Microbiome
24.7 Our current understanding of relationships between the human microbiome and health status is derived from complementary studies of select human study groups and experimental manipulation of animal models, in particular the mouse. Human studies are limited by incomplete control of lifestyle and genetic factors influencing the microbiome. Animal studies are limited by differences from humans in microbiology, anatomy, and physiology.
Q What are the major anatomical differences between mouse and human gastrointestinal systems, and how might those differences influence microbial composition?
24.8 The newborn gut microbial community evolves into a more complex adultlike community over the first 2–3 years of life. Initial colonization is influenced by vaginal or C-section delivery and by feeding of breast milk or formula. The unique oligosaccharides in human breast milk suppress pathogen colonization and select for beneficial microorganisms such as Bifidobacterium species. Changes in the gut microbial community with age have been associated with frailty in the elderly.
Q When a child is transitioned from breast milk to solid food, what are the associated changes in the gut microbial community and what physiological differences do those changes reflect?
IV Disorders Attributed to the Human Microbiome
24.9 The two major disorders associated with changes in the human gut microbial community are inflammatory bowel disease and obesity. Inflammatory bowel disease is associated with dysbiosis, a breakdown in the normal interactions between the human host and its intestinal microbiota reflected by reduced diversity of microbial genes in the diseased state. Obesity is associated with the microbial community composition of the gut, which influences host energy recovery, but obesity is likely not determined by a single contributing microbial or host factor.
Q How are short-chain fatty acids associated with good health as well as with inflammatory bowel disease and obesity?
24.10 Dental caries and periodontitis are diseases of the mouth caused by the combined activities of multiple microbial species, not a single pathogen. Strain variation among Propionibacterium species is associated with ability to cause acne, a common cutaneous inflammatory disease of adolescents. The vaginal community is dominated by Lactobacillus species that limit colonization by other organisms through lowering pH by production of lactic acid. Although generally very resilient to perturbation, the vaginal community transitions to a more diverse community during menstruation and during incidents of vaginitis or vaginosis.
**Q Why might a therapy based on colonization of the skin by selected Propionibacterium strains be effective in preventing the development of acne?**
V Modulation of the Human Microbiome
24.11 Antibiotic therapy may predispose an individual to infection by Clostridiodes difficile, a toxigenic bacterium that causes severe intestinal disease, because oral antibiotics reduce competition from the normal microbiota of the gut. Infections that cannot be resolved by standard or repeated antibiotic therapy have been shown to respond to fecal transplants, which introduce fecal material from a healthy donor into the gut of the infected individual.
**Q Why has the success of fecal transplants in treating C. difficile infection encouraged the development of therapies based on microbial ecology for other disorders associated with the human microbiome?**
24.12 Probiotics are live microorganisms that, when administered in adequate amounts, confer a health benefit on the host. New understanding of the mechanisms by which organisms such as Clostridium scindens and Bacillus species suppress pathogen colonization show that targeted probiotics can likely be developed to prevent or treat certain diseases. Prebiotics are nutritional supplements thought to promote the growth of beneficial gut microbes. Synbiotics combine a prebiotic with a probiotic to enhance the efficacy of the probiotic.
**Q What is the mechanism by which certain Bacillus species suppress colonization by pathogenic S. aureus?**
Application Question
You are told that you must be placed on a high dose of a broad-spectrum antibiotic to treat a serious infection. You are concerned that this therapy will seriously disrupt your intestinal microbiota, possibly leading to intestinal disease such as that caused by Clostridiodes difficile. In advance of the treatment, what might you do to ensure that your normal intestinal microbiota is restored after extended antibiotic therapy? Discuss two ways this restoration could be accomplished, one that would return your exact microbiota and one that would return some representative species.
Chapter Glossary
an alteration or imbalance of an individual’s microbiome relative to the normal, healthy state, primarily observed in the microbiota of the digestive tract or the skin Fecal transplant
the transfer of microbiota from the colon of one individual into the colon of another Host
an organism that can harbor pathogenic or beneficial (micro)organisms Human microbiome
the total microbial content in and on the human body Lower respiratory tract
a functional collection of different microbes in an environmental system such as the human body Microbiota
the types of organisms present in an environmental habitat, such as the human skin or the human gastrointestinal tract Mucin
a secretion from specialized epithelial cells containing water-soluble glycoproteins and proteins, forming the mucus that retains moisture and impedes microbial invasion on mucosal surfaces Normal microbiota
microorganisms that are usually found associated with healthy body tissue Prebiotic
food additive that promotes the growth and activity of beneficial microorganisms, usually in the gastrointestinal tract Probiotic
a live microorganism that, when administered in adequate amounts, confers a health benefit on the host Short-chain fatty acids (SCFAs)
also called volatile fatty acids or VFAs, these are the major fatty acids (acetate, propionate, and butyrate) produced during fermentation in the large intestine of monogastric animals and the rumen or cecum of herbivores Synbiotic
a mixture of probiotic and prebiotic that beneficially affects the host by altering the microbiome Upper respiratory tract
the sinuses, nasal and oral cavities, and structures of the throat—the pharynx, tonsils, and larynx Virome
the entire population of viruses, including animal, microbial, and plant viruses, in an environmental system such as in and on the human body